

Commercial recombinant SAA variants contaminated with bacterial products do not reflect the inherent biological characteristics of endogenous SAA. In reliable experimental settings (e.g. using pure SAA and SAA knockout mice), a role for SAA in innate immunity has been evidenced, that is recruitment of leucocytes, regulation of T‐cell responses, mono(cyto)poiesis and antimicrobial activity. In leucocyte mobilization, the signalling of SAA via the FPR2 receptor has been confirmed, but the implication of other receptors (CD36, RAGE, SR‐BI/II and P2RX7) potentially involved in SAA activities requires further validation.
![]()
Keywords: chemotaxis, immunity, inflammation, knockout, serum amyloid A
Summary
Serum amyloid A (SAA) is an acute‐phase protein (APP) to which multiple immunological functions have been attributed. Regardless, the true biological role of SAA remains poorly understood. SAA is remarkably conserved in mammalian evolution, thereby suggesting an important biological function. Since its discovery in the 1970s, the majority of researchers have investigated SAA using recombinant forms made available through bacterial expression. Nevertheless, recent studies indicate that these recombinant forms of SAA are unreliable. Indeed, commercial SAA variants have been shown to be contaminated with bacterial products including lipopolysaccharides and lipoproteins. As such, biological activities and receptor usage (TLR2, TLR4) revealed through the use of commercial SAA variants may not reflect the inherent nature of this APP. Within this review, we discuss the biological effects of SAA that have been demonstrated through more solid experimental approaches. SAA takes part in the innate immune response via the recruitment of leucocytes and executes, through pathogen recognition, antimicrobial activity. Knockout animal models implicate SAA in a range of functions, such as regulation of T‐cell‐mediated responses and monopoiesis. Moreover, through its structural motifs, not only does SAA function as an extracellular matrix protein, but it also binds extracellular matrix proteins. Finally, we here also provide an overview of definite SAA receptor‐mediated functions and highlight those that are yet to be validated. The role of FPR2 in SAA‐mediated leucocyte recruitment has been confirmed; nevertheless, SAA has been linked to a range of other receptors including CD36, SR‐BI/II, RAGE and P2RX7.
Abbreviations
- AAA
Abdominal aortic aneurysm
- AngII
Angiotensin II
- APP
Acute‐phase protein
- APR
Acute‐phase response
- A‐SAA
Acute‐phase serum amyloid A
- CAF
Cancer‐associated fibroblast
- CLP
Caecal ligation and puncture sepsis model
- ConA
Concanavalin
- CRP
C‐reactive protein
- C‐SAA
Constitutive serum amyloid A
- DCs
Dendritic cells
- DSS
Dextran sodium sulphate
- EAE
Experimental autoimmune encephalomyelitis
- ECM
Extracellular matrix
- erhSAA
Eukaryotic‐source recombinant human serum amyloid A
- FPR
Formyl peptide receptor
- GAG
Glycosaminoglycan
- HCV
Hepatitis C virus
- HDL
High‐density lipoprotein
- HFHSC
High‐fat, high‐sucrose diet with 0.15% added cholesterol
- HMGB1
High‐mobility group box 1
- HO‐1
Haem oxygenase‐1
- IDP
Intrinsically disordered protein
- IL
Interleukin
- KO
Knockout
- LPL
Lipoprotein lipase
- LPS
Lipopolysaccharide
- LT
Lethal toxin
- MOG
Myelin oligodendrocyte glycoprotein
- NI
Not indicated
- OmpA
Outer membrane protein A
- P2RX7
P2X purinoceptor 7
- PBMCs
Peripheral blood mononuclear cells
- PDAC
Pancreatic ductal adenocarcinoma
- PTH
Parathyroid hormone
- PTX
Pentraxin
- RAGE
Receptor for advanced glycation end products
- RBP
Retinol‐binding protein
- rhSAA
Recombinant human serum amyloid A
- rmSAA
Recombinant murine serum amyloid A
- ROS
Reactive oxygen species
- rSAA
Recombinant serum amyloid A
- SAA
Serum amyloid A
- SFB
Segmented filamentous bacteria
- sPRR
Soluble pathogen recognition receptor
- SR‐BI
Scavenger receptor class B type I
- TLR
Toll‐like receptor
- TNF
Tumour necrosis factor
- WTD
Western‐type diet
INTRODUCTION
Acute and chronic inflammatory conditions such as infection, trauma, tissue infarction and inflammatory disorders are accompanied by a systemic phenomenon, collectively termed the acute‐phase response (APR). The APR is characterized by changes in the concentration of liver‐derived plasma proteins, referred to as acute‐phase proteins (APPs). APPs constitute a range of functionally diverse proteins such as complement system proteins, coagulation and fibrinolytic proteins, antiproteases and transport proteins to name a few. Of particular interest is serum amyloid A (SAA), which is characterized as a positive APP; that is, its serum level is elevated by at least 25% during the APR. Owing to its momentous upregulation during the APR, aside from C‐reactive protein (CRP), SAA is perhaps the most prominent APP. 1 Indeed, during the APR 2.5% of protein synthesis in the liver is diverted towards the production of SAA. 2 The APR is largely regulated by members of the interleukin‐6 (IL‐6) family and IL‐1‐like cytokines, though other cytokines might also influence the process, such as interferon (IFN)‐γ and transforming growth factor‐β. 1 In line with this, hepatic expression of SAA is mainly upregulated in response to the inflammatory cytokines IL‐1β, IL‐6 and tumour necrosis factor (TNF)‐α. 3 In the present paper, we highlight and discuss current dilemmas pertaining to the study of SAA. Recent reports have highlighted the unreliability of recombinantly expressed SAA forms, which have been utilized in the majority of studies to investigate the biological function of SAA. 4 , 5 , 6 As such, we give a critical overview of the biological activities of SAA that are thought to be sound and have been demonstrated through the use of more reliable experimental approaches.
THE DISCOVERY OF SAA AND THE DEVELOPMENT OF ITS RECOMBINANT COUNTERPART
Serum amyloid A, first discovered in the 1970s, 7 was initially thought to be comprised of one member, the precursor protein of amyloid A plaques, which drive the pathogenesis of secondary amyloid A amyloidosis during chronic inflammation. 8 Nevertheless, the amyloid A precursor was later found to belong to a four‐membered family of proteins. 3 SAA is highly conserved throughout evolution within a wide range of species signifying an important biological role. 9 With the rise of recombinant protein expression, research into the biological function of SAA saw a peak in the late 1990s that has carried on to current times. Initially, a hybrid form of two human SAA isoforms (SAA1.1 and SAA2.2), which does not occur in nature, was developed (rhSAA, Table 1) containing a residual amino‐terminal methionine following bacterial expression. In 2009, a recombinant form of human SAA1 was developed also containing a residual amino‐terminal methionine (rhSAA1.1). Using these recombinant SAA (rSAA) forms of bacterial (namely Escherichia coli) origin, researchers attributed multiple functions to SAA (reviewed in Ref. 3). Human SAA1 of eukaryotic origin (erhSAA1.1), containing a myc‐tag at its carboxy‐terminus, has been recently made available; however, it has been scarcely utilized during the characterization of SAA.
TABLE 1.
Amino acid sequence of commercially available recombinant SAA preparations versus naturally occurring endogenous SAAs
| Amino acid sequence | |
|---|---|
| Recombinant human SAA1 (rhSAA1.1; PeproTech a ) | M b RS FFSFL GEAFD GARDM WRAYS DMREA NYIGS DKYFH ARGNY DAAKR GPGGV WAAEA ISDAR ENIQR FFGHG AEDSL ADQAA NEWGR SGKDP NHFRP AGLPE KY |
| Recombinant human SAA c (rhSAA; PeproTech) | M b RS FFSFL GEAFD GARDM WRAYS DMREA NYIGS DKYFH ARGNY DAAKR GPGGV WAAEA ISNAR ENIQR FFGRG AEDSL ADQAA NEWGR SGKDP NHFRP AGLPE KY |
| Recombinant human SAA1 d (erhSAA1.1; OriGene e ) | RS FFSFL GEAFD GARDM WRAYS DMREA NYIGS DKYFH ARGNY DAAKR GPGGV WAAEA ISDAR ENIQR FFGHG AEDSL ADQAA NEWGR SGKDP NHFRP AGLPE KYEQK LISEE DL |
| Natural human SAA1.1 | RS FFSFL GEAFD GARDM WRAYS DMREA NYIGS DKYFH ARGNY DAAKR GPGGV WAAEA ISDAR ENIQR FFGHG AEDSL ADQAA NEWGR SGKDP NHFRP AGLPE KY |
| Natural human SAA2.2 | RS FFSFL GEAFD GARDM WRAYS DMREA NYIGS DKYFH ARGNY DAAKR GPGGA WAAEV ISNAR ENIQR LTGRG AEDSL ADQAA NKWGR SGRDP NHFRP AGLPE KY |
| Natural murine SAA1.1 | G FFSFV HEAFQ GAGDM WRAYT DMKEA NWKNS DKYFH ARGNY DAAQR GPGGV WAAEK ISDGR EAFQE FFGRG HEDTI ADQEA NRHGR SGKDP NYYRP PGLPD KY |
| Natural murine SAA2.2 | G FFSFI GEAFQ GAGDM WRAYT DMKEA GWKDG DKYFH ARGNY DAAQR GPGGV WAAEK ISDAR ESFQE FFGRG HEDTM ADQEA NRHGR SGKDP NYYRP PGLPA KY |
| Natural murine SAA3 | R WVQFM KEAGQ GSRDM WRAYS DMKKA NWKNS DKYFH ARGNY DAARR GPGGA WAAKV ISDAR EAVQK FTGHG AEDSR ADQFA NEWGR SGKDP NHFRP AGLPK RY |
Rocky Hill, NJ, USA.
Residual methionine following recombinant synthesis in Escherichia coli.
Recombinant hybrid form of human SAA1.1 and SAA2.2 in which position 61 is occupied by asparagine derived from SAA2.2 and position 72 is occupied by arginine derived from SAA2.2 replacing the aspartic acid and histidine residues in SAA1.1, respectively.
Recombinant human SAA1.1 expressed in human embryonic kidney cells containing a myc‐tag at the carboxy‐terminus.
Rockville, MD, USA.
Modifications compared to the primary amino acid sequence of natural SAA occurring in recombinantly expressed forms are indicated in bold.
THE CURRENT PREDICAMENT IN THE STUDY OF SAA
Several claims regarding the biological effects of SAA have been strongly contested, particularly those revealed through the use of rSAA. Indeed, an increasing number of studies have reported a discrepancy between rSAA of bacterial origin, rSAA of eukaryotic origin and endogenous SAA that has been purified from inflammatory plasma. Björkman et al. and Christenson et al. 10 , 11 observed a lack of inflammatory capacity with endogenous SAA in comparison with rSAA of a bacterial origin. Endogenous SAA lacked the capacity to activate neutrophils, demonstrated through the absence of CXCL8 production and L‐selectin shedding. More recently, Burgess et al. compared rSAA of bacterial and eukaryotic origins and observed that rSAA from eukaryotic origin lacks certain biological activities observed with rSAA of bacterial origin, such as induction of cytokine and chemokine expression (IL‐1β, IL‐6, TNF‐α and CXCL8) in peripheral blood mononuclear cells (PBMCs) and Th17 polarization of CD4+ T cells mediated through the activation of dendritic cells (DCs). Furthermore, the composition of bacterial rhSAA1.1 of commercial origin was analysed and was shown to consist of an additional 91 bacterial proteins of which 15 were predicted to be lipoproteins. Indeed, treatment of rhSAA1.1 with lipoprotein lipase (LPL) downregulated the cytokine‐inductive capacity. Taking into account the activation of Toll‐like receptor 2 (TLR2) by lipoproteins, the authors concluded that the TLR2‐mediated effects of rSAA are due to contamination with bacterial lipoproteins rather than being an inherent effect of SAA. 4 Furthermore, TLR4 activation by recombinantly expressed SAA has been linked to lipopolysaccharide (LPS) contamination 6 (SAA receptor utilization is discussed in the Section "Receptors that recognize SAA").
Another potential issue introduced through the use of rSAA variants is the presence of additional amino acids as a consequence of recombinant expression, whether that is an amino‐terminal methionine or a carboxyl‐terminal expression tag (Table 1). The presence of a single additional amino acid may alter the structural conformation of a protein. Chaudhuri et al. 12 demonstrated a loss of stability and altered electric properties due to the presence of an amino‐terminal methionine in α‐lactalbumin following recombinant synthesis in E. coli. Reversed‐phase purified, homogenous rhSAA1.1 and chemically synthesized carboxyl‐terminal SAA peptides share neutrophil‐activating properties, 5 , 13 , 14 but one cannot rule out the tampering of an extra amino‐terminal methionine, and certainly not a 10 amino acid carboxyl‐terminal tag, with the stability and folding of rSAA variants. As such, biological functions that have been reported through the use of rSAA are of limited validity and need to be confirmed through an alternative approach.
SAA GENE AND PROTEIN STRUCTURE
Gene structure
The SAA gene cluster is located on the short arm of chromosome 11, within a 150 Kb area, in the p15.1 region. Humans have four different genes coding for SAA, giving rise to SAA1, SAA2, SAA3 and SAA4. SAA1 and SAA2 possess several polymorphic alleles: SAA1.1, SAA1.2, SAA1.3, SAA1.4, SAA1.5 and SAA2.1 and SAA2.2, respectively. 3 SAA proteins are classified based on their induced expression and fall within two categories: acute‐phase SAAs (A‐SAAs; SAA1 and SAA2) and constitutive SAA (C‐SAA; SAA4). A‐SAAs contain NF‐IL‐6 and NF‐kB binding sites in their promoter regions, thus allowing their induction by IL‐1, IL‐6 and TNF‐α during the APR. On the other hand, the C‐SAA promoter region lacks the NF‐IL‐6 binding site and contains a truncated NF‐kB binding region. 15 Owing to the presence of a premature stop codon, human SAA3 was assumed to be a pseudogene. However, nearly three decades following the discovery of SAA, transcription of the human SAA3 gene was detected by RT‐PCR in cultures of mammary epithelial cells treated with LPS or prolactin. 16 Later on, a human SAA2‐SAA3 fusion transcript was detected in mammary gland epithelial cells following stimulation with IL‐1β, IL‐6 and dexamethasone. 17 Conversely, SAA3 is a fully functional gene in other mammals, including mice, where its expression is upregulated in response to inflammatory cytokines. 3 , 18
Protein structure
Mature A‐SAA and SAA4 proteins are 104 and 112 amino acids long, respectively. 3 Based on the transcriptional expression of SAA3 in mammary gland epithelial cells, the SAA3 gene is predicted to give rise to a protein that would be 42 amino acids long in its mature form. 16 Early studies by McCubbin et al. 19 have proposed the secondary structure of murine SAA to be largely helical in nature. However, it has been only recently confirmed by Lu et al. who revealed, through the use of multiwavelength anomalous dispersion, that human SAA1.1 is composed of a four up‐down/up‐down α‐helix bundle, corresponding to the amino acid residues 1–27, 32–47, 50–69 and 73–88, arranged into a cone shape. The C‐terminal tail, spanning amino acids 89–104, provides structural stability through the formation of multiple salt bridges and hydrogen bonds with residues in three of the four α‐helices. Furthermore, Lu et al. demonstrated the propensity of this four‐helix bundle to form oligomers in solution, most notably a hexamer composed of a dimer of trimers. Nevertheless, human SAA1.1 was found to also exist as a monomer and dodecamer in solution. 20 In line with this, the capacity of murine SAA2.2 to exist in several oligomeric states, namely as an octamer and a more stable hexamer, has been demonstrated. 21 , 22 However, it is currently unclear whether SAA carries out its biological functions as a single monomer or rather through a higher oligomeric state. One proposed hypothesis is that SAA is capable of inducing several diverse effects due to its range of oligomeric forms, which may activate distinct signalling pathways. To further complicate the matter, SAA has been described as an intrinsically disordered protein (IDP). The C‐terminal tail of murine SAA2.2 was found to lack any stable secondary structure, which was revealed through the use of PROF, a profile‐based neural network algorithm. 21 Furthermore, whereas murine SAA2.2 forms stable hexamers at 20°C, once exposed to near‐physiological conditions (37°C) this murine variant of SAA dissociates into its monomeric form and displays a significant loss of its α‐helical structure. 23 Similar findings have been observed with human SAA1.1. 24 As such, more research needs to be undertaken to identify the structural organization of SAA in vivo and its influence on the biological activity of SAA.
BIOLOGICAL ACTIVITY OF SAA
Relevant biological activity
Leucocyte attraction
The in vitro and in vivo chemotactic potential of rhSAA towards monocytes and polymorphonuclear leucocytes was initially described by Badolato et al. 25 Over the years, the chemotactic capacity of SAA has been extended to multiple cell subsets, including DCs, mast cells, T cells, endothelial cells, fibroblasts and smooth muscle cells. 26 Whether or not the cell recruiting capacity of SAA is due to an indirect effect of contamination with bacterial products is not yet carefully examined for all these cell types. We recently demonstrated through the use of rhSAA1.1, purified to homogeneity, that SAA1 does indeed possess the capacity to induce in vivo recruitment of neutrophils and mononuclear cells following intra‐articular injection. 5 Nevertheless, the wide range of cell subsets recruited in response to SAA remains questionable. Professional chemo‐attractants such as chemokines display unique receptor specificity profiles which are in turn differentially expressed on distinct cell subsets, thereby conferring tight regulation of recruited subsets. 27 By the same token, further research is required to ascertain the target cell spectrum of SAA. Interestingly, not only has SAA been found to display a direct effect on cell recruitment but it also co‐operates with chemokines such as the neutrophil‐specific chemokine CXCL8 to enhance neutrophil recruitment in a formyl peptide receptor (FPR) 2‐dependent manner, 28 an effect that was confirmed through the use of homogenous rhSAA1.1. 5 Similarly, recombinant murine SAA3 (rmSAA3), purified to homogeneity, displayed a synergistic effect with human CXCL8 during the activation of human neutrophils. 6
The role of SAA in innate immunity
The innate immune system represents the first line of defence against invading pathogens. A number of APPs regulate innate immune responses, such as CRP, pentraxin (PTX) 3 and serum amyloid P. 29 Amongst these immune modulatory APPs is SAA, as it protects against invading micro‐organisms of variable origin. The following section gives an overview of SAA‐mediated immune responses. The antimicrobial effects of SAA are summarized in Figure 1.
FIGURE 1.
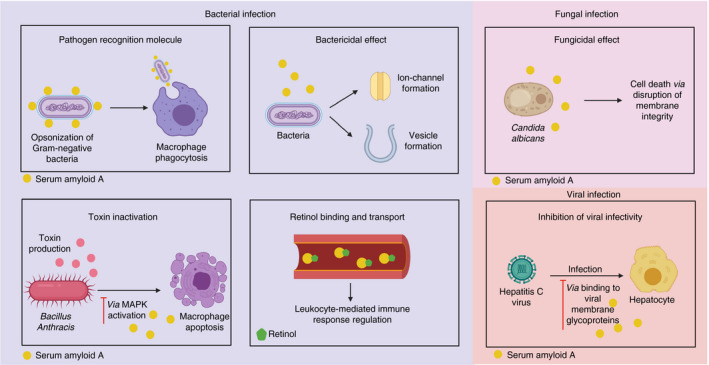
An overview of the antimicrobial properties of SAA. Created with Biorender.com
Antibacterial effect
Through the use of animal models, several studies have demonstrated upregulated expression of SAA upon bacterial infection. 30 , 31 Whether or not SAA expression occurs in vivo directly in response to TLR ligands or via upregulated inflammatory cytokines is unknown. Nevertheless, in vitro studies have demonstrated the expression of SAA by human hepatocytes in response to the bacterial product LPS. 32 SAA contributes towards bacterial elimination through multiple pathways. Firstly, serum‐derived human SAA1 functions as a pattern recognition molecule. SAA1 binds a range of Gram‐negative organisms including E. coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Salmonella typhimurium, Shigella flexneri and Vibrio cholerae through its interaction with outer membrane protein A (OmpA). 33 Shah et al. demonstrated enhanced antimicrobial effects, including neutrophil‐mediated oxidative burst and macrophage phagocytosis, following the interaction between natural serum‐derived human SAA1 and OmpA on E. coli. This enhanced phagocytic effect, mediated by SAA1, is reportedly due to this APP functioning as an opsonizing agent rather than as a direct leucocyte activator. 34 Secondly, SAA plays a role in the inactivation of bacterial toxins. Recombinant human SAA1 protects against the Gram‐positive bacteria Bacillus anthracis via the inactivation of B. anthracis’ lethal toxin (LT), which inactivates macrophages, thus contributing to disease progression. It was demonstrated that SAA1 inhibits LT‐induced cytotoxicity via the activation of the p38 MAP kinase pathway. 35 Thirdly, in congruence with other amyloid‐forming peptides, Hirakura et al. demonstrated the capacity of rhSAA1.1 to induce dose‐dependent ion channel formation within phospholipid membranes. Moreover, recombinant expression of human SAA1.1 in E. coli induced cell lysis, evidenced through absorbance measurement at 590 nm, a measure of cell viability, and increased viscosity of the culture medium, reflecting cell lysis. This bactericidal effect was conserved within other mammalian A‐SAAs. Interestingly, conductance induced by rhSAA suggests that SAA relays this function in an oligomeric form rather than as a monomer. 36 Fourthly, endogenous and recombinant murine A‐SAA displays the capacity to bind both Gram‐positive and Gram‐negative bacterial membrane phospholipids. This interaction leads to the formation of vesicles and therefore disrupts the integrity of the bacterial membrane, causing leakage of cellular components and cell death. This particular effect was also observed with recombinant human A‐SAAs. 30 Finally, through a rather indirect effect, SAA regulates the immune response via its interaction with retinol. 37 Pino‐Lagos et al. have described retinol as indispensable during host defence against not only invading bacteria but also protozoa and viruses. Indeed, retinol regulates both the innate and adaptive immune responses with functions including, but not limited to, regulation of monocyte/macrophage activity, DC function and phenotype and mediating the balance between the Th1/Th2 responses. Owing to its polarity, retinol must be transported by specialized transport proteins, namely cellular retinol‐binding proteins and serum retinol‐binding proteins (RBPs). 38 However, the levels of RBPs have been shown to decline under inflammatory conditions. 39 Nevertheless, murine A‐SAAs associate with retinol in vivo, thus superseding the role of RBP. 37
Apart from its antibacterial function, SAA seems to reduce the inflammatory burden brought about by LPS, through directly interacting with this bacterial product. Indeed, transgenically expressed human SAA1 downregulates LPS‐induced cytokine expression and neutrophil recruitment while enhancing LPS uptake by macrophages in mouse models of inflammation. 40 The binding of SAA to other lipophilic bacterial products such as lipoproteins 4 promotes speculation on whether SAA also confers protection against other bacterial products causing inflammatory‐mediated tissue injury.
Antifungal effect
In addition to its antibacterial function, SAA also relays antifungal activity. In a study by Gong et al., systemic SAA1 levels were shown to be upregulated in mice following infection with Candida albicans. Recombinant forms of mouse and human SAA1 were utilized to demonstrate the capacity of SAA1 to bind to and mediate the killing of both yeast form and filamentous C. albicans through compromising the cell membrane integrity in a dose‐dependent manner. In addition to its effects on C. albicans, rSAA1 displayed activity against its closely related species C. dubliniensis but not C. glabrata, C. parapsilosis or Saccharomyces cerevisiae, thereby indicating a species‐dependent effect. Because both mouse and human SAA1, which show 74% sequence homology, display antifungal activity, it was suggested that the antifungal effects of SAA are rather conserved between different species. 41
Antiviral effect
Upregulated expression of SAA has been described in a range of viral infections in both animals and humans. 42 However, the role of SAA during viral infections has been seldom studied and has only been investigated in the context of hepatitis C virus (HCV) infection, where rSAA has been demonstrated to bind HCV particles through its interaction with viral membrane glycoproteins, thereby impeding cellular entry 43 , 44 (reviewed in Ref. 42).
The role of SAA during inflammation
The inflammatory response is fundamental to both physiological and pathological processes. Considerable progress has been made in understanding the underlying molecular aspects of inflammation revealing the involvement of a range of molecules such as cytokines, reactive oxygen species (ROS), cell adhesion molecules, proteases, vasoactive amines and eicosanoids 45 of which many have been reported to be modulated by SAA. 26 , 46 , 47 , 48 Nevertheless, the regulation of inflammatory mediators by SAA has been largely demonstrated through recombinantly expressed variants. Thus, many of these effects may not reflect the inherent biological function of SAA. In fact, the TLR‐mediated expression of cytokines and chemokines by PBMCs in response to rSAA stimulation has been recently disputed. 4 , 5 , 6 Furthermore, the production of ROS and release of matrix metalloproteinase‐9 (MMP‐9) by rhSAA1.1‐stimulated monocytes have also been recently negated. 5
Following an inflammatory trigger of a particular nature, a specific cellular response is initiated to protect the host and/or repair the tissue and re‐establish homeostasis. Thus, depending on the inflammatory trigger, multiple distinct cell subsets are implicated within the inflammatory response. 45 The functions attributed to SAA during inflammation appear to extend beyond its induction of inflammatory mediators; namely, SAA has been described to regulate cell populations that are central to the local inflammatory response. For instance, using rhSAA1.1 purified to homogeneity, the capacity of SAA1 to promote neutrophil and monocyte survival was confirmed. 5 , 49 , 50 Furthermore, SAA was suggested to modulate macrophage polarization in vitro with the majority of studies suggesting either a mixed M1/M2 or an M2 phenotype in response to rSAA stimulation, 51 , 52 , 53 a function that was disproven with homogenous rhSAA1.1. 5 Nevertheless, one cannot rule out the possibility that SAA may co‐operate with other mediators to regulate the phenotype of macrophages in vivo. Indeed, by neutralizing SAA in vivo through the use of specific anti‐SAA antibodies, Wang et al. demonstrated the generation of a more fibrogenic macrophage phenotype implicating SAA in the process of macrophage polarization. 54 Alternatively, recombinant human SAA displays an inhibitory effect on GM‐CSF‐induced DC differentiation through downregulation of the GM‐CSF receptor in an FPR2‐dependent manner. 55 Apart from the aforementioned functions, additional roles that have been ascribed to SAA during inflammation include angiogenesis 47 , 56 , 57 , 58 , 59 , 60 and inflammasome activation. 61 , 62 , 63 , 64 Further research using pure SAA is required to confirm these findings.
Animal models
The use of animal models is perhaps the most reliable method currently available to study the biological function of SAA. As mentioned in the Section "Protein structure", SAA has been described as an IDP. Thus, it is currently unclear which structural organization is acquired by SAA in vivo. Due to their conformational plasticity, IDPs are thought to regulate cellular signalling pathways through their allosteric binding to their physiological partner. Furthermore, the presence of disorder within a protein allows it to potentially bind multiple targets using distinct structural organizations. Such IDPs often undergo a disorder‐to‐order transition upon binding to their specific target. 65 Within living organisms, there exist a number of factors that influence protein folding, which are difficult to mimic using in vitro experimental approaches. 66 As such, in vivo experiments provide an advantage to study SAA in the conformation that it naturally acquires. Moreover, as we currently lack a recombinantly expressed SAA variant with an amino acid sequence perfectly corresponding to the naturally occurring molecule and lacking any modifications as a result of said recombinant expression, the use of animals, through gene knockout (KO) or transgenic expression, is the best approach to study SAA. Current data on the in vivo functions of SAA obtained through the use of SAA KO animals and transgenic expression are summarized in Tables 2 and 3 (Table 2, SAA KO; Table 3, transgenic expression of SAA).
TABLE 2.
In vivo functions relayed by SAA revealed through SAA KO in mice
| SAA KO | Disease model | Role | Reference |
|---|---|---|---|
| SAA a | Pseudomonas aeruginosa infection | Suppression of neutrophil activity | 92 |
| SAA1 + 2 | Staphylococcus aureus infection | Bacterial clearance | 30 |
| Salmonella typhimurium infection | Retinol binding | 37 | |
| SFB colonization | Induction of Th17 response | 93 | |
| Apical periodontitis | Myeloid cell recruitment | 94 | |
| Inflammation (LPS)‐induced atherosclerosis | HDL binding to vascular proteoglycans | 95 | |
| DSS‐induced colitis | Dampening disease activity | 96 | |
| Allergen‐induced AHR | sPRR mediating type II immunity | 97 | |
| AngII‐induced AAA | Increased disease activity | 98 | |
| WTD‐induced hyperlipidaemia |
Atherosclerosis Regulation of monopoiesis |
99, 100 | |
| SAA1 + 2 + 3 |
Helicobacter hepaticus‐induced colitis MOG‐induced EAE |
Induction of Th17 response | 101 |
| SAA1 + 2 + 3 + 4 | Salmonella typhimurium infection | Retinol binding and transport | 102 |
| SAA3 | H1N1 influenza A virus infection | Regulation of the CD4+ and CD8+ T‐cell‐mediated adaptive immune response | 103 |
| Pseudomonas aeruginosa‐induced lung injury |
Bacterial clearance Resolution of inflammation |
31 | |
| Apical periodontitis | Myeloid cell recruitment | 94 | |
| LPS‐induced neuroinflammation | Restraining tau hyperphosphorylation | 104 | |
| LPS administration | Development of the innate immune response | 105 | |
| DSS‐induced colitis | Dampening of disease activity | 106 | |
| HFHSC diet | Regulation of lipoprotein metabolism | 107 | |
| Continuous PTH infusion | Suppression of PTH‐induced bone anabolism | 108 | |
| PDAC | Protumorigenic effect on CAFs | 109 | |
| No treatment/infection | Regulation of weight and metabolic homeostasis | 105 |
Abbreviations: AAA, abdominal aortic aneurysm; AHR, airway hyperresponsiveness; AngII, angiotensin II; CAF, cancer‐associated fibroblasts; DSS, dextran sodium sulphate; EAE, experimental autoimmune encephalomyelitis; HDL, high‐density lipoprotein; HFHSC, high‐fat, high‐sucrose diet with 0.15% added cholesterol; LPS, lipopolysaccharide; MOG, myelin oligodendrocyte glycoprotein; PDAC, pancreatic ductal adenocarcinoma; PTH, parathyroid hormone; SFB, segmented filamentous bacteria; sPRR, soluble pathogen recognition receptor; WTD, western‐type diet.
SAA KO was performed in zebrafish.
TABLE 3.
In vivo functions relayed by SAA revealed through transgenic expression of SAA in mice
| SAA expressed | Tissue | Disease model | Effect | Reference |
|---|---|---|---|---|
| Human SAA1 | Systemic | CLP‐ and LPS‐induced acute lung injury | Protection against LPS‐induced tissue injury through LPS binding | 40 |
| Human SAA1 | Systemic | No treatment/infection | Enhanced atherosclerosis | 110 |
| Murine SAA1 | Liver | No treatment/infection | Psoriasis‐like phenotype | 111 |
| Murine SAA1 | Liver | ConA‐induced hepatitis | Increased hepatic injury | 112 |
Abbreviations: CLP, caecal ligation and puncture sepsis model; ConA, concanavalin A; LPS, lipopolysaccharide.
In addition to inhibiting or upregulating SAA expression in vivo, several researchers have opted to study the effects of SAA through the use of neutralizing antibodies. Yu et al. demonstrated the role of SAA in imiquimod‐induced psoriasis in mice. Neutralization of SAA resulted in a reduction in epidermal hyperplasia, the number of recruited inflammatory cells and cytokine expression (IL‐17A and CCL20). 67 While SAA may not play a direct role in cytokine expression, it may contribute towards increased cytokine levels through the recruitment of cytokine‐expressing inflammatory cells. Moreover, SAA contributes towards in vivo macrophage polarization, an effect demonstrated through the neutralization of SAA (vide supra). 54
The link between SAA protein structure and its biological activity
The following section provides an overview of the roles relayed by SAA and their link to the SAA protein structure. The role of particular structural motifs within the primary structure of mature human SAA1 is indicated in Figure 2.
FIGURE 2.

The biological role of structural motifs within the primary structure of human SAA1.1. Created with Biorender.com
SAA and the extracellular matrix (ECM): a dual function relayed by SAA
Preciado‐Patt et al. were the first to describe the presence of ECM protein‐like motifs within the sequence of human SAA1, in particular RGN(39–41) and YIGSD(29–33) similar to the cell‐binding motifs of fibronectin (RGD) and laminin (YIGSR), respectively. 68 Through the use of adhesion assays, an inhibitory effect of SAA‐derived peptides RGNY and YIGSD on the binding of activated T lymphocytes to immobilized fibronectin and laminin was demonstrated, suggesting that SAA possesses similar functionality as ECM proteins. 68 Binding of SAA to platelets is mediated through the integrin receptor αIIbβ3, whose ligands encompass a range of ECM proteins amongst which is fibronectin. 69 , 70 SAA has been shown to bind αIIbβ3 through the residues 29–42 (YIGSDKYFHARGNY) that comprise the RGN motif. 70 The RGD adhesion sequence, initially identified in fibronectin, has later been identified in several other integrin‐binding proteins and forms the basis of attachment. Whereas for many proteins, the RGD motif is required for integrin binding, variations of this motif in proteins possessing integrin‐binding capacity have been described. A common denominator between many of these different integrin‐binding motifs is the presence of a negatively charged aspartic acid residue or its biochemically similar counterpart glutamic acid in the third position. 71 However, Urieli‐Shoval et al. 70 suggested that the presence of an isoelectric amino acid such as asparagine in the third position may contribute towards αIIbβ3 binding through the formation of hydrogen bonds.
Through its binding to αIIbβ3, fibronectin keeps the balance between haemostasis and thrombosis. 72 Zimlichman et al. 73 demonstrated an inhibitory effect of intact endogenous SAA on the thrombin‐induced platelet aggregation while having no effect on collagen or adenosine diphosphate‐induced platelet aggregation. However, the underlying mechanism involving the inhibitory effect of full‐size SAA on thrombin‐induced platelet aggregation is unclear. The SAA peptide 25–76 relayed an inhibitory effect on collagen‐induced platelet activation. 74 But, the RGNY SAA‐derived peptide displayed no effect on collagen‐induced platelet aggregation. 68 As such, it is unknown whether the inhibitory effect of the 25–76 SAA peptide on collagen‐induced platelet aggregation is relayed through its RGN motif binding to αIIbβ3 or rather through another mechanism. Perhaps the conformational organization of amino acids surrounding the RGN motif plays a role herein.
Regarding ECM function, SAA seems to possess a dual role. Besides mimicking ECM protein function, rhSAA can also directly bind ECM proteins such as laminin and fibronectin. The exact residues within rhSAA involved in ECM protein binding have not yet been identified. However, they are thought to reside within its N‐terminal (2–82) amyloid A fragment. In addition, endogenous murine A‐SAAs bind laminin through the residues 17–23 and 24–103. 75 Interestingly, through its RDG‐like motif rhSAA can bind T lymphocytes via integrin interaction forming a laminin–rhSAA‐T‐lymphocyte complex. Thus, apart from its direct chemotactic effect, SAA plays an indirect role in regulating leucocyte recruitment. Furthermore, SAA‐ECM protein interaction may regulate the inflammatory response by fine‐tuning the concentration of SAA at the site of inflammation. 76
In addition to binding platelets, intact rhSAA and human SAA1‐derived peptide 77–104 also bind neutrophils in their free form, as well as when complexed with high‐density lipoprotein (HDL). This interaction probably involves the A‐SAA receptor FPR2 expressed on neutrophils, which facilitates neutrophil chemotaxis (vide infra). Furthermore, it has been hypothesized that SAA–neutrophil binding may facilitate cleavage of SAA by neutrophilic enzymes, thereby generating peptides that regulate leucocyte migration or platelet function. 77 Indeed, incubation of intact rSAA with polymorphonuclear leucocyte‐derived proteases generated a peptide corresponding to the amino acids 28–36, containing the YIGSD motif, which displayed an inhibitory effect on the binding of T lymphocytes to laminin. 68 Moreover, neutrophilic MMPs process SAA‐producing C‐terminal peptides that regulate chemokine‐mediated leucocyte recruitment. 13
SAA as a glycosaminoglycan (GAG)‐binding protein
Several GAG‐binding consensus sequences have been identified in GAG‐binding proteins namely XBBXBX and XBBBXXBX, where X is a hydrophobic amino acid and B is a basic amino acid. 78 However, across species, SAA seems to lack a conserved GAG‐binding consensus within its primary structure. Nevertheless, Ancsin et al. have identified several basic amino acid residues within the C‐terminus of murine A‐SAA that are involved in the binding of A‐SAA to heparin and heparan sulphate. Specifically R86, R95 and K102 are phylogenetically conserved in humans (corresponding to R87, R96 and K103 in human SAA1). 79 Having said that, according to the three‐dimensional structure of SAA1.1 proposed by Lu et al. the C‐terminal tail of human SAA1.1 is involved in the stabilization of the structure through the formation of hydrogen bonds and salt bridges and thus is not free to bind GAGs. As such, another model was put forward. Through the formation of a hexamer, human SAA1.1 monomers display two positively charged clusters formed by R15, R19 and R47 and R1, R62 and H71, respectively, that display heparin‐binding capacity. 20 Nevertheless, taking into account the propensity of SAA to function as an IDP under physiological conditions, the suggestion that the C‐terminal basic residues are involved in SAA‐GAG binding may still hold to be true. The biological relevance of SAA‐GAG binding has not been clearly defined. However, it has been postulated that competitive binding of GAGs to HDL‐associated SAA serves to free SAA in order to execute biological functions that are precluded by its association to HDL. In addition, the binding of SAA to GAGs seems to be a requisite for the occurrence of amyloid A amyloidosis. 80
The hydrophobic motif of SAA
A‐SAAs function as apolipoproteins mainly binding HDL. 42 Early studies suggested that the N‐terminal region of human SAA1.1 is involved in its binding to HDL, particularly the amino acids corresponding to 1–11 (RSFFSFLGEAF). Indeed, in comparison with the intact form, a mutant form of SAA1.1 lacking the first 11 amino acids showed a decline in its capacity to bind HDL. Furthermore, the substitution of glycine in position 8 with aspartic acid in intact SAA1.1 abated the SAA‐HDL binding capacity, thereby signifying the role of the SAA N‐terminus during HDL binding. 81 More recently, SAA‐HDL binding was investigated in the context of amyloidosis. 20 To form amyloid fibrils, SAA must dissociate from its binding to HDL. 82 A model has been proposed by which GAGs and HDL share a binding site and competitive binding to GAGs releases SAA from HDL. Mutational studies revealed the presence of a shared GAG and HDL binding site within one of the positively charged GAG‐binding clusters formed by R1, R62 and R71 in the SAA hexamer. 20 Having said that, crossing‐linking studies demonstrated that in order to bind HDL, hexameric recombinant murine SAA2.2 first dissociates into its monomeric form. 83 Consequently, an alternative HDL binding model was suggested by Frame et al. 84 In line with previous studies, 81 hydrophobicity profiles revealed that in addition to helix 3, the N‐terminal region of helix 1 displayed the highest hydrophobicity within human SAA1. Frame et al. suggested that helices 1 and 3 are arranged to form a concave polar surface that can bind an HDL particle. 84
The antibacterial function of SAA is somewhat intertwined to its hydrophobic motif. As previously mentioned, SAA can serve as a RBP, thereby regulating the immune response (vide supra). Derebe et al. identified the probable retinol‐binding site in murine SAA3, which displays 70% similarity to human SAA1 in terms of amino acid sequence. Whereas the SAA3 structure varies from prototypical RBPs, this murine SAA isoform assembles into a tetramer forming a hydrophobic central channel composed primarily of non‐polar residues held in helix 3, the proposed retinol‐binding site. 37 In addition, the previously discussed bactericidal effect of A‐SAA conveyed through its binding to bacterial membrane phospholipids is mediated through its hydrophobic N‐terminal region (amino acids 1–20). 30 Finally, the binding with LPS occurs through residues residing within the amino acids 32–47 40 and might confer protection against inflammation‐mediated tissue injury brought about by the pro‐inflammatory TLR4 ligand (vide supra).
RECEPTORS THAT RECOGNIZE SAA
To relay its diverse functions, SAA has been described to activate a number of receptors. SAA is thought to be recognized by the G protein‐coupled receptor FPR2, scavenger receptor class B type I (SR‐BI) and its splice variant SR‐BII, CD36, receptor for advanced glycation end products (RAGE) and the P2X purinoceptor 7 (P2RX7) (non‐contested functions, Table 4; postulated functions, Table S1). Despite no activity having been assigned to the complex of SAA and CD55, otherwise known as decay‐accelerating factor, Artl et al. 85 have shown that SAA‐enriched HDL comigrates with CD55 upon Western blot of lysates from macrophages preincubated with SAA. Also Tanis, a membrane‐bound receptor associated with insulin resistance has been linked to SAA via surface plasmon resonance analysis, 86 without demonstrated biological implication for this interaction. However, SAA has been linked to weight gain and insulin resistance. 87 Hepatic expression of both SAA and Tanis was found to be upregulated in rats with induced insulin resistance, suggesting that SAA might contribute towards the inflammatory aspect of type II diabetes via interaction with Tanis. 88
TABLE 4.
Non‐contested receptor‐mediated functions of SAA
| Receptor | Cell type | Function | Effective dose (µg/ml) a | Reference |
|---|---|---|---|---|
| FPR2 | Transfected cell line | Chemotaxis | 0.125–125 µg/ml | 113, 114, 115 |
| Ca2+ signalling | 3.1–125 µg/ml | 113, 114, 115 | ||
| Neutrophil | Enhanced CXCL8‐mediated cell recruitment | 0.3–3 µg/ml | 5, 13, 28 | |
| Macrophage | Chemotaxis | 3.75–12.5 µg/ml | 116, 117 | |
| Monocyte | Ca2+ signalling | 12.5 µg/ml | 114 | |
| Epithelial cells | IL−33 induction b | NI | 97 |
Abbreviation: NI, not indicated.
Receptor‐mediated functions are arranged in order of increasing effective dose.
The induction of IL‐33 by epithelial cells overexpressing murine SAA1 has been demonstrated in the presence of house dust mite allergens where SAA1 functions as a soluble pathogen recognition receptor forming an immune complex that activates FPR2 leading to cytokine expression.
A large proportion of SAA functions have been attributed to its interaction with TLRs 89 ; nonetheless, taking into account the recently published data, this concept is one that is likely to be disowned. 4 , 5 , 6 Indeed, as mentioned, Burgess et al. demonstrated that the widely reported TLR2‐mediated activities of SAA are conveyed through bacterial lipoprotein contamination of rSAA preparations. rhSAA1.1 of a bacterial source induced chemokine (CXCL8) expression in TLR1/2‐transfected HEK cells, whereas mouse and human SAA1 expressed in HEK cells failed to do so. Furthermore, treatment of rhSAA1.1 with LPL induced a partial, but dose‐dependent, downregulation of rhSAA1.1 activity on macrophages. 4 A plausible explanation behind the incomplete downregulation of rhSAA1.1 activity, following LPL treatment, is the presence of other bacterial contaminants. We regularly detected the presence of LPS in commercial recombinant murine SAA3 (rmSAA3) protein. Very low levels of LPS are still biologically active. Indeed, the treatment of monocytes with LPS levels as low as 70 pg/ml induced CXCL8 levels of physiological relevance. To determine whether LPS contributes towards some of the described SAA‐mediated inflammatory effects, we investigated in vivo neutrophil recruitment in response to homogenously purified rmSAA3, an effect shown to be mediated in a TLR4‐dependent manner. Whereas the purchased SAA3 originally recruited neutrophils, upon reversed‐phase purification we observed a loss of neutrophil chemoattraction. In addition, purified rmSAA3 failed to induce in vitro CXCL8 and CXCL2 expression in human monocytes and murine peritoneal cells, respectively. 6 We concluded that SAA probably is not a TLR ligand.
One may also question whether the reported activation of FPR2 by SAA is due to contamination with formylated bacterial peptides, of which fMLF is considered the prototypical variant. While fMLF activates FPR2, its affinity for FPR1 activation is much higher. In fact, in order to activate FPR2, higher fMLF concentrations of approximately 10−6‐fold are required in comparison with FPR1 activation. 90 To exclude the presence of formylated bacterial peptides within the preparation of rhSAA1.1, FPR1‐transfected HEK cells were stimulated with rhSAA1.1 in which it failed to induce Ca2+ mobilization. In addition, rhSAA1.1 did not desensitize FPR1‐transfected HEK cells against fMLP‐mediated Ca2+ mobilization. Furthermore, using rhSAA1.1 purified to homogeneity we confirmed FPR2 activation by SAA1. Firstly, we observed desensitization of Ca2+ signalling by the FPR2 ligand CCL23(46–137) upon pretreatment with homogenous rhSAA1.1. Secondly, we were able to inhibit synergy between homogenous rhSAA1.1 and CXCL8 using a selective FPR2 antagonist. 5 Additional evidence supporting the use of FPR2 by SAA1 is provided through the use of chemically synthesized human SAA1 C‐terminal peptides, which display an FPR2‐mediated synergistic effect with CXCL8 during neutrophil recruitment. 13 , 14 Regarding SAA‐RAGE interaction, several biological effects have been demonstrated such as the induction of high‐mobility group box 1 (HMGB1), tissue factor, haem oxygenase‐1 (HO‐1) and cytokines (Table S1). Nevertheless, Wang et al. demonstrated the role of RAGE in LPS‐induced NF‐κB activation in endothelial cells. 91 Thus further research is required to ascertain the SAA‐RAGE interaction and the corresponding downstream effect following receptor activation. FPR2 and additional receptors including CD36, SR‐BI and SR‐BII are reported to relay biological effects that have recently been disputed, such as cytokine induction, the release of MMP‐9 and ROS production (Table S1). Indeed, the latter biological effects of SAA are also induced downstream of TLR2 or TLR4 and were lost upon further purification of the commercial rhSAA1.1. Thus, further research is required to ascertain whether homogenous rhSAA, devoid of contamination, still activates the receptors indicated in Table S1.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Table S1. Postulated receptor‐mediated functions of SAA.
ACKNOWLEDGEMENTS
This work was supported by the Research Foundation of Flanders (FWO‐Vlaanderen) and C1 funding (C1 Project number C16/17/010) of the KU Leuven. Mieke Gouwy is a research expert funded by the Rega Foundation.
Senior author: Sofie Struyf.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Gabay C, Kushner I. Acute‐phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–54. [DOI] [PubMed] [Google Scholar]
- 2. Morrow JF, Stearman RS, Peltzman CG, Potter DA. Induction of hepatic synthesis of serum amyloid A protein and actin. Proc Natl Acad Sci USA. 1981;78:4718–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Buck M, Gouwy M, Wang JM, Van Snick J, Opdenakker G, Struyf S, et al. Structure and expression of different serum amyloid A (SAA) variants and their concentration‐dependent functions during host insults. Curr Med Chem. 2016;23:1725–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burgess EJ, Hoyt LR, Randall MJ, Mank MM, Bivona JJ, Eisenhauer PL, et al. Bacterial lipoproteins constitute the TLR2‐stimulating activity of serum amyloid A. J Immunol. 2018;201:2377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abouelasrar Salama S, De Bondt M, De Buck M, Berghmans N, Proost P, Oliveira VLS, et al. Serum amyloid A1 (SAA1) revisited: restricted leukocyte‐activating properties of homogeneous SAA1. Front Immunol. 2020;11:843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abouelasrar Salama S, De Bondt M, Berghmans N, Gouwy M, Oliveira VLS, Oliveira SC, et al. Biological characterization of commercial recombinantly expressed immunomodulating proteins contaminated with bacterial products in the year 2020: the SAA3 case. Mediators Inflamm. 2020;2020:6087109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosenthal CJ, Franklin EC, Frangione B, Greenspan J. Isolation and partial characterization of SAA‐an amyloid‐related protein from human serum. J Immunol. 1976;116:1415–8. [PubMed] [Google Scholar]
- 8. Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute‐phase reactant. Eur J Biochem. 1999;265:501–23. [DOI] [PubMed] [Google Scholar]
- 9. Santiago‐Cardona PG, Berríos CA, Ramírez F, García‐Arrarás JE. Lipopolysaccharides induce intestinal serum amyloid A expression in the sea cucumber holothuria glaberrima. Dev Comp Immunol. 2003;27:105–10. [DOI] [PubMed] [Google Scholar]
- 10. Björkman L, Raynes JG, Shah C, Karlsson A, Dahlgren C, Bylund J. The proinflammatory activity of recombinant serum amyloid A is not shared by the endogenous protein in the circulation. Arthritis Rheum. 2010;62:1660–5. [DOI] [PubMed] [Google Scholar]
- 11. Christenson K, Björkman L, Ahlin S, Olsson M, Sjöholm K, Karlsson A, et al. Endogenous acute phase serum amyloid A lacks pro‐inflammatory activity, contrasting the two recombinant variants that activate human neutrophils through different receptors. Front Immunol. 2013;4:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chaudhuri TK, Horii K, Yoda T, Arai M, Nagata S, Terada TP, et al. Effect of the extra N‐terminal methionine residue on the stability and folding of recombinant α‐lactalbumin expressed in Escherichia coli . J Mol Biol. 1999;285:1179–94. [DOI] [PubMed] [Google Scholar]
- 13. Gouwy M, De Buck M, Abouelasrar Salama S, Vandooren J, Knoops S, Pörtner N, et al. Matrix metalloproteinase‐9‐generated COOH‐, but not NH2‐terminal fragments of serum amyloid A1 retain potentiating activity in neutrophil migration to CXCL8, with loss of direct chemotactic and cytokine‐inducing capacity. Front Immunol. 2018;9:1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Buck M, Gouwy M, Berghmans N, Opdenakker G, Proost P, Struyf S, et al. COOH‐terminal SAA1 peptides fail to induce chemokines but synergize with CXCL8 and CCL3 to recruit leukocytes via FPR2. Blood 2018;131:439–49. [DOI] [PubMed] [Google Scholar]
- 15. Steel DM, Sellar GC, Uhlar CM, Simon S, DeBeer FC, Whitehead AS. A constitutively expressed serum amyloid a protein gene (SAA4) is closely linked to, and shares structural similarities with, an acute‐phase serum amyloid a protein gene (SAA2). Genomics 1993;16:447–54. [DOI] [PubMed] [Google Scholar]
- 16. Larson MA, Wei SH, Weber A, Weber AT, McDonald TL. Induction of human mammary‐associated serum amyloid A3 expression by prolactin or lipopolysaccharide. Biochem Biophys Res Commun. 2003;301:1030–7. [DOI] [PubMed] [Google Scholar]
- 17. Tomita T, Ieguchi K, Sawamura T, Maru Y. Human serum amyloid A3 (SAA3) protein, expressed as a fusion protein with SAA2, binds the oxidized low density lipoprotein receptor. PLoS One 2015;10:e0118835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang JH, Liao WS‐L. Synergistic induction of mouse serum amyloid A3 promoter by the inflammatory mediators IL‐1 and IL‐6. J Interferon Cytokine Res. 1999;19:1403–11. [DOI] [PubMed] [Google Scholar]
- 19. McCubbin WD, Kay CM, Narindrasorasak S, Kisilevsky R. Circular‐dichroism studies on two murine serum amyloid A proteins. Biochem J. 1988;256:775–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu J, Yu Y, Zhu L, Cheng Y, Sun PD. Structural mechanism of serum amyloid A‐mediated inflammatory amyloidosis. Proc Natl Acad Sci USA. 2014;111:5189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L, Lashuel HA, Walz T, Colón W. Murine apolipoprotein serum amyloid A in solution forms a hexamer containing a central channel. Proc Natl Acad Sci USA. 2002;99:15947–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y, Srinivasan S, Ye Z, Javier Aguilera J, Lopez MM, Colón W. Serum amyloid A 2.2 refolds into a octameric oligomer that slowly converts to a more stable hexamer. Biochem Biophys Res Commun. 2011;407:725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang L, Lashuel HA, Colón W. From hexamer to amyloid: marginal stability of apolipoprotein SAA2.2 leads to in vitro fibril formation at physiological temperature. Amyloid 2005;12:139–48. [DOI] [PubMed] [Google Scholar]
- 24. Takase H, Tanaka M, Miyagawa S, Yamada T, Mukai T. Effect of amino acid variations in the central region of human serum amyloid A on the amyloidogenic properties. Biochem Biophys Res Commun. 2014;444:92–7. [DOI] [PubMed] [Google Scholar]
- 25. Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, et al. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Buck M, Gouwy M, Wang JM, Van Snick J, Proost P, Struyf S, et al. The cytokine‐serum amyloid A‐chemokine network. Cytokine Growth Factor Rev. 2016;30:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018; 285:2944–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Buck M, Berghmans N, Pörtner N, Vanbrabant L, Cockx M, Struyf S, et al. Serum amyloid A1α induces paracrine IL‐8/CXCL8 via TLR2 and directly synergizes with this chemokine via CXCR2 and formyl peptide receptor 2 to recruit neutrophils. J Leukoc Biol. 2015;98:1049–60. [DOI] [PubMed] [Google Scholar]
- 29. Bottazzi B, Inforzato A, Messa M, Barbagallo M, Magrini E, Garlanda C, et al. The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. J Hepatol. 2016;64:1416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng H, Li H, Zhang J, Fan H, Jia L, Ma W, et al. Serum amyloid A exhibits pH dependent antibacterial action and contributes to host defense against Staphylococcus aureus cutaneous infection. J Biol Chem. 2020;295:2570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan Y, Zhang G, Vong CT, Ye RD. Serum amyloid A3 confers protection against acute lung injury in Pseudomonas aeruginosa‐infected mice. Am J Physiol Lung Cell Mol Physiol. 2020;318:L314–22. [DOI] [PubMed] [Google Scholar]
- 32. Migita K, Abiru S, Nakamura M, Komori A, Yoshida Y, Yokoyama T, et al. Lipopolysaccharide signaling induces serum amyloid A (SAA) synthesis in human hepatocytes in vitro. FEBS Lett. 2004;569:235–9. [DOI] [PubMed] [Google Scholar]
- 33. Hari‐Dass R, Shah C, Meyer DJ, Raynes JG. Serum amyloid A protein binds to outer membrane protein A of gram‐negative bacteria. J Biol Chem. 2005;280:18562–7. [DOI] [PubMed] [Google Scholar]
- 34. Shah C, Hari‐Dass R, Raynes JG. Serum amyloid A is an innate immune opsonin for gram‐negative bacteria. Blood 2006;108:1751–7. [DOI] [PubMed] [Google Scholar]
- 35. Rose K, Long P, Shankar M, Ballard JD, Webb CF. Serum amyloid A protects murine macrophages from lethal toxin‐mediated death. Cell Immunol. 2012;272:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirakura Y, Carreras I, Sipe JD, Kagan BL. Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense. Amyloid 2002;9:13–23. [DOI] [PubMed] [Google Scholar]
- 37. Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, et al. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. Elife. 2014;3:e03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pino‐Lagos K, Guo Y, Noelle RJ. Retinoic acid: a key player in immunity. BioFactors 2010;36:430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosales FJ, Ritter SJ, Zolfaghari R, Smith JE, Ross AC. Effects of acute inflammation on plasma retinol, retinol‐binding protein, and its mRNA in the liver and kidneys of vitamin A‐sufficient rats. J Lipid Res. 1996;37:962–71. [PubMed] [Google Scholar]
- 40. Cheng N, Liang Y, Du X, Ye RD. Serum amyloid A promotes LPS clearance and suppresses LPS‐induced inflammation and tissue injury. EMBO Rep. 2018;19:e45517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gong J, Wu J, Ikeh M, Tao L, Zhang Y, Bing J, et al. Antifungal activity of mammalian serum amyloid A1 against Candida albicans . Antimicrob Agents Chemother. 2019;64:e01975–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abouelasrar Salama S, Lavie M, De Buck M, Van Damme J, Struyf S. Cytokines and serum amyloid A in the pathogenesis of hepatitis C virus infection. Cytokine Growth Factor Rev. 2019;50:29–42. [DOI] [PubMed] [Google Scholar]
- 43. Lavie M, Voisset C, Vu‐Dac N, Zurawski V, Duverlie G, Wychowski C, et al. Serum amyloid A has antiviral activity against hepatitis C virus by inhibiting virus entry in a cell culture system. Hepatology 2006;44:1626–34. [DOI] [PubMed] [Google Scholar]
- 44. Cai Z, Cai L, Jiang J, Chang K‐S, van der Westhuyzen DR, Luo G. Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J Virol. 2007;81:6128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Medzhitov R. Origin and physiological roles of inflammation. Nature 2008;454:428–35. [DOI] [PubMed] [Google Scholar]
- 46. De Buck M, Gouwy M, Struyf S, Opdenakker G, Van Damme J. The ectoenzyme‐side of matrix metalloproteinases (MMPs) makes inflammation by serum amyloid A (SAA) and chemokines go round. Immunol Lett. 2019;205:1–8. [DOI] [PubMed] [Google Scholar]
- 47. Connolly M, Rooney PR, McGarry T, Maratha AX, McCormick J, Miggin SM, et al. Acute serum amyloid A is an endogenous TLR2 ligand that mediates inflammatory and angiogenic mechanisms. Ann Rheum Dis. 2016;75:1392–8. [DOI] [PubMed] [Google Scholar]
- 48. Hatanaka E, Dermargos A, Armelin HA, Curi R, Campa A. Serum amyloid A induces reactive oxygen species (ROS) production and proliferation of fibroblast. Clin Exp Immunol. 2011;163:362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Christenson K, Björkman L, Tängemo C, Bylund J. Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7‐sensitive pathway independent of formyl peptide receptor‐like 1. J Leukoc Biol. 2008;83:139–48. [DOI] [PubMed] [Google Scholar]
- 50. El Kebir D, József L, Khreiss T, Pan W, Petasis NA, Serhan CN, et al. Aspirin‐triggered lipoxins override the apoptosis‐delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol. 2007;179:616–22. [DOI] [PubMed] [Google Scholar]
- 51. Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor‐2. Oncogene 2011;30:3887–99. [DOI] [PubMed] [Google Scholar]
- 52. Sun L, Zhou H, Zhu Z, Yan Q, Wang L, Liang Q, et al. Ex vivo and in vitro effect of serum amyloid A in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. J Immunol. 2015;194:4891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Anthony D, McQualter JL, Bishara M, Lim EX, Yatmaz S, Seow HJ, et al. SAA drives proinflammatory heterotypic macrophage differentiation in the lung via CSF‐1R‐dependent signaling. FASEB J. 2014;28:3867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Y, Huang H, Sun R, Chen B, Han F, Li Q, et al. Serum amyloid a induces M2b‐like macrophage polarization during liver inflammation. Oncotarget. 2017;8:109238–46. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Kim JC, Jung YS, Lee HY, Park JS, Bae YS. Serum amyloid a inhibits dendritic cell differentiation by suppressing GM‐CSF receptor expression and signaling. Exp Mol Med. 2017;49:e369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Connolly M, Marrelli A, Blades M, McCormick J, Maderna P, Godson C, et al. Acute serum amyloid A induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol. 2010;184:6427–37. [DOI] [PubMed] [Google Scholar]
- 57. Connolly M, Veale DJ, Fearon U. Acute serum amyloid A regulates cytoskeletal rearrangement, cell matrix interactions and promotes cell migration in rheumatoid arthritis. Ann Rheum Dis. 2011;70:1296–303. [DOI] [PubMed] [Google Scholar]
- 58. Hong C, Shen C, Ding H, Huang S, Mu Y, Su H, et al. An involvement of SR‐B1 mediated p38 MAPK signaling pathway in serum amyloid A‐induced angiogenesis in rheumatoid arthritis. Mol Immunol. 2015;66:340–5. [DOI] [PubMed] [Google Scholar]
- 59. Lee M‐S, Yoo S‐A, Cho C‐S, Suh P‐G, Kim W‐U, Ryu SH. Serum amyloid A binding to formyl peptide receptor‐like 1 induces synovial hyperplasia and angiogenesis. J Immunol. 2006;177:5585–94. [DOI] [PubMed] [Google Scholar]
- 60. Mullan RH, Bresnihan B, Golden‐Mason L, Markham T, O’Hara R, FitzGerald O, et al. Acute‐phase serum amyloid A stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an NF‐κB–dependent signal transduction pathway. Arthritis Rheum. 2006;54:105–14. [DOI] [PubMed] [Google Scholar]
- 61. Niemi K, Teirilä L, Lappalainen J, Rajamäki K, Baumann MH, Öörni K, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B‐sensitive pathway. J Immunol. 2011;186:6119–28. [DOI] [PubMed] [Google Scholar]
- 62. Ather JL, Ckless K, Martin R, Foley KL, Suratt BT, Boyson JE, et al. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol. 2011;187:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu N, Liu S, Yi X, Zhang S, Ding Y. Serum amyloid A induces interleukin‐1β secretion from keratinocytes via the NACHT, LRR and PYD domains‐containing protein 3 inflammasome. Clin Exp Immunol. 2015;179:344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Migita K, Izumi Y, Jiuchi Y, Kozuru H, Kawahara C, Nakamura M, et al. Serum amyloid A induces NLRP‐3‐mediated IL‐1β secretion in neutrophils. PLoS One 2014;9:e96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hingorani KS, Gierasch LM. Comparing protein folding in vitro and in vivo: foldability meets the fitness challenge. Curr Opin Structl Biol. 2014;24:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yu N, Zhang S, Lu J, Li Y, Yi X, Tang L, et al. Serum amyloid A, an acute phase protein, stimulates proliferative and proinflammatory responses of keratinocytes. Cell Prolif. 2017;50:e12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Preciado‐Patt L, Levartowsky D, Prass M, Hershkoviz R, Lider O, Fridkin M. Inhibition of cell adhesion to glycoproteins of the extracellular matrix by peptides corresponding to serum amyloid A: toward understanding the physiological role of an enigmatic protein. Eur J Biochem. 1994;223:35–42. [DOI] [PubMed] [Google Scholar]
- 69. Warltier DC, Kam PCA, Egan MK. Platelet glycoprotein IIb/IIIa antagonists. Anesthesiology 2002;96:1237–49. [DOI] [PubMed] [Google Scholar]
- 70. Urieli‐Shoval S, Shubinsky G, Linke RP, Fridkin M, Tabi I, Matzner Y. Adhesion of human platelets to serum amyloid A. Blood 2002;99:1224–9. [DOI] [PubMed] [Google Scholar]
- 71. Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. [DOI] [PubMed] [Google Scholar]
- 72. Wang Y, Ni H. Fibronectin maintains the balance between hemostasis and thrombosis. Cell Mol Life Sci. 2016; 73:3265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zimlichman S, Danon A, Nathan I, Mozes G, Shainkin‐Kestenbaum R. Serum amyloid A, an acute phase protein, inhibits platelet activation. J Lab Clin Med. 1990;116:180–6. [PubMed] [Google Scholar]
- 74. Syversen PV, Sæter U, Cunha‐Ribeiro L, Ørvim U, Sletten K, Husby G, et al. The effect of serum amyloid protein A fragment‐SAA25‐76 on blood platelet aggregation. Thromb Res. 1994;76:299–305. [DOI] [PubMed] [Google Scholar]
- 75. Ancsin JB, Kisilevsky R. Laminin interactions with the apoproteins of acute‐phase HDL: preliminary mapping of the laminin binding site on serum amyloid A. Amyloid 1999;6:37–47. [DOI] [PubMed] [Google Scholar]
- 76. Preciado‐Patt L, Hershkoviz R, Fridkin M, Lider O. Serum amyloid A binds specific extracellular matrix glycoproteins and induces the adhesion of resting CD4+ T cells. J Immunol. 1996;156:1189–95. [PubMed] [Google Scholar]
- 77. Preciado‐Patt L, Pras M, Ati FM. Binding of human serum amyloid a (hSAA) and its high‐density Iipoprotein3 complex (hSAA‐HDL3) to human neutrophils. Possible implication to the function of a protein of an unknown physiological role. Int J Pept Protein Res. 1996;48:503–13. [DOI] [PubMed] [Google Scholar]
- 78. Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan‐protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. BioEssays 1998;20:156–67. [DOI] [PubMed] [Google Scholar]
- 79. Ancsin JB, Kisilevsky R. The heparin/heparan sulfate‐binding site on apo‐serum amyloid A: implications for the therapeutic intervention of amyloidosis. J Biol Chem. 1999;274:7172–81. [DOI] [PubMed] [Google Scholar]
- 80. Aguilera JJ, Zhang F, Beaudet JM, Linhardt RJ, Colón W. Divergent effect of glycosaminoglycans on the in vitro aggregation of serum amyloid A. Biochimie 2014;104:70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Patel H, Bramall J, Waters H, De Beer MC, Woo P. Expression of recombinant human serum amyloid A in mammalian cells and demonstration of the region necessary for high‐density lipoprotein binding and amyloid fibril formation by site‐directed mutagenesis. Biochem J. 1996;318:1041–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Noborn F, Ancsin JB, Ubhayasekera W, Kisilevsky R, Li JP. Heparan sulfate dissociates serum amyloid A (SAA) from acute‐phase high‐density lipoprotein, promoting SAA aggregation. J Biol Chem. 2012;287:25669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang L, Colón W. The interaction between apolipoprotein serum amyloid A and high‐density lipoprotein. Biochem Biophys Res Commun. 2004;317:157–61. [DOI] [PubMed] [Google Scholar]
- 84. Frame NM, Gursky O. Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. FEBS Lett. 2016;590:866–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Artl A, Marsche G, Lestavel S, Sattler W, Malle E. Role of serum amyloid A during metabolism of acute‐phase HDL by macrophages. Arterioscler Thromb Vasc Biol. 2000;20:763–72. [DOI] [PubMed] [Google Scholar]
- 86. Walder K, Kantham L, McMillan JS, Trevaskis J, Kerr L, De Silva A, et al. Tanis: a link between type 2 diabetes and inflammation? Diabetes 2002;51:1859–66. [DOI] [PubMed] [Google Scholar]
- 87. de Oliveira EM, Ascar TP, Silva JC, Sandri S, Migliorini S, Fock RA, et al. Serum amyloid A links endotoxaemia to weight gain and insulin resistance in mice. Diabetologia 2016;59:1760–8. [DOI] [PubMed] [Google Scholar]
- 88. Liu J, Tang H, Niu L, Xu Y. Upregulation of tanis mRNA expression in the liver is associated with insulin resistance in rats. Tohoku J Exp Med. 2009;219:307–10. [DOI] [PubMed] [Google Scholar]
- 89. Ye RD, Sun L. Emerging functions of serum amyloid A in inflammation. J Leukoc Biol. 2015;98:923–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Le Y, Murphy PM, Wang JM. Formyl‐peptide receptors revisited. Trends Immunol. 2002;23:541–8. [DOI] [PubMed] [Google Scholar]
- 91. Wang L, Wu J, Guo X, Huang X, Huang Q. RAGE plays a role in LPS‐induced NF‐κB activation and endothelial hyperpermeability. Sensors 2017;17:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Murdoch CC, Espenschied ST, Matty MA, Mueller O, Tobin DM, Rawls JF. Intestinal serum amyloid A suppresses systemic neutrophil activation and bactericidal activity in response to microbiota colonization. PLoS Pathog. 2019;15:e1007381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL‐23R/IL‐22 Circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell 2015;163:381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hirai K, Furusho H, Kawashima N, Xu S, de Beer MC, Battaglino R, et al. Serum amyloid A contributes to chronic apical periodontitis via TLR2 and TLR4. J Dent Res. 2019;98:117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chiba T, Chang MY, Wang S, Wight TN, McMillen TS, Oram JF, et al. Serum amyloid A facilitates the binding of high‐density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler Thromb Vasc Biol. 2011;31:1326–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Eckhardt ER, Witta J, Zhong J, Arsenescu R, Arsenescu V, Wang Y, et al. Intestinal epithelial serum amyloid A modulates bacterial growth in vitro and pro‐inflammatory responses in mouse experimental colitis. BMC Gastroenterol. 2010;10:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Smole U, Gour N, Phelan J, Hofer G, Köhler C, Kratzer B, et al. Serum amyloid A is a soluble pattern recognition receptor that drives type 2 immunity. Nat Immunol. 2020;21:756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Webb NR, De Beer MC, Wroblewski JM, Ji A, Bailey W, Shridas P, et al. Deficiency of endogenous acute‐phase serum amyloid A protects apoE‐/‐ mice from angiotensin II‐induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2015;35:1156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Krishack PA, Bhanvadia CV, Lukens J, Sontag TJ, De Beer MC, Getz GS, et al. Serum amyloid A facilitates early lesion development in Ldlr‐/‐ mice. J Am Heart Assoc. 2015;4:e001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Krishack PA, Sontag TJ, Getz GS, Reardon CA. Serum amyloid A regulates monopoiesis in hyperlipidemic Ldlr−/− mice. FEBS Lett. 2016; 590:2650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lee J‐Y, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, et al. Serum amyloid A proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell 2020;180:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hu Z, Bang YJ, Ruhn KA, Hooper LV. Molecular basis for retinol binding by serum amyloid A during infection. Proc Natl Acad Sci USA. 2019;116:19077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ather JL, Dienz O, Boyson JE, Anathy V, Amiel E, Poynter ME. Serum amyloid A3 is required for normal lung development and survival following influenza infection. Sci Rep. 2018;8:16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liu J, Wang D, Li S‐Q, Yu Y, Ye RD. Suppression of LPS‐induced tau hyperphosphorylation by serum amyloid A. J Neuroinflammation. 2016;13:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ather JL, Poynter ME. Serum amyloid A3 is required for normal weight and immunometabolic function in mice. PLoS One 2018;13:e0192352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhang G, Liu J, Wu L, Fan Y, Sun L, Qian F, et al. Elevated expression of serum amyloid A 3 protects colon epithelium against acute injury through TLR2‐dependent induction of neutrophil IL‐22 expression in a mouse model of colitis. Front Immunol. 2018;9:1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Den Hartigh LJ, Wang S, Goodspeed L, Ding Y, Averill M, Subramanian S, et al. Deletion of serum amyloid A3 improves high fat high sucrose diet‐induced adipose tissue inflammation and hyperlipidemia in female mice. PLoS One 2014;9:e108564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Choudhary S, Santone E, Yee SP, Lorenzo J, Adams DJ, Goetjen A, et al. Continuous PTH in male mice causes bone loss because it induces serum amyloid A. Endocrinology 2018;159:2759–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Djurec M, Graña O, Lee A, Troulé K, Espinet E, Cabras L, et al. Saa3 is a key mediator of the protumorigenic properties of cancer‐associated fibroblasts in pancreatic tumors. Proc Natl Acad Sci USA. 2018;115:E1147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Thompson JC, Jayne C, Thompson J, Wilson PG, Yoder MH, Webb N, et al. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J Lipid Res. 2015;56:286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Choi M, Kim MO, Lee J, Jeong J, Sung Y, Park S, et al. Hepatic serum amyloid A1 upregulates interleukin‐17 (IL‐17) in γδ T cells through Toll‐like receptor 2 and is associated with psoriatic symptoms in transgenic mice. Scand J Immunol. 2019;89:e12764. [DOI] [PubMed] [Google Scholar]
- 112. Ji YR, Kim HJ, Bae KB, Lee S, Kim MO, Ryoo ZY. Hepatic serum amyloid A1 aggravates T cell‐mediated hepatitis by inducing chemokines via Toll‐like receptor 2 in mice. J Biol Chem. 2015;290:12804–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Liang TS, Wang J‐M, Murphy PM, Gao J‐L. Serum Amyloid A is a chemotactic agonist at FPR2, a low‐affinity N‐formyl peptide receptor on mouse neutrophils. Biochem Biophys Res Commun. 2000;270:331–5. [DOI] [PubMed] [Google Scholar]
- 114. Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ, et al. A seven‐transmembrane, G protein‐coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chen M, Zhou H, Cheng N, Qian F, Ye RD. Serum amyloid A1 isoforms display different efficacy at Toll‐like receptor 2 and formyl peptide receptor 2. Immunobiology 2014;219:916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chen X, Zhuo S, Zhu T, Yao P, Yang M, Mei H, et al. FPR2 deficiency alleviates diet‐induced insulin resistance through reducing body weight gain and inhibiting inflammation mediated by macrophage chemotaxis and M1 polarization. Diabetes 2019;68:1130–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Dufton N, Hannon R, Brancaleone V, Dalli J, Patel HB, Gray M, et al. Anti‐Inflammatory role of the murine formyl‐peptide receptor 2: ligand‐specific effects on leukocyte responses and experimental inflammation. J Immunol. 2010;184:2611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Postulated receptor‐mediated functions of SAA.
Data Availability Statement
Not applicable.