

Abstract
Presented herein are two complementary approaches to the synthesis of the core N-glycan pentasaccharide. The first, a traditional manual approach in solution, makes use of the H-bond-mediated aglycone delivery method for the highly diastereoselective introduction of the β-mannosidic linkage at room temperature. The synthesis of the core pentasaccharide was also accomplished using an high-performance liquid chromatography-assisted automated approach. The overall assembly was swift (8 h) and efficient (31%).
Graphical Abstract

INTRODUCTION
Glycans and glycosylated proteins (glycoproteins) play important roles in various biological processes, and the general interest in these compounds and their functions has rapidly increased in recent years.1-6 Among a plethora of structures, N-glycans and other oligosaccharides are covalently attached to proteins at asparagine (Asn) residues by an N-glycosidic bond. N-Glycans are also found on the surface of a variety of pathogens and are involved in the mediation of the pathogenesis of cancers,7 AIDS,8 Alzheimer’s disease,9 and other processes.10,11 All N-glycans are classified into three general classes: high-mannose, hybrid, and complex,2 but all share Manα1 → 6(Manα1 → 3)Manβ1 → 4GlcNAcβ1 → 4GlcNAcβ1 → Asn, the common core sequence (Figure 1).
Figure 1.

Core pentasaccharide sequence of N-glycans.
The construction of the core structure as the key intermediate toward a diverse library of N-glycans has been a vibrant area of research. Chemical and chemoenzymatic syntheses of the N-glycan core pentasaccharide have been reported by Ogawa,12 Unverzagt,13-15 Ito,16-18 Schmidt,19,20 Seeberger,21,22 Danishefsky,7,8,23,24 Boons,5,25 Wang,26 Fukase,27,28 Wong,29 Wang,30 among others. Nevertheless, the construction of these glycan sequences remains a notable challenge, which slows biomedical studies related to understanding the roles of glycoproteins and the creation of N-glycan-derived pharmaceuticals. Described herein are two distinct approaches to the synthesis of the N-linked core pentasaccharide 1 (Scheme 1). One approach was based on the traditional linear sequential synthesis in the solution, whereas the second approach was based on the convergent assembly using the high-performance liquid chromatography (HPLC)-based oligosaccharide synthesizer developed in our labs.
Scheme 1.

Retrosynthetic Analysis for the Synthesis of the Target Pentasaccharide 1 from Building Blocks 2–4
RESULTS AND DISCUSSION
One of the challenges of the chemical synthesis of N-glycans is the introduction of the Manβ(1 → 4)GlcNAc unit present at the branching point of the core sequence.31,32 A strong anomeric effect in mannosides and the impossibility of using the neighboring group participation to aid in the synthesis of the β-linkage in the manno series make this bond one of the most difficult steps in the synthesis.32 Some promising methods have been established by Crich33-39 and others,40-42 and the approach developed by us also allows the achievement of the excellent stereocontrol in β-mannosylation.43 Our approach relies on the use of picoloyl (Pico) protecting groups at remote positions capable of controlling the stereoselectivity via the H-bond-mediated Aglycone Delivery (HAD).44-46 According to this approach, the hydroxyl of the glycosyl acceptor forms the hydrogen bond with the picoloyl nitrogen of the glycosyl donor. As a result, the nucleophile is delivered from the same (syn) face of the sugar ring in respect to the picoloyl group. For compounds of the d-manno series, an excellent stereocontrol could be achieved with picoloyl groups at C-3 and/or C-6 positions, and a series of β-mannosides have been obtained with high stereoselectivity and yields.43 Hence, we decided to investigate whether this approach would also be suitable for the synthesis of the core pentasaccharide sequence of N-glycans.
For this synthesis, three building blocks (2–4) were selected in accordance with the retrosynthetic analysis of the pentasaccharide target 1 depicted in Scheme 1. Among these, building block 447 was chosen for introducing the two glucosamine units at the reducing end of the sequence. Building block 343 equipped with two picoloyl protecting groups at positions C-3 and C-6 was selected to carry out the HAD-assisted synthesis of the Manβ(1 → 4)GlcNAc linkage. Since the picoloyl groups can be selectively removed in the presence of many other protecting groups used in carbohydrate chemistry, this 3,6-di-O-Pico substitution would offer straightforward access to subsequent α-mannosylation. For the purpose of introducing the two terminal α-mannosyl residues, building block 248 was selected. Finally, we chose to equip our target molecule with a 4-azidobutyl spacer to offer convenient access to the selective conjugation or surface immobilization of the final product.
In accordance with the retrosynthetic analysis, the assembly of the target pentasaccharide 1 started with the universal precursor 4 that was used to introduce both glucosamine units. First, compound 447 was converted into glycosyl donor 5 that was reacted with 4-azidobutanol49,50 to give the functionalized monosaccharide 6 in 82% yield (Scheme 2). Second, the glycosylation of acceptor 6 with donor 4 led to the formation of disaccharide 7 in 87% yield. The subsequent reductive opening of the benzylidene acetal afforded compound 8 in 94% yield. Disaccharide 8 was then used as the glycosyl acceptor for β-mannosylation.
Scheme 2.
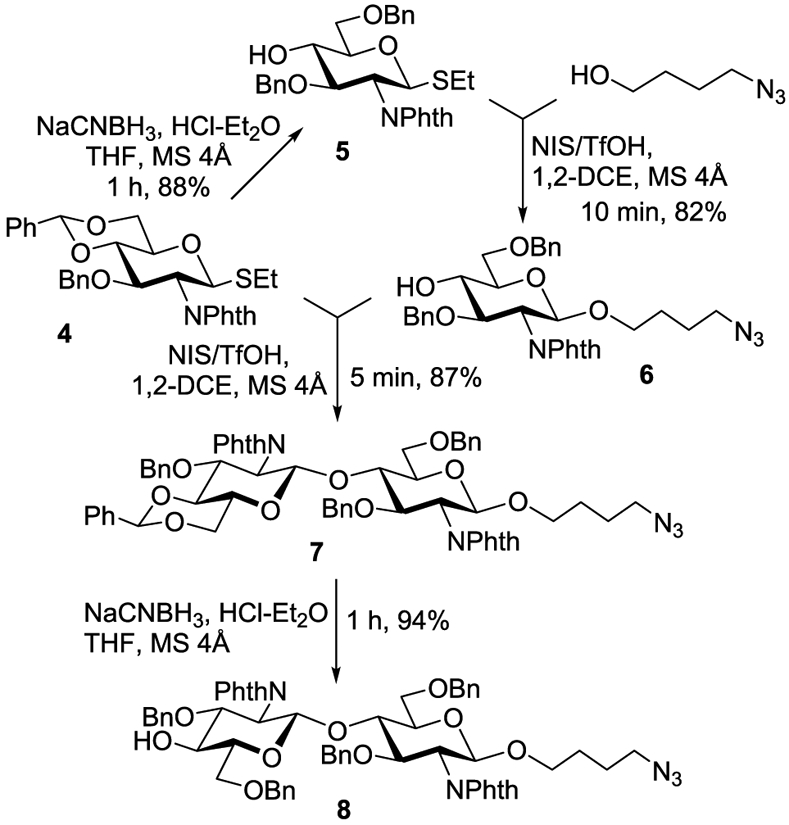
Synthesis of Disaccharide Acceptor 8
The next synthetic step, the introduction of the β-linked mannose unit, represents the key step in the entire synthesis of the N-glycan pentasaccharide. Somewhat unexpectedly, all attempts to glycosidate 3,6-di-O-Pico ethylthio glycosyl donor 3 with acceptor 8 by varying the amount of the donor as well as type and amount of the promoter have been unsuccessful (entries 1–5, Table 1). None of the attempts led to the formation of the desired trisaccharide intermediate 9. Hence, next we decided to utilize another known donor, 3-O-Pico ethylthio glycoside 1043 for the introduction of the β-linked mannose unit. Initially, our attempts were unsuccessful, and conventional reaction conditions for the HAD reaction43 did not work (entries 6 and 7). Success was achieved when we decided “to push” the reaction and added a large excess of N-iodosuccinimide (NIS, 6 equiv in respect to the donor). Under these reaction conditions, the coupling of glycosyl donor 10 and acceptor 8 produced trisaccharide 11 in 50% yield with excellent stereoselectivity (α/β = 1:11.2, entry 8). Encouraged by this progress, we continued optimizing the reaction conditions and discovered that using 3 equiv of donor 10 along with 3 equiv of NIS (in respect to the donor) seemed necessary to bring the reaction to completion. As a result, trisaccharide 11 was obtained in a high yield of 92% and commendable stereoselectivity (α/β = 1:12, entry 10). It should be noted that this reaction was performed at an ambient temperature, which offers an important experimental advantage over other known methods for β-mannosylation that require very low temperatures.32,38,51-53
Table 1.
Optimization of the β-Mannosylation of Disaccharide Acceptor 8
 | |||
|---|---|---|---|
| entry | donor (equiv to acceptor) |
promoter (equiv to donor) |
product (yield, α/β ratio) |
| 1 | 3 (1.1) | NIS (2.0), TfOH (0.2) | 9, no reaction |
| 2 | 3 (1.3) | NIS (2.0), TfOH (0.2) | 9, no reaction |
| 3 | 3 (3.0) | NIS (3.0), TfOH (0.3) | 9, no reaction |
| 4 | 3 (1.3) | DMTST (2.0) | 9, no reaction |
| 5 | 3 (1.3) | MeOTf (4.5) | 9, no reaction |
| 6 | 10 (1.1) | NIS (2.0), TfOH (0.2) | 11, no reaction |
| 7 | 10 (1.1) | DMTST (2.0) | 11, no reaction |
| 8 | 10 (1.1) | NIS (6.0), TfOH (0.3) | 11 (50%, α/β = 1:11.2) |
| 9 | 10 (1.3) | NIS (6.0), TfOH (0.3) | 11 (56%, α/β = 1:10.0) |
| 10 | 10 (1.6) | NIS (4.0), TfOH (0.3) | 11 (61%, α/β = 1:13.1) |
| 11 | 10 (3.0) | NIS (3.0), TfOH (0.3) | 11 (92%, α/β = 1:12.0) |
With trisaccharide 11 in hand, we continued the synthesis of the target pentasaccharide core, and our altered synthetic plan is depicted in Scheme 3. Thus, 3″-O-picoloyl group in 11 was removed with NaOMe in MeOH─CH2Cl2. The resulting trisaccharide acceptor 12 was glycosylated with mannosyl donor 2 in the presence of NIS/TfOH to afford tetrasaccharide 13 in 91% yield. The latter was subjected to acid hydrolysis of the 4″,6″-O-benzylidene acetal to afford tetrasaccharide acceptor 14 in 88% yield. The latter was regioselectively glycosylated with mannosyl donor 2 to afford the protected N-linked core pentasaccharide 15 in 60% yield.
Scheme 3.

Final Assembly of the N-glycan Pentasaccharide Core 15
Having achieved the linear synthesis of the core pentasaccharide in the solution, we decided to investigate whether the HPLC-assisted oligosaccharide synthesis, an automated approach developed in our labs,54,55 would also be a suitable mode for obtaining this challenging branched structure. The traditional solution phase synthesis is an important method to obtain oligosaccharides, but also the solid-phase automated approach has become a viable means to obtain glycan sequences.56 Following the pioneering work by Seeberger and co-workers, who have been the first to develop an automated solid-phase oligosaccharide synthesizer,57-62 several other research groups63-72 have been developing other platforms. These efforts have already demonstrated that the automation is a viable alternative for obtaining complex glycans.56 Hence, the synthesis of the N-linked core glycan appealed to us as an interesting challenge, particularly since our method has not yet been applied to the synthesis of branched glycan sequences.
Since the pentasaccharide target contains a challenging Manβ(1 → 4)GlcNAc linkage, we initially performed a number of model β-mannosylations on the solid phase. For this purpose, we obtained the resin-bound glycosyl acceptor 19, as depicted in Scheme 4. Known glucosamine derivative 547 was coupled with spacer 16 in the presence of NIS/TfOH to afford glucosamine-spacer intermediate 17 in 92% yield. Benzoyl deprotection followed by the treatment with succinic anhydride in pyridine allowed us to obtain building block 18. The latter was coupled with the amine of JandaJel resin in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP) to afford building block 19. The loading capacity of JandaJel-function-alized acceptor 19 was determined to be 0.11 mmol/g by cleaving off and quantifying the loaded compound. Although we managed to obtain very high yields in glycosylation of 19 with phosphate donor 20, ultimately, these attempts were deemed unsuccessful due to the poor stereoselectivity observed in these reactions. As a result, disaccharide 21 was obtained in 86% yield albeit no stereoselectivity.
Scheme 4.

Preliminary Attempt to Obtain Manβ(1 → 4)GlcNAc-Linked Disaccharide 21 on the Solid Phase Using the HPLC Synthesizer
Since the direct stereoselective formation of the β-mannosidic linkage on the solid phase has failed, we explored an alternative approach wherein the Manβ(1 → 4)GlcNAc linkage was presynthesized in the solution, and the resulting disaccharide building block was used for the indirect introduction of the β-mannosidic unit on a solid support. Thus, our subsequent plan to assemble the N-glycan core pentasaccharide 22 using the HPLC-assisted automated approach involved building blocks 19, 23, and 24 depicted in Scheme 5.
Scheme 5.

Retrosynthetic Analysis of the Solid-Supported Pentasaccharide Target 22
As shown in Scheme 6, the synthesis of disaccharide building block 24 began by the glycosylation of acceptor 2573 with donor 10. To ensure the completeness of the reaction, we had to use an excess of the donor and promoter, as shown in Scheme 6. Under these conditions, we were able to obtain disaccharide 26 in an excellent yield of 95% and commendable β-manno stereoselectivity (α/β = 1:6.0). Picoloyl deprotection was affected using a 1 M solution of NaOMe in MeOH to afford compound 27. This step was necessary to ensure the regioselectivity of the subsequent reductive opening of the benzylidene acetal that otherwise was nonregioselective. As a result, disaccharide 28 was obtained with a high regioselectivity and 77% isolated yield. The later was 3,6-di-picoloylated to give 29 in 95% yield. Compound 29 was then treated with tetrabutylammonium fluoride (TBAF) to afford hemiacetal 30 in 60% yield. Finally, compound 30 was converted into trichloroacetimidate donor 24 in 83% yield by a reaction with trichloroacetonitrile in the presence of 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU).
Scheme 6.

Synthesis of Disaccharide Donor 24 for the Automated Assembly
The automated assembly shown in Scheme 7 followed typical cycles of glycosylation and deprotection steps with the interim washing off the excess reagent, similarly to that used in our previously described automated oligosaccharide synthesis.55 JandaJel polymeric resin functionalized with glycosyl acceptor 19 was packed in Omnifit glass chromatography column. The column was integrated into the standard Agilent Infinity 1260 HPLC system equipped with a quad-pump and an autosampler, and the automated assembly was programmed as follows. To affect the initial washing and swelling of the resin containing acceptor 19, pump D was programmed to deliver dichloromethane. After that pump C was programmed to deliver donor 18 in CH2Cl2, and the resulting solution was recirculated for 10 min. The delivery of the donor was monitored with the UV detector that showed a plateau at the end of the recirculation. The autosampler was programmed to deliver a solution of trimethylsilyl trifluoromethanesulfonate (TMSOTf) in CH2Cl2 (3 injections), and the resulting reaction mixture was recirculated for 90 min. The glycosylation was monitored with the UV detector that showed a steady change in the absorbance of the reaction mixture due to the consumption of glycosyl donor 18. When the UV-monitoring showed no further change in the absorbance of the eluate passing through the detector, the system was washed with CH2Cl2 (pump D). Subsequent deprotection of the two picoloyl groups from positions C-3″ and C-6″ was then performed by delivering the deprotection reagent copper(II) acetate in methanol/dichloromethane using pump A for 20 min. The deprotection was monitored with the UV detector that showed a steady change in the absorbance of the reaction mixture due to the release of the Pico protecting groups. When the UV-monitoring showed no further change in the absorbance of the eluate passing through the detector, the system was washed with CH2Cl2 (pump D for 10 min). The final branching via dimannosylation was conducted by delivering mannosyl donor 17 (pump C). This monitored reaction was repeated two times with the interim washing (pump D) to ensure the complete glycosylation of both hydroxyls of the acceptor. At the end of this automated sequence, the immobilized pentasaccharide 16 was cleaved from the polymer support by delivering a mixture containing a 1 M solution of NaOMe/MeOH in CH2Cl2 and MeOH with pump B for 10 min. The eluate was collected, neutralized, concentrated, and the residue was acetylated with Ac2O in pyridine to afford pentasaccharide 20 in 31% yield overall.
Scheme 7.

Automated Assembly of Pentasaccharide 22 and Its Cleavage from the Solid Support to Produce Compound 31
With similarly protected pentasaccharides 15 and 20 in hand, we performed the global deprotection of compound 15, as shown in Scheme 8. O-Acyl protecting groups were removed by the treatment with NaOMe/MeOH. The resulting crude product was then subjected to the treatment with 1,2-ethylendiamine in n-butanol–toluene to affect the removal of the N-phthaloyl groups. The crude deprotected product was then per-acetylated with acetic anhydride in the presence of pyridine to afford compound 21 in 90% yield over three steps. The latter was subjected to the Birch conditions (Na/liq. NH3) that affected the global deprotection by removing benzyls, acetates and reducing the azide group into amine. The fully deprotected N-glycan pentasaccharide core 1 was isolated in 66% yield. It should be noted that the deprotection of 20 can be performed similarly and will require essentially the same sequence of synthetic steps.
Scheme 8.

Deprotection of Compound 15 to Afford the Target N-glycan 1
CONCLUSIONS
N-Linked glycan core pentasaccharide was assembled using the solution-based manual and solid-phase automated approaches. Both syntheses produced the desired core structure with high efficiency and comparable outcome for the assembly stage. This was our first attempt to obtain the branched oligosaccharide using the automated approach, and the overall assembly was swift (8 h). High diastereoselectivity for β-mannosylation was achieved at room temperature (rt) using the H-bond-mediated aglycone delivery (HAD) method in solution. The HAD method on solid phase yielded no stereoselectivity for β-mannosylation, which has been bypassed by using the convergent approach. According to this approach, we obtained the disaccharide building block containing challenging Manβ(1 → 4)GlcNAc bond that was then integrated in our HPLC-based automated oligosaccharide assembly.
EXPERIMENTAL SECTION
General Methods.
Column chromatography was performed on silica gel 60 (70–230 mesh), reactions were monitored by thin layer chromatography on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 and ClCH2CH2Cl (1,2-DCE) were distilled from CaH2 directly prior to the application. Pyridine was dried by refluxing with CaH2 and then distilled and stored over molecular sieves (3 Å). Molecular sieves (3 Å or 4 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to the application. Optical rotations were measured at the “Jasco P-1020” polarimeter. Unless noted otherwise, 1H NMR spectra were recorded in CDCl3 at 300 or 600 MHz, 13C{1H} NMR spectra were recorded in CDCl3 at 75 or 150 MHz. High-resolution mass spectrometry determinations were made with the use of a mass spectrometer with fast atom bombardment (FAB) ionization and ion-trap detection.
Manual Synthesis of Pentasaccharide 1. Ethyl 2,4-Di-O-benzyl-3,6-di-O-picoloyl-1-thio-α-d-mannopyranoside (3).
The title compound was synthesized according to the reported procedure, and its analytical data was essentially the same, as reported previously.43
4-Azidobutyl 3,6-Di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (6).
A mixture of ethyl 3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β -d- glucopyranoside (5,47 0.36 g, 0.66 mmol), 4-azidobutanol49,50 (0.09 g, 0.80 mmol), and freshly activated molecular sieves (4 Å, 0.9 g) in CH2Cl2 (15 mL) was stirred under argon for 1 h at rt. N-Iodosuccinimide (NIS, 0.30 g, 1.30 mmol) and TfOH (12 μL, 0.13 mmol) were added, and the resulting mixture was stirred for 10 min at rt. After that the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~100 mL) was washed with sat. aq Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (0.322 g, 82%) as a colorless foam. Analytical data for 6: Rf = 0.46 (ethyl acetate/hexanes, 1:1.5, v/v); [α]d20 +58.2 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.23–1.46 (m, 4H, 2 × CH2), 2.92 (d, 1H, J = 2.4 Hz, OH), 3.01 (t, 2H, J = 6.8 Hz, NCH2), 3.35–3.39 (m, 1H, OCH2a), 3.63 (m, 1H, H-6a), 3.73–3.84 (m, 4H, H-4, 5, 6b, OCH2b), 4.09–4.24 (m, 2H, H-2, 3), 4.52 (d, 1H, 2J = 12.2 Hz, 1/2 CH2Ph), 4.46 (d, 2H, 2J = 8.7 Hz, CH2Ph), 4.73 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 5.12 (d, 1H, J1,2 = 8.0 Hz, H-1), 6.92–6.96 (m, 3H, aromatic), 7.03–7.05 (m, 2H, aromatic), 7.24–7.35 (m, 5H, aromatic), 7.61–7.85 (m, 4H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 27.9, 29.1, 53.5, 57.9, 71.3, 73.4, 76.0, 76.4, 76.9, 77.2, 81.2, 100.9, 130.1 (×2), 130.4 (×2), 130.5 (×4), 130.6 (×2), 130.8 (×4), 131.1 (×2), 131.2 (×2), 140.2, 140.8 ppm; HR-FAB MS [M + Na]+ calcd for C32H34N4O7Na 609.2339, found 609.2309.
4-Azidobutyl O-(3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (7).
A mixture of ethyl 3-O-benzyl-4,6-O-benzylidene-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (4,47 0.59 g, 1.10 mmol), acceptor 6 (0.50 g, 0.80 mmol), and freshly activated molecular sieves (4 Å, 1.50 g) in CH2Cl2 (24 mL) was stirred under argon for 1 h at rt. N-Iodosuccinimide (NIS, 0.50 g, 2.20 mmol) and TfOH (20 μL, 0.20 mmol) were added, and the resulting mixture was stirred for 5 min at rt. After that the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~200 mL) was washed with sat. aq Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (1.00 g, 87%) as a colorless foam. Analytical data for 7: Rf = 0.68 (ethyl acetate/hexanes, 1:1 v/v); [α]d20 +52.8 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.38–1.44 (m, 4H, 2 × CH2), 3.01–3.05 (m, 2H, J = 6.8 Hz, NCH2), 3.31–3.63 (m, 6H, H-4, 5, 5′, 6a, 6a′, OCH2a), 3.71–3.81 (m, 2H, H-4′, OCH2b), 4.13–4.32 (m, 5H, H-2, 2′, 3, 6b, 6b′), 4.44–4.57 (m, 5H, H-3′, 2 × CH2Ph), 4.85 (dd, 2H, 2J = 8.5 Hz, CH2Ph), 5.00 (d, 1H, J1,2 = 8.0 Hz, H-1), 5.43 (d, 1H, J1,2 = 8.3 Hz, H-1′), 5.57 (s, 1H, CHPh), 6.94–7.13 (m, 10H, aromatic), 7.33–7.57 (m, 10H, aromatic), 7.70–8.10 (m, 8H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 14.3, 21.1, 25.3, 26.4, 50.9, 55.7, 56.6, 60.4, 65.8, 68.5, 72.8, 74.6, 83.2, 97.8, 98.1, 101.3, 123.3, 136.1, 126.2 (×3), 127.2, 127.4 (×3), 127.5, 127.6, 127.9 (×3), 128.1 (×10), 128.2 (×3), 128.3 (×10), 129.1, 131.6, 133.9, 137.5, 138.0, 138.3, 138.6 ppm; HR-FAB MS [M + Na]+ calcd for C60H57N5O13Na 1078.3851, found 1078.3838.
4-Azidobutyl O-(3,6-Di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (8).
Sodium cyanoborohydride (NaCNBH3, 0.80 g, 12.9 mmol) was added to a solution of compound 7 (1.00 g, 0.96 mmol) and molecular sieves (3 Å, 3.0 g) in dry tetrahydrofuran (THF) (30 mL) at rt. A 2 M solution of HCl in diethyl ether (~6.4 mL) was added dropwise under vigorous stirring until the gas evolution ceased. The resulting mixture was stirred for 1 h at rt. After that the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~100 mL) was washed with water (10 mL), NaHCO3 (10 mL), and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (0.94 g, 94%) as a white amorphous solid. Analytical data for 8: Rf = 0.43 (ethyl acetate/hexanes, 1:1.5 v/v); [α]d20 +29.4 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ, 1.35–1.41 (m, 4H, 2 × CH2), 3.01 (t, 2H, J = 6.8 Hz, NCH2), 3.17 (d, 1H, J = 1.2 Hz, OCH2a), 3.32–3.34 (m, 2H, H-4, 6a), 3.43–3.46 (m, 2H, H-5′, 6b′) 3.33–3.59 (m, 2H, H-6a′, 6b), 3.71–3.74 (m, 2H, H-5, OCH2b), 3.85 (dd, 1H, J4,5 = 4.8 Hz, H-4′), 4.12–4.21 (m, 3H, H-2, 2′, 3), 4.28 (dd, 1H, J3,4 = 5.0 Hz, H-3′), 4.49–4.59 (m, 6H, 3 × CH2Ph), 4.82 (dd, 2H, 2J = 7.3 Hz, CH2Ph), 4.97 (d, 1H, J1,2 = 4.7 Hz, H-1), 5.33 (d, 1H, J1′,2′ = 4.9 Hz, H-1′), 6.87–7.06 (m, 10H, aromatic), 7.33–7.39 (m, 10H, aromatic), 7.55–7.80 (m, 8H, aromatic) ppm; 13C{1H} NMR (125 MHz, CDCl3): δ, 25.7, 26.7, 51.2, 56.0, 56.5, 68.5, 68.8, 71.3, 73.0, 73.2, 74.1, 74.6, 74.7, 74.9, 75.8, 76.1, 78.7, 97.4, 98.4, 123.5, 124.0, 127.3, 127.7 (×3), 127.8, 128.1 (×6), 128.2 (×6), 128.3 (×3), 128.5 (×3), 128.6 (×3), 128.9 (×4), 132.0, 132.2, 134.3, 134.4, 138.7 (×2), 139.0, 168.8, 168.8 ppm; HR-FAB MS [M + Na]+ calcd for C60H59N5O13Na 1080.4007, found 1080.4017.
4-Azidobutyl O-(2-O-Benzyl-4,6-O-benzylidene-3-O-picoloyl-β-d-mannopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (11).
A mixture of ethyl 2-O-benzyl-4,6-O-benzylidene-3-O-picoloyl-1-thio-α-d-mannopyranoside43 (10, 0.452 g, 0.90 mmol), acceptor 8 (0.313 g, 0.30 mmol), and freshly activated molecular sieves (4 Å, 1.50 g) in 1,2-dichloroethane (90 mL) was stirred under argon for 1 h at rt. N-Iodosuccinimide (NIS, 0.60 g, 2.60 mmol) and TfOH (230 μL, 0.26 mmol) were added, and the resulting mixture was stirred for 10 min at rt. After that the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~200 mL) was washed with sat. aq Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on the silica gel (acetone–toluene) to afford the title compound (0.425 g, 95%) as a white amorphous solid. Analytical data for 11: Rf = 0.34 (acetone/toluene, 1:10, v/v); [α]d20 −18.3 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.12–1.21 (m, 4H, 2 × CH2), 2.85 (t, 2H, J = 6.8 Hz, NCH2), 3.08–3.21 (m, 4H, H-5′, 6a, 6a′, OCH2a), 3.28–3.44 (m, 4H, H-5″, 6a″, 6b, 6b″), 3.52–3.58 (m, 2H, H-6b′, OCH2b) 3.97–4.14 (m, 10H, H-1″, 2, 2′, 2″, 3, 3′, 4, 4′, 4″, 5), 4.14–4.56 (m, 6H, 3 × CH2Ph), 4.65–4.82 (m, 5H, H-1, 2 × CH2Ph), 4.99 (d, 1H, J2,3 = 3.1 Hz, H-3″), 5.17 (d, 1H, J1,2 = 9.0 Hz, H-1′), 5.33 (s, 1H, CHPh), 6.65–7.01 (m, 14H, aromatic), 7.14–7.66 (m, 26H, aromatic), 7.89 (d, 1H, aromatic), 8.71 (d, 1H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 25.4, 26.5, 51.0, 55.8, 56.7, 67.3, 67.9, 68.4, 68.5, 68.6, 72.8, 73.5, 73.9, 74.5, 74.7 (×2), 75.7, 75.8, 76.0, 76.6, 76.9, 77.2, 77.4, 79.2, 97.2, 98.3, 101.1, 101.7, 123.3, 123.8, 125.6 (×2), 126.3, (×2), 127.0 (×2), 127.1 (×2), 127.3 (×2), 127.4 (×2), 127.5 (×2), 127.7 (×2), 127.8 (×2), 127.0 (×5), 128.2 (×3), 128.3 (×2), 128.4 (×10), 128.5 (×2), 128.8 (×2), 129.1, 131.8, 133.8, 134.04, 137.0, 137.4, 137.9, 138.2, 138.6, 138.8, 139.0, 147.7, 150.2, 164.4 ppm; HR-FAB MS [M + Na]+ calcd for C86H82N6O19Na 1525.5532, found 1525.5530.
4-Azidobutyl O-(2-O-Benzyl-4,6-O-benzylidene-β-d-mannopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (12).
A 1 M solution of NaOMe in MeOH (2.0 mL) was added to a solution of trisaccharide 11 (0.480, 0.319 mmol) in CH2Cl2 (2.0 mL) and MeOH (18 mL), and the resulting mixture was stirred for 1 h at rt. DOWEX (H+) was added until pH = 6, the resin was filtered off, and rinsed successively with CH2Cl2 (5 × 5.0 mL) and MeOH (5 × 5.0 mL). The combined filtrate (~70 mL) was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to give the title compound as a white amorphous solid in 86% yield (380 mg, 0.272 mmol). Analytical data for 12: Rf = 0.55 (ethyl acetate/hexane gradient elution, 1:1, v/v); [α]d20 +0.64 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.20–1.44 (m, 4H, 2 × CH2), 2.33 (d, 1H, OH), 2.97 (t, 2H, CH2), 3.10–3.33 (m, 4H), 3.39–3.70 (m, 8H), 4.06–4.24 (m, 7H), 4.36–4.68 (m, 9H), 4.83–5.00 (m, 4H), 5.27 (d, 1H, J1′,2′ = 3.2 Hz, H-1′), 5.40 (s, 1H, CHPh), 6.76–6.98 (m, 11H, aromatic), 7.23–7.43 (m, 19H, aromatic), 7.64–7.68 (m, 8H, aromatic), ppm; 13C{1H} NMR (75 MHz, CDCl3): δ 25.4, 26.5, 50.9 (×2), 55.8, 56.6, 66.9, 67.9, 68.3, 68.5, 68.6, 71.0, 72.7, 73.5, 74.4, 74.6, 74.7 (×3), 75.9, 76.8, 79.0, 79.2, 79.5, 97.1, 98.2, 102.0 (×2), 123.2, 126.4 (×3), 127.0, 127.1, 127.4 (×5), 127.6 (×5), 127.9 (×5), 128.0 (5), 128.1 (×5), 128.3 (×6), 128.6 (×3), 128.7 (×3), 129.1, 131.7, 133.7, 137.3, 137.8, 138.3, 138.5, 138.7, 138.9 ppm; HR-FAB MS [M + Na]+ calcd for C80H79N5O18Na 1420.5318, found 1420.5320.
4-Azidobutyl O-(2,3,4,6-Tetra-O-benzoyl-α-d-mannopyranosyl)-(1 → 4)-O-(2-O-benzyl-4,6-O-benzylidene-β-d-mannopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (13).
A mixture of ethyl 2,3,4,6-tetra-O-benzoyl-1-thio-α-d-mannopyranoside48 (2, 0.524 g, 0.81 mmol), trisaccharide 12 (0.880 g, 0.62 mmol), and freshly activated molecular sieves (4 Å, 1.50 g) in 1,2-dichloroethane (40 mL) was stirred under argon for 1 h at rt. N-Iodosuccinimide (NIS, 0.364 g, 1.60 mmol) and TfOH (14 μL, 0.16 mmol) were added, and the resulting mixture was stirred for 20 min at rt. After that the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~200 mL) was washed with sat. aq Na2SO4 (20 mL) and water (3 × 20 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (acetone–toluene gradient elution) to afford the title compound (1.10 g, 91%) as a white amorphous solid. Analytical data for 13: Rf = 0.60 (acetone/toluene, 1:10, v/v); [α]d21 −3.8 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.30–1.51 (m, 4H, 2 × CH2), 3.08 (t, 2H, J = 6.8 Hz, NCH2), 3.14 (m, 1H), 3.34–3.37 (m, 3H), 3.39–3.60 (m, 4H) 3.73–3.76 (m, 2H), 3.87 (m, 1H), 3.95 (m, 1H), 4.16–4.76 (m, 19H), 4.91–5.10 (m, 4H), 5.39 (d, 1H, J1′,2′ = 8.1 Hz, H-1′) 5.54–5.57 (m, 2H), 5.95 (m, 1H), 6.03–6.13 (m, 2H), 7.01–7.29 (m, 11H, aromatic), 7.30–8.08 (m, 47H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 25.4 (×2), 26.5 (×2), 51.0 (×2), 55.9, 56.7, 62.6, 63.0, 65.5, 66.8, 67.3, 67.8, 68.6, 69.6, 71.1, 70.6, 72.9, 73.5, 74.6, 74.7, 74.8, 75.5, 75.7, 76.3, 78.3, 78.4, 79.3 (×2), 82.9, 97.4, 98.2, 99.0, 101.1, 123.3, 125.4, 126.0, 127.0, 127.2 (×5), 127.5 (×2), 127.6 (×2), 128.0 (×2), 128.1 (×5), 128.2 (×4), 128.4 (×4), 128.6 (×8), 128.7 (×7), 128.8, 129.0, 129.1 (×2), 129.2 (×4), 129.3, 129.5, 129.9 (×4), 130.0 (×4), 133.3, 133.4, 133.6, 133.7, 134.6, 135.9, 137.9 (×2), 138.0, 138.6, 138.7, 138.8, 165.1, 165.5, 165.6 (×2), 165.8, 166.3 ppm; HR-FAB MS [M + Na]+ calcd for C114H105N5O27Na 1998.6895, found 1998.6915.
4-Azidobutyl O-(2,3,4,6-Tetra-O-benzoyl-α-d-mannopyranosyl)-(1 → 3)-O-(2-O-benzyl-β-d-mannopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (14).
Water (350 μL) and trifluoroacetic acid (3.5 mL) were added to a stirring mixture of tetrasaccharide 13 (1.80 g, 0.60 mmol) in CH2Cl2 (35 mL), and the resulting mixture was stirred for 1 h at rt. After that the reaction mixture was neutralized with Et3N (~4 mL), diluted with CH2Cl2 (~200 mL), and washed with cold water (20 mL), sat. aq NaHCO3 (20 mL), and cold water (3 × 20 mL). The organic phase was separated, dried with magnesium sulfate, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound as a white amorphous solid in 88% yield (0.98 g, 0.52 mmol). Analytical data for 14: Rf = 0.33 (ethyl acetate/hexane, 1:1, v/v); [α]d21 −7.2 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.20–1.34 (m, 4H, 2 × CH2), 2.96 (t, 2H, J = 6.8 Hz, NCH2), 3.10 (m, 1H), 3.24–3.33 (m, 3H), 3.40–3.46 (m, 3H) 3.51–3.54 (m, 2H), 3.64–3.69 (m, 3H), 3.81–3.82 (m, 1H), 4.04–4.45 (m, 12H), 4.49–4.58 (m, 5H), 4.73–4.94 (m, 4H), 5.09–5.13 (d, 2H, J =12 Hz), 5.29 (d, 1H, J = 7.4 Hz) 5.38 (s, 1H), 5.73 (s, 1H), 5.90 (dd, 1H, J = 2.8 Hz), 6.08 (dd, 1H, J = 9.9 Hz), 6.74–6.76 (m, 3H, aromatic), 6.88–6.97 (m, 8H, aromatic), 7.16–7.67 (m, 33H, aromatic), 7.82–7.84 (m, 5H, aromatic), 7.98–8.01 (m, 4H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 50.8, 55.7, 56.5, 62.5, 62.8, 66.6, 67.1, 67.7, 68.2, 68.4, 69.5, 69.9, 70.4, 70.5, 72.7, 73.3, 74.4, 74.5 (×2), 74.6, 75.5, 76.1, 78.3, 78.4, 82.7, 97.2, 98.1, 98.8, 100.9, 123.1, 123.7, 126.9, 127.0 (×2), 127.1, 127.2 (×2), 127.3 (×3), 127.4 (×2), 127.5 (×2), 127.6, 127.8 (×2), 127.9 (×2) 128.0 (×4) 128.1 (×2), 128.2 (×3), 128.3 (×2), 128.4 (×6), 128.5 (×2), 128.6 (×4), 128.9, 129.0, 129.2, 129.6, 129.7 (×2), 129.8 (×4), 129.9 (×2), 131.4, 131.7, 133.2, 133.3, 133.4, 133.5, 133.6, 133.9, 134.1, 137.7, 138.4, 138.5, 138.6, 138.7, 165.4 (×2), 165.6, 166.1, 167.6 (×2), 168.6 ppm; HR-FAB MS [M + Na]+ calcd for C107H101N5O27Na 1910.6582, found 1910.6584.
4-Azidobutyl Di-O-(2,3,4,6-tetra-O-benzoyl-α-d-mannopyranosyl)-(1 → 3, 1 → 6)-O-(2-O-benzyl-β-d-mannopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (15).
A mixture of donor 2 (0.365 g, 0.570 mmol), tetrasaccharide 14 (0.980 g, 0.519 mmol), and freshly activated molecular sieves (4 Å, 1.50 g) in 1,2-dichloromethane (70 mL) was stirred under argon for 1 h at rt. N-Iodosuccinimide (NIS, 0.256 g, 1.14 mmol) and TfOH (10 μL, 0.114 mmol) were added, and the resulting mixture was stirred for 10 min at rt. After that the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~200 mL) was washed with sat. aq Na2SO4 (20 mL) and water (3 × 20 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (0.740 g, 60%) as a white amorphous solid. Analytical data for 15: Rf = 0.51 (ethyl acetate/hexane, 1:1, v/v); [α]d21 −1.4 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.15–1.50 (m, 4H, 2 × CH2), 22.96 (t, 2H, J = 6.8 Hz, NCH2), 3.17–3.60 (m, 8H), 3.61–3.77 (m, 2H), 3.80–3.95 (m, 2H) 3.97–4.27 (m, 8H), 4.27–4.55 (m, 10H), 4.57–4.72 (m, 4H), 4.76–5.37 (m, 4H), 5.10–5.20 (m, 2H), 5.28 (d, 1H, J = 7.7 Hz), 5.37 (d, 1H, J = 1.3 Hz), 5.79 (dd, 1H), 5.90 (dd, 1H), 6.10 (dd, 1H), 6.57–7.11 (m, 12H, aromatic), 7.12–7.69 (m, 42H, aromatic), 7.70–8.20 (m, 19H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 14.0, 20.9, 25.1 (×2), 26.2 (×2), 50.6 (×2), 55.5, 56.3, 60.2, 62.4, 62.5, 66.5, 66.7, 67.3, 67.8, 68.0, 68.2, 68.7, 69.3, 69.8, 69.9, 70.0, 70.3, 72.4, 73.0, 74.2 (×2), 74.3, 74.4, 74.7, 79.3, 82.8, 97.0, 97.8, 97.9, 99.0, 101.0, 122.9 (×2), 127.3 (×4), 127.5 (×4), 127.6 (×8), 127.8 (×4), 127.9 (×4), 128.0 (×4), 128.1 (×4), 128.2 (×4), 128.3 (×4), 128.4 (×3), 128.7 (×3), 128.8, 128.9, 129.1 (×3), 129.6 (×4), 129.7 (×4), 131.3 (×2), 131.6 (×2), 133.2 (×2), 133.3 (×2), 133.6 (×2), 137.7 (×2), 138.2 (×2), 138.4 (×2), 138.5 (×4), 164.7 (×2), 165.0 (×2), 165.1 (×2), 165.2 (×2), 165.3 (×2), 165.4 (×2), 165.9 (×2), 166.0 (×2), 167.2, 167.4, 168.1 ppm; HR-FAB MS [M + Na]+ calcd for C141H127N5O36Na 2488.8158, found 2488.8135.
4-Azidobutyl Di-O-(2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl)-(1 → 3, 1 → 6)-O-(2-O-benzyl-β-d-mannopyranosyl)-(1 → 4)-O-(2-acetamido-3,6-di-O-benzyl-2-deoxy-β-d-glucopyranosyl)-(1 → 4)-2-acetamido-3,6-O-benzyl-β-d-glucopyranoside (32).
A 1 M solution of NaOMe in MeOH (4.0 mL) was added to a solution of pentasaccharide 15 (0.460 mg, 0.186 mmol) in CH2Cl2 (3.8 mL) and MeOH (38 mL), and the resulting mixture was kept for 12 h at rt. DOWEX (H+) was added until pH = 6, the resin was filtered off and washed with MeOH (7 × 10 mL). The combined filtrate (~120 mL) was concentrated under the reduced pressure. The crude residue was dissolved in toluene (6.1 mL) and n-BuOH (12 mL), 1,2-ethylenediamine (3.7 mL) was added, and the resulting mixture was stirred for 12 h at 90 °C. The volatiles were removed in vacuo, and the residue was co-evaporated with toluene (2 × 10 mL). The crude residue was dissolved in pyridine (10 mL), Ac2O (5.0 mL) was added, and the resulting mixture was kept for 12 h at rt. Methanol (~10 mL) was added, the volatiles were evaporated in vacuo, and the residue was co-evaporated with toluene (2 × 10 mL). The residue was purified by column chromatography on silica gel (acetone–toluene) to afford the title compound (0.309 g, 90%) as a white amorphous solid. Analytical data for 32: Rf = 0.44 (acetone/toluene, 4:6, v/v); [α]d21 −2.4 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.50–1.56 (m, 4H, 2 × CH2), 1.75–2.14 (m, 33H, 11 × COCH3), 3.19–3.42 (m, 7H), 3.52–3.86 (m, 13H), 3.94–4.04 (m, 5H), 4.19 (dd, 1H, J = 8.2 Hz), 4.32 (dd, 1H, J =12 Hz), 4.40–4.60 (m, 6H), 4.64–4.69 (m, 4H), 4.72–4.97 (m, 3H), 5.10 (m 1H), 5.16–5.30 (m, 6H), 6.40 (d, 1H), 7.13–7.38 (m, 25H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 20.8 (×4), 20.9 (×2), 21.0 (×2), 23.3, 23.6, 25.7, 26.8, 51.3, 55.5, 62.4, 65.6, 66.1, 68.1, 68.5 (×2), 68.6, 68.9, 69.1, 69.4, 69.8, 72.3, 73.0, 73.5, 73.7, 73.9, 74.1, 74.5, 74.7, 74.9, 78.6, 79.6, 97.3, 99.0, 100.1, 100.2, 100.5, 127.3 (×3), 127.5, 127.7 (×3), 127.9, 128.0, 128.1 (×3), 128.2 (×3) 128.3 (×3) 128.4 (×3), 128.5 (×7), 128.6 (×3), 128.8 (×3), 137.9, 138.1, 138.3, 138.7, 138.8, 169.6, 169.7, 169.9, 170.1, 170.2 (×2), 170.4 (×2), 170.5, 170.7, 171.0 ppm; HR-FAB MS [M + Na]+ calcd for C91H113N5O35Na 1858.7135, found 1858.7153.
4-Aminobutyl Di-O-(α-d-mannopyranosyl)-(1 → 3, 1 → 6)-O-(β-d-mannopyranosyl)-(1 → 4)-O-(2-acetamido-2-deoxy-β-d-glucopyranosyl)-(1 → 4)-2-acetamido-2-deoxy-β-d-glucopyranoside (1).
A three-necked flask equipped with a Dewar-type condenser was cooled to −78 °C and charged with predried (Na) liquid ammonia (150 mL) and anhydrous THF (15 mL). Sodium (0.250 g, 0.01 mmol) was added, and the resulting mixture was stirred for 1 h at −78 °C. A suspension of pentasaccharide 32 (0.300 g, 0.19 mmol) in THF (15 mL) was added, and the resulting mixture was stirred for 6 h at −78 °C. After that NH4Cl (0.30 g) was added, and the resulting mixture was allowed to warm to rt. The volatiles were removed by passing a stream of nitrogen (16 h). The residue was dissolved in water, purified by passing through a short column of Sephadex G-10, and lyophilized to provide the title compound (0.105 g, 66%) as a white amorphous solid. Analytical data for 1: Rf = 0.55 (methanol/dichloromethane, 3:7, v/v); [α]d21 +0.46 (c 1.0, CHCl3); 1H NMR (600 MHz, CD3OD): δ, 1.52–1.56 (m, 4H, 2 × CH2), 1.94, 1.98 (2 s, 6H, 2 × OCH3), 2.97 (t, 2H, CH2), 3.41–3.87 (m, 4H), 3.96 (m, 1H), 4.15 (m, 1H), 4.38 (d, 1H), 4.50 (d, 1H), 4.81 (d, 1H), 5.08 (d, 1H) ppm; 13C{1H} NMR (150 MHz, CD3OD): δ, 21.4, 22.0, 22.4, 23.1 (×2), 25.6, 30.7, 40.6, 56.5, 61.4 (×2), 63.0, 63.2, 67.3, 67.9, 68.9, 69.1, 70.1, 71.2, 71.3, 72.0 (×2), 72.4, 73.4, 74.4, 75.1, 76.3, 76.4, 81.2 (×2), 82.5, 92.1, 101.1, 102.0, 102.9 (×2), 104.1 ppm; HR-FAB MS [M + Na]+ calcd for C38H67N3O26Na 1004.3910, found 1004.3929.
Synthesis of the Polymer Support-Bound Acceptor 19. N-(p-Benzoyloxymethyl)benzyloxycarbonyl-4-benzylaminobutyl 3,6-O-Benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (17).
A mixture of ethyl 3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside47 (5, 0.50 g, 0.94 mmol), N-(p-benzoyloxymethyl) benzyloxycarbonyl-4-benzylaminobutanol (16, 0.45 g, 1.13 mmol), and freshly activated molecular sieves (4 Å, 1.5 g) in CH2Cl2 (40 mL) was stirred under argon for 1 h at rt. N-Iodosuccinimide (NIS, 0.42 g, 1.88 mmol) and TfOH (17 μL, 0.18 mmol) were added, and the resulting mixture was stirred for 10 min at rt. After that the solids were filtered off through a pad of Celite and washed successively with CH2Cl2. The combined filtrate (~150 mL) was washed with sat. aq Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (0.75 g, 92%) as a colorless syrup. Analytical data for 17: Rf = 0.48 (ethyl acetate/hexanes, 1:1.5, v/v); [α]d21 +13.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.21–1.33 (m, 4H, 2 × CH2), 2.95–3.04 (m, 2H, 1/2 NCH2, OH), 3.25–3.31 (m, 1H, H-5), 3.60–3.64 (m, 1H, 1/2 NCH2), 3.62–3.78 (m, 3H, H-4, 6a, 6b), 4.06–4.28 (m, 4H, OCH2, H-2, 3), 4.48–4.63 (m, 3H, 1 1/2 CH2Ph), 4.72 (d, 1H, 2J = 12.2 Hz, 1/2 CH2Ph), 5.03–5.07 (m, 3H, H-1, CH2Ph), 5.33 (s, 2H, CH2Ph), 6.91–7.61 (m, 26H, aromatic), 8.04–8.06 (d, 2H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 27.0, 55.5, 66.6, 74.0 (×2), 78.8, 98.5, 107.5 (×2), 127.5, 128.0 (×4), 128.1 (×8), 128.2 (×4), 128.3 (×4), 128.5 (×4), 128.6 (×4), 128.7 (×8), 129.9 (×4), 130.5, 133.2 (×2) 137.7, 138.0 ppm; HR-FAB MS [M + Na]+ calcd 941.3625 for C55H54N2O11Na, found 941.3635.
N-(p-Succinoyloxymethyl)benzyloxycarbonyl-4-benzylaminobutyl 3,6-Di-O-Benzyl-2-deoxy-2-phthalimidoβ-d-glucopyranoside (18).
A 1 M solution of NaOMe in MeOH (2.0 mL) was added to a solution of compound 17 (0.60 g, 0.69 mmol) in MeOH (25 mL) and CH2Cl2 (5.0 mL), and the resulting mixture was stirred for 1 h at rt. DOWEX (H+) was added until pH = 6, the resin was filtered off, and rinsed successively with CH2Cl2 (5 × 5.0 mL) and MeOH (5 × 5.0 mL). The combined filtrate (~80 mL) was concentrated and dried in vacuo for 6 h. The crude residue was dissolved in pyridine (20 mL), succinic anhydride (0.11 g, 1.10 mmol) was added, and the resulting mixture was stirred under argon for 18 h at 65 °C. After that the volatiles were removed under the reduced pressure, the residue was co-evaporated with toluene (3 × 10 mL), and purified by column chromatography on silica gel (methanol–dichloromethane) to afford the title compound (0.52 g, 78%) as a colorless foam. Analytical data for 18: Rf = 0.41 (methanol/dichloromethane, 1:10, v/v); [α]d21 +16.7 (c 1.0, CHCl3); 1H NMR (300 MHz, C5Cl3): δ, 1.19–1.31 (m, 4H, 2 × CH2), 2,26 (s, 4H, 2 × CH2), 2.95–3.04 (m, 2H, 1/2 NCH2, OH), 3.40–3.43 (m, 1H, H-5), 3.58–3.64 (m, 1H, 1/2 NCH2), 3.72–3.77 (m, 3H, H-4, 6a, 6b), 4.07–4.23 (m, 4H, OCH2, H-2, 3), 4.49–4.63 (m, 3H, 1 1/2 CH2Ph), 4.73 (d, 1H, 2J = 12.2 Hz, 1/2 CH2Ph), 5.01–5.10 (m, 5H, H-1, 2 × CH2Ph), 6.90–7.74 (m, 26H, aromatic), 8.59–8.60 (d, 2H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 24.9, 25.4, 27.4, 28.9, 55.5 (×2), 65.5, 66.4, 70.4, 73.9, 74.2, 74.5, 75.0, 82.3, 65.5, 66.4, 70.4, 73.9, 75.0, 78.9, 98.5, 127.6 (×4), 128.0 (×4), 128.1 (×4), 128.3 (×5), 128.4, 128.7 (×8), 133.1, 137.7, 148.0, 157.3, 172.2 ppm; HR-FAB MS [M + Na]+ calcd 937.3524 for C52H54N2O13Na, found 937.3514.
Resin-Bound Acceptor 19.
JandaJel amine resin (1% cross-linked polystyrene, 400 mg, 0.20 mmol) was added to a solution of compound 18 (270 mg, 0.29 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, 169 mg, 0.88 mmol), and DMAP (43 mg, 0.35 mmol) in CH2Cl2 (10.0 mL), and the resulting suspension was agitated under argon for 18 h at rt. After that the resin was filtered off, washed with CH2Cl2 (3 × 20 mL), methanol (3 × 20 mL), and CH2Cl2 (3 × 20 mL). The combined filtrate was concentrated and dried in vacuo for 4 h. The loading (0.11 mmol/g) of the acceptor on the solid-phase support was determined by cleaving off the linker from 0.05 g of the resin.
Synthesis of Mannosyl Phosphate Donors 20 and 23. 2,4-Di-O-benzyl-3,6-di-O-picoloyl-α-d-mannopyranosyl Dibutyl Phosphate (20).
A mixture of ethyl 2,4-di-O-benzyl-3,6-di-O-picoloyl-1-thio-α-d-mannopyranoside43 (3, 490 mg, 0.79 mmol), dibutyl phosphate (0.43 mL, 2.39 mmol), and freshly activated molecular sieves (4 Å, 1.5 g) in CH2Cl2 (10 mL) was stirred under argon for 1 h at rt. After that NIS (227 mg, 1.03 mmol) and TfOH (9 μL, 0.10 mmol) were added, and the resulting mixture was stirred for 1 h at rt. The solid was filtered off and rinsed successively with CH2Cl2. The combined filtrate (~40 mL) was washed with sat. aq Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (0.61 g, 99%) as a clear syrup. Analytical data for 20: Rf = 0.29 (ethyl acetate/hexanes, 1:1.5, v/v); [α]d21 +20.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.84–0.92 (m, 6H, 2 × CH3), 1.34–1.41 (m, 4H, 2 × CH2), 1.53–1.65 (m, 4H, 2 × CH2), 3.89–4.12 (m, 5H, H-2, 2 × CH2), 4.22–4.25 (m, 1H, H-5), 4.38 (dd, 1H, J4,5 = 9.7 Hz, H-4), 4.55–4.70 (m, 5H, H-6a, 6b, 1 1/2 CH2Ph), 4.82 (d, 1H, 2J = 10.8 Hz, 1/2 CH2Ph), 5.57 (dd, 1H, J3,4 = 9.5 Hz, H-3), 5.80 (d, 1H, J1,2 = 6.4 Hz, H-1), 7.09–8.03 (m, 16H, aromatic), 8.03–8.78 (m, 2H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 13.6 (×2), 18.6 (×2), 29.6, 32.2, 63.7, 68.0 (×2), 71.7, 72.0, 73.0, 74.5, 75.0, 75.3, 95.2, 125.3, 125.4, 126.9, 127.1, 127.8 (×4), 128.2 (×2), 128.3 (×4), 136.9, 137.0, 137.3, 147.5, 147.6, 150.0 (×2), 164.2, 164.5, 178.0 ppm; HR-FAB MS [M + Na]+ calcd 785.2815 for C40H47O11N2PNa, found 785.2830.
2,3,4,6-Tetra-O-benzoyl-α-d-mannopyranosyl Dibutyl Phosphate (23).
A mixture of ethyl 2,3,4,6-tetra-O-benzoyl-1-thio-α-d-mannopyranoside48 (2, 1.30 g, 2.0 mmol), dibutyl phosphate (1.18 mL, 6.0 mmol), and freshly activated molecular sieves (4 Å, 3 g) in CH2Cl2 (25 mL) was stirred under argon for 20 min at rt. After that NIS (572 mg, 2.6 mmol) and TfOH (23 μL, 0.26 mmol) were added, and the resulting mixture was stirred for 18 h at rt. The solid was then filtered off and rinsed successively with CH2Cl2. The combined filtrate (~40 mL) was washed with sat. aq Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (1.50 g, 95%) as a clear syrup. Analytical data for 23: Rf = 0.33 (ethyl acetate/hexanes, 3:7, v/v); [α]d21 −49.2 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.87–0.89 (m, 6H, 2 × CH3), 1.35–1.44 (m, 4H, 2 × CH2), 1.64–1.70 (m, 4H, 2 × CH2), 4.07–4.19 (m, 4H, 2 × OCH2), 4.47 (dd, 1H, J = 3.9 Hz, H-6a), 4.60–4.68 (m, 2H, H-5, 6a), 5.75 (d, 1H, J = 2.2 Hz, H-1), 5.84–5.89 (m, 1H, H-2), 5.71 (d, 1H, J = 3.2 Hz, H-3), 6.18 (dd, 1H, J = 10.1 Hz, H-4), 7.21–7.57 (m, 12H, aromatic), 7.78–8.08 (m, 8H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 13.5 (×2), 13.6 (×2), 18.6, 32.1, 32.2, 62.2, 66.0, 68.2, 68.3, 69.3, 69.7, 70.5, 94.9, 128.3 (×3), 128.4 (×3), 128.6 (×3), 128.7 (×3), 128.8, 129.7 (×3), 129.8 (×3), 133.1, 133.3, 133.5, 133.6, 164.9, 165.2, 165.3, 165.7 ppm; HR-FAB MS [M + Na]+ calcd 811.2451 for C42H45O13PNa, found 811.2493.
Nonstereoselective Synthesis of Disaccharide 21 on the Solid Phase and the Solution Phase Synthesis of Disaccharide Building Block 24. N-(p-Acetoxymethyl)benzyloxycarbonyl-4-benzylaminobutyl O-(4,6-Di-O-acetyl-2,3-O-benzyl-α/β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (21).
Functionalized JandaJel resin 19 (50 mg, 0.011 mmol) was packed in an Omnifit glass chromatography column, and the latter was integrated into the HPLC system. Pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 5 min (5 mL, step 1). The system was then switched to the recirculation mode, and the delivery of CH2Cl2 continued for 30 min at 1.0 mL/min (swelling, step 2). After that pump D was stopped, and pump C was programmed to deliver a solution of donor 20 (83 mg, 0.11 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 3). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (20 μL, 0.11 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min, and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in the absorbance of the eluate. After that pump C was stopped, and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 4). After that pump D was stopped, and pump B was programmed to deliver a 0.1 M solution of NaOMe in CH3OH/CH2Cl2 (10 mL, 0.04:1:1, v/v/v) that was recirculated at 1.0 mL/min for 20 min (step 5). Pump B was stopped, and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min for 10 min, and the combined eluate was neutralized with Dowex (H+) resin. The resin was filtered off, washed successively with CH2Cl2 and CH3OH, and the combined filtrate was concentrated in vacuo to afford the crude residue that was dissolved in pyridine (2.0 mL), Ac2O (73 μL, 0.771 mmol) was added dropwise, and the resulting mixture was stirred for 16 h at rt. The reaction mixture was quenched with CH3OH (~1.0 mL), and the volatiles were removed under the reduced pressure. The residue was diluted with CH2Cl2 (20 mL) and washed with 1 N HCl (2 × 10 mL), water (20 mL), sat. aq NaHCO3 (20 mL), and water (2 × 20 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford disaccharide 21 in 86% yield (α/β = 1.0:1). Selected analytical data for 21: Rf = 0.52 (ethyl acetate/hexane, 1:1, v/v); 1H NMR (300 MHz, CDCl3): δ 1.29–1.55 (m, 4H, CH2), 1.89–2.07 (m, 9H, 3 × CH3), 2.90–3.12 (m, 2H, CH2), 3.20–3.40 (m, 2H), 3.50–4.10 (m, 7H), 4.12–4.59 (m, 8H, 4 × CH2Ph), 5.05–5.12 (m, 6H), 6.87–7.34 (m, 28H, aromatic), 7.58–7.82 (m, 5H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ 25.7, 26.7, 51.2, 56.0, 56.5, 68.5, 68.8, 71.3, 73.0, 73.2, 74.1, 74.6, 74.7, 74.9, 75.8, 76.1, 78.7, 97.4, 98.4, 123.5, 124.0, 127.3, 127.7 (×3), 127.8, 128.1 (×6), 128.2 (×6), 128.3 (×3), 128.5 (×3), 128.6 (×3), 128.9 (×4), 132.0, 132.2, 134.3, 134.4, 138.7 (×2), 139.0, 168.8 (×2) ppm; HR-FAB MS [M + Na]+ calcd for C74H78N2O18Na 1305.5148, found 1305.5157.
tert-Butyldimethylsilyl O-(2-O-Benzyl-4,6-O-benzylidene-3-O-picoloyl-β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (23).
A mixture of ethyl 2-O-benzyl-4,6-O-benzylidene-3-O-picoloyl-1-thio-α-d-mannopyranoside43 (10, 977 mg, 1.92 mmol), t-butyldimethylsilyl 3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside73 (25, 830 mg, 1.37 mmol), and freshly activated molecular sieves (4 Å, 3 g) in CH2Cl2 (307 mL) was stirred under argon for 60 min at rt. After that NIS (1.271 g, 5.78 mmol) and TfOH (105 μL, 0.57 mmol) were added, and the resulting mixture was stirred for 6 h at rt. The solid was then filtered off and rinsed successively with CH2Cl2. The combined filtrate (~800 mL) was washed with sat. aq Na2SO4 (50 mL) and water (3 × 50 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound (3.2 g, 95%, α/β = 1:6.0) as a clear syrup. The β-anomer 26 was separated by column chromatography on silica gel (ethyl acetate/hexane, slow gradient elution). Analytical data for 26: Rf = 0.32 (ethyl acetate/dichloromethane, 1:19, v/v); [α]d21 −70.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, −0.28 (s, 3H, CH3), −0.27 (s, 3H, CH3), 0.46 (s, 9H, C(CH3)3), 3.10–3.15 (m, 1H, H-5′), 3.34–3.42 (m, 2H, H-5, 6a), 3.50–3.54 (m, 2H, H-1′, 6b), 3.89–4.15 (m, 4H, H-2, 3, 4, 6a′), 4.29 (d, 1H, 2J = 12.3 Hz, 1/2 CH2Ph), 4.40 (d, 1H, 2J = 12.1 Hz, 1/2 CH2Ph), 4.46–4.73 (m, 4H, 2 × CH2Ph), 4.97 (dd, 1H, J = 3.2 Hz, H-3′), 5.16 (d, 1H, J1,2 = 8.0 Hz, H-1), 5.31 (s, 1H, CHPh), 6.68–7.61 (m, 26H, aromatic), 7.82–7.84 (m, 1H, aromatic), 8.59–8.60 (m, 1H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 4.01, 5.01, 17.7, 25.5 (×3), 58.0, 67.4, 68.7 (×2), 73.7, 73.9, 74.5, 74.9, 75.6, 75.8, 76.5, 76.8, 79.5, 93.6, 101.6, 101.7, 125.6, 126.3 (×3), 127.1 (×2), 127.7, 127.9 (×6), 128.0 (×3), 128.1, 128.4 (×9), 128.7 (×3), 129.1, 137.0, 137.4, 138.0, 138.1, 139.0, 147.7, 150.2, 164.3 ppm; HR-FAB MS [M + Na]+ calcd for C60H64N2O13NaSi 1071.4074, found 1071.4064.
tert-Butyldimethylsilyl O-(2-O-Benzyl-4,6-O-benzylidene-β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (27).
A 1 M solution of NaOMe in MeOH (3.0 mL) was added to a solution of disaccharide 26 (2.37 g, 2.26 mmol) in CH2Cl2 (10 mL) and MeOH (20 mL), and the resulting mixture was stirred for 1 h at rt. DOWEX (H+) was added until pH = 6, the resin was filtered off, and rinsed successively with CH2Cl2 (5 × 10 mL) and MeOH (5 × 10 mL). The combined filtrate (~140 mL) was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to give the title compound as a white amorphous solid in 98% yield (2.10 g, 2.20 mmol). Analytical data for 27: Rf = 0.73 (ethyl acetate/hexane gradient elution, 1:1, v/v); [α]d21 +11.5 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, −0.24 (s, 3H, CH3), −0.08 (s, 3H, CH3), 0.51 (s, 9H, C(CH3)3), 2.20 (d, 1H, J = 8.52 Hz, OH), 3.03–3.08 (m, 1H, H-5′), 3.49–3.65 (m, 6H, H-2′, 3′, 4′, 5, 6a, 6b), 3.90–4.16 (m, 5H, H-2, 3, 4, 6a′, 6b′), 4.30 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 4.38 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 4.51–4.62 (m, 2H, CH2Ph), 4.72 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 4.89 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 5.19 (d, 1H, J = 9.0 Hz, H-1), 5.30 (s, 1H, CHPh), 6.72–6.82 (m, 5H, aromatic), 7.15–7.33 (m, 14H, aromatic), 5.52–7.53 (m, 5H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 4.08, 5.34, 17.7, 25.4 (×3), 57.9, 67.0, 68.6, 68.7, 71.0, 73.8, 74.5, 75.8, 79.0, 79.3, 79.8, 93.6, 102.0, 102.2, 126.4 (×3), 127.1, 127.8 (×3), 127.9 (×3), 128.0 (×19), 128.1, 128.3 (×3), 128.6 (×3), 128.7 (×3), 129.1, 137.3, 137.8, 138.2, 138.9 ppm; HR-FAB MS [M + Na]+ calcd for C54H61NO12NaSi 966.3860, found 966.3871.
tert-Butyldimethylsilyl O-(2,4-Di-O-benzyl-β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (28).
Copper(II) trifluoromethanesulfonate (80 mg, 0.22 mmol) was added to a solution of 27 (2.10 g, 2.20 mmol) in a 1 M soln of BH3 in THF (11 mL), and the resulting mixture was stirred under argon for 1 h at rt. The reaction mixture was cooled to 0 °C and quenched with triethylamine (~1 mL) until pH = 7, and the volatiles were removed in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to give the title compound as a white amorphous solid in 77% yield (1.62 g, 1.71 mmol). Analytical data for 28: Rf = 0.4 (ethyl acetate/hexane, 1:1, v/v); [α]D21 +18.5 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, −0.22 (s, 3H, CH3), −0.07 (s, 3H, CH3), 0.55 (s, 9H, C(CH3)3), 1.89 (dd, 1H, J = 6.9 Hz, OH), 2.19 (dd, 1H, J = 9.4 Hz, OH), 3.06–3.09 (m, 1H, H-5′), 3.33–3.65 (m, 9H, H-1′, 2′, 3′, 4′, 5, 6a, 6a′, 6b, 6b′), 3.92 (dd, 1H, J = 9.0 Hz, H-4), 4.02 (d, 1H, J = 8.0 Hz, H-2), 4.18 (dd, 1H, J = 9.7 Hz, H-3), 4.33 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 4.40–4.60 (m, 4H, 2 × CH2Ph), 4.75 (dd, 2H, 2J = 12.0 Hz, CH2Ph), 4.92 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 5.23 (d, 1H, J = 9.0 Hz, H-1), 6.77–6.84 (m, 5H, aromatic), 7.13–7.25 (m, 15H, aromatic), 7.53–7.56 (m, 4H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3); δ, 4.7, 5.2, 17.7, 25.5 (×2), 58.3, 62.5, 68.7, 73.8, 74.4, 74.5, 74.9, 75.0, 75.3, 77.0, 78.4, 79.2, 93.6, 101.5, 107.5, 127.3 (×2), 127.5 (×2), 128.0 (×6), 128.1 (×6), 128.6 (×4), 128.7 (×4), 128.8 (×4), 137.9, 138.3, 138.4, 138.5 ppm; HR-FAB MS [M + Na]+ calcd for C54H63NO12NaSi 968.4016, found 968.4003.
tert-Butyldimethylsilyl O-(2,4-Di-O-benzyl-3,6-O-picoloyl-β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (29).
Picolinic acid (2.08 g, 16.9 mmol), 3-(ethyliminomethyleneamino)-N,N-dimethylpropan-1-amine (EDC, 3.23 g, 1.69 mmol), and DMAP (237 mg, 1.94 mmol) were added to a solution of 28 (1.22 g, 1.67 mmol) in CH2Cl2 (70 mL), and the resulting mixture was stirred under argon for 20 min at rt. The reaction mixture was diluted with CH2Cl2 (~100 mL) and was washed with cold water (10 mL), sat. aq NaHCO3 (10 mL), and water (10 mL). The organic phase was separated, dried with magnesium sulfate, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to give the title compound white amorphous solid in 95% yield (1.67 g, 1.44 mmol). Analytical data for 29: Rf = 0.25 (ethyl acetate/hexane, 1:1, v/v); [α]D21 −36.5 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, −0.13, 0.02 (2 s, 6H, 2 × CH3), 0.64 (s, 9H, C(CH3)3), 3.51–3.72 (m, 4H, H-5, 5′, 6a, 6b), 4.09–4.29 (m, 5H, H-2, 3, 4, 4′, 6b′), 4.41 (dd, 1H, J = 6.0 Hz, H-6a′), 4.48–4.89 (m, 10H, H-1′, 2′, 4 × CH2Ph,), 5.16 (dd, 1H, J = 6.0 Hz, H-3′), 5.29 (d, 1H, J = 9.0 Hz, H-1), 6.62–6.77 (m, 5H, aromatic), 7.09–7.80 (m, 23H, aromatic), 7.96–8.01 (m, 2H, aromatic), 8.61–8.63 (m, 1H, aromatic), 8.78–82 (m, 1H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3); 4.0, 5.3, 17.7, 25.5 (×3), 58.1, 64.5, 68.6, 73.3, 73.4, 73.7, 74.5, 74.8, 74.9, 75.0, 75.7, 76.7, 80.1, 93.6, 101.3, 107.5, 125.4, 125.7, 126.9 (×2), 127.2, 127.7 (×3), 127.9, (×4), 128.0 (×5), 128.2 (×8), 128.3 (×2), 128.5 (×2), 128.7 (×2), 137.1, 137.6, 137.7, 137.9, 138.4, 138.9, 147.7, 147.8, 149.9, 150.3, 164.3, 164.7 ppm; HR-FAB MS [M + Na]+ calcd for C66H69N3O14NaSi 1178.4447 found 1178.4457
O-(2,4-Di-O-benzyl-3,6-O-picoloyl-β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranose (30).
A 1 M soln of tetrabutylammonium fluoride (TBAF, 4.7 mmol) in THF (1.36 mL) was added to a solution of compound 29 (1.65 g, 1.40 mmol) in dry THF (45 mL), and the resulting mixture was stirred for 2 h at rt. The volatiles were removed under the reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the title compound contaminated with a tetrabutylammonium salt. To simplify the separation, the mixture was dissolved in pyridine (10 mL), acetic anhydride (5 mL) was added, and the resulting mixture was stirred for 18 h at rt. The reaction mixture was quenched with CH3OH (~1.0 mL), and the volatiles were removed under the reduced pressure. The residue was diluted with CH2Cl2 (10 mL) and washed with 1 N HCl (2 × 5 mL), water (10 mL), sat. aq NaHCO3 (10 mL), and water (2 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford the acetylated intermediate. The latter was dissolved in dimethylformamide (10 mL), hydrazine acetate (80 mg) was added, and the resulting mixture was stirred for 1 h. The reaction mixture was concentrated by a continuous flashing with a stream of air. The residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to give the title compound as a white amorphous solid in 60% yield (850 mg, 0.84 mmol). Analytical data for 30: Rf = 0.42 (ethyl acetate/hexane, 1:1.5, v/v); [α]D22 +28.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 3.52–3.71 (m, 4H, H-5, 5′, 6a, 6b), 3.98–4.17 (m, 4H, H-2, 4, 4′, 6a′), 4.27 (dd, 1H, J = 9.0 Hz, H-3), 4.41–4.72 (m, 9H, H-1′, 2, 6b′, 3 × CH2Ph), 4.85 (dd, 2H, 2J = 12.0 Hz, CH2Ph), 5.04 (dd, 1H, J = 3.0 Hz, H-3′), 5.28 (d, 1H, J = 9.0 Hz, H-1), 6.60–6.75 (m, 4H, aromatic), 7.07–7.99 (m, 26H, aromatic), 8.62–8.94 (m, 1H, aromatic), 8.78–8.79 (m, 1H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3); 57.3, 64.0, 68.3, 72.7, 73.0, 74.3, 74.4, 74.6, 74.7, 75.6, 79.5, 92.7, 100.8, 123.1, 125.1, 125.4, 126.6 (×2), 127.0 (×2), 127.4 (×6), 127.5 (×3), 127.8 (×6), 129.9 (×5), 128.1 (×2), 128.5 (×2), 131.4 (×2), 133.5, 136.8, 137.1, 137.3, 137.4, 138.0, 138.5, 147.2, 147.3, 149.6, 150.0, 162.5, 163.9, 164.2 ppm; HR-FAB MS [M + Na]+ calcd for C60H55N3O14Na 10643581, found 1064.3549
O-(2,4-Di-O-benzyl-3,6-O-picoloyl-β-d-mannopyranosyl)-(1 → 4)-3,6-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl Trichloroacetimidate (24).
CCl3CN (80 μL, 4.0 mmol) and DBU (3.0 μL, 0.02 mmol) were added to a solution of 30 (200 mg, 0.2 mmol) in CH2Cl2 (5.0 mL), and the resulting mixture was stirred for 2 h at rt. After that the volatiles were removed in vacuo, and the residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to give the title compound in 83% yield (192 mg, 0.165 mmol) as a white amorphous solid. Analytical data for 24: Rf = 0.45 (ethyl acetate/hexane, 6:4, v/v); [α]D22 +33.6 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 3.54–3.67 (m, 1H, H-5′), 3.68–3.82 (m, 3H, H-2′, 4, 4′), 4.29–4.89 (m, 11H, H-1′, 2, 3, 6a′, 6b′, 3 × CH2Ph), 4.86 (dd, 2H, 2J = 12.0 Hz, CH2Ph), 5.05 (dd, 1H, J = 9.0 Hz, H-3′), 6.37 (d, 1H, J = 9.0 Hz, H-1), 6.60–6.76 (m, 5H, aromatic), 7.09–8.03 (m, 26H, aromatic), 8.51 (s, 1H, NH), 8.64–8.66 (d, 1H, aromatic), 8.78–8.80 (d, 1H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3): δ, 14.2, 21.0, 54.5, 60.3, 64.2, 67.9, 72.9, 73.1, 73.5, 74.6, 74.7, 74.8, 75.5, 75.6, 79.1, 90.3, 94.0, 100.8, 123.2, 125.2, 125.6, 126.7, 126.8, 127.1, 127.5 (×2), 127.6 (×2), 127.8 (×2), 128.8 (×3), 127.9 (×3), 128.0 (×4), 128.1 (×2), 128.3 (×3), 128.6 (×2), 131.3, 133.7, 136.9, 137.3, 137.4, 137.5, 138.1, 138.5, 147.4, 147.5, 149.8, 150.1, 160.8, 164.1, 164.4 ppm; HR-FAB MS [M + Na]+ calcd 1207.2678 for C62H55Cl3N4O14Na, found 1207.2658
Automated Assembly of Pentasaccharide 31. N-(p-Acetoxymethyl)benzyloxycarbonyl-4-benzylaminobutyl Di-O-(2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl)-(1 → 3,1 → 6)-O-(2-O-benzyl-β-d-mannopyranosyl)-(1 → 4)-O-(3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (31).
Functionalized JandaJel resin 19 (50 mg, 0.011 mmol) was packed in an Omnifit glass chromatography column, and the latter was integrated into the HPLC system. Pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 5 min (5 mL, step 1). The system was then switched to the recirculation mode, and the delivery of CH2Cl2 continued for 30 min at 1.0 mL/min (swelling, step 2). Pump D was stopped, and pump C was programmed to deliver a solution of donor 24 (128 mg, 0.11 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 3). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (20 μL, 0.11 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min, and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in the absorbance of the eluate. Pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 4). After that pump D was stopped, and pump A was programmed to deliver a solution of Cu(OAc)2 (50 mg) in MeOH/CH2Cl2 (1/10, v/v) for 20 min at 1.0 mL/min (step 5). This step was monitored by the integrated UV detector (λmax = 254 nm). Pump A was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 6). Pump D was stopped, and pump C was programmed to deliver a solution of donor 23 (87 mg, 0.11 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 7). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (20 μL, 0.11 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min, and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in the absorbance of the eluate. After that pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 8). Pump D was stopped, and pump C was programmed to deliver a solution of donor 23 (87 mg, 0.11 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 11). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (20 μL, 0.11 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min, and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in the absorbance of the eluate. Pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 12). At this stage, the resin contains the immobilized pentasaccharide 22. Pump D was stopped, and pump B was programmed to deliver a 0.1 M solution of NaOMe in CH3OH/CH2Cl2 (10 mL, 0.04:1:1, v/v/v) that was recirculated at 1.0 mL/min for 20 min (step 13). Pump B was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min for 10 min, and the combined eluate was neutralized with Dowex (H+) resin. The resin was filtered off, washed successively with CH2Cl2 and CH3OH, and the combined filtrate was concentrated in vacuo. The crude residue was dissolved in pyridine (2.0 mL), Ac2O (73 μL, 0.771 mmol) was added dropwise, and the resulting mixture was stirred for 16 h at rt. The reaction mixture was quenched with CH3OH (~1.0 mL), and the volatiles were removed under the reduced pressure. The residue was diluted with CH2Cl2 (20 mL) and washed with 1 N HCl (2 × 10 mL), water (20 mL), sat. aq NaHCO3 (20 mL), and water (2 × 20 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The crude residue was purified by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford pentasaccharide 31 in 31% yield. Analytical data for 31: Rf = 0.6 (ethyl acetate/hexane, 6:4, v/v); [α]D22 +40.7 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): 1.19–1.24 (m, 4H, 2 × CH2), 1.79–2.12 (m, 27H, 9 × CH3), 2.90–2.99 (m, 2H), 3.24–3.37 (m, 5H), 3.44–3.87 (m, 13H), 4.07–4.20 (m, 10H), 4.37–4.63 (m, 8H), 4.71–4.89 (m, 6H), 5.00–5.16 (m, 6H), 5.20–5.35 (m, 7H), 6.69–6.93 (m, 12H, aromatic), 7.17–7.54 (m, 36H, aromatic) ppm; 13C{1H} NMR (75 MHz, CDCl3); δ, 14.3, 20.6, 20.7 (×3), 20.8 (×5), 20.9, 21.1 (×2), 26.5, 31.0, 55.7, 56.4, 60.4, 62.4, 62.5, 65.8, 66.0 (×3), 66.1, 66.7, 66.8, 68.5, 68.9, 69.0, 62.2, 69.5 (×2), 72.7, 73.4, 74.1, 74.4, 74.5, 74.6, 74.8, 75.0, 75.1 (×2), 76.7, 77.8, 78.6, 82.1, 97.4, 97.5, 98.1, 99.7, 101.1, 123.2, 123.5, 126.1, 127.1 (×2), 127.2 (×2), 127.3, 127.4 (×2), 127.5 (×4), 127.6, 127.7 (×3), 127.8 (×3), 127.9 (×3), 128.0 (×10), 128.2 (×2), 128.3 (×3), 128.4 (×6), 128.5 (×4), 128.7, 131.5, 131.9, 133.7, 134.0, 135.6, 136.9 (×2), 137.8, 137.9, 138.0, 138.5, 138.7 (×2), 167.5, 168.4, 169.6, 169.7, 169.8, 170.0, 170.5, 170.7, 170.9 ppm; HR-FAB MS [M + Na]+ calcd 2352.8520 for C126H135N3O40Na found 2352.8545.
Supplementary Material
Acknowledgments
Funding
This work was supported by grants from the National Institute of General Medical Sciences (GM111835 and GM120673).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b03056.
1H and 13C{1H} NMR spectra for all new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Varki A Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 1993, 3, 97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dwek RA Glycobiology: toward understanding the function of sugars. Chem. Rev 1996, 96, 683–720. [DOI] [PubMed] [Google Scholar]
- (3).Moremen KW; Tiemeyer M; Nairn AV Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol Cell. Biol 2012, 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Helenius A; Aebi M Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [DOI] [PubMed] [Google Scholar]
- (5).Wang Z; Chinoy ZS; Ambre SG; Peng W; McBride R; de Vries RP; Glushka J; Paulson JC; Boons GJ A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science 2013, 341, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gamblin DP; Scanlan EM; Davis BG Glycoprotein synthesis: an update. Chem. Rev 2009, 109, 131–163. [DOI] [PubMed] [Google Scholar]
- (7).Walczak MA; Hayashida J; Danishefsky SJ Building biologics by chemical synthesis: practical preparation of di- and triantennary N-linked glycoconjugates. J. Am. Chem. Soc 2013, 135, 4700–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Aussedat B; Vohra Y; Park PK; Fernandez-Tejada A; Alam SM; Dennison SM; Jaeger FH; Anasti K; Stewart S; Blinn JH; Liao HX; Sodroski JG; Haynes BF; Danishefsky SJ Chemical synthesis of highly congested gp120 V1V2 N-glycopeptide antigens for potential HIV-1-directed vaccines. J. Am. Chem. Soc 2013, 135, 13113–13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Futakawa S; Nara K; Miyajima M; Kuno A; Ito H; Kaji H; Shirotani K; Honda T; Tohyama Y; Hoshi K; Hanzawa Y; Kitazume S; Imamaki R; Furukawa K; Tasaki K; Arai H; Yuasa T; Abe M; Arai H; Narimatsu H A unique N-glycan on human transferrin in CSF: a possible biomarker for iNPH. Neurobiol. Aging 2012, 33, 1807–1815. [DOI] [PubMed] [Google Scholar]
- (10).Feizi T Glycobiology of Aids. In Carbohydrates in Chemistry and Biology; Ernst B, Hart GW, Sinay P, Eds.; Wiley-VCH: Weinheim, New York, 2000; Vol. 2; pp 851–863. [Google Scholar]
- (11).Coss KP; Byrne JC; Coman DJ; Adamczyk B; Abrahams JL; Saldova R; Brown AY; Walsh O; Hendroff U; Carolan C; Rudd PM; Treacy EP IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol. Genet. Metab 2012, 105, 212–220. [DOI] [PubMed] [Google Scholar]
- (12).Ogawa T; Sugimoto M; Kitajima T; Sadozai KK; Nukada T Total synthesis of a nudecasaccharide: a typical carbohydrate sequence for the complex type of glycan chains of a glycoprotein. Tetrahedron Lett. 1986, 27, 5739–5742. [Google Scholar]
- (13).Unverzagt C Building blocks for glycoproteins: synthesis of the ribonuclease B fragment 21-25 containing an undecasaccharide N-glycan. Tetrahedron Lett. 1997, 38, 5627–5630. [Google Scholar]
- (14).Eller S; Schuberth R; Gundel G; Seifert J; Unverzagt C Synthesis of pentaantennary N-glycans with bisecting GlcNAc and core fucose. Angew. Chem., Int. Ed 2007, 46, 4173–4175. [DOI] [PubMed] [Google Scholar]
- (15).Unverzagt C; Eller S; Mezzato S; Schuberth R A double regio- and stereoselective glycosylation strategy for the synthesis of N-glycans. Chem. - Eur. J 2008, 14, 1304–1311. [DOI] [PubMed] [Google Scholar]
- (16).Ito Y; Ohnishi Y Oligosaccharides: Synthesis: Stereoselective synthesis of b-manno glycosides. In Glycoscience: Chemistry and Chemical Biology; Fraser-Reid B, Tatsuta K, Thiem J, Eds.; Springer: Berlin, Heidelberg, New York, 2001; Vol. 2, pp 1589–1620. [Google Scholar]
- (17).Matsuo I; Kashiwagi T; Totani K; Ito Y First chemical synthesis of triglucosylated tetradecasaccharide (Glc3Man9GlcNAc2), a common precursor of asparagine-linked oligosaccharides. Tetrahedron Lett. 2005, 46, 4197–4200. [Google Scholar]
- (18).Matsuo I; Totani K; Tatami A; Ito Y Comprehensive synthesis of ER related high-mannose-type sugar chains by convergent strategy. Tetrahedron 2006, 62, 8262–8277. [Google Scholar]
- (19).Wu X; Grathwohl M; Schmidt RR Efficient solid-phase synthesis of a complex, branched N-glycan hexasaccharide: use of a novel linker and temporary-protecting-group pattern. Angew. Chem., Int. Ed 2002, 41, 4489–4493. [DOI] [PubMed] [Google Scholar]
- (20).Jonke S; Liu K.-g.; Schmidt RR Solid-phase oligosaccharide synthesis of a small library of N-glycans. Chem. - Eur. J 2006, 12, 1274–1290. [DOI] [PubMed] [Google Scholar]
- (21).Ratner DM; Plante OJ; Seeberger PH A Linear synthesis of branched high-mannose oligosaccharides from the HIV-1 viral surface envelope glycoprotein gp120. Eur. J. Org. Chem 2002, 2002, 826–833. [Google Scholar]
- (22).Ratner DM; Swanson ER; Seeberger PH Automated Synthesis of a Protected N-Linked Glycoprotein Core Pentasaccharide. Org. Lett 2003, 5, 4717–4720. [DOI] [PubMed] [Google Scholar]
- (23).Geng X; Dudkin VY; Mandal M; Danishefsky SJ Angew. Chem., Int. Ed 2004, 43, 2562–2565. [DOI] [PubMed] [Google Scholar]
- (24).Wu B; Hua Z; Warren JD; Ranganathan K; Wan Q; Chen G; Tan Z; Chen J; Endo A; Danishefsky SJ Synthesis of the fucosylated biantennary N-glycan of erythropoietin. Tetrahedron Lett. 2006, 47, 5577–5579. [Google Scholar]
- (25).Watt GM; Boons G-J A convergent strategy for the preparation of N-glycan core di-, tri-, and pentasaccharide thioaldoses for the site-specific glycosylation of peptides and proteins bearing free cysteines. Carbohydr. Res 2004, 339, 181–193. [DOI] [PubMed] [Google Scholar]
- (26).Huang W; Wang D; Yamada M; Wang LX Chemoenzymatic synthesis and lectin array characterization of a class of N-glycan clusters. J. Am. Chem. Soc 2009, 131, 17963–17971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tanaka K; Fujii Y; Tokimoto H; Mori Y; Tanaka S; Bao GM; Siwu ER; Nakayabu A; Fukase K Synthesis of a sialic acid containing complex-type N-glycan on a solid support. Chem. - Asian J 2009, 4, 574–580. [DOI] [PubMed] [Google Scholar]
- (28).Nagasaki M; Manabe Y; Minamoto N; Tanaka K; Silipo A; Molinaro A; Fukase K Chemical Synthesis of a Complex-Type N-Glycan Containing a Core Fucose. J. Org. Chem 2016, 81, 10600–10616. [DOI] [PubMed] [Google Scholar]
- (29).Shivatare SS; Chang SH; Tsai TI; Ren CT; Chuang HY; Hsu L; Lin CW; Li ST; Wu CY; Wong CH Efficient convergent synthesis of bi-, tri-, and tetra-antennary complex type N-glycans and their HIV-1 antigenicity. J. Am. Chem. Soc 2013, 135, 15382–15391. [DOI] [PubMed] [Google Scholar]
- (30).Li L; Liu Y; Ma C; Qu J; Calderon AD; Wu B; Wei N; Wang X; Guo Y; Xiao Z; Song J; Sugiarto G; Li Y; Yu H; Chen X; Wang PG Efficient chemoenzymatic synthesis of an N-glycan isomer library. Chem. Sci 2015, 6, 5652–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gridley JJ; Osborn HMI Recent advances in the construction of b-D-mannose and b-D-mannosamine linkages. J. Chem. Soc., Perkin Trans 1 2000, 1471–1491. [Google Scholar]
- (32).Crich D Chemistry of glycosyl triflates: synthesis of b-mannopyranosides (Reprinted from Glycochemistry: Principles, Synthesis, and Applications, pg 53-75, 2001). J. Carbohydr. Chem 2002, 21, 667–690. [Google Scholar]
- (33).Crich D Methodology development and physical organic chemistry: a powerful combination for the advancement of glycochemistry. J. Org. Chem 2011, 76, 9193–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Crich D Mechanism of a chemical glycosylation reaction. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- (35).Crich D; Sharma I Is Donor–Acceptor Hydrogen Bonding Necessary for 4,6-O-Benzylidene-directed β-Mannopyranosylation? Stereoselective Synthesis of β-C-Mannopyranosides and α-C-Glucopyranosides. Org. Lett 2008, 10, 4731–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Crich D; Wu B; Jayalath P Convergent synthesis of a b-(1–3)-mannohexaose. J. Org. Chem 2007, 72, 6806–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Crich D; Banerjee A; Yao Q Direct chemical synthesis of the b-D-mannans: the b-(1-2) and b-(1-4) series. J. Am. Chem. Soc 2004, 126, 14930–14934. [DOI] [PubMed] [Google Scholar]
- (38).Crich D; Sun S Direct formation of b-mannopyranosides and other hindered glycosides from thioglycosides. J. Am. Chem. Soc 1998, 120, 435–436. [Google Scholar]
- (39).Crich D; Sun S Formation of b-mannopyranosides of primary alcohols using the sulfoxide method. J. Org. Chem 1996, 61, 4506–4507. [DOI] [PubMed] [Google Scholar]
- (40).Baek JY; Choi TJ; Jeon HB; Kim KS A highly reactive and stereoselective b-mannosylation system: mannosyl 4-pentenoate/PhSeOTf. Angew. Chem., Int. Ed 2006, 45, 7436–7440. [DOI] [PubMed] [Google Scholar]
- (41).El Ashry ESH; Rashed N; Ibrahim ESI Strategies of synthetic methodologies for constructing b-mannosidic linkage. Curr. Org. Synth 2005, 2, 175–213. [Google Scholar]
- (42).De Meo C; Kamat MN; Demchenko AV Remote participation-assisted synthesis of b-mannosides. Eur. J. Org. Chem 2005, 706–711. [Google Scholar]
- (43).Pistorio SG; Yasomanee JP; Demchenko AV Hydrogen bond-mediated aglycone delivery: focus on β-mannosylation. Org. Lett 2014, 16, 716–719. [DOI] [PubMed] [Google Scholar]
- (44).Yasomanee JP; Demchenko AV The effect of remote picolinyl and picoloyl substituents on the stereoselectivity of chemical glycosylation. J. Am. Chem. Soc 2012, 134, 20097–20102. [DOI] [PubMed] [Google Scholar]
- (45).Yasomanee JP; Demchenko AV Hydrogen bond-mediated aglycone delivery: the synthesis of linear and branched α-glucans. Angew. Chem., Int. Ed 2014, 53, 10453–10456. [DOI] [PubMed] [Google Scholar]
- (46).Yasomanee JP; Demchenko AV Hydrogen bond-mediated aglycone delivery (HAD): a highly stereoselective synthesis of 1,2-cis α-D-glucosides from common glycosyl donors in the presence of bromine. Chem. - Eur. J 2015, 21, 6572–6581. [DOI] [PubMed] [Google Scholar]
- (47).Nagorny P; Fasching B; Li X; Chen G; Aussedat B; Danishefsky SJ Toward Fully Synthetic Homogeneous β-Human Follicle-Stimulating Hormone (β-hFSH) with a Biantennary N-Linked Dodecasaccharide. Synthesis of β-hFSH with Chitobiose Units at the Natural Linkage Sites. J. Am. Chem. Soc 2009, 131, 5792–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sail D; Kovac P Benzoylated ethyl 1-thioglycosides: direct preparation from per-O-benzoylated sugars. Carbohydr. Res 2012, 357, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Alewood PF; Benn M; Reinfried R Cyclizations of Azidoformates to Tetrahydro-1,3-oxazin-2-ones and Oxazolidin-2-ones. Can. J. Chem 1974, 52, 4083–4089. [Google Scholar]
- (50).Khiar N; Werner S; Mallouk S; Lieder F; Alcudia A; Fernandez I Enantiopure sulforaphane analogues with various substituents at the sulfinyl sulfur: asymmetric synthesis and biological activities. J. Org. Chem 2009, 74, 6002–6009. [DOI] [PubMed] [Google Scholar]
- (51).Crich D; Jayalath P; Hutton TK Enhanced diastereoselectivity in B-mannopyranosylation through the use of sterically minimal propargyl ether protecting groups. J. Org. Chem 2006, 71, 3064–3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Adero PO; Furukawa T; Huang M; Mukherjee D; Retailleau P; Bohe L; Crich D Cation Clock Reactions for the Determination of Relative Reaction Kinetics in Glycosylation Reactions: Applications to Gluco- and Mannopyranosyl Sulfoxide and Trichloroacetimidate Type Donors. J. Am. Chem. Soc 2015, 137, 10336–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Sasaki K; Tohda K Recent topics in β-stereoselective mannosylation. Tetrahedron Lett. 2018, 59, 496–503. [Google Scholar]
- (54).Ganesh NV; Fujikawa K; Tan YH; Stine KJ; Demchenko AV HPLC-assisted automated oligosaccharide synthesis. Org. Lett 2012, 14, 3036–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pistorio SG; Nigudkar SS; Stine KJ; Demchenko AV HPLC-assisted automated oligosaccharide synthesis: the implementation of the autosampler as a mode of the reagent delivery. J. Org. Chem 2016, 81, 8796–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Panza M; Pistorio SG; Stine KJ; Demchenko AV Automated Chemical Oligosaccharide Synthesis: Novel Approach to Traditional Challenges. Chem. Rev 2018, 118, 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Plante OJ; Palmacci ER; Seeberger PH Automated solid-phase synthesis of oligosaccharides. Science 2001, 291, 1523–1527. [DOI] [PubMed] [Google Scholar]
- (58).Palmacci ER; Plante OJ; Hewitt MC; Seeberger PH Automated synthesis of oligosaccharides. Helv. Chim. Acta 2003, 86, 3975–3990. [Google Scholar]
- (59).Seeberger PH Automated oligosaccharide synthesis. Chem. Soc. Rev 2008, 37, 19–28. [DOI] [PubMed] [Google Scholar]
- (60).Kröck L; Esposito D; Castagner B; Wang C-C; Bindschadler P; Seeberger PH Streamlined access to conjugation-ready glycans by automated synthesis. Chem. Sci 2012, 3, 1617–1622. [Google Scholar]
- (61).Seeberger PH The logic of automated glycan assembly. Acc. Chem. Res 2015, 48, 1450–1463. [DOI] [PubMed] [Google Scholar]
- (62).Hahm HS; Schlegel MK; Hurevich M; Eller S; Schuhmacher F; Hofmann J; Pagel K; Seeberger PH Automated glycan assembly using the Glyconeer 2.1 synthesizer. Proc. Natl. Acad. Sci. U.S.A 2017, 114, E3385–E3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Tanaka H; Matoba N; Tsukamoto H; Takimoto H; Yamada H; Takahashi T Automated parallel synthesis of a protected oligosaccharide library based upon the structure of dimeric Lewis X by one-pot sequential glycosylation. Synlett 2005, 824–828. [Google Scholar]
- (64).Jaipuri FA; Pohl NL Toward solution-phase automated iterative synthesis: fluorous-tag assisted solution-phase synthesis of linear and branched mannose oligomers. Org. Biomol. Chem 2008, 6, 2686–2691. [DOI] [PubMed] [Google Scholar]
- (65).Machida K; Hirose Y; Fuse S; Sugawara T; Takahashi T Development and application of a solution-phase automated synthesizer, ‘ChemKonzert’. Chem. Pharm. Bull 2010, 58, 87–93. [DOI] [PubMed] [Google Scholar]
- (66).Hsu CH; Hung SC; Wu CY; Wong CH Toward automated oligosaccharide synthesis. Angew. Chem., Int. Ed 2011, 50, 11872–11923. [DOI] [PubMed] [Google Scholar]
- (67).Nokami T; Hayashi R; Saigusa Y; Shimizu A; Liu C-Y; Mong K-KT; Yoshida J.-i. Automated solution-phase synthesis of oligosaccharides via iterative electrochemical assembly of thioglycosides. Org. Lett 2013, 15, 4520–4523. [DOI] [PubMed] [Google Scholar]
- (68).Nokami T; Isoda Y; Sasaki N; Takaiso A; Hayase S; Itoh T; Hayashi R; Shimizu A; Yoshida J.-i. Automated electrochemical assembly of the protected potential TMG-chitotriomycin precursor based on rational optimization of the carbohydrate building block. Org. Lett 2015, 17, 1525–1528. [DOI] [PubMed] [Google Scholar]
- (69).Tang S-L; Linz LB; Bonning BC; Pohl NLB Automated solution-phase synthesis of insect glycans to probe the binding affinity of pea enation mosaic virus. J. Org. Chem 2015, 80, 10482–10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Tang S-L; Pohl NLB Automated solution-phase synthesis of β-1,4-mannuronate and β-1,4-mannan. Org. Lett 2015, 17, 2642–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Nagy G; Peng T; Kabotso DEK; Novotny MV; Pohl NLB Protocol for the purification of protected carbohydrates: toward coupling automated synthesis to alternate-pump recycling high-performance liquid chromatography. Chem. Commun 2016, 52, 13253–13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Geert Volbeda A; van Mechelen J; Meeuwenoord N; Overkleeft HS; van der Marel GA; Codee JDC Cyanopivaloyl Ester in the Automated Solid-Phase Synthesis of Oligorhamnans. J. Org. Chem 2017, 82, 12992–13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Chang R; Moquist P; Finney NS Chemical synthesis of UDP-4-O-methyl-GlcNAc, a potential chain terminator of chitin synthesis. Carbohydr. Res 2004, 339, 1531–1536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.