

Abstract
In pancreatic ductal adenocarcinoma (PDAC), early onset of hypoxia triggers remodeling of the extracellular matrix, epithelial-to-mesenchymal transition, increased cell survival, the formation of cancer stem cells, and drug resistance. Hypoxia in PDAC is also associated with the development of collagen-rich, fibrous extracellular stroma (desmoplasia), resulting in severely-impaired drug penetration. To overcome these daunting challenges, we created polymer nanoparticles (polymersomes) that target and penetrate pancreatic tumors, reach the hypoxic niches, undergo rapid structural destabilization, and release the encapsulated drugs. In-vitro studies indicated a high cellular uptake of the polymersomes and increased cytotoxicity of the drugs under hypoxia compared to unencapsulated drugs. The polymersomes decreased tumor growth by nearly 250% and significantly increased necrosis within the tumors by 60% in mice compared to untreated controls. We anticipate that these polymer nanoparticles possess considerable translational potential for delivering drugs to solid hypoxic tumors.
Keywords: Polymersomes, tumor-penetrating, hypoxia-responsive, pancreatic cancer, nanoparticles
Graphical Abstract
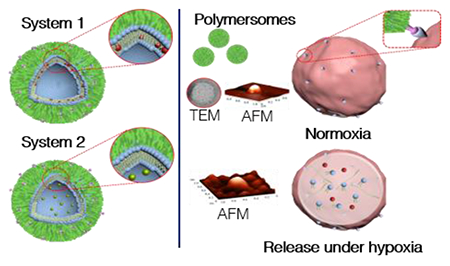
Introduction.
Solid tumors account for roughly 85% of all human cancers.1 While surgical resection is often the most effective treatment, it is not always possible or practical, depending on the type and location of the tumor. Thus, an anticancer drug(s) is the primary treatment option. The success of each drug largely depends on efficient delivery to the tumor site, ideally with as little offsite toxicity as possible. Not surprisingly, even the best clinical chemotherapy agents are ineffective if they are unable to reach their intended site of action. For example, a drug given intravenously will concentrate in areas with the best blood supply. However, in solid tumors, sufficient blood supply is restricted to the periphery, and 90% of cancer cells receive little or no drug.2 While this may result in an initial rapid reduction of the tumor volume, the death of only the outer cells often leads to continued inner growth, and eventually an increase in tumor volume as cancer cells become drug-resistant or tumor growth outpaces cell death.
Hypoxia (reduced oxygen partial pressure) further adds to the conundrum of solid tumors. Koong and colleagues were the first to measure the oxygenation of pancreatic tumors in humans. Examining seven different human patient samples, they found extreme hypoxia in pancreatic tumor tissues. The partial pressure of oxygen (pO2) in these tumors ranged from 0 to 5.3 mm Hg, while a normal pancreas range is 9.3 to 92.7 mm Hg. Multiple tumors showed a pO2 of less than 2.5 mm Hg, indicating significant levels of hypoxia.3 Studies in glioblastoma, sarcomas, head and neck carcinomas, breast, cervix, and prostate cancers have shown similar results for solid tumors.4–10 Cancer cells can adapt to this hostile microenvironment through the transcriptional activity of hypoxia-inducible factors (HIF1 and HIF2). Hypoxia, along with HIFs, play intricate roles in regulating the expression of multiple genes involved in glucose metabolism, angiogenesis, cell invasion, and metastasis, which can significantly increase risk of cancer mortality.11–15
The tumor’s microenvironment plays a crucial role in pancreatic cancer. Many cell types including fibroblasts, adipocytes, endothelial cells, and others, create the heterogeneity in tumors.16–18 This heterogeneity leads to a unique environment with significant fibrosis and a dense extracellular matrix (desmoplasia). Cancer stem cells (CSCs) or tumor-initiating cells are present within this microenvironment and possess the capacity to self-renew. While often representing less than 1% of total cells, the CSCs significantly contribute to chemotherapy resistance, metastasis, and relapse.19–21 The signal transducer and activator of transcription (STAT3) gene encodes a transcription factor pivotal in stem cell self-renewal. Persistent activation of STAT3 is observed in multiple gastrointestinal cancers including hepatocellular, pancreas, and colon carcinomas.22–24 Evidence shows that STAT3 interacts with CD44, upregulates NANOG, and induces CSC like properties in cancer cells.25–28
Pancreatic cancer cells overexpress the neuropilin-1 receptor.29 We recently demonstrated that pancreatic CSCs also overexpress this receptor.30 The small circular peptide iRGD (named for its specific amino acid sequence and internalizing property) has been reported as a tumor-targeting and penetrating ligand.31 Recent advances have used this RGD sequence as a diagnostic tool along with PET/CT scanning for tumor detection and conjugation for oncolytic virotherapy.32,33 The iRGD peptide binds to αvβ3 and αvβ5 integrins on the cell surface.34–36 Subsequently, proteolytic enzymes cleave the iRGD peptide, altering its specificity towards the neuropilin-1 receptor. This binding induces a transcytosis mechanism, allowing increased tissue penetration and cargo delivery deep into the tumors.31,37,38 The increased expression of these receptors on pancreatic cancer cells allows for active targeting of our polymersomes, followed by tumor penetration.
These hypoxia-induced changes, resilient CSCs, and the lack of sufficient vasculature necessitate the need for deep-penetrating drug carriers that target the hypoxic niches of solid tumors. Herein, we report a tumor-penetrating, hypoxia-responsive, polymeric carrier (polymersome), which releases encapsulated drugs in the reducing microenvironment of the hypoxic-niches inside solid pancreatic tumors. Previous use of these hypoxia-responsive polymersomes in our laboratory using gemcitabine and an epidermal growth factor receptor inhibitor (erlotinib) showed increased response to treatment in spheroidal cultures of BxPC-3 pancreatic cancer cells.39 Here, we encapsulated a first-line chemotherapy treatment option for pancreatic cancer (gemcitabine) along with a drug currently in clinical trials as a STAT3 inhibitor (napabucasin). Napabucasin impairs tumor self-renewal by CSC as well as induce apoptosis in this cellular subset in a variety of solid tumors.40–43 We selected a drug with well-documented clinical efficacy in pancreatic cancer (gemcitabine) that effects all malignant cells and napabucasin, which has increased toxicity towards resilient CSCs. We hypothesized a more significant effect on tumor suppression and prevention of tumor self-renewal from inside the tumor core.42,44
Materials and Methods
Materials.
The amino acids and resin for peptide synthesis were purchased from Peptides International. Polymers for the synthesis of diblock copolymers were purchased from Biochempeg. The fluorescent lipid 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl ammonium salt was purchased from Avanti Polar Lipids. Pancreatic cancer cell line (BxPC-3) was obtained from ATCC and cell culture media, serum, and antibiotics from VWR International. The mice were purchased from Envigo.
Synthesis and Characterization of Hypoxia Responsive Polymer PEG-azobenzene-PLA.
Polymer m-PEG2000–NH2 was conjugated to azobenzene-4,4′-dicarboxylic acid by following a previously published protocol.45 The PEG–diphenylazacarboxylate (50 mg, 0.023 mmol) was dissolved in pyridine (1.25 mL). To this solution, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC.HCl; 6.7 mg, 0.0345 mmol) and N-hydroxysuccinimide (NHS; 4 mg, 0.0345 mmol) were added followed by excess 3-aminopropanol (8.75 mg, 0.116 mmol). The reaction mixture was stirred at room temperature overnight. The solvent was then evaporated under reduced pressure. The residue obtained was dissolved in dichloromethane (10 mL) and washed with water three times, the bottom organic layer was dried under vacuum, yielding 34 mg (67%) of a yellow solid product. 1H NMR (400 MHz, chloroform-d) δ ppm: 8.00-8.23(CH=CH-CH, m, 8 H), 3.67 ((CH2-CH2-O), t, 4 H), 3.40 ((CH3-O), s, 3 H), 0.88 ((NH-CH2-CH2-CH2-OH), m, 2 H).
Synthesis of the block copolymer.
The product obtained from the previous step (100 mg, 0.05 mmol), D, L-lactide (500 mg, 3.5mmol) and tin(II) ethoxyhexanoate (3 μL, 0.009 mmol) were added to anhydrous toluene (5 mL) in a 35 mL glass high-pressure vessel, then purged with nitrogen gas. The solution was stirred at 120° C under nitrogen for 24 hours. After cooling to room temperature, the reaction mixture was added dropwise to cold ether. The top clear supernatant was decanted, and the precipitate was rewashed with ether, dried under vacuum. The orange solid product obtained (305 mg, 51%) was analyzed by FTIR, 1H NMR, 13C spectroscopy and gel permeation chromatography. 1H NMR (400 MHz, chloroform-d) δ ppm: 5.19 ((-CH-C=O), q, 1 H), 3.67 ((CH2-CH2-O), t, 4 H), 1.59 ((CH3-CH-C=O), d, 3 H) (Figure S2–S5).
Determination of Copolymer Composition.
The repeating monomer number m of PLA was also estimated from 1H NMR by comparing (-CH-C=O) (δ ppm = 5.19 from the PLA block) to –(OCH2CH2)n– (δ ppm = 3.67 from the PEG block), m is the number of protons of -CH-C=O; 4n is the number of protons of –(OCH2CH2)n–
m/4n = a1/a2 : a1 = area of peak at 5.19 ppm a2 = area of peak at 3.67 ppm
Mw of PEG = 2000, n = 46, 4n = 184, then m = 188a1/a2, then Mw (PLA) = 5900.
Synthesis and Characterization of the iRGD Peptide.
Microwave-assisted, solid-phase peptide synthesis was carried out using a Liberty Blue (CEM Corporation) synthesizer. The resin used was a Rink amide (purchased from CEM Corporation). The sequence hexynoic acid-Cys(Acm)-Arg(Pbf)-Gly-Asp(OBtu)-Lys(Boc)-Gly-Pro-Asp(OBtu)-Cys(Acm)-OH was synthesized without the final deprotection step. To cyclize, 0.1 mmol thallium trifluoroacetate in DMF (5 mL) was stirred with the peptide-resin conjugate for 1 hour. The resin was then washed with DMF and dichloromethane 3 times. Next, the peptide was cleaved from the resin using trifluoroacetic acid (19 mL), distilled water, (0.5 mL), and triisopropylsilane (0.5 mL) for 2 hours. Whatman Grade 1 qualitative filter paper was used to collect the peptide in a 50 mL centrifuge tube to which 30 mL of ice-cold diethyl ether was added. The precipitate was collected and dried in a vacuum desiccator overnight. The dried product was then characterized by MALDI-ToF mass spectrometry (observed mass: 1041.42, expected mass: 1042.43, elemental composition C41H64N14O14S2, Figure S6)
Synthesis of iRGD Peptide–Polymer Conjugate.
The polymer PLA-PEG-N3 was reacted with the alkyne (hexynoic acid) moiety of the iRGD peptide using Click chemistry (1:2 molar ratio peptide to polymer). The copper complex was made by mixing copper (II) sulfate with N, N, N’, N’, N”–pentamethyl diethylenetriamine (PMDETA) for 2 hours. Ascorbic acid solution (1.4 μmol) was prepared in distilled water. The reaction mixture was then stirred for 24 hours at room temperature. The mixed solution was then transferred to a 1000 kD dialysis bag and dialyzed against water for 72 hours to remove the catalyst (PMDETA and ascorbic acid) as well as unreacted iRGD peptide. The product was then analyzed by CD spectroscopy (Figure S7).
Preparation of Polymersomes.
PEG-azobenzene-PLA and iRGD polymer conjugate were dissolved (10 mg/mL) in acetone, and lissamine rhodamine lipid (0.01 mg/mL) in chloroform. First, the lissamine rhodamine lipid was added to a clean glass vial and air-dried. Next, polymers were added dropwise in an 85:10:5 ratio of PEG-azobenzene-PLA, iRGD-PEG-PLA, and lissamine respectively to HEPES buffer (10 mM and pH 7.4). The resulting mixture was stirred for 1 hour, bubbled air for 45 minutes (to evaporate acetone), and then sonicated in a water bath sonicator (VWR Symphony ultrasonic cleaner, Model: 97043-936) for 1 hour at 35 kHz. The polymersomes were then passed through a Sephadex G100 size exclusion column. The eluted polymersomes were collected and used for the studies. The encapsulation of drugs was achieved by passive or active loading. For passive loading, napabucasin was added to the polymer mixture and added to the HEPES buffer. For active loading, we utilized a citrate buffer (100 mM and pH 4) instead of HEPES. Once collected through the size exclusion column, gemcitabine was added, then pH was adjusted to 8 by adding a 1 M sodium bicarbonate solution dropwise and testing the pH. This mixture was then stirred for 8 hours before passing through the size exclusion column again to remove the unencapsulated drug.
Determination of critical aggregation concentration.
From a stock solution of 0.1 mM pyrene in dichloromethane, 10 μL aliquots were taken in different vials, and the dichloromethane was allowed to evaporate in air. To each of these vials, various measured amounts of the polymer were added (stock solution concentration 10 mg/mL in a 1:5 acetone: deionized water solution by volume) so that the concentrations varied from 5 mg/mL to 9.8 μg/mL, and the final concentration of pyrene in each vial was 1 μM. The vials were sonicated at 35 kHz using a VWR Symphony ultrasonic cleaner (Model: 97043-936) for 45 minutes and then allowed to stand for 2 hours before recording the fluorescence spectra. The fluorescence emission spectra were recorded at an excitation wavelength of 337 nm with a bandwidth of 2.5 nm (for both excitation and emission). The ratio of the intensities at 373 nm and 393 nm were plotted against the concentration of the polymer, and the inflection point of the curve was used to determine the critical aggregation concentration (Figure S2).46,47
Size Analysis of the polymersomes.
The hydrodynamic diameter of the polymersomes was measured using dynamic light scattering, using a Malvern Zetasizer instrument and Zetasizer software version 7.02. One mL of the sample was placed in a Sarstedt Cuvette. The sample was given 120 second equilibration time followed by three measurements, each with ten runs. Atomic force microscopy was carried out in noncontact mode at a scanning rate of 0.7 Hz and a resonance frequency of 145 kHz using an NT-MDT NTEGRA (NT-MDT America). The cantilever was made of silicon nitride and was 100 μm long. Scanning area was 5 x 5 or 20 x 20 μm2 at a resolution of 512 or 1024 points per line, respectively. For electron microscopic imaging, copper TEM grids (300-mesh, Formvar-carbon coated, Electron Microscopy Sciences) was prepared by applying a drop of 0.01% poly(l-lysine), allowing it to stand for 30 s, wicking off the liquid with torn filter paper, and allowing the grids to air-dry. A drop of the suspension diluted 1:100 was placed on a prepared grid for 30 s, and wicked off; grids were allowed to air-dry again. Phosphotungstic acid, 0.1% pH adjusted to 7–8, was dropped onto the grid containing the sample, allowed to stand for 2 min, and wicked off. After the grids had dried, images were obtained using a JEOL JEM-2100 LaB6 transmission electron microscope (JEOL USA, Peabody, Massachusetts) running at 200 keV.
Encapsulation of 5(6)-carboxyfluorescein.
5(6)-Carboxyfluorescein was encapsulated by the following procedure. Briefly, 10 mg of the polymer and 1 mg of carboxyfluorescein were dissolved in 250 μL DMSO and was then added dropwise to a 750 μL PBS buffer solution under magnetic stirring. This was left stirring for an hour at room temperature followed by dialysis (MWCO 1-1. 5 kDa) against 800 mL PBS buffer with regular media change till no further discoloration of the media was observed. After the release experiments, 20 μL of Triton was added to disintegrate the polymersome and the fluorescence emission intensity was measured for total release after disintegration. The cumulative percentage release was plotted versus elapsed time. Percentage release was calculated using the following equation:
In-Vitro Uptake and Internalization.
Polymersomes were prepared using our standard protocol, but in place of lissamine rhodamine B lipid, a FITC-PEG-PLA polymer was used at 5% concentration. Uptake was analyzed using an Accuri C6 flow cytometer with subsequent data analysis using FlowJo software (FlowJo, LLC).
Cell Viability Studies in Monolayer Culture.
BxPC-3 cells (5,000) were seeded in each well of 96-well clear bottom plate. The cells were allowed 24 hours for attachment before placing them in either a normal oxygen incubator (20% oxygen) or a hypoxia chamber (2% oxygen). The cells then grew for 24 hours before being subjected to their respective treatments. After 3 days, the media was removed, and cells were washed three times to remove any remaining polymersomes or drugs. Subsequently, 20 μL of Alamar Blue (Invitrogen, a cell health indicator that uses the reducing power of living cells to measure viability quantitatively) and 180 μL of fresh medium was added. The fluorescence was then measured, and viability was calculated.
Cell Viability Study in Spheroid Cultures.
Spheroid scaffolds were prepared by adding agarose to a silicone mold (Microtissues) following the manufacturer’s protocol using 96,000 cells/75 μL to produce a spheroid diameter of 200 μm. The seeded scaffolds were incubated for 7 days, changing the standard cell culture media every 2 days. The scaffolds were then placed in either normoxic (20%) or hypoxic (2%) conditions for 24 hours before being exposed to their respective treatments for 72 hours. After treatment, the scaffolds were washed with phosphate-buffered saline before viability was analyzed by Celltiter-Glo 3D cell viability assay (Promega).
Animals.
All mice were housed under standard housing conditions at the Animal Studies Core Facility of North Dakota State University (NDSU). All animal procedures were reviewed and approved by the Institute of Animal Care and Use Committee at the NDSU, protocol number A18037.
Orthotopic Tumor Implantation.
Mice were first anesthetized by isoflurane (3% in 1 L/min 100% oxygen for induction and 2% in 1 L/min oxygen for maintenance). A cell suspension of 106 BxPC-3 cells in 25 μL sterile saline was injected into the pancreas using a 28G needle. The peritoneum was sutured back together using Ethicon Chromic Gut dissolvable sutures. The skin was then sutured with an Ethicon nylon suture. A topical antibiotic and tissue glue were used over the wound to prevent infection and help close the wound. The mice then received buprenorphine subcutaneous injection in the scruff of the neck for post-surgical pain. Mice were given welfare checks daily post-surgery.
Ultrasound and Photoacoustic Imaging of Tumor Volume and Oxygenation.
A VevoLAZR system equipped with LZ250 linear array transducer (VISUALSONICS) was used to measure tumor size, and tumor oxygenation status (oxygenated/deoxygenated hemoglobin) A tunable laser with a pulse repetition rate of 10 Hz was used to excite the sample from 680-970 nm. Images were obtained under identical conditions using Vevo’s 3D ultrasound imaging (B-mode) and Photoacoustics (PA) modes, respectively. Subsequent PA measurements of oxygenated/deoxygenated hemoglobin (750/850 nm absorption, respectively) were obtained in triplicate from the tumor core, edge, and associated healthy adjacent pancreas and subsequently quantified using Vevo’s OxyZated analysis software.
In-Vivo Biodistribution.
Athymic nude mice were used to establish subcutaneous tumors. Tumor-bearing mice were injected with iRGD containing polymersomes and imaged using a Kodak In-Vivo Multispectral Imaging System FX after two hours. Organ images were taken from mice lacking subcutaneous tumors. The mice were injected with the iRGD polymersome formulation via tail vein and were then euthanized at their respective time points (5 minutes, 2 hours, and 6 hours) and imaged using the Kodak camera at excitation 590 and emission 615.
In-Vivo Circulation Time.
iRGD polymersomes encapsulated with 12 μM ICG were injected (200 μL) via tail vein into 3 nude athymic mice. Blood draws (200 μL) were taken at 1 hour, 24 hours, and 72 hours post-injection. The fluorescence was measured at excitation 820 and emission 850. The total remaining ICG was estimated by extrapolating the signal from 200 μL of blood to the total blood volume of the mouse using the equation total blood volume = 58.5 mL/kg.48 A standard curve was made using various amounts of ICG and percent remaining was calculated use the initial 200 μL injection as a reference signal.49
In-Vivo Animal Study.
Six-week-old nude athymic mice were purchased from Envigo. BxPC-3 cells (1.5 x 106) were suspended in Matrigel and injected in the right flank of the mice. Once tumors had reached a significant size (greater than 30 mm3) as measured by calipers, the 36 mice were randomly allocated to each of the six treatment groups with three males and three females within each group. The control group received 200 μL of sterile saline, while the drug treatment groups received gemcitabine at 30 mg/kg and/or napabucasin at 20 mg/kg. Injections were given via the tail vein using a 26G needle twice weekly for four weeks. Mice were weighed, and tumor volume measured using calipers before each injection. One week of observation continued after the treatment ended, and tumor volume and weight were again measured twice during this time.
Measurement by Caliper.
The estimated tumor volume was measured using an external caliper. The greatest length and greatest width were selected and measured (mm). The total tumor volume was calculated by the modified ellipsoidal formula.50,51
Human Pancreatic Cancer Samples.
All human tissue samples were obtained from the University of Nebraska Medical Center (UNMC) Rapid Autopsy pancreatic program and associated UNMC tissue bank.
Western Blot.
The normal pancreas, pancreatic cancer tissue, and metastatic tumor tissue (5 mg each) were exposed to 300 μL ice-cold RIPA lysis buffer containing 0.1% Sodium dodecyl sulfate (SDS) and protease inhibitor and were homogenized on ice for two hours. Then, protein contents were collected by centrifuging tissue samples at 13,000 g for 20 min at 4 °C. The supernatant was collected, and protein concentration in each sample was measured using the Bio-Rad DC protein assay kit. Protein samples were separated by SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked by 5% non-fat milk for 2 hours, washed 3 times, and then incubated with Neuropilin-1, STAT-3, and GAPDH primary antibodies (1:1000 dilution) overnight. Subsequently, the membrane was incubated with a secondary antibody (1:1000 dilution) for two hours. The signals on the membrane were developed by ECL reagent (Pierce, Rockford, IL) and exposed to X-ray films. The quantitative density of the bands for Neuropilin-1/GAPDH and STAT-3/GAPDH ratios was analyzed by NIH ImageJ software.
Results.
Synthesis of hypoxia-responsive copolymers (PLA-Azo-PEG), tumor-penetrating peptide-polymer conjugate (PLA-PEG-iRGD), and preparation of tumor penetrating polymersomes.
In our polymersome design, a 4,4’-diazobenzene is used for linking the hydrophilic polyethylene glycol (PEG) and the hydrophobic polylactic acid (PLA) of the diblock copolymer (Figure 1A). The PEG to whole polymer ratio is 25%. Bilayer vesicles (polymersomes) are formed when the ratio of PEG to the whole polymer ranges from 20-40%.52,53 Micelles would form if using a higher ratio (45-70%) PEG.52,53 Biological reducing environments are amplified under hypoxic conditions.54,55 The addition of the reduction-sensitive 4,4’-diazobenzene functional group allows for the triggered release of the encapsulated payload in hypoxic regions.39,56 Both PEG and PLA polymer blocks are approved for human use by the US Food and Drug Administration (FDA) and are classified into the “generally regarded as safe” category. We recrystallized commercially available DL-lactide and carefully controlled the reaction conditions to reduce the polydispersity of the polymers. We synthesized the copolymer by ring-opening polymerization of DL-lactide using Sn(octanoate)2 as the catalyst. Hence, the resultant copolymer is racemic. The purified hypoxia-responsive polymer PLA600–diazobenzene–PEG2000 was characterized by infrared, 1H, and 13C NMR spectroscopy and gel-permeation chromatography (GPC, Supporting Information). Analysis of the GPC data showed a polydispersity index of 1.12 for the hypoxia-responsive copolymer.
Figure 1.

Polymersome components. (A) diblock copolymer consisting of polyethylene glycol linked to polylactic acid by the hypoxia-sensitive 4,4’-diazobenzene linker (red), and (B) iRGD peptide conjugated to the PLA-PEG copolymer.
The synthesis of the PLA–PEG–iRGD polymer (Figure 1B) was a two-step process. First, we synthesized PLA6000–PEG2000–N3 polymer following our previously reported protocol.57 Next, we conjugated the synthesized hexynoic acid-iRGD to PLA6000–PEG2000–N3 using the Cu2+-catalyzed Click chemistry ([2+3]-cycloaddition). Further experimental details can be found in the Materials and Methods section and characterization by MALDI-TOF and circular dichroism in the Supporting Information (Figure S6–7). We determined the critical aggregation concentration of the synthesized hypoxia-responsive polymer to be 0.86 ± 0.05 mg/mL (107 ± 6 μM) employing fluorescence spectroscopy of added pyrene (Figure S1).46,47 We prepared the polymersomes from the hypoxia-responsive polymer (85%), the PLA-PEG-iRGD polymer (10%), and a fluorescent reporter, 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-lissamine-rhodamine B sulfonyl ammonium salt (5%) employing the reported solvent-exchange method.58
The effectiveness of a nanoparticle formulation can be dramatically changed by its characteristics.59,60 We determined the size and shape of the polymersomes by transmission electron microscopy (TEM), atomic force microscopy (AFM), and dynamic light scattering (DLS). The enhanced permeability and retention effect, which arises due to leaky vasculature, allows nanoparticles less than 200 nm to rapidly accumulate at the tumor site.61–63 Based on our characterization, our nanoparticle formulations should be able to exploit this leaky vasculature and reach the tumor site.
In addition to TEM and AFM (Figures 2A, B, C) images, the hydrodynamic diameter, polydispersity index, and zeta-potential of the polymersomes were determined by dynamic light scattering (Table S1). The average size of 107, 117, and 188 nm for control polymersomes (containing iRGD and HEPES buffer), iGem (gemcitabine encapsulated iRGD polymersomes), and iNap (napabucasin encapsulated iRGD polymersomes), respectively were measured. Entrapment efficiency and drug loading content were also calculated for each formulation (Table S1). Gemcitabine is a hydrophilic drug and resides in the aqueous core of the polymersomes. Napabucasin, a hydrophobic molecule, partitions into the bilayer of the polymer vesicles, causing an increase in vesicle size. These polymersomes fit into a size window where they are large enough to escape renal filtration (>10 nm), yet, small enough (<200 nm) to potentially avoid significant uptake by mononuclear phagocytes and the reticuloendothelial system.64–67 The exact pharmacodynamics and pharmacokinetics of this formulation are outside the scope of this study and are reserved for future investigation. Next, we demonstrated the polymersomes’ ability to release the encapsulated contents under hypoxic conditions (2% oxygen). A fluorescent dye, carboxyfluorescein, was encapsulated into the polymersomes as a model for the hydrophilic gemcitabine. We used human liver microsomes that contain cytochrome P450 enzymes. Reduction of the azobenzene linker requires a hydrogen donor source. The microsomes act to oxidize the NADPH, which, in turn, provides the hydrogens needed to reduce the azobenzene group to two aniline molecules (Scheme S1).68 A hypoxia chamber set to 2% oxygen was to mimic hypoxic conditions further.39 The release profile was monitored over time by measuring the fluorescence intensity of the solution. Our polymersome formulation released 81% (± 3) of its contents under hypoxic conditions compared to only 21% (± 2) under normoxia after 60 minutes (Figure 2D). A similar strategy, but using HPLC was used to measure the release of napabucasin which showed 86% (± 7) release over 60 minutes.
Figure 2.

Characterization of polymersomes. (A-C) Hypoxia-responsive iRGD peptide conjugated polymersomes’ size and shape, as shown by transmission electron microscopy (A) and atomic force microscopy under normoxic (B) and hypoxic conditions (C), respectively. (D) Carboxyfluorescein release under hypoxic and normoxic conditions with NADPH and liver microsomes. (E) Napabucasin Release under hypoxic and normoxic conditions with NADPH and liver microsomes.
Scheme 1.
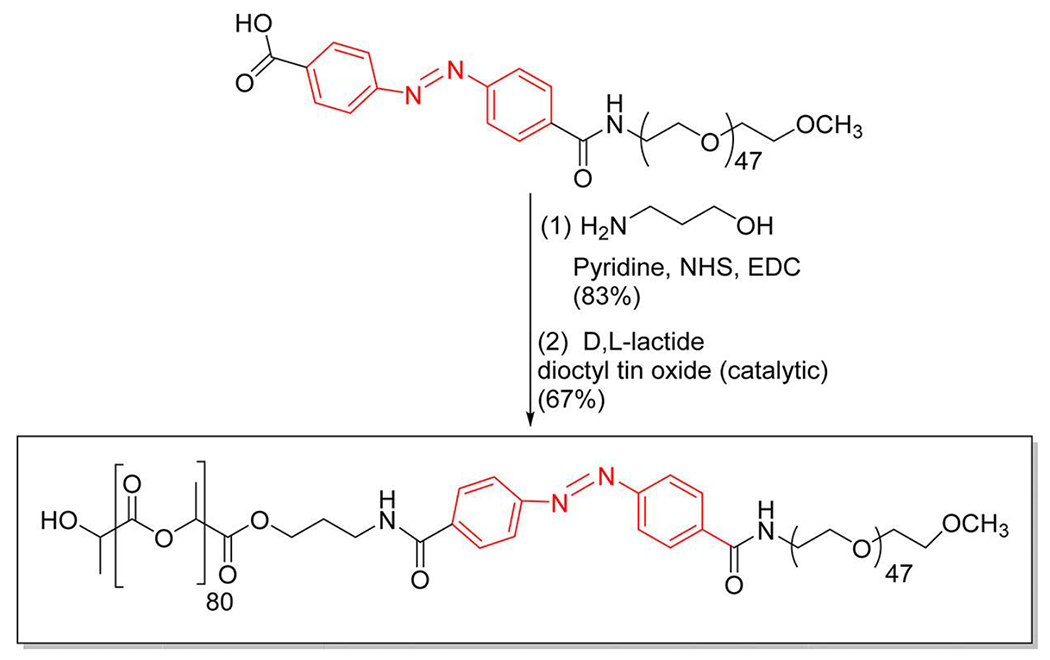
Synthesis of the hypoxia-responsive copolymer.
The stability of polymersomes was investigated by incubation in human serum under normoxia. The polymersomes average size increased by about 20 nm within 48 hours of incubation with serum and they then maintained that average size for up to 120 hours (Figure S8). The increase in size is likely due to adhesion of molecules within the serum to the polymersomes. Further size monitoring under hypoxic conditions was conducted using glutathione as a reducing agent. Concentrations of 50 μM simulate the extracellular tumor environment, while 1 mM mimics those concentrations within the cytosol.69 While under 50 μM, the polymersomes remain mostly intact as indicated by a lack of change in size. The 1 mM concentration breaks the azobenzene linker causing a significant reduction in size as broken polymers attempt to reform (Figure S9).
In-Vitro Cytotoxicity.
Cytotoxicity was measured in monolayer cultures of BxPC-3 pancreatic cancer cells (Figure 3A). Previous studies demonstrated that hypoxia induces biochemical and genetic changes in pancreatic cancer cells resulting in a more gemcitabine-resistant phenotype. 13,14,70–72 We observed that 73% (± 4) of cancer cells cultured in a hypoxic environment were viable after treatment with free gemcitabine compared to 48% (±3) viability after encapsulated (iGem) treatment. A similar result was observed with napabucasin that showed 74% (± 4) and 52% (± 3) viability between unencapsulated free drug and the encapsulated polymersome formulation, respectively. iRGD polymersomes (iGem, iNap, and the drug combination iPsome) were able to either increase cellular toxicity or mitigate the resistant phenotype developed under hypoxia, possibly due to increased uptake of the vesicles, resulting in higher drug concentrations inside the cells (Figure 3C). Next, we tested iRGD polymersomes using a three-dimensional (3D) model allowing gradients in oxygen, nutrients, and metabolites, creating a more heterogeneous population along with physiological cell-cell interactions.50, 51 Cell spheroids were prepared using 3D Petri Dish® from Microtissues®. We then tested free Gem and free Nap against the iRGD polymersomes encapsulating these drugs. In normoxia, we observed free napabucasin having significant cellular toxicity. This is likely due to its ease of diffusion inside the spheroids due to small molecular size. A high chemoresistance (10-30% greater viability) under hypoxia was observed for the free drugs due to increased diffusion barriers preventing penetration of the drug.75,76 This chemoresistance was mitigated using the iRGD polymersomes (Figure 4A). Cell viability of 76% (± 4) and 71% (± 3) for unencapsulated free drugs gemcitabine and napabucasin, while the iRGD-polymersome formulations of these drugs were 60% (± 3) and 38% (± 4), respectively, after 72 hours treatment in hypoxic conditions. The polymersomes were likely better able to penetrate the dense spheroids as compared to the free drugs leading to an overall decreased cell viability. This possibility was previously examined by our laboratory, using an ex-vivo penetration apparatus that showed a penetration depth of 2.2 mm for iRGD-polymersomes.56
Figure 3.
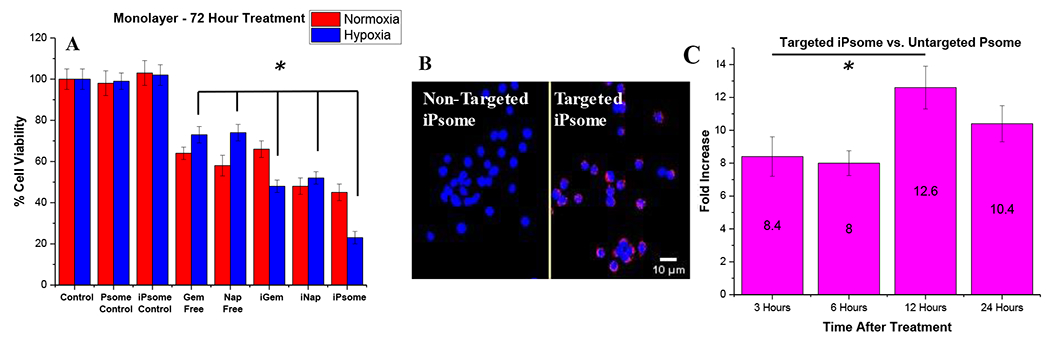
In vitro monolayer studies on cell viability. (A) Viability of BxPC-3 cells in monolayer cultures under normoxic and hypoxic (2% O2) conditions. Control treatments lack encapsulated drugs (n = 8). (B) Cellular uptake after 2 hours of iRGD conjugated hypoxia-responsive polymersomes in monolayer cultures of BxPC-3 cells. Control represents polymersomes without iRGD. (C) Cellular uptake using a FITC conjugated polymer and flow cytometry, showing fold increase in cellular uptake at selected time points (n = 3). * p-value <0.05.
Figure 4.
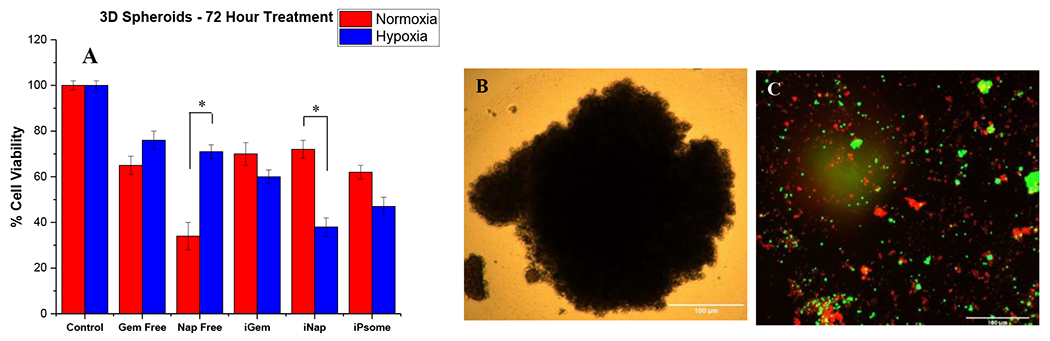
In vitro 3D spheroids. (A) Viability of BxPC-3 cells in 3D spheroid cultures under normoxic and hypoxic (2% O2) conditions. (B) Image of a single spheroid pre-treatment. (C) Spheroid after 72-hour treatment with iRGD-polymersomes, using live (green) and dead (red) staining. * p-value <0.05, n = 8.
In-Vitro Uptake and Internalization.
The increased cellular toxicity of the drug-encapsulated polymersome formulations is likely due to a higher accumulation of the drugs inside the cancer cells. To validate this hypothesis, uptake and internalization of fluorescently labeled polymersomes (incorporating a FITC-PEG-PLA polymer) into pancreatic cancer cells (BxPC-3) was studied using flow cytometry. The iRGD-targeted polymersomes showed 12-fold greater uptake after 12 hours of incubation with the BxPC-3 cells compared to the non-targeted polymersomes lacking the iRGD peptide (Figure 3C). A qualitative fluorescence microscopy experiment supported the significant difference in uptake of the iRGD-polymersomes in the pancreatic cancer cells compared to the non-targeted (no iRGD peptide) polymersomes after 6 hours of incubation (Figure 3B). The BxPC-3 cells are reported to overexpress the neuropilin-1 receptor on the surface.77,78 The macropinocytosis mechanism induced by NRP-1 activation produces vacuoles in the range of 0.2 – 3 μm in size that engulf the surrounding/bound polymersomes.79 The enhanced uptake is likely due to the integrin and neuropilin-1 receptor targeting by the iRGD peptide.
Bio-distribution and accumulation of the polymersomes.
One of the crucial properties of drug delivery vehicles is a longer retention time in the circulation. To evaluate the circulation time of the iRGD-polymersomes, we injected fluorescently labeled iRGD-polymersomes into non-tumor bearing NOD skid gamma (NSG) mice. The iRGD-polymersomes encapsulating the near-infrared dye indocyanine green (ICG; 10 μM, excitation: 820 nm; emission 850 nm) suspended in 200 μL of phosphate-buffered saline were injected in the mice through the tail vein.49 Blood (100 μL) was drawn at various time points (1 h, 24 h, 72 h), and the fluorescence intensity of indocyanine green was measured at excitation 820 nm and emission 850 nm. A right shift in excitation and emission maximum was observed compared to unencapsulated ICG (780/810, respectively). We observed that 20% of the injected ICG signal remained after 72 hours (Figure 5B). The PEG polymer on the exterior of the polymersomes provides the stealth needed to delay uptake of the iRGD-polymersomes by the reticuloendothelial system.80 While we lack the ability for standard pharmacokinetic testing using clearance and volume of distribution, the circulation times we observed put these nanoparticles in the range of other FDA approved PEGylated nanoparticles with half-lives from 20 hours to 5 days.80–85 Further studies are needed to confirm this theory.
Figure 5.
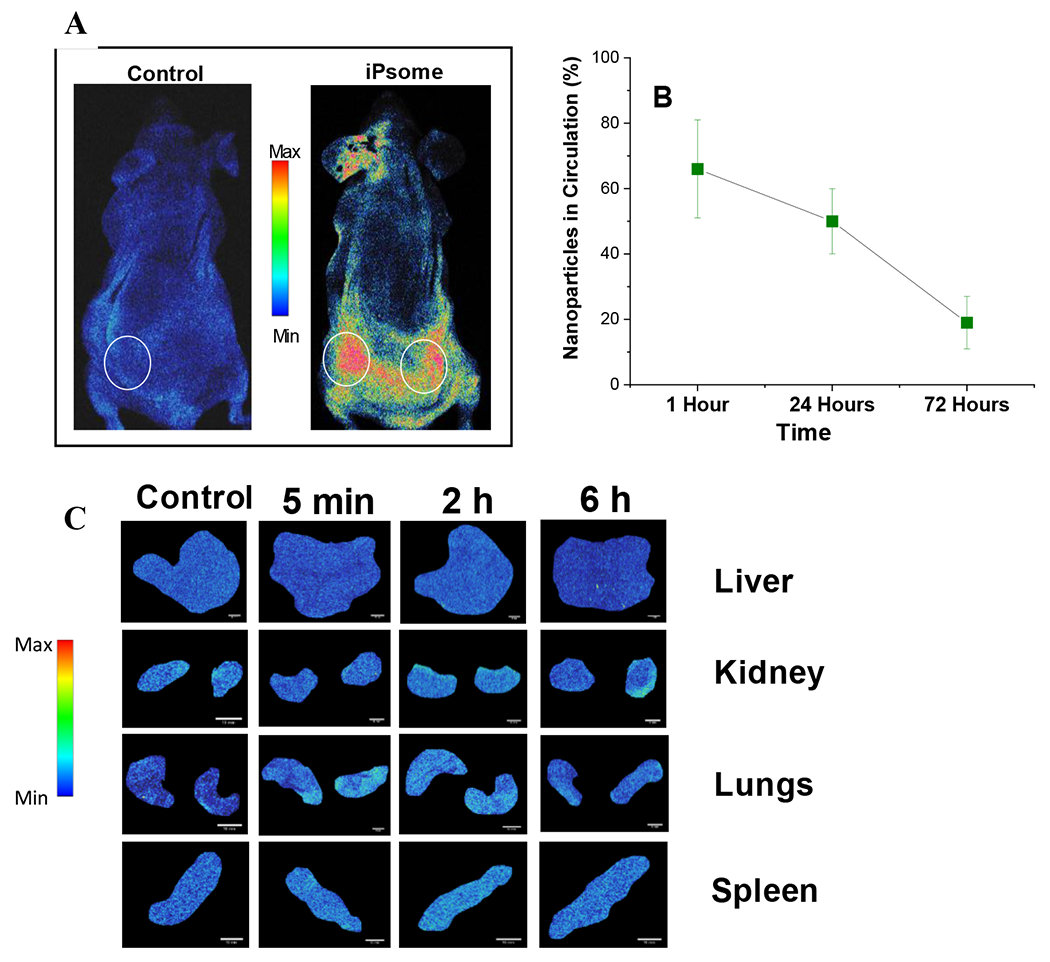
In-Vivo Bio-Distribution. (A) Mice with subcutaneous tumors (circled in white) injected with iRGD-polymersomes and imaged after two hours showing accumulation at the tumor site. *Ear fluorescence from permanent marker used to label mouse. (B) Circulation time of iRGD-polymersomes based on the remaining fluorescence signal in the blood. (C) Biodistribution to organs at 5 minutes, 2 hours, and 6 hours after tail vein injection of iRGD-polymersomes containing indocyanine green dye. (n = 3) (scale bar = 10 mm).
To demonstrate tumor accumulation, BxPC-3 tumor-bearing mice were injected with iRGD-polymersomes, and live animal images were recorded. Images were acquired 2 hours after injecting (tail vein) either a control (saline) or iRGD-polymersomes (no encapsulated drugs). Even when a mouse had two flank tumors, we observed a high accumulation of the polymersomes in both tumors (Figure 5A). The organs were also examined for the accumulation of polymersomes at various time points (Figure 5C). We found no significant organ accumulation after either 2 hours or 6 hours post tail-vein injection (Figure S11).
Hemoglobin and Oxygenation Saturation in a Mouse Model of PDAC:
BxPC-3 cells (106) were injected into the pancreas of four athymic nude mice. Once tumors reached an average size of 500 mm3 (measured by calipers), ultrasound and photoacoustic imaging were performed to verify tumor size and oxygenation, respectively. Hemoglobin oxygenation was determined by averaging three measurements from three different sites (normal pancreas, tumor edge, and tumor core). The average percentage of saturated oxygen (sO2) was significantly different within these locations (F(2,9) = 7.85, p < 0.05, Figure 6A). Specifically, sO2 was significantly reduced in the tumor core, relative to both the tumor edge (p < 0.05) and normal adjacent pancreas (p < 0.01). Likewise, total hemoglobin (Figure 6B) was significantly different across locations (F(2,9) = 4.43, p < 0.05) with the tumor core showing significantly less hemoglobin than the tumor edge and normal adjacent pancreas (p < 0.05). Notably, these initial results resemble those observed in patient-derived PDAC tumors.3 The observed low levels of sO2 indicate the presence of a hypoxic environment facilitating drug release from the polymersomes.
Figure 6.
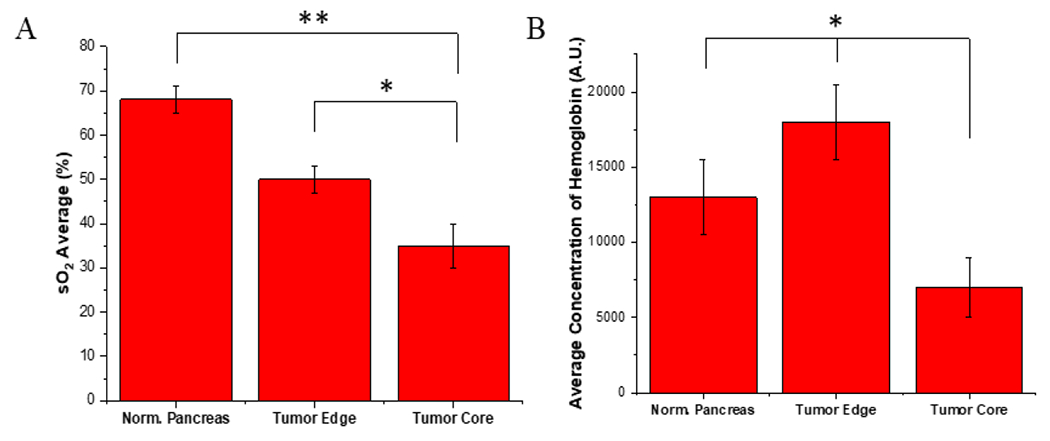
In-Vivo Saturated Oxygen and Hemoglobin. (A) Average saturated oxygen levels using photoacoustic imaging across all tumors. (B) The average concentration of hemoglobin across all tumors. * p-value <0.05, ** p-value <0.01, n = 4.
A pilot study was then performed on these mice to better understand if the unencapsulated drug or the polymersome is penetrating the tumor. A right shift in fluorescence of encapsulated ICG vs. free ICG allowed for unmixing of photoacoustic images and ability to see accumulation of the released dye and encapsulated dye simultaneously (Figure S10). After just 1-hour post tail vein injection, we observed penetration into the tumor. Due to the density of the tumor core, we were unable to visualize deeper polymersome penetration.
Efficacy of polymersomes in a subcutaneous mouse model of pancreatic cancer.
Athymic nude mice were injected with 106 BxPC-3 cells suspended in 200 μL of Matrigel® into the left flank, and the tumors were allowed to grow for four weeks. Three male and three female mice were randomly assigned to each treatment group. Gemcitabine was dosed at 30 mg/kg, napabucasin was dosed at 20 mg/kg either as free drugs or encapsulated into the polymersomes.86,87 The iPsome formulation was a volumetric mixture of both iGem and iNap. The amount of polymer injected with each dose varied slightly depending on the drug and entrapment of that batch of polymersomes. The average amount of polymer per injection ranged from 2.5 mg (iNap) to 5 mg (iGem). One week after the last treatment, we euthanized the mice and harvested the tumors. Significant suppression of tumor growth as compared to control and free drug treatments were observed for all iRGD polymersome groups. The end-stage tumor size was reduced by 242%, 254%, and 260% compared to no drug control for iGem, iNap, and iPsome, respectively (Figure 7A). Suppression of growth was also noted in the free drug groups of 113%, and 118% reduced size for free gemcitabine and free napabucasin, but this was not significant compared to control. Interestingly, we observed that the iRGD-treatments rendered the tumors physically soft compared to the solid and dense control tumors. The resected tumors showed sunken fluid-filled cores due to vastly enhanced necrosis of the tumors from inside. In a few instances, the inside necrosis was so much that the overall tumor resembled a donut-like structure. In general, the mice had no obvious treatment complications. Two mice from the iPsome group were removed from the study after two weeks due to a decrease in body weight of greater than 15% as well as one mouse from the control and free gem group that was removed due to excessive tumor size affecting ambulation. A graphical breakdown of weight changes overtime can be found in supporting information (Figures S12 and 13).
Figure 7.
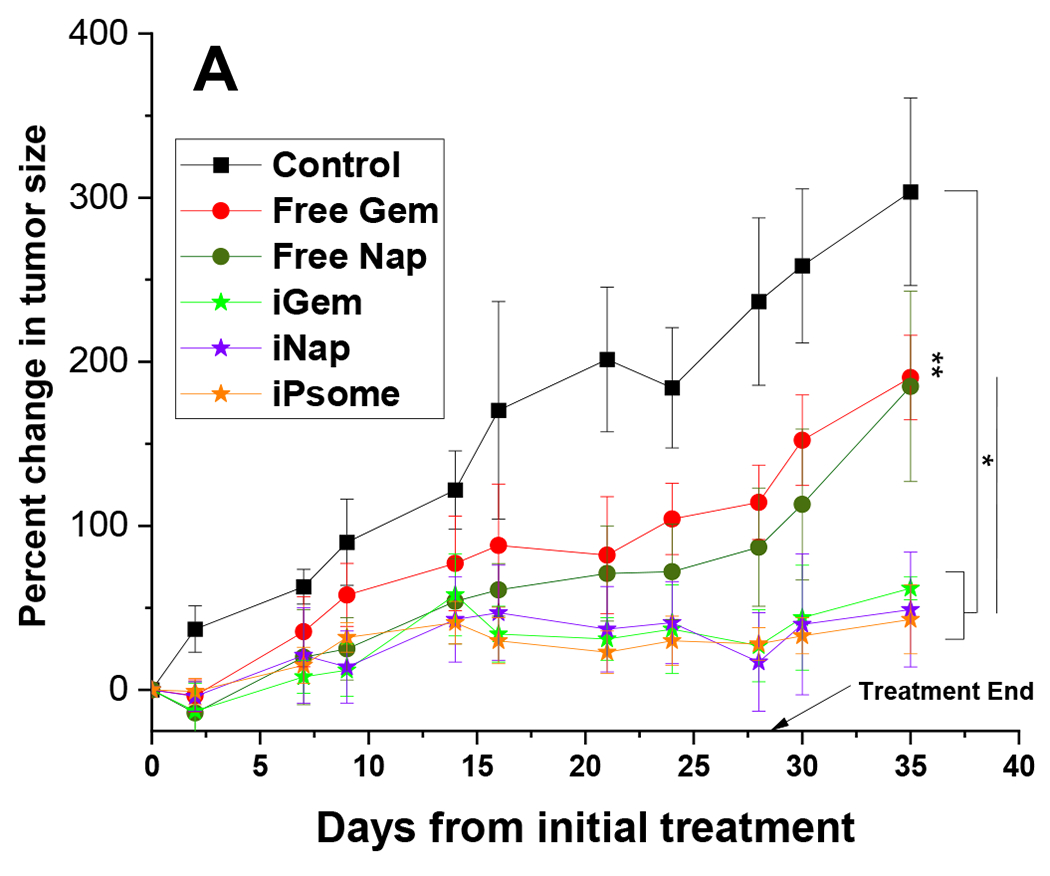
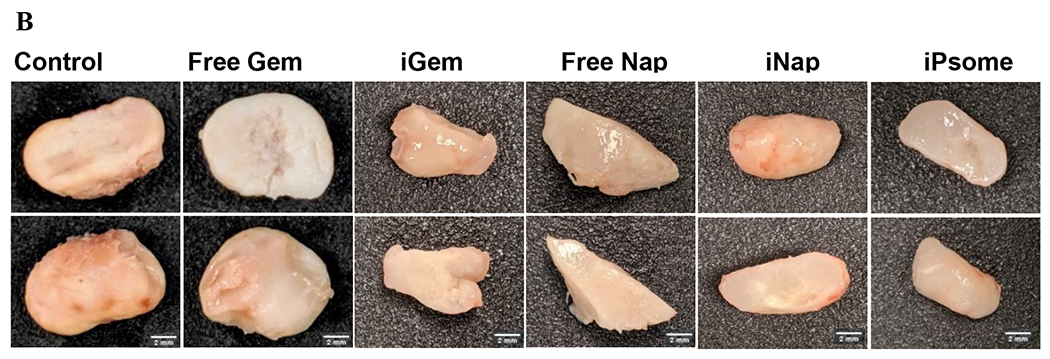
In-Vivo Animal Study. (A) Graphical representation of the change in tumor size throughout the treatment. A significant difference in the end-stage tumor size between control and all iPsome groups. (B) Excised tumors outer surface (bottom) and tumor core (top).
* p-value <0.05; N = 5 (Free Nap, Free Gem, iGem, iNap) (Control = 4, iPsome = 4)
Histology Analysis.
Tumors were collected after study (day 35), bisected, fixed, and embedded in paraffin wax. Tumor slices (10 μm thick) from the cut edge (tumor core) were placed on microscope slides, stained with hematoxylin and eosin, and analyzed using NIH ImageJ software. The area of necrosis was compared to the total area of tissue for that slide. The average area of necrosis was then compared between all treatment groups. The iRGD polymersome groups showed a significantly higher area of necrosis compared to control and free drug groups (Figure 8B), with 25% more necrosis seen in iGem and iNap groups compared to free drug. Visually, the necrotic areas can be seen in the scanned slides with a significant difference between free drug and the respective polymersome treatment group (Figure 8A).
Figure 8.
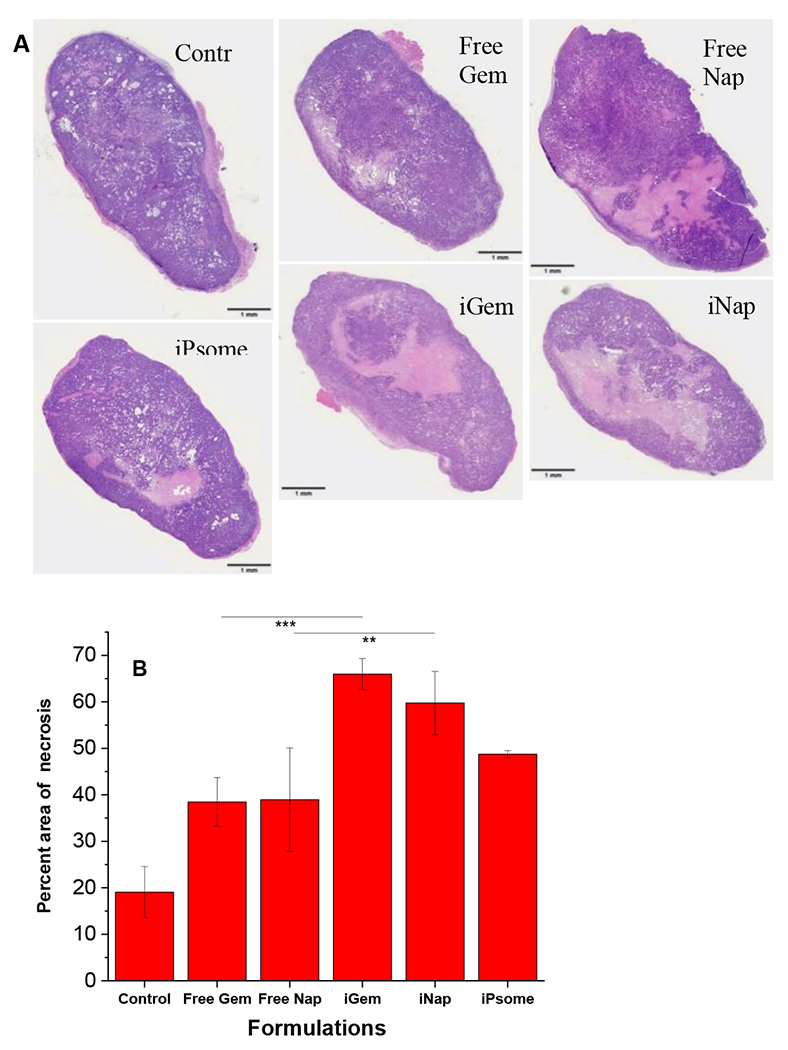
Tumor Core Necrosis. (A) Excised tumors were sliced in half, formalin-fixed, embedded in paraffin wax, and stained using hematoxylin and eosin. (B) Calculated and averaged total necrotic area for each tissue section. **p-value < 0.01 ***p-value <0.001
Human Pancreatic Cancer Samples.
To further assess the translational potential, we examined the tissue samples from two pancreatic cancer patients. Patient samples included a primary pancreatic tumor, unaffected pancreas tissue, and lung metastasis all from the same patient. Inflammation is a significant factor in the development of pancreatic cancer.88 The release of growth factors and cytokines by tumor associated immune cells in response to inflammation further promotes the activation of STAT3.89 We saw a 4-5 fold increased expression of both NRP-1 and STAT3 proteins in the primary tumor as well as the metastatic liver site (Figure 9). High expression of NRP-1 in PDAC is associated with other clinicopathologic characteristics such as lymph node involvement, advanced stage, and decreased survival time.90 New evidence shows NRP-1 promotes proliferation through activation of VEGF and EGF receptors and downstream activation of Akt leading to downregulation of p27.91 The p27 normally suppresses activity of the CDK2/cyclin E complex, which leads to DNA replication.92,93 When downregulated, p27 will not effectively suppress CDK2 and further proliferation will ensue. Insinuating that the most lethal PDAC tumors may have high NRP-1 levels and better targets for our nanoparticle therapy.
Figure 9.

Western Blot of Human Pancreatic Cancer Samples. (A) Western blot of the unaffected pancreas, primary pancreatic tumor, lung metastatic site, and liver metastatic site all from the same patient. (B) NRP-1 to GAPDH expression ratio. (C) STAT3 to GAPDH expression ratio.
Discussion
Pancreatic cancer patients often have minimal clinical success. The 5-year survival rate has remained below 10% for over 40 years, and only in the last 10 years have these rates been above 5%.94 Potentially, targeting the hypoxic tumor core, instead of the outside, may pose a strategic advantage to these patients. Utilizing polymeric nanoparticles with a tumor penetrating iRGD peptide and a hypoxia-responsive group, increased toxicity and uptake to cancer cells to combat hypoxia-induced resistance. These polymersomes were able to increase cellular toxicity by as much as 50% compared to unencapsulated drugs. The incorporation of the iRGD peptide allowed for a 12-fold increase in cellular uptake compared to polymersomes lacking this peptide. These initial results compelled us to further investigate the therapeutic potential of an inside-out treatment strategy. Upon completion of our animal study using a mouse model of pancreatic cancer, we found intriguing results. First, the apparent tumor size and volume was suppressed by over 200% compared to untreated controls. Interestingly, tumor volume bounced back up immediately when the treatment had stopped in the case of the free drug group, whereas the tumor volume stayed relatively steady when treated with iRGD polymersome encapsulated drugs. This result potentially suggests that iRGD polymersome encapsulated drugs decreased the number of free drug-resistant cancer cells and delays cancer recurrence. Second, and possibly most alluring, were the resected tumors. The iRGD polymersome groups had subjectively squishy and less dense tumors as compared to either the free drug or control tumors. The final finding of increased necrosis in the tumor core was another indication that our tumor penetrating polymersomes were effective in delivering their payload deep inside the tumor tissue.
To our knowledge this is the first reported use of iRGD conjugated polymersomes encapsulating gemcitabine or napabucasin in an animal model of pancreatic cancer. Previous studies have shown significant tumor reduction by co-administering free drug and free iRGD peptide separately to induce increased cellular uptake.95,96 Our polymersome system, adds the benefit of enhanced uptake, longer circulation time, and lower likelihood of toxicity compared to delivery of free drugs.
Our future studies may look at utilizing these hypoxia-responsive nanoparticles as a vascular normalization strategy. Development of an improved vascular system within the hypoxic niches of the tumor may decrease the ability of stem cells to self-renew and provide a channel for chemotherapy agents to reach their target.97,98 Additional studies are also needed to determine organ toxicity and better understand the pharmacokinetics of these nanoparticles.
The polymersomes were able to suppress tumor growth, but in the end, the tumors did not achieve complete remission. Optimization of the treatment protocol or a possible combination of these polymersomes with non-penetrating chemotherapy may have a better chance of achieving complete remission.
Hypoxia-responsive targeted polymers were successfully synthesized and self-assembled polymersomes were formed. The inclusion of the iRGD peptide significantly increased cellular uptake and led to increased cellular toxicity in monolayer, spheroids, and an animal model of pancreatic cancer. The inclusion of a hypoxia-responsive azo-benzene linker allows for selective release of encapsulated cargo deep inside the tumor’s hypoxic niches. By penetrating the outer layers of the tumor, we were able to induce significant necrosis to the tumor’s core that led to a reduction in tumor size. Our experiments provide a rational a treatment strategy that works to destroy the tumor from the inside-out.
Supplementary Material
Acknowledgments
This research was supported by NIH grant 1 R01GM 114080 (NIGMS) and a Ready-to-Go award from the DaCCoTA Center (NIGMS U54 GM128729) to SM. SM also acknowledges support from the Grand Challenge Initiative and the Office of the Dean, College of Health Professions, North Dakota State University. The Animal Core Facility is partly-supported by the NIH COBRE award 1P20 GM109024.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.0c00247
Proposed mechanism of azobenzene reduction under hypoxia, polymersome characterization by DLS, critical aggregation concentration determination plots, 1H, 13C NMR, IR spectra and GPC chromatogram of the hypoxia-responsive polymer, mass and CD spectra of the synthesized iRGD peptide, polymersome stability plot in human serum, size of the polymersomes (by DLS) as a function of time in hypoxia and normoxia, penetration of ICG encapsulated polymersomes in mice orthotopic pancreatic tumors, accumulation of polymersomes in various organs in mice, changes in mouse weights during treatments with polymersomes encapsulated drugs, changes in mouse weights during treatments with the free drugs, raw data for tumor volumes, and the associated calculations.
References
- (1).Jain RK Normalization of Tumor Vasculature: An Emerging Concept in Anti angiogenic Therapy. Science 2005, 307 (5706), 58–62. 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- (2).Sands H; Jones PL; Shah SA; Palme D; Vessella RL; Gallagher BM Correlation of Vascular Permeability and Blood Flow with Monoclonal Antibody Uptake by Human Clouser and Renal Cell Xenografts. Cancer Res. 1988, 48 (1), 188–193. [PubMed] [Google Scholar]
- (3).Koong AC; Mehta VK; Le QT; Fisher GA; Terris DJ; Brown JM; Bastidas AJ; Vierra M Pancreatic Tumors Show High Levels of Hypoxia. Int. J. Radiat. Oncol 2000, 48 (4), 919–922. 10.1016/S0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- (4).Rampling R; Cruickshank G; Lewis AD; Fitzsimmons SA; Workman P Direct Measurement of pO2 Distribution and Bioreductive Enzymes in Human Malignant Brain Tumors. Int. J. Radiat. Oncol 1994, 29 (3), 427–431. 10.1016/0360-3016(94)90432-4. [DOI] [PubMed] [Google Scholar]
- (5).Brizel DM; Scully SP; Harrelson JM; Layfield LJ; Bean JM; Prosnitz LR; Dewhirst MW Tumor Oxygenation Predicts for the Likelihood of Distant Metastases in Human Soft Tissue Sarcoma. Cancer Research 1996, 56, 941–943; http://cancerres.aacrjournals.org/content/56/5/941.short. [PubMed] [Google Scholar]
- (6).Nordsmark M; Overgaard M; Overgaard J Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother. Oncol 1996, 41, 31–39; 10.1016/S0167-8140(96)91811-3. [DOI] [PubMed] [Google Scholar]
- (7).Okunieff P; Hoeckel M; Dunphy EP; Schlenger K; Knoop C; Vaupel P Oxygen Tension Distributions Are Sufficient to Explain the Local Response of Human Breast Tumors Treated with Radiation Alone. Int. J. Radiat. Oncol 1993, 26 (4), 631–636. 10.1016/0360-3016(93)90280-9. [DOI] [PubMed] [Google Scholar]
- (8).Höckel M; Knoop C; Schlenger K; Vorndran B; Bauβnann E; Mitze M; Knapstein PG; Vaupel P Intratumoral PO2 Predicts Survival in Advanced Cancer of the Uterine Cervix. Radiother. Oncol 1993, 26 (1), 45–50. 10.1016/0167-8140(93)90025-4. [DOI] [PubMed] [Google Scholar]
- (9).Fyles AW; Milosevic M; Wong R; Kavanagh M-C; Pintilie M; Sun A; Chapman W; Levin W; Manchul L; Keane TJ; Hill RP Oxygenation Predicts Radiation Response and Survival in Patients with Cervix Cancer. Radiother. Oncol 1998, 48 (2), 149–156. 10.1016/S0167-8140(98)00044-9. [DOI] [PubMed] [Google Scholar]
- (10).Movsas B; Chapman JD; Horwitz EM; Pinover WH; Greenberg RE; Hanlon AL; Iyer R; Hanks GE Hypoxic Regions Exist in Human Prostate Carcinoma. Urology 1999, 53 (1), 11–18. 10.1016/S0090-4295(98)00500-7. [DOI] [PubMed] [Google Scholar]
- (11).Vaupel P; Höckel M; Mayer A Detection and Characterization of Tumor Hypoxia Using PO2 Histography. Antioxid. Redox Signal 2007, 9 (8), 1221–1235. 10.1089/ars.2007.1628. [DOI] [PubMed] [Google Scholar]
- (12).Bertout JA; Patel SA; Simon MC The Impact of O2 Availability on Human Cancer. Nat. Rev. Cancer 2008, 8 (12), 967–975. 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Finger EC; Giaccia AJ Hypoxia, Inflammation, and the Tumor Microenvironment in Metastatic Disease. Cancer Metastasis Rev. 2010, 29 (2), 285–293. 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Semenza GL Defining the Role of Hypoxia-Inducible Factor 1 in Cancer Biology and Therapeutics. Oncogene 2010, 29 (5), 625–634. 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Semenza GL Molecular Mechanisms Mediating Metastasis of Hypoxic Breast Cancer Cells. Trends Mol. Med 2012, 18 (9), 534–543. 10.1016/j.molmed.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Alizadeh AA; Aranda V; Bardelli A; Blanpain C; Bock C; Borowski C; Caldas C; Califano A; Doherty M; Elsner M; Esteller M; Fitzgerald R; Korbel JO; Lichter P; Mason CE; Navin N; Pe’er D; Polyak K; Roberts CWM; Siu L; Snyder A; Stower H; Swanton C; Verhaak RGW; Zenklusen JC; Zuber J; Zucman-Rossi J Toward Understanding and Exploiting Tumor Heterogeneity. Nat. Med 2015, 21 (8), 846–853. 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Marusyk A; Almendro V; Polyak K Intra-Tumour Heterogeneity: A Looking Glass for Cancer? Nat. Rev. Cancer 2012, 12 (5), 323–334. 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- (18).Meacham CE; Morrison SJ Tumour Heterogeneity and Cancer Cell Plasticity. Nature 2013, 501 (7467), 328–337. 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bailey JM; Alsina J; Rasheed ZA; McAllister FM; Fu Y-Y; Plentz R; Zhang H; Pasricha PJ; Bardeesy N; Matsui W; Maitra A; Leach SD DCLK1 Marks a Morphologically Distinct Subpopulation of Cells with Stem Cell Properties in Preinvasive Pancreatic Cancer. Gastroenterology 2014, 146 (1), 245–256. 10.1053/j.gastro.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ohara Y; Oda T; Sugano M; Hashimoto S; Enomoto T; Yamada K; Akashi Y; Miyamoto R; Kobayashi A; Fukunaga K; Morishita Y; Ohkohchi N Histological and Prognostic Importance of CD44(+) /CD24(+) /EpCAM(+) Expression in Clinical Pancreatic Cancer. Cancer Sci. 2013, 104 (8), 1127–1134. 10.1111/cas.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li L; Borodyansky L; Yang Y Genomic Instability En Route to and from Cancer Stem Cells. Cell Cycle Georget. Tex 2009, 8 (7), 1000–1002. 10.4161/cc.8.7.8041. [DOI] [PubMed] [Google Scholar]
- (22).Corcoran RB; Contino G; Deshpande V; Tzatsos A; Conrad C; Benes CH; Levy DE; Settleman J; Engelman JA; Bardeesy N STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res. 2011, 71 (14), 5020–5029. 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rebouissou S; Amessou M; Couchy G; Poussin K; Imbeaud S; Pilati C; Izard T; Balabaud C; Bioulac-Sage P; Zucman-Rossi J Frequent In-Frame Somatic Deletions Activate Gp130 in Inflammatory Hepatocellular Tumours. Nature 2009, 457 (7226), 200–204. 10.1038/nature07475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Putoczki TL; Thiem S; Loving A; Busuttil RA; Wilson NJ; Ziegler PK; Nguyen PM; Preaudet A; Farid R; Edwards KM; Boglev Y; Luwor RB; Jarnicki A; Horst D; Boussioutas A; Heath JK; Sieber OM; Pleines I; Kile BT; Nash A; Greten FR; McKenzie BS; Ernst M Interleukin-11 Is the Dominant IL-6 Family Cytokine during Gastrointestinal Tumorigenesis and Can Be Targeted Therapeutically. Cancer Cell 2013, 24 (2), 257–271. 10.1016/j.ccr.2013.06.017. [DOI] [PubMed] [Google Scholar]
- (25).Su YJ; Lai HM; Chang YW; Chen GY; Lee CJ Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 2011, 30, 3186–3199; 10.1038/emboj.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lee TKW; Castilho A; Cheung VCH; Tang KH; Ma S; Ng IOL CD24+ Liver Tumor-Initiating Cells Drive Self-Renewal and Tumor Initiation through STAT3-Mediated NANOG Regulation. Cell Stem Cell 2011, 9 (1), 50–63. 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- (27).Schroeder A; Hermann A; Cherryholmes G; Kowolik C; Buettner R; Pal S; Yu H; Muller-Newen G; Jove R; Loss of Androgen Receptor Expression Promotes a Stem-like Cell Phenotype in Prostate Cancer through STAT3 Signaling. Cancer Res. 2013, 74(4), 1227–1237; 10.1158/0008-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Panni RZ; Sanford DE; Belt BA; Mitchem JB; Worley LA; Goetz BD; Mukheijee P; Wang-Gillam A; Link DC; DeNardo DG; Goedegebuure SP; Linehan DC Tumor-Induced STAT3 Activation in Monocytic Myeloid-Derived Suppressor Cells Enhances Sternness and Mesenchymal Properties in Human Pancreatic Cancer. Cancer Immunol. Immunother 2014, 63 (5), 513–528. 10.1007/s00262-014-1527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Fukahi K; Fukasawa M; Neufeld G; Itakura J; Korc M Aberrant Expression of Neuropilin-1 and -2 in Human Pancreatic Cancer Cells. Clin. Cancer Res 2004, 10 (2), 581–590. 10.1158/1078-0432.CCR-0930-03. [DOI] [PubMed] [Google Scholar]
- (30).Karandish F; Froberg J; Borowicz P; Wilkinson JC; Choi Y; Mallik S Peptide-Targeted, Stimuli-Responsive Polymersomes for Delivering a Cancer Sternness Inhibitor to Cancer Stem Cell Microtumors. Colloids Surf. B Biointerfaces 2018, 163, 225–235. 10.1016/j.colsurfb.2017.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yin H; Yang J; Zhang Q; Yang J; Wang H; Xu J; Zheng J IRGD as a Tumor-penetrating Peptide for Cancer Therapy (Review). Mol. Med. Rep 2017, 15 (5), 2925–2930. 10.3892/mmr.2017.6419. [DOI] [PubMed] [Google Scholar]
- (32).Liu J; Yuan S; Wang L; Sun X; Hu X; Meng X; Yu J Diagnostic and Predictive Value of Using RGD PET/CT in Patients with Cancer: A Systematic Review and Meta-Analysis https://www.hindawi.com/journals/bmri/2019/8534761/ (accessed May 21, 2020). 10.1155/2019/8534761. [DOI] [PMC free article] [PubMed]
- (33).Martínez-Vélez N; Garcia-Moure M; Marigil M; González-Huarriz M; Puigdelloses M; Gallego Pérez-Larraya J; Zalacaín M; Marrodán L; Varela-Guruceaga M; Laspidea V; Aristu JJ; Ramos LI; Tejada-Solís S; Díez-Valle R; Jones C; Mackay A; Martínez-Climent JA; García-Barchino MJ; Raabe E; Monje M; Becher OJ; Junier MP; El-Habr EA; Chneiweiss H; Aldave G; Jiang H; Fueyo J; Patiño-García A; Gomez-Manzano C; Alonso MM The Oncolytic Virus Delta-24-RGD Elicits an Antitumor Effect in Pediatric Glioma and DIPG Mouse Models. Nat. Commun 2019, 10. 10.1038/s41467-019-10043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sugahara KN; Teesalu T; Karmali PP; Kotamraju VR; Agemy L; Girard OM; Hanahan D; Mattrey RF; Ruoslahti E Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16 (6), 510–520. 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li Z; Huang P; Zhang X; Lin J; Yang S; Liu B; Gao F; Xi P; Ren Q; Cui S RGD-conjugated Dendrimer-Modified Gold Nanorods for in Vivo Tumor Targeting and Photothermal Therapy. Mol. Pharm 2010, 7, 94–104. 10.1021/mp9001415. [DOI] [PubMed] [Google Scholar]
- (36).Xu W; Luo T; Li P; Zhou C; Cui D; Pang B; Ren Q; Fu S RGD-Conjugated Gold Nanorods Induce Radiosensitization in Melanoma Cancer Cells by Downregulating Avβ3 Expression. Int. J. Nanomedicine 2012, 7, 915–924. 10.2147/IJN.S28314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Simón-Gracia L; Hunt H; Scodeller P; Gaitzsch J; Kotamraju VR; Sugahara KN; Tammik O; Ruoslahti E; Battaglia G; Teesalu T iRGD Peptide Conjugation Potentiates Intraperitoneal Tumor Delivery of Paclitaxel with Polymersomes. Biomaterials 2016, 104, 247–257. 10.1016/j.biomaterials.2016.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kadonosono T; Yamano A; Goto T; Tsubaki T; Niibori M; Kuchimaru T; Kizaka-Kondoh S Cell Penetrating Peptides Improve Tumor Delivery of Cargos through Neuropilin-1-Dependent Extravasation. J. Controlled Release 2015, 201, 14–21. 10.1016/j.jconrel.2015.01.011. [DOI] [PubMed] [Google Scholar]
- (39).Kulkarni P; Haldar MK; You S; Choi Y; Mallik S Hypoxia-Responsive Polymersomes for Drug Delivery to Hypoxic Pancreatic Cancer Cells. Biomacromolecules 2016, 77(8), 2507–2513. 10.1021/acs.biomac.6b00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Luo J; Yan R; He X; He J Constitutive Activation of STAT3 and Cyclin D1 Overexpression Contribute to Proliferation, Migration and Invasion in Gastric Cancer Cells. Am. J. Transl. Res 2017, 9 (12), 5671–5677. [PMC free article] [PubMed] [Google Scholar]
- (41).Li Y; Rogoff HA; Keates S; Gao Y; Murikipudi S; Mikule K; Leggett D; Li W; Pardee AB; Li CJ Suppression of Cancer Relapse and Metastasis by Inhibiting Cancer Sternness. Proc. Natl. Acad. Sci 2015, 112 (6), 1839–1844. 10.1073/pnas.1424171112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hubbard JM; Grothey A Napabucasin: An Update on the First-in-Class Cancer Sternness Inhibitor. Drugs 2017, 77, 1091–1103. 10.1007/s40265-017-0759-4 (accessed Oct 1, 2019). [DOI] [PubMed] [Google Scholar]
- (43).Zhang Y; Jin Z; Zhou H; Ou X; Li H; Liu C Li B Suppression of prostate cancer progression by cancer cell sternness inhibitor napabucasin. Cancer Med. 2016, 5(6), 1251–1258. 10.1002/cam4.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sonbol MB; Ahn DH; Bekaii-Saab T Therapeutic Targeting Strategies of Cancer Stem Cells in Gastrointestinal Malignancies. Biomedicines 2019, 7 (1). 10.3390/biomedicines7010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Perche F; Biswas S; Wang T; Zhu L; Torchilin VP Hypoxia-Targeted SiRNA Delivery. Angew. Chem. Int. Ed Engl 2014, 53 (13), 3362–3366. 10.1002/anie.201308368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dan K; Bose N; Ghosh S Vesicular Assembly and Thermo-Responsive Vesicle-to-Micelle Transition from an Amphiphilic Random Copolymer. Chem. Commun 2011, 47 (46), 12491–12493. 10.1039/C1CC15663B. [DOI] [PubMed] [Google Scholar]
- (47).Ray P; Confeld M; Borowicz P; Wang T; Mallik S; Quadir M PEG-b-poly (carbonate)-derived nanocarrier platform with pH-responsive properties for pancreatic cancer combination therapy. Colloids Surf. B: Biointerfaces 2019, 774, 126–135. 10.1016/j.colsurfb.2018.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Mouse: Decision tree for blood sampling | NC3Rs https://nc3rs.org.uk/mouse-decision-tree-blood-sampling (accessed May 30, 2019). [Google Scholar]
- (49).Saxena V; Sadoqi M; Shao J Polymeric Nanoparticulate Delivery System for Indocyanine Green: Biodistribution in Healthy Mice. Int. J. Pharm 2006, 308 (1), 200–204. 10.1016/j.ijpharm.2005.11.003. [DOI] [PubMed] [Google Scholar]
- (50).Euhus DM; Hudd C; LaRegina MC; Johnson FE Tumor Measurement in the Nude Mouse. J. Surg. Oncol 1986, 31 (4), 229–234. [DOI] [PubMed] [Google Scholar]
- (51).Tomayko MM; Reynolds CP Determination of Subcutaneous Tumor Size in Athymic (Nude) Mice. Cancer Chemother. Pharmacol 1989, 24 (3), 148–154. [DOI] [PubMed] [Google Scholar]
- (52).Won Y-Y; Brannan AK; Davis HT; Bates FS Cryogenic Transmission Electron Microscopy (Cryo-TEM) of Micelles and Vesicles Formed in Water by Poly(Ethylene Oxide)-Based Block Copolymers. J. Phys. Chem. B 2002, 106 (13), 3354–3364. 10.1021/jp013639d. [DOI] [Google Scholar]
- (53).Won Y-Y; Davis HT; Bates FS Giant Wormlike Rubber Micelles. Science 1999, 283 (5404), 960–963. 10.1126/science.283.5404.960. [DOI] [PubMed] [Google Scholar]
- (54).Loscalzo J Adaptions to Hypoxia and Redox Stress: Essential Concepts Confounded by Misleading Terminology. Circ. Res 2016, 119 (4), 511–513. 10.1161/CIRCRESAHA.116.309394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jubb AM; Buffa FM; Harris AL Assessment of Tumour Hypoxia for Prediction of Response to Therapy and Cancer Prognosis. J. Cell. Mol. Med 2010, 14 (1–2), 18–29. 10.1111/j.1582-4934.2009.00944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kulkarni P; Haldar MK; Karandish F; Confeld M; Hossain R; Borowicz P; Gange K; Xia L; Sarkar K; Mallik S Tissue-Penetrating, Hypoxia-Responsive Echogenic Polymersomes For Drug Delivery To Solid Tumors. Chem. Eur. J 2018, 24 (48), 12490–12494. 10.1002/chem.201802229. [DOI] [PubMed] [Google Scholar]
- (57).Anajafi T; Yu J; Sedigh A; Haldar MK; Muhonen WW; Oberlander S; Wasness H; Froberg J; Molla MS; Katti KS; Choi Y; Shabb JB; Srivastava DK; Mallik S Nuclear Localizing Peptide-Conjugated, Redox-Sensitive Polymersomes for Delivering Curcumin and Doxorubicin to Pancreatic Cancer Microtumors. Mol. Pharm 2017, 14 (6), 1916–1928. 10.1021/acs.molpharmaceut.7b00014. [DOI] [PubMed] [Google Scholar]
- (58).Nahire R; Haldar MK; Paul S; Ambre AH; Meghnani V; Layek B; Katti KS; Gange KN; Singh J; Sarkar K; Mallik S Multifunctional Polymersomes for Cytosolic Delivery of Gemcitabine and Doxorubicin to Cancer Cells. Biomaterials 2014, 35 (24), 6482–6497. 10.1016/j.biomaterials.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Toy R; Peiris PM; Ghaghada KB; Karathanasis E Shaping Cancer Nanomedicine: The Effect of Particle Shape on the in Vivo Journey of Nanoparticles. Nanomed. 2014, 9 (1), 121–134. 10.2217/nnm.13.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Nagayasu A;Uchiyama U; Kiwada H The Size of Liposomes: A Factor Which Affects Their Targeting Efficiency to Tumors and Therapeutic Activity of Liposomal Antitumor Drugs. Adv. Drug Deliv. Rev 1999, 40 (1–2), 75–87. [DOI] [PubMed] [Google Scholar]
- (61).Maeda H The Enhanced Permeability and Retention (EPR) Effect in Tumor Vasculature: The Key Role of Tumor-Selective Macromolecular Drug Targeting. Adv. Enzyme Regul 2001, 41 (1), 189–207. 10.1016/S0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- (62).Acharya S; Sahoo SK PLGA Nanoparticles Containing Various Anticancer Agents and Tumour Delivery by EPR Effect. Adv. Drug Deliv. Rev 2011, 63 (3), 170–183. 10.1016/j.addr.2010.10.008. [DOI] [PubMed] [Google Scholar]
- (63).Maeda H; Nakamura H; Fang J The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging in Vivo. Adv. Drug Deliv. Rev 2013, 65 (1), 71–79. 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- (64).Hoshyar N; Gray S; Han H; Bao G The Effect of Nanoparticle Size on in Vivo Pharmacokinetics and Cellular Interaction. Nanomed. 2016, 11 (6), 673–692. 10.2217/nnm.16.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Albanese A; Tang PS; Chan WCW The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng 2012, 14, 1–16. 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- (66).Dreaden EC; Austin LA; Mackey MA; El-Sayed MA Size Matters: Gold Nanoparticles in Targeted Cancer Drug Delivery. Ther. Deliv 2012, 3 (4), 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kulkarni SA; Feng S-S Effects of Particle Size and Surface Modification on Cellular Uptake and Biodistribution of Polymeric Nanoparticles for Drug Delivery. Pharm. Res 2013, 30 (10), 2512–2522. 10.1007/s11095-012-0958-3. [DOI] [PubMed] [Google Scholar]
- (68).Kiyose K; Hanaoka K; Oushiki D; Nakamura T; Kajimura M; Suematsu M; Nishimatsu H; Yamane T; Terai T; Hirata Y; Nagano T Hypoxia-Sensitive Fluorescent Probes for in Vivo Real-Time Fluorescence Imaging of Acute Ischemia. J. Am. Chem. Soc 2010, 132 (45), 15846–15848. 10.1021/ja105937q. [DOI] [PubMed] [Google Scholar]
- (69).Karandish F; Haldar MK; You S; Brooks AE; Brooks BD; Guo B; Choi Y; Mallik S Prostate-Specific Membrane Antigen Targeted Polymersomes for Delivering Mocetinostat and Docetaxel to Prostate Cancer Cell Spheroids. ACS Omega 2016, 1 (5), 952–962. 10.1021/acsomega.6b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Gilkes DM; Semenza GL; Wirtz D Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat. Rev. Cancer 2014, 14 (6), 430–439. 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Cangul H; Salnikow K; Yee H; Zagzag D; Commes T; Costa M Enhanced Overexpression of an HIF-1/Hypoxia-Related Protein in Cancer Cells. Environ. Health Perspect 2002, 110 Suppl 5, 783–788. 10.1289/ehp.02110s5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Zhong H; De Marzo AM; Laughner E; Lim M; Hilton DA; Zagzag D; Buechler P; Isaacs WB; Semenza GL; Simons JW Overexpression of Hypoxia-Inducible Factor lalpha in Common Human Cancers and Their Metastases. Cancer Res. 1999, 59 (22), 5830–5835. [PubMed] [Google Scholar]
- (73).Pampaloni F; Reynaud EG; Stelzer EHK The Third Dimension Bridges the Gap between Cell Culture and Live Tissue. Nat. Rev. Mol. Cell Biol 2007, 8 (10), 839–845. 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- (74).Baker BM; Chen CS Deconstructing the Third Dimension: How 3D Culture Microenvironments Alter Cellular Cues. J. Cell Sci 2012, 125 (Pt 13), 3015–3024. 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Thomas D; Radhakrishnan P Tumor-Stromal Crosstalk in Pancreatic Cancer and Tissue Fibrosis. Mol. Cancer 2019, 18. 10.1186/s12943-018-0927-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Cannon A; Thompson C; Hall BR; Jain M; Kumar S; Batra SK Desmoplasia in Pancreatic Ductal Adenocarcinoma: Insight into Pathological Function and Therapeutic Potential. Genes Cancer 2018, 9 (3–4), 78–86. 10.18632/genesandcancer.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Matkar PN; Singh KK; Rudenko D; Kim YJ; Kuliszewski MA; Prud’homme GJ; Hedley DW; Leong-Poi H Novel Regulatory Role of Neuropilin-1 in Endothelial-to-Mesenchymal Transition and Fibrosis in Pancreatic Ductal Adenocarcinoma. Oncotarget 2016, 7(43), 69489–69506. 10.18632/oncotarget.11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Zhou R; Curry JM; Roy LD; Grover P; Haider J; Moore LJ; Wu S; Kamesh A; Yazdanifar M; Ahrens WA; Leung T; Mukheijee P A Novel Association of Neuropilin-1 and MUC1 in Pancreatic Ductal Adenocarcinoma: Role in Induction of VEGF Signaling and Angiogenesis. Oncogene 2016, 35 (43), 5608–5618. 10.1038/onc.2015.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Falcone S; Cocucci E; Podini P; Kirchhausen T; Clementi E; Meldolesi J Macropinocytosis: Regulated Coordination of Endocytic and Exocytic Membrane Traffic Events. J. Cell Sci 2006, 119 (22), 4758–4769. 10.1242/jcs.03238. [DOI] [PubMed] [Google Scholar]
- (80).Suk JS; Xu Q; Kim N; Hanes J; Ensign LM PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Deliv. Rev 2016, 99 (Pt A), 28–51. 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).PEG-Intron (Peginterferon alfa-2b) Package Insert https://www.accessdata.fda.gov/drugsatfda_docs/label/2001/pegsche080701LB.htm (accessed May 27, 2020).
- (82).PLEGRIDY (Peginterferon Beta-la) [Package Insert] https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125499s011lbl.pdf (accessed May 27, 2020).
- (83).Oncaspar (Pegaspargase) https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/103411s5126lbl.pdf (accessed May 27, 2020)
- (84).Working PK; Newman MS; Huang SK; Mayhew E; Vaage J; Lasic DD Pharmacokinetics, Biodistribution and Therapeutic Efficacy of Doxorubicin Encapsulated in Stealth® Liposomes (Doxil®). J. Liposome Res 1994, 4 (1), 667–687. 10.3109/08982109409037065. [DOI] [Google Scholar]
- (85).Stinchcombe TE; Socinski MA; Walko CM; O’Neil BH; Collichio FA; Ivanova A; Mu H; Hawkins MJ; Goldberg RM; Lindley C; Claire Dees E Phase I and Pharmacokinetic Trial of Carboplatin and Albumin-Bound Paclitaxel, ABI-007 (Abraxane®) on Three Treatment Schedules in Patients with Solid Tumors. Cancer Chemother. Pharmacol 2007, 60 (5), 759–766. 10.1007/s00280-007-0423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Espey MG; Chen P; Chalmers B; Drisko J; Sun AY; Levine M; Chen Q Pharmacologic Ascorbate Synergizes with Gemcitabine in Preclinical Models of Pancreatic Cancer. Free Radic. Biol. Med 2011, 50 (11), 1610–1619. 10.1016/j.freeradbiomed.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Tauchi T; Katagiri S; Okabe S; Tanaka Y; Ohyashiki K Activity of the Sternness Inhibitor, BBI608, on the Self-Renewal of BCR-ABL1 Positive Leukemia Cells: Molecular Mechanisms. Blood 2014, 124 (21), 905–905. [Google Scholar]
- (88).Yadav D; Lowenfels AB The Epidemiology of Pancreatitis and Pancreatic Cancer. Gastroenterology 2013, 144(6), 1252–1261. 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Daddacha WB; Koyen AE; Schlafstein AJ; Yu DS Role of STAT3 in Pancreatic Cancer: A Target for Therapy. In Role of Transcription Factors in Gastrointestinal Malignancies; Nagaraju GP, Bramhachari PV, Eds.; Springer: Singapore, 2017; pp 341–350. 10.1007/978-981-10-6728-0_24. [DOI] [Google Scholar]
- (90).Ben Q; Zheng J; Fei J; An W; Li P; Li Z; Yuan Y High Neuropilin 1 Expression Was Associated With Angiogenesis and Poor Overall Survival in Resected Pancreatic Ductal Adenocarcinoma: Pancreas 2014, 43 (5), 744–749. 10.1097/MPA.0000000000000117. [DOI] [PubMed] [Google Scholar]
- (91).Ma L; Zhai B; Zhu H; Li W; Jiang W; Lei L; Zhang S; Qiao H; Jiang X; Sun X The MiR-141/Neuropilin-1 Axis Is Associated with the Clinicopathology and Contributes to the Growth and Metastasis of Pancreatic Cancer. Cancer Cell Int. 2019, 19 (1), 248. 10.1186/s12935-019-0963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Wander SA; Zhao D; Slingerland JM P27: A Barometer of Signaling Deregulation and Potential Predictor of Response to Targeted Therapies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 2011, 17(1), 12–18. 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Yoshida A; Shimizu A; Asano H; Kadonosono T; Kondoh SK; Geretti E; Mammoto A; Klagsbrun M; Seo MK VEGF-A/NRP1 Stimulates GIPC1 and Syx Complex Formation to Promote RhoA Activation and Proliferation in Skin Cancer Cells. Biol. Open 2015, 4 (9), 1063–1076. 10.1242/bio.010918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Pancreatic Cancer - Cancer Stat Facts https://seer.cancer.gov/statfacts/html/pancreas.html (accessed May 30, 2020).
- (95).Akashi Y; Oda T; Ohara Y; Miyamoto R; Kurokawa T; Hashimoto S; Enomoto T; Yamada K; Satake M; Ohkohchi N Anticancer Effects of Gemcitabine Are Enhanced by Co-Administered IRGD Peptide in Murine Pancreatic Cancer Models That Overexpressed Neuropilin-1. Br. J. Cancer 2014, 110 (6), 1481–1487. 10.1038/bjc.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Zhang Q; Zhang Y; Li K; Wang H; Li H; Zheng J A Novel Strategy to Improve the Therapeutic Efficacy of Gemcitabine for Non-Small Cell Lung Cancer by the Tumor-Penetrating Peptide IRGD. PLoS ONE 2015, 10 (6). 10.1371/journal.pone.0129865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Siemann DW; Horsman MR Modulation of the Tumor Vasculature and Oxygenation to Improve Therapy. Pharmacol. Ther 2015, 153, 107–124. 10.1016/j.pharmthera.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Viallard C; Larrivée B Tumor Angiogenesis and Vascular Normalization: Alternative Therapeutic Targets. Angiogenesis 2017, 20 (4), 409–426. 10.1007/s10456-017-9562-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.