

Abstract
Background
Clinical outcomes in neuromyelitis optica spectrum disorders (NMOSD) vary across different regions.
Objective
To describe clinical profiles in Japanese and German NMOSD patients.
Methods
Medical records of aquaporin-4-immunoglobulin G (AQP4-IgG) positive NMOSD patients from Japan (n = 54) and Germany (n = 38) were retrospectively analyzed.
Results
The disability status was similar between both cohorts, although Japanese patients had a longer disease duration (13.3 ± 11.1 vs. 8.1 ± 6.9 years, p = 0.018) but similar relapse rates. Optic neuritis and myelitis were the most frequent attacks in both cohorts. Brain attacks occurred more frequently in Japanese patients (40.7% vs. 15.8%, p = 0.020). The time from disease onset (median [interquartile range] 2.3 [0.3-10.1] vs. 0.6 [0.2-1.9] years, p = 0.009) and the number of attacks (2.5 [1-7] vs. 2 [1-3], p = 0.047) until start of the first immunotherapy were higher in the Japanese cohort. Rituximab was the most common drug in the German cohort (52.6%) and not given in the Japanese cohort (p < 0.001), where oral prednisolone was the most common drug (92.6% vs. 15.8%, p < 0.001). The frequency of autoimmune comorbidities was higher in the German cohort (39.5% vs. 18.5%, p = 0.047).
Conclusion
Compared with Japanese NMOSD patients, German patients presented with similar disability despite shorter disease duration and earlier and more frequent immunosuppressive therapy.
Keywords: Anti-aquaporin 4 antibodies, autoimmune diseases, ethnicity, immunotherapy, neuromyelitis optica spectrum disorders
Introduction
Neuromyelitis optica spectrums disorders (NMOSD) are a group of autoimmune conditions of the CNS that share an overlapping clinical phenotype including optic neuritis (ON), longitudinally extensive transverse myelitis (LETM), encephalitis, and brainstem involvement.1,2 In most cases, attacks occur recurrently and clinical symptoms are severe, recover only partially, and lead to chronic disability.2 Consequently, patients suffer from visual deficits up to blindness, sensorimotor dysfunction, sphincter and bladder disturbances, pain, depression, fatigue and cognitive impairment, leading to a significantly reduced quality of life.3–8
Disease remission is rare and symptomatic treatment can be difficult.2,9 About 80% of patients with a clinical phenotype of NMOSD have serum immunoglobulin G antibodies against aquaporin-4 (AQP4-IgG), a water channel expressed in the astrocytic end-feet at the blood-brain-barrier.10 Causing astrocytopathy with secondary demyelination, AQP4-IgG are of pathogenic relevance.1
In a subgroup of AQP4-IgG negative patients with a clinical phenotype of NMOSD, antibodies targeting myelin oligodendrocyte glycoprotein (MOG) have been detected.2,11 The present study focuses only on AQP4-IgG-positive NMOSD. Data on Anti-MOG antibody-associated disorders from the same cohorts have recently been published separately.12
In some frequent autoimmune disorders like MS, epidemiology and disease presentation differ substantially between different regions of the world.13 In NMOSD, data on prevalence and incidence are scarce. A few reports suggest that its global distribution is relatively similar,14 while some studies reported that NMOSD prevalence is higher in non-white than in white populations15,16 and higher in individuals with Asian ancestry than in other ethnicities.17 Asian, Afro-American, Afro-Caribbean, and Afro-European patients have a lower age of onset than Caucasian patients, a higher prevalence of brain attacks and more frequent brain abnormalities on MRI.13,18–20 Clinically, Afro-American and Afro-European patients are more likely to have severe attacks, a higher likelihood of visual disability13,18 and worse motor deficits than Asian and Caucasian patients.18,21 Compared to Asian patients, Caucasian and Afro-Caribbean patients have a younger age at disease onset, worse onset attacks with a higher risk of visual impairment and a more severe disease course with a higher relapse frequency and greater disability, despite earlier immunosuppression.13
The aim of this study was to compare demographic and clinical features as well as treatment strategies of patients with AQP4-IgG-positive NMOSD from Germany and Japan.
Patients and methods
Patients
Data were obtained from adult AQP4-IgG-positive patients with NMOSD treated at outpatient and inpatient clinics at Chiba University Hospital, Japan, and from an ongoing observational study following patients with AQP4-IgG-positive NMOSD and related disorders at Charité - Universitätsmedizin Berlin, Germany. All patients gave written informed consent to study participation. The study was approved by the Ethics Committees of the participating centers. Patients were included at any time during their disease course. All data were acquired during remission.
Databases at the respective study centers were screened for eligibility of AQP4-IgG-positive patients. Inclusion criteria for the study was a diagnosis of AQP4-IgG-positive NMOSD according to the 2015 international consensus diagnostic criteria for NMOSD.1 Serostatus was tested in Germany using a cell-based assay (CBA) (Euroimmun, Lübeck, Germany) and in Japan using flow cytometry-CBA, as described previously.22 Although the methodology in the centers was different, the high specificity and sensitivity of both methods have already been demonstrated.23 Of note, all patients included tested negative for MOG-IgG.
Patient data assessment
All data was retrospectively analyzed. We studied demographics (sex, ethnicity), age at disease onset, number and type of attacks (categorized as ON, transverse myelitis, brainstem, area postrema, cerebral and mixed attacks), disease duration at last follow-up, attack treatment, previous and current immunotherapy, annualized relapse rate (ARR), ARR before any treatment and during current treatment, Expanded Disability Status Scale (EDSS) score including functional system scores (FSS) at last follow-up, EDSS increase per attack, recovery from first attack, first ON and first myelitis, concomitant autoimmune diseases, presence/absence of CSF-specific oligoclonal bands and presence/absence of long spinal cord lesions at any time during the disease course, as well as persistent myelitis, brainstem/area postrema and cerebral lesions on 1.5-Tesla (Japan) and 3-Tesla (Germany) MRI at last follow-up. As all data were acquired outside of acute attacks, no contrast agent was given during MRI.
Statistics
Statistical analysis was performed in R version 3.4.4 with R Studio Version 1.1.442 (R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, www.r-project.org/). Continuous variables are given as mean±standard deviation (SD) and were compared with t-tests in the case of a normal distribution. Non-parametric data are presented as median and interquartile range (IQR) and were tested with Wilcoxon-Mann-Whitney U test. Categorical data was tested using Pearson’s Chi-Square test. Kaplan-Meier curves with cox proportional hazard models were used for estimating the time to the second attack. For the latter analysis, we included only patients with a follow-up period of at least two years, avoiding bias from patients without a second attack due to short follow-up. Patients were censored after seven years due to a low number of cases with a longer follow-up. P-values ≤0.05 were considered to indicate statistical significance. Due to the exploratory nature of this study no adjustments for multiple testing were made.
Results
Demographics and clinical characteristics
We included 92 AQP4-IgG-positive NMOSD patients from Germany (n = 38, Caucasian) and Japan (n = 54, East-Asian). One patient from Germany had to be excluded because of incomplete clinical data. Table 1 provides an overview of the demographic and clinical findings. There were no differences regarding sex distribution, age at disease onset or age at last follow-up between the groups. A histogram analysis revealed two peaks of disease onset in both cohorts: 1) around 20 years of age, 2) around 40 years of age (Figure 1). There was no difference regarding disease onset in summer (April to September) or winter (October to March) between both cohorts (p = 0.324), nor between onset in summer (Germany: p = 0.447, Japan: p = 0.574) or winter (Germany t: p = 0.553, Japan: p = 0.426) within the cohorts. Nine patients from Japan and four patients from Germany had only one attack at time of last follow-up. The frequency of autoimmune comorbidities was higher in the German cohort (p = 0.047). Six German patients (15.8%) but no Japanese patient (p = 0.010) had concomitant systemic lupus erythematosus (SLE) (Table 1).
Table 1.
Demographic and clinical characteristics of AQP4-IgG-positive Japanese and German NMOSD patients included in this study.
| AQP4-IgG-positive NMOSD | German (Berlin) (n = 38) | Japanese (Chiba) (n = 54) | P |
|---|---|---|---|
| Age at last follow-up, years: mean ± SD | 50.61 ± 14.00 | 55.30 ± 13.13 | 0.104 |
| Sex: n female/male (female %) | 35/3 (92.1%) | 48/6 (88.9%) | 0.877 |
| Age at disease onset, years: mean ± SD | 42.50 ± 15.32 | 41.96 ± 14.96 | 0.867 |
| Early/late disease onset: n (%) | 0.858 | ||
| <30 years | 7 (18.4%) | 12 (22.2%) | |
| 30–50 years | 13 (34.2%) | 16 (29.6%) | |
| >50 years | 18 (47.4%) | 26 (48.1%) | |
| Disease duration at last follow-up, years: mean ± SD | 8.11 ± 6.90 | 13.33 ± 11.08 | 0.018 |
| EDSS at last follow-up: median [IQR] | 4 [2.12, 5.25] | 4.00 [2.00, 5.75] | 0.784 |
| Visual FSS: median [IQR] | 1.00 [0.00, 3.00] | 1.00 [1.00, 2.00] | 0.807 |
| Brainstem FSS: median [IQR] | 0.00 [0.00, 1.00] | 0.00 [0.00, 0.00] | <0.001 |
| Pyramidal FSS: median [IQR] | 1.00 [0.00, 3.00] | 0.00 [0.00, 0.00] | <0.001 |
| Cerebellar FSS: median [IQR] | 1.00 [0.00, 2.00] | 2.00 [0.00, 3.00] | 0.155 |
| Sensory FSS: median [IQR] | 2.00 [1.00, 3.00] | 0.50 [0.00, 2.00] | 0.001 |
| Bowel and bladder FSS: median [IQR] | 1.00 [0.00, 2.00] | 2.00 [0.00, 5.00] | 0.037 |
| Cerebral FSS: median [IQR] | 0.50 [0.00, 1.00] | 0.00 [0.00, 0.00] | <0.001 |
| Ambulation FSS: median [IQR] | 4.50 [0.00, 4.88] | 1.00 [0.00, 5.25] | 0.669 |
| Severe disability at last follow-up: n (%) | 8 (21.1%) | 14 (25.9%) | 0.771 |
| EDSS increase per attack: median [IQR] | 0.95 [0.67, 1.56] | 0.63 [0.48, 1.24] | 0.059 |
| CSF-specific oligoclonal bands: n (%) | 6/25(19.4%) | 8/27 (22.9%) | 0.964 |
| Autoimmune comorbidities: n (%) | 15 (39.5%) | 10 (18.5%) | 0.047 |
| – Sjoergen syndrome: n (%) | 3 (7.9%) | 6 (11.1%) | 0.877 |
| – Hashimoto disease: n (%) | 5 (13.2%) | 3 (5.6%) | 0.369 |
| – Rheumatoid arthritis: n (%) | 1 (2.6%) | 1 (1.9%) | >0.999 |
| – Myasthenia gravis: n (%) | 3 (7.9%) | 1 (1.9%) | 0.379 |
| – Systemic lupus erythematosus: n (%) | 6 (15.8%) | 0 (0.0%) | 0.010 |
| – Raynaud’s syndrome: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| – Mixed connective tissue disease: n (%) | 2 (5.3%) | 0 (0.0%) | 0.328 |
| – Secondary antiphospholipid syndrome: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
AQP4-IgG: Aquaporin 4-immunoglobulin G; EDSS: expanded disability status scale; FSS: functional system score; IQR: interquartile range; n = number; NMOSD: neuromyelitis optica spectrum disorders, SD: standard deviation.
Note that these group comparisons were performed using t-test for current age, age at disease onset, and disease duration, Chi-square-test for categorial variables, severe disability at last follow-up (defined as an EDSS ≥6), and Wilcoxon-Mann-Whitney test for EDSS and functional system scores and EDSS increase per attack. Significant p-values are indicated in bold.
Figure 1.
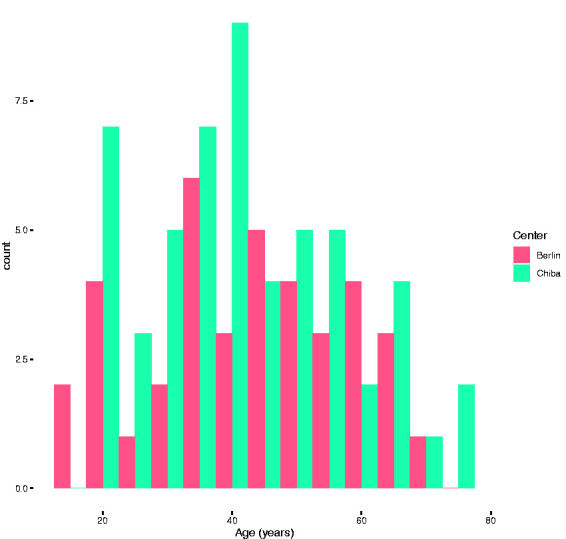
Histogram for age at onset for both centers. The histogram reveals two peaks of disease onset (1) around 20 years of age and (2) around 40 years of age.
Disease course
At the time of assessment, Japanese patients had a longer disease duration than German patients (mean±SD: 13.33 ± 11.08 vs. 8.11 ± 6.90, p = 0.018), although the number of attacks was similar (median [IQR]: 3.00 [2.00, 5.00] vs. 5.00 [2.00, 9.75], p = 0.115).
Table 2 provides details about the disease course and the type of attacks. A comparable number of patients had experienced at least one myelitis. There was no difference between Japanese and German patients in the frequency of ON. However, brain attacks - including area postrema, brainstem and cerebrum - were more frequent in Japanese patients (p = 0.020). Most brain attacks were area postrema and/or brainstem attacks (German cohort n = 8 (100%), Japanese cohort n = 22 (61.1%)). Cerebral attacks occurred only in the Japanese cohort (n = 14 (38.9%)).
Table 2.
Disease course and the type of attacks in AQP4-IgG-positive Japanese and German NMOSD patients.
| AQP4-IgG-positive NMOSD | German (Berlin) (n = 38) | Japanese (Chiba) (n = 54) | P | |
|---|---|---|---|---|
| Entire disease course | ||||
| Total number of attacks: median [IQR] | 3.00 [2.00, 5.00] | 5.00 [2.00, 9.75] | 0.115 | |
| Optic neuritis: n (%) | 24 (63.2%) | 40 (74.1%) | 0.373 | |
| Bilateral optic neuritis: n (%) | 11 (28.9%) | 22 (40.7%) | 0.347 | |
| Myelitis: n (%) | 34 (89.5%) | 43 (79.6%) | 0.331 | |
| Long spinal cord lesiona: n (%) | 31 (86.1%) | 36 (66.7%) | 0.068 | |
| Brain attack (including area postrema, brainstem and cerebral attacks): n (%) | 6 (15.8%) | 22 (40.7%) | 0.020 | |
| Brainstem attack: n (%) | 6 (15.8%) | 12 (22.2%) | 0.618 | |
| Area postrema attack: n (%) | 2 (5.3%) | 7 (13.0%) | 0.386 | |
| Cerebral attack: n (%) | 0 (0.0%) | 9 (16.7%) | 0.022 | |
| Annualized relapse rate: median [IQR] | 0.50 [0.38, 0.70] | 0.41 [0.31, 0.86] | 0.403 | |
| Presentation at onset | ||||
| Optic neuritis at onset: n (%) | 18 (47.4%) | 25 (46.3%) | >0.999 | |
| Myelitis at onset: n (%) | 18 (47.4%) | 22 (40.7%) | 0.676 | |
| Brain attack (including area postrema, brainstem and cerebral attacks) at onset: n (%) | 5 (13.2%) | 11 (20.4%) | 0.536 | |
| Area postrema attack at onset: n (%) | 0 (0.0%) | 5 (9.3%) | 0.144 | |
| Brainstem attack at onset: n (%) | 5 (13.2%) | 5 (9.3%) | 0.801 | |
| Cerebral attack at onset: n (%) | 0 (0.0%) | 1 (1.9%) | >0.999 | |
| Presentation at second attack | ||||
| Optic neuritis at second attack: n (%) | 16 (42.1%) | 22 (40.7%) | >0.999 | |
| Myelitis at second attack: n (%) | 20 (52.6%) | 21 (38.9%) | 0.274 | |
| Brain attack at second attack: n (%) | 1 (2.6%) | 4 (7.4%) | 0.598 | |
| Recovery | ||||
| First attack (any type) | Full: n (%) | 8 (25.8%) | 7 (14.3) | 0.229 |
| Partial: n (%) | 21 (67.7%) | 41 (83.7%) | ||
| None: n (%) | 2 (6.5%) | 1 (2.0%) | ||
| First myelitis | Full: n (%) | 8 (25.8%) | 9 (18.4%) | 0.541 |
| Partial: n (%) | 17 (54.8%) | 29 (59.2%) | ||
| None: n (%) | 2 (6.5%) | 1 (2.0%) | ||
| First optic neuritis | Full: n (%) | 6 (19.4%) | 7 (14.3%) | 0.497 |
| Partial: n (%) | 11 (35.5%) | 26 (53.1%) | ||
| None: n (%) | 2 (6.5%) | 2 (4.1%) | ||
AQP4-IgG: Aquaporin 4-immunoglobulin G; CSF: cerebro-spinal fluid; IQR: interquartile range; n: number; NMOSD: neuromyelitis optica spectrum disorders; SD: standard deviation.
Note that these group comparisons were performed using Chi-Square test to compare categorial variables and Wilcoxon-Mann-Whitney test for continuous variables. Please note, that Figure 3 provides details on the occurrence of combined syndromes during the first attack. a) Lesion extension of ≥3 contiguous vertebral segments. Significant p-values are indicated in bold.
ARR did not differ between both groups. Kaplan-Meier statistics with Cox regression showed that there was no significant difference in the risk for a second attack, though with a trend for a lower risk in the Japanese cohort cohort (Hazard ratio =0.461, confidence interval (CI) 0.195-1.09, p = 0.078) (Figure 2). EDSS at last follow-up did not differ between both groups (4.00 [2.00, 5.75] (Japan) vs. 4.00 [2.12, 5.25] (Germany), p = 0.784). However, brainstem, pyramidal, sensory, and cerebral FSS were lower in the Japanese than in the German cohort. Only bowel and bladder FSS was higher in the Japanese cohort. Also the EDSS increase per attack showed a tendency to higher values in the German cohort. Severe disability at last follow-up defined as an EDSS ≥6 was similar in both cohorts (Table 1).
Figure 2.

Comparison of relapse risk in the German and the Japanese cohorts. Kaplan–Meier plot with Cox regression comparing show that there is no significant difference in the risk for a relapse between both cohorts. Please note that the analysis includes only patients with a follow-up period of at least two years, and that patients were censored after seven years as only few patients had a follow-up beyond that.
Onset attack and relapses
The clinical presentation at disease onset was similar in both cohorts (Table 2, Figure 3).
Figure 3.

Type of onset attack in the German and the Japanese cohorts. The chart shows the respective type of onset attacks. AP: area postrema syndrome; BS: brainstem syndrome; CB: cerebral syndrome; MY: myelitis; ON: optic neuritis.
The median time to the second attack after the onset attack was 0.65 years (IQR: 0.23-1.65) in the German cohort and 1.21 years (IQR: 0.34-2.48) in the Japanese cohort (p = 0.087) (Table 2). The recovery from the first attack, the first ON and the first myelitis was similar in both cohorts (Table 2).
Treatment
Acute attacks were mainly treated with intravenous high-dose corticosteroids. In the German cohort, escalation of therapy ensued in 23.8% of all attacks after a first treatment course. Only one attack within the Japanese cohort received escalation therapy. Table 4 provides details about acute attack treatment.
Table 4.
Treatment history in AQP4-IgG-positive Japanese and German NMOSD patients.
| AQP4-IgG-positive NMOSD | German (Berlin) (n = 38) | Japanese (Chiba) (n = 54) | P |
|---|---|---|---|
| Immunomodulatory therapy during the entire disease course | |||
| Ever treated: n (%) | 37 (97.4%) | 51 (94.4%) | 0.874 |
| Rituximab: n (%) | 24 (63.2%) | 0 (0.0%) | <0.001 |
| Azathioprine: n (%) | 24 (63.2%) | 14 (25.9%) | 0.001 |
| Cyclophosphamide: n (%) | 4 (10.5%) | 0 (0.0%) | 0.055 |
| Mitoxantron: n (%) | 4 (10.5%) | 0 (0.0%) | 0.055 |
| Tacrolimus: n (%) | 0 (0.0%) | 1 (1.9%) | >0.999 |
| Belimumab: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Cyclosporin A: n (%) | 0 (0.0%) | 2 (3.7%) | 0.636 |
| Mycophenolate mofetil: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Methotrexat: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Eculizumab: n (%) | 0 (0.0%) | 1 (1.9%) | >0.999 |
| Tocilizumab: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Attack treatment during the entire disease coursea | |||
| IVMP: n of attacks (%) | 111 (85.4%) | 311 (84.5%) | 0.923 |
| Plasmapheresis: n of attacks (%) | 6 (4.6%) | 6 (1.6%) | 0.115 |
| Immunoadsorption: n of attacks (%) | 1 (0.8%) | 29 (7.9%) | 0.115 |
| Intravenous immunoglobulins: n of attacks (%) | 1 (0.8%) | 1 (0.3%) | >0.999 |
| No treatment: n of attacks (%) | 11 (8.5%) | 21 (5.7%) | 0.372 |
| No information: n of attacks (%) | 34 (20.7%) | 18 (4.7%) | <0.001 |
| Escalation therapyb | |||
| Plasmapheresis: n of attacks (%) | 10 (8.2%) | 0 (0.0%) | - |
| IVMP: n of attacks (%) | 14 (11.5%) | 0 (0.01%) | - |
| Immunoadsorption: n of attacks (%) | 2 (1.6%) | 1 (0.0%) | - |
| Intravenous immunoglobulines: n of attacks (%) | 3 (2.5%) | 0 (0.0%) | - |
AQP4-IgG: Aquaporin 4-immunoglobulin G; n: number; NMOSD: neuromyelitis optica spectrum disorders. Note that these group comparisons were performed using Chi-Square test. Significant p-values are indicated in bold.aAttack treatment information was available for 498 out of 550 attacks, 130 attacks in the German cohort and 368 attacks in the Japanese cohort, respectively.bDue to the small numbers no p-values are provided for escalation therapy.
Only a minority of patients received preventive treatment after the onset attack (German cohort: n = 13 (34.2%), Japanese cohort: n = 16 (29.6%)). The time between disease onset and start of first continuous treatment was significantly longer in the Japanese cohort (Median [IQR]=2.34 years [0.28-10.08]) than in the German cohort (Median [IQR]=0.55 years [0.15-1.88], p = 0.009). Of note, four German patients had already received immunotherapy before the onset of NMOSD-related symptoms because of comorbidities. In these cases, the time to first treatment was set to zero. Patients from the Japanese cohort had on average more attacks before first treatment (Median [IQR] =2.5 [1-7]) than German patients (Median [IQR] =2 [1-3], p = 0.047).
Most patients were receiving continuous immunotherapy at last follow-up (German cohort: 89.5%, Japanese cohort 92.6%, p = 0.883) (Figure 4/Table 3). While rituximab was the most commonly used drug in the German cohort, it had not been prescribed to any Japanese patient (p < 0.001) (Table 3). In the Japanese cohort, in contrast, oral prednisolone monotherapy was the most common medication (Figure 4). Among Japanese patients with oral prednisolone as mono- or add-on therapy, 49 out of 50 received a dose of ≥7.5 mg per day, while only 1 out of 6 German patients received a dose of ≥7.5 mg per day (p < 0.001). The frequency of azathioprine was similar in both cohorts, though in the Japanese cohort it was combined with continuous prednisolone treatment (Figure 4). In both groups there were no major changes in treatment strategies over the disease course (Table 4). The ARR before any treatment (Germany: Median [IQR] =2.48 [1.10-5.37], Japan: 1.88 [0.58-6.97]) was higher compared to the ARR during treatment at last follow-up (Germany: Median [IQR] =0.00 [0.00-0.20], Japan: 0.11 [0.00-0.29], p < 0.001) and did not differ between the German and the Japanese cohort (Table 3). Treatment duration and dosage of the respective medication are given in Table 3.
Figure 4.

Current treatment strategies in the German and Japanese cohorts. The chart shows the type of current medication in the Japanese and German cohorts. AZA: azathioprine, monoclonal AB: monoclonal antibody, PSL: prednisolone, other includes cyclophosphamide (n = 1) and glatirameracetate (n = 1).
Table 3.
Treatment strategies at last follow-up in AQP4-IgG-positive Japanese and German NMOSD patients.
| AQP4-IgG-positive NMOSD | German (Berlin) (n = 38) | Japanese (Chiba) (n = 54) | P |
|---|---|---|---|
| Currently treated: n (%) | 34 (89.5%) | 50 (92.6%) | 0.883 |
| Oral prednisolone: n (%) | 6 (15.8%) | 50 (92.6%) | <0.001 |
| Glatirameracetate: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Rituximab: n (%) | 20 (52.6%) | 0 (0.0%) | <0.001 |
| Azathioprine: n (%) | 9 (23.7%) | 12 (22.2%) | >0.999 |
| Tacrolimus: n (%) | 0 (0.0%) | 1 (1.9%) | >0.999 |
| Belimumab: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Mycophenolate mofetil: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Eculizumab: n (%) | 0 (0.0%) | 1 (1.9%) | >0.999 |
| Tocilizumab: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Cyclophosphamide: n (%) | 1 (2.6%) | 0 (0.0%) | 0.859 |
| Time on current treatment: median in years [IQR] | 4.48 [1.95, 5.70] | 4.58 [2.13, 7.38] | 0.517 |
| Time on prednisolone monotherapy: median in years [IQR] | – | 5.22 [2.27, 8.18] | – |
| Time on rituximab monotherapy: median in years [IQR] | 4.48 [1.50, 5.64] | – | – |
| Time on azathioprine monotherapy: median in years [IQR] | 6.53 [4.33, 7.32] | – | |
| Time on azathioprine plus prednisolone: median in years [IQR] | 20.27 [10.78, 29.76] | – | |
| Number of attacks during current treatmenta: median [IQR] | 0.00 [0.00, 1.00] | 1.00 [0.00, 1.75] | 0.273 |
| ARR before any treatmenta: median [IQR] | 2.48 [1.10, 5.37] | 1.88 [0.58, 6.97] | 0.360 |
| ARR on current treatmenta: median [IQR] | 0.00 [0.00, 0.20] | 0.11 [0.00, 0.29] | 0.242 |
AQP4-IgG: Aquaporin 4-immunoglobulin G; IQR: interquartile range; n: number; n.a.: not available; NMOSD: neuromyelitis optica spectrum disorders.
Note that these group comparisons were performed using Chi-Square test for categorial variables and Wilcoxon-Mann-Whitney test for continuous variables.aFor 31 patients from Germany and 46 patients from Japan with at least one year of treatment duration at last follow-up; Dosage of oral prednisolone: 2 mg–20 mg/d, glatirameracetate: 20 mg/d, rituximab: 500-2000 mg/6 months, azathioprine: 50 to 150 mg/d, tacrolimus 3 mg/d, mycophenolate mofetil: 1500 mg/d, eculizumab: 1200 mg every 2 weeks, tocilizumab: 400 mg/month, cyclophosphamide: n.a. Significant p-values are indicated in bold.
MRI at last follow-up
MRI at last follow-up showed chronic myelitis lesions in 26 patients (70.3%) from the German cohort and in 33 patients (63.5%) from the Japanese cohort (p = 0.658). Chronic area postrema and/or brainstem lesions were present in 17 patients (47.2%) from the German and in 15 patients (27.8%) from the Japanese cohort. Chronic cerebral lesions were more frequent in the German cohort (n = 33 (89.2%) vs. n = 37 (68.5%, p = 0.041).
Discussion
This comparative study of German and Japanese patients with AQP-IgG-positive NMOSD compared commonalities and differences in demographics, clinical presentation, disease course, autoimmune comorbidities, and immunotherapy. Both cohorts show the expected female predominance with a female-to-male ratio of 11.7:1 in the German cohort, and of 8:1 in the Japanese cohort.2 In contrast to previous data, where Japanese and Korean patients had a younger age at disease onset, mean age at disease onset was slightly above forty years in both cohorts, as previously described in German cohorts.13,18,24 Consequently, ethnicity alone does not predict the age of disease onset, which may differ between different Asian populations.18
In line with the literature,2 the most frequent syndromes at disease onset in the German and Japanese cohorts were ON in 47% and 46%, and myelitis in 47% and 41%, respectively. Only 2% (Germany) and 3% (Japan) of the patients exhibited an eponymous neuromyelitis optica syndrome with simultaneous myelitis and ON. Forty-one percent of Japanese patients suffered from area postrema, brainstem or cerebral symptoms. Brain attacks during disease course occurred more frequently in the Japanese cohort. Interestingly, MRI at last follow-up showed more residual cerebral lesions in the German cohort. This discrepancy is probably due to different MRI tools (1.5-Tesla in Chiba vs. 3-Tesla in Berlin) with higher resolution MRI showing higher numbers of lesions. Of note, most cerebral lesions were asymptomatic, unspecific white matter lesions, which occur frequently in NMOSD.25
Differences between both cohorts mainly concerned the degree of disability and long-term treatment strategies, suggesting a lower disease activity in Japanese than in German patients. Though the total EDSS was similar in both cohorts, Japanese patients had a longer disease duration at the time of assessment and brainstem, cerebral, pyramidal, and sensory FSS were lower than in German patients. Moreover, the EDSS increase per attack showed a tendency to higher values in the German cohort. Only bowel and bladder FSS was higher in the Japanese cohort. These findings confirm previous data showing that Japanese patients have more frequent brain manifestations than German patients but in general less severe disability scores.13,18,19,25,26
In both cohorts, acute attacks were mainly treated with intravenous high-dose corticosteroid therapy. Only a small subset of attacks was treated with immunoadsorption therapy – more frequently used in the Japanese cohort – and plasmapheresis – more frequently used in the German cohort. In German patients, escalation of therapy ensued in 23.8% of all attacks after a first treatment course, mainly with high-dose corticosteroid therapy or intravenous immunoglobulins. Only one attack in the Japanese cohort received escalation therapy. This indicates again a higher attack severity among German patients. The final degree of recovery from relapses was similar in both cohorts.
In the Japanese cohort, relapses were more frequent before the start of the first treatment. However, as previously described in Japanese NMOSD cohorts,13,18 the time between onset attack and first treatment was about two years in the Japanese cohort but significantly shorter – also compared with other Caucasian cohorts18 – in the German cohort. These findings support the hypothesis that Japanese patients presenting with a brain-dominant disease manifestation may have a better clinical prognosis than German patients.27
An overall lower attack severity in the Japanese cohort could explain the physicians’ choice for slower treatment induction and less aggressive medication. A more disabling attack severity in the German cohort however, would necessitate an early and more aggressive treatment induction, similarly to what has been previously described.13 First and current treatment differed considerably between the two cohorts. At the time of assessment, most Japanese patients were treated with oral prednisolone, often in high doses. In the German cohort, in contrast, 50% of patients were treated with rituximab and 97.4% had a history of current or previous immunosuppressive therapy. Rituximab is a monoclonal antibody against the CD20 molecule expressed on B-lymphocytes. It has a significant effect on disease activity and is one of the most frequently used attack-preventive immunotherapies for NMOSD in Europe.2 Currently there are no controlled trials comparing the effect of long-time therapy with oral corticosteroids and rituximab in AQP4-IgG-positive NMOSD. Moreover, at the time of assessment, no drug was approved for the treatment on NMOSD and treatment options have relied on off-label use and empiric drugs with immunosuppressive and B-cell-depleting effect (azathioprine, rituximab, tocilizumab etc.).28 Since then, the C5 complement inhibitor eculizumab, the CD19 inhibitor inebilizumab and the interleukin-6 receptor inhibitor satralizumab have been approved for the therapy of AQP4-IgG-positive NMOSD.29,30
A different usage of immunotherapies is very likely related to different reimbursement policies of Japanese and German healthcare systems and explains a higher bar in Japan than in Germany but also compared to Korea24,31 to use rituximab as off-label therapy. Recently, the efficacy of rituximab in AQP4-IgG-positive NMOSD was confirmed in a first randomized, double-blinded, placebo-controlled Japanese trial (RIN-1).32 These results may affect the practice of treating NMOSD in Japan. As there have been few clinical trials for NMOSD to give clear guidelines,2,33 there is an important need for future multicenter studies.
In contrast to previous studies,13 the ARR was similar in both cohorts. This is presumably the consequence of early and effective immunotherapy in the German cohort, reducing the relatively higher disease activity. These results might be biased as the German cohort is observed and treated in a tertiary referral center for NMOSD. Therefore, the treatment strategies in the German cohort might not be representative of the average German clinical practice. Still, the median EDSS in both cohorts was above three, confirming the rarity of benign NMOSD,34 despite early treatment and significant reduction of the ARR under immunotherapy. These findings correspond to previous data from a Korean cohort.24
NMOSD often coexists with other autoimmune diseases, including most frequently thyroiditis, myasthenia gravis, SLE and Sjogren’s syndrome.35,36 Interestingly, SLE was the most frequent autoimmune comorbidity in the German cohort, but was not reported in the Japanese cohort. This is in contrast to the general population, where SLE has a higher incidence in Asia than in Europe.37 This variation could reflect differences in the genetic background of affected patients with a higher susceptibility to concomitant autoimmune diseases in German NMOSD patients. In line with a previous study on NMOSD in Japanese patients,27 Sjogren's Syndrome was the most frequent autoimmune comorbidity in the Japanese cohort. In general, autoimmune comorbidities occurred considerably more often in the German cohort and the frequency in the Japanese cohort was similar than previously reported in Korean patients with AQP4-autoimmunity.24
The main limitations of our study are the retrospective design and, due to the rarity of AQP4-IgG-positive NMOSD, the relatively small number of patients. Unfortunately, our data do not allow any conclusion about the incidence of AQP4-IgG-positive NMOSD in Germany and Japan: First study participation was voluntary in both centers. Second, the Berlin cohort included patients from all over Germany, but the center is not the only referral center in the country. The Chiba cohort included mainly patients who lived in Chiba Prefecture. The assay method for anti-AQP4-IgG was different between both cohorts and a more consistent detection method may lead to more accurate diagnosis of antibody serostatus, although the high specificity and sensitivity of both had been previously well established.23 Further, MRI results are based on written reports, were not centrally reviewed and may include inter-rater discrepancies. Additionally, the interval between the onset of clinical symptoms and the MRI can influence the MRI-presentation of LETM lesions, which could not be controlled for in the retrospective study design. As all data were acquired outside of acute attacks, no Gadolinium-based contrast agent was administered. Therefore, we were unable to adjudicate whether clinical attacks were accompanied by Gadolinium-enhancing lesions on MRI.
The main strengths of our study are the well-defined patient samples of Caucasian and East-Asian ethnicity, enabling us to question whether clinical differences are due to treatment effects.
In conclusion, we show that patients from the Japanese cohort have more brain attacks, less autoimmune comorbidities, and lower brainstem, cerebral, pyramidal, and sensory FSS than German patients. Conversely, German patients received immunotherapies earlier and more frequently. This may result in a similar presentation of overall disease activity and relapse rate despite a presumably higher underlying disease activity in the German cohort. Further research is necessary to clarify why Asian patients have more frequent brain attacks. Moreover, our findings emphasize the necessity for prospective, international multicenter studies in order to evaluate the efficacy of the respective medication and to develop optimized treatment guidelines for AQP4-IgG-positive NMOSD with regard to the patients’ ethnicity. The data presented here might help design future interventional clinical trials and treatment guidelines that should take the ethnicity of patients with NMOSD into consideration.
Acknowledgement
We acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Fund of Charité – Universitätsmedizin Berlin.
Authors’ disclosures
SA received a travel grant from Celgene and speaker’s honorary from Alexion, Bayer and Roche.
JBS has received travel grants and speaking honoraria from Bayer Healthcare, Biogen Idec, Merck Serono, Sanofi Genzyme, Teva Pharmaceuticals, Roche, and Novartis, none of them related to preparing this manuscript.
KR received research support from Novartis, Merck Serono, German Ministry of Education and Research, European Union (821283-2), Stiftung Charité (BIH Clinical Fellow Program) and Arthur Arnstein Foundation; received speaker honoraria and travel grants from Bayer, Biogen Idec, Merck Serono, sanofi-aventis/Genzyme, Teva, Roche, Novartis, and Guthy Jackson Charitable Foundation.
NS received travel funding from Sanofi-Aventis/Genzyme and speaker honoraria from Bayer AG.
GC received speaker honoraria from Merck and Bayer unrealted to this project.
AD has received a speaker honorarium from Roche.
CC received honoraria for lecturing from Bayer, and research funding from Novartis.
FP serves as an Associate Editor for Neurology: Neuroimmunology & Neuroinflammation, reports research grants and speaker honoraria from Bayer, Teva, Genzyme, Merck, Novartis, MedImmune and is member of the steering committee of the OCTIMS study (Novartis).
AUB is cofounder and sharehoulder of motognosis and nocturne. He is named as inventor on several patent applications regarding MS serum biomarkers, OCT image analysis and perceprive visual computing.
SK serves as the Deputy Editor of the Journal of Neurology, Neurosurgery, and Psychiatry and is an Editorial Board member of the Journal of the Neurological Sciences.
HZ received research grants from Novartis and speaking honoraria from Bayer Healthcare.
Funding: This work was supported in part by the Health and Labour Sciences Research Grant on Intractable Diseases (Neuroimmunological Diseases) and the Research Grant 16B-1 for Nervous and Mental Disorders from the Ministry of Health, Labour and Welfare of Japan; and by NeuroCure Clinical Research Center (NCRC), funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germanýs Excellence Strategy – EXC-2049 – 390688087 and by the SFB-TR128 and by the Research Grant “CC-Neuro” from the German Federal Ministry of Education and Research.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ORCID iDs: Susanna Asseyer https://orcid.org/0000-0001-6289-1791
Masahiro Mori https://orcid.org/0000-0002-8767-255X
Graham Cooper https://orcid.org/0000-0001-8383-6476
Claudia Chien https://orcid.org/0000-0001-8280-9513
Kazuo Sugimoto https://orcid.org/0000-0002-7585-1278
Ryohei Ohtani https://orcid.org/0000-0001-7075-341X
Alexander U Brandt https://orcid.org/0000-0002-9768-014X
Contributor Information
Susanna Asseyer, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine and Charite-Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany NeuroCure Clinical Research Center, Charite- Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
Hiroki Masuda, Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan.
Masahiro Mori, Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan.
Judith Bellmann-Strobl, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
Klemens Ruprecht, Department of Neurology, Charité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
Nadja Siebert, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
Graham Cooper, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charité – UUniversitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Einstein Center for Neurosciences, Berlin, Germany; Department of Experimental Neurology and Center for Stroke Research, Charité – Universitätsmedizin, Berlin, Germany.
Claudia Chien, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
Ankelien Duchow, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
Jana Schließeit, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
Jia Liu, Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan; Department of Neurology, The Second Hospital of Hebei Medical University, Shijiazhuang, Hebei, China.
Kazuo Sugimoto, Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan; Department of Neurology, Dongzhimen Affiliated Hospital, Beijing University of Chinese Medicine, Beijing, China.
Akiyuki Uzawa, Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan.
Ryohei Ohtani, Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan.
Friedemann Paul, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Einstein Center for Neurosciences, Berlin, Germany.
Alexander U Brandt, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; Department of Neurology, University of California Irvine, CA, USA.
*Satoshi Kuwabara, Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan.
*Hanna G Zimmermann, Experimental and Clinical Research Center, Max Delbrück Center for Molecular Medicine, and Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; NeuroCure Clinical Research Center, Charite – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany.
References
- 1.Wingerchuk DM, Banwell B, Bennett JL, International Panel for NMO Diagnosis et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borisow N, Mori M, Kuwabara S, et al. Diagnosis and treatment of NMO spectrum disorder and MOG-encephalomyelitis. Front Neurol 2018; 9:888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarius S, Wildemann B, Paul F. Neuromyelitis optica: clinical features, immunopathogenesis and treatment. Clin Exp Immunol 2014; 176: 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asseyer S, Schmidt F, Chien C, et al. Pain in AQP4-IgG-positive and MOG-IgG-positive neuromyelitis optica spectrum disorders. Mult Scler J - Exp Transl Clin 2018; 4(3): 2055217318796684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chavarro VS, Mealy MA, Simpson A, et al. Insufficient treatment of severe depression in neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 2016; 3: e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oertel FC, Kuchling J, Zimmermann H, et al. Microstructural visual system changes in AQP4-antibody-seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm 2017; 4: e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papadopoulou A, Oertel FC, Gaetano L, et al. Attack-related damage of thalamic nuclei in neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry 2019; 90: 1156–1159. [DOI] [PubMed] [Google Scholar]
- 8.Oertel FC, Schließeit J, Brandt AU, et al. Impairment in neuromyelitis optica spectrum disorders: a review of clinical and neuroradiological features. Front Neurol 2019; 10: 34–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleiter I, Gahlen A, Borisow N, on behalf of NEMOS (Neuromyelitis Optica Study Group), et al. Apheresis therapies for NMOSD attacks a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm 2018; 5: e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jarius S, Paul F, Weinshenker BG, et al. Neuromyelitis optica. Nat Rev Dis Primers 2020; 6: 1–32. [DOI] [PubMed] [Google Scholar]
- 11.Reindl M, Schanda K, Woodhall M, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm 2020; 7: e674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Mori M, Zimmermann H, et al. Anti-MOG antibody–associated disorders: differences in clinical profiles and prognosis in Japan and Germany. J Neurol Neurosurg Psychiatry 2020; jnnp-2020-324422 [DOI] [PubMed] [Google Scholar]
- 13.Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012; 135: 1834–1849. [DOI] [PubMed] [Google Scholar]
- 14.Mori M, Kuwabara S, Paul F. Worldwide prevalence of neuromyelitis optica spectrum disorders. J Neurol Neurosurg Psychiatry 2018; 89: 555–556. [DOI] [PubMed] [Google Scholar]
- 15.Mealy MA, Wingerchuk DM, Greenberg BM, et al. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol 2012; 69: 1176–1180. [DOI] [PubMed] [Google Scholar]
- 16.Flanagan E, Cabre P, Weinshenker B, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Physiol Behav 2017; 176: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hor JY, Asgari N, Nakashima I, et al. Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front Neurol 2020; 11:501 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SH, Mealy MA, Levy M, et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology 2018; 91: e2089–e2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palace J, Lin DY, Zeng D, et al. Outcome prediction models in AQP4-IgG positive neuromyelitis optica spectrum disorders. Brain 2019; 142: 1310–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mealy MA, Kessler RA, Rimler Z, et al. Mortality in neuromyelitis optica is strongly associated with African ancestry. Neurol Neuroimmunol Neuroinflamm 2018; 5: e468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sepúlveda M, Armangué T, Sola-Valls N, et al. Neuromyelitis optica spectrum disorders: comparison according to the phenotype and serostatus. Neurol Neuroimmunol Neuroinflamm 2016; 3: e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalluri SR, Illes Z, Srivastava R, et al. Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Arch Neurol 2010; 67: 1201–1208. [DOI] [PubMed] [Google Scholar]
- 23.Waters P, Reindl M, Saiz A, et al. Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica. J Neurol Neurosurg Psychiatry 2016; 87: 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim SH, Kim W, Li XF, et al. Clinical spectrum of CNS aquaporin-4 autoimmunity. Neurology 2012; 78: 1179–1185. [DOI] [PubMed] [Google Scholar]
- 25.Kim HJ, Paul F, Lana-Peixoto MA, Guthy-Jackson Charitable Foundation NMO International Clinical Consortium & Biorepository et al. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology 2015; 84: 1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kremer L, Mealy M, Jacob A, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler J 2014; 20: 843–847. [DOI] [PubMed] [Google Scholar]
- 27.Nagaishi A, Takagi M, Umemura A, et al. Clinical features of neuromyelitis optica in a large Japanese cohort: comparison between phenotypes. J Neurol Neurosurg Psychiatry 2011; 82: 1360–1364. [DOI] [PubMed] [Google Scholar]
- 28.Duchow A, Chien C, Paul F, et al. Emerging drugs for the treatment of neuromyelitis optica. Expert Opin Emerg Drugs 2020; 25: 285–297. [DOI] [PubMed] [Google Scholar]
- 29.Traboulsee A, Greenberg BM, Bennett JL, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol 2020; 19: 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019; 394: 1352–1363. [DOI] [PubMed] [Google Scholar]
- 31.Kim SH, Jeong IH, Hyun JW, Joung AR, et al. Treatment outcomes with rituximab in 100 patients with neuromyelitis optica: influence of FCGR3A polymorphisms on the therapeutic response to rituximab. JAMA Neurol 2015; 72: 989–995. [DOI] [PubMed] [Google Scholar]
- 32.Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2020; 19: 298–306. [DOI] [PubMed] [Google Scholar]
- 33.Trebst C, Jarius S, Berthele A, Neuromyelitis Optica Study Group (NEMOS) et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the neuromyelitis optica study group (NEMOS). J Neurol 2014; 261: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uzawa A, Mori M, Muto M, et al. Benign neuromyelitis optica is rare in Japanese patients. Mult Scler 2015; 21: 1204–1208. [DOI] [PubMed] [Google Scholar]
- 35.Shahmohammadi S, Doosti R, Shahmohammadi A, et al. Autoimmune diseases associated with neuromyelitis optica spectrum disorders: a literature review. Mult Scler Relat Disord 2019; 27: 350–363. [DOI] [PubMed] [Google Scholar]
- 36.Beekman J, Keisler A, Pedraza O, et al. Neuromyelitis optica spectrum disorder: Patient experience and quality of life. Neurol Neuroimmunol Neuroinflamm. 2019; 6:e580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rees F, Doherty M, Grainge MJ, et al. The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatology 2017; 56: 1945–1961. [DOI] [PubMed] [Google Scholar]