

ABSTRACT
There has been little understanding of the molecular pathogenesis of pediatric thyroid cancers. Most of them are histologically classified as papillary thyroid carcinoma (PTC). Ionizing radiation is the most important environmental factor to induce PTC, especially in children. Particularly, radiation-related pediatric PTCs after the Chernobyl accident provided invaluable information. In addition, the recent accumulation of sporadic pediatric PTC cases, partly due to advances in diagnostic imaging, has also provided insight into their general pathogenesis. In PTC development, basically two types of genetic alterations, fusion oncogenes, mainly RET/PTC, and a point mutation, mainly BRAFV600E, are thought to play a key role as driver oncogenes. Their frequencies vary depending on patient age. The younger the age, the more prevalent the fusion oncogenes are. Higher incidence of fusion oncogenes was also observed in cases exposed to radiation. In short, fusion oncogenes are associated with both age and radiation and are not evidence of radiation exposure. The type of driver oncogene is shifted toward BRAFV600E during adolescence in sporadic PTCs. However, until about this age, fusion oncogenes seem to still confer dominant growth advantages, which may lead to the higher discovery rate of the fusion oncogenes. It has been postulated that RET/PTC in radiation-induced PTC is generated by ionizing radiation; however, there is an interesting hypothesis that thyroid follicular cell clones with pre-existing RET/PTC were already present, and radiation may play a role as a promoter/progressor but not initiator. Telomerase reverse transcriptase gene (TERT) promoter mutations, which are the strongest marker of tumor aggressiveness in adult PTC cases, have not been detected in pediatric cases; however, TERT expression without the mutations may play a role in tumor aggressiveness. In this paper, the recent information regarding molecular findings in sporadic and radiation-associated pediatric PTCs is summarized.
INTRODUCTION
Thyroid cancer is the most frequent malignant tumor in the endocrine system. Its incidence is increasing worldwide and was 14.3 per 100 000 people in 2017 in Japan [Cancer Registry and Statistics. Cancer Information Service, National Cancer Center, Japan (Ministry of Health, Labour and Welfare, National Cancer Registry)]. Thyroid cancer in the young population is rare, and its incidence rates (per 100 000) in 2015 in Japan are estimated to be 0.039 (boys) and 0 (girls) for ages 0–4, 0.110 (boys) and 0.385 (girls) for ages 5–9, 0.417 (boys) and 0.547 (girls) for ages 10–14, and 0.771 (boys) and 2.787 (girls) for ages 15–19, in total 1.353 under 19 years old [Cancer Registry and Statistics. Cancer Information Service, National Cancer Center, Japan (Monitoring of Cancer Incidence in Japan)]. Histologically, thyroid cancer is classified into papillary thyroid carcinoma (PTC), follicular thyroid carcinoma (FTC), poorly differentiated thyroid carcinoma (PDTC), anaplastic thyroid carcinoma (ATC) and medullary thyroid carcinoma (MTC). PTC and FTC are sometimes referred to as differentiated thyroid carcinoma (DTC). The only exception regarding originating cells is MTC, which is derived from parafollicular C cells, while the others are from thyroid follicular cells. The majority of cases are PTCs, accounting for ~90% in Japan.
Ionizing radiation is well known to induce thyroid cancer, and the histological type of radiation-induced thyroid cancer is also PTC. Children are far more susceptible to ionizing radiation, the risk sharply decreased with increasing age-at-exposure, and at >20 years old there was little increase in risk [1, 2]. Therefore, this review focuses on molecular pathogenesis of PTC, especially among pediatric and adolescent cases.
DRIVER ONCOGENES
First, we describe driver oncogenes that are thought to play a major role in carcinogenesis of PTC. Regardless of age of onset, PTC has a particular series of driver oncogenes leading to the activation of the mitogen-activated protein kinase (MAPK) signaling pathway, which is implicated in cell growth and dedifferentiation [3]. The major ones are: the BRAFV600E mutation and RET/PTC and ETV6/NTRK3 fusion genes, and these account cumulatively for >90% in Japan.
BRAF is a serine/threonine kinase and a member of the MAPK pathway and transmits a signal from RAS to MEK. Most of the BRAF mutations found in PTCs are BRAFV600E. The V600E mutation is a thymine-to-adenine transversion at nucleotide 1799, resulting in a valine-to-glutamic acid substitution at codon 600 (p.V600E), which converts the kinase into a constitutively active form without stimulus form RAS. This BRAFV600E mutation is the most prevalent genetic change in adult PTCs. Its prevalence seems to be dependent on regions: generally lower (40–60%) in Western countries and higher (60–80%) in Asian countries including Japan [4]. It has been reported that this mutation frequency is associated with the amount of iodine intake [5], but this is still controversial [6], and its cause remains unclear. It also should be noted that regarding the pathological diagnosis of PTC, there is considerable inter-observer variation, even among thyroid pathology experts, especially when they work in different countries [7]. Pathologists in the USA tend to diagnose a tumor with slight papillary nuclear features as a PTC, and such a tumor may be diagnosed as a benign follicular adenoma (FA) in Japan. This difference probably affects the mutation frequency described above. Another important difference among different regions is the association between the BRAFV600E mutation and clinicopathological aggressiveness. In Western countries, there are many reports demonstrating that PTCs harboring this mutation show higher frequencies of extrathyroidal extension, recurrence and advanced stages [8]. On the other hand, most reports from Japan suggest no such correlations [9–11]. The reason for this difference is also unknown.
RET is a transmembrane receptor tyrosine kinase and is usually not expressed in thyroid follicular cells. However, by chromosomal rearrangement, the C-terminal kinase domain of RET is fused to the N-terminal domain of a partner gene, and its promoter drives transcription of the fusion gene, which is called RET/PTC when this fusion is found in PTC [12]. So far, at least 19 different types of RET/PTC chimeric genes that differ according to partner genes have been identified [13]. RET/PTC1 and RET/PTC3 are the most common types, both of which are generated by intrachromosomal inversion in chromosome 10, and account for >90% of all RET/PTCs. The N-terminal domain from the partner genes usually has a portion promoting dimerization, such as the coiled-coil domain, leading to dimerization and autophosphorylation of key tyrosine residues located in the RET kinase domain in a ligand-independent manner, then resulting in constitutive activation of the kinase. The frequency of RET/PTCs in adult sporadic PTCs is ~5–15% [12, 14].
The ETV6/NTRK3 fusion also has a similar activation mechanism, between the ETV6 gene on chromosome 12 and the NTRK3 gene on chromosome 15. NTRK3 is also a transmembrane receptor tyrosine kinase that is involved in neuronal cellular processes. The N-terminal of ETV6 has the SAM domain, promoting dimerization and resulting in constitutive activation of the kinase function of NTRK3 as well. The frequency of this fusion in adult sporadic PTCs is usually a few percent [14–16].
As described above, RAS is also a member of the MAPK signaling pathway; however, in thyroid follicular cells, mutant RAS seems to preferentially activate the PI3K-AKT pathway rather than the MAPK pathway. The activation of the PI3K-AKT pathway generally drives development of another type of DTC, FTC. Therefore, the RAS mutations are frequently detected in FTC and also in FA, albeit with lower prevalence [3]. The frequency of the RAS mutations in PTC is very low, up to a few percent in Japan (N. Mitsutake and V. Saenko, unpublished results), mainly in a follicular variant of PTC. However, its frequency in PTC in the USA is 10–15% [14], presumably because of the inter-observer variation described above. RAS consists of three isoforms: KRAS, NRAS and HRAS, and the most prevalent mutation in thyroid carcinoma is NRAS at codon 61. Mutations in RAS render RAS proteins insensitive to GTPase-activating proteins, leading to constitutive activation of RAS.
These driver oncogenes are mutually exclusive [14, 17]. Since RET and NTRK3 activate not only the MAPK signaling pathway but also PI3K and PLCγ [18, 19], this mutually exclusive event provides strong genetic evidence that the activation of the MAPK pathway is a key to development of PTC. The difference between the receptor tyrosine kinase fusion genes and BRAFV600E is the magnitude of output of the MAPK signaling. This is due to insensitivity of the mutant BRAF to negative feedback of the MAPK pathway, and the mutant BRAF leads to more elevated expression of ETS/MYC/FOS compared with receptor tyrosine kinases [20]. It is assumed that this difference may be one of the reasons that tumors with BRAFV600E are more aggressive, although this is not the case in Japan. The activation of the MAPK pathway is also associated with dedifferentiation, and impairment of iodine metabolism causes resistance to radioiodine therapy [18, 21, 22].
The frequency of the above oncogenes, especially BRAFV600E and RET/PTC, apparently varies with patient age. As described above, in adult PTCs, the BRAFV600E mutation is the most prevalent, accounting for 50–85%. On the other hand, in pediatric sporadic PTCs, it has been reported to be 0–63%, although the numbers of analyzed cases were limited in most of the studies [23, 24]. Regarding RET/PTC, the opposite trend is observed. In adult sporadic PTCs, its frequency is generally 5–15%, while in pediatric cases, it is 25–65% [24].
Recent improvement in detection methods, including sharing information regarding primers/probes, may change sensitivity/specificity of detection of the oncogenes. Hence, recent papers (in the last 5 years) reporting the prevalence of the oncogenes, especially BRAFV600E and/or RET/PTC in pediatric PTCs, are summarized in Table 1 and Fig. 1 [25–39]. Their overall frequencies in the 15 publications are: BRAFV600E, 28.4% and RET/PTC, 24.6%, while they were 13 and 41%, respectively, in the previous review published in 2015 [24]. In terms of the BRAFV600E mutation, its frequency still seems to be lower than that in adult cases analyzed at the same time in three studies (Fig. 1). There is an impression that the data in recent years have been a bit closer to that of adult PTCs. This trend might be due to the recent spread and advances of ultrasound imaging systems, and small and slow-growing tumors may be detected earlier. However, there is no clear explanation so far.
Table 1.
Recent publications demonstrating the prevalence of major oncogenes in pediatric PTCs
| PMID | Year | Author | Country | No. of PTCs | Agea | BRAF | RET/PTC | ETV6/NTRK3 | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 32495721 | 2020 | Pekova | Czech | 93 | 14.5 (6–20) | 18 (19.4%) | 19 (20.4%) | 10 (10.8%) | 39 |
| 31456750 | 2019 | Galuppini | Italy | 54 | 14.4 (6.4–17.8) | 8/50 (16.0%) | 14 (25.9%) | 38 | |
| 30924609 | 2019 | Sisdelli | Brazil | 80 | 12.7 (3–18) | 12 (15.0%) | 37 | ||
| 28646474 | 2017 | Geng | China | 48 | (3–13) | 16 (35.4%) | 36 | ||
| 28521635 | 2017 | Hardee | USA | 59 | 24/50 (48.0%) | 35 | |||
| 28209747 | 2017 | Vanden Borre | USA | 12 | 12.8 (7–19) | 2 (16.7%) | 4 (33.3%) | 34 | |
| 28176151 | 2017 | Oishi | Japan | 81 | 17.4 (6–20) | 44 (54.3%) | 33 | ||
| 28077340 | 2017 | Poyrazoğlu | Turkey | 75 | 12.4 (1.3–17.8) | 14/56 (25.0%) | 32 | ||
| 27849443 | 2017 | Cordioli | Brazil | 35 | 11.8 (4–18) | 3 (8.6%) | 14 (40.0%) | 3 (8.6%) | 31 |
| 27824297 | 2017 | Alzahrani | Saudi Atabia | 72 | 15.5b (8–18) | 19 (26.4%) | 30 | ||
| 26951110 | 2016 | Onder | Turkey | 50 | 14.7 (6–18) | 15 (30.0%) | 29 | ||
| 26910217 | 2016 | Gertz | USA | 13 | 13 | 4 (30.8%) | 28 | ||
| 26784937 | 2016 | Prasad | USA | 28 | (6–18) | 13 (46.4%) | 6 (21.4%) | 5 (17.9%) | 27 |
| 26711586 | 2016 | Alzahrani | Saudi Atabia | 53 | 16b (9–18) | 12 (22.6%) | 26 | ||
| 26649796 | 2016 | Nikita | USA | 28 | 14.7 (7.9–18.4) | 9 (32.1%) | 6 (17.6%) | 25 |
aMean (range);
bmedian.
Fig. 1.
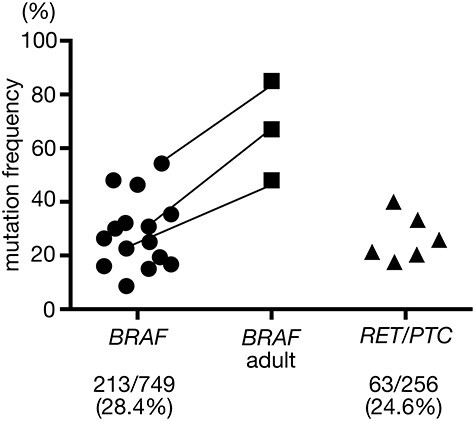
The frequencies of BRAFV600E (left) and RET/PTCs (right) in sporadic pediatric PTC cases in 15 papers published after 2016. Three studies also examined the prevalence of BRAFV600E in adult cases simultaneously (middle); same studies are connected by lines. Total numbers and percentages of each oncogene are also shown below.
TERT PROMOTER MUTATION
Recently, point mutations in the promoter region of the telomerase reverse transcriptase gene (TERT) have been found in many types of cancers including PTCs [40–42]. There are two recurrent locations, called C228T (derived from chromosomal location, chr5: 1 295 228C > T) and C250T (chr5: 1 295 250C > T). They are mutually exclusive, and C228T is more frequent in PTCs [42]. The prevalence of either mutation in adult sporadic PTCs is ~10%. These mutations create a binding site for the ETS family transcription factors [43, 44]. When the ETS family members are activated (e.g. by BRAFV600E), TERT transcription is further upregulated. Indeed, the coexistence of both the TERT promoter mutation and the BRAFV600E mutation has a strong negative impact on PTC aggressiveness and prognosis [42]. Unlike the BRAFV600E mutation, no regional differences have been recognized in this correlation so far. Another important link between these mutations and clinical parameter is age dependence. These mutations are rarely detected in cases <45 years old. According to our data of Japanese PTC patients, its frequency increases with age, and in cases >70 years old, about half of the cases carry this mutation [10].
Very recently, it has been found that there are some PTC cases that show TERT mRNA expression even in the absence of the TERT promoter mutations [11]. If cases with very small amount of expression are included, nearly 40% of the mutation-negative cases are positive for TERT expression. Interestingly, high TERT expression has been reported to be associated with worse prognosis as well. Unlike the TERT promoter mutations, the tumors with high TERT expression are found among relatively younger cases, implying that TERT expression may be a good molecular marker to predict disease outcome in young cases. We detected TERT expression in cases <20 years old but the number of analyzed cases was very limited, and further study is needed.
CHERNOBYL RADIATION-INDUCED PTC
After the accident at the Chernobyl nuclear power plant in 1986, a dramatic increase in the incidence of childhood thyroid cancer was observed, starting from around 1990. This was due to exposure to radioiodine (131I) released from the reactor. Since the half-life of 131I is ~8 days, the children born after the accident were virtually unaffected [45]. The higher incidence was observed in children of younger ages, particularly <5 five years old at the time of the accident [46]. Therefore, they were basically childhood PTCs. However, since, once exposed, the elevated risk of cancer development may continue for their lifetime, there may be radiation-induced adult PTC cases these days, although it is difficult to distinguish them from sporadic cases.
Regarding driver oncogenes, in cases with short latency (<7–10 years after the accident), a very high rate of RET/PTC (65–86%) and no BRAFV600E mutations were observed [47]. In cases with long latency (>9–10 years after the accident), the frequencies of RET/PTC and the BRAFV600E mutation were 50–60% and 4–16%, respectively [47], which is not much different from those of pediatric sporadic PTC cases [24]. The cases that were exposed after birth and developed PTC ~5 years after the exposure, which were certainly radiogenic, were very young, which is almost never seen in sporadic PTCs. Therefore, it is still not clear and controversial whether this genetic pattern (the very high rate of RET/PTC and no BRAFV600E) is due to radiation exposure or just age of onset. Powell et al. examined the presence of RET/PTC and BRAFV600E in 27 exposed cases (median age 13.8 years, range 10.3–15.7) and 8 unexposed cases (median age 11.9 years, range 7.9–15.1) from Ukraine, and found no difference in the frequency of both oncogenes [48]. On the other hand, Ricarte-Filho et al. examined 26 radiation-associated PTCs from Ukraine and 27 age-matched sporadic PTCs from the same regions and compared their pattern of oncogenes [49]. Although their median ages of the radiation and sporadic groups were 17.8 and 16.6 years, respectively, fusion oncogenes including RET/PTC were significantly more prevalent in the radiation-exposed cases (84.6%) compared with the sporadic cases (33.3%). Another study by Efanov et al. has demonstrated an association between radiation dose and the prevalence of fusion oncogenes in 65 Ukrainian cases [50]. The mean 131I dose in cases with fusion oncogenes was significantly higher than that in point mutation-positive cases. Even in multivariate analysis, the adjusted odds ratio for cases with fusion oncogenes relative to cases with point mutations still significantly increased with 131I dose. The latter two studies investigated not only RET/PTC1 and 3, which are the most prevalent fusion oncogenes in PTCs, but also other fusion oncogenes such as relatively rare RET/PTCs and other fusions with NTRK and BRAF. These indicate that the frequency of fusion oncogenes may be affected by both radiation exposure and younger age. It should also be noted that the radiation-exposed cases described above may contain actual sporadic cases. So far, there is no specific genetic alteration that is exclusive to radiation-induced PTC.
FUKUSHIMA PEDIATRIC AND ADOLESCENT THYROID CANCERS
After the accident at the Fukushima Daiichi Nuclear Power Plant in March 2011, a large amount of radioiodine was released into the environment. Although estimated thyroid doses in Fukushima were very low, a thyroid ultrasound screening program for all children aged 0–18 years old at the time of the accident was started in October 2011. In the first round of the screening, the so-called baseline survey, ~100 thyroid cancers per 300 000 children were found. These cancers were not thought to be radiogenic because of low thyroid doses and too short latency.
Among these, we investigated the driver oncogenes in 63 PTC cases (mean age at operation: 17.7 years) [51]. We detected BRAFV600E in 43 cases (68.3%), RET/PTC in 7 (11.1%) and ETV6/NTRK3 in 4 (6.3%). The TERT promoter mutations were not found. The frequencies of BRAFV600E and RET/PTC were the highest and the lowest, respectively, compared with previous reports regarding pediatric PTCs. This oncogenic profile is similar to adult sporadic cases. Note that almost all of these tumors were asymptomatic and discovered by the mass screening. Therefore, if they had not been screened, they would be latent until a later age. This logically explains why they had the oncogenic profile described.
Another interesting finding in this study is that tumors with the BRAFV600E mutation were significantly smaller in size than those without this mutation, most of which presumably harbor fusion genes [51]. This implies that in this age group, the BRAFV600E mutation does not confer a dominant growth advantage on PTC compared with other fusion oncogenes. On the other hand, it may be considered that tumors with fusion oncogenes are more aggressive. As described earlier, there are many publications demonstrating that tumors with the BRAFV600E mutation display more aggressive clinicopathological features in adult sporadic PTCs, and this is the opposite result. Again, note that this aggressiveness is absent in Japanese cases. This finding also supports the above oncogenic profile. Without screening, only children having a symptom due to a large tumor visit a hospital, and such tumors have a fusion oncogene. Therefore, in previous reports about pediatric PTCs, fusion oncogenes, including RET/PTC, were more prevalent than BRAFV600E [24]. In recent years, there are some consistent data. Pekova et al. have demonstrated that fusion-positive PTCs were significantly associated with extrathyroidal extension, higher T status, and distant metastasis compared with fusion-negative PTCs [39]. Geng et al. have also reported that the presence of the BRAFV600E mutation was associated with lower risk scores (MACIS/AMES) and concluded that the mutation negatively correlated with aggressiveness of pediatric PTCs [36]. However, Onder et al. have shown adverse correlation between the BRAFV600E mutation and disease-free survival, although this correlation was mainly due to histological variants [29]. These findings suggest that different types of driver oncogenes (BRAFV600E or fusion oncogenes) may have different roles compared with those in adult cases (Table 2).
Table 2.
Current hypothesis on crucial driver oncogenes in different etiologies and age groups
| Age at diagnosis | Radiation-induced (Chernobyl) | Sporadic | |
|---|---|---|---|
| 5–10 years (Pediatric) | Initiating event frequency | Fusion | Very rare |
| Importance in growth advantage | Fusion | ||
| 10–20 years (Adolescent) | Initiating event frequency | Fusion ≫ BRAF | Fusion < BRAFa |
| Importance in growth advantage | Fusion ≫ BRAF | Fusion ≫ BRAF | |
| 20+ years (adult) | Initiating event frequency | Fusion >? BRAF | Fusion ≪ BRAF |
| Importance in growth advantage | Fusion ? BRAF | Fusion <? BRAF |
aIncluding latent cases.
?: still not clear
A HYPOTHESIS OF PRE-EXISTING FUSION ONCOGENES
It has been thought that RET/PTC in radiation-induced pediatric PTCs around Chernobyl are generated by ionizing radiation because two breakpoints involved in generation of RET/PTC are physically close in the nucleus of interphase human thyroid cells (but not other type of cells) and also have sequence homology [52, 53]. Therefore, it may be possible that a single radiation track can produce two double strand breaks and generate RET/PTC. In addition, radiation exposure to normal human thyroid cells produced RET/PTC in vitro in a dose-dependent manner [54], albeit at very high dose. However, the latent period of the first childhood PTC cases that developed in ~1990, which were most likely radiogenic, was very short, which means that it took only 4 or 5 years since the first hit (initiation) in a single cell.
Nakamura proposed an interesting hypothesis regarding radiation-related leukemia [55]. Generation of fusion genes specific to acute lymphocytic leukemia (ALL) occurs much more frequently than actual ALL cases. In addition, clonal expansion of the cells harboring the fusion gene is often found in individuals who do not develop ALL. Hence, it was postulated that the risk for radiation-induced ALL may be attributable to a small number of individuals who already had the clone that expanded to a certain size.
There is no such evidence for fusion oncogenes in PTCs such as RET/PTC; however, RET/PTC was reported to be present in benign adenomas and also in Hashimoto thyroiditis tissues [12, 56], although these findings still remain controversial. Some of the above studies might have detected RET/PTC in normal untransformed cells. Assuming that there already were thyroid follicular cells with RET/PTC, radiation may play a role as a promoter/progressor, not initiator, enabling short latency within 4–5 years. However, this is just a hypothesis, and further study is definitely necessary.
CONCLUSION
In PTCs, the major difference in molecular pathogenesis between childhood and adult cases lies in the difference between fusion genes and point mutations, mainly BRAFV600E, as a driver oncogene. The younger the age, the more important the fusion oncogenes are in the development of PTC. In teenagers or older cases, it is assumed that BRAFV600E becomes a major player if latent cases are included. Regarding aggressiveness, in childhood and adolescent PTCs, fusion oncogenes may have a higher growth impact compared with BRAFV600E, although this is not the case in adult PTCs. In radiation-associated PTCs, the frequency of fusion oncogenes is certainly high. However, the frequency itself depends on both radiation exposure and younger age. It is apparent that radiation increases the risk for PTC development in children; however, its molecular mechanisms including how fusion genes are generated still remain to be elucidated.
SUPPLEMENT FUNDING
This work was supported by the Program of the Network-type Joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University, and Fukushima Medical University.
CONFLICT OF INTEREST
None declared.
REFERENCES
- 1. Ron, E, Lubin, JH, Shore, RE et al. Thyroid cancer after exposure to external radiation: A pooled analysis of seven studies. Radiat Res. 1995; 141(3): 259–77. [PubMed] [Google Scholar]
- 2. Furukawa, K, Preston, D, Funamoto, S et al. Long-term trend of thyroid cancer risk among Japanese atomic-bomb survivors: 60 years after exposure. Int J Cancer. 2013; 132(5): 1222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kondo, T, Ezzat, S, Asa, SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006; 6(4): 292–306. [DOI] [PubMed] [Google Scholar]
- 4. Song, YS, Lim, JA, Park, YJ. Mutation profile of well-differentiated thyroid cancer in Asians. Endocrinol Metab (Seoul). 2015; 30(3): 252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guan, H, Ji, M, Bao, R et al. Association of high iodine intake with the T1799A BRAF mutation in papillary thyroid cancer. J Clin Endocrinol Metab. 2009; 94(5): 1612–7. [DOI] [PubMed] [Google Scholar]
- 6. Vuong, HG, Kondo, T, Oishi, N et al. Genetic alterations of differentiated thyroid carcinoma in iodine-rich and iodine-deficient countries. Cancer Med. 2016; 5(8): 1883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elsheikh, TM, Asa, SL, Chan, JK et al. Interobserver and intraobserver variation among experts in the diagnosis of thyroid follicular lesions with borderline nuclear features of papillary carcinoma. Am J Clin Pathol. 2008; 130(5): 736–44. [DOI] [PubMed] [Google Scholar]
- 8. Xing, M. BRAF mutation in papillary thyroid cancer: Pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007; 28(7): 742–62. [DOI] [PubMed] [Google Scholar]
- 9. Ito, Y, Yoshida, H, Maruo, R et al. BRAF mutation in papillary thyroid carcinoma in a Japanese population: Its lack of correlation with high-risk clinicopathological features and disease-free survival of patients. Endocr J. 2009; 56(1): 89–97. [DOI] [PubMed] [Google Scholar]
- 10. Matsuse M, Yabuta T, Saenko V et al. TERT promoter mutations and Ki-67 labeling index as a prognostic marker of papillary thyroid carcinomas: Combination of two independent factors. Sci Rep. 2017;7:41752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanaka, A, Matsuse, M, Saenko, V et al. TERT mRNA expression as a novel prognostic marker in papillary thyroid carcinomas. Thyroid. 2019; 29(8): 1105–14. [DOI] [PubMed] [Google Scholar]
- 12. Nikiforov, YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol. 2002; 13(1): 3–16. [DOI] [PubMed] [Google Scholar]
- 13. Yakushina, VD, Lerner, LV, Lavrov, AV. Gene fusions in thyroid cancer. Thyroid. 2018; 28(2): 158–67. [DOI] [PubMed] [Google Scholar]
- 14. Cancer Genome Atlas Research , N. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014; 159(3): 676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leeman-Neill, RJ, Kelly, LM, Liu, P et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer. 2014; 120(6): 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bastos, AU, de Jesus, AC, Cerutti, JM. ETV6-NTRK3 and STRN-ALK kinase fusions are recurrent events in papillary thyroid cancer of adult population. Eur J Endocrinol. 2018; 178(1): 83–91. [DOI] [PubMed] [Google Scholar]
- 17. Kimura, ET, Nikiforova, MN, Zhu, Z et al. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003; 63(7): 1454–7. [PubMed] [Google Scholar]
- 18. Knauf, JA, Kuroda, H, Basu, S et al. RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene. 2003; 22(28): 4406–12. [DOI] [PubMed] [Google Scholar]
- 19. Lange, AM, Lo, HW. Inhibiting TRK proteins in clinical cancer therapy. Cancers (Basel). 2018; 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pratilas, CA, Taylor, BS, Ye, Q et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009; 106(11): 4519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitsutake, N, Knauf, JA, Mitsutake, S et al. Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid PCCL3 cells. Cancer Res. 2005; 65(6): 2465–73. [DOI] [PubMed] [Google Scholar]
- 22. Caronia, LM, Phay, JE, Shah, MH. Role of BRAF in thyroid oncogenesis. Clin Cancer Res. 2011; 17(24): 7511–7. [DOI] [PubMed] [Google Scholar]
- 23. Henke, LE, Perkins, SM, Pfeifer, JD et al. BRAF V600E mutational status in pediatric thyroid cancer. Pediatr Blood Cancer. 2014; 61(7): 1168–72. [DOI] [PubMed] [Google Scholar]
- 24. Cordioli, MI, Moraes, L, Cury, AN et al. Are we really at the dawn of understanding sporadic pediatric thyroid carcinoma? Endocr Relat Cancer. 2015; 22(6): R311–24. [DOI] [PubMed] [Google Scholar]
- 25. Nikita, ME, Jiang, W, Cheng, SM et al. Mutational analysis in Pediatric thyroid cancer and correlations with age, ethnicity, and clinical presentation. Thyroid. 2016; 26(2): 227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alzahrani, AS, Qasem, E, Murugan, AK et al. Uncommon TERT promoter mutations in Pediatric thyroid cancer. Thyroid. 2016; 26(2): 235–41. [DOI] [PubMed] [Google Scholar]
- 27. Prasad, ML, Vyas, M, Horne, MJ et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in Northeast United States. Cancer. 2016; 122(7): 1097–107. [DOI] [PubMed] [Google Scholar]
- 28. Gertz, RJ, Nikiforov, Y, Rehrauer, W et al. Mutation in BRAF and other members of the MAPK pathway in papillary thyroid carcinoma in the Pediatric population. Arch Pathol Lab Med. 2016; 140(2): 134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Onder, S, Ozturk Sari, S, Yegen, G et al. Classic architecture with multicentricity and local recurrence, and absence of TERT promoter mutations are correlates of BRAF (V600E) Harboring Pediatric papillary thyroid carcinomas. Endocr Pathol. 2016; 27(2): 153–61. [DOI] [PubMed] [Google Scholar]
- 30. Alzahrani, AS, Murugan, AK, Qasem, E et al. Single point mutations in Pediatric differentiated thyroid cancer. Thyroid. 2017; 27(2): 189–96. [DOI] [PubMed] [Google Scholar]
- 31. Cordioli, MI, Moraes, L, Bastos, AU et al. Fusion oncogenes are the main genetic events found in sporadic papillary thyroid carcinomas from children. Thyroid. 2017; 27(2): 182–8. [DOI] [PubMed] [Google Scholar]
- 32. Poyrazoglu, S, Bundak, R, Bas, F et al. Clinicopathological characteristics of papillary thyroid cancer in children with emphasis on pubertal status and association with BRAF(V600E) mutation. J Clin Res Pediatr Endocrinol. 2017; 9(3): 185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oishi, N, Kondo, T, Nakazawa, T et al. Frequent BRAF (V600E) and absence of TERT promoter mutations characterize sporadic Pediatric papillary thyroid carcinomas in Japan. Endocr Pathol. 2017; 28(2): 103–11. [DOI] [PubMed] [Google Scholar]
- 34. Vanden Borre, P, Schrock, AB, Anderson, PM et al. Pediatric, adolescent, and young adult thyroid carcinoma Harbors frequent and diverse targetable genomic alterations, including kinase fusions. Oncologist. 2017; 22(3): 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hardee, S, Prasad, ML, Hui, P et al. Pathologic characteristics, natural history, and prognostic implications of BRAF(V600E) mutation in Pediatric papillary thyroid carcinoma. Pediatr Dev Pathol. 2017; 20(3): 206–12. [DOI] [PubMed] [Google Scholar]
- 36. Geng, J, Wang, H, Liu, Y et al. Correlation between BRAF (V600E) mutation and clinicopathological features in pediatric papillary thyroid carcinoma. Sci China Life Sci. 2017; 60(7): 729–38. [DOI] [PubMed] [Google Scholar]
- 37. Sisdelli, L, Cordioli, M, Vaisman, F et al. AGK-BRAF is associated with distant metastasis and younger age in pediatric papillary thyroid carcinoma. Pediatr Blood Cancer. 2019; 66(7): e27707. [DOI] [PubMed] [Google Scholar]
- 38. Galuppini F, Vianello F, Censi S et al. Differentiated thyroid carcinoma in Pediatric age: Genetic and clinical scenario. Front Endocrinol (Lausanne). 2019;10:552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pekova B, Sykorova V, Dvorakova S et al. RET, NTRK, ALK, BRAF, and MET fusions in a large cohort of Pediatric papillary thyroid carcinomas. Thyroid. 2020. [DOI] [PubMed] [Google Scholar]
- 40. Landa, I, Ganly, I, Chan, TA et al. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab. 2013; 98(9): E1562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vinagre J, Almeida A, Populo H et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. [DOI] [PubMed] [Google Scholar]
- 42. Liu, R, Xing, M. TERT promoter mutations in thyroid cancer. Endocr Relat Cancer. 2016; 23(3): R143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang, FW, Hodis, E, Xu, MJ et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013; 339(6122): 957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin, HY, Chin, CY, Huang, HL et al. Crystalline inorganic frameworks with 56-ring, 64-ring, and 72-ring channels. Science. 2013; 339(6121): 811–3. [DOI] [PubMed] [Google Scholar]
- 45. Shibata, Y, Yamashita, S, Masyakin, VB et al. 15 years after Chernobyl: New evidence of thyroid cancer. Lancet. 2001; 358(9297): 1965–6. [DOI] [PubMed] [Google Scholar]
- 46. Ron, E. Thyroid cancer incidence among people living in areas contaminated by radiation from the Chernobyl accident. Health Phys. 2007; 93(5): 502–11. [DOI] [PubMed] [Google Scholar]
- 47. Nikiforov, YE. Radiation-induced thyroid cancer: What we have learned from Chernobyl. Endocr Pathol. 2006; 17(4): 307–17. [DOI] [PubMed] [Google Scholar]
- 48. Powell, N, Jeremiah, S, Morishita, M et al. Frequency of BRAF T1796A mutation in papillary thyroid carcinoma relates to age of patient at diagnosis and not to radiation exposure. J Pathol. 2005; 205(5): 558–64. [DOI] [PubMed] [Google Scholar]
- 49. Ricarte-Filho, JC, Li, S, Garcia-Rendueles, ME et al. Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J Clin Invest. 2013; 123(11): 4935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Efanov, AA, Brenner, AV, Bogdanova, TI et al. Investigation of the relationship between radiation dose and gene mutations and fusions in post-Chernobyl thyroid cancer. J Natl Cancer Inst. 2018; 110(4): 371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mitsutake N, Fukushima T, Matsuse M et al. BRAF(V600E) mutation is highly prevalent in thyroid carcinomas in the young population in Fukushima: A different oncogenic profile from Chernobyl. Sci Rep. 2015;5:16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nikiforov, YE, Koshoffer, A, Nikiforova, M et al. Chromosomal breakpoint positions suggest a direct role for radiation in inducing illegitimate recombination between the ELE1 and RET genes in radiation-induced thyroid carcinomas. Oncogene. 1999; 18(46): 6330–4. [DOI] [PubMed] [Google Scholar]
- 53. Nikiforova, MN, Stringer, JR, Blough, R et al. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000; 290(5489): 138–41. [DOI] [PubMed] [Google Scholar]
- 54. Caudill, CM, Zhu, Z, Ciampi, R et al. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to gamma-radiation: A model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab. 2005; 90(4): 2364–9. [DOI] [PubMed] [Google Scholar]
- 55. Nakamura, N. A hypothesis: Radiation-related leukemia is mainly attributable to the small number of people who carry pre-existing clonally expanded preleukemic cells. Radiat Res. 2005; 163(3): 258–65. [DOI] [PubMed] [Google Scholar]
- 56. Sadetzki, S, Calderon-Margalit, R, Modan, B et al. Ret/PTC activation in benign and malignant thyroid tumors arising in a population exposed to low-dose external-beam irradiation in childhood. J Clin Endocrinol Metab. 2004; 89(5): 2281–9. [DOI] [PubMed] [Google Scholar]