

Summary
Dysfunction of the immune system underlies a plethora of human diseases, requiring the development of immunomodulatory therapeutic intervention. To date, most strategies employed have been focusing on the modification of T lymphocytes, and although remarkable improvement has been obtained, results often fall short of the intended outcome. Recent cutting‐edge technologies have highlighted macrophages as potential targets for disease control. Macrophages play central roles in development, homeostasis and host defence, and their dysfunction and dysregulation have been implicated in the onset and pathogenesis of multiple disorders including cancer, neurodegeneration, autoimmunity and metabolic diseases. Recent advancements have led to a greater understanding of macrophage origin, diversity and function, in both health and disease. Over the last few years, a variety of strategies targeting macrophages have been developed and these open new therapeutic opportunities. Here, we review the progress in macrophage reprogramming in various disorders and discuss the potential implications and challenges for macrophage‐targeted approaches in human disease.
Keywords: macrophages, polarization, reprogramming
Macrophages play roles at the heart of normal tissue physiology and in many diseases and show impressive capacity to adapt to their environment. Strategies to reprogramme macrophages represent new treatment options to be explored and exploited.

Abbreviations
- AAV
adeno‐associated virus
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- BBB
blood–brain barrier
- BMDM
bone marrow‐derived macrophage
- CIA
collagen‐induced arthritis
- CNS
central nervous system
- DC
dendritic cell
- EAE
experimental autoimmune encephalomyelitis
- EGFR
epidermal growth factor receptor
- FOXO3
forkhead box transcription factor family O 3
- GA
glatiramer acetate
- GM‐CSF
granulocyte–macrophage CSF
- HDAC
histone deacetylase
- ID3
inhibitor of DNA 3
- IFN‐γ
interferon‐gamma
- IL
interleukin
- IRF5
interferon regulatory factor 5
- KC
Kupffer cell
- LXR‐α
liver X receptor‐alpha
- mAB
monoclonal antibody
- MBC
metastatic breast cancer
- MCRPC
metastatic castration‐resistant prostate cancer
- M‐CSF
macrophage colony‐stimulating factor
- M‐CSFR
M‐CSF receptor
- MHCI
major histocompatibility class I
- MI
myocardial infarction
- mo‐KC
monocyte‐derived KC
- MS
multiple sclerosis
- MФ
macrophage
- NAFLD
non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- NO
nitric oxide
- NRF2
nuclear factor erythroid 2‐related factor 2
- PDAC
pancreatic ductal adenocarcinoma
- PGM
proneural glioblastoma multiforme
- RA
rheumatoid arthritis
- scRNAseq
single‐cell RNA sequencing
- TAM
tumour‐associated macrophages
- TGF‐β1
transforming growth factor beta 1
- TLR
Toll‐like receptor
- TNBC
triple negative breast cancer
- TREM2
triggering receptor expressed on myeloid cells‐2
- TTP
tristetraprolin
- VEGF
vascular endothelial growth factor
- Ywhaz
tyrosine 3‐monooxygenase/tryptophan 5‐monooxygenase activation protein zeta
Introduction
Historically considered as broad‐range phagocytes playing a relatively ‘passive’ role within the immune system, macrophages (MФ) have since benefited from more recent intensive characterization. Present in almost every tissue, MФ have been divided into two broad subclasses: those derived from an embryonic progenitor and those from adult monocytes. Many tissue‐resident MФ have a prenatal origin (yolk sac‐ or fetal liver‐derived), their development is dependent on at least one essential tissue‐specific transcription factor, and they maintain themselves by self‐renewal, while others are recruited from the peripheral monocyte pool. 1 The origins and replenishment of MФ populations have been the subject of numerous reviews2, 3, 4, 5, 6 and are not the focus of the present one. Tissue‐resident MФ are the predominant type of MФ present during steady state and are thought to monitor tissues, and maintain homeostasis, cellular communication and immune surveillance.6, 7 They also participate in developmental processes during embryogenesis.8, 9, 10, 11 Upon inflammation, whether induced by infection or injury, MФ are recruited in large numbers from circulating monocytes to the tissue and are often loosely classified as pro‐ or anti‐inflammatory. Previously, based on the actions of interferon‐gamma (IFN‐γ) and interleukin (IL)‐4 on MФ activation and with analogy to the Th1/2 T‐cell subsets, MФ were divided into two subtypes: ‘M1’ MФ, ‘classically activated’ by IFN‐γ; and ‘M2’ MФ, ‘alternatively activated’ by type 2 anti‐inflammatory cytokines such as IL‐10. Those two subtypes of MФ exhibit distinct metabolic function, the ‘M1’ having an anaerobic profile, based on glycolysis and production of nitric oxide (NO), whereas the ‘M2’ MФ have an aerobic one, based on oxidative phosphorylation and production of arginase.12, 13, 14 However, with current knowledge on MФ origin, diversity and significant plasticity,15, 16 the acceptance of this dogma has diminished. Recent cutting‐edge technological developments such as single‐cell RNA sequencing (scRNAseq), advanced animal genetic modification and intravital microscopy17, 18, 19, 20 have allowed for a greater appreciation of cellular diversity than previously acknowledged. MФ not only have impressive variability in their gene expression but they are also phenotypically plastic, allowing them to adapt to their environment and ensure appropriate responses. Tissue‐specific transcriptional programmes, instigated by local signals, enable phenotypic specialization in discrete microenvironmental niches controlled by differential transcription factor usage.21, 22 Dysfunction of MФ behaviour or phenotype has been associated with the development of many conditions such as neurodegeneration, arthritis, chronic inflammation, atherosclerosis and cancer,23, 24 and the identification of distinct subpopulations of MФ may be key in disease understanding and treatment.25, 26, 27
All MФ rely on specific cytokine availability for survival, proliferation and phenotype, including macrophage colony‐stimulating factor (M‐CSF), granulocyte–macrophage CSF (GM‐CSF), IL‐34 and transforming growth factor beta 1 (TGF‐β1), as single factor or in combination.6, 28, 29, 30 MФ programming strongly depends on the environment, which profoundly affects the cells at a transcriptomic and epigenetic level.21, 22, 31 MФ also demonstrate remarkable immune memory or ‘trained immunity’ capacities32, 33, 34, 35 based on epigenetic modification following stimulation and long‐term priming or ‘imprinting’, as seen with apoptotic cell recognition leading to a relatively stable tolerant state.36, 37, 38 This immune memory appears critical during transplant, when monocytes and MФ acquire specific memory to major histocompatibility class I (MHCI). 39 However, once isolated for ex vivo study, the characteristic gene signature of tissue‐resident MФ, including epigenetic modification,22, 40, 41 is often lost, indicating the need to explore their function and reprogramme them preferentially in vivo, or with the in vivo context considered.
The aim of this review was to have a broad overview of current research and possible treatments directed at MФ in a selection of disease contexts. We have confined the focus to specific organs and conditions detailed below, purely as exemplars of the kinds of approaches that are being considered. However, the potential for MФ targeting should not be considered restricted to the provided examples.
Methods to reprogramme macrophages
MФ exhibit a high degree of plasticity in response to environmental signals, many of which are tissue‐ and context‐specific. This results in a variety of MФ subtypes with different origins, which may play distinct roles in human disease and potentially provide unique opportunities for targeted therapies.26, 27, 42 Before considering detailed examples of therapeutic approaches in specific tissue and disease contexts, we briefly introduce some common methods used to reprogramme MФ, from conventional approaches, such as targeted antibody treatments and small molecule drugs to cutting‐edge technology of gene expression modification using viral vectors, artificial DNA carriers, naked DNA and cell therapy (Figure 1).
Figure 1.
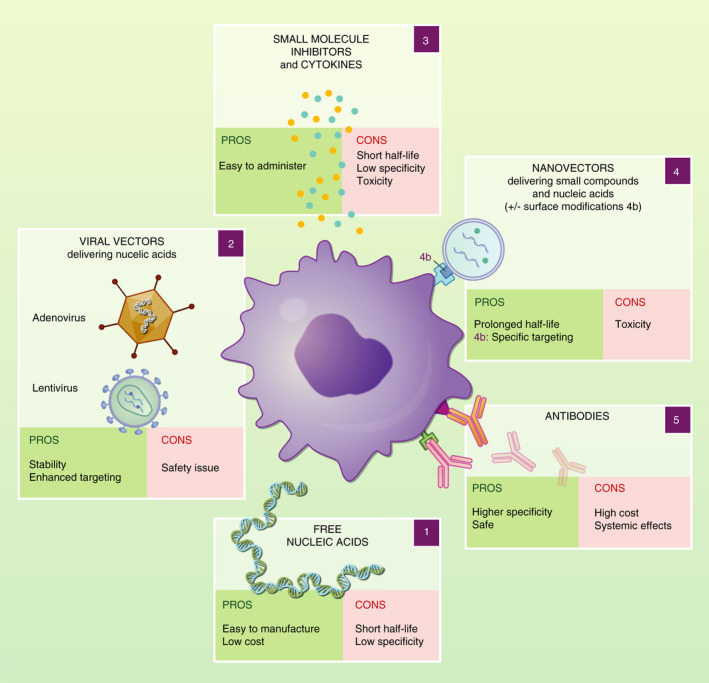
Summary of macrophage manipulation techniques for therapeutic purpose. These strategies can directly be applied in vivo, as well as in vitro followed by adaptive transfer of manipulated MФ. Free nucleic acids (1) can be manufactured easily and are very successfully used in some tissues including lungs and skeletal muscle. However, they lack MФ specificity and are rapidly cleared from the environment, mostly by circulating enzymes and kidney. Viral vectors (2) can be employed to deliver nucleic acids, preventing clearance from the system. Depending on the type of vector, gene manipulation can be long term (lentivirus) or transient (adenovirus). Viral vectors are highly efficient and can be modified to improve MФ targeting. However, they do entail safety considerations for patients and manufacturing staff. Free small molecules and cytokines (3) are known to act on MФ polarization. They are easy to administer but prone to degradation. They are also often not MФ specific and can cause off‐target effects and toxicity. Encapsulation of nucleic acids, small molecules and cytokines into nanovectors (4) prolongs their half‐life in the organism, while surface modifications (4b) allow targeting of specific cell types. Antibodies (5) can manipulate MФ polarization by directly binding Fc or other cell surface receptors. While they are generally safe, high doses are often required for therapeutic efficacy translating into high costs
Targeted antibody treatments are among the easiest and most efficient methods to target not only MФ surface receptors involved in the regulation of immune responses 43 but also circulating cytokines/growth factors, preventing their interaction. 44 As a result, antibodies can alter MФ activation status. However, this technique is mostly systemic and can lead to numerous off‐target effects.
Gene therapy aims to alter specific gene expression by inserting genetic material into the target cell. Free nucleic acids can be directly injected in vivo. While this is generally considered safe, 45 detection by MФ can induce inflammatory signalling. Although possibly advantageous when reprogramming MФ into pro‐inflammatory phenotypes, it may counter anti‐inflammatory states and raises concern of off‐target effects. Free nucleic acids also lack cell‐targeting specificity, an issue that can be solved by attachment to carrier molecules, such as coupling to peptides directly targeting MФ cell surface receptors. 46
Nucleic acids can also be introduced using modified viral vectors that lack the genes necessary for replication. Lentiviral vectors stably integrate genetic material into the host cell genome, while adenoviruses and adeno‐associated viruses (AAVs) only cause transient gene expression. Despite their high efficacy, viral vectors are associated with significant disadvantages: from a manufacturing perspective, viral vector production is costly and requires specific safety measurements. 47 From a clinical point of view, random lentiviral RNA insertion into the genome could cause tumour suppressor gene disruption triggering malignancy. 48 Also, viral vectors bear the risk of potentially high immunogenicity.49, 50 Adenoviruses’ triggering of immune responses could, however, be exploited for the use in tumour contexts where immune activation could be beneficial.
Another option for delivery of nucleic acids are non‐immunogenic nanoparticles (<100 nm), and organic (e.g. liposome, polymers) or inorganic (e.g. gold, silica) particles widely used in clinical applications 51 that are readily ingested by MФ. Different types of nanovectors vary in their advantages in clinical applications 52 and can inherently favour polarization towards either end of the MФ activation spectrum. 53
The use of nanoparticles in not restricted to nucleic acid delivery. MФ can also be targeted with compounds acting on signalling pathways to promote polarization such as cyclooxygenase‐2 inhibitors, 54 receptor tyrosine kinase inhibitors 55 and histone deacetylase (HDAC) inhibitors, 56 as well as small molecule Toll‐like receptor (TLR) agonists 57 and possibly cytokines. 58 Free small molecules can be associated with systemic side effects, while unprotected molecules and cytokines are relatively unstable in vivo. 59 Such limitations can be overcome by encapsulation into nanovectors.60, 61 In general, nanoparticles are well tolerated, small enough to cross physiological barriers including the blood–brain barrier (BBB) and easily modified to allow cell‐targeted delivery. They do, however, need to be carefully manufactured considering possible toxicity associated with different materials and delivery routes, as well as inflammatory responses associated with uptake by MФ. 62
Blood and bone marrow‐derived MФ (BMDM) can be reprogrammed ex vivo by the same aforementioned methods and adoptively transferred to individuals. This may help alleviate some of the off‐target limitations of directly targeting MФ in vivo. 63 To retain polarization stability, MФ can be genetically engineered ex vivo to over‐ or under‐express factors associated with polarization phenotypes. An intriguing option was proposed by Shields and colleagues: ex vivo attachment of IFN‐γ‐loaded phagocytosis‐resistant ‘backpacks’ to BMDM enabled slow release of IFN‐γ in vivo, allowing injected cells to maintain a pro‐inflammatory phenotype while simultaneously polarizing tumour‐infiltrating MФ. 64
The type of protocol used for therapeutic intervention will depend on the tissue, MФ subtype and pathology to be treated. Recently, a first‐in‐human phase 1 dose–escalation trial confirmed the safety of autologous MФ therapy in end‐stage liver disease, which was well tolerated and is currently undergoing efficacy measures in an ongoing phase 2 randomized controlled trial. 65
Tissue‐specific consideration in macrophage reprogramming
Cardiac macrophages
Mouse cardiac MФ are composed of four different subsets, distinguishable by their cell surface expression of typical markers such as MHCII, Ly6C, CCR2, CD11c, MerTK, CD206 and CD64, and are derived from yolk sac and fetal liver. 66 Their self‐renewal capacity decreases with age, and they are thought to be gradually replenished by monocyte‐derived MФ over time. 67 Cardiac MФ play important roles in tissue homeostasis, angiogenesis and vascular remodelling during embryogenesis, as well as in the action potential propagation by being electronically coupled with cardiomyocytes.68, 69, 70 During inflammation or following injury such as myocardial infarction (MI), resident cardiac MФ become activated and high numbers of monocyte‐derived MФ are recruited to the injured site where their heterogeneity is thought to impact MI outcome.71, 72, 73 Recruited cardiac MФ participate in all phases of MI, from the first acute inflammatory wave to the reparative phase and the promotion of angiogenesis and muscle regeneration.74, 75 The repair after MI is associated with scar formation and fibrosis, decreasing cardiac functionality. 76 Interestingly, the resident cardiac MФ seem to limit adverse remodelling 77 and Gata6+ pericardial MФ have recently been shown to enter the site of injury and prevent fibrosis. 78 MI is a leading cause of death worldwide. 79 Clinical trials mainly focus on decreasing systemic inflammation by suppressing the immune system and inflammation with corticosteroid, 80 methotrexate (phase 3, completed and recruiting) or blocking pro‐inflammatory molecules such as IL‐6 (tocilizumab, phase 2, active) and IL‐1β (Anakinra, completed 81 ). However, the use of broad‐range immunosuppressors is questionable, as a controlled first phase of inflammation has been shown to be essential to tissue regeneration. 82 In this line, recent work has shown that a located injury such as MI not only modifies the local MФ pool, but also affects the number and molecular signature of off‐site MФ throughout the organism (liver, lung and kidney), 83 placing the effect of systemic drugs in an even more central question. Recent research focusing on the modulation of cardiac MФ phenotype and activity in animal models shows promising results encouraging further human clinical trials. Using nanoparticle‐delivered siRNA, Courties et al. successfully silenced interferon regulatory factor 5 (IRF5) in cardiac MФ in a mouse model of MI, which improved healing. 84 Similarly, the targeting of miRNA‐21 in cardiac MФ with nanoparticles containing mimics (intravenously injected) or adenovirus particles overexpressing miR‐21 (intramyocardially injected) reduced inflammation, fibrosis and cardiac dysfunction.85, 86 Finally, the intramyocardial transplantation of in vitro M‐CSF‐ and IL‐4‐primed ‘reparative’ MФ following MI in mice showed beneficial effects over transplantation of non‐primed bone marrow mononuclear cells. 87 While human assays to modify MФ in vivo has been less prominent, recent work explored the MФ heterogeneity and identified at least two subsets (CCR2+ and CCR2‐) present in human heart which are essential for tissue function. 88
Macrophages in the central nervous system
Resident myeloid cells in the central nervous system (CNS) include parenchymal microglia and different types of non‐parenchymal MФ. 89 Additionally, inflammatory conditions can trigger the influx of monocyte‐derived MФ 90 through disruption of the BBB. Under physiological conditions, this layer of endothelial cells restricts the crossing of cells and compounds into the CNS as previously reviewed. 91 Both dysregulated resident and infiltrating MФ have been linked to the pathophysiology of several neurological disorders including Alzheimer's disease (AD),92, 93 multiple sclerosis (MS) 94 and amyotrophic lateral sclerosis (ALS). 95 Currently, no cure exists for those conditions and potential preventative and early treatment options face the difficulty of drug delivery.
Multiple sclerosis
MS is an autoimmune, demyelinating disease of the brain and spinal cord. It affects young adults causing progressive neurological deterioration with common symptoms including numbness, burning sensations, visual impairment, loss of balance, bladder dysfunction, fatigue and depression. 96 While the aetiology of MS is incompletely understood, it is believed that CNS‐infiltrating pro‐inflammatory phagocytes are key drivers in tissue destruction. 94 Indeed, in MS patients such cells have been found to express reduced levels of SHP‐1 causing increased activation of STAT1, STAT6 and NF‐κB. 97 This was associated with a pro‐inflammatory phenotype characterized by elevated proteinases including ADAM8, which could disrupt the BBB and contribute to demyelination, as well as increased molecules associated with antigen presentation and costimulation. 98 This view is supported by studies of experimental autoimmune encephalomyelitis (EAE), the murine model for MS. 99 Depletion of infiltrating phagocytes protected mice form axonal damage, 100 while blocking microglial release of nitrite and pro‐inflammatory chemokines and cytokines significantly reduced clinical signs in the EAE model. 101 The polarization of microglia by molecular control of cytokines and costimulatory molecules by nuclear receptor has been shown to be essential for the onset and development of EAE. 102 EAE has limitations concerning translation to human disease but does still play an important role in drug development. 103
Glatiramer acetate (GA) is a polymer approved for the treatment of relapsing–remitting MS. While its mode of action is attributed to T‐cell manipulation, GA also increases microglial phagocytic activity and IL‐10 production while decreasing TNF in vitro. 104 GA likely does not cross the BBB on its own. However, it can be taken up by dendritic cells (DCs) and released in the CNS, with GA uptake promoting trans‐endothelial migration of DCs. 105 Alternatively, GA could enter the CNS when the BBB is disrupted in active MS lesions. Studies report GA‐mediated reductions in relapse rates; 106 however, it is still unclear whether its effect on microglia contributes to this and whether GA actually slows disease progression. 107 As GM‐CSF has been implicated in disease induction in the EAE model, MOR103, a monoclonal antibody (mAb) directed against GM‐CSF, has been tested in MS patients and was found to be well tolerated, although no assessment regarding efficacy was conducted at this stage (NCT01517282).
While infiltrating pro‐inflammatory phagocytes are potentially detrimental in MS, microglial production of pro‐inflammatory factors, especially TNF, might support remyelination, 108 calling for a specific targeting of peripheral MФ. Indeed, preventing peripheral MФ entry into the CNS was beneficial in EAE. 109
Therapeutics aiming to interfere with peripheral MФ need to be carefully considered to not affect microglia populations. In EAE, scRNAseq recently identified eight different monocyte subsets, one of which expressing CXCL10+ was associated with a pathogenic gene signature. This subset had also been observed in other inflammatory models such as pathogen infection, and its depletion in EAE was associated with clinical improvement. 110 Whether and how this finding translates into the human context remains to be investigated.
Alzheimer's disease
AD is a chronic neurodegenerative disorder associated with CNS inflammation and the main cause of dementia. Genome‐wide association studies have uncovered a variety of loci increasing susceptibility to the development of late‐onset Alzheimer’s disease, many of which are associated with immunity. 93 In addition, rare coding variants, such as that of triggering receptor expressed on myeloid cells 2 (TREM2), have strongly been linked to increased risk of developing AD. 111 In line with this, in a murine AD model expressing a TREM2 risk variant (R47H), anti‐human TREM2 mAb increased microglial proliferation, reduced neuroinflammation and was associated with cognitive benefits. 112 The same antibody was well tolerated in a phase I clinical trial (NCT03635047).
Activated microglia are thought protective early in disease but over time become dysfunctional causing inflammatory injury. 113 Importantly, scRNAseq identified distinct microglia subsets in AD, including a type associated with neuroprotection and increased phagocytosis also found in a mouse model of ALS. 25
An important regulator in microglial differentiation is the M‐CSF receptor (M‐CSFR): depending on the murine model and dose of inhibitor used, inhibition of M‐CSFR signalling resulted in blockade of microglial proliferation114, 115 or microglial depletion 116 inducing an anti‐inflammatory state and cognitive improvements not associated with alterations in amyloid plaques. Together, these and other studies suggest M‐CSFR inhibition as a viable option for clinical trials. Importantly, M‐CSFR expression is not restricted to microglia but important for all macrophages. In line with this, M‐CSFR inhibition was shown to also affect circulating and tissue‐resident macrophages and lymphocytes in other organs in a mouse model. 117 In this paper, the M‐CSFR inhibitor PLX5622 was administered through diet. While not investigated by the authors, it is likely that such a method of drug delivery would also affect the gut microbiome. Considering the link between intestinal dysbiosis and neurodegeneration, 118 such a method could affect treatment outcomes and a more localized way of drug delivery, potentially by intranasal 119 or direct intraventricular/intrathecal administration, 120 could minimize off‐target effects.
Amyotrophic lateral sclerosis
ALS is characterized by motoneuron degeneration typically causing paralysis and death within five years of diagnosis. In murine models, the number of resident microglia increases throughout disease progression concomitant with a switch from an anti‐ to a pro‐inflammatory phenotype. 121 Simultaneously, peripheral monocytes from ALS patients have been suggested to be more readily activated into pro‐inflammatory phenotypes compared with those of healthy individuals, further driving neuroinflammation after CNS infiltration. 122
Vascular endothelial growth factor (VEGF) has been suggested to play a neuroprotective role by reducing motor neuron death through downregulation of pro‐inflammatory cytokine production. 123 Intrathecal injection of adeno‐associated virus containing VEGF expressing plasmid induced VEGF expression in motor neurons in a mouse model of ALS. This prolonged survival of the mice by two modes of action: increasing anti‐apoptotic factors such as Bcl‐2 and decreasing pro‐apoptotic ones such as Bax, caspase‐3 and caspase‐9 in neurons and a switch in the inflammation balance (reduction in the pro‐inflammatory TNF, IL‐1β and CD68 while increase in anti‐inflammatory TGF‐β released by microglia). 124 Unfortunately, phase II clinical trials for intracerebroventricular administration of the common splicing isoform VEGF165 were terminated due to a lack of favourable benefit–risk ratio (NCT01384162).
The involvement of pro‐inflammatory MФ and microglia in the pathophysiology of neuroinflammatory conditions strongly suggests the targeting of CNS myeloid cells for re‐education as a treatment option. However, to date no agent designed to manipulate MФ activation states in the CNS has been approved for the treatment of a neurological condition. This is partly due to poor animal models, which do not fully mimic human pathophysiology. It is essential to understand the heterogeneity of myeloid populations and individual contributions to neurological pathologies in the CNS, to specifically target disease‐associated cell types. Imaging mass cytometry is already used to characterize key players in neurological diseases by applying antibodies to post‐mortem sample. 125 Additionally, scRNAseq of human microglia from brain autopsy samples could also uncover cell‐specific targets for intervention that could aid the development of therapeutics. With potential targets being discovered, it is vital to also reconsider options for drug delivery: compounds delivered systemically can eventually reach the brain but will also affect cells in other tissues and organs. In particular, in the case of macrophage manipulation, many receptors and targets are shared by different tissue‐resident macrophage populations making it difficult to prevent off‐target effects. This issue could be overcome by exploring intracerebral, 126 interstitial 127 or intranasal 128 delivery methods.
Liver macrophages
The murine liver comprises two distinct populations of tissue‐resident MФ, the Kupffer cells (KCs) that occupy the sinusoidal vascular space, and the phenotypically distinct liver capsular MФ, which reside in the hepatic capsule. 129 Additionally, the liver may also contain monocyte‐derived and peritoneal MФ, 86 which are recruited following inflammatory events or injury.130, 131, 132 Liver MФ heterogeneity was recently reviewed.130, 131 In mice, fetal liver monocytic precursors give rise to embryonic KCs, which at steady‐state maintain the KC pool through self‐renewal, independent of BM‐derived progenitors.1, 133, 134 In contrast, liver capsular MФ arise entirely from adult circulating monocytes. 129 More recently however, BM‐derived monocytes were shown to populate the KC niche during postnatal liver development, contributing significantly to the adult KC population. 135 Following the loss of KCs after infection, 136 or experimental depletion,135, 137, 138 the KC pool is repopulated through proliferation of surviving KCs, as well as the recruitment and differentiation of blood monocytes into KCs (mo‐KCs). These mo‐KCs were shown to be highly functionally and transcriptionally homologous to their embryonic counterparts 135 after 30 days post‐depletion, although other studies have shown that embryonically derived KCs may exhibit some phenotypic and functional differences to their monocyte‐derived counterparts.137, 139 The replenishment of the depleted KC niche by either mechanism however, appears to be context‐dependent. Indeed, in an acute liver injury model, the depleted KC niche was replenished through proliferation of the surviving KCs without input from circulating monocytes. 140 Similar to the development and maintenance of other tissue‐resident MФ populations, recent studies have highlighted the critical role of transcription factors in governing KC development and identity, with the loss of inhibitor of DNA 3 (ID3) 141 or liver X receptor‐alpha (LXR‐α) 142 resulting in KC deficiency in mice. Two recent studies in mice demonstrated that liver‐derived signals orchestrate monocyte recruitment 143 and initiate and maintain KC identity through the induction of lineage‐determining factors including ID3 and LXR‐α by acting on pre‐existing but poised enhancers143, 144. Additionally, leveraging of scRNAseq technology for profiling human liver MФ has revealed distinct subpopulations of KCs with discrete gene expression signatures26, 145, 146, 147, but our understanding of their biology is still restricted compared with mouse liver MФ.
Liver MФ play a key role in the pathogenesis of acute and chronic liver diseases, including acute liver failure, alcoholic liver disease, non‐alcoholic fatty liver disease (NAFLD), viral hepatitis and hepatocellular carcinoma. They are also critically required for the restoration of tissue homeostasis and resolution of liver disease. 148 These seemingly contrasting roles for liver MФ highlight the importance to identify and understand the relative contributions that distinct subsets have in disease progression, as well as tissue repair, to help the development of improved‐targeted therapeutics, as well as the definition of biomarkers indicating either disease progression or regression (for a recent review, see Ref 131).
A recent study described a subset of Trem2hi MФ enriched in mouse models of non‐alcoholic steatohepatitis (NASH), as well as in human NASH livers, correlating with disease severity. 149 Interestingly, in a diet‐induced mouse model of NASH, KC identity was significantly altered by the NASH diet. NASH‐induced changes in KC enhancers and gene expression were driven by activator protein 1 and early growth response protein 1 inducing a scar‐associated MФ phenotype, with increased expression of both Trem2 and Cd9. 150 Interestingly, an independent study also identified a scar‐associated TREM2+CD9+ subpopulation of MФ in humans, which differentiate from circulating monocytes and expand during liver fibrosis. 26 In another study, a subset of MФ (MerTK+HLA‐DRhigh) was reported to expand during the resolution phase of acute liver disease, with a comparable population identified in mice during the resolution phase of an acute liver injury model. 151 Further studies are needed to fully define liver MФ subsets and their contributions to disease progression, as well as in tissue repair and the restoration of homeostasis. However, some approaches to target MФ in liver disorders are already under investigation.
Glucocorticoid and antibody–drug conjugate
Glucocorticoid receptor signalling modulates inflammation in KCs, suggesting that glucocorticoid treatment could serve as a potential MФ‐directed treatment for liver diseases. 152 Liposomal delivery of dexamethasone was shown to significantly reduce liver injury and fibrosis in experimental models of both acute and chronic liver injuries. 153 Direct targeting of MФ with an antibody–drug conjugate composed of dexamethasone linked to an antibody against CD163 (a scavenger receptor highly expressed in KCs and infiltrating monocytes/MФ) was shown to reduce inflammation, hepatocyte ballooning and fibrosis in a mouse model of NASH, while having no apparent systemic side effects. 154
Cytokines modulation
The pro‐inflammatory cytokine TNF plays a major part in the development of steatosis, inflammation and fibrosis in NAFLD. KCs have been identified as the main cellular source of TNF in mouse models of NAFLD. Mannose‐modified trimethyl chitosan–cysteine‐conjugated nanoparticles were used to deliver siRNA targeting TNF to MФ and protected mice from inflammation‐driven liver damage and lethality in an acute liver injury model. 155
Gene expression modification
Oxidative stress and the associated damage have been suggested to link obesity and liver disease. Recently, Azzimato and colleagues 156 showed that oxidative stress was triggered by obesity in mouse and human livers. In parallel, nuclear factor erythroid 2‐related factor 2 (NRF2), a transcription factor regulating the antioxidant response, was reduced, leading to an impaired antioxidant response. miR‐144 was greatly upregulated in the liver of obese and insulin‐resistant mice and humans and was shown to target NRF2. Consequently, delivery of an antagomiR targeting miR‐144 expression to MФ in vivo with glucan‐encapsulated RNA interference particle technology increased NRF2 protein levels, reduced oxidative stress and improved hepatic metabolism in insulin‐resistant mice.
Small molecule inhibitors
Galectin‐3 is a pleiotropic protein highly expressed by MФ in the liver and upregulated in models of liver disease.157, 158 Galectin‐3 deficiency in mice was protective in a concanavalin A‐induced liver injury model, reducing pro‐inflammatory cytokines, as well as attenuating fibrosis. 159 Similarly, pharmacological inhibition of Galectin‐3 in liver injury models significantly reduced fibrosis and led to a reversal in cirrhosis and is now under evaluation for NASH in clinical trials.157, 158
The dual CCR2/CCR5 inhibitor cenicriviroc was shown to reduce monocyte recruitment and liver injury in an acute liver failure model, 160 as well as ameliorating hepatic inflammation and fibrosis in experimental models of NASH, 161 and is also currently under evaluation in clinical trials for the treatment of liver fibrosis in NASH (NCT03059446 and NCT03028740). 162
Adoptive transfer
In an experimental model of acute liver injury, adoptive transfer of ex vivo IL‐4/IL‐13‐polarized BMDM rapidly reduced liver injury and several mediators of inflammation. 163 Of note, the adoptive transfer of primary human‐polarized monocyte‐derived MФ partially recapitulated the therapeutic effect observed with polarized mouse BMDM in the same mouse model. 163 Similarly, injection of BMDM or embryonic stem cell‐derived MФ reduced both fibrosis and improved liver regeneration in a hepatic injury and fibrosis model.164, 165 Clinical trials in humans have demonstrated the safety of administration of large and frequent infusions of autologous MФ. A recent first‐in‐human trial evaluated the safety of a single peripheral infusion of autologous MФ in end‐stage liver disease, which was well tolerated and led to a reduction in clinical scoring, and is currently undergoing efficacy measures in an ongoing phase 2 randomized controlled trial. 65 However, this study did not determine whether the infused MФ migrated to and engrafted in the liver. Previous studies in mice, as well as a case study in humans, suggest that the administration via peripheral or central veins MФ traffic from the pulmonary vasculature via the blood before engrafting in the liver and spleen.164, 166, 167, 168 Significant challenges remain for the adoption of autologous MФ therapies, such as their scalability, as well ensuring that engrafted cells maintain the intended phenotype, which can be greatly impacted by the tissue microenvironment.
Macrophages in arthritis
Rheumatoid arthritis (RA) is a chronic, systemic inflammatory autoimmune disorder that primarily affects synovial joints, leading to irreversible bone and cartilage destruction. Multiple studies have implicated monocytes and MФ in the initiation and progression of RA. 169 MФ are the most abundant immune cell and are a source of pro‐inflammatory cytokines associated with RA pathogenesis including TNF, IL‐6 and IL‐1β, 170 as well as chemo‐attractants and metalloproteinases. Conversely, synovial tissue MФ in healthy and RA patients in sustained remission suggest that MФ have a fundamental role in maintaining and/or reinstating synovial homeostasis.171, 172 In a murine model of sterile inflammatory arthritis, non‐classical Ly6C‐ but not Ly6C+ monocytes were reported to be crucial for the initiation of arthritis, while tissue‐resident synovial MФ restricted the development of arthritis. 173 Murine synovial lining CX3CR1+ MФ form an immunological barrier in the lining layer of the synovium of healthy joints. 174 Depleting them in a mouse model of arthritis disrupted barrier function and accelerated the onset and magnitude of arthritis, whereas depletion of CSF1R+ monocytes and MФ expedited the resolution of inflammation. Interestingly, comparison of scRNAseq data with human data sets from RA patients revealed significant overlap, suggesting cells similar to synovial lining MФ may also exist in humans. 174 A recent study described synovial MФ subsets enriched in active RA (MerTK‐ CD206‐) or sustained remission (MerTK+ CD206+), which are thought to contribute to RA pathogenesis or remission through the production of pro‐inflammatory cytokines or lipid mediators, respectively. 175 Similarly, IL1B+ pro‐inflammatory monocytes were enriched in synovial tissue from patients with RA, whereas NUPR1+ monocytes were inversely correlated with tissue inflammation. 176 Another study recently identified HBEGF+ inflammatory MФ enriched in the human RA tissues, 177 which promote synovial fibroblast invasiveness in an epidermal growth factor receptor (EGFR)‐dependent manner. Interestingly, a previous study showed that EGFR inhibition reduced the severity of established RA in mice. 178 Collectively, these studies demonstrate that distinct monocyte and MФ subsets have defined roles in RA. Strategies are currently being developed to target MФ and improve RA pathology.
Cytokines
Several lines of evidence suggest that MФ and GM‐CSF strongly influence the development and progression of RA.179, 180 In patients with RA, GM‐CSF is elevated in plasma, synovial fluid and synovial tissues, while administration of recombinant GM‐CSF has been reported to exacerbated RA disease activity. 181 Depletion of GM‐CSF or blockade of GM‐CSFRα in a mouse model of RA significantly reduced the number of MФ in the inflamed synovium, decreased synovial inflammation and joint destruction.182, 183 Therapeutic antibodies targeting GM‐CSF and its receptor have been developed and evaluated in clinical trials.181, 184 Collectively, these studies have shown that treatment is associated with rapid and sustained improvements in measures of RA disease outcomes, as well as being well tolerated in safety studies. 185 Of note, a phase III clinical trial is currently ongoing evaluating otilimab, a fully human anti‐GM‐CSF monoclonal antibody, in RA patients who have had an inadequate response to disease‐modifying anti‐rheumatic drugs and/or Janus kinase inhibitors (NCT04134728).
Gene therapy
In the collagen‐induced arthritis (CIA) mouse model, Kong and colleagues identified three key pro‐resolving factors that were elevated in mouse synovial tissues during the resolution phase of CIA. 186 Among them, tyrosine 3‐monooxygenase/tryptophan 5‐monooxygenase activation protein zeta (Ywhaz) was also shown to be elevated in rat synovial tissue in a CIA model during the resolution phase compared with the peak phase, as well as in RA patients who responded well to treatment with anti‐rheumatic drugs. Ywhaz is highly expressed in Tregs and MФ and is one of the most common proteins found in exosomes 187 from Tregs, although no data were shown for MФ in this study. Interestingly, treatment of thioglycollate‐elicited peritoneal MФ with recombinant Ywhaz reduced the expression of Tnf and Il6, while enhancing Il10 expression. 186 Furthermore, siRNA knockdown of Ywhaz or treatment with an anti‐Ywhaz antibody enhanced the expression of Tnf and Il6 following LPS stimulation, supporting a role of Ywhaz in modulating pro‐inflammatory cytokine production in MФ. 186 Intra‐articular delivery of adenovirus expressing Ywhaz suppressed the production of pro‐inflammatory cytokines and significantly reduced synovial inflammation and joint destruction in mice. 186 Ywhaz is a member of the 14‐3‐3 protein family, which modulate the activity of binding partners by controlling protein localization, stability, conformation and activity mainly through phosphoserine/threonine motifs. 188 Previous studies have suggested that Ywhaz can modulate the activity of forkhead box transcription factor family O 3 (FOXO3), a transcription factor regulating the expression of cytokines, 189 and tristetraprolin (TTP), 190 which regulates cytokine production by destabilizing target mRNA molecules. 191 However, further studies are needed to understand how Ywhaz regulates the production of cytokines and the subsequent resolution of arthritis. In arthritic rats, MФ phenotype has been manipulated in vivo by tuftsin‐modified nanoparticle‐mediated delivery of plasmid encoding IL‐10, leading to a significant reduction in pro‐inflammatory cytokine production, diminishing inflammation and preventing the progression of joint damage. 192
Tumour‐associated macrophages
Cancers are heterogeneous tissues with tumour‐infiltrating immune cells playing key roles in disease progression. 193 Tumour‐associated macrophages (TAMs) are major components of the immune cells infiltrating solid tumours. They are a heterogeneous population often coexpressing pro‐ and anti‐inflammatory markers. 194 While pro‐inflammatory functions could aid tumour cell elimination, 195 immune‐suppressive TAMs are linked to worse prognosis due to their contribution to tumour growth and metastasis.196, 197 TAMs repolarization to produce inflammatory mediators is currently being evaluated in a number of clinical trials, examples of which are summarized in Table 1.198, 199, 200, 201, 202, 203, 204, 205, 206 Several strategies to target TAMs have been adopted, and we will describe common ones below.
Table 1.
Selective examples of TAMs reprogramming compounds currently undergoing clinical trials updated
| Compound | Target | Clinical phase | Clinicaltrials.gov identifier | Status | Results | Type of malignancy |
|---|---|---|---|---|---|---|
| Imiquimod, cyclophosphamide and radiotherapy | TLR7 | Phase II | NCT01421017 | Complete | No results available | Skin metastasis in breast cancer |
| Imiquimod together with Abraxane | TLR7 | Phase II | NCT00821964 | Complete | Pathologic clinical response in 71·4% of patients | Advanced breast cancer 200 |
| Imiquimod | TLR7 | Phase II | NCT00031759 | Complete | No impact on recurrence of cervical dysplasia | Cervical cancer 201 |
| Imiquimod | TLR7 | Phase III | NCT00941252 | Complete | Histologic regression in 73% of patients | Cervical intraepithelial neoplasia 202 |
| Imiquimod | TLR7 | Phase III | NCT01861535 | Active, not recruiting | ‐ | Vulvar intraepithelial neoplasia |
| Imiquimod | TLR7 | Phase III | NCT02394132 | Recruiting | ‐ | Complex lentigo maligna |
| Imiquimod | TLR7 | Phase IV | NCT01161888 | Complete | No results available | Lentigo malignant of the face |
| Resiquimod | TLR7/8 | Phase I/II | NCT01676831 | Complete | Significant improvements of treated lesions in 75% of patients, clearing of all treated lesions in 30% | Cutaneous T‐cell lymphoma 203 |
| 852A | TLR7 | Phase I | NCT00095160 | Complete | No results available | Refractory solid organ tumours |
| 852A | TLR7 | Phase II | NCT00319748 | Complete | Evidence of immune activation as evaluated by cytokine production | Breast, ovarian, endometrial and cervical cancers |
| 852A | TLR7 | Phase II | NCT00189332 | Complete | No results available | Metastatic cutaneous melanoma |
| Imo‐2055 | TLR9 | Phase II | NCT00729053 | Complete | Treatment‐emergent adverse events observed in > 90% of patients | Renal cell carcinoma |
| CD40 mAb CP‐870,893 | CD40 | Phase I | NCT02225002 | Complete | No results available | Advanced solid tumours |
| CD40 mAb CP‐870,893 and gemcitabine | CD40 | Phase I | NCT01456585 | Complete | No results available | Pancreatic ductal adenocarcinoma 204 |
| CD40 mAb CP‐870,893 and chemotherapy | CD40 | Phase I | NCT00711191 | Complete | Partial response in 4/21 patients, stable diseases in 11/21 patients | Advanced cancer of the pancreas 205 |
| Vorinostat, gefitinib | HDAC | Phase I | NCT02151721 | Unknown | ‐ | EGFR mutant lung cancer |
| IPI‐549 alone and with nivolumab | PI3Kγ | Phase I | NCT02637531 | Recruiting | ‐ | Advanced solid tumours |
| IPI‐549 with Tecentriq and Abraxane/Avastin | PI3Kγ | Phase II | NCT03961698 | Recruiting | ‐ | Breast cancer, renal cell carcinoma |
| IPI‐549 | PI3Kγ | Phase II | NCT03795610 | Recruiting | ‐ | Locally advanced HPV + and HPV‐ head and neck squamous cell carcinoma |
| IPI‐549 with nivolumab | PI3Kγ | Phase II | NCT03980041 | Recruiting | ‐ | Advanced urothelial carcinoma |
| BLZ945 monotherapy or combination with PDR001 | M‐CSFR (+/‐ PD‐1 blockade) | Phase I/II | NCT02829723 | Recruiting | ‐ | Advanced solid tumours |
|
PLX3397 + radiation therapy + temozolomide |
M‐CSFR (+ cKit, Flt3) | Phase Ib/II | NCT01790503 | Complete | Stable disease in 24/50 patients, complete response in 2/50, partial response in 5/50 patients | Glioblastoma |
| PLX3397 and sirolimus | M‐CSFR (+ cKit, Flt3) | Phase I/II | NCT02584647 | Recruiting | ‐ | Unresectable sarcoma and malignant peripheral nerve sheath tumours |
| MCS110 with carboplatin and gemcitabine | M‐CSF | Phase II | NCT02435680 | Complete | No results available | Advanced triple negative breast cancer with high TAMs |
| MCS110 with PDR001 | M‐CSFR (+/‐ PD‐1 blockade) | Phase Ib/II | NCT02807844 | Complete | Partial response in 1/48, stable disease in 9/48 patients | Advanced malignancies 206 |
| LY3022855 | M‐CSFR | Phase I | NCT02265536 | Complete | Stable disease in 5/22 MBC and 3/7 MCPRC patients |
Metastatic breast cancer (MBC) Metastatic castration‐resistant prostate cancer (MCRPC) 207 |
Receptor targeting
Just as in other tissues, the M‐CSFR axis is an attractive target in the tumour context: in many pre‐clinical models, the use of M‐CSFR inhibitors is associated with MФ depletion and tumour regression in a T‐cell‐dependent manner. 207 However, blocking M‐CSFR signalling in murine proneural glioblastoma multiforme (PGM) and hepatocellular carcinoma caused tumour regression without TAM depletion, with persisting TAMs associated with functional alterations.208, 209 M‐CSFR signalling can be blocked by the use of antibodies targeting either M‐CSF or its receptor. While in some clinical trials antibody treatment achieved disease stabilization, 210 other studies report TAM reductions without anti‐tumour activity. 211
Another option is the use of small molecule inhibitors acting on the receptor tyrosine kinase domain (e.g. PLX3397, BLZ945). Those inhibitors also interfere with other receptors expressed in myeloid and tumour cells, which could enhance their efficacy. As it has been shown in a PGM model that tumours can acquire resistance to BLZ945‐mediated inhibition, 212 the combination of M‐CSFR inhibitors with other immune‐ or chemotherapeutic agents could be beneficial and is being evaluated in clinical trials.
TLRs are involved in immune surveillance, and their agonists can drive pro‐inflammatory mediator release. 213 Imiquimod (TLR7 agonist) is already approved for topical therapy in squamous and basal cell carcinoma, and clinical trials are ongoing for several other TLR agonists. Imiquimod’s mechanism of action likely affects not only MФ but also dendritic cells and neutrophils. 214 in vivo, many TLR agonists have short half‐lives, and some free agonists have been associated with toxicity. Such issues could be prevented by encapsulation into nanovectors. While no such strategies are currently in clinical trials, murine models support the efficacy and safety of resiquimod‐loaded nanoparticles 61 and ferumoxytol‐linked Poly(I:C). 215
Another example of receptor targeting exploits CD40, a costimulatory molecule expressed on the surface of myeloid cells and B cells, with key roles in immune regulation. 216 Ligation with an agonist CD40 mAb triggered T‐cell‐dependent anti‐tumour immunity in murine models of pancreatic ductal adenocarcinoma (PDAC) associated with increased MHCII, CD80 and CD86 expression on MФ and elevated serum levels of IL‐12 and TNF. 204 Depletion of MФ in this context prevented tumour regression. In a clinical trial, CD40 mAb combined with chemotherapy showed a trend of increased overall survival, even though the sample size was too small (n = 21) for conclusive results. 204 Several other clinical trials that investigate the effect of targeting CD40 in different types of tumours have already been summarized. 217 While some of these therapeutics are associated with positive tumour outcomes, MФ‐specific effects of treatment are generally not investigated.
In a novel particle‐based strategy, Shields and colleagues injected MФ equipped with ‘backpacks’ of biodegradable polymers loaded with IFN‐γ into mice bearing 4T1 tumours; they showed that ex vivo‐polarized MФ carrying these backpacks were able to not only maintain a pro‐inflammatory phenotype in the tumour microenvironment, but also induce the same phenotype in resident TAMs, associated with reduced tumour growth, lung metastasis and improved survival. 64 As backpacks could also be filled with cytokines promoting anti‐inflammatory polarization, such an approach could be useful in other disease contexts.
Intracellular pathway targeting
TAMs can also be targeted with compounds that interfere with internal signalling, including modifications of nucleic acids, cellular kinases or HDAC.
Vorinostat, a small molecule inhibitor of HDAC, is approved for cutaneous T‐cell lymphoma due to its growth‐inhibiting effect on tumour cells. 218 Vorinostat also affects TAMs in murine models: encapsulating vorinostat and a chemotherapeutic drug within liposomes that target both non‐small‐cell lung cancer cells and CD206‐expressing MФ suppressed tumour growth via the upregulation of iNOS, CD86 and TNF while downregulating CD206, arginase and IL‐10 in MФ. 219
Another internal target for influencing MФ signalling is PI3Kγ, a phosphoinositide 3‐kinases subunit mainly expressed by myeloid cells. In a murine model of PDAC, PI3Kγ blockade with the PI3Kγ/δ inhibitor TG100‐115 reduced MФ expression of Arginase1, TGF‐ß and IL‐10 and increased IL‐12 and IFN‐γ. 220 This induced tumour suppression and prevented metastasis. MФ‐specific effects were replicated in a clinical trial of the selective PI3Kγ inhibitor IPI‐549 as monotherapy and in combination with chemotherapy, although the study is still ongoing and no results regarding survival and tumour regression are available yet. 221
In mouse models of ovarian cancer, melanoma and glioblastoma, nanoparticle‐mediated delivery of in vitro‐transcribed mRNAs encoding IRF5 together with its activating kinase IKKβ increased pro‐inflammatory myeloid cells and caused tumour clearance in some animals. 222 Similar results were found with nanoparticles delivering siRNA targeting growth factors including VEGF and placental growth factor, which reduced MФ CD206 expression and IL‐10 production while increasing IL‐12 and IFN‐y in tumour tissues. 223 Recently, siRNA‐loaded nanoparticle mediated silencing of signal transducer and activator of transcription 3 and hypoxia‐inducible factor 1 α prevented TAM‐mediated angiogenesis and decreased tumour size. 224 In vitro, repolarization has also been achieved with nanoparticle delivery of plasmid DNA encoding the IL‐12 gene 225 and with overexpression of microRNA‐155 in triple negative breast cancer TAMs. 226 Another approach delivers CRISPR‐Cas9 gene editing machinery into MФ: knock out of signal regulatory protein α, which engages CD47 on cancer cells and prevents phagocytosis, significantly increased MФ targeting of osteosarcoma cells in vitro. 227
TAMs re‐education into pro‐inflammatory phenotypes can be beneficial to overcome tumour‐induced immune suppression. Reprogramming strategies can be combined with existing immunotherapies, chemotherapy and radiation, which has been shown to be successful in a variety of clinical trials. Importantly, TAMs targeting should be localized to cancerous areas to prevent systemic inflammation, and only affect pro‐tumoral MФ types. To achieve this, further investigation into TAMs heterogeneity and careful design and delivery of potential therapeutics are required.
Discussion
The recent development of individual cell‐based technologies has led to an explosion of knowledge about MФ ontogeny and diversity. The identification of their subpopulations in homeostatic and disordered contexts is a constantly growing field that highlights a broad phenotypic repertoire consistent with the variety of stimuli they are exposed to. Their impact on disease development is still under intense investigation and seems to be subtype‐dependent. While some subsets are thought to directly contribute to disease progression, others have been found to be protective. However, most investigation is performed in animal models and the translation of this to human patients is often less clear.
Past approaches to MФ reprogramming did not appreciate MФ heterogeneity and plasticity, and how microenvironmental niche influences the phenotype. Early adoptive transfer studies had limited/no efficacy because of the impact of the in vivo microenvironment on MФ phenotype once transferred. 228 Novel experimental avenues, however, have succeeded in maintaining the phenotype of adoptively transferred cells despite environmental factors. 64 Other strategies have focused on direct in vivo modulation of MФ phenotypes, also giving promising results. While different approaches to MФ reprogramming have been described, each is associated with pitfalls and benefits influencing their utility for specific tissues. Where possible, future therapeutic approaches should consider tailoring of strategy towards a specific tissue microenvironment, as well as a specific disease‐associated subset of cells, to improve efficacy and minimize off‐target effects.
As MФ reprogramming is a recent approach to therapy, important questions still need to be answered: How stably can different subsets of MФ be re‐educated? Is long‐term reprogramming of long‐lived resident MФ safe? As MФ are in constant interaction with their environment and other immune cells, including other MФ subtypes, will modifying one particular subtype have secondary unintended impacts on the tissue? An alternative approach may be secondary targeting of MФ populations via manipulation of their environmental cues through alteration of the communication between tissue and MФ. This could be achieved by targeting the cells that support MФ growth and/or polarization, for example. Either way, it is clear that further growth in understanding of MФ phenotypic heterogeneity in a given microenvironmental/disease context is required to appreciate the potential of targeting the MФ in disease and capitalize on the advances that have begun to be made in this area.
Competing interest statement
The authors declare no conflict of interest.
Acknowledgments
P.R.T is a Wellcome Trust Investigator Award (107964/Z/15/Z) and an MRC UK Dementia Research Institute Professor. R.J.P was an employee of GlaxoSmithKline at the time of writing. R.J.P is currently an employee of the University of Cambridge.
VMTB and RJP contributed equally as co‐first author.
PRT and NI contributed equally as co‐last author.
Contributor Information
Valentina M. T. Bart, Email: bartvm@cardiff.ac.uk.
Philip R. Taylor, Email: TaylorPR@cardiff.ac.uk.
Natacha Ipseiz, Email: ipseizn@cardiff.ac.uk.
Data Availability Statement
Not applicable.
References
- 1. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue‐resident macrophages self‐maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013;38:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue‐resident macrophages. Nat Immunol. 2013;14:986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davies LC, Taylor PR. Tissue‐resident macrophages: then and now. Immunology 2015;144:541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ginhoux F, Guilliams M. Tissue‐resident macrophage ontogeny and homeostasis. Immunity 2016;44:439–49. [DOI] [PubMed] [Google Scholar]
- 5. Gordon S, Martinez‐Pomares L. Physiological roles of macrophages. Pflugers Arch. 2017;469:365–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kierdorf K, Prinz M, Geissmann F, Gomez PE. Development and function of tissue resident macrophages in mice. Semin Immunol. 2015;27:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo . Science 2005;308(5726):1314–8. [DOI] [PubMed] [Google Scholar]
- 8. Michaelson MD, Bieri PL, Mehler MF, Xu H, Arezzo JC, Pollard JW, et al. CSF‐1 deficiency in mice results in abnormal brain development. Development 1996;122:2661–72. [DOI] [PubMed] [Google Scholar]
- 9. DeFalco T, Bhattacharya I, Williams AV, Sams DM, Capel B. Yolk‐sac‐derived macrophages regulate fetal testis vascularization and morphogenesis. Proc Natl Acad Sci U S A. 2014;111:E2384–E2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diez‐Roux G, Argilla M, Makarenkova H, Ko K, Lang RA. Macrophages kill capillary cells in G1 phase of the cell cycle during programmed vascular regression. Development 1999;126:2141–7. [DOI] [PubMed] [Google Scholar]
- 11. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M‐1/M‐2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–73. [DOI] [PubMed] [Google Scholar]
- 13. Modolell M, Corraliza IM, Link F, Soler G, Eichmann K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow‐derived macrophages by TH1 and TH2 cytokines. Eur J Immunol. 1995;25:1101–4. [DOI] [PubMed] [Google Scholar]
- 14. Viola A, Munari F, Sanchez‐Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre‐Bosquet N, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome‐based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014;40:274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McArdle S, Mikulski Z, Ley K. Live cell imaging to understand monocyte, macrophage, and dendritic cell function in atherosclerosis. J Exp Med. 2016;213:1117–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tay TL, Mai D, Dautzenberg J, Fernandez‐Klett F, Lin G, Sagar , et al. A new fate mapping system reveals context‐dependent random or clonal expansion of microglia. Nat Neurosci. 2017;20:793–803. [DOI] [PubMed] [Google Scholar]
- 19. Li C, Menoret A, Farragher C, Ouyang Z, Bonin C, Holvoet P, Vella AT, Zhou B. Single‐cell transcriptomics–based MacSpectrum reveals macrophage activation signatures in diseases. JCI Insight. 2019;4:10. 10.1172/jci.insight.126453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z, et al. Deciphering human macrophage development at single‐cell resolution. Nature 2020;582:571–6. [DOI] [PubMed] [Google Scholar]
- 21. Lavin Y, Winter D, Blecher‐Gonen R, David E, Keren‐Shaul H, Merad M, et al. Tissue‐resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014;159:1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue‐specific macrophage identities. Cell 2014;159:1327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013;496:445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schultze JL, Schmieder A, Goerdt S. Macrophage activation in human diseases. Semin Immunol. 2015;27:249–56. [DOI] [PubMed] [Google Scholar]
- 25. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell 2017;169:1276–90 e17. [DOI] [PubMed] [Google Scholar]
- 26. Ramachandran P, Dobie R, Wilson‐Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single‐cell level. Nature 2019;575:512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor‐associated macrophage and monocyte transcriptional landscapes reveal cancer‐specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 2019;35:588–602 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Dev Comp Immunol. 2004;28:509–54. [DOI] [PubMed] [Google Scholar]
- 29. Hamilton JA. GM‐CSF‐dependent inflammatory pathways. Front Immunol. 2019;10:2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamilton JA. Colony‐stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–44. [DOI] [PubMed] [Google Scholar]
- 31. T'Jonck W, Guilliams M, Bonnardel J. Niche signals and transcription factors involved in tissue‐resident macrophage development. Cell Immunol. 2018;330:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte‐to‐macrophage differentiation and trained innate immunity. Science 2014;345:1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Foster SL, Hargreaves DC, Medzhitov R. Gene‐specific control of inflammation by TLR‐induced chromatin modifications. Nature. 2007;447:972–8. [DOI] [PubMed] [Google Scholar]
- 34. Netea MG, Dominguez‐Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. 2020;20:375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018;556:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF‐beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weavers H, Evans IR, Martin P, Wood W. Corpse engulfment generates a molecular memory that primes the macrophage inflammatory response. Cell 2016;165:1658–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gordon S, Pluddemann A. Macrophage clearance of apoptotic cells: a critical assessment. Front Immunol. 2018;9:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dai H, Lan P, Zhao D, Abou‐Daya K, Liu W, Chen W, et al. PIRs mediate innate myeloid cell memory to nonself MHC molecules. Science 2020;368:1122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shemer A, Grozovski J, Tay TL, Tao J, Volaski A, Suss P, et al. Engrafted parenchymal brain macrophages differ from microglia in transcriptome, chromatin landscape and response to challenge. Nat Commun. 2018;9:5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. Diverse requirements for microglial survival, specification, and function revealed by defined‐medium cultures. Neuron 2017;94(4):759–73 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mould KJ, Jackson ND, Henson PM, Seibold M, Janssen WJ. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight. 2019;4:5. 10.1172/jci.insight.126556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pellizzari G, Hoskin C, Crescioli S, Mele S, Gotovina J, Chiaruttini G, et al. IgE re‐programs alternatively‐activated human macrophages towards pro‐inflammatory anti‐tumoural states. EBioMedicine. 2019;43:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Degboe Y, Rauwel B, Baron M, Boyer JF, Ruyssen‐Witrand A, Constantin A, et al. Polarization of rheumatoid macrophages by TNF targeting through an IL‐10/STAT3 mechanism. Front Immunol. 2019;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herweijer H, Wolff JA. Progress and prospects: naked DNA gene transfer and therapy. Gene Ther. 2003;10:453–8. [DOI] [PubMed] [Google Scholar]
- 46. Kim SS, Ye C, Kumar P, Chiu I, Subramanya S, Wu H, et al. Targeted delivery of siRNA to macrophages for anti‐inflammatory treatment. Mol Ther. 2010;18:993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Collins DE, Reuter JD, Rush HG, Villano JS. Viral vector biosafety in laboratory animal research. Comp Med. 2017;67:215–21. [PMC free article] [PubMed] [Google Scholar]
- 48. Hacein‐Bey‐Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, et al. Efficacy of gene therapy for X‐linked severe combined immunodeficiency. N Engl J Med. 2010;363:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gregory SM, Nazir SA, Metcalf JP. Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 2011;6:357–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Annoni A, Gregori S, Naldini L, Cantore A. Modulation of immune responses in lentiviral vector‐mediated gene transfer. Cell Immunol. 2019;342:103802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med. 2019;4:e10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu G, Guo M, Xu J, Wu F, Fan J, Huang Q, et al. Nanoparticles targeting macrophages as potential clinical therapeutic agents against cancer and inflammation. Front Immunol. 2019;10:1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reichel D, Tripathi M, Perez JM. Biological effects of nanoparticles on macrophage polarization in the tumor microenvironment. Nanotheranostics. 2019;3:66–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake‐Kawano R, Kanda K, et al. COX‐2 inhibition alters the phenotype of tumor‐associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis 2011;32:1333–9. [DOI] [PubMed] [Google Scholar]
- 55. Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer. 2019;18:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Moreira JD, Koch BEV, van Veen S, Walburg KV, Vrieling F, Guimaraes MPD, et al. Functional inhibition of host histone deacetylases (HDACs) enhances in vitro and in vivo anti‐mycobacterial activity in human macrophages and in zebrafish. Front Immunol. 2020;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8‐agonist‐loaded nanoparticles promote the polarization of tumour‐associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2:578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shahbazi MA, Sedighi M, Bauleth‐Ramos T, Kant K, Correia A, Poursina N, et al. Targeted reinforcement of macrophage reprogramming toward M2 polarization by IL‐4‐loaded hyaluronic acid particles. ACS Omega. 2018;3:18444–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zidek Z, Anzenbacher P, Kmonickova E. Current status and challenges of cytokine pharmacology. Br J Pharmacol. 2009;157:342–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kwon D, Cha BG, Cho Y, Min J, Park EB, Kang SJ, et al. Extra‐large pore mesoporous silica nanoparticles for directing in vivo M2 macrophage polarization by delivering IL‐4. Nano Lett. 2017;17:2747–56. [DOI] [PubMed] [Google Scholar]
- 61. Thauvin C, Widmer J, Mottas I, Hocevar S, Allemann E, Bourquin C, et al. Development of resiquimod‐loaded modified PLA‐based nanoparticles for cancer immunotherapy: a kinetic study. Eur J Pharm Biopharm. 2019;139:253–61. [DOI] [PubMed] [Google Scholar]
- 62. Chenthamara D, Subramaniam S, Ramakrishnan SG, Krishnaswamy S, Essa MM, Lin FH, et al. Therapeutic efficacy of nanoparticles and routes of administration. Biomater Res. 2019;23:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Parsa R, Andresen P, Gillett A, Mia S, Zhang XM, Mayans S, et al. Adoptive transfer of immunomodulatory M2 macrophages prevents type 1 diabetes in NOD mice. Diabetes 2012;61:2881–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shields CWT, Evans MA, Wang LL, Baugh N, Iyer S, Wu D, et al. Cellular backpacks for macrophage immunotherapy. Sci Adv. 2020;6:eaaz6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moroni F, Dwyer BJ, Graham C, Pass C, Bailey L, Ritchie L, et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat Med. 2019;25:1560–5. [DOI] [PubMed] [Google Scholar]
- 66. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult‐derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, et al. Progressive replacement of embryo‐derived cardiac macrophages with age. J Exp Med. 2014;211:2151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell 2017;169:510–22 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao‐Cortes D, et al. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively‐activated macrophage profile. PLoS One 2012;7:e36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. Primitive embryonic macrophages are required for coronary development and maturation. Circ Res. 2016;118:1498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ruparelia N, Godec J, Lee R, Chai JT, Dall'Armellina E, McAndrew D, et al. Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur Heart J. 2015;36:1923–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54:130–8. [DOI] [PubMed] [Google Scholar]
- 73. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, et al. Differential contribution of monocytes to heart macrophages in steady‐state and after myocardial infarction. Circ Res. 2014;115:284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Frodermann V, Nahrendorf M. Macrophages and cardiovascular health. Physiol Rev. 2018;98:2523–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mouton AJ, DeLeon‐Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, et al. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol. 2018;113:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dick SA, Macklin JA, Nejat S, Momen A, Clemente‐Casares X, Althagafi MG, et al. Self‐renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Deniset JF, Belke D, Lee WY, Jorch SK, Deppermann C, Hassanabad AF, et al. Gata6(+) pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 2019;51:131–40 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics‐2018 update: a report from the American Heart Association. Circulation 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 80. Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE. Meta‐analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol. 2003;91:1055–9. [DOI] [PubMed] [Google Scholar]
- 81. Abbate A, Trankle CR, Buckley LF, Lipinski MJ, Appleton D, Kadariya D, et al. Interleukin‐1 blockade inhibits the acute inflammatory response in patients with ST‐segment‐elevation myocardial infarction. J Am Heart Assoc. 2020;9:e014941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu J, Wang H, Li J. Inflammation and inflammatory cells in myocardial infarction and reperfusion injury: a double‐edged sword. Clin Med Insights Cardiol. 2016;10:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hoyer FF, Naxerova K, Schloss MJ, Hulsmans M, Nair AV, Dutta P, et al. Tissue‐specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity 2019;51:899–914 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J, et al. in vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2014;63:1556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bejerano T, Etzion S, Elyagon S, Etzion Y, Cohen S. Nanoparticle delivery of miRNA‐21 mimic to cardiac macrophages improves myocardial remodeling after myocardial infarction. Nano Lett. 2018;18:5885–91. [DOI] [PubMed] [Google Scholar]
- 86. Yang L, Wang B, Zhou Q, Wang Y, Liu X, Liu Z, et al. MicroRNA‐21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7. Cell Death Dis. 2018;9:769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Podaru MN, Fields L, Kainuma S, Ichihara Y, Hussain M, Ito T, et al. Reparative macrophage transplantation for myocardial repair: a refinement of bone marrow mononuclear cell‐based therapy. Basic Res Cardiol. 2019;114:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24:1234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lopez‐Atalaya JP, Askew KE, Sierra A, Gomez‐Nicola D. Development and maintenance of the brain's immune toolkit: Microglia and non‐parenchymal brain macrophages. Dev Neurobiol. 2018;78:561–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Garre JM, Yang G. Contributions of monocytes to nervous system disorders. J Mol Med (Berl). 2018;96:873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Banks WA. Characteristics of compounds that cross the blood‐brain barrier. BMC Neurol. 2009;9(Suppl 1):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. International Genomics of Alzheimer's Disease C . Convergent genetic and expression data implicate immunity in Alzheimer's disease. Alzheimer's Dementia. 2015;11:658–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chu F, Shi M, Zheng C, Shen D, Zhu J, Zheng X, et al. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol. 2018;318:1–7. [DOI] [PubMed] [Google Scholar]
- 95. Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019;18:211–20. [DOI] [PubMed] [Google Scholar]
- 96. Goldenberg MM. Multiple sclerosis review. P T. 2012;37:175–84. [PMC free article] [PubMed] [Google Scholar]
- 97. Christophi GP, Panos M, Hudson CA, Christophi RL, Gruber RC, Mersich AT, et al. Macrophages of multiple sclerosis patients display deficient SHP‐1 expression and enhanced inflammatory phenotype. Lab Invest. 2009;89:742–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL, Lassmann H. Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain 2017;140:1900–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Locatelli G, Theodorou D, Kendirli A, Jordao MJC, Staszewski O, Phulphagar K, et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat Neurosci. 2018;21:1196–208. [DOI] [PubMed] [Google Scholar]
- 100. Moreno MA, Burns T, Yao P, Miers L, Pleasure D, Soulika AM. Therapeutic depletion of monocyte‐derived cells protects from long‐term axonal loss in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2016;290:36–46. [DOI] [PubMed] [Google Scholar]
- 101. Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–52. [DOI] [PubMed] [Google Scholar]
- 102. Rothe T, Ipseiz N, Faas M, Lang S, Perez‐Branguli F, Metzger D, et al. The nuclear receptor Nr4a1 acts as a microglia rheostat and serves as a therapeutic target in autoimmune‐driven central nervous system inflammation. J Immunol. 2017;198:3878–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164:1079–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Pul R, Moharregh‐Khiabani D, Skuljec J, Skripuletz T, Garde N, Voss EV, et al. Glatiramer acetate modulates TNF‐alpha and IL‐10 secretion in microglia and promotes their phagocytic activity. J Neuroimmune Pharmacol. 2011;6:381–8. [DOI] [PubMed] [Google Scholar]
- 105. Liu J, Johnson TV, Lin J, Ramirez SH, Bronich TK, Caplan S, et al. T cell independent mechanism for copolymer‐1‐induced neuroprotection. Eur J Immunol. 2007;37:3143–54. [DOI] [PubMed] [Google Scholar]
- 106. Wynn DR. Enduring clinical value of copaxone(R) (Glatiramer Acetate) in multiple sclerosis after 20 years of use. Mult Scler Int. 2019;2019:7151685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Boster A, Bartoszek MP, O'Connell C, Pitt D, Racke M. Efficacy, safety, and cost‐effectiveness of glatiramer acetate in the treatment of relapsing‐remitting multiple sclerosis. Ther Adv Neurol Disord. 2011;4:319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, et al. Identification of a microglia phenotype supportive of remyelination. Glia 2012;60:306–21. [DOI] [PubMed] [Google Scholar]
- 109. Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, van Vreden C, et al. Therapeutic inflammatory monocyte modulation using immune‐modifying microparticles. Sci Transl Med. 2014;6:219ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Giladi A, Wagner LK, Li H, Dorr D, Medaglia C, Paul F, et al. Cxcl10(+) monocytes define a pathogenic subset in the central nervous system during autoimmune neuroinflammation. Nat Immunol. 2020;21:525–34. [DOI] [PubMed] [Google Scholar]
- 111. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013;368:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, et al. Anti‐human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med. 2020;217:e20200785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. 2012;3:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Olmos‐Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas‐Caballero M, et al. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's‐like pathology. Brain 2016;139(Pt 3):891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, et al. Colony‐stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg‐AD mice. J Neuroinflammation. 2015;12:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton‐Jones M, et al. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid‐beta pathology. Brain 2016;139(Pt 4):1265–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lei F, Cui N, Zhou C, Chodosh J, Vavvas DG, Paschalis EI. CSF1R inhibition by a small‐molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc Natl Acad Sci USA. 2020;117:23336–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Quigley EMM. Microbiota‐brain‐gut axis and neurodegenerative diseases. Curr Neurol Neurosci Rep. 2017;17:94. [DOI] [PubMed] [Google Scholar]
- 119. Erdo F, Bors LA, Farkas D, Bajza A, Gizurarson S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res Bull. 2018;143:155–70. [DOI] [PubMed] [Google Scholar]
- 120. Atkinson AJ Jr. Intracerebroventricular drug administration. Transl Clin Pharmacol. 2017;25:117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D'Ambrosi N. The dual role of microglia in ALS: mechanisms and therapeutic approaches. Front Aging Neurosci. 2017;9:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Du Y, Zhao W, Thonhoff JR, Wang J, Wen S, Appel SH. Increased activation ability of monocytes from ALS patients. Exp Neurol. 2020;328:113259. [DOI] [PubMed] [Google Scholar]
- 123. Wang H, Wang Y, Li D, Liu Z, Zhao Z, Han D, et al. VEGF inhibits the inflammation in spinal cord injury through activation of autophagy. Biochem Biophys Res Commun. 2015;464:453–8. [DOI] [PubMed] [Google Scholar]
- 124. Wang Y, Duan W, Wang W, Di W, Liu Y, Liu Y, et al. scAAV9‐VEGF prolongs the survival of transgenic ALS mice by promoting activation of M2 microglia and the PI3K/Akt pathway. Brain Res. 2016;1648(Pt A):1–10. [DOI] [PubMed] [Google Scholar]
- 125. Ramaglia V, Sheikh‐Mohamed S, Legg K, Park C, Rojas OL, Zandee S, et al. Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. Elife. 2019;8:e48051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yurek D, Hasselrot U, Sesenoglu‐Laird O, Padegimas L, Cooper M. Intracerebral injections of DNA nanoparticles encoding for a therapeutic gene provide partial neuroprotection in an animal model of neurodegeneration. Nanomedicine. 2017;13:2209–17. [DOI] [PubMed] [Google Scholar]
- 127. Hu N, Shi X, Zhang Q, Liu W, Zhu Y, Wang Y, et al. Special interstitial route can transport nanoparticles to the brain bypassing the blood‐brain barrier. Nano Research. 2019;12:2760–5. [Google Scholar]
- 128. Ali J, Ali M, Baboota S, Sahani JK, Ramassamy C, Dao L, et al. Potential of nanoparticulate drug delivery systems by intranasal administration. Curr Pharm Des. 2010;16:1644–53. [DOI] [PubMed] [Google Scholar]
- 129. Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, et al. A liver capsular network of monocyte‐derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity 2017;47:374–88 e6. [DOI] [PubMed] [Google Scholar]
- 130. Bleriot C, Ginhoux F. Understanding the heterogeneity of resident liver macrophages. Front Immunol. 2019;10:2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. 2019;3:730–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 2016;165:668–78. [DOI] [PubMed] [Google Scholar]
- 133. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013;38:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C‐Myb(+) erythro‐myeloid progenitor‐derived fetal monocytes give rise to adult tissue‐resident macrophages. Immunity 2015;42:665–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow‐derived monocytes give rise to self‐renewing and fully differentiated Kupffer cells. Nat Commun. 2016;7:10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver‐resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type‐2‐mediated tissue repair during bacterial infection. Immunity 2015;42:145–58. [DOI] [PubMed] [Google Scholar]
- 137. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida RF, et al. Bone marrow‐derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatol. 2016;65:758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology 2016;151:1176–91. [DOI] [PubMed] [Google Scholar]
- 139. Soysa R, Lampert S, Yuen S, Douglass AN, Li W, Pfeffer K, et al. Fetal origin confers radioresistance on liver macrophages via p21(cip1/WAF1). J Hepatol. 2019;71:553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zigmond E, Samia‐Grinberg S, Pasmanik‐Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte‐derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193:344–53. [DOI] [PubMed] [Google Scholar]
- 141. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue‐resident macrophages during organogenesis. Science 2016;353:aaf4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Scott CL, T'Jonck W, Martens L, Todorov H, Sichien D, Soen B, et al. The transcription factor ZEB2 is required to maintain the tissue‐specific identities of macrophages. Immunity 2018;49(2):312–25 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Bonnardel J, T'Jonck W, Gaublomme D, Browaeys R, Scott CL, Martens L, et al. Stellate cells, hepatocytes, and endothelial cells imprint the kupffer cell identity on monocytes colonizing the liver macrophage niche. Immunity 2019;51:638–54 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver‐derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity 2019;51:655–70 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. 2018;9:4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Aizarani N, Saviano A, Sagar ML, Durand S, Herman JS, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019;572:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhao J, Zhang S, Liu Y, He X, Qu M, Xu G, et al. Single‐cell RNA sequencing reveals the heterogeneity of liver‐resident immune cells in human. Cell Discov. 2020;6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single‐cell secretome gene analysis. Mol Cell. 2019;75:644–60 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche‐specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity 2020;52:1057–74 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018;67:333–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Robert O, Boujedidi H, Bigorgne A, Ferrere G, Voican CS, Vettorazzi S, et al. Decreased expression of the glucocorticoid receptor‐GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J Hepatol. 2016;64:916–24. [DOI] [PubMed] [Google Scholar]
- 153. Bartneck M, Scheyda KM, Warzecha KT, Rizzo LY, Hittatiya K, Luedde T, et al. Fluorescent cell‐traceable dexamethasone‐loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 2015;37:367–82. [DOI] [PubMed] [Google Scholar]
- 154. Svendsen P, Graversen JH, Etzerodt A, Hager H, Roge R, Gronbaek H, et al. Antibody‐directed glucocorticoid targeting to CD163 in M2‐type macrophages attenuates fructose‐induced liver inflammatory changes. Mol Ther Methods Clin Dev. 2017;4:50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. He C, Yin L, Tang C, Yin C. Multifunctional polymeric nanoparticles for oral delivery of TNF‐alpha siRNA to macrophages. Biomaterials 2013;34:2843–54. [DOI] [PubMed] [Google Scholar]
- 156. Azzimato V, Jager J, Chen P, Morgantini C, Levi L, Barreby E, et al. Liver macrophages inhibit the endogenous antioxidant response in obesity‐associated insulin resistance. Sci Transl Med. 2020;12:eaaw9709. [DOI] [PubMed] [Google Scholar]
- 157. Traber PG, Chou H, Zomer E, Hong F, Klyosov A, Fiel MI, et al. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide‐induced liver disease. PLoS One 2013;8:e75361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Traber PG, Zomer E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS One 2013;8:e83481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Volarevic V, Milovanovic M, Ljujic B, Pejnovic N, Arsenijevic N, Nilsson U, et al. Galectin‐3 deficiency prevents concanavalin A‐induced hepatitis in mice. Hepatology 2012;55:1954–64. [DOI] [PubMed] [Google Scholar]
- 160. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C‐C motif) receptor 2‐positive monocytes aggravate the early phase of acetaminophen‐induced acute liver injury. Hepatology 2016;64:1667–82. [DOI] [PubMed] [Google Scholar]
- 161. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270–83. [DOI] [PubMed] [Google Scholar]
- 162. Anstee QM, Neuschwander‐Tetri BA, Wong VW, Abdelmalek MF, Younossi ZM, Yuan J, et al. Cenicriviroc for the treatment of liver fibrosis in adults with nonalcoholic steatohepatitis: AURORA Phase 3 study design. Contemp Clin Trials. 2020;89:105922. [DOI] [PubMed] [Google Scholar]
- 163. Starkey Lewis P, Campana L, Aleksieva N, Cartwright JA, Mackinnon A, O'Duibhir E, et al. Alternatively activated macrophages promote resolution of necrosis following acute liver injury. J Hepatol. 2020;73:349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon‐Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology 2011;53:2003–15. [DOI] [PubMed] [Google Scholar]
- 165. Haideri SS, McKinnon AC, Taylor AH, Kirkwood P, Starkey Lewis PJ, O'Duibhir E, et al. Injection of embryonic stem cell derived macrophages ameliorates fibrosis in a murine model of liver injury. NPJ Regen Med. 2017;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, et al. Cutting Edge: Immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. 2011;187:2072–8. [DOI] [PubMed] [Google Scholar]
- 167. Bird TG, Lu WY, Boulter L, Gordon‐Keylock S, Ridgway RA, Williams MJ, et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage‐mediated TWEAK signaling. Proc Natl Acad Sci U S A. 2013;110:6542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Sharkey J, Starkey Lewis PJ, Barrow M, Alwahsh SM, Noble J, Livingstone E, et al. Functionalized superparamagnetic iron oxide nanoparticles provide highly efficient iron‐labeling in macrophages for magnetic resonance‐based detection in vivo . Cytotherapy. 2017;19:555–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol. 2016;12:472–85. [DOI] [PubMed] [Google Scholar]
- 170. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis ‐ shaping the immunological landscape. Nat Rev Rheumatol. 2016;12(1):63–8. [DOI] [PubMed] [Google Scholar]
- 171. Smith MD. The normal synovium. Open Rheumatol J. 2011;5:100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Alivernini S, Tolusso B, Petricca L, Bui L, Di Sante G, Peluso G, et al. Synovial features of patients with rheumatoid arthritis and psoriatic arthritis in clinical and ultrasound remission differ under anti‐TNF therapy: a clue to interpret different chances of relapse after clinical remission? Ann Rheum Dis. 2017;76:1228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, et al. Nonclassical Ly6C(‐) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 2014;9:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Culemann S, Gruneboom A, Nicolas‐Avila JA, Weidner D, Lammle KF, Rothe T, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019;572:670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med 2020;26:1295–306. [DOI] [PubMed] [Google Scholar]
- 176. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single‐cell transcriptomics and mass cytometry. Nat Immunol. 2019;20:928–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Kuo D, Ding J, Cohn IS, Zhang F, Wei K, Rao DA, et al. HBEGF(+) macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci Transl Med. 2019;11:eaau8587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Swanson CD, Akama‐Garren EH, Stein EA, Petralia JD, Ruiz PJ, Edalati A, et al. Inhibition of epidermal growth factor receptor tyrosine kinase ameliorates collagen‐induced arthritis. J Immunol. 2012;188:3513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Cook AD, Pobjoy J, Steidl S, Durr M, Braine EL, Turner AL, et al. Granulocyte‐macrophage colony‐stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis Res Ther. 2012;14:R199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Hamilton JA. GM‐CSF as a target in inflammatory/autoimmune disease: current evidence and future therapeutic potential. Expert Rev Clin Immunol. 2015;11:457–65. [DOI] [PubMed] [Google Scholar]
- 181. Hamilton JA, Cook AD, Tak PP. Anti‐colony‐stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov. 2016;16:53–70. [DOI] [PubMed] [Google Scholar]
- 182. Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen‐induced arthritis in granulocyte‐macrophage colony‐stimulating factor‐deficient mice. J Immunol. 1998;161:3639–44. [PubMed] [Google Scholar]
- 183. Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen‐induced arthritis post‐onset by antibody to granulocyte‐macrophage colony‐stimulating factor (GM‐CSF): requirement for GM‐CSF in the effector phase of disease. Arthritis Res. 2001;3:293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Hamilton JA. GM‐CSF in inflammation. J Exp Med. 2020;217:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Cook AD, Hamilton JA. Investigational therapies targeting the granulocyte macrophage colony‐stimulating factor receptor‐alpha in rheumatoid arthritis: focus on mavrilimumab. Ther Adv Musculoskelet Dis. 2018;10:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kong JS, Park JH, Yoo SA, Kim KM, Bae YJ, Park YJ, et al. Dynamic transcriptome analysis unveils key proresolving factors of chronic inflammatory arthritis. J Clin Invest. 2020;130:3974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics 2009;9:4997–5000. [DOI] [PubMed] [Google Scholar]
- 188. Kaplan A, Bueno M, Fournier AE. Extracellular functions of 14‐3‐3 adaptor proteins. Cell Signal. 2017;31:26–30. [DOI] [PubMed] [Google Scholar]
- 189. Dobson M, Ramakrishnan G, Ma S, Kaplun L, Balan V, Fridman R, et al. Bimodal regulation of FoxO3 by AKT and 14‐3‐3. Biochim Biophys Acta. 2011;1813:1453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, et al. MK2‐induced tristetraprolin:14‐3‐3 complexes prevent stress granule association and ARE‐mRNA decay. EMBO J. 2004;23:1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, et al. Tristetraprolin is required for full anti‐inflammatory response of murine macrophages to IL‐10. J Immunol. 2009;183:1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Jain S, Tran TH, Amiji M. Macrophage repolarization with targeted alginate nanoparticles containing IL‐10 plasmid DNA for the treatment of experimental arthritis. Biomaterials 2015;61:162–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32:1267–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Helm O, Held‐Feindt J, Grage‐Griebenow E, Reiling N, Ungefroren H, Vogel I, et al. Tumor‐associated macrophages exhibit pro‐ and anti‐inflammatory properties by which they impact on pancreatic tumorigenesis. Int J Cancer. 2014;135:843–61. [DOI] [PubMed] [Google Scholar]
- 195. Bucana C, Hoyer LC, Hobbs B, Breesman S, McDaniel M, Hanna MG Jr. Morphological evidence for the translocation of lysosomal organelles from cytotoxic macrophages into the cytoplasm of tumor target cells. Cancer Res. 1976;36:4444–58. [PubMed] [Google Scholar]
- 196. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor‐associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Liu Y, Cao X. The origin and function of tumor‐associated macrophages. Cell Mol Immunol. 2015;12:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, et al. Topical TLR7 agonist imiquimod can induce immune‐mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. 2012;18:6748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Salazar LG, Lu H, Reichow JL, Childs JS, Coveler AL, Higgins DM, et al. Topical imiquimod plus nab‐paclitaxel for breast cancer cutaneous metastases: a phase 2 clinical trial. JAMA Oncol. 2017;3:969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Pachman DR, Barton DL, Clayton AC, McGovern RM, Jefferies JA, Novotny PJ, et al. Randomized clinical trial of imiquimod: an adjunct to treating cervical dysplasia. Am J Obstet Gynecol. 2012;206:42 e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Grimm C, Polterauer S, Natter C, Rahhal J, Hefler L, Tempfer CB, et al. Treatment of cervical intraepithelial neoplasia with topical imiquimod: a randomized controlled trial. Obstet Gynecol. 2012;120:152–9. [DOI] [PubMed] [Google Scholar]
- 202. Rook AH, Gelfand JM, Wysocka M, Troxel AB, Benoit B, Surber C, et al. Topical resiquimod can induce disease regression and enhance T‐cell effector functions in cutaneous T‐cell lymphoma. Blood 2015;126:1452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, et al. A phase I study of an agonist CD40 monoclonal antibody (CP‐870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011;331:1612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Calvo A, Joensuu H, Sebastian M, Naing A, Bang Y‐J, Martin M, et al. Phase Ib/II study of lacnotuzumab (MCS110) combined with spartalizumab (PDR001) in patients (pts) with advanced tumors. J Clin Oncol 2018;36(15_suppl):3014. [Google Scholar]
- 206. Autio KA, Klebanoff CA, Schaer D, Kauh JS, Slovin SF, Blinder VS, et al. Phase 1 study of LY3022855, a colony‐stimulating factor‐1 receptor (CSF‐1R) inhibitor, in patients with metastatic breast cancer (MBC) or metastatic castration‐resistant prostate cancer (MCRPC). J Clin Oncol 2019;37(15_suppl):2548.31246526 [Google Scholar]
- 207. Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, et al. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor‐associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. 2013;2:e26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF‐1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Ao JY, Zhu XD, Chai ZT, Cai H, Zhang YY, Zhang KZ, et al. Colony‐stimulating factor 1 receptor blockade inhibits tumor growth by altering the polarization of tumor‐associated macrophages in hepatocellular carcinoma. Mol Cancer Ther. 2017;16:1544–54. [DOI] [PubMed] [Google Scholar]
- 210. Gomez‐Roca CA, Cassier PA, Italiano A, Cannarile M, Ries C, Brillouet A, et al. Phase I study of RG7155, a novel anti‐CSF1R antibody, in patients with advanced/metastatic solid tumors. J Clin Oncol 2015;33(15_suppl):3005. [Google Scholar]
- 211. Gomez‐Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D'Angelo SP, et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2‐like macrophages. Ann Oncol. 2019;30:1381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF‐1R inhibition in gliomas. Science 2016;352:aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Kawai T, Akira S. Signaling to NF‐kappaB by Toll‐like receptors. Trends Mol Med. 2007;13:460–9. [DOI] [PubMed] [Google Scholar]
- 214. Schon MP, Schon M. Imiquimod: mode of action. Br J Dermatol. 2007;157(Suppl 2):8–13. [DOI] [PubMed] [Google Scholar]
- 215. Zhao J, Zhang Z, Xue Y, Wang G, Cheng Y, Pan Y, et al. Anti‐tumor macrophages activated by ferumoxytol combined or surface‐functionalized with the TLR3 agonist poly (I : C) promote melanoma regression. Theranostics. 2018;8:6307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. van Kooten C, Banchereau J. CD40‐CD40 ligand. J Leukoc Biol. 2000;67:2–17. [DOI] [PubMed] [Google Scholar]
- 217. Piechutta M, Berghoff AS. New emerging targets in cancer immunotherapy: the role of Cluster of Differentiation 40 (CD40/TNFR5). ESMO Open. 2019;4(Suppl 3):e000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Bubna AK. Vorinostat‐an overview. Indian J Dermatol. 2015;60:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Peng H, Chen B, Huang W, Tang Y, Jiang Y, Zhang W, et al. Reprogramming tumor‐associated macrophages to reverse EGFR(T790M) resistance by dual‐targeting codelivery of gefitinib/vorinostat. Nano Lett. 2017;17:7684–90. [DOI] [PubMed] [Google Scholar]
- 220. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov. 2016;6:870–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Sullivan RJ, Hong DS, Tolcher AW, Patnaik A, Shapiro G, Chmielowski B, et al. Initial results from first‐in‐human study of IPI‐549, a tumor macrophage‐targeting agent, combined with nivolumab in advanced solid tumors. J Clin Oncol 2018;36(15_suppl):3013. [Google Scholar]
- 222. Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, et al. Genetic programming of macrophages to perform anti‐tumor functions using targeted mRNA nanocarriers. Nat Commun. 2019;10:3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Song Y, Tang C, Yin C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi‐functionalized nanoparticles to tumor‐associated macrophages and breast cancer cells. Biomaterials 2018;185:117–32. [DOI] [PubMed] [Google Scholar]
- 224. Shobaki N, Sato Y, Suzuki Y, Okabe N, Harashima H. Manipulating the function of tumor‐associated macrophages by siRNA‐loaded lipid nanoparticles for cancer immunotherapy. J Control Release. 2020;325:235–48. [DOI] [PubMed] [Google Scholar]
- 225. He XY, Liu BY, Xu C, Zhuo RX, Cheng SX. A multi‐functional macrophage and tumor targeting gene delivery system for the regulation of macrophage polarity and reversal of cancer immunoresistance. Nanoscale. 2018;10:15578–87. [DOI] [PubMed] [Google Scholar]
- 226. Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, et al. Re‐polarization of tumor‐associated macrophages to pro‐inflammatory M1 macrophages by microRNA‐155. J Mol Cell Biol. 2012;4:341–3. [DOI] [PubMed] [Google Scholar]
- 227. Ray M, Lee YW, Hardie J, Mout R, Yesilbag Tonga G, Farkas ME, et al. CRISPRed macrophages for cell‐based cancer immunotherapy. Bioconjug Chem. 2018;29:445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Andreesen R, Hennemann B, Krause SW. Adoptive immunotherapy of cancer using monocyte‐derived macrophages: rationale, current status, and perspectives. J Leukoc Biol. 1998;64:419–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.