

Abstract
Bacterial expression of β-lactamase enzymes that hydrolyze β-lactam antibiotics contributes to the growing threat of antibacterial drug resistance. Metallo-β-lactamases, such as NDM-1, use catalytic zinc ions in their active sites and hydrolyze nearly all clinically available β-lactam antibiotics. Inhibitors of metallo-β-lactamases are urgently needed to overcome this resistance mechanism. Zinc-binding compounds are promising leads for inhibitor development, as many NDM-1 inhibitors contain zinc-binding pharmacophores. Here, we evaluated thirteen chelating agents containing benzimidazole and benzoxazole scaffolds as NDM-1 inhibitors. Six of the compounds showed potent inhibitory activity with IC50 values as low as 0.38 μM, and several compounds restored meropenem susceptibility of NDM-1-expressing E. coli. Spectroscopic and docking studies suggest ternary complex formation as the mechanism of inhibition, making these compounds promising for development as NDM-1 inhibitors.
Keywords: metallo-β-lactamase, chelator, antibiotic resistance, benzimidazole, benzoxazole, zinc
Graphical Abstract
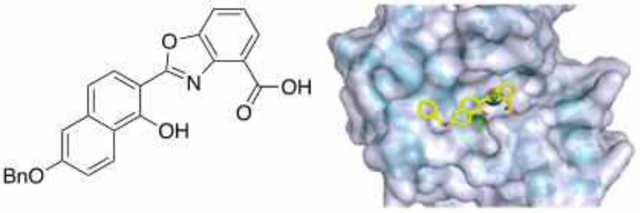
Zinc-binding benzimidazoles and benzoxazoles were investigated for inhibition of the clinically important metallo-β-lactamase NDM-1, which inactivates nearly all β-lactam antibiotics. Several potent inhibitors were identified with IC50 values as low as 0.38 μM. Top compounds restored susceptibility of NDM-1-expressing E. coli to meropenem. Spectroscopic and molecular docking studies suggest ternary complex formation as the inhibition mechanism.
Introduction
β-Lactamases are bacterial enzymes that inactivate β-lactam antibiotics via hydrolysis, leading to drug resistance.[1] The metallo-β-lactamases, of which NDM-1[2][3] is a prototypical example, pose a particular clinical threat because of their broad substrate scope and insensitivity to most existing β-lactamase inhibitors, which inactivate serine active sites but not the zinc-containing active sites of metallo-β-lactamases.[4] Many of the known metallo-β-lactamase inhibitors are chelators that bind to the active site zinc ions, although none of these inhibitors are currently approved by the US FDA as β-lactamase inhibitors. Inhibitors of metallo-β-lactamases are urgently needed for β-lactam antibiotics to continue to be useful against highly resistant pathogens.
Zinc-binding compounds are a promising starting point in the search for inhibitors of metallo-β-lactamases, since most reported inhibitors interact with the active site zinc ions as their main inhibitory mechanism.[5] These zinc-binding inhibitors can be divided into two groups: inhibitors that remove zinc from the active site, and inhibitors that form a ternary complex with the zinc in the active site. Zinc-removing inhibitors include strong metal chelators such as EDTA; these compounds are effective inhibitors but interact with other metals and metalloproteins nonspecifically due to their high metal binding affinities and lack of features that specifically target metallo-β-lactamases. Inhibitors that form ternary complexes include sulfur-containing compounds such as d-captopril. In principle, this type of inhibitor can be more specific for the metallo-β-lactamase active site and less likely to cause the same undesirable off-target effects as metal-removing inhibitors.
Here, we investigated the NDM-1-inhibitory activity of a set of benzoxazole- and benzimidazole-containing compounds related to the bacterial natural product UK-1 (Figure 1). UK-1 and truncated analogs such a 1 exhibit broad-spectrum anticancer activity, potentially by inhibiting topoisomerase II;[6] they also inhibit replication of the hepatitis C virus and inhibit the viral NS3 helicase.[7] Importantly, a number of these compounds have been shown to bind a variety of divalent metal ions, including zinc.[7b] Because the ability to bind zinc is a feature found in many reported NDM-1 inhibitors, we hypothesized that these compounds may exhibit NDM-1 inhibitory activity as well. We found that many of these chelators inhibit NDM-1 and restore the activity of a carbapenem antibiotic in NDM-1-expressing E. coli. Analysis of the structure-activity relationship and absorption spectra of an NDM-1-inhibitor complex suggests a ternary complex forming mechanism of inhibition, and docking studies provide information about likely binding modes. These compounds, especially the most potent inhibitor compound 9 with a sub-micromolar IC50, provide promising leads for the development of NDM-1 inhibitors.
Figure 1.
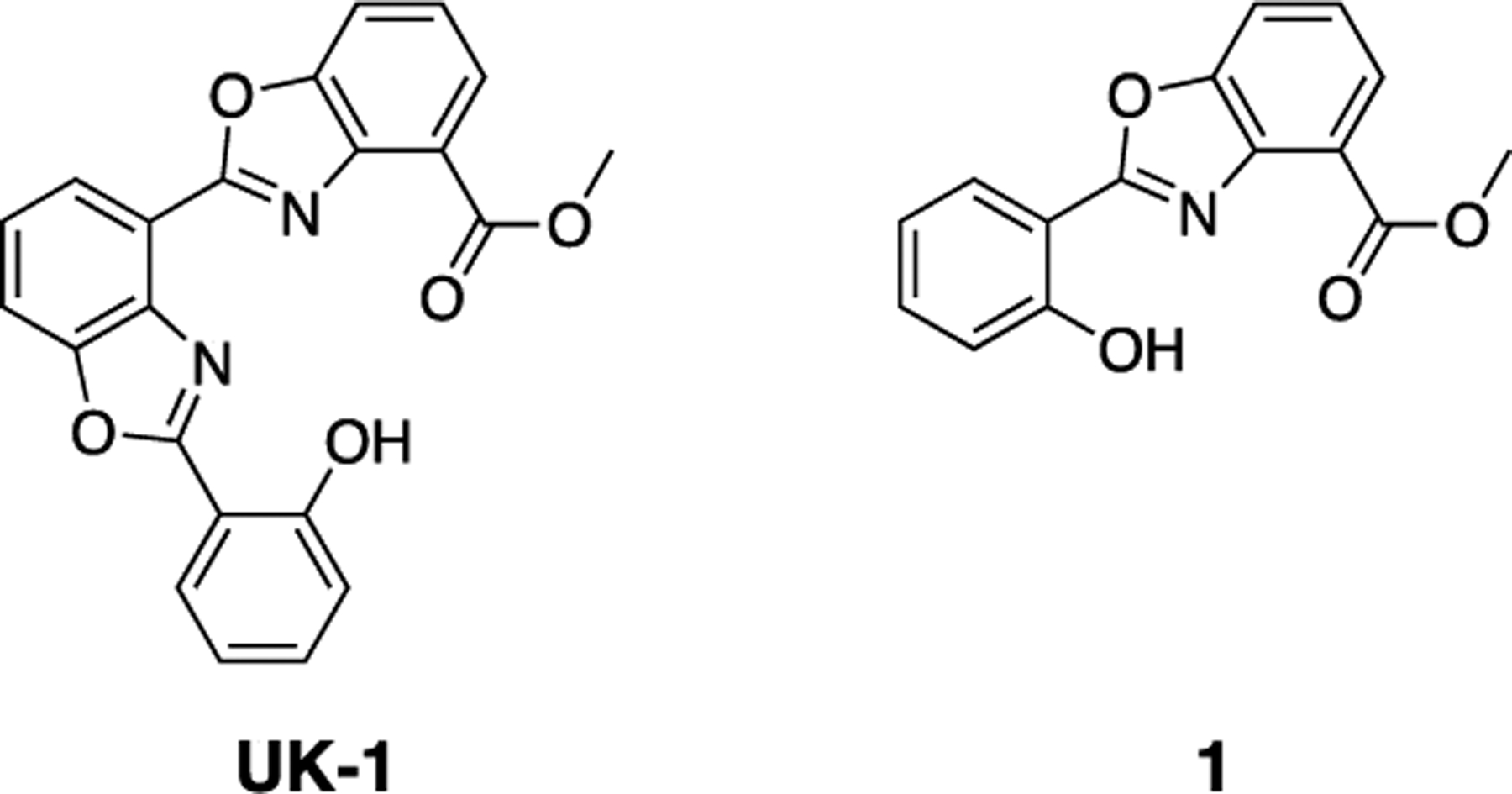
The bacterial natural product UK-1 and truncated analog 1.
Results and Discussion.
Design and synthesis of metal-binding benzimidazoles and benzoxazoles.
The structures of the chelators evaluated as NDM-1 inhibitors are shown in Figure 2. Each compound has a core based on a benzimidazole or benzoxazole ester, acid, or amide linked to a phenol, napthol, or quinoline to form a tridentate zinc-binding motif consisting of a combination of oxygen and nitrogen coordinating atoms.
Figure 2.
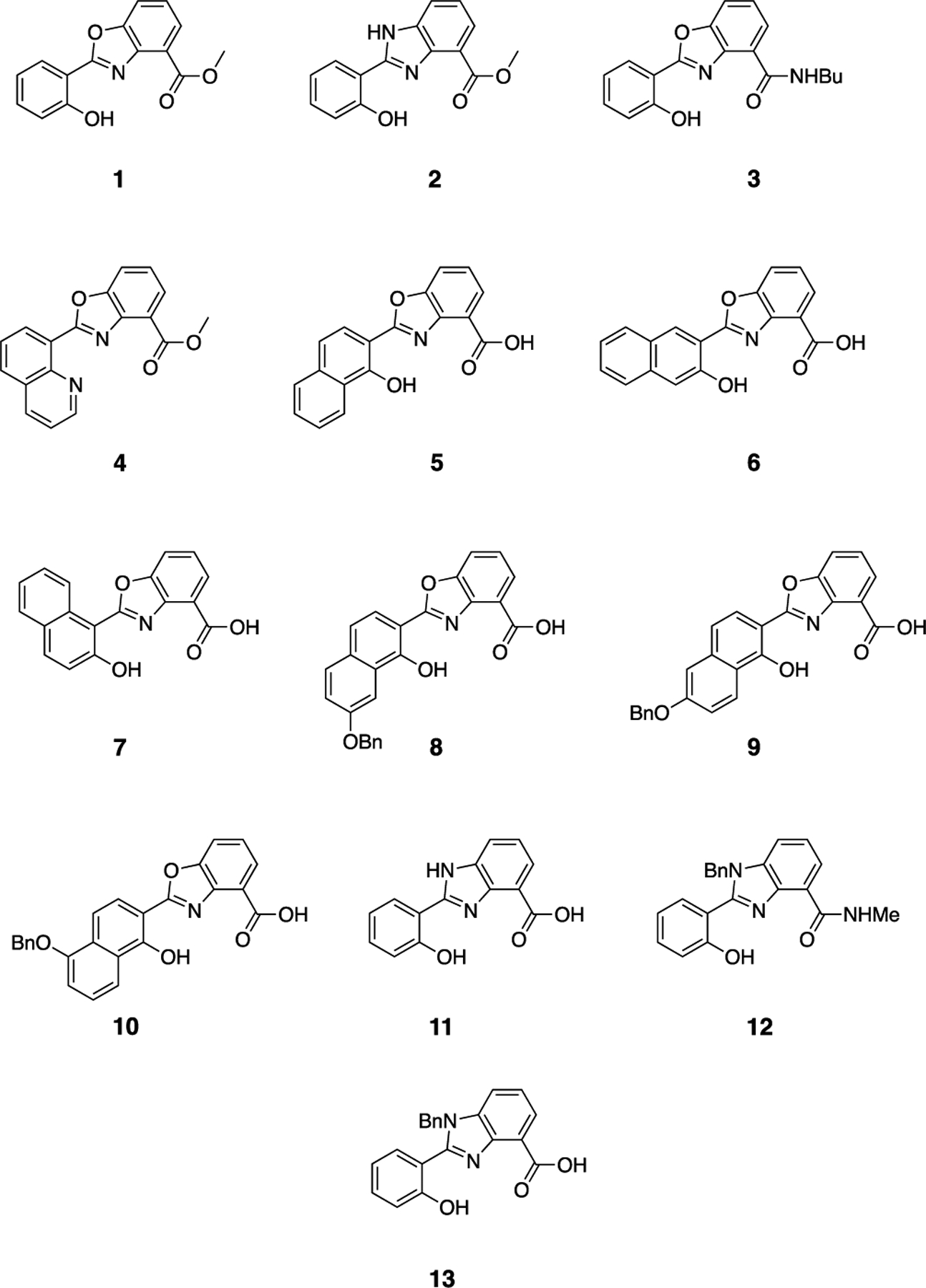
Structures of benzimidazole and benzoxazole zinc chelators synthesized and evaluated as NDM-1 inhibitors.
Compounds 1–4 and 11–13 have been previously reported.[6c, 7b] Compounds 5–8 and 10 were prepared by hydrolysis of the previously reported methyl esters.[7] Compound 9 was prepared by reaction of 6-benzyloxy-1-hydroxy-2-naphthoic acid (14) with methyl 3-hydroxyanthranilate, acid-catalyzed cyclodehydration of the resulting amide (15) to form the benzoxazole ring, and hydrolysis of the ester (Scheme 1 and Scheme S1).
Scheme 1.
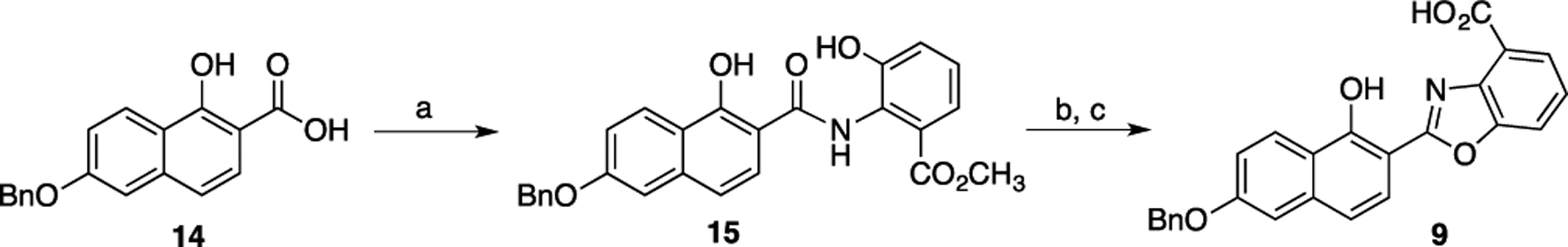
Reagents and conditions: (a) carbonyldiimidazole, methyl 3-hydroxyanthranilate, THF; (b) pyridinium p-toluenesulfonate, toluene, reflux, 26%, 2 steps; (c) NaOH, water/THF, reflux, 72%.
In vitro inhibition of NDM-1 by compounds 1–13.
We determined the ability of the compounds to inhibit the metallo-β-lactamase NDM-1 with an in vitro assay using purified NDM-1 enzyme. IC50 values were determined for the inhibition of enzyme-catalyzed turnover of the colorimetric β-lactamase substrate nitrocefin.[8] Table 1 shows IC50 values for the inhibition of nitrocefin turnover by NDM-1. (For inhibition curves used to calculate IC50 values, see Figure S1.) Six of the compounds showed good inhibitory activity with IC50 values less than 10 μM, and 9 had the best activity with an IC50 of 0.38 μM. The presence of either a benzimidazole or benzoxazole core was not predictive of inhibitory activity, but a free carboxylic acid was essential. Interestingly, compounds with similar structures differing only in the position of the naphthalene group (compounds 5, 6, and 7) or placement of the benzyloxy group (compounds 8, 9, and 10) had different levels of inhibitory activity against NDM-1. If these chelators act by removing the zinc ions from the NDM-1 active site, as many reported NDM-1 inhibitors do, we would expect that the shape of the compounds and position of non-coordinating functional groups positioned away from the zinc coordinating moieties would not have a dramatic effect on inhibitory activity, since the zinc affinity of these isomeric compounds should be similar. Based on our observation that isomeric compounds inhibited differently, we hypothesized that inhibition depends on both chelating and non-chelating interactions within the protein active site to form a ternary complex between the protein, the metals, and the inhibitor, rather than a mechanism that relies on removal of zinc from the active site.
Table 1.
IC50 values for the inhibition of NDM-1-catalyzed turnover of nitrocefin by compounds and computed mean binding energy (MBE) from docking studies to the NDM-1 active site.
| Compound | IC50 (μM) | 95% CI (μM)[a] | Computed MBE[b] (kCal/mol) |
|---|---|---|---|
| 1 | >40 | - | −6.98 |
| 2 | >20[c] | - | −6.56 |
| 3 | >20[c] | - | −7.94 |
| 4 | >40 | - | −7.69 |
| 5 | 0.74 | 0.44–1.1 | −9.70 |
| 6 | 9.8 | 8.6–11 | −9.50 |
| 7 | 8.5 | 7.5–9.6 | −9.20 |
| 8 | 2.3 | 1.6–3.2 | −10.16 |
| 9 | 0.38 | 0.22–0.55 | −9.99 |
| 10 | >40 | - | −9.79 |
| 11 | >40 | - | −8.31 |
| 12 | >40 | - | −8.14 |
| 13 | 5.4 | 4.4–6.4 | −8.57 |
95% CI = 95% confidence interval for the IC50.
MBE = mean binding energy.
Compounds 2 and 3 were not soluble above 20 μM and did not inhibit activity by more than 50% at 20 μM.
Top compounds restore carbapenem susceptibility of NDM-1-expressing E. coli.
The top five compounds from the IC50 screen (5, 7, 8, 9, and 13) were evaluated in bacterial culture to determine whether they could restore antibacterial activity of the carbapenem antibiotic meropenem, which itself has poor activity (MIC ≥ 64 μg/mL) in E. coli MG1655 and UTI89 strains expressing NDM-1 due to inactivation by hydrolysis. To measure the effects of the compounds on the antibacterial activity of meropenem, a fixed concentration of each compound (100 μM) was added to a broth microdilution assay measuring the minimum inhibitory concentration (MIC) of meropenem. Importantly, none of the compounds tested inhibited growth of either bacterial strain on their own when treated at 100 μM.
Compounds 5, 8, 9, and 13 decreased the MIC of meropenem by at least 16-fold to 4 μg/mL or lower (Table 2). Only compound 7, which had the least favorable IC50 of the top five compounds, failed to restore meropenem susceptibility. Compound 9 had the best activity in this assay, with meropenem’s MIC restored to less than 0.5 μg/mL in both strains. These results are consistent with our enzyme inhibition assay, as many of the most potent NDM-1 inhibitors showed the best recovery of meropenem, with 9 once again showing the best activity. This assay was repeated with compound 9 at a range of concentrations to demonstrate the dose dependence of its ability to restore meropenem susceptibility and determine its growth-inhibitory effects on its own at different concentrations. Compound 9 reduced the MIC of meropenem at least 4-fold even when treated at only 25 μM and did not show any antibacterial activity on its own even when treated at 200 μM in either strain.
Table 2.
Recovery of meropenem susceptibility in E. coli MG1655 and UTI89 expressing NDM-1.
| Treatment | Meropenem MIC (μg/mL) | |
|---|---|---|
| E. coli MG1655 | E. coli UTI89 | |
| Meropenem alone | 64 | >64 |
| Meropenem + 100 μM 5 | 4 | ≤0.5 |
| Meropenem + 100 μM 7 | 64 | 64 |
| Meropenem + 100 μM 8 | 2 | 4 |
| Meropenem + 100 μM 13 | 2 | 1 |
| Meropenem + 25 μM 9 | 16 | 4–8[a] |
| Meropenem + 50 μM 9 | 1–4[a] | ≤0.5 |
| Meropenem + 100 μM 9 | ≤0.5 | ≤0.5 |
| Meropenem + 200 μM 9 | ≤0.5 | ≤0.5 |
Ranges are given where differences were consistently observed between biological replicates.
UV-Visible spectroscopy of NDM-1 treated with 9 suggests ternary complex mechanism.
As discussed earlier, we hypothesized that 9 is forming a ternary complex with the NDM-1 active site and not removing zinc by chelation. UV-Visible spectroscopy was used to examine the interaction of NDM-1 with the top compound 9 to probe its mechanism of inhibition of NDM-1. If 9 is removing metal from the active site, we expect the absorption spectrum of 9 mixed with NDM-1 to resemble the sum of the spectra of zinc-bound 9 and apo-NDM-1. If a unique spectral signature is produced that is not present in the spectra of 9, its zinc complex, or NDM-1, this would suggest ternary complex formation as the inhibition mechanism.
Upon addition of one equivalent of 9 to NDM-1 and incubation at 37 °C in HEPES (50 mM, pH 7.2), a new feature at approximately 430 nm (indicated by the arrow in Figure 3) appeared that was not present in the absorption spectra of 9, its zinc complex, or NDM-1. In comparison, this spectral feature is not present when the assay is repeated with apo-NDM-1. This result is supportive of a stable ternary complex between 9 and the NDM-1 zinc active site rather than zinc removal.
Figure 3.
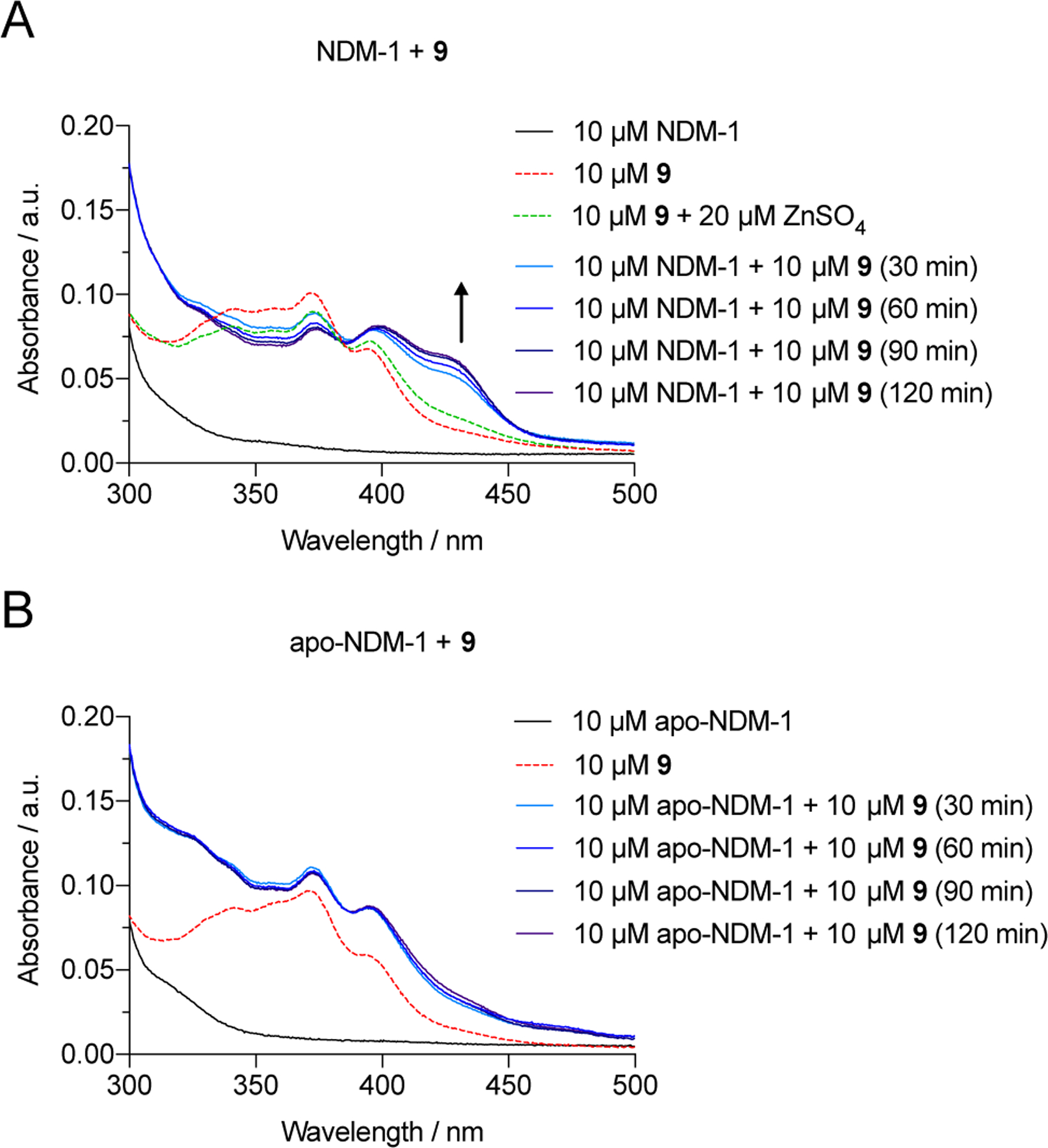
UV-Visible spectrum of (A) NDM-1 treated with 9, compared to the spectrum of 9 and its zinc complex and (B) apo-NDM-1 treated with 9.
Docking studies reveal key enzyme-inhibitor interactions.
To better characterize the interactions between compounds 1–13 and NDM-1, we performed computational docking studies of these molecules with a crystal structure of NDM-1. Of several structures of NDM-1 published with small molecules bound to the active site, we selected the crystal structure of NDM-1 in complex with hydrolyzed ampicillin at pH 7.3 (PDB 5ZGR[9]) as the most attractive for these docking simulations because of its good resolution (1.15 Å), the presence of the hydrolyzed product of a common antibiotic, and the use of physiological pH to grow the crystals. The 5ZGR structure lacks the first 28 residues of the N-terminal signaling sequence.
To validate our docking methods, we attempted to re-dock the hydrolyzed ampicillin to this NDM-1 structure using five separate approaches, all of which have been previously used in numerous studies to screen databases for NDM-1 inhibitors: (i) Autodock Vina; (ii) Autodock (AD) with default Gastieiger charges for the receptor; (iii) AD with the charge of receptor zinc atoms set to +2 eV; (iv) AD with charge polarization of receptor zinc-coordinating atoms (zinc depolarization +1.6 eV), and (v) Autodock4Zn (AD4Zn). Autodock Vina has been used to identify antibiotic natural product inhibitors,[10] while method (ii) has been used to identify binding conformations of β-lactams[11] and antibacterial natural products that inhibit NDM-1.[12] Neither of these methods could be validated in our study so we moved on to method (iii), which was employed to emphasize zinc-ligand electrostatic interactions.[13] However, method (iii) returned unrealistically short zinc-ligand coordination bond lengths in our simulations, so we used method (iv) to examine a depolarized zinc ion model, and method (v) which is specific for zinc-binding proteins. Methods (i), (iii), and (v) were directly compared previously in the development of AD4Zn.[14] AD4Zn has also been previously used to determine the structure-activity relationship of propionic sulfhydryl compounds to NDM-1.[15]
Of the five methods we examined, AD4Zn, method (v), showed the smallest deviation from the position of the hydrolyzed ampicillin in the X-ray crystal structure (Figure S2). AD4Zn was specifically developed to better account for zinc-coordinating compounds.[14] It uses an updated potential that was calibrated using zinc-containing crystal structures and implements directionality with the use of a pseudoatom and updated forcefield parameters. In our modeling, AD4Zn correctly oriented the hydrolyzed ampicillin in the NDM-1 active site with appropriate zinc coordinating atom bond lengths and only minor variation to the position of the non-coordinating phenyl group. Thus, we consider our implementation of AD4Zn to be validated for this study. Other docking methods returned alternate ligand orientations, positions, or unrealistic Zn–ligand coordination bond lengths.
Results of our docking studies between compounds 1–13 and NDM-1 using our validated AD4Zn protocol are found in Table 1, Figure 4, and Figures S3 and S4. The mean binding energies from docking correlated well with the IC50 values that were determined experimentally, with more inhibitory compounds returning a lower calculated docking binding energy. Every active compound was computed to have a mean binding energy less than −8.5 kCal/mol, which could be used as a cutoff for further in silico development of these compounds. Compound 9, which had the best activity in inhibition assays, also had one of the best docking scores, with a computed binding energy of −9.99 kCal/mol.
Figure 4.
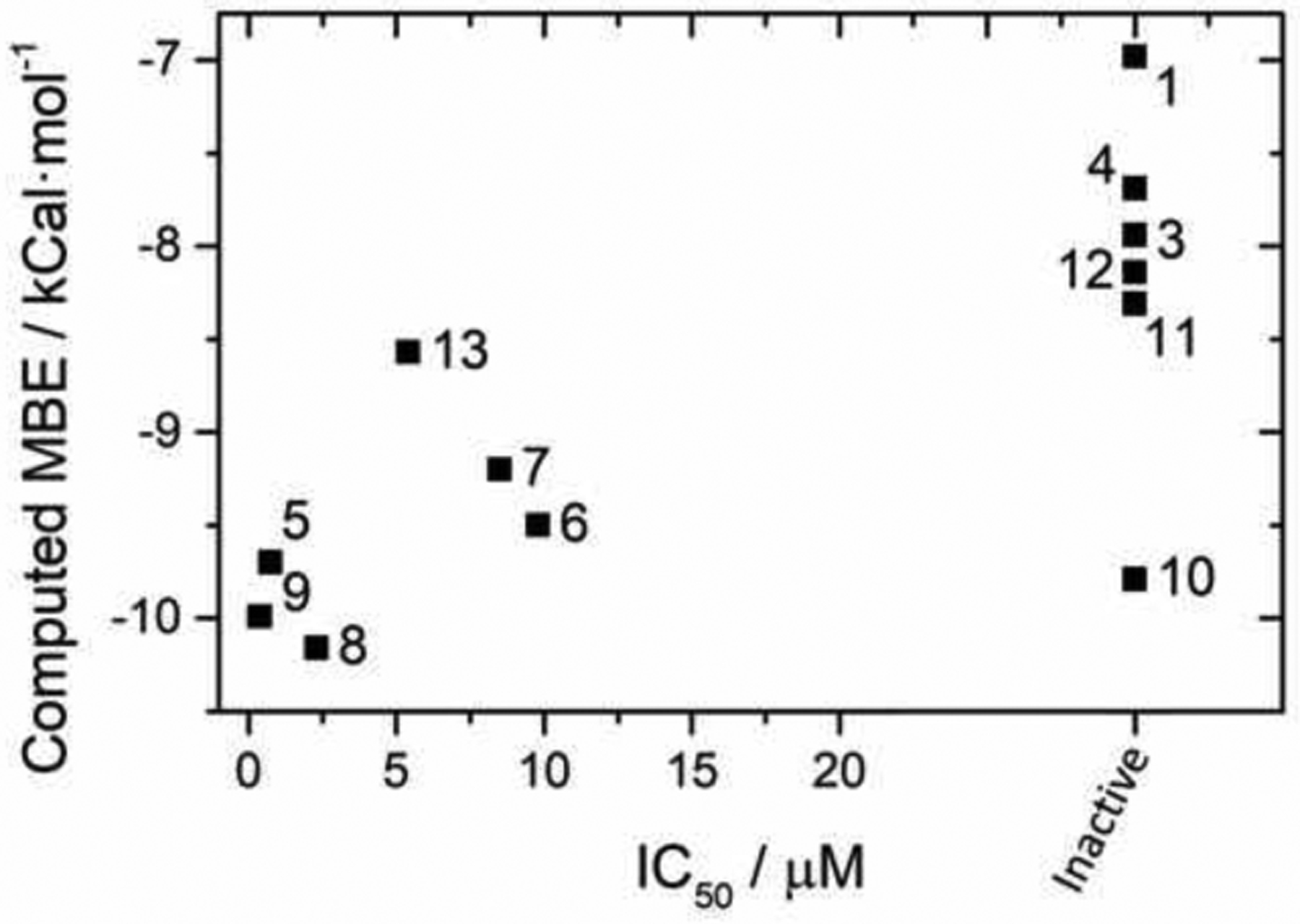
Comparison of experimentally determined IC50 values with computed mean binding energy (MBE) from the lowest energy cluster using the AD4Zn method. The docking score is shown as the mean cluster binding energy of the lowest energy cluster from AD4Zn analysis (see methods). Compounds labeled inactive showed no activity or were insoluble at ligand concentrations ≥40 μM (see Table 1).
There are two notable exceptions to this trend. The docking results suggest that compound 10 would be much more active than we observed experimentally and that compound 13 would be less active. Compounds 9 and 10 are structurally identical except for the position of the benzyloxy group on the naphthol ring; its position in compound 9 makes the compound linear in shape, which leads to favorable interactions within the active site cleft. Compound 10, however, is predicted to coordinate to the zinc in a similar manner, and suitable poses and energies were predicted from the docking. Comparing the binding poses of compounds 9 and 10 (Figure 5) we see nearly identical positioning of the coordinating moieties. Compound 13, with an extra benzyl group on the benzimidazole ring, is a much more sterically bulky ligand, likely impacting the available coordination modes and resulting docking score.
Figure 5.
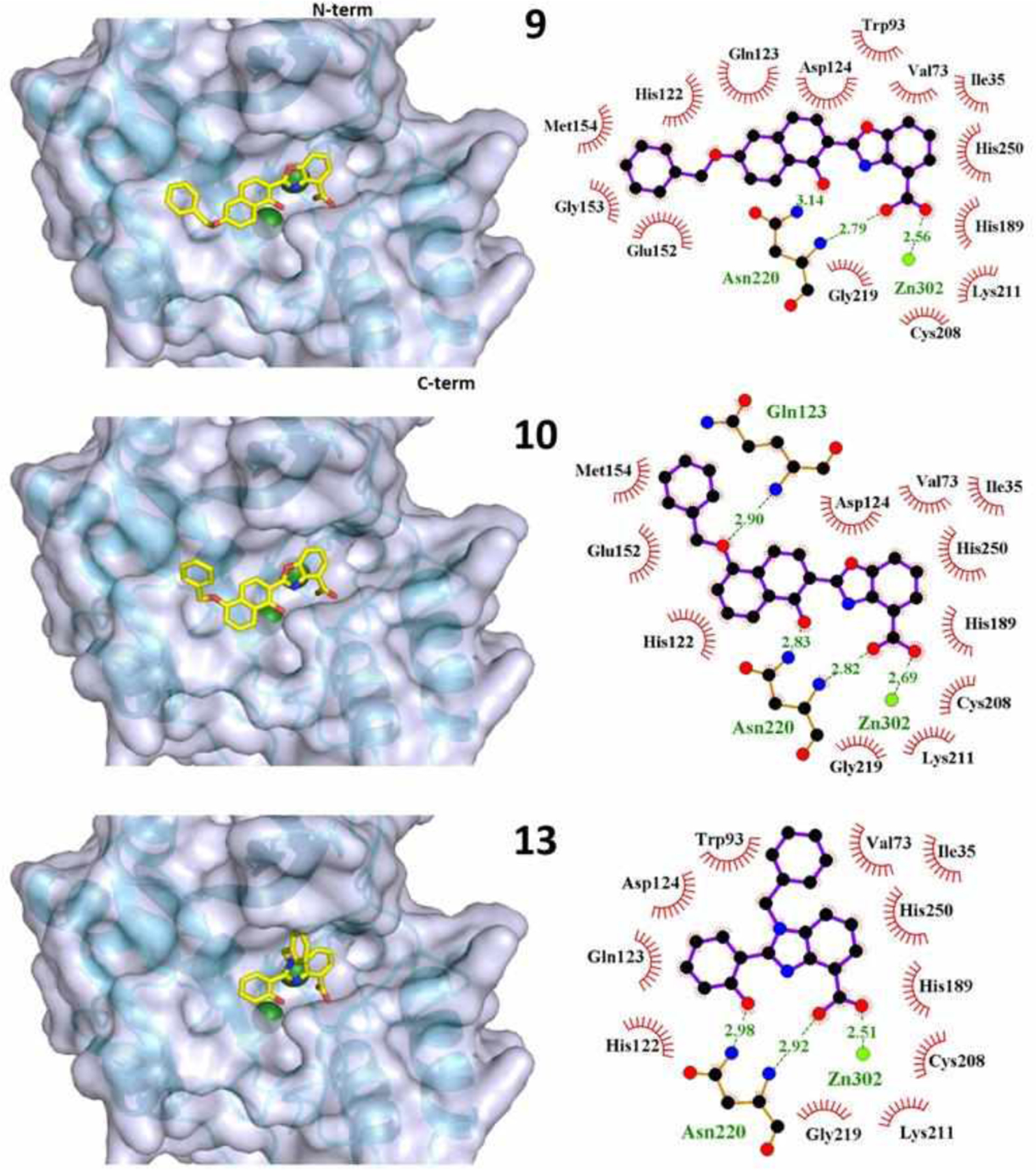
Binding poses of compounds 9, 10, and 13 shown in 3D (left) and as LigPlots (right). The protein backbone is shown as a cyan ribbon model with the N-terminus at the top and C-terminus at the bottom of each and the active site cleft facing forward. The two zinc ions in the 5ZGR active site are shown as green spheres. The compounds are colored by element type with carbon atoms shown in yellow. For LigPlots: Compounds are shown with purple bonds and amino acid residues are shown with orange bonds. Hydrophobic interactions are indicated with residues labelled in black. Hydrogen bonds, bonds to zinc, and bonding residues are shown and labeled in green. Zn302 in the LigPlots represents the Zn2 site by Asp, Cys, His. The complete series of binding pose LigPlots are provided in the Supporting Information (Figures S3 and S4).
The docked poses suggest that NDM-1-inhibitor interactions occur in the same general area of the active site cleft for all compounds (Figure S5). Tables 3 and S1 list the LigPlot-identified interactions between each compound and NDM-1 residues, and Figure S6 shows the position of the residues involved in binding compound 9. The dinuclear active site is composed of Zn1 coordinated by two surface-exposed His residues (His122, 189) and a buried His120, and Zn2 (labeled Zn302 in the crystal structure), coordinated by Asp124, Cys208, and His 250 (Figure S6). All of the active compounds are observed to bind to this Zn2 site via monodentate carboxylate coordination with computed Zn–O distance of 2.4–2.8 Å. This result corroborates the functional assays indicating a free carboxylic acid to be essential for activity. The active inhibitors also display Zn–N interactions at approximately 3 Å between the benzoxazole or benzimidazole nitrogen and Zn2, suggesting direct binding. In principle the phenolate O of these compounds could also serve in metal coordination, although the 4.5–5 Å O–Zn1 distance measured from the docked poses suggests it would be weak. Rather, the most active compounds docked with the phenolate hydrogen-bonded at ~3 Å to Asn220 (Figure S4), a conserved residue in B1 and B2 metallo-β-lactamases that is located at the edge of the active site pocket in proximity to the Zn1 His3 site (Figure S6). The exceptions to this trend were 6 and 7, the docked poses of which positioned the phenolate away from both Zn sites and Asn220. The less favorable activity of 6 and 7 compared to 5, 8, 9, and 13 reveals a structure-activity correlation regarding positioning of the phenolate within the active site.
Table 3.
Interactions between active compounds and NDM-1 residues from docked structures by LigPlot+. The complete table for all compounds is provided in the Supporting Information (Table S1).
| Compound | H-bonding interactions | Hydrophobic interactions |
|---|---|---|
| 5 | Asn220, Zn302 | Ile35, Val73, Trp93, His122, Gln123, Asp124, Glu152, His189, Cys208, Lys211, Gly219, His250 |
| 6 | Asp124, Cys208, Asn220, Zn302 | Val73, Trp93, His122, Gln123, Glu152, His189, Lys211, Gly219, His250 |
| 7 | Ans220, Zn302 | Ile35, His189, Cys208, Lys211, Asp212, Ala215, Ser217, Gly219, His250, Ser251 |
| 8 | Asn220, Zn302 | Ile35, Val73, Trp93, His122, Gln123, Asp124, Glu152, His189, Cys208, Lys211, Gly219, His250 |
| 9 | Asn220, Zn302 | Ile35, Val73, Trp93, His122, Gln123, Asp124, Glu152, Gly153, Met154, His189, Cys208, Lys211, Gly219, His250 |
| 13 | Asn220, Zn302 | Ile35, Val73, Trp93, His122, Gln123, Asp124, His189, Cys208, Lys211, Gly219, His250 |
The inhibitors also have in common numerous hydrophobic interactions, notably with non-buried zinc-coordinating residues that form both sites (Asp124, Cys208, and His250 of Zn1 and His122 and His 189 of Zn2). Interactions within the deep pocket of the active site formed by Trp93, Gln123, Asp124, and His250 are also common to nearly all of the active compounds.[16] Interactions with Trp93 and Gly219, both shown to be important for activity and conserved in all class B1 metallo-β-lactamases, are also common.[17] Finally, every compound is docked with hydrogen bonding between the compound and Asn220 and hydrophobic interactions with Lys211, residues that are critical for substrate binding and catalysis.[18] It has been suggested that many NDM-1-inhibtor interactions alter the hydrogen bonding within the protein, reducing recognition of NDM-1 for β-lactams.[15] Our docking studies demonstrate the complementary nature of in silico molecular docking approaches with in vitro experiments to reveal structure-activity relationships. While the positioning of the phenolate, benzoxazole, and carboxylate within the active site is clearly favorable for both activity and docking, the lack of inhibitory activity for compounds 10 and 11, the docked structures of which display all three of these favorable interactions, suggests additional factors are also important.
Conclusion
Here we have reported that benzimidazole- and benzoxazole-containing zinc chelators have inhibitory activity against the clinically important metallo-β-lactamase NDM-1. The top compound, 9, had an IC50 of 0.38 μM against purified NDM-1 enzyme. Compound 9 as well as several other hits restored the susceptibility of two NDM-1-expressing E. coli strains to meropenem, a carbapenem antibiotic that is usually not effective against carbapenemase-producing bacterial strains. Spectroscopic evidence suggests that ternary complex formation is the primary mechanism used by 9 to inhibit NDM-1, rather than the metal removal mechanism displayed by many other NDM-1-inhibiting chelators. This conclusion is consistent with the structure-activity relationship observed for these compounds wherein non-zinc-coordinating functional group positioning influenced inhibitory activity, suggesting that metal affinity is not the only determinant of inhibition. This mechanism is more desirable in inhibitors than metal removal, which is often nonspecific. Docking studies revealed likely binding modes of compounds to the active site, supporting an inhibition mechanism involving both zinc coordination and interactions with residues within and near the active site. Computed mean binding energies from docking analysis correlated well with experimentally determined IC50 values. The low inherent antibacterial activity of the compounds, their efficacy in restoring meropenem activity, and their use of a ternary complex mechanism to inhibit NDM-1 make these compounds favorable for development into specific metallo-β-lactamase inhibitors.
Experimental Section
Materials.
Purified NDM-1 enzyme was prepared[19] and generously provided by Professor Bo Li and her research group (University of North Carolina – Chapel Hill). Protein purity and identity was confirmed using ESI-MS (Agilent 6460 Triple Quadrupole LC-MS with Waters Atlantis T3 3 μm 3.0×100 mm column). UV-Visible spectra were acquired with a Cary 50 UV-Vis spectrometer.
Synthesis
General.
Thin layer chromatography (TLC) was done using EMD 60F glass-backed silica gel plates. Fisher Scientific silica gel 60 (230–400 mesh) was used for flash chromatography. 1H NMR and 13C NMR spectra were collected using a JEOL ECX 400 MHz NMR spectrometer or a Bruker 500 MHz Avance III HD spectrometer. Chemical shifts are reported in ppm. High-resolution mass spectra were acquired with an Agilent G6224 LCMS-TOF using an electrospray ionization source and a Series 1200 HPLC. Samples were analyzed on a Phenomenex C18 column using water/MeCN/0.3% formic acid as the mobile phase, a 0–100% gradient, a flow rate of 0.5 mL/min, UV detection at 254 nm, and positive ion MS. Purity of compounds 1–13 was assessed by HPLC using integration of 254-nm UV absorbance chromatograms (Figure S7) and found to be >85% for 1, >90% for 3–7, and >95% for 2 and 8–13 by this method, which over-estimates the contribution of strongly-absorbing impurities that come from the cyclization reactions. By NMR, all compounds were >95% pure (Figures S8–S26).
Compounds 5–7[7b], 8, and 10[7a] were prepared by hydrolysis of the previously reported methyl esters:
General method for hydrolysis of methyl esters.
Methanol (1.0mL) and 5M NaOH (0.50 mL) were added to the methyl ester (0.03–0.035 mmol) and the mixture was heated to reflux for 18–22 h. The resulting mixture was cooled to rt and water (1.0 mL) and 6M HCl (1.0 mL) were added. After stirring to obtain a uniform suspension, the solid was collected by filtration, washing with water.
2-(1-hydroxynaphthalen-2-yl)benzoxazole-4-carboxylic acid (5).
1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 8.3 Hz, 1H), δ 8.11 (dd, J = 7.8 Hz, J = 0.9 Hz, 1H), 8.01–7.95 (m, 3H), 7.71–7.54 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 166.2, 164.9, 157.5, 149.9, 139.1, 136.4, 129.8, 128.4, 127.8, 127.0, 125.6, 124.6, 123.6, 122.5, 122.1, 120.1, 115.7, 103.4. HRMS calc. for C18H12NO4 [M+H]+ 306.0761; found 306.0768.
2-(3-hydroxynaphthalen-2-yl)benzoxazole-4-carboxylic acid (6).
1H NMR (400 MHz, DMSO-d6) δ 11.55 (bs, 1H), δ 8.76 (s, 1H), 8.13 (dd, J = 8.2 Hz, J = 0.9 Hz, 1H), 8.05 (d, J = 7.8 Hz, 1H), 7.99 (dd, J = 7.8 Hz, J = 0.9 Hz, 1H), 7.81 (d, J = 8.3 Hz, 1H), 7.61–7.57 (m, 1H), 7.56–7.52 (m, 1H), 7.49 (s, 1H), 7.40–7.36 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 166.1, 163.8, 154.3, 150.3, 139.4, 136.9, 129.9, 129.5, 127.8, 127.6, 126.8, 126.3, 124.8, 122.6, 115.9, 112.7, 111.7. HRMS calc. for C18H11NO4 [M+H]+ 306.0761; found 306.0769.
2-(2-hydroxynaphthalen-1-yl)benzoxazole-4-carboxylic acid (7).
1H NMR (400 MHz, DMSO-d6) δ 8.75 (d, J = 8.7 Hz, 1H), 8.19 (dd, J = 8.2 Hz, J = 0.9 Hz, 1H), 8.10 (d, J = 8.7 Hz, 1H), 8.00–7.95 (m, 2H), 7.68–7.64 (m, 1H), 7.59–7.55 (m, 1H), 7.47–7.44 (m, 1H), 7.35 (d, J = 8.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.2, 165.2, 160.7, 150.2, 138.4, 135.7, 131.2, 129.7, 129.2, 128.5, 127.7, 125.6, 124.5, 124.4, 121.9, 119.6, 116.0, 103.0. HRMS calc. for C18H11NO4 [M+H]+ 306.0761; found 306.0767.
2-(7-benzyloxy-1-hydroxynaphthalen-2-yl)benzoxazole-4-carboxylic acid (8).
1H NMR (400 MHz, DMSO-d6) δ 8.11 (d, J = 8.2 Hz, 1H), δ 7.99 (d, J = 7.8 Hz, 1H), 7.90 (d, J = 9.2 Hz, 1H), 7.85 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 2.3 Hz, 1H), 7.57–7.51 (m, 4H), 7.42–7.38 (m, 3H), 7.34–7.30 (m, 1H). 13C NMR (125 MHz, DMSO-d6) δ 166.6, 164.9, 157.7, 149.9, 139.2, 137.3, 131.8, 130.2, 129.0, 128.8, 128.4, 128.3, 127.6, 126.0, 125.4, 123.3, 122.0, 120.4, 119.4, 115.1, 103.7, 69.9 HRMS calc. for C25H18NO5 [M+H]+ 412.1180; found 412.1187.
2-(5-benzyloxy-1-hydroxynaphthalen-2-yl)benzoxazole-4-carboxylic acid (10).
1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 7.3 Hz, 1H), δ 8.00–7.94 (m, 3H), 7.84 (d, J = 8.7 Hz, 1H), 7.57–7.52 (m, 4H), 7.44–7.40 (m, 2H), 7.36–7.33 (m, 1H), 7.28 (d, J = 7.8 Hz, 1H), 5.32 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 166.1, 164.8, 157.2, 154.3, 150.0, 139.1, 137.4, 129.1, 128.5, 128.2, 128.1, 127.8, 127.4, 125.8, 125.7, 122.0, 121.9, 115.9, 115.7, 114.0, 109.9, 104.0, 70.3. HRMS calc. for C25H18NO5 [M+H]+ 412.1180; found 412.1188.
Ethyl 1,2,3,4-tetrahydro-2-bromo-6-benzyloxy-1-oxo-2-naphthalenecarboxylate (17).[20]
To compound 16 (0.9807 g, 3.02 mmol) was added acetic acid (6.0 mL) and sodium acetate (0.25 g, 3.0 mmol). The mixture was cooled over ice and pyridinium tribromide (0.967 g, 3.02 mmol) was added in small portions. The mixture was allowed to come to rt with stirring for an additional 22 h, then transferred to a separatory funnel with CH2Cl2 (100 mL), and washed with water (50 mL), saturated NaHCO3 (2 × 50mL), water (50 mL), and brine (50 mL). The organic phase was dried with MgSO4 and the solvent was removed in vacuo to give crude 17 as a dark brown oil (1.14 g, 93%). δ 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.8 Hz, 1H), 7.43–7.32 (m, 4H), 7.35–7.27 (m, 1H), 6.90 (dd, J = 8.8, 2.5 Hz, 1H), 6.78 (d, J = 2.6 Hz, 1H), 5.08 (s, 2H), 4.26 (q, J = 7.2 Hz, 2H), 3.15 (ddd, J = 17.8, 9.7, 4.5 Hz, 1H), 2.99–2.85 (m, 2H), 2.54–2.44 (m, 1H), 1.26 (t, J = 7.2 Hz, 3H).
Ethyl 6-benzyloxy-1-hydroxy-2-naphthalenecarboxylate (18).
Diazabicycloundecane (0.85 mL, 5.6mmol) in 2.0 mL of THF was added dropwise over 20 min to a solution of crude 17 (1.13 g, 2.81 mmol) in 9 mL of THF. The reaction mixture was stirred for 22 h at rt under inert atmosphere, poured into 25 g of ice water, and acidified by adding 20 mL of 6M HCl. The resulting solid was collected by filtration, washed with water, and crystallized from methanol/water, giving 18 as brown crystals (0.5136 g, 57%). 1H NMR (400 MHz, CDCl3) δ 12.04 (s, 1H), 8.33 (d, J = 9.1 Hz, 1H), 7.75 (d, J = 8.8 Hz, 1H), 7.52–7.46 (m, 2H), 7.46–7.31 (m, 3H), 7.23 (dd, J = 9.1, 2.4 Hz, 1H), 7.15 (dd, J = 5.5, 3.2 Hz, 2H), 5.20 (s, 2H), 4.44 (q, J = 7.0 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H).
6-Benzyloxy-1-hydroxy-2-naphthalenecarboxylic acid (14).
4M aqueous KOH (1.0mL) was added to a solution of 18 (0.4744 g, 1.47 mmol in 4 mL of ethanol). The resulting solution was heated to reflux for 20 h, allowed to cool, and acidified with 50 mL of 2M HCl. The resulting solution was extracted with ethyl acetate (3 × 50 mL) and the combined organic phases were washed with brine (2 × 50mL), dried over MgSO4, and filtered. Removal of the solvent provided 14 as a dark brown solid (0.3012 g, 70%). 1H NMR (400 MHz, DMSO-d6) δ 8.19 (d, J = 9.2 Hz, 1H), 7.70 (d, J = 8.7 Hz, 1H), 7.55–7.48 (m, 2H), 7.46–7.39 (m, 3H), 7.39–7.31 (m, 1H), 7.30–7.19 (m, 2H), 5.26 (s, 2H).
Methyl 2-((6-benzyloxy-1-hydroxynaphthalene-2-carboxyl)amino)-3-hydroxybenzoate (15).
To a solution of carbonyldiimidazole (0.20 g, 1.2 mmol in 6 mL of THF) was added 14 (0.30 g, 1.0 mmol) and the resulting solution was stirred at rt under nitrogen for 15 minutes. Methyl 3-hydroxyanthranilate (0.20 g, 1.2 mmol) was then added, the reaction mixture was stirred for 10 min and then heated to reflux for 24 h. The resulting mixture was cooled, diluted with water (50 mL) and extracted with ethyl acetate (3 × 50 mL). The combined organic phases were washed with water (50 mL) and brine (50 mL), dried over MgSO4, filtered and the solvent removed to provide crude 15 (0.46 g), which was used in the next step without further purification.
Methyl 2-(6-benzyloxy-1-hydroxynaphthalen-2-yl)benzoxazole-4-carboxylate (16).
Toluene (11 mL) was added to a flask containing crude 15 (0.223 g, 0.503 mmol) and pyridinium p-toluenesulfonate (0.253 g, 1.01 mmol) and the flask was fitted with a Dean Stark trap and refluxed under nitrogen for 20 h. After cooling, the mixture was transferred to a separatory funnel with 40 mL of CH2Cl2 and 40 mL of saturated NaHCO3 and separated. The aqueous phase was extracted with additional CH2Cl2 (2 × 40mL) and the combined organic phases were washed with water (75 mL) and brine (75 mL), dried over MgSO4, and filtered. After removal of the solvent, the dark brown solid obtained was purified by flash chromatography[21] using 100% CH2Cl2 as the eluent. Fractions containing a spot with Rf = 0.39 were combined and the solvent was removed to provide 16 as a light brown solid (55.9 mg, 26%, two steps). 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 9.2 Hz, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 8.3 Hz, 1H), δ 7.51–7.20 (m, 9H), 5.22 (s, 2H), 4.09 (s, 3H).
2-(6-benzyloxy-1-hydroxynaphthalen-2-yl)benzoxazole-4-carboxylic acid (9).
To solution of 16 (30.0 mg, 0.0705 mmol in 0.44 mL of THF) was added 5M NaOH (0.24 mL) and the resulting mixture was refluxed for 27 h. The reaction was cooled to rt and the heterogeneous mixture was acidified with 5 mL of 1M HCl in a separatory funnel with 10 mL of ethyl acetate; the phases were separated, and the aqueous phase was extracted with additional ethyl acetate (2 × 10 mL). The combined organic phases were washed with brine (25 mL), dried over MgSO4, and the solvent removed to provide 9 as a yellow solid (208 mg, 72%). 1H NMR (400 MHz, DMSO-d6) δ 8.28 (d, J = 9.2 Hz, 1H), 8.09 (d, J = 8.3 Hz, 1H), 7.98–7.93 (m, 2H), 7.54–7.30 (m, 9H), 5.26 (s, 2H). 13C NMR (125 MHz, DMSO-d6) δ 166.2, 165.1, 159.5, 157.6, 149.8, 139.3, 138.4, 137.1, 129.0, 128.5, 128.4, 127.7, 125.6, 125.4, 123.3, 121.6, 119.4, 119.3, 119.1, 115.7, 108.6, 101.7, 70.0. HRMS calc. for C25H18NO5 [M+H]+ 412.1180; found 412.1187.
Enzyme inhibition assays.
Compounds were serially diluted in 96-well plates in HEPES buffer (50 mM, pH 7.2) and incubated with NDM-1 (1 nM) for ten minutes at rt. Nitrocefin (20 μM) was added and a PerkinElmer Victor3 V multilabel plate reader was used to monitor the absorbance at 492 nm over 30 min at rt. The slope of the absorbance increase was normalized to the maximum slope of all concentrations tested of each compound. This activity was plotted against log(concentration) of the compounds and GraphPad Prism software was used to calculate IC50 values and confidence intervals.
Microbiology.
Preparation of E. coli strains expressing β-lactamases was described previously.[22] Prior to all experiments, E. coli carrying the appropriate plasmids was streaked onto LB agar containing 100 μg/mL ampicillin and 50 μg/mL kanamycin. A single colony was used to inoculate 2 mL of Mueller-Hinton broth containing 100 μg/mL ampicillin and 50 μg/mL kanamycin, which was then incubated at 37 °C, 200 rpm, for 16–18 h. This overnight culture was diluted 1:500 in fresh broth and used as the working culture. Meropenem was serially diluted 2-fold in Mueller-Hinton broth to final concentrations ranging from 0 to 64 μg/mL in clear, flat-bottomed 96-well plates. The working culture, spiked with inhibitors at concentrations of 25–200 uM or no additional treatment was then added to the 96-well plate, giving a final inoculum dilution of 1:1000 (5 × 105 to 1 × 106 CFU/mL) and final volume of 100 μL per well. Wells containing media only were included to verify sterility. Plates were incubated for 20 h at 37 °C, 200 rpm. Plates were covered with AeraSeal film (Sigma-Aldrich) during incubation to minimize evaporation. Bacterial growth was evaluated by measuring the optical density at 600 nm (OD600) using a PerkinElmer Victor3 V multilabel plate reader at 0 and 20 h. The 0-h time point data were subtracted from the 20-h data to remove background signal from the media and absorbance of compounds. The minimum inhibitory concentration (MIC) was defined as the concentration at which no detectable growth occurred after 20 h of incubation (background subtracted, normalized OD600 < 0.010). Three or four biological replicates were performed for each MIC determination. If a different MIC was determined for different replicates, the most common value was used. Rarely, if MICs varied by more than 2-fold between replicates or a single most common value was not observed, the MIC was represented as a range of values.
Docking
Ligand and receptor preparation.
Molecular structures of each compound were drawn, 3D optimized in ChemSketch, and saved as PDB files. These structures were adjusted for the correct protonation state at pH 7.4 and geometry optimized (MMFF94) in Avogadro (v 1.1[23]). Ligand PDB files were then converted to AutoDock PDBQT format using the prepare_ligand4.py script of AutoDockTools (ADT, v1.5.7rc1[24]). The crystal structure of NDM-1 in complex with hydrolyzed ampicillin at pH 7.3 (PDB 5ZGR[9]) was imported into PyMol (v2.5[25]), where all Chain B atoms and non-catalytic water molecules were removed. The hydrolyzed ampicillin was extracted as a validation ligand. The prepare_receptor4.py script of ADT with the deleteAltB option was used to remove lower occupancy residue positions from the 5ZGR X-ray crystal structure.
Docking methods.
Five different docking methods were examined: i) Autodock Vina[26]; ii) Autodock (AD, v4.2.6[27]) with no modification to Gasteiger charges; iii) AD with zinc atoms set at +2 eV; iv) AD with charge distribution to ligating zinc atoms (+0.2 eV charge added to atoms: HIS120 Nε, HIS122 Nδ, ASP124 O, HIS189 Nε, Cys208 S, and His250 Nε), with Zn charge set to +1.6 eV to account for zinc depolarization, and v) Autodock4Zn[14] (AD4Zn). Dockings were performed in ADT with a grid box (90 × 90 × 60) centered above and between the two active site zinc (−5, −20, 27) with spacing set to 0.375 Å. Autodock4Zn docking used the same PDBQT input files and gridbox with the addition of the Tz pseudo atoms and updated Autodock4Zn potentials. New grids were recalculated for each method, ligand starting coordinates were randomly set and docked with the Lamarckian genetic algorithm[28] pseudo-Solis & Wets local search, using default settings from ADT except for the number of runs (ga_run) was set to 50 for AD and AD4Zn and the exhaustiveness was set to 64 for Vina. Cluster analysis was performed to group docked solutions within 2 Å RMSD, and the mean cluster energy and the binding pose with the lowest energy solution in the lowest energy cluster were used for the accepted solution.[29] The lowest energy binding pose of the docked solutions were visualized and figures generated in PyMol and LigPlot+ (v2.2[30]).
Supplementary Material
Acknowledgements
This project was supported in part with funds from Duke University and the National Institutes of Health (GM084176) to K.J.F and through an Undergraduate Research Award to A.B-A. and D.P. from the UMBC Division of Undergraduate Academic Affairs. A.C.J. acknowledges an NSF Graduate Research Fellowship (DGE 1644868). We thank Dr. Peter Silinski (Duke University Department of Chemistry Shared Instrument Facility) for help with obtaining LC-MS data, Prof. Bo Li and Dr. Andrew N. Chan (UNC - Chapel Hill) for generously providing purified NDM-1 enzyme, Bradford Becken III (Duke University) for transforming E. coli strains, and Vignesh Dhanasekaran for writing scripts to expedite data analysis.
Abbreviations
- AD
Autodock
- EDTA
ethylenediaminetetraacetic acid
- IC50
half maximal inhibitory concentration
- MIC
minimum inhibitory concentration
- MBE
mean binding energy
- THF
tetrahydrofuran
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- [1].a) Fisher JF, Meroueh SO, Mobashery S, Chem. Rev 2005, 105(2), 395–424; [DOI] [PubMed] [Google Scholar]; b) Livermore DM, Clin. Microbiol. Rev 1995, 8(4), 557–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. NDM-1 is an abbreviation for New Delhi metallo β-lactamase 1. This name associates bacterial disease with a specific geographical region and therefore a specific group of people who live there. Naming of diseases and disease-associated proteins or other agents in this way has the potential to lead to discrimination and xenophobia. Since efforts to rename NDM-1 have not been successful (see ref. 3), we recommend use of the abbreviation “NDM-1” only and avoidance of use of the full name of the enzyme.
- [3].a) Singh AR, Mens sana monographs 2011, 9(1), 294–319; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mohapatra PR, Indian J Med. Res 2013, 137(1), 213–215. [PMC free article] [PubMed] [Google Scholar]
- [4].a) Bush K, Bradford PA, Nat. Rev. Microbiol 2019, 17(5), 295–306; [DOI] [PubMed] [Google Scholar]; b) Palzkill T, Ann. N. Y. Acad. Sci 2013, 1277(1), 91–104; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nordmann P, Poirel L, Walsh TR, Livermore DM, Trends Microbiol. 2011, 19(12), 588–595; [DOI] [PubMed] [Google Scholar]; d) Crowder MW, Spencer J, Vila AJ, Acc. Chem. Res 2006, 39(10), 721–728. [DOI] [PubMed] [Google Scholar]
- [5].a) Yan YH, Li G, Li GB, Med. Res. Rev 2020; [DOI] [PubMed] [Google Scholar]; b) Ju L-C, Cheng Z, Fast W, Bonomo RA, Crowder MW, Trends Pharmacol. Sci 2018, 39(7), 635–647; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rotondo CM, Wright GD, Curr. Opin. Microbiol 2017, 39, 96–105; [DOI] [PubMed] [Google Scholar]; d) Fast W, Sutton LD, Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2013, 1834(8), 1648–1659; [DOI] [PubMed] [Google Scholar]; e) Linciano P, Cendron L, Gianquinto E, Spyrakis F, Tondi D, ACS Infect. Dis 2019, 5(1), 9–34. [DOI] [PubMed] [Google Scholar]
- [6].a) Wang BB, Maghami N, Goodlin VL, Smith PJ, Bioorg. Med. Chem. Lett 2004, 14(12), 3221–3226; [DOI] [PubMed] [Google Scholar]; b) Reynolds MB, DeLuca MR, Kerwin SM, Bioorg. Chem 1999, 27(4), 326–337; [Google Scholar]; c) Kumar D, Jacob MR, Reynolds MB, Kerwin SM, Biorg. Med. Chem 2002, 10(12), 3997–4004; [DOI] [PubMed] [Google Scholar]; d) Huang S-T, Hsei IJ, Chen C, Biorg. Med. Chem 2006, 14(17), 6106–6119. [DOI] [PubMed] [Google Scholar]
- [7].a) Ward DN, Talley DC, Tavag M, Menji S, Schaughency P, Baier A, Smith PJ, Bioorg. Med. Chem. Lett 2014, 24(2), 609–612; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Talley DC, Delang L, Neyts J, Leyssen P, Smith PJ, Bioorg. Med. Chem. Lett 2016, 26(4), 1196–1199. [DOI] [PubMed] [Google Scholar]
- [8].O’Callaghan CH, Morris A, Kirby SM, Shingler AH, Antimicrob. Agents Chemother 1972, 1(4), 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang H, Ma G, Zhu Y, Zeng L, Ahmad A, Wang C, Pang B, Fang H, Zhao L, Hao Q, Antimicrob. Agents Chemother 2018, 62(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Wang X, Yang Y, Gao Y, Niu X, Int. J. Mol. Sci 2020, 21(10), 3567; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Khan AU, Ali A, Srivastava G, Sharma A, Sci. Rep 2017, 7(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yuan Q, He L, Ke H, Antimicrob. Agents Chemother 2012, 56(10), 5157–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Thakur PK, Kumar J, Ray D, Anjum F, Hassan MI, J. Nat. Sci. Biol. Med 2013, 4(1), 51; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chetia H, Sharma DK, Sarma R, Verma A, Int. J. Pharm. Pharm. Sci 2014, 6(4), 299–303. [Google Scholar]
- [13].a) Stote RH, Karplus M, Proteins: Structure, Function, and Bioinformatics 1995, 23(1), 12–31; [DOI] [PubMed] [Google Scholar]; b) Hu X, Shelver WH, J. Mol. Graph. Model 2003, 22(2), 115–126. [DOI] [PubMed] [Google Scholar]
- [14].Santos-Martins D, Forli S, Ramos M. J. o., Olson AJ, J. Chem. Inf. Model 2014, 54(8), 2371–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Duan H, Liu X, Zhuo W, Meng J, Gu J, Sun X, Zuo K, Luo Q, Luo Y, Tang D, Mol. Simul 2019, 45(9), 694–705. [Google Scholar]
- [16].Guo Y, Wang J, Niu G, Shui W, Sun Y, Zhou H, Zhang Y, Yang C, Lou Z, Rao Z, Protein & cell 2011, 2(5), 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Ali A, Kumar R, Iquebal MA, Jaiswal S, Kumar D, Khan AU, Phys. Chem. Chem. Phys 2019, 21(32), 17821–17835; [DOI] [PubMed] [Google Scholar]; b) Zhang g., Hao Q, The FASEB Journal 2011, 25(8), 2574–2582; [DOI] [PubMed] [Google Scholar]; c) Khan AU, Rehman MT, Antimicrob. Agents Chemother 2016, 60(1), 356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Linciano P, Cendron L, Gianquinto E, Spyrakis F, Tondi D, ACS Infect. Dis 2018, 5(1), 9–34. [DOI] [PubMed] [Google Scholar]
- [19].Chan AN, Shiver AL, Wever WJ, Razvi SZA, Traxler MF, Li B, Proc. Natl. Acad. Sci. USA 2017, 114(10), 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rao AVR, Yadav JS, Reddy KK, Upender V, Tetrahedron Lett. 1991, 32(38), 5199–5202. [Google Scholar]
- [21].Still WC, Kahn M, Mitra A, J. Org. Chem 1978, 43(14), 2923–2925. [Google Scholar]
- [22].Zaengle-Barone JM, Jackson AC, Besse DM, Becken B, Arshad M, Seed PC, Franz KJ, ACS Infect. Dis 2018, 4(6), 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR, J. Cheminform 2012, 4(1), 17; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Avogadro: an open-source molecular builder and visualization tool. Version 1.1 http://avogadro.cc/. [Google Scholar]
- [24].Dallakyan S, MGLTools 1.5.7 RC 1, Molecular Graphics Laboratory, The Scripps Research Institute., 2013. [Google Scholar]
- [25].Schrodinger, LLC, The PyMOL Molecular Graphics System, Version 2.5, 2015.
- [26].Trott O, Olson AJ, J. Comput. Chem 2010, 31(2), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, J. Comput. Chem 2009, 30(16), 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ, J. Comput. Chem 1998, 19(14), 1639–1662. [Google Scholar]
- [29].Huey R, Morris GM, Olson AJ, Goodsell DS, J. Comput. Chem 2007, 28(6), 1145–1152. [DOI] [PubMed] [Google Scholar]
- [30].Laskowski RA, Swindells MB, J. Chem. Inf. Model 2011, 51(10), 2778–2786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.