

Summary
Single-cell RNA sequencing (scRNA-seq) of peripheral blood mononuclear cells (PBMCs) allows in-depth assessment of transcriptional changes in immune cells of patients with COVID-19. However, collecting, processing, and analyzing samples from patients with COVID-19 pose many challenges because blood samples may contain infectious virus, identification of immune cell subtypes can be difficult, and biological interpretation of analytical results is complex. To address these issues, we describe a protocol for sample processing, sorting, methanol fixation, and scRNA-seq analysis of PBMCs from frozen buffy coat samples from patients with COVID-19.
For complete details on the use and execution of this protocol, please refer to (Yao et al., 2021).
Subject areas: Bioinformatics, Sequence analysis, Immunology
Graphical abstract
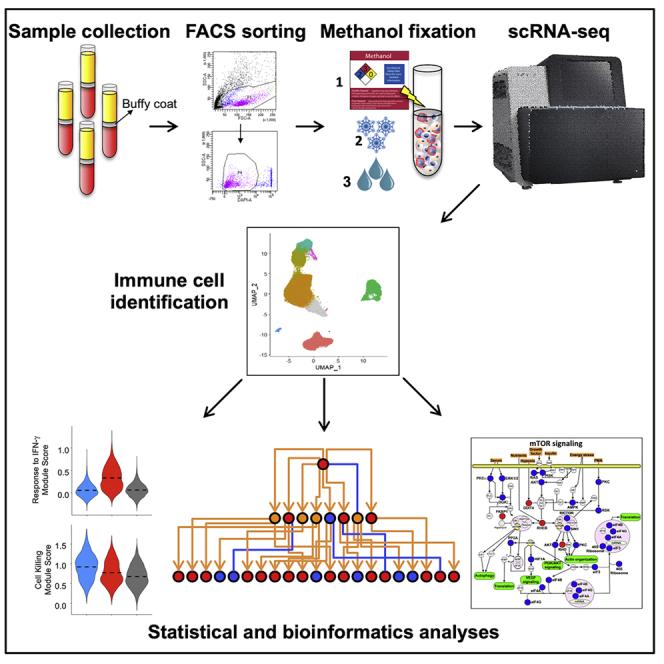
Highlights
-
•
scRNA-seq analysis profiles circulating immune cell subsets in patients with COVID-19
-
•
Reveals cell-specific gene expression differences across spectrum of disease severity
-
•
Pathway analysis provides mechanistic insights
-
•
Methanol fixation mitigates safety concerns about virus in samples
Single-cell RNA sequencing (scRNA-seq) of PBMCs allows in-depth assessment of transcriptional changes in immune cells of patients with COVID-19. However, collecting, processing, and analyzing samples from patients with COVID-19 pose many challenges because blood samples may contain infectious virus, identification of immune cell subtypes can be difficult, and biological interpretation of analytical results is complex. To address these issues, we describe a protocol for sample processing, sorting, methanol fixation, and scRNA-seq analysis of PBMCs from frozen buffy coat samples from patients with COVID-19.
Before you begin
Timing: allow 6–8 h to process samples through methanol fixation.
Blood collection
-
1.
Collect venous blood in EDTA-coated tubes.
-
2.
Centrifuge at 1000 × g for 10 min with brake.
-
3.
Collect buffy coat and add to 1.8 mL cryopreservation media.
-
4.
Store cells in cryopreservation media at –80°C.
Note: Patient samples should be collected in accordance with institutional review board (IRB) rules, including patient consent, and handled with strict adherence to safety rules, including the use of appropriate PPE. Samples must be processed as quickly as possible to maximize viability, so careful advanced planning and coordination between clinical staff and research facilities is recommended. For this study, vials with a suggested draw volume of 10 mL were used. For 10 mL of starting sample, approximately 1 mL of buffy coat should be recoverable.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human adult PBMCs from patients positive for COVID-19 | NA | NA |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Corning | Cat.# 10-013-CV |
| DMSO | Sigma | Cat.# D2650 |
| DAPI | Thermo Fischer Scientific | Cat.# D1306 |
| DPBS | Corning | Cat.# 45000-434 |
| Fetal bovine serum | Fischer Scientific | Cat.# SH3007103 |
| Methanol | Millipore Sigma | 34860 |
| Protector RNAse Inhibitor | Millipore Sigma | 335399001 |
| SSC Buffer 20× Concentrate | Millipore Sigma | S6639-1L |
| DL-Dithiothreitol solution | Millipore Sigma | 43816 |
| Dulbecco's phosphate-buffered saline (DPBS), no calcium, no magnesium | Thermo Fisher Scientific | 14190144 |
| UltraPure BSA | Thermo Fisher Scientific | AM2616 |
| Critical commercial assays | ||
| Single Cell 3' Next GEM V3.1 | 10x Genomics | PN-1000121 |
| KAPA Library Quantification Kit | Roche | KK4824 |
| Deposited data | ||
| COVID-19 raw and analyzed data | Yao et al., 2021 | GEO: GSE154567 |
| Healthy PBMC data sets | Chen et al., 2018 | GEO: GSE112845 |
| SARS-CoV-2 genome | https://www.ncbi.nlm.nih.gov/nuccore/MT246667.1 | GenBank: MT246667.1 |
| Human reference transcriptome GRCh38 | https://www.gencodegenes.org/human/ | GRCh38.p13 |
| Software and algorithms | ||
| CellRanger v3.0.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| R package Seurat v.3.1.5 | Stuart et al., 2019 | https://github.com/satijalab/seurat |
| R package SoupX | Young and Behjati, 2020 | https://github.com/constantAmateur/SoupX |
| Harmony | Korsunsky et al., 2019 | https://github.com/immunogenomics/harmony |
| R package MAST | Finak et al., 2015 | https://github.com/RGLab/MAST |
| WebGestalt v2019 | Liao et al., 2019 | http://www.webgestalt.org |
| Ingenuity Pathway Analysis | QIAGEN | https://digitalinsights.qiagen.com/product-login/ |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| FACS Aria III | BD Biosciences | N/A |
| 10X Chromium Controller | 10x Genomics | Cat.# 1000202 |
| NovaSeq 6000 | Illumina | N/A |
| 70 μm Cell strainer (optional) | Falcon | Cat.# 08-771-2 |
| 40 μm Flowmi Cell Strainer | Bel-Art | H13680-0040 |
Materials and equipment
Cryopreservation media (sterile, temperature ∼20°C)
| Reagent | Final concentration | Stock concentration | Amount (mL) |
|---|---|---|---|
| DMEM | n/a | n/a | 400 |
| FBS | 10% v/v | n/a | 50 |
| DMSO | 10% v/v | 10% v/v | 50 |
Buffy coat wash solution (temperature ∼20°C)
| Reagent | Final concentration | Stock concentration | Amount (mL) |
|---|---|---|---|
| Sterile DPBS | n/a | n/a | 450 |
| FBS | 10% v/v | n/a | 50 |
Excess solutions that contain FBS should be stored at 4°C. All other reagents in this protocol are listed in the key resources table.
Step-by-step method details
FACS sorting
Note: Sorting time can vary considerably depending on the concentration of viable cells recovered after thawing. Clogs may occur which will also lengthen sorting time. Cell clumps can be removed by cell filtering using 70 μm strainers, and if available, multiple sorters can be used simultaneously to decrease the sorting time.
This section covers thawing, washing, and sorting live cells from frozen buffy coat.
-
1.
Collect frozen buffy coat samples and immediately place them on dry ice until ready for sorting.
-
2.
Thaw one sample at a time unless multiple sorters are available that can be used simultaneously. Thaw samples by swirling gently in a 37°C water bath, until one or two small ice crystals remain.
-
3.
Add 15 mL of buffy coat wash solution to each sample and centrifuge at 350 rcf at ambient temperature for 5 min. Discard the supernatant, and resuspend the cell pellets in a minimum of 500 μL of buffy coat wash solution.
-
4.
Add 3 μM DAPI and incubate for 5 min at room temperature.
-
5.
After DAPI staining, proceed immediately to sorting. Sort live (DAPI-negative) singlet cells for scRNA-seq analysis. The gating strategy for FACS sorting is illustrated in Figure 1.
Note:
Buffy coats will likely have erythrocyte contamination. If blood clots are present, they may decrease cell recovery and filtering sample through a 70 μm cell strainer is recommended.
Access to multiple, simultaneous sorting may not be an option without a dedicated core facility. While it may introduce a batch effect, sample preparation may be spread over multiple days if necessary.
Viability of thawed buffy coat is lower than viability of PBMCs isolated from fresh blood samples; expect 60%–75% viability. After thawing and sorting, an average cell yield of 200–250,000 cells can be expected.
Figure 1.

Gating strategy for FACS sorting
Sequential gating is utilized to obtain live PBMCs for subsequent scRNA-seq.
Methanol fixation and 10X processing
This part of the protocol is modified based on the 10x Genomics protocol “Methanol Fixation of Cells for Single Cell RNA Sequencing” CG000136 (https://support.10xgenomics.com/single-cell-gene-expression/sample-prep/doc/demonstrated-protocol-methanol-fixation-of-cells-for-single-cell-rna-sequencing).
-
6.Buffer preparation before starting:
-
a.3X SSC Buffer: Prepare 3× SSC buffer in nuclease free water using 20× SSC stock solution.
3× SSC Buffer 15 mL 20× SSC Buffer diluted with 85 mL nuclease free water -
b.Wash-Resuspension buffer: Prepare wash resuspension buffer with 0.04% BSA, 1 mM DTT, 0.2 U/μL RNase inhibitor in 3× SSC buffer.Wash-Resuspension buffer
Reagent Final concentration Amount 3× SSC Buffer n/a 49.3 mL 1M DTT 1 mM 50 μL 5% BSA 0.04% 0.4 mL 40 U/μL 0.2 U/μL 250 μL Total 50 mL Note:Keep prepared buffers at 4°C.Timing is critical at this stage. Any delay may result in RNA loss.Start with as many cells as possible (ideally, more than a million) because there will be significant cell loss at the rehydration step.
-
a.
-
7.
Collect FACS sorted cells by centrifuging at 500 rcf for 5 min at 4°C.
Note: generally 300 rcf is suggested, but we recommend 500 rcf for efficient precipitation.
-
8.
Carefully remove the supernatant and do not disturb the pellet.
-
9.
Add 1 mL chilled 1× DPBS with a wide-bore pipette tip (e.g., Thermo Scientific™2079G), gently pipette mix, centrifuge at 500 rcf at 4°C for 5 min.
-
10.
Carefully remove the supernatant and do not disturb the pellet.
-
11.
Resuspend the pellet in 200 μL chilled DPBS with a wide-bore pipette tip.
-
12.
Add 800 μL chilled 100% methanol drop by drop to the cell suspension. Gently stir the cell suspension during the process. Scale up the volumes of 1× DPBS and methanol if using more than 1 million cells.
-
13.
Incubate at −20°C for 30 min.
-
14.
The fixed cells can be stored at −20°C or −80°C for up to 6 weeks, or immediately proceed to rehydration.
Rehydration ∼10–15 min
-
15.
Before starting the rehydration, prepare the 10X Genomics Single Cell kit for loading into the controller as soon as the cells are rehydrated.
-
16.
Equilibrate the methanol-fixed cells on ice for 5 min.
-
17.
Centrifuge at 1000 rcf for 5 min at 4°C.
-
18.
Transfer the supernatant to a new tube without disrupting the cell pellet (save the supernatant in case not enough cells were pelleted at this step).
-
19.
Resuspend the cell pellet with an appropriate volume of wash-resuspension buffer based on the starting cell concentration and assuming ∼50% cell loss (refer to the 10X Genomics user manual Cell Suspension Volume Calculator Table for more information). Gently mix with a regular-bore pipette tip.
-
20.
Filter the sample through a 40 μm Flowmi Cell Strainer if there are large clumps.
-
21.
Count cells using a hemocytometer or automated cell counter to determine cell concentration. Proceed to scRNA-seq with the 10x Genomics Single Cell protocols.
Single-cell RNA-seq
Note:Figure 2 summarizes the steps in the bioinformatics pipeline detailed below for analysis of the scRNAseq data
Figure 2.

Overview of scRNA-seq data analysis
A step-wise approach for systematic data processing, cell type identification, differential gene expression and pathway analysis is shown, as well as recommended software programs for each step.
-
22.
Capture single cells using a 10X Chromium Controller (10X Genomics) and prepare libraries as described in the Single Cell 3’ Next GEM V3.1 Reagent Kits User Guide (10X Genomics, https://support.10xgenomics.com/single-cell-gene-expression/library-prep/doc/user-guide-chromium-single-cell-3-reagent-kits-user-guide-v31-chemistry).
-
23.
Quantify the barcoded sequencing libraries by quantitative PCR using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA).
-
24.
Sequence the libraries using a Novaseq 6000 (Illumina) with custom sequencing settings of 28 bp and 91 bp for read 1 and 2, respectively, to obtain a sequencing depth of ∼5 × 104 reads per cell.
Alignment, 10X cell barcode demultiplexing, quality control, and batch correction
Alignment and cell demultiplexing
-
25.
Use CellRanger v3.0.0 software with the default settings for demultiplexing cells according to their 10X cell barcodes and for aligning reads with STAR software to the human GRCh38 transcriptome reference downloaded from https://www.gencodegenes.org/ (contains all protein-coding and long non-coding RNA genes based on human GENCODE version 33 annotation) and the SARS-Cov2 virus genome MT246667.1, https://www.ncbi.nlm.nih.gov/nuccore/MT246667.1.
Note: We built a custom reference genome using the CellRanger software mkref function (see GEO: GSE154567).
QC and filtering
-
26.
Remove ambient RNA derived from lysed cells using the SoupX package (Young and Behjati, 2020).
-
27.
Use the single cell analysis R package Seurat v3.1.5 for data analysis (Stuart et al., 2019).
-
28.
For quality control and filtering out low quality cells, select only cells expressing more than 200 genes (defined as genes detected in at least 3 cells), UMI counts more than 500 and fewer than 20% mitochondrial genes. To minimize doublet contamination for each dataset, remove cells with a high number of genes detected using a fit model generated from the suggested “multiplet rate”/ “number of cells recovered” ratio as in the 10X Genomics user manual (Carraro et al., 2020).
Note:
Figure 3 shows an example of ambient RNA removal (Figures 3A and 3B), and filtering of low-quality cells (Figure 3C).
In our data, three patient groups were used: moderate COVID-19 (CM), severe COVID-19 (CS), and recovering (CR).
Selection of cutoff range for filtering cells with mitochondrial content can vary from 1% to 20%. In our case, despite using a permissive value, very few cells had large mitochondrial gene content.
The following codes have been deposited in GitHub using R markdown (https://github.com/ivonyao/Cell-Type-Specific-Immune-Dysregulation-in-Severely-Ill-COVID-19-Patients/tree/main/r%20code%20and%20markdowns).
Figure 3.

Single cell demultiplex, quality control, and batch correction
(A and B) HBA2 and HBB expression before and after ambient RNA clean up with SoupX.
(C) Before and after quality control for nFeature_RNA (gene number per cell), nCount_RNA (UMI) and percent.mt (percentage of mitochondrial genes).
Sample code (Please note: assign your own data directory to DataDir):
library(Seurat)
library(ggplot2)
library(SoupX)
library(DropletUtils)
library(dplyr)
### SoupX procedure
DataDir = c('∼/covid19scRNAseq/CM1_2/outs', '∼/covid19scRNAseq/CM1_2/outs/filtered_feature_bc_matrix')
#Load 10X Data to SoupX and Seurat
sc = load10X(DataDir[1])
## Loading raw count data
## Loading cell-only count data
## Loading extra analysis data where available
seu <- Read10X(DataDir[2])
seu <- CreateSeuratObject(counts = seu, project = "CM1_2")
seu <- SCTransform(object = seu, verbose = T)
seu <- RunPCA(object = seu, verbose = T)
seu <- RunUMAP(object = seu, dims = 1:30,verbose = T)
seu <- FindNeighbors(object = seu, dims = 1:30, verbose = T)
seu <- FindClusters(object = seu, resolution = 0.5, verbose = T)
#Check gene expression to ensure minimal RBC ambient RNA contamination
FeaturePlot(seu, features=c("HBA2", "HBB", "HBD"), max.cutoff="q90")
Clusters <- seu$seurat_clusters
sc = setClusters(sc,Clusters)
sc = autoEstCont(sc)
out = adjustCounts(sc)
#check correction quality
CM1_2 <- CreateSeuratObject(counts = out, project = "CM1_2")
CM1_2 <- SCTransform(object = CM1_2, verbose = T)
CM1_2 <- RunPCA(object = CM1_2, verbose = T)
CM1_2 <- RunUMAP(object = CM1_2, dims = 1:30,verbose = T)
CM1_2 <- FindNeighbors(object = CM1_2, dims = 1:30, verbose = T)
CM1_2 <- FindClusters(object = CM1_2, resolution = 0.5, verbose = T)
FeaturePlot(CM1_2, features=c("HBA2", "HBB", "HBD"), max.cutoff="q90")
write10xCounts('∼/covid19scRNAseq/CM1_2/outs/desoup2', out)
##QC process
CM1_2=Read10x(∼/covid19scRNAseq/CM1_2/outs/desoup2')
CM1_2 <- CreateSeuratObject(counts = CM1_2, project = "CM1_2")
CM1_2[["percent.mt"]] <- PercentageFeatureSet(CM1_2, pattern = "ˆMT-")
VlnPlot(CM1_2, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
ncells <- c(500,1000,2000,3000,4000,5000,6000,7000,8000,9000,10000)
multiplets <- c(0.4,0.8,1.6,2.3,3.1,3.9,4.6,5.4,6.1,6.9,7.6)
curva <- data.frame(multiplets,ncells)
- ggplot(curva, aes(multiplets, ncells)) +
- geom_point() +
- geom_smooth(method = "lm")
fit <- lm(multiplets ∼ ncells, curva)
model <- function(x){
0.0007589∗x + 0.0527214
}
perc <- model(ncol(CM1_2))
q = (100 - perc)/100
feature_limit <- quantile(CM1_2$nFeature_RNA, q)
VlnPlot(object=CM1_2,c("nFeature_RNA", "nCount_RNA", "percent.mt"), pt.size = 0.1)
CM1_2 <- subset(CM1_2, subset = nFeature_RNA > 200 & nCount_RNA>500 &nFeature_RNA < feature_limit & percent.mt < 20)
CM1_2$group="Moderate"
CM1_2$patient=”CM1_2”
##QC check
VlnPlot(CM1_2, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
save(CM1_2, file="CM1_2.rds")
Batch correction and identification of major immune cell types
-
29.
Normalize the data using default normalization and data scaling from the Seurat package, which is a log normalization and linear model for data scaling.
-
30.
The batch correction package Harmony with Seurat 3 wrapper can be used for data integration due to consideration of computation power (Korsunsky et al., 2019). Process the batch correction with PCA (Principal Component Analysis) using the 5000 most variable genes, and use the first 20–30 independent components for downstream unbiased clustering with a resolution of 0.4.
-
31.
Use the UMAP (Uniform Manifold Approximation and Projection) method for visualization of unsupervised clustering. Use the Seurat RunUMAP function for UMAP reduction using the first 20 harmonized dimensions for immune cell types and 30 harmonized dimensions for immune cell subsets. Apply default settings embedded in the Seurat RunUMAP function, with min.dist of 0.3 and n_neighbors of 30.
Note:
For batch correction, the Harmony package requires less computing power compared to the Seurat Integration vignette.
The number of PCs, genes, and resolution used can vary depending on sample quality. Generally, 20–30 PCs and 2000–5000 most variable genes will suffice, but testing with user-specific data is recommended. An estimation of the number of PCs can be calculated using the JackStrawPlot() or ElbowPlot() functions in Seurat, and in our dataset, 20 or 30 were appropriate, however, users should test their data to determine the optimal number. Please see https://satijalab.org/seurat/articles/pbmc3k_tutorial.html#determine-the-dimensionality-of-the-dataset-1.
Sample code:
library(Seurat)
library(cowplot)
library(harmony)
library(dplyr)
library(ggplot2)
load("CM1_2.rds")
load("CM3_4.rds")
load("CM5.rds")
load("CR13_14.rds")
load("CR15_16.rds")
load("CR17_18.rds")
load("CS7_8.rds")
load("CS9_10.rds")
load("CS11_12.rds")
covid=merge(x=CM1_2, y=c(CM3_4, CM5, CR13_14, CR15_16, CR17_18, CS7_8, CS9_10, CS11_12))
- covid<- covid%>%
- Seurat::NormalizeData(verbose = T) %>%
- FindVariableFeatures(selection.method = "vst", nfeatures = 5000) %>%
- ScaleData(verbose = T, features = rownames(covid)) %>%
- RunPCA(pc.genes = covid@var.genes, npcs = 20, verbose = T)
- covid<- covid%>%
- RunHarmony("patient", plot_convergence = TRUE)
- covid<- covid%>%
- RunUMAP(reduction = "harmony", dims = 1:20) %>%
- FindNeighbors(reduction = "harmony", dims = 1:20) %>%
- FindClusters(resolution = 0.4) %>%
- identity()
DimPlot(covid, reduction="umap", label=T)
save(covid, file="covidharmony.rds")
Identification of major immune cell types
-
32.Identify major immune cell types using the key genes presented in Table 1 and illustrated in Figure 4, and verify cluster identification using at least one other method (see also Notes below this step).
-
a.T cells can be identified as clusters expressing CD3G and either CD4 (CD4 T cells) or CD8A and CD8B (CD8 T cells) (Figure 4A).
- b.
-
c.Monocytes share expression of TYROBP with NK cells, but they cluster separately from NK cells and T cells (Figure 4B). They also express CD14 and/or FCGR3A.
- d.
-
e.B cells are identified as cells expressing MS4A1 (encodes CD20), although antibody-producing plasma cells lack MS4A1 expression (Figure 4D).
- f.
-
a.
Note:
Contaminating erythrocytes (HBA2) and platelets (PF4) can be identified and excluded along with any other undesired or undefined populations (Figures 4E and 4F).
After major immune cell types have been defined, further dimensionality reduction analyses can be performed to identify subsets of immune cells (see Table 2 for key genes). For instance, performing dimensionality reduction of B and plasma cells permits identification of immature, naïve, activated, memory and plasma cells (Figure 5), and classical and non-classical monocytes can also be distinguished by dimensionality reduction of monocytes and DCs (Figure 6).
Investigators should not rely exclusively on the marker genes indicated here but should also check other differentially expressed genes between clusters (e.g., top 10 or top 20) to confirm cell identities. Comparison to Human Cell Atlas (Immune System) or other immune subset RNAseq datasets can be helpful. Cell type identification can also be performed using computational packages such as SingleR, which applies cell identification labels from reference datasets to the test dataset based on gene expression profiles. However, it is important that appropriate reference datasets are used, and the expression of key markers should be verified. Whichever method is used, caution should be exercised to verify that subsets are correctly identified.
Table 1.
Marker genes for identification of major peripheral blood cell type
| Key cluster genes | Other cluster genes | Genes not expressed | |
|---|---|---|---|
| CD4 T cells | CD3G, CD4 | IL7R, TRAT1 | CD8A, CD8B |
| CD8 T cells | CD3G, CD8A, CD8B |
CCL5, GZMA, GZMH, NKG7, CTSW |
CD4 |
| NK cells | TYROBP, FCGR3A |
CCL5, GZMA, GZMH, NKG7, CTSW |
CD14 |
| Monocytes and cDCs | TYROBP, CD14/FCGR3A/HLA genes | LYZ, S100A8, FCN1 | |
| pDCs |
JCHAIN, SERPINF1, LILRA4 |
PLD4, ITM2C, MZB1 | |
| B cells | MS4A1 |
CD79A, BANK1, IGHD, IGHM |
|
| Plasma cells | JCHAIN | ITM2C, MZB1 |
MS4A1, SERPINF1, LILRA4 |
| Erythrocytes | HBA1, HBA2, HBB | CA1 | |
| Platelets | PF4 | PPBP |
Figure 4.

Identification of major blood cell subsets
UMAP of all cells. (A–E) Major immune cell subsets were identified as: A) CD4 and CD8 T cells (CD3G and either CD4 or CD8B); (B) NK cells (cells expressing TYROBP and FCGR3A that cluster with CD8 T cells), monocytes (cells expressing CD14 and/or FCGR3A that cluster together), and cDCs (express high HLA genes and cluster near monocytes); (C) proliferating lymphocytes (MKI67 clustering with lymphocytes); (D) B cells (MS4A1), plasma cells (express JCHAIN but lack MS4A1 and SERPINF1), and pDCs (JCHAIN and SERPINF1); and E) erythrocytes (HBA2) and platelets (PF4) cells. (F) Identified immune cell subsets are indicated. Undefined and undesired subsets such as erythrocytes and platelets can be excluded from subsequent analysis. UMAPs before and after exclusion are shown. Figure modified from (Yao et al., 2021).
Table 2.
Identification of immune cell subsetsa
| UMAP clusters | Subset | Key cluster genes |
|---|---|---|
| B cells and plasma cells | immature | IL7R |
| mature and activated | IGHM, IGHD, IL4R | |
| activated | CD69 | |
| plasma cells | CD27, CD38, JCHAIN | |
| memory | AIM2 | |
| Monocytes and DCs | classical monocytes | CD14 |
| non-classical monocytes | FCGR3A | |
| cDCs (DC2) | high HLA genes and CD74 plus CD1C, FCER1A, CLEC10A (DC2 markers) | |
| pDCs | SERPINF1, LILRA4 |
Note that the immune cell subsets are representative examples and are not meant to be comprehensive.
Figure 5.

Identification of B and plasma cell subsets
UMAP of B and plasma cells only.
(A) B and plasma cell clusters were identified as immature B cells (IL7R), naïve and activated B cells (IGHM, IGHD, IL4R), activated B cells (CD69), plasma cells (CD27, CD38, JCHAIN) and memory B cells (AIM2).
(B) Identified subsets are indicated.
Figure 6.

Identification of monocyte and DC subsets
UMAP of monocytes and DCs only.
(A) Monocyte clusters and pDCs were identified as classical monocytes (CD14), non-classical monocytes (FCGR3A), and pDCs (SERPINF1, LILRA4).
(B) cDCs are predominantly DC2 cells (HLA-DRB1, CD1C, FCER1A, CLEC10 and low or no monocyte gene expression).
(C) Identified subsets are indicated.
Statistical analyses
Differential gene expression (DEG)
Once major cell types have been identified, up- or downregulated genes in a cell type of interest from COVID-19 patient groups can be determined.
-
33.
Calculate differentially expressed genes between groups for each cell type using Mode-based Analysis of Single-cell transcriptomics (MAST) (Finak et al., 2015). Comparisons are made between two patient groups at a time, for example CD8 T cells from Severe vs Moderate COVID-19 or Severe vs. Recovering.
Sample code:
load("CD8.rds")
library(ggplot2)
library(Seurat)
library(cowplot)
library(dplyr)
library(ggpubr)
library(calibrate)
library(MAST)
Idents(CD8)="group"
DimPlot(CD8, label=T, split.by="group")
levels(CD8)=c("Moderate", "Severe", "Recovering")
CD8$celltype=Idents(CD8)
CD8_Moderate_vs_Severe=FindMarkers(CD8, ident.1="Moderate", ident.2="Severe", test.use="MAST")
write.csv(CD8_Moderate_vs_Severe, "CD8_Moderate_vs_Severe.csv")
-
34.
Transfer CSV files to Excel and sort the data into genes that are upregulated or downregulated between groups, with an adjusted P-value (FDR) cutoff of < 0.01 (or other thresholds as deemed appropriate).
Note: Comparisons in gene expression are made based on what is defined as “ident.1” and “ident.2”. If the average log fold change is positive, this indicates gene expression is increased in ident.1 compared to ident.2. If the average log fold change is negative, gene expression is decreased in ident.1 vs ident.2. Because lists of DEGs are loaded into other analysis platforms later, it is important to name DEG lists in such a way that it is clear what comparison is being made based on what is defined as ident.1 and ident.2.
Functional enrichment analysis
-
35.
Upload gene lists of up- or downregulated DEGs (FDR < 0.01) into Webgestalt (Liao et al., 2019) to perform over-representation analysis using the Gene Ontology (GO) database (http://geneontology.org) for non-redundant biological processes in each immune cell type. This allows identification of biological processes that are over-represented from user provided DEGs between severity groups in each cell type. Results are based on the hypergeometric test to evaluate enrichment P-values for GO biological processes which are then adjusted for multiple comparisons (FDR < 0.05). Enriched gene sets provide an overview of altered biological processes in circulating immune cell types between patient groups.
-
36.
Download gene sets for further analysis such as calculation of pathway module scores.
Note: Many alternative programs for functional enrichment analysis are available, such as DAVID (https://david.ncifcrf.gov/tools.jsp), EnrichR (https://maayanlab.cloud/Enrichr/), and Gene Set Enrichment Analysis (GSEA, https://www.gsea-msigdb.org/gsea/index.jsp).
Pathway module analysis
-
37.
Use enriched gene sets for biological processes of interest returned from functional enrichment analysis to define pathway modules for each cell type.
-
38.
Use the union of genes listed in the enriched gene sets in all COVID-19 groups for biological processes of interest to determine pathway modules within each immune compartment. For example, if “response to type I interferon” in CD8 T cells is enriched in Moderate vs Severe groups, and in Severe vs Recovering groups, the union of both gene sets forms the pathway module.
-
39.
Calculate pathway module scores using the AddModuleScore function of the Seurat package, which calculates the average expression of each gene signature list subtracted from the aggregated expression of control feature sets. All analyzed features are binned based on averaged expression, and the control features are randomly selected from each bin. Pathway module scores can be calculated for single cells and individual patients within groups.
Sample code for pathway module scores for the biological process “response to type I interferon” in single cells:
load("CD8.rds")
library(ggplot2)
library(Seurat)
library(cowplot)
library(dplyr)
library(future)
library(ggpubr)
library(calibrate)
Idents(CD8)="group"
levels(CD8)=c("Moderate", "Severe", "Recovering")
CD8$group=Idents(CD8)
Idents(CD8)="subtype"
#Define list for module
responseT1IFN<-list(c("MX1", "ISG15", "IFI6", "IRF7", "MX2", "STAT1", "ISG20", "XAF1", "IFITM1", "BST2", "SP100", "IFIT3", "IFIT1", "OAS1", "OAS2", "IFITM2", "IFI35", "ADAR", "SAMHD1", "HSP90AB1", "STAT2", "IFNAR1", "SETD2", "OASL", "PTPN11", "C19orf66", "PSMB8", "IRF9", "EIF2AK2", "NDUFA13", "PLSCR1"))
#Module score and stats
my_comparisons <- list(c("Moderate", "Severe"), c("Moderate", "Recovering"), c("Recovering", "Severe"))
Method <- "kruskal.test"
Test <- "wilcox.test"
CD8=AddModuleScore(object = CD8, features = responseT1IFN, ctrl = 100, name = "responseT1IFN1")
Idents(CD8)="group"
#Clean graph, no stats
- VlnPlot(object = CD8, features = c("responseT1IFN11"), pt.size = 0.0000, cols=c("#03A0FF", "#FF2600", "#7E7D7E")) +
- stat_summary(fun = mean, geom='errorbar',aes(ymax = ..y.., ymin = ..y.., group = factor("group")),
- width = 0.75, linetype = "dashed", colour="black", size=1.5)+xlab("Patient Group")+
- ylab("Response to type I interferon")+
- ggtitle(label = element_blank())+theme(axis.text.x = element_text(angle = 30))
#Use this to plot default meta data, which is cells
responseT1IFN.score3=CD8@meta.data
#Makes dot plot of cells with stats
- ggdotplot(responseT1IFN.score3, "group", "responseT1IFN11", order=c("Moderate", "Severe", "Recovering"),
- color="black", legend="none", xlab="Patient Group",
- ylab="response to type I IFN", binwidth=0.03)+stat_compare_means(comparisons=my_comparisons)+
stat_compare_means(method=Method, paired=FALSE, size=6)+
- stat_summary(fun = mean, geom='errorbar',aes(ymax = ..y.., ymin = ..y.., group = factor("group")),
- width = 0.5, linetype = "dashed", colour="black", size=.75)+geom_boxplot()
Sample code for pathway module scores for the biological process “response to type I interferon in individual patients:
#Module score and stats
my_comparisons <- list(c("Moderate", "Severe"), c("Moderate", "Recovering"), c("Recovering", "Severe"))
Method <- "kruskal.test"
Test <- "wilcox.test"
CD8=AddModuleScore(object = CD8, features = responseT1IFN, ctrl = 100, name = "responseT1IFN1")
Idents(CD8)="group"
responseT1IFN.score<-aggregate(CD8$responseT1IFN11, by=list(CD8$patient), FUN=mean)
responseT1IFN.dot<-CD8@meta.data
responseT1IFN.dot$group<-factor(responseT1IFN.dot$group, levels = c("Moderate", "Severe", "Recovering"))
responseT1IFN.dot1<-responseT1IFN.dot[,c( "group", "patient", "responseT1IFN11")]
responseT1IFN.dot2<-aggregate(responseT1IFN.dot1$responseT1IFN 1, by=list(responseT1IFN.dot1$patient), FUN=mean)
colnames(responseT1IFN.dot2)<-c("patient", "responseT1IFN11")
responseT1IFN.dot2<-merge(responseT1IFN.dot2, unique(responseT1IFN.dot1[,c("patient", "group")]))
#Clean graph, no stats
ggdotplot(responseT1IFN.dot2, "group", "responseT1IFN11", order=c("Moderate", "Severe", "Recovering"), color="black", fill=c("#03A0FF", "#03A0FF", "#03A0FF", "#03A0FF", "#03A0FF", "#FF2600", "#FF2600", "#FF2600", "#FF2600", "#FF2600", "#FF2600","#7E7D7E", "#7E7D7E","#7E7D7E","#7E7D7E", "#7E7D7E","#7E7D7E"), legend="none", xlab="Patient Group", ylab="response to type I interferon", binwidth=0.03)+stat_summary(fun = mean, geom='errorbar',aes(ymax = ..y.., ymin = ..y.., group = factor("group")), width = 0.5, linetype = "dashed", colour="black", size=.75)
#Dot plot with stats
- ggdotplot(responseT1IFN.dot2, "group", " responseT1IFN11", order=c("Moderate", "Severe", "Recovering"), color="black", fill=c("#03A0FF", "#03A0FF", "#03A0FF", "#03A0FF", "#03A0FF", "#FF2600", "#FF2600", "#FF2600", "#FF2600", "#FF2600", "#FF2600","#7E7D7E", "#7E7D7E","#7E7D7E","#7E7D7E", "#7E7D7E","#7E7D7E"), legend="none", xlab="Patient Group", ylab="response to type I interferon", binwidth=0.02)+stat_compare_means(comparisons = my_comparisons)+stat_compare_means(method=Method, paired=FALSE, size=6)+
- stat_summary(fun = mean, geom='errorbar',aes(ymax = ..y.., ymin = ..y.., group = factor("group")), width = 0.5, linetype = "dashed", colour="black", size=.75)
Note: Sample code includes plots with and without statistical information; depending on how graphs are formatted it may be preferable to generate graphs in R and add statistical values in a different program better suited for editing.
Canonical pathway, upstream regulator, and causal network analyses
Ingenuity Pathway Analysis (IPA, Qiagen) is commercial software that provides a number of computational methods for functional enrichment and network analysis of transcriptomics (and other omics) data via a graphical user interface. Alternative, non-commercial options for gene product interaction visualization include STRING (https://string-db.org/) as well as Cytoscape (https://cytoscape.org/).
-
40.Use IPA for the following analyses:
-
a.Canonical pathway analysis: Most commonly used functional enrichment algorithms (such as DAVID, Webgestalt, GSEA) designate a pathway or process as being “over-represented” or “enriched” based on the same pattern of expression within genes (i.e., up- or down-regulation). However, activation of a pathway may require concurrent up-regulation of key genes as well as down-regulation of others (such as an inhibitor). IPA’s canonical pathway analysis is based on manually curated and highly granular information on over 700 pathways that incorporates the differential expression pattern of member genes when calculating activation scores. Canonical pathway analysis can be performed on all major immune cell types by inputting differentially expressed genes (FDR < 0.01) for each cell type (classical monocytes, NK cells, CD8 T cells, CD4 T cells, and B cells) in pairwise comparisons of patient conditions (Moderate, Severe, Recovering). Note that for each comparison (e.g., Severe vs. Moderate in a specific cell type) differentially up and down-regulated genes (FDR < 0.01) and their log2[fold change] values should be imported into IPA and a core analysis performed. Enrichment of canonical pathways is determined using Fisher’s exact test with Benjamini Hochberg adjusted P-values (FDR). Simultaneously, pathway activation status is assessed to determine whether significantly enriched pathways (FDR < 0.01) were activated or inhibited based on IPA’s knowledgebase of expected expression and phosphorylation patterns of gene products in a given canonical pathway using a z-score statistic: a positive z-score indicating activation and negative z-score indicating inhibition. A summary bubble chart of canonical pathway analysis is shown in Figure 7.Note:A larger positive or negative z-score implies more confidence in the activation/inhibition status of a given pathway, but not its enrichment.Not every significantly enriched canonical pathway has a z-score due to insufficient information on the expected pattern of expression among its member genes in IPA’s knowledgebase.Non-commercial methods for building gene regulatory networks such as SCENIC (Van de Sande et al., 2020) and NicheNet (Browaeys et al., 2020), are alternatives to IPA.
-
b.Upstream regulator/mechanistic network and causal network analyses: These can be performed as part of IPA’s core analysis using differentially expressed genes (FDR < 0.01) for each immune cell type in pairwise comparisons of COVID-19 conditions. The goal of this analysis is to identify regulators whose activation or inhibition is expected to result in gene expression patterns similar to those from the user’s inputted data. The direction of expression of the differentially expressed genes is compared to IPA’s knowledgebase using a statistical model (Kramer et al., 2014) to identify key putative regulators and construct a mechanistic regulatory network. An overlap P-value is used to measure enrichment of network-regulated genes in the dataset and an activation z-score is calculated to identify likely regulating molecules based on statistically significant patterns of up- and downregulation as well as the expected activation state (activated or inhibited) of each regulator.
-
a.
Notes:
In our experience, interpretation of the results from this analysis can be challenging. For the upstream regulator/mechanistic network analysis, one issue we faced was identification of a very large number of candidate regulators in each comparison (e.g., Severe vs. Moderate in NK cells), which was further compounded by the many different comparisons being performed. To overcome this challenge, we focused on the Severe vs. Moderate COVID-19 comparison and selected one representative upstream regulator, IRF7, to build a mechanistic regulatory network (Figure 8). IRF7 was chosen because it was consistently identified as one of the most significantly enriched upstream regulators across cell types (except monocytes). In addition, although a regulator’s own differential expression in the inputted data is not considered when calculating its significance by IPA, IRF7 was highly differentially expressed across immune cell types in our data, further increasing confidence in its selection as a critical coordinator of the host immune response in COVID-19.
Similar challenges can be encountered when applying the causal network analysis in IPA. While resembling upstream regulator analysis in some respects, causal network analysis builds relationships based on identifying a “master regulator” that modulates a number of “intermediate regulators” whose downstream targets correspond to the gene expression patterns inputted by the user (Kramer et al., 2014). Once again, this analysis yielded many potential master regulators in our dataset given the many cell types and comparison groups. Since a unique feature of this project was inclusion of patients recovering from severe SARS-CoV-2 infection, which provided an opportunity to investigate development of humoral immunity in COVID-19, we focused on B cell transcriptional responses between Recovering vs. acute Severe patients by performing causal network analysis on differentially upregulated genes in Recovering patients. Furthermore, similar to the upstream analysis, we selected the most significant candidate master regulator that was itself differentially expressed in the scRNA-seq B cell data—SYK. The resulting causal network’s structure was built on SYK as its “root” and was connected to 40 other regulators which in turn modulated 139 DEGs in B cells of Recovering vs. Severe COVID-19 patients (Figure 9).
Figure 7.

Canonical pathway analysis
For each immune cell type, differentially expressed genes between Severe vs. Moderate and Severe vs. Recovering COVID-19 patients were imported into IPA for core analysis. Significantly enriched canonical pathways between disease group comparisons across cell types were identified using FDR < 0.01. The activation/inhibition state of a given pathway was determined using z-scores. The bubble chart depicts a select number of canonical pathways that were significantly enriched in most cell types between patient groups. Figure modified from (Yao et al., 2021).
Figure 8.

Upstream regulator analysis
(A) Differentially expressed gene patterns for immune cells were leveraged to identify IRF7 as a putative master regulator in Severe vs. Moderate COVID-19. The regulatory network, with IRF7 as the key orchestrator, was constructed based on the overlap between the patterns of differential gene expression and IPA’s knowledgebase across cell types as assessed by Fisher's exact test P-value and a z-score, with a positive z-score indicating activation and negative z-score indicating inhibition. Note that each member of this network is itself a regulator of other gene targets in each cell type.
(B) Heatmap highlighting whether a given member of the regulatory network is expected to be activated or inhibited in each immune cell population in Severe versus Moderate groups. This analysis implies that the IRF7 network is inhibited in monocytes, but activated in other immune cells. Figure modified from (Yao et al., 2021).
Figure 9.

Causal network analysis
IPA causal network analysis was applied to B cell transcriptional profiles of Recovering vs. Severe groups to identify putative mechanistic relationships between regulators. One of the most significantly enriched “master regulators” was SYK, which was itself differentially upregulated, and orchestrated a causal network comprised of 40 other regulators. This analysis suggests that activation of SYK-regulated pathways is a key driver of humoral responses in patients recovering from severe SARS-Cov2 infection. Figure modified from (Yao et al., 2021).
Expected outcomes
Following these step-wise guidelines should provide investigators with the necessary tools to successfully extract single cell-level transcriptional information from peripheral blood immune cells of COVID-19 patients. Below we describe additional issues, how to address them, and the expected outcomes for this protocol.
Using this protocol, it is possible to identify major immune cell types, including CD4 and CD8 T cells, NK cells, B and plasma cells, monocytes, cDCs and pDCs (Figure 4). Undefined or undesired cells, such as contaminating erythrocytes and platelets, can be excluded from subsequent analysis (Figure 4F).
It may be possible to identify subsets of major immune cell types when performing dimensionality reduction with all PBMCs. Alternatively, additional dimensionality reduction analyses can be performed to facilitate subset identification, such as analysis of B and plasma cells to reveal naïve, activated and memory B cells and plasma cells (Figure 5), and monocytes and DCs to more clearly visualize clusters of classical versus non-classical monocytes, cDCs and pDCs (Figure 6). As expected, we observed only small proportions of cDCs and pDCs (Figures 4 and 6). Expression of FCER1A and CLEC10 with low or no expression of monocyte genes indicated that the cDCs were predominantly DC2 cells (Figure 6C).
Limitations
Isolating, processing and performing scRNA-seq in circulating immune cells of COVID-19 patients is a complex and time-consuming task. This protocol summarizes our experiences, challenges and solutions for this project. Several important limitations and suggested solutions are discussed below.
Neutrophils are very sensitive to freezing, so they were not present in the illustrated dataset, but it may be possible to preserve and detect neutrophils if fresh rather than frozen samples are used. We have not, however, evaluated the impact of methanol fixation on neutrophils.
Viability of frozen buffy coat cells (60%–70%) is lower than PBMCs isolated from fresh blood samples using a density medium such as Ficoll or Leukopak. Erythrocyte contamination can also be expected, but erythrocytes can be reduced by FACS sorting. Thus, buffy coats are a good alternative if PBMC isolation is not available with your preferred bio-banking center.
We attempted to multiplex our samples using cell hashing (Stoeckius et al., 2018) by staining the cells with TotalSeq cell hashing antibodies (BioLegend) immediately after FACS sorting, but unfortunately, we found that this approach was incompatible with methanol fixation and we were unable to demultiplex the combined samples. Therefore, if methanol-fixed samples need to be analyzed separately, they must be captured separately, and if samples are combined for capture, they must be from the same patient group. Alternative methods that might be useful are also discussed in the troubleshooting section.
Immune cell subsets that have not been well characterized by scRNA-seq may be difficult to identify. Cell annotation packages such as SingleR (https://bioconductor.org/packages/release/bioc/html/SingleR.html) can be useful but should be used with caution because reliability will depend on the reference datasets used to define them (Aran et al., 2019). Cell activation states may also be difficult to distinguish if they exist as a continuum rather than discrete populations of naïve vs. activated vs. memory cells, but pseudotime trajectory analysis may be useful in these cases (Cano-Gamez et al., 2020).
Pathway analysis using IPA can often result in identification of many upstream regulators and causal networks. While this may be helpful from an exploratory standpoint, it is often difficult to digest the large amount of information generated. We opted to limit our analysis to the most statistically significant candidates that were themselves differentially expressed in our data.
Troubleshooting
Problem 1
Cell clumps and clogs during sorting (before step 1).
Potential solution
Add 2–5 mM EDTA to the cell suspension prior to sorting to reduce cell clumping. Filter cells before sorting using 70 μm strainers. Utilize multiple sorters to reduce the time needed to sort cells.
Problem 2
Limited sorter availability (before step 1).
Potential solution
If it is not possible sort all the samples in one day, sorting can be spread out over a period of time. This has the potential to introduce batch effects, however, so if simultaneous sorting with multiple sorters is not an option, care should be taken to randomize samples, use the same reagents and procedures throughout all the sample processing steps, and check that batch correction successfully removes batch effects during the data analysis.
Problem 3
Erythrocyte contamination (step 5).
Potential solution
Remove erythrocytes by FACS sorting and/or by excluding erythrocytes identified during scRNA-seq analysis.
Problem 4
Cell hashing didn’t work for methanol-fixed cells (before step 6).
Potential solution
We only evaluated cell hashing with oligonucleotide-barcoded antibodies before methanol fixation; we did not evaluate antibody labeling after methanol fixation. Oligonucleotide-barcoded lipids, which can also be used to label whole cells and nuclei (McGinnis et al., 2019) are now available as CellPlex from 10X Genomics, although it is unclear whether methanol fixation would also interfere with lipid labeling. Alternatively, a recent report demonstrated sample demultiplexing by computational analysis of SNPs (Kang et al., 2018), which might be an option if it isn’t possible to hash the samples for multiplexing.
Problem 5
Too many regulator candidates identified in IPA (step 37).
Potential solution
This is a common issue when using IPA’s upstream and master regulator features. Refining significant candidates will depend on the aim of the project; for example, it is possible to limit identified regulators to therapeutic drugs or small molecules if that is the primary goal. In our case, we selected a regulator by requiring it to be the highest ranked candidate identified by IPA that was itself significantly differentially expressed in our dataset. Our primary motivation was to define a limited set of upstream and master regulators that were highly likely to coordinate the observed transcriptional response of immune cells in COVID-19. However, a limitation of this approach is that potentially important regulators can be post-transcriptionally modified and therefore not be differentially expressed. Finally, alternative methods for identifying regulatory networks in scRNA-seq data are freely available (Browaeys et al., 2020; Van de Sande et al., 2020) and can be compared to IPA.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Sina Gharib (sagharib@uw.edu).
Materials availability
This study did not generate new unique materials.
Data and code availability
All code used for data analysis not included in this manuscript is available upon request.
Acknowledgments
We thank the staff of the Biobank & Translational Research Core, the Flow Cytometry Core, and the Applied Genomics, Computation & Translational Core for their help in this project. This study was funded by the Parker B. Francis Foundation Fellowship (to C.Y.), Plum Foundation Funding (to P.C.), UCLA CTSI KL2- NCATS KL2TR001882 (to C.Y.), and NIH grants T32HL134637 (to S.A.B.), R01AI134987 (to H.S.G.), R01AI137111 (to S.A.G.), and R01HL137076 (to P.C.).
Author contributions
Conceptualization, C.Y., S.A.B., S.A.G., H.S.G., and P.C.; methodology and formal analysis, C.Y., S.A.B., S.A.G, and H.S.G.; data curation, C.Y., S.A.B., H.S.G., and P.C.; writing – original draft, C.Y., S.A.B., H.S.G., and S.A.G.; visualization, C.Y., S.A.B., S.A.G., H.S.G., and P.C.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Changfu Yao, Email: changfu.yao@csmc.edu.
Helen S. Goodridge, Email: helen.goodridge@csmc.edu.
Sina A. Gharib, Email: sagharib@uw.edu.
References
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- Cano-Gamez E., Soskic B., Roumeliotis T.I., So E., Smyth D.J., Baldrighi M., Wille D., Nakic N., Esparza-Gordillo J., Larminie C.G.C. Single-cell transcriptomics identifies an effectorness gradient shaping the response of CD4(+) T cells to cytokines. Nat. Commun. 2020;11:1801. doi: 10.1038/s41467-020-15543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro G., Mulay A., Yao C., Mizuno T., Konda B., Petrov M., Lafkas D., Arron J.R., Hogaboam C.M., Chen P. Single-cell reconstruction of human basal cell diversity in normal and idiopathic pulmonary fibrosis lungs. Am. J. Respir. Crit. Care Med. 2020;202:1540–1550. doi: 10.1164/rccm.201904-0792OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Cheung F., Shi R., Zhou H., Lu W., Consortium C.H.I. PBMC fixation and processing for Chromium single-cell RNA sequencing. Journal of Translational Medicine. 2018;16 doi: 10.1186/s12967-018-1578-4. In this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A., Green J., Pollard J., Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis C.S., Patterson D.M., Winkler J., Conrad D.N., Hein M.Y., Srivastava V., Hu J.L., Murrow L.M., Weissman J.S., Werb Z. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods. 2019;16:619–626. doi: 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M., Zheng S., Houck-Loomis B., Hao S., Yeung B.Z., Mauck W.M., 3rd, Smibert P., Satija R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19:224. doi: 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande B., Flerin C., Davie K., De Waegeneer M., Hulselmans G., Aibar S., Seurinck R., Saelens W., Cannoodt R., Rouchon Q. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020;15:2247–2276. doi: 10.1038/s41596-020-0336-2. [DOI] [PubMed] [Google Scholar]
- Yao C., Bora S.A., Parimon T., Zaman T., Friedman O.A., Palatinus J.A., Surapaneni N.S., Matusov Y.P., Cerro Chiang G., Kassar A.G. Cell-type-specific immune dysregulation in severely Ill COVID-19 patients. Cell Rep. 2021;34:108590. doi: 10.1016/j.celrep.2020.108590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young M.D., Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience. 2020;9:giaa151. doi: 10.1093/gigascience/giaa151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All code used for data analysis not included in this manuscript is available upon request.