

Abstract
Short circulation time and off-target toxicity are the main challenges faced by small-molecule chemotherapeutics. To overcome these shortcomings, an albumin-binding peptide conjugate of chemotherapeutics is developed that binds specifically to endogenous albumin and harnesses its favorable pharmacokinetics and pharmacodynamics for drug delivery to tumors. A protein-G-derived albumin-binding domain (ABD) is conjugated with doxorubicin (Dox) via a pH-sensitive linker. One to two Dox molecules are conjugated to ABD without loss of aqueous solubility. The albumin-binding ABD–Dox conjugate exhibits nanomolar affinity for human and mouse albumin, and upon administration in mice, shows a plasma half-life of 29.4 h, which is close to that of mouse albumin. Additionally, 2 h after administration, ABD–Dox exhibits an approximately 4-fold higher concentration in the tumor than free Dox. Free Dox clears quickly from the tumor, while ABD–Dox maintains a steady concentration in the tumor for at least 72 h, so that its relative accumulation at 72 h is ≈120-fold greater than that of free Dox. The improved pharmacokinetics and biodistribution of ABD–Dox result in enhanced therapeutic efficacy in syngeneic C26 colon carcinoma and MIA PaCa-2 pancreatic tumor xenografts, compared with free Dox and aldoxorubicin, an albumin-reactive Dox prodrug currently in clinical development.
Keywords: albumin binding, cancer, doxorubicin, drug delivery, peptides
1. Introduction
Small-molecule chemotherapeutics, although in routine use for cancer treatment, suffer from a short circulation half-life and indiscriminate accumulation in healthy tissues that result in systemic toxicities and hence limit their maximum dose. These limitations inhibit accumulation of chemotherapeutics in tumors at therapeutic levels and limit their clinical application. Efforts in past decades have been focused on developing macromolecular[1] and nanoparticulate[2] drug formulations that prevent first-pass elimination in kidneys and allow for selective accumulation in tumors via the enhanced permeation and retention effect.[3] However, the interaction of these macromolecule and nanoparticle carriers with serum proteins and immune system is not well understood and is affected by several factors such as interfacial chemistry, size, shape, and stability,[4,5] which makes optimization challenging.
There has been interest in recent years in harnessing endogenous proteins that circulate for extended periods of time in blood, as carriers of drugs.[6] Albumin—the most abundant endogenous protein in blood—has been the focus of these efforts,[7] with several albumin-based nanoparticles and drug conjugates currently in clinic or clinical trials.[8] This is because albumin has the longest half-life of all serum proteins, as its molecular weight of ≈ 66 kDa, which is above the glomerular filtration cutoff, and its high negative charge (−19) prevent its renal excretion,[9] and its trafficking via the FcRn recycling pathway protects it from lysosomal degradation within endothelial cells that take up albumin and other serum proteins.[10] As albumin is an endogenous protein, it is also well tolerated, and has no toxicity or immunogenicity.[11] In addition, the leaky vasculature and the impaired lymphatic system of tumors lead to albumin extravasation and accumulation. Tumor cells them-selves also ingest albumin by pinocytosis as a source of amino acids to fuel their rapid growth.[12,13] Albumin is hence a promising carrier to extend the biological half-life of drugs and to selectively deliver them to tumor cells.
Albumin can be exploited ex vivo or in vivo for drug delivery. The ex vivo approach requires extraction of albumin by fractionation of blood plasma and involves extra processing steps for drug conjugation or encapsulation that can alter the structure and conformation of albumin, leading to potential immunogenicity.[14] In addition, the emergence of new infectious diseases continues to create concern regarding the safety of blood products such as serum-derived albumin, necessitating strict screening of blood donors and the development of new techniques to inactivate emerging blood-borne pathogens.[15–17] High-yield recombinant synthesis of albumin and albumin fusions requires a specialized yeast expression system that is not readily available to most researchers.[18–21] To address these limitations of the ex vivo albumin approach, drugs can, alternatively, be conjugated to albumin-binding moieties that bind albumin in vivo.[22–26] The small size and lower viscosity of these albumin-binding drug conjugates compared to albumin conjugates and fusions can also provide for easier formulation and administration.
Aldoxorubicin (AlDox) is a promising product in the in vivo albumin approach category.[27,28] Formerly known as INNO-206, it is an albumin-binding prodrug of the anticancer drug doxorubicin (Dox), in which Dox is derivatized at its C-13 keto position with the thiol-binding 3,3′-N-(ε-maleimidocaproic acid) hydrazide (EMCH) linker. Upon intravenous administration, AlDox primarily—but not exclusively—binds to the free thiol on cysteine-34 of endogenous albumin via maleimide chemistry and piggybacks on it to tumor sites.[27] Its chemical selectivity for albumin arises from the high, approximately millimolar, concentration of albumin in blood,[29] and the hyper-reactivity of the exposed thiol group on the solvent-accessible Cys34 residue in albumin.[30] Upon cellular uptake into tumor cells, the acid-sensitive hydrazone bond between Dox and EMCH is cleaved in the tumor microenvironment that has a weakly acidic pH of 6.5–6.9[31]—if trapped there for long enough—or in the acidic environment of endosomes (pH 5.5–6.5)[32] upon intracellular uptake, releasing free Dox within cells.[33]
In a phase III clinical trial of advanced soft tissue sarcomas (STS) in a relapsed or refractory setting (2014, NCT02049905), AlDox showed superior progression-free survival (PFS) compared to the standard of care regimen and showed minimal cardiotoxicity[28]—the major dose-limiting safety concern associated with free Dox.[34,35] However, in a phase II clinical trial in patients with previously untreated STS (2012, NCT01514188), despite superior PFS and no evidence of cardiotoxicity in the AlDox arm, the overall survival was not considerably different between AlDox and the free Dox arms. Therefore, the clinical efficacy of endogenous albumin delivery systems, such as AlDox, though promising, has considerable room for improvement.
Here, we report on an albumin-binding conjugate of doxorubicin that consists of Dox molecules that are site-specifically conjugated to an albumin-binding protein domain (ABD) via an acid-sensitive linker. We hypothesized that ABD–Dox may have several potential advantages over AlDox. First, besides the cysteine-34 on albumin, the maleimide group on AlDox can also react with cysteines and lysines on other blood proteins and cells[36–38] such as the lysine-430 on erythrocyte band 3 protein that is known as the binding site in the eosin-5′-maleimide-binding test for detection of erythrocyte membrane disorder.[39] Therefore, interaction of AlDox with plasma proteins is not specific and is the probable cause of off-site toxicities, such as anemia and neutropenia.[39] ABD, in contrast, binds specifically with albumin, which should limit the off-site toxicity of ABD–Dox. Second, AlDox binds albumin via a stable thioether bond. ABD, in contrast, binds albumin via noncovalent interactions. Therefore, the interaction of ABD with albumin, though very strong, is still weaker than the covalent thioether bond of AlDox, so that the ABD can dissociate from albumin, and hence show better penetration into the tumor than AlDox.[40,41]
2. Results and Discussion
2.1. Design and Synthesis of ABD–Dox
The ABD used here is a 46–amino acid, three-helix protein domain from streptococcal protein G (Figure 1A). It shows a high stability with respect to both temperature (Tm ≈ 72 °C)[42] and pH,[43] has high aqueous solubility, and can be easily produced recombinantly at a high level by overexpression in Escherichia coli.[44,45] In addition, it binds to albumin from different species allowing for a plethora of animal models to be used for preclinical studies. All of these properties make the ABD desirable for pharmaceutical formulations. In addition, ABD does not contain any cysteine residues and therefore does not interfere with the C-terminal cysteine-containing drug conjugation site used in our synthesis scheme.
Figure 1.
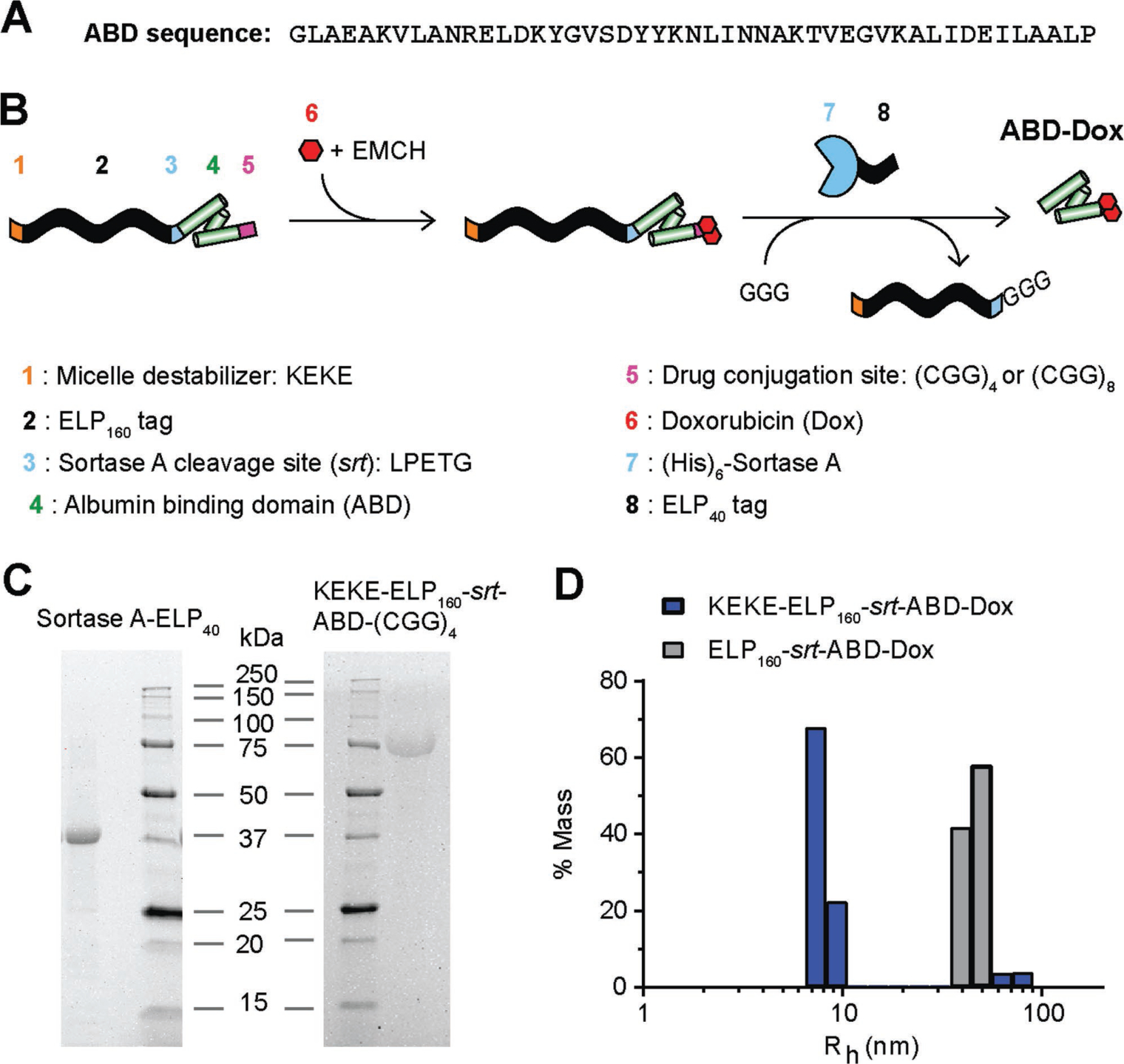
ABD–Dox synthesis. A) Amino acid sequence of ABD. B) Schematic illustration of the design and synthesis steps. ELP was used as a purification tag for ABD purification from bacteria and was removed following drug conjugation using sortase A. The KEKE peptide was included at the N-terminus to disrupt micelle self-assembly upon Dox conjugation, and to enable the subsequent sortase A cleavage of the ELP from the ABD–Dox conjugate. C) SDS-PAGE analysis confirmed successful purification of KEKE–ELP160–srt–ABD–(GGC)4 and (His)6–sortase A–ELP40 with theoretical molecular weights of 69.4 and 34.4 kDa, respectively. D) Hydrodynamic radius of ELP160–srt–ABD–Dox conjugates was measured with DLS. ELP160–srt–ABD–Dox conjugates without N-terminal KEKE segment self-assembled into micelles with Rh of 40–50 nm, whereas conjugates containing the N-terminal KEKE segment were mostly (≈90%) unimers with Rh < 10 nm.
The design of our construct from the N- to C-terminus is as follows: 1) a peptide with the sequence KEKE; 2) an elastin-like polypeptide (ELP); 3) a sortase A cleavage site with the amino acid sequence—LPETG—that is named srt; 4) the ABD; and 5) a (GGC)4 or (GGC)8 peptide for site-specific conjugation of Dox (Figure 1B). ELPs are recombinant polypeptides composed of repeats of the pentapeptide (Val–Pro–Gly–Xaa–Gly)n, where Xaa is the guest residue and can be any amino acid except proline, and n is the number of pentapeptide repeats. ELPs are thermally responsive polypeptides and undergo a lower critical solution temperature phase transition. ELPs retain this behavior when fused to other peptides and proteins. We chose an ELP because it can promote high levels of soluble protein expression,[46] and importantly an ELP can be used as a purification tag to isolate recombinant ELP-fused peptides and proteins from cell contaminants by cycling the soluble fraction of the cell lysate through the insoluble and soluble phases by a chromatography-free method known as inverse transition cycling.[47] The ELP used here consists of 160 repeats of the VPGAG pentapeptide (denoted here as ELP160).
Because the final product that we sought is an ABD–Dox conjugate, the srt sequence was added between ELP160 and ABD to remove the ELP160 tag by cleavage with sortase A. We chose two drug conjugation segments downstream of the ABD—(CGG)4 and (CGG)8—to provide four or eight cysteine residues as Dox attachment sites. In early studies, we found that attachment of Dox to the ELP–ABD fusions led to the formation of micelles (Figure 1D), which is undesirable because micelle formation buries the sortase cleavage site within the micelle, making cleavage with sortase A inefficient. The alternative strategy of first cleaving the ELP from the fusion, prior to Dox conjugation, was problematic as the insufficient size and physicochemical differences between ABD–Dox and Dox make their separation by centrifugal ultrafiltration or chromatography difficult.
To prevent the self-assembly of ELP–ABD–Dox conjugates into micelles, a zwitterionic KEKE peptide was incorporated at the N-terminus of the construct, as we had previously—seren-dipitously—discovered it is a potent destabilizer of ELP micelles (unpublished data). Following gene synthesis, the plasmid encoding KEKE–ELP160–srt–ABD–(CGG)4/8 was transformed into E. coli BL21(DE3) cells, The cells were allowed to grow for 6–8 h at 37 °C, following which the fusion was expressed by addition of 1 × 10−3 M isopropyl-β-d-thiogalactopyranoside (IPTG). The fusion was purified from the soluble fraction of the E. coli lysate by two rounds of inverse transition cycling[48] (Figure 1C) at a yield of 75–100 mg protein per liter of culture.
To remove the ELP tag from ELP160–ABD, sortase A was fused to an N-terminal tag comprised of six histidine residues (His tag) and a C-terminal ELP tag comprised of 40 repeats of a VPGAG pentapeptide (ELP40) (Figure 1B). The ELP was appended at the C-terminus of sortase A to increase its molecular weight from 18 to 34 kDa to facilitate its separation from the ABD–Dox product that has a molecular weight of 8 kDa, following the sortase cleavage reaction by size exclusion chromatography (SEC). The (His)6–sortase A–ELP40 fusion was transformed into and expressed in E. coli BL21(DE3) cells and was successfully isolated from cell contaminants by immobilized metal affinity chromatography with a yield of ≈100 mg per liter of culture.
For drug conjugation, Dox was conjugated with EMCH and the resulting Dox–EMCH conjugate, which is chemically identical to AlDox, was reacted with the free thiols of the Cys residues of the (CGG)4/8 segment at the C-terminus of KEKE–ELP160–srt–ABD–(CGG)4/8. Free Dox was removed by ultracentrifugation (Figure 2A). Dynamic light scattering (DLS) confirmed that the KEKE–ELP160–srt–ABD–Dox conjugates were mostly unimers (Rh < 10 nm) so that the sortase cleavage site should be accessible to sortase A, whereas ELP160–srt–ABD–Dox without the N-terminal KEKE segment self-assembled into micelles with an Rh of 40–50 nm (Figure 1D) with the ABD–Dox and the srt sortase cleavage site presumably buried in the core of the micelle.
Figure 2.
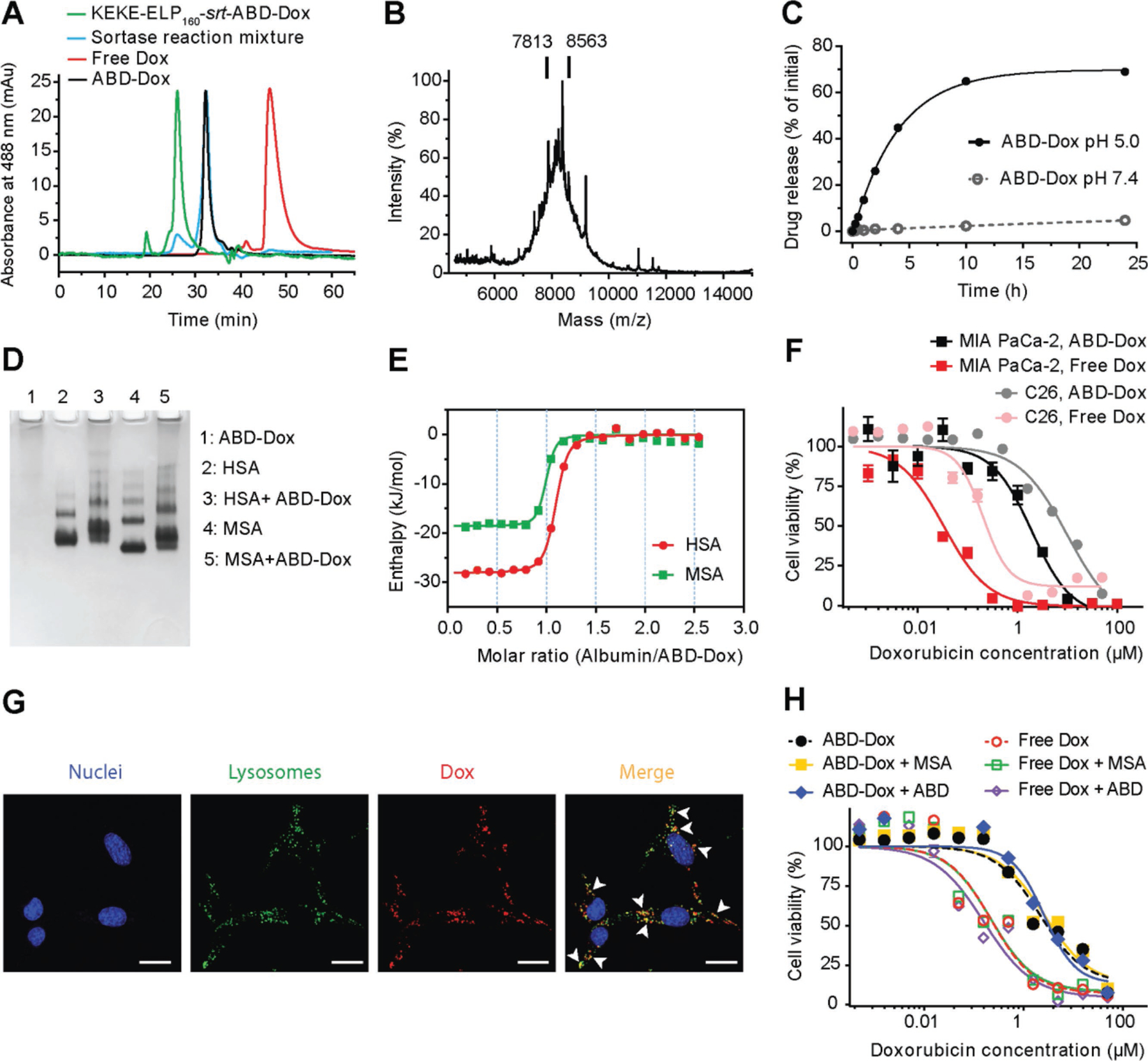
In vitro characterization of ABD–Dox. A) HPLC chromatograms of the KEKE–ELP160–srt–ABD–Dox, sortase reaction solution after 24 h incubation, and SEC-purified ABD–Dox. B) MALDI-MS spectra of ABD–Dox and vertical bars showing the mass of conjugates with one and two drug molecules. C) In vitro drug release from ABD–Dox conjugate as a function of time at pH 5.0 and pH 7.4. Dox was released at pH 5.0 corresponding to the pH in late endosomes, and showed a low, basal level of release from the conjugate at pH 7.4, corresponding to the pH in vascular and extracellular space of normal tissues. Data are presented as mean ± SEM, n = 3. D) Native PAGE of the interaction of ABD–Dox with HSA and MSA. For lanes 3 and 5, ABD–Dox was mixed at a 1:1 molar ratio with HSA and MSA, respectively. E) Isothermal titration calorimetry thermograms of the interaction of ABD–Dox with HSA and MSA. The enthalpy data were fit to an independent binding site model, and the thermodynamic parameters (n, KD, ΔH, and ΔS) were calculated as shown in Table 1. F) In vitro cytotoxicity of ABD–Dox and free Dox against C26 and MIA PaCa-2 cancer cells after 72 h incubation. The IC50 of ABD–Dox and free Dox were computed as 6.43 × 10−6 and 0.51 × 10−6 M, respectively, for C26 and as 1.41 × 10−6 and 0.04 × 10−6 M, respectively, for MIA PaCa-2 cells. Data are presented as mean ± SEM, n = 3. G) Cellular uptake of ABD–Dox. C26 cells were incubated for 4 h with ABD–Dox (red) at 37 °C, followed by incubation with CytoPainter LysoDeep (green) to stain lysosomes and Hoechst 33342 (blue) to stain nuclei. Arrowheads indicate regions of colocalized ABD–Dox and CytoPainter LysoDeep (yellow). Scale bar is 20 μm. H) In vitro cytotoxicity of ABD–Dox and free Dox against C26 cancer cells in the presence of MSA or ABD. The IC50 values of ABD–Dox and free Dox were 2.17 × 10−6 and 0.23 × 10−6 M, respectively, in the absence of MSA and ABD, 2.69 × 10−6 M (ABD–Dox) and 0.22 × 10−6 M (Dox) in the presence of MSA, and 2.62 × 10−6 M (ABD–Dox) and 0.16 × 10−6 M (Dox) in the presence of ABD. Data are presented as mean ± SEM, n = 3.
To remove the ELP160 tag, the KEKE–ELP160–srt–ABD–Dox conjugate was buffer exchanged into sortase reaction buffer and was reacted with (His)6–sortase A–ELP40 in the presence of a 50-fold molar excess of triglycine (GGG) (Figure 1B). The efficiency of the sortase reaction, defined as the percent conversion of ELP–ABD–Dox substrate to ABD–Dox, was determined by size exclusion high-performance liquid chromatography (HPLC) and was calculated to be ≥85% (Figure 2A). The ABD–Dox product was then separated from (His)6–sortase A–ELP40, the cleaved KEKE–ELP160 tag, and unreacted KEKE–ELP160–srt–ABD–Dox in the sortase reaction solution by preparative SEC. The number of Dox molecules conjugated per ABD (Dox/ABD molar ratio) was measured before the sortase reaction by dis-solving the lyophilized KEKE–ELP160–srt–ABD–Dox conjugate in phosphate-buffered saline (PBS) and calculating the moles of KEKE–ELP160–srt–ABD–Dox and Dox gravimetrically and spectrophotometrically at 488 nm, respectively. Three to four Dox molecules were attached to each KEKE–ELP160–srt–ABD–(CGG)8. However, following the removal of the ELP160 tag, the resulting ABD–Dox conjugate was too hydrophobic and insoluble in pH 7.4 PBS because of the high number of conjugated Dox molecules. In contrast, only one to two Dox molecules were attached to KEKE–ELP160–srt–ABD–(CGG)4, and this ABD–Dox conjugate showed aqueous solubility up to 4 mg Dox equivalent (equiv.) mL−1 in pH 7.4 PBS and was hence used for further studies. We also note that our site-specific ABD–Dox conjugate has a better solubility profile compared with AlDox that shows poor solubility at physiological pH and requires an acidic pH for solubility in aqueous solutions[27] at concentrations as low as 0.5 mg Dox equiv. mL−1. Formulation of AlDox in acidic pH is undesirable as it can trigger the premature—in vitro—degradation of the hydrazone bond between Dox and EMCH, converting AlDox back to free Dox.
2.2. In Vitro Characterization of ABD–Dox
The attachment of one to two Dox molecules to ABD–(CGG)4 was confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOFMS) that gave a broad peak centered around 7813 and 8563 m/z that corresponds to the theoretical molecular weight of ABD–(CGG)4–Dox with one and two conjugated Dox molecules, respectively. We refer to this conjugate simply as ABD–Dox in the remainder of the paper.
The interaction of ABD–Dox with albumin was confirmed qualitatively by native polyacrylamide gel electrophoresis (PAGE) and quantitatively by isothermal titration calorimetry. Human serum albumin (HSA) and mouse serum albumin (MSA) are negatively charged[49] and migrate toward the cathode in native PAGE. ABD–Dox, however, does not migrate on the native PAGE gel, and when mixed with HSA and MSA, hindered their mobility resulting in higher and smeared bands compared with free HSA and MSA bands (Figure 2D). This mobility shift confirms binding of ABD–Dox to HSA and MSA. The thermodynamics of the interaction was studied by isothermal titration calorimetry, and the binding isotherms were fit using an independent binding site model and yielded an equilibrium dissociation constant (KD) of 125 × 10−9 and 73.8 × 10−9 M, and a stoichiometry (n) of 1.03 and 0.94 for HSA and MSA, respectively (Figure 2E, Table 1). These nanomolar dissociation constants indicate that ABD–Dox has a high affinity for both HSA and MSA. Given the high concentration of albumin in plasma (≈0.6 × 10−3 M),[50] it suggests that the ABD–Dox conjugates will largely exist as an albumin-bound complex in murine or human circulation.
Table 1.
Thermodynamic parameters of the binding of ABD–Dox to HSA and MSA.
| n | KD [× 10−9 M] | ΔH [kJ mol−1] | ΔS [J mol−1 K−1] | |
|---|---|---|---|---|
| ABD–Dox:HSA | 1.03 | 125 | −28.1 | 41.5 |
| ABD–Dox:MSA | 0.94 | 73.8 | −18.6 | 76.6 |
The release of Dox from the ABD–Dox conjugate was studied at the physiological pH of 7.4 and the late endosomal pH of 5.0.[51] Dox release was negligible at pH 7.4, whereas at pH 5.0, about 70% of the drug was released over 24 h (Figure 2C), which can be attributed to the pH-sensitive hydrolysis of the hydrazone bond between Dox and EMCH linker.[52] The release data at pH 5.0 were fit to a first-order release model, from which 50% of the total release (t1/2) was calculated to occur within 2.8 h at pH 5.0. The pH-dependent release of Dox from ABD–Dox prevents systemic toxicity of ABD–Dox as it prevents premature release of Dox in the blood and the extracellular space of healthy tissues where the pH is 7.4.
To explore the mechanism of ABD–Dox internalization, cells were visualized by confocal fluorescence microscopy after incubation with ABD–Dox for 4 h. As expected, ABD–Dox colocalized with the lysosomal marker (Figure 2G) suggesting that ABD–Dox conjugates enter the cells via the endocytic pathway that exposes them to the acidic environments of late endosomes/lysosomes resulting in the release and activation of the drug.
We also studied the stability of ABD–Dox in serum and in the presence of serum proteases. ABD–Dox was diluted 1:1 into mouse serum, incubated at 37 °C for different time periods, and analyzed by SEC at 488 nm. In the absence of mouse serum, ABD–Dox elutes at around 30 min (Figure S1A, Supporting Information). At 280 nm, mouse serum showed one prominent peak eluting at around 25 min (Figure S1I, Supporting Information) that is consistent with the elution time of MSA (Figure S1J, Supporting Information) and several small peaks related to other serum proteins. ABD–Dox mixed with mouse serum before incubation at 37 °C showed one prominent peak at 30 min, as expected, but also a smaller peak at 25 min (Figure S1B, Supporting Information) demonstrating that even in the presence of the organic solvent used in the HPLC mobile phase, a small fraction of ABD–Dox is still bound to albumin and elutes with it. The intensity of the albumin-bound ABD–Dox increased with incubation time (Figure S1B–G, Supporting Information). This is speculated to be due to disulfide bond formation between the remaining free cysteines on ABD–Dox and the free cysteine on albumin over time, which is a covalent bond and does not break in the organic mobile phase of HPLC. This speculation was confirmed by treating the 96 h time point sample with the reducing agent tris(2-carboxyethyl)phosphine (TCEP) before HPLC analysis, which led to a decreased intensity of the albumin-bound peak at 25 min (Figure S1H, Supporting Information). Furthermore, other than the ABD–Dox and albumin-bound ABD–Dox peaks, no major peaks were observed in the SEC chromatograms up to 144 h after incubation confirming the stability of ABD–Dox in the presence of serum proteases.
2.3. In Vivo Performance of ABD–Dox
Next, the intravenous (i.v.) pharmacokinetics of ABD–Dox was compared with that of AlDox and free drug. ABD–Dox, AlDox, and Dox were injected i.v. at a dose of 5 mg Dox equiv. per kg of body weight (BW) into healthy BALB/c mice and the Dox fluorescence was measured from blood samples collected at different time points following i.v. injection to quantify the Dox equiv. concentration in plasma as a function of time. Free Dox has an in vivo elimination half-life on the order of minutes, with a systemic clearance that is so rapid that 15 min after i.v. injection, the concentration of Dox in blood was less than 2% of the initial concentration (Figure 3A). In contrast, ABD–Dox has a much longer elimination half-life of 29.4 ± 0.8 h (Table 2) that is close to the previously reported 35 h half-life of MSA,[53] and the drug was detectable in plasma even 96 h after injection (Figure 3B). The elimination half-life of AlDox is 21.3 ± 0.6 h that is shorter than the 29.4 ± 0.8 h half-life of ABD–Dox (P < 0.001, Student’s t-test). The impact of this difference in the half-life on pharmacokinetics is relevant, for as soon as 15 min after i.v. injection, the concentration of AlDox was approximately 2-fold lower than that of ABD–Dox (P < 0.05, one-way ANOVA and Tukey’s test) (Figure 3A), which could be the result of the reaction of AlDox with thiols and lysines on blood cells leading to some loss from the plasma compartment of blood. Finally, total plasma exposure—as defined by the area under the curve (AUC)—of ABD–Dox was (2263.6 ± 44.7) × 10−6 M h, which is 3-fold higher than that of AlDox ((801.2 ± 23.7) × 10−6 M h) (Table 2) and is statistically different (P < 0.001, Student’s t-test), demonstrating that the use of a peptide that binds albumin via molecular recognition is superior to a Dox derivative—aldoxorubicin—that piggybacks onto albumin through its preferential chemical reactivity with albumin.
Figure 3.
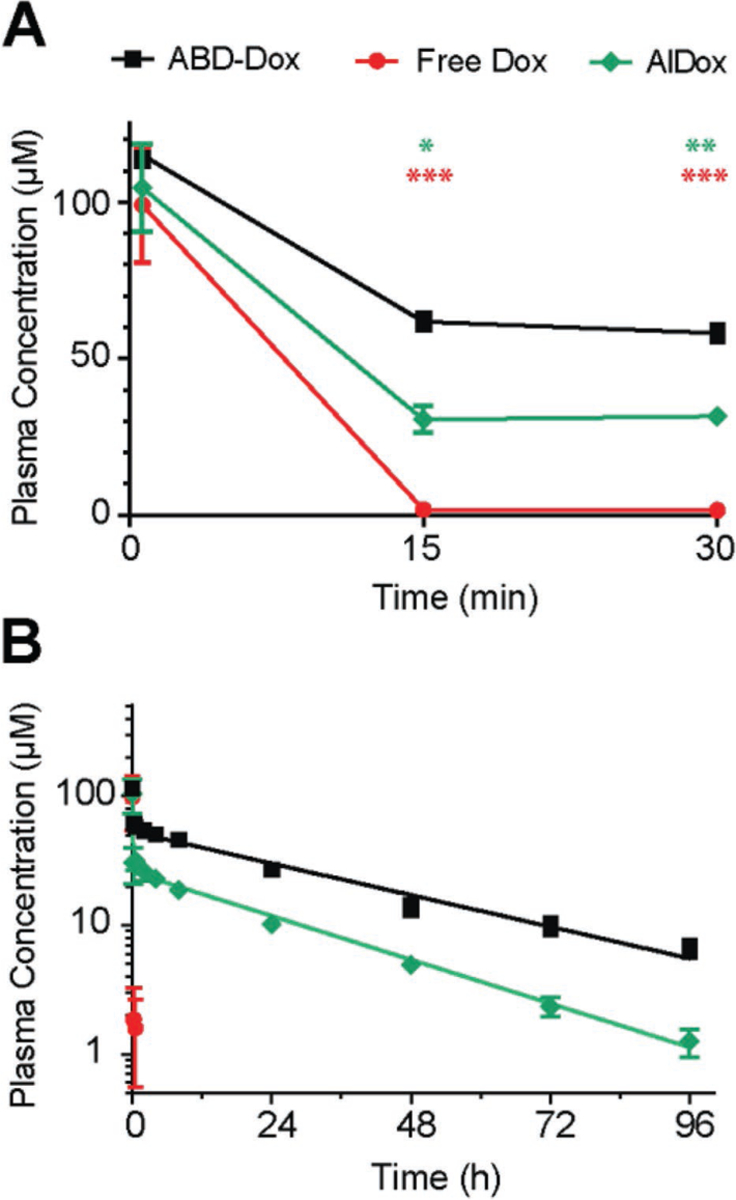
Pharmacokinetics of ABD–Dox. Plasma Dox concentration was measured over 96 h after administration of ABD–Dox via tail vein to BALB/c mice and fit to a two-compartment model from which the elimination half-life and the plasma drug exposure (area under the curve) were estimated, and are reported in Table 2: A) 0–30 min view; B) 0–96 h view. Data are presented as mean ± SEM, n = 5–6, one-way ANOVA and Tukey’s test, *P < 0.05, **P < 0.01, and ***P < 0.001.
Table 2.
Pharmacokinetic parameters of ABD–Dox. Values are shown as mean ± SD.
| Pharmacokinetic parameter | ABD–Dox | AIDox |
|---|---|---|
| Elimination half-life (h) | 29.4 ± 0.8 | 21.3 ± 0.6 |
| Area under curve (× 10−6 m h) | 2263.6 ± 44.7 | 801.2 ± 23.7 |
To study the biodistribution and tumor localization of ABD–Dox, tumor and major tissues were harvested at 2, 24, and 72 h post injection and the concentration of the Dox equiv. at each time point was measured and compared with animals injected with the same dose of free Dox. Within 2 h after administration, the concentration of ABD–Dox was 6.2% of injected dose (ID) g−1 of Dox equiv. in the tumor, which is approximately 4- and 2-fold greater than the accumulation of free Dox (P < 0.001, two-way ANOVA and Tukey’s test) and AlDox (P < 0.01, two-way ANOVA and Tukey’s test), respectively. In the longer term, at 24 and 72 h, the tumor accumulation of ABD–Dox reached ≈8% ID g−1. In contrast to ABD–Dox, which showed steady levels of Dox accumulation over 72 h, the accumulation of free Dox decreased over 72 h, with a maximum of 1.6% ID g−1 at 2 h and reaching a minimum of 0.07% ID g−1, indicating that much so the initial accumulation of free drug is transient. This level of accumulation of ABD–Dox, especially at 72 h, is notable, as it is ≈120- and ≈7-fold greater than that of the free drug and AlDox, respectively (Figure 4A).
Figure 4.
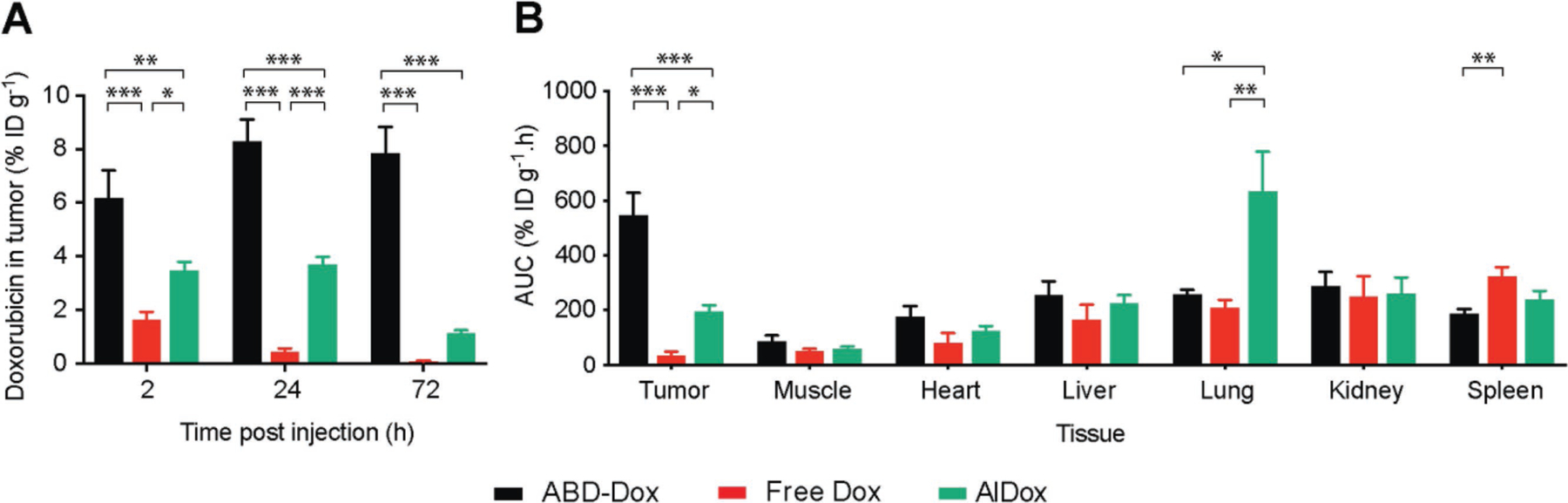
Tissue biodistribution of ABD–Dox. A) Concentration of ABD–Dox, free Dox, and AlDox in tumor at 2, 24, and 72 h post administration. ABD–Dox, free Dox, and AlDox were injected into C26 tumor-bearing mice, and Dox concentration was measured in tumor and normal tissues at 2, 24, and 72 h post administration. B) Total ABD–Dox exposure in different tissues. Total drug exposure was assessed by calculating the AUC of the drug concentration–time graph from 2 to 72 h post administration in different tissues. Data are presented as mean ± SEM, n = 5–7, two-way ANOVA and Tukey’s test, *P < 0.05, **P < 0.01, and ***P < 0.001.
At 2 h post administration, ABD–Dox accumulated at a lower concentration than free Dox in the liver (P < 0.01, two-way ANOVA and Tukey’s test), spleen (P < 0.001, two-way ANOVA and Tukey’s test), kidneys (P < 0.01, two-way ANOVA and Tukey’s test), and muscle (P < 0.01, two-way ANOVA and Tukey’s test), and at a lower concentration than AlDox in the lungs (P < 0.01, two-way ANOVA and Tukey’s test), liver (P < 0.05, two-way ANOVA and Tukey’s test), spleen (P < 0.01, two-way ANOVA and Tukey’s test), and kidneys (P < 0.001, two-way ANOVA and Tukey’s test) (Figure S2, Supporting Information). Furthermore, compared with tumor, where ABD–Dox continued to accumulate for at least 72 h (Figure 4A), the concentration of ABD–Dox in other tissues—with the exception of spleen and muscle—decreased over time. At 24 h after administration, compared to free Dox, ABD–Dox had a lower concentration than free Dox in the spleen (P < 0.001, two-way ANOVA and Tukey’s test), and a lower concentration than AlDox in the lungs and spleen (P < 0.05, two-way ANOVA and Tukey’s test), but a higher concentration than free Dox in the heart (P < 0.01, two-way ANOVA and Tukey’s test) and muscle (P < 0.05, two-way ANOVA and Tukey’s test) (Figure S2, Supporting Information). At 72 h, the accumulation of ABD–Dox was higher than free Dox and AlDox in the muscle (P < 0.001 for free Dox, P < 0.01 for AlDox, two-way ANOVA and Tukey’s test) (Figure S2, Supporting Information), but lower than AlDox in the lungs (P < 0.001, two-way ANOVA and Tukey’s test).
As the biodistribution data for ABD–Dox and free Dox (control) were collected at three time points spanning 2–72 h post administration, it allowed the total exposure of different tissues over time to be determined by calculating the AUC of the Dox equiv. concentration for each formulation. ABD–Dox showed significantly higher—16- and 2.8-fold—AUC in tumor compared to free Dox and AlDox, respectively, 0.5-fold lower AUC in spleen compared to free Dox, and 2.5-fold lower AUC in lungs compared to AlDox. No significant difference was observed between the AUC of ABD–Dox and free Dox in other tissues (Figure 4B). The lower AUC of ABD–Dox than free Dox in the spleen is consistent with the ability of albumin to reduce opsonization,[54,55] which reduces subsequent uptake by mac-rophages that are present at a high level in the spleen, while also directing cellular uptake of albumin and its conjugates via FcRn receptors in the liver and spleen[56–58] that then recycle albumin in these organs back into the systemic circulation. The markedly high accumulation of AlDox in lungs compared with ABD–Dox is likely, we speculate, due to the its nonspecific binding to the cysteines and lysines on blood cells such as red blood cells that have been previously reported to increase the accumulation of nanocarriers absorbed on their surface into the lungs.[59,60] Nonspecific binding to blood cells also decreases the plasma availability of AlDox, which is consistent with the pharmacokinetic data, and could explain its lower accumulation in tumors compared with ABD–Dox.
The significantly greater drug exposure solely in the tumor afforded by ABD–Dox compared with free Dox and AlDox is of clinical significance as it shows that that the long circulating ABD–Dox conjugate targets the tumor to a far higher level than the free drug, and suggested that this formulation may achieve therapeutic levels of the drug in the tumor. The biodistribution results also suggested that this formulation should not intensify cardiotoxicity, hepatotoxicity, and nephrotoxicity, which are the major side effects associated with free Dox.[61–63]
With these encouraging biodistribution results in hand, we next investigated tumor regression by ABD–Dox in vivo. Prior to launching a study with implanted tumors, the maximum tolerated dose (MTD) of ABD–Dox was established in healthy mice. To do so, a range of doses of ABD–Dox were injected into healthy BALB/c mice and body weight loss and mortality of the mice were monitored over a 2-week period to assess systemic toxicity. The MTD was defined as the highest dose that did not cause mortality or body weight loss greater than 15% in any of the mice. Using these criteria, the MTD was determined to be 20 mg Dox equiv. kg−1 for ABD–Dox (Figure S3, Supporting Information) and 10 mg kg−1 BW for free Dox (Figure S4, Supporting Information).
Next, tumor regression studies were performed in a syngeneic model of murine colon carcinoma (C26) and in a xenograft model of human pancreatic adenocarcinoma (MIA PaCa-2). We chose these tumor lines for several reasons. First, we wanted to include one human-derived cancer cell line as it is clinically more relevant, but this requires using immunocompromised mice for tumor implantation, and one mouse-derived cancer cell line that allows using immunocompetent mice as our previous studies showed that host immunity can contribute to the therapeutic efficacy of doxorubicin formulations.[64] Second, both C26 and MIA PaCa-2 are known to respond poorly to doxorubicin treatment in vivo, so that they are a stringent test of the utility of the ABD–Dox formulation in enhancing the efficacy of the drug.[65] Third, in an in vitro scouting study to identify the most promising pancreatic cancer for treatment by ABD–Dox, of the three human pancreatic cell lines—MIA PaCa-2, AsPC-1, and BxPC-3—treated with ABD–Dox, MIA PaCa-2 was the most sensitive to ABD–Dox (Figure 2F; Figure S5, Supporting Information) and was therefore selected as a candidate for in vivo studies.
Prior to in vivo regression studies, the efficacy of ABD–Dox against C26 and MIA PaCa-2 tumor cells was first investigated in vitro. The half-maximal inhibitory concentration (IC50) of ABD–Dox was 6.4 × 10−6 and 1.4 × 10−6 M for C26 and MIA PaCa-2 cells, respectively. In contrast, the IC50 of free Dox was 0.5 × 10−6 and 0.04 × 10−6 M for C26 and MIA PaCa-2 cells, respectively (Figure 2F).[66] Although the in vitro cytotoxicity of ABD–Dox was 10–30-fold lower than that of free Dox, based on our experience with Dox loaded into nanoparticles,[67] this level of cytotoxicity is significant and acceptable, as we hypothesized that the extended half-life and greater tumor accumulation of ABD–Dox compared with free Dox would more than compensate for its lower cytotoxicity, in vivo. Furthermore, to study the effect of albumin binding on the cellular uptake and toxicity of ABD–Dox, we assessed the cytotoxicity of ABD–Dox mixed 1:1 with MSA. In addition, since the cell culture medium contains bovine albumin and considering that ABD also binds to bovine albumin, though with a much weaker affinity compared with MSA and HSA,[68] we also conducted a competitive cytotoxicity study by adding 0.5 × 10−3 M ABD to the cell culture medium to saturate the ABD-binding sites on the bovine albumin before adding the drug formulations. No difference was observed in the cytotoxicity of ABD–Dox and free Dox in the presence of either MSA or ABD indicating that binding to albumin does not affect the cellular uptake and tumoricidal activity of ABD–Dox.
For in vivo tumor regression studies, a single dose of ABD–Dox was injected via tail vein at 10 mg Dox equiv. kg−1 BW (MTD of free Dox) and 20 mg Dox equiv. kg−1 BW (MTD of ABD–Dox) in separate cohorts of BALB/c mice with subcutaneous C26 tumors or nude mice with subcutaneous MIA PaCa-2 tumors. Free Dox was injected at its MTD of 10 mg kg−1 BW, and AlDox was injected at 20 mg Dox equiv. kg−1 BW, a dose that was reported to be safe in previous studies.[69] All animals were injected when the tumors were 75–100 mm3 in size for C26 tumors and 100–125 mm3 for MIA PaCa-2 tumors. Tumor growth was monitored over 100 days after treatment, with predetermined endpoints for euthanasia: if the tumor volume exceeded 2000 m3, or body weight loss exceeded 15%, or if animals showed signs of morbidity including hunching, limb paralysis, and fur ruffling, as examined and determined by the veterinary staff at the Duke Cancer Center Isolation Facility.
In the colon carcinoma C26 tumor model, ABD–Dox at both 10 and 20 mg Dox equiv. kg−1 BW doses showed superior efficacy to free Dox (dose of 10 mg kg−1 BW) with respect to both tumor regression (P < 0.001, paired one-way ANOVA and Tukey’s test) (Figure 5A) and survival (P < 0.001, log-rank (Mantel-Cox) test) (Figure 5B). At 20 mg Dox equiv. kg−1 BW, both ABD–Dox and AlDox treatments resulted in complete eradication of C26 tumors, without recurrence of the primary tumor in any mice. At 10 mg Dox equiv. kg−1 BW, ABD–Dox showed complete tumor disappearance within 2 weeks after injection, but tumor recurrence was observed in 33% of mice with eradicated tumors. 25% and 89% of the mice in the ABD–Dox and AlDox arms (both at 20 mg Dox equiv. kg−1 BW), respectively, did not survive over the 100-day course of study (Figure 5B) due to either poor body conditions or body weight loss exceeding 15%, presumably caused by metastasis of the primary tumor.[39] Therefore, the therapeutic efficacy of ABD–Dox was equivalent to AlDox with respect to tumor regression, but higher with respect to overall survival (P < 0.05, log-rank (Mantel–Cox) test).
Figure 5.
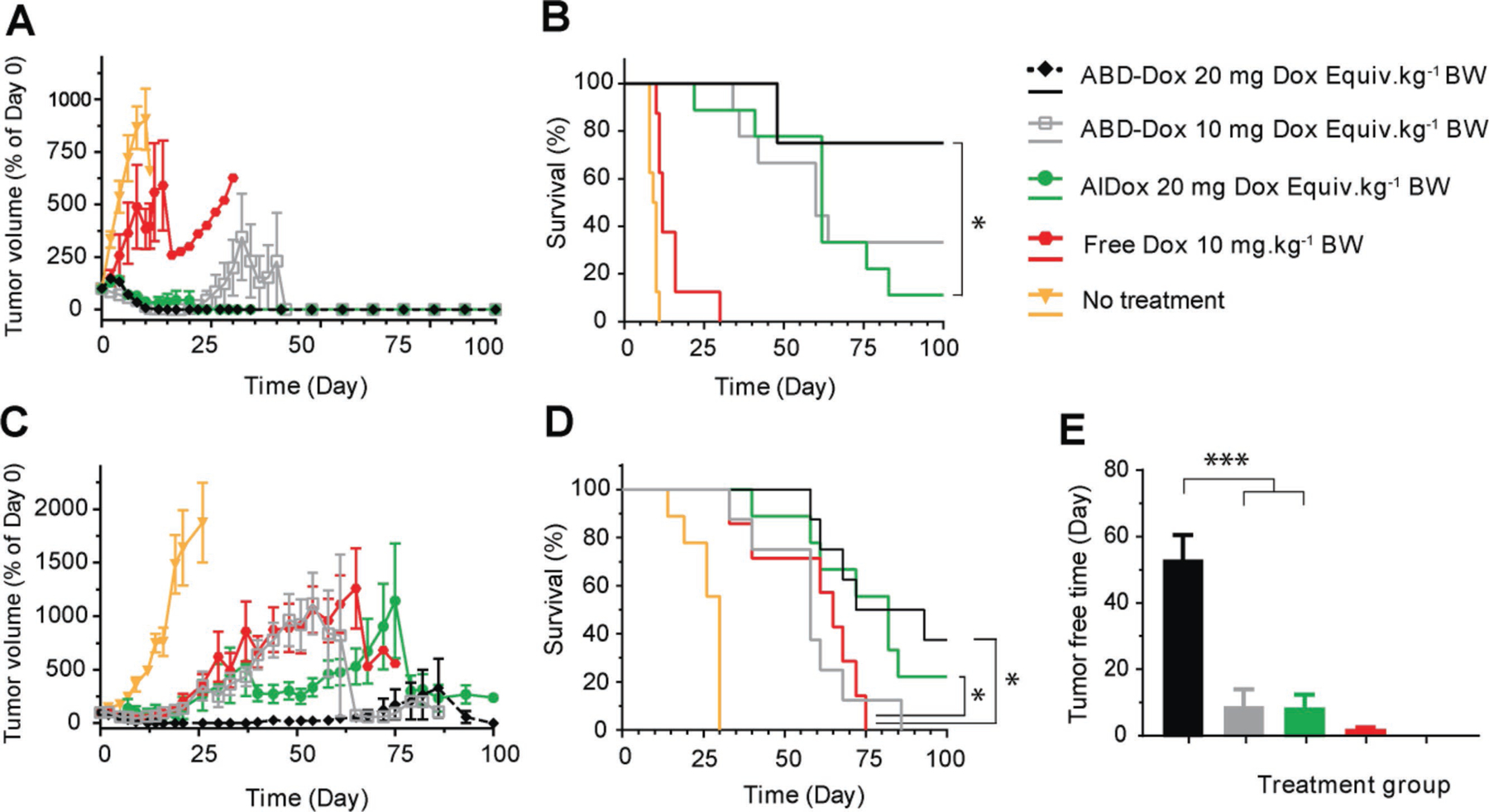
Antitumor activity of ABD–Dox. BALB/c and nude BALB/c mice bearing A,B) syngeneic C26 and C–E) xenograft MIA PaCa-2 tumors, respectively, were treated on day 0 with ABD–Dox (10 and 20 mg Dox equiv. kg−1 BW), AlDox (20 mg Dox equiv. kg−1 BW), and free Dox (10 mg kg−1 BW). A,C) Tumor volume over 100 days after treatment. B,D) Kaplan–Meier cumulative survival of mice over 100 days post-treatment. E) Tumor-free time during the 100-day span after the treatment of mice with MIA PaCa-2 xenografts. Data are presented as mean ± SEM, n = 7–9, A,C) paired one-way ANOVA and Tukey’s test, B,D) log-rank (Mantel–Cox) test, E) one-way ANOVA and Tukey’s test, *P < 0.05, **P < 0.01, and ***P < 0.001.
In the pancreatic MIA PaCa-2 tumor model, ABD–Dox treatment at 20 mg Dox equiv. kg−1 BW resulted in a significantly longer survival compared with treatment with free Dox (P < 0.05, log-rank (Mantel–Cox) test). No significant difference was found between ABD–Dox and free Dox at 10 mg Dox equiv. kg−1 BW dose and between ABD–Dox and AlDox arms at 20 mg Dox equiv. kg−1 BW dose (Figure 5D). ABD–Dox, however, showed superior tumor regression efficacy (P < 0.05, paired one-way ANOVA and Tukey’s test) (Figure 5C) and a longer tumor-free period (P < 0.001, one-way ANOVA and Tukey’s test) (Figure 5E) compared with AlDox at the same dose of 20 mg Dox equiv. kg−1 BW. By day 15 after treatment, all of ABD–Dox-treated mice, but only 50% for AlDox-treated mice showed complete tumor disappearance. Furthermore, by day 50, tumors recurred in all of the tumor-eradicated mice in the AlDox arm, while in the 20 mg Dox equiv. kg−1 BW ABD–Dox treatment group, tumors recurred in only 25% of the tumor-eradicated mice throughout the 100-day course of study.
Our results highlight—what should now be commonly accepted knowledge—that in vitro tumor cytotoxicity does not predict in vivo therapeutic efficacy, as it does not account for the important role of physiological transport barriers in vivo that control tumor exposure, and hence efficacy. In fact, despite a 10–30-fold lower antitumor cytotoxicity compared with free Dox in vitro, ABD–Dox showed far superior tumoricidal efficacy in vivo. In addition, ABD–Dox was more efficacious in the C26 model that showed less sensitivity to ABD–Dox and free Dox in vitro than MIA PaCa-2 and resulted in significant tumor regression and a higher survival rate compared with free Dox in this tumor model even at the lower dose of 10 mg Dox equiv. kg−1 BW. The higher therapeutic efficacy of ABD–Dox in the C26 allografts compared with MIA PaCa-2 xenografts is likely due to a cellular immune response triggered by treatment with ABD–Dox in this immunocompetent model, resulting in immunogenic cell death in addition to the direct tumor cytotoxicity caused by the drug.[64,70,71] Further studies are, however, required to explore this hypothesis and determine the under-lying mechanisms that drive the differences in the therapeutic efficacy between these two tumor models.
3. Conclusion
In aggregate, our results show that ABD–Dox has superior therapeutic efficacy to AlDox in the syngeneic C26 colon carcinoma model in BALB/c mice and in the MIA PaCa-2 pancreatic adenocarcinoma xenograft model in nude mice, and that both formulations are superior to free Dox. To the best of our knowledge, this is the first report showing that a conjugate of doxorubicin with an albumin-binding protein domain provides a 16-fold greater drug exposure in tumor than the free drug formulation, a 2-fold higher maximum tolerated dose, and results in complete tumor eradication and prolonged survival with a single injection. We note that there is one published report of a somewhat different system that consists of doxorubicin conjugated to an albumin-binding domain fused to tumor-pen-etrating sequences,[72] but despite the apparent sophistication of the design, it failed to exhibit any tumor eradication, highlighting the fact that careful design of a delivery system is critical to its success.
The albumin-binding strategy developed in this work could be readily applied to other small-molecule anticancer drugs such as gemcitabine, paclitaxel, and fluorouracil whose clinical application is limited by their poor pharmacokinetic or pharmacodynamic profiles using conjugation methods we have previously demonstrated.[73–75] In addition, small active targeting protein domains such as affibodies[76] or Fn3 domains[77] that are readily overexpressed in E. coli can be recombinantly fused to the ABD to yield actively targeted, long-circulating drug conjugates to further enhance tumor localization and therapeutic efficacy.[22,26]
One potential concern for our system is that, because of its bacterial origin, the native ABD sequence used in this work creates a potential risk of immunogenicity.[78] In preclinical studies, however, ABD variants were well tolerated in repeated administrations and ABD fusions have even been shown to reduce the immunogenicity of the fused protein.[79] Never-theless, to address any potential immunogenicity concerns, a deimmunized ABD variant (ABD094) has been generated by mutagenesis of the ABD sequence[80] by substituting the T-cell epitope residues located within the ABD. In T-cell proliferation assays, this variant showed a very weak immunogenic response (Affibody AB, unpublished data).[81]
In conclusion, the system demonstrated here allows site-specific conjugation of chemotherapeutics to an ABD that is readily overexpressed in E. coli and purified with high yield and purity without the need for chromatography. This system should also enable other functional elements—such as targeting protein domains—to be easily incorporated at the gene level to yield a modular and flexible system for delivery of small-molecule drugs to tumors for cancer therapy.
4. Experimental Section
Materials:
pET24+ plasmid was purchased from Novagen (Madison, WI). Oligodeoxynucleotides and gBlocks encoding sequences of interest were purchased from Integrated DNA Technologies (Coralville, IA). BL21(DE3) and EB5α chemically competent E. coli cells, and all molecular biology enzymes were purchased from New England Biolabs (Ipswich, MA). DNA extraction kits, DNA gel purification kits, and PCR purification kits were purchased from Qiagen (Germantown, MD). PBS tablets were purchased from MilliporeSigma (St Louis, MO). All E. coli cultures were grown in 2xYT medium, comprised of 16 g L−1 of tryptone (Becton, Dickinson and Co., Franklin Lakes, NJ), 10 g L−1 of yeast extract (Becton, Dickinson and Co., Franklin Lakes, NJ), and 5 g L−1 of NaCl (Alfa Aesar, Ward Hill, MA), with 1 μL mL−1 kanamycin sulfate (MilliporeSigma, St Louis, MO). IPTG was purchased from Gold Biotechnology (St. Louis, MO). Doxorubicin·HCl was purchased from Carbosynth LLC (San Diego, CA). Aldoxorubicin was purchased from MedKoo Biosciences (Morrisville, NC). Protein molecular weight marker (Precision Plus Protein unstained standards) and AnykD TGX Stain-Free gels, and Laemmli’s sample buffer were purchased from Bio-Rad Laboratories (Hercules, CA). SimplyBlue stain solution was purchased from Invitrogen (Carlsbad, CA). TCEP hydrochloride and Detoxi-Gel resin, HisPur cobalt spin columns were purchased from Thermo Fisher Scientific (Waltham, MA). N-(ε-Maleimidocaproic acid) hydrazide, trifluoroacetic acid salt, anhydrous methanol, Amicon Ultra-15 Amicon ultrafiltration spin columns (3 and 10 kDa molecular weight cutoff (MWCO)), and mouse and human serum albumin were purchased from Sigma-Aldrich (St. Louis, MO). All salts used for the preparation of sortase reaction buffer (50 × 10−3 M Tris, 150 × 10−3 M NaCl, 10 × 10−3 M CaCl2, pH 7.5), conjugation buffer (0.1 M NaPO4, 1 × 10−3 M EDTA, 3 × 10−3 M TCEP–HCl, pH 7.0), SDS-PAGE running buffer (25 × 10−3 M Tris, 192 × 10−3 M glycine, 0.1% SDS, pH 8.3), and native PAGE running buffer (25 × 10−3 M Tris, 192 × 10−3 M glycine, 0.1% SDS, pH 8.3) were purchased from Alfa Aesar (Ward Hill, MA). CellTiter-96 aqueous cell proliferation assay containing 3-(4,5-dimethythizol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent was purchased from Promega (Madison, WI). 96-well plates and 75 and 150 cm2 tissue culture flasks were purchased from Corning Inc. (Corning, NY). C26 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 4.5 g L−1 D-glucose (all from Sigma, St. Louis, MO), 10 × 10−3 M HEPES, 1 × 10−3 M sodium pyruvate (both from Invitrogen, Carlsbad, CA), and 100 U mL−1 penicillin–streptomycin (Gibco, Grand Island, NY). MIA PaCa-2 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2.5% horse serum (Sigma, St. Louis, MO), and 100 U mL−1 penicillin–streptomycin. The materials used for the confocal microscopy were supplied as follows: Hank’s balanced salt solution (HBSS) from Thermo Fisher Scientific (Waltham, MA), 1× CytoPainter LysoDeep Red Indicator Reagent from Abcam (Cambridge, MA), Hoechst 33342 from Life Technologies (Carlsbad, CA), and μ-Dish (35 mm, high) with four-well Culture-Insert from Ibidi (Madison, WI).
Cell Lines and Animals:
The C26 cell line was generously provided by Dr. Francis C. Szoka (University of California, San Francisco) and the BxPC-3, AsPC-1, and MIA PaCa-2 cell lines were purchased from the Duke University Cell Culture Facility (Durham, NC). BALB/c mice (5–6 weeks, female) and BALB/c athymic nude mice (nu/nu, 5–6 weeks, male) were purchased from Charles River Laboratories (Wilmington, MA) and Duke University Cancer Center Isolation Facility (Durham, NC), respectively. All animal studies were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals under protocols approved by the Duke Institutional Animal Care and Use Committee. Mice were group-housed under a 12 h:12 h light:dark cycle with ad libitum access to food and water.
Gene Synthesis and Expression:
EB5α competent cells were used for cloning and gene assembly. Genes encoding ABD, ELP40, ELP160, (CGG)4, (CGG)8, KEKE, (His)6–sortase A, and srt, all in a modified pET24+ plasmid, were available from previous studies, and were used to assemble the genes encoding the KEKE–ELP160–srt–ABD–(CGG)4/8 and (His)6–sortase A–ELP40 fusions by the method of recursive directional ligation by plasmid reconstruction,[82] as described elsewhere. For protein expression, plasmids encoding the genes of interest were transformed into BL21(DE3) competent cells and were expressed in 2xYT medium. 50 mL of the medium was inoculated with the frozen DMSO bacterial stock and were grown overnight. Cultures were then inoculated in 1 L 2xYT medium and were incubated for 6–8 h at 37 °C and 200 rpm, following which IPTG was added at 1 × 10−3 M final concentration and the culture was incubated for an additional 10 h. Cells were harvested by centrifugation at 4 °C and 3400 rcf for 15 min, and were resuspended in PBS.
Protein Purification:
KEKE–ELP160–srt–ABD–(CGG)4, KEKE–ELP160–srt-ABD–(CGG)8, and (His)6–sortase A–ELP40 fusions were purified by inverse transition cycling (ITC) and immobilized metal affinity chromatography (IMAC), respectively. Cells were lysed by sonication (Q500 sonicator; QSonica, Newtown, CT) with 10 s on and 40 s off pulses for a total of 3 min. Nucleic acids and other negatively charged cellular debris were precipitated by addition of 2 mL of 20% v/v polyethyleneimine (PEI) per liter of culture and removed by centrifugation at 24 000 rcf for 15 min at 4 °C. The KEKE–ELP160–srt–ABD–(CGG)4/8 constructs were then purified from the supernatant using two cycles of ITC, as described elsewhere with slight modifications.[48] Briefly, KEKE–ELP160–srt–ABD–(CGG)4/8 went through multiple “bakeout” cycles consisting of 10 min incubation at 60 °C to transition the KEKE–ELP160–srt–ABD–(CGG)4/8 fusion and precipitate contaminant proteins, 10 min incubation on ice to resolubilize the KEKE–ELP160–srt–ABD–(CGG)4/8 constructs, and 10 min centrifugation at 24 000 rcf and 4 °C (“cold spin”) to remove the insoluble contaminants. The KEKE–ELP160–srt–ABD–(CGG)4/8 were then aggregated by adding NaCl to a final concentration of 2.5 M and centrifugation at 24 000 rcf for 10 min at 35 °C (“hot spin”). The pellet was resuspended in 30 × 10−3 M TCEP, pH 7, followed by one more cold spin to complete the first ITC cycle. The second ITC cycle was carried out similarly to the first one but using NaCl at 0.5 M final concentration for the hot spin. (His)6–sortase A–ELP40 was purified from the clear supernatant, after PEI precipitation of DNA and centrifugation, using HisPur cobalt IMAC spin columns according to the manufacturer’s instructions. Briefly, supernatant was added to the columns and was incubated for 1 h at 4 °C. Impurities were removed by washing the resin multiple times with PBS. (His)6–sortase A–ELP40 was then eluted by addition of PBS with 150 × 10−3 M imidazole and dialyzed against sortase reaction buffer. For in vivo experiments, endotoxin was removed from the fusions using Detoxi-Gel endotoxin removal gel, according to the manufacturer’s protocol. SDS-PAGE analysis was performed to confirm the expression and purity of the fusions. 30 μg of proteins were diluted in Laemmli’s buffer, heated for 10 min at 95 °C, chilled on ice, loaded onto a stain-free gel, and run at 200 V using SDS-PAGE buffer following which the gel was UV-activated and imaged using the Gel Doc imaging system (Bio-Rad, Hercules, CA).
ABD–Dox Synthesis:
Dox was conjugated to cysteine residues at the C-terminus of the KEKE–ELP160–srt–ABD–(CGG)4 and KEKE–ELP160–srt–ABD–(CGG)8 via a pH-sensitive hydrazone bond in a two-step reaction. In the first step, Dox (4.25 M) was reacted with EMCH (3.4 M) in anhydrous methanol supplemented with 0.1% v/v TFA with stirring at room temperature for 16 h. The activated Dox–EMCH conjugate was concentrated to 17 M by rotary evaporation and was added dropwise to the ABD–ELP fusion (1.7 M) in conjugation buffer with a methanol:buffer volume ratio of 2:1. The reaction mixture was stirred for 24 h at room temperature. Unreacted Dox was removed using 10 kDa MWCO Amicon ultrafiltration spin columns and with PBS:acetonitrile (70:30 v/v) as the eluent. The purified KEKE–ELP160–srt–ABD–Dox was then buffer exchanged using 10 kDa MWCO Amicon ultrafiltration spin columns into sortase reaction buffer in preparation for sortase-catalyzed cleavage of the ELP160 tag. To calculate the conjugation efficiency, a small fraction of KEKE–ELP160–srt–ABD–Dox was buffer exchanged into Milli-Q water, lyophilized, weighed, and dissolved in PBS. The conjugation efficiency was defined as the number of Dox molecules per KEKE–ELP160–srt–ABD molecule, and was determined by measuring the concentration of the KEKE–ELP160–srt–ABD by gravimetry of the lyophilized conjugate, and that of Dox by spectrophotometry. Following drug conjugation, the ELP160 tag was removed by cleavage with the (His)6–sortase A–ELP40 fusion. KEKE–ELP160–srt–ABD–Dox was mixed with (His)6–sortase A–ELP40 in the presence of triglycine (GGG) at a KEKE–ELP160–srt–ABD–Dox:(His)6–sortase A–ELP40:GGG molar ratio of 1:5:250, and final KEKE–ELP160–srt-ABD–Dox concentration of 500 × 10−6 M. The reaction solution was incubated for 24 h at room temperature in the dark, following which the ABD–Dox product was purified from (His)6–sortase A–ELP40, cleaved ELP160 tag, and unreacted KEKE–ELP160–srt–ABD–Dox by size exclusion chromatography using a Superdex 75 column (HiLoad 16/60 Superdex 75 prep grade) at 4 °C on an AKTA Purifier system (both from GE Healthcare, Waukesha, WI) equipped with a photodiode detector set at 280 and 488 nm and using H2O:PBS:acetonitrile (35:35:30 v/v) as the mobile phase.
Characterization of Dox Conjugates:
The hydrodynamic radii (Rh) of the ELP160–srt–ABD–Dox and KEKE–ELP160–srt–ABD–Dox conjugates were determined by DLS (DynaPro; Wyatt Technology, Santa Barbara, CA) at 25 °C using a single detector at 90°. Samples were prepared in sortase reaction buffer at 25 × 10−6 M, filtered through 0.22 μm Millex-GV filters (Sigma-Aldrich, St. Louis, MO), and measured using a DynaPro plate reader (Wyatt Technology, Santa Barbara, CA). The data were analyzed with a regularization fit of the autocorrelation function using a Rayleigh sphere model. The purity of the drug conjugates (KEKE–ELP160–ABD–Dox and ABD–Dox) and efficiency of the sortase enzymatic reaction were assessed by size exclusion HPLC. Samples were injected into a LC10 HPLC (Shimadzu Scientific Instruments, Columbia, MD) with a Shodex OHPak SB-804 column (New York, NY) and PBS:acetonitrile (70:30 v/v) as the mobile phase at an isocratic flow rate of 0.3 mL min−1. Eluting peaks were detected with a UV–vis detector set at 488 nm. Mass spectrometry analysis of the ABD–Dox conjugate was performed on a Bruker Autoflex Speed MALDI-TOFMS (Bruker Daltonics, Billerica, MA) using succinic acid matrix and porcine insulin as the internal standard.
Drug Release:
Dox release from the ABD–Dox conjugate was measured at pH 7.4 and 5.0, corresponding to the physiological pH and late endosomal pH, respectively. ABD–Dox in PBS (400 × 10−6 M Dox equiv.) was diluted with an equal volume of either 0.1 M Na-PO4, pH 7.4 buffer or 0.1 M Na-acetate, pH 5.0 buffer, incubated at 37 °C for 0, 0.25, 0.5, 1, 2, 4, 10, and 24 h, and neutralized using an equal volume of 0.1 M NaPO4, pH 7.4 buffer. Samples were then analyzed by size exclusion HPLC using the same chromatography conditions as described above for the characterization of Dox conjugates and the percentage of released Dox was calculated by integrating the area under the peak corresponding to unbound Dox. The cumulative percent released Dox (F%,released) versus time was fit to a first-order release model: F%,released = a[1 − exp(−ln(2)t/t1/2)], where t is the time (h) after incubation, t1/2 is the half-maximal release time (h), and a (%) is the maximum extent of drug release, using GraphPad Prism software (GraphPad, San Diego, CA).
Stability of ABD–Dox in Serum:
Stability of ABD–Dox in serum and in the presence of serum proteases was studied by incubation of ABD–Dox in the mouse serum at 37 °C. ABD–Dox (500 × 10−6 M) in PBS was mixed 1:1 (v/v) with non-heat inactivated mouse serum (Abcam, Cambridge, MA) and was incubated at 37 °C for 6, 24, 48, 96, and 144 h. Samples were then diluted 10-fold with PBS and fractionated by size exclusion HPLC using the same chromatography conditions as described above for the characterization of Dox conjugates.
Interaction of ABD–Dox with Human and Mouse Albumin:
The affinity of ABD–Dox for MSA and HSA was studied qualitatively by native PAGE electrophoresis and quantitatively by isothermal titration calorimetry. For native PAGE, ABD–Dox was mixed with an equal number of moles of MSA and HSA, and incubated for 30 min at room temperature, prior to carrying out native PAGE. ABD–Dox, HSA, MSA, and mixtures of ABD–Dox with HSA and MSA were mixed with the native PAGE buffer, loaded onto stain-free gels, and run at 180 V for 45 min using native PAGE running buffer. The gel was stained with SimplyBlue stain according to the manufacturer’s instructions and imaged using the Gel Doc imaging system (Bio-Rad, Hercules, CA). Isothermal titration calorimetry was done using a standard volume Nano ITC from TA Instruments (Lindon, Utah). 250 μL of HSA or MSA at a 500 × 10−6 M concentration in PBS was injected as 10 μL aliquots into the calorimeter cell containing 1 mL of 45 × 10−6 M ABD–Dox in PBS at 37 °C with stirring. The heat of dilution was measured by injecting HSA or MSA in PBS into the PBS buffer without ABD–Dox under the same titration conditions and was subtracted from the heat of titration of ABD–Dox with HSA or MSA. The calorimetric data were analyzed by NanoAnalyze software (TA Instruments, Lindon, UT) and were fit to an independent binding site model to compute the thermodynamic parameters, including dissociation constant (KD), binding stoichiometry (n), enthalpy (ΔH), and entropy (ΔS).
Cellular Uptake of ABD–Dox:
To study the intracellular trafficking of ABD–Dox, 5000 C26 cells were seeded in 110 μL of cell culture medium into each of the four chambers of a μ-Dish and allowed to adhere overnight. Cell medium was replaced with complete medium containing ABD–Dox at 10 × 10−6 M Dox equiv. concentration and cells were incubated for 4 h at 37 °C. After treatment, the medium was removed and replaced with complete medium containing 1× CytoPainter Reagent to stain lysosomal compartments for 30 min at 37 °C. The medium was then gently removed and replaced with medium containing 2 × 10−6 M Hoechst 33342 to stain cell nuclei for 10 min at 37 °C. Cells were then rinsed twice with HBSS and maintained in complete medium until imaging on an Andor Dragonfly Spinning Disk 500 series confocal on a LeicaDMi8 microscope stand (Oxford Instruments, Abingdon, UK) with a 63× water immersion objective and equipped with a Zyla 4.2 series camera. Dox was detected with a 488 nm excitation laser and 525/50 nm emission filter, Hoechst 33342 dye with a 400 nm excitation filter and 450/50 nm emission filter, and CytoPainter with a 637 nm excitation laser and 700/75 nm emission filter in CF40 imaging mode.
Pharmacokinetics and Biodistribution:
Pharmacokinetics and biodistribution experiments were performed to study the concentration of ABD–Dox in blood and tissues over time following i.v. administration. ABD–Dox and Dox in PBS and AlDox in 10 × 10−3 M Na-PO4 buffer, 5% d-(+)-glucose, pH 5.8, were injected at 5 mg Dox equiv. kg−1 BW into healthy BALB/c mice for the pharmacokinetic study and C26 tumor-bearing BALB/c mice for the biodistribution study. 10 μL blood samples were collected at 40 s, 15 and 30 min, and 2, 4, 8, 24, 48, 72, and 96 h post injection and were diluted into 100 μL heparinized PBS (1 kU mL−1), and centrifuged at 4000 rcf, 10 min, 4 °C to collect the plasma. 10 μL of the plasma samples were then diluted in 490 μL of acidified isopropanol (0.075 M HCl, 10% water) and incubated overnight at 4 °C. At 2, 24, and 72 h post injection, mice were sacrificed and tumor, heart, lung, liver, spleen, kidney, and muscle tissues were dissected, weighed, and homogenized in acidified isopropanol using a Mini-Beadbeater (Biospec, Bartlesville, OK) with 2 mm diameter zirconia beads for 1–2 min, and were incubated overnight at 4 °C. Following overnight incubation in acidified isopropanol, plasma and tissue samples were centrifuged at 16 000 rcf, 4 °C for 10 min and the supernatants were loaded onto black 96-well plates in duplicate and the Dox fluorescence was measured using a Victor3 microplate reader (Perkin Elmer, Waltham, MA) with fluorescence excitation at 485 nm and emission at 590 nm. Tissues were also dissected from three untreated mice, processed in the same manner, serially diluted, and loaded on the plates to prepare a standard curve of fluorescence intensity versus the weight for each tissue from which the background tissue fluorescence was calculated and subtracted for each sample. Serial dilution of free Dox in acidified isopropanol was used as standard to quantify the Dox concentration as micromoles per liter of plasma (×10−6 M) and as the percent of the injected dose per gram (%ID g−1) in the tumor and other tissues. Plasma concentrations of Dox equiv. of ABD–Dox versus time were fit to a two-compartment model using SAAM II software (SAAM Institute, Seattle, WA) and the elimination half-life and the area under curve were computed. The tissue concentrations of Dox equiv. of ABD–Dox, AlDox, and free Dox at different time points were compared in GraphPad Prism software (GraphPad, San Diego, CA) by two-way ANOVA followed by Tukey-Kramer (Tukey’s) post-hoc test with P < 0.05 considered statistically significant.
In Vitro Cytotoxicity:
C26 murine colon carcinoma cells and three different pancreatic cancer cell lines—BxPC-3, AsPC-1, and MIA PaCa-2—were used to study the in vitro cytotoxicity of ABD–Dox and Dox. Cells were cultured and grown as monolayers at 37 °C with CO2 in a humidified incubator and were passaged at ≈80% confluency using 0.05% trypsin–EDTA (Invitrogen, Carlsbad, CA). C26, BxPC-3, AsPC-1, and MIA PaCa-2 cells were seeded in 96-well plates (5000 cells per well) and incubated overnight. After this incubation, the medium was removed and 3-fold serial dilutions of the drug, prepared in the cell culture medium, were added to the wells in triplicate and also to control wells (medium with no cells) to correct for the background Dox absorbance. To assess the effect of albumin binding on the cytotoxicity of ABD–Dox, in a separate experiment, C26 cells were treated with the serial dilutions of the drug that were mixed with equimolar MSA or prepared in the cell culture medium containing 0.5 × 10−3 M ABD. After 72 h treatment, 20 μL MTS reagent was added to each well and was incubated for 3 h at 37 °C, and the absorbance at 490 nm was measured using a Victor3 plate reader (Perkin Elmer, Boston, MA). Cell viability (%) was defined as the percentage of the absorbance of the drug-treated cells relative to buffer-treated cells and IC50 values were determined using logistic nonlinear regression analysis with GraphPad Prism software. The IC50 was defined as the Dox equiv. concentration of formulations resulting in 50% growth inhibition compared with a negative control—buffer-treated cells.
In Vivo Tumor Regression:
For in vivo tumor implantation, C26 cells at 5 × 105 cells in 30 μL Minimum Essential Medium (MEM) (Invitrogen, Carlsbad, CA) and MIA PaCa-2 cells at 2 × 106 cells in 50 μL 10:90 v/v MEM:Matrigel matrix (Corning Inc., Corning, New York) were inoculated in the right flank of normal and athymic nude BALB/c mice, respectively. Tumor cells were allowed to grow until the tumor volume—calculated as π/6 × length × width2—reached 75–100 mm3 for C26 tumors and 100–125 mm3 for MIA PaCa-2 tumors. Tumor-bearing mice were then treated with a single i.v. injection of ABD–Dox at 10 and 20 mg Dox equiv. kg−1 BW, AlDox at 20 mg Dox equiv. kg−1 BW, and free Dox at 10 mg kg−1 BW. Tumor volumes and body weights were monitored for 100 days after treatment and mice displaying excessive tumor volume (> 2000 mm3), body weight loss (> 15%), or ill health were euthanized. Tumor growth and survival data were analyzed by GraphPad Prism software. Survival curves were generated with the Kaplan–Meier method and compared with the log-rank (Mantel–Cox) test. Tumor growth data were compared with a paired one-way ANOVA followed by Tukey–Kramer (Tukey’s) post-hoc test. P < 0.05 was considered as statistically significant.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge the support of the National Institutes of Health through grants R01EB000188 and R01EB007205 to A.C. P.Y. thanks Jonathan R. McDaniel for helpful discussions and assistance with the design of the conjugate. P.Y. and A.C. conceived the idea and designed the experiments, P.Y. performed the experiments, and P.Y. and A.C. analyzed the data and wrote the manuscript. L.A. and J.T. assisted with synthesis of the drug material, S.S. performed the MALDI-MS analysis, S.A.C. assisted with the confocal microscopy study, J.J.B. assisted with designing the sortase enzymatic reaction, and X.L. assisted with in vivo experiments.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/smll.201804452.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Torchilin V, Adv. Drug Delivery Rev 2011, 63, 131. [DOI] [PubMed] [Google Scholar]
- [2].Singh R, Lillard JW Jr., Exp. Mol. Pathol 2009, 86, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maeda H, Seymour LW, Miyamoto Y, Bioconjugate Chem. 1992, 3, 351. [DOI] [PubMed] [Google Scholar]
- [4].Kudgus RA, Walden CA, McGovern RM, Reid JM, Robertson JD, Mukherjee P, Sci. Rep 2014, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang J, Mao W, Lock LL, Tang J, Sui M, Sun W, Cui H, Xu D, Shen Y, ACS Nano 2015, 9, 7195. [DOI] [PubMed] [Google Scholar]
- [6].Yousefpour P, Chilkoti A, Biotechnol. Bioeng 2014, 111, 1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Larsen MT, Kuhlmann M, Hvam ML, Howard KA, Mol. Cell. Ther 2016, 4, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kratz F, J. Controlled Release 2014, 190, 331. [DOI] [PubMed] [Google Scholar]
- [9].Alvarez B, Carballal S, Turell L, Radi R, Methods Enzymol 2010, 473, 117. [DOI] [PubMed] [Google Scholar]
- [10].Sockolosky JT, Szoka FC, Sciences T, Francisco S, Adv. Drug Delivery Rev 2015, 91, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kratz F, J. Controlled Release 2008, 132, 171. [DOI] [PubMed] [Google Scholar]
- [12].Stehle G, Sinn H, Wunder A, Schrenk HH, Stewart JC, Hartung G, Maier-Borst W, Heene DL, Crit. Rev. Oncol./Hematol 1997, 26, 77. [DOI] [PubMed] [Google Scholar]
- [13].Merlot AM, Kalinowski DS, Richardson DR, Front. Physiol 2014, 5, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shang L, Wang Y, Jiang J, Dong S, Langmuir 2007, 23, 2714. [DOI] [PubMed] [Google Scholar]
- [15].Alter HJ, Stramer SL, Dodd RY, Semin. Hematol 2007, 44, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Willyard C, Nature 2017, 549, S19. [DOI] [PubMed] [Google Scholar]
- [17].Jimenez A, Shaz BH, Bloch EM, Transfus. Med. Rev 2017, 31, 1. [DOI] [PubMed] [Google Scholar]
- [18].Yu Z, Fu Y, US8084021B2, 2011. [Google Scholar]
- [19].Pukac L, Adar L, Barash S, Clark S, Liu P, Bock J, Shen WD, Blood 2013, 122, 4854. [Google Scholar]
- [20].Christensen B, Hjort C, Poulsen TA, Bach P, EP2169067A1, 2009. [Google Scholar]
- [21].Chen Z, He Y, Shi B, Yang D, Biochim. Biophys. Acta: Gen. Subj 2013, 1830, 5515. [DOI] [PubMed] [Google Scholar]
- [22].Orlova A, Jonsson A, Rosik D, Lundqvist H, Lindborg M, Abrahmsen L, Ekblad C, Frejd FY, Tolmachev V, J. Nucl. Med 2013, 54, 961. [DOI] [PubMed] [Google Scholar]
- [23].Marre M, Shaw J, Brändle M, Bebakar WMW, Kamaruddin NA, Strand J, Zdravkovic M, Le Thi TD, Colagiuri S, LSU Study Group, Diabetic Med. 2009, 26, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shan L, Zhuo X, Zhang F, Dai Y, Zhu G, Yung BC, Fan W, Zhai K, Jacobson O, Kiesewetter DO, Ma Y, Gao G, Chen X, Theranostics 2018, 8, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu Y, Wang G, Zhang H, Ma Y, Lang L, Jacobson O, Kiesewetter DO, Zhu L, Gao S, Ma Q, Chen X, Bioconjugate Chem. 2016, 27, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Andersen JT, Pehrson R, Tolmachev V, Daba MB, Abrahmsén L, Ekblad C, J. Biol. Chem 2011, 286, 5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kratz F, Expert Opin. Invest. Drugs 2007, 16, 855. [DOI] [PubMed] [Google Scholar]
- [28].Gong J, Yan J, Forscher C, Hendifar A, Drug Des. Dev. Ther 2018, 12, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Busher JT, in Clin Methods Hist Phys Lab Exam (Eds: Walker HK, Hall WD, Hurst JW), Butterworth-Heinemann, Boston, USA, 1990, pp. 497–499. [Google Scholar]
- [30].Turell L, Radi R, Alvarez B, Free Radical Biol. Med 2013, 65, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ, Cancer Res. 2013, 73, 1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hu Y-B, Dammer EB, Ren R-J, Wang G, Transl. Neurodegener 2015, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kratz F, Top. Curr. Chem 2008, 283, 73. [DOI] [PubMed] [Google Scholar]
- [34].Lenaz L, Page JA, Cancer Treat. Rev 1976, 3, 111. [DOI] [PubMed] [Google Scholar]
- [35].Zhang Y-W, Shi J, Li Y-J, Wei L, Arch. Immunol. Ther. Exp 2009, 57, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sharpless NE, Flavin M, Biochemistry 1966, 5, 2963. [DOI] [PubMed] [Google Scholar]
- [37].Kedar PS, Colah RB, Kulkarni S, Ghosh K, Mohanty D, Clin. Lab. Haematol 2003, 25, 373. [DOI] [PubMed] [Google Scholar]
- [38].Sahaf B, Heydari K, Herzenberg LA, Herzenberg LA, Proc. Natl. Acad. Sci 2003, 100, 4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Christensen RD, Agarwal AM, Nussenzveig RH, Heikal N, Liew MA, Yaish HM, J. Perinatol 2015, 35, 357. [DOI] [PubMed] [Google Scholar]
- [40].Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, Marks JD, Weiner LM, Cancer Res. 2001, 61, 4750. [PubMed] [Google Scholar]
- [41].Wall JS, Williams A, Richey T, Stuckey A, Huang Y, Wooliver C, Macy S, Heidel E, Gupta N, Lee A, Rader B, Martin EB, Kennel SJ, PLoS One 2013, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rozak DA, Orban J, Bryan PN, Biochim. Biophys. Acta: Proteins Proteomics 2005, 1753, 226. [DOI] [PubMed] [Google Scholar]
- [43].Johansson MU, de Chateau M, Bjorck L, Forsen S, Drakenberg T, Wikstrom M, FEBS Lett. 1995, 374, 257. [DOI] [PubMed] [Google Scholar]
- [44].Nygren P, Eliasson M, Abrahmsén L, Uhlén M, Palmcrantz E, J. Mol. Recognit 1988, 1, 69. [DOI] [PubMed] [Google Scholar]
- [45].Sjölander A, Nygren PA, Stahl S, Berzins K, Uhlen M, Perlmann P, Andersson R, Immunol J. Methods 1997, 201, 115. [DOI] [PubMed] [Google Scholar]
- [46].Gilroy CA, Roberts S, Chilkoti A, J. Controlled Release 2018, 277, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hassouneh W, Christensen T, Chilkoti A, Curr. Protoc. Protein Sci 2010, Chapter 6, Unit 6.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].MacEwan SR, Hassouneh W, Chilkoti A, J. Vis. Exp 2014, 88, e51583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fogh-Andersen N, Bjerrum PJ, Siggaard-Andersen O, Clin. Chem 1993, 39, 48. [PubMed] [Google Scholar]
- [50].Qi Y, Simakova A, Ganson NJ, Li X, Luginbuhl KM, Ozer I, Liu W, Hershfield MS, Matyjaszewski K, Chilkoti A, Nat. Biomed. Eng 2017, 1, 0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huotari J, Helenius A, EMBO J. 2011, 30, 3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sonawane SJ, Kalhapure RS, Govender T, Eur. J. Pharm. Sci 2017, 99, 45. [DOI] [PubMed] [Google Scholar]
- [53].Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, Anderson CL, J. Exp. Med 2003, 197, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gao H, He Q, Expert Opin. Drug Delivery 2014, 11, 409. [DOI] [PubMed] [Google Scholar]
- [55].Kummitha CM, Malamas AS, Lu Z-R, Int. J. Nanomed 2012, 7, 5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chen N, Wang W, Fauty S, Fang Y, Hamuro L, Hussain A, Prueksaritanont T, mAbs 2014, 6, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cataldi M, Vigliotti C, Mosca T, Cammarota M, Capone D, Int. J. Mol. Sci 2017, 18, 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Akilesh S, Christianson GJ, Roopenian DC, Shaw AS, J. Immunol 2007, 179, 4580. [DOI] [PubMed] [Google Scholar]
- [59].Anselmo AC, Gupta V, Zern BJ, Pan D, Zakrewsky M, Muzykantov V, ACS Nano 2013, 7, 11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Brenner JS, Pan DC, Myerson JW, Marcos-Contreras OA, Villa CH, Patel P, Hekierski H, Chatterjee S, Tao JQ, Parhiz H, Bhamidipati K, Uhler TG, Hood ED, Kiseleva RY, Shuvaev VS, Shuvaeva T, Khoshnejad M, Johnston I, Gregory JV, Lahann J, Wang T, Cantu E, Armstead WM, Mitragotri S, Muzykantov V, Nat. Commun 2018, 9, 2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yasuda K, Park H-C, Ratliff B, Addabbo F, Hatzopoulos AK, Chander P, Goligorsky MS, Am. J. Pathol 2010, 176, 1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Damodar G, Smitha T, Gopinath S, Vijayakumar S, Rao YA, Ann. Med. Health Sci. Res 2014, 4, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chatterjee K, Zhang J, Honbo N, Karliner JS, Cardiology 2010, 115, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mastria EM, Cai LY, Kan MJ, Li X, Schaal JL, Fiering S, Gunn MD, Dewhirst MW, Nair SK, Chilkoti A, J. Controlled Release 2018, 269, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Schultz RM, Merriman RL, Toth JE, Zimmermann JE, Hertel LW, Andis SL, Dudley DE, Rutherford PG, Tanzer LR, Grindey GB, Oncol. Res. Featuring Preclin. Clin. Cancer Ther 1993, 5, 223. [PubMed] [Google Scholar]
- [66].Rydzewski RM, Real World Drug Discovery: A Chemist’s Guide to Biotech and Pharmaceutical Research, Elsevier, Oxford; 2008. [Google Scholar]
- [67].MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A, Nat. Mater 2009, 8, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Johansson MU, Frick I-M, Nilsson H, Kraulis PJ, Hober S, Jonasson P, Linhult M, Nygren P-Å, Uhlén M, Björck L, Drakenberg T, Forsén S, Wikström M, J. Biol. Chem 2002, 277, 8114. [DOI] [PubMed] [Google Scholar]
- [69].Graeser R, Esser N, Unger H, Fichtner I, Zhu A, Unger C, Kratz F, Invest. New Drugs 2010, 28, 14. [DOI] [PubMed] [Google Scholar]
- [70].Zhang Z, Yu X, Wang Z, Wu P, Huang J, Cancer Lett. 2015, 369, 331. [DOI] [PubMed] [Google Scholar]
- [71].Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ, Cancer Res. 2011, 71, 4809. [DOI] [PubMed] [Google Scholar]
- [72].Liu L, Zhang C, Li Z, Wang C, Bi J, Yin S, Wang Q, Yu R, Liu Y, Su Z, Mol. Pharm 2017, 14, 3739. [DOI] [PubMed] [Google Scholar]
- [73].Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li X, Lin C-C, Chi J-TA, Chilkoti A, Nat. Commun 2015, 6, 7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bhattacharyya J, Weitzhandler I, Ho SB, McDaniel JR, Li X, Tang L, Liu J, Dewhirst M, Chilkoti A, Adv. Funct. Mater 2017, 27, 1605421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bhattacharyya J, Ren X-R, Mook RA, Wang J, Spasojevic I, Premont RT, Li X, Chilkoti A, Chen W, Nanoscale 2017, 9, 12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, Frejd FY, FEBS Lett. 2010, 584, 2670. [DOI] [PubMed] [Google Scholar]
- [77].Jacobs S, in Protein Engineering (Ed: Kaumaya KOE-P), IntechOpen, Rijeka: 2012. [Google Scholar]
- [78].Goetsch L, Haeuw JF, Champion T, Lacheny C, N’Guyen T, Beck A, Corvaia N, Clin. Vaccine Immunol 2003, 10, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Frejd F, in Therapeutic Proteins: Strategies to Modulate Their Plasma Half-Lives (Ed: Kontermann R), Wiley-Blackwell, Weinheim: 2012. [Google Scholar]
- [80].Erickson M, Litzinger DC, Ghosh SS, Guo Z, Ekbland C, Roth JD, US9879063B2, 2014. [Google Scholar]
- [81].Nilvebrant J, Hober S, Comput. Struct. Biotechnol. J 2013, 6, e201303009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].McDaniel JR, MacKay JA, Quiroz FG, Chilkoti A, Biomacro-molecules 2010, 11, 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.