

Abstract
Pre-exposure prophylaxis (PrEP) using antiretroviral oral drugs is effective at preventing HIV transmission when individuals adhere to the dosing regimen. Tenofovir alafenamide (TAF) is a potent antiretroviral drug, with numerous long-acting (LA) delivery systems under development to improve PrEP adherence. However, none has undergone preventive efficacy assessment. Here we show that LA TAF using a novel subcutaneous nanofluidic implant (nTAF) confers partial protection from HIV transmission. We demonstrate that sustained subcutaneous delivery through nTAF in rhesus macaques maintained tenofovir diphosphate concentration at a median of 390.00 fmol/106 peripheral blood mononuclear cells, 9 times above clinically protective levels. In a non-blinded, placebo-controlled rhesus macaque study with repeated low-dose rectal SHIVSF162P3 challenge, the nTAF cohort had a 62.50% reduction (95% CI: 1.72% to 85.69%; p=0.068) in risk of infection per exposure compared to the control. Our finding mirrors that of tenofovir disoproxil fumarate (TDF) monotherapy, where 60.00% protective efficacy was observed in macaques, and clinically, 67.00% reduction in risk with 86.00% preventive efficacy in individuals with detectable drug in the plasma. Overall, our nanofluidic technology shows potential as a subcutaneous delivery platform for long-term PrEP and provides insights for clinical implementation of LA TAF for HIV prevention.
Keywords: Nanofluidics, HIV pre-exposure prophylaxis, tenofovir alafenamide, implantable devices, drug delivery
Graphical Abstract
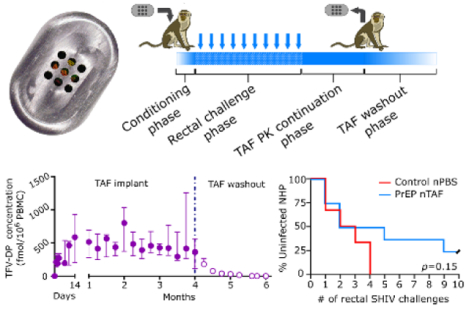
The Grattoni group performed the first HIV pre-exposure prophylaxis (PrEP) assessment of an implantable long-acting antiretroviral platform. In this foremost study, the partial protection of simian HIV with tenofovir alafenamide (TAF) delivered by a nanofluidic implant was demonstrated in nonhuman primates.
1. Introduction
The approval of Descovy® (200 mg emtricitabine [FTC]/25 mg tenofovir alafenamide [TAF]) as the second HIV pre-exposure prophylaxis (PrEP) medication, following Truvada® (200 mg FTC/300 mg tenofovir disoproxil fumarate [TDF]) is fueling global efforts to end the AIDS pandemic by 2030.[1] Compared to Truvada®, Descovy® offers safety advantages with lower systemic tenofovir (TFV) concentrations without compromising overall preventive efficacy in men who have sex with men (NCT02842086).[2] The efficacy of these agents to prevent sexual HIV infection is exceptional, provided that individuals strictly adhere to the dosing regimen.[3–5] According to the iPrEx study, seven doses of Truvada® per week correlated with 99% PrEP efficacy, whereas the rate dropped to 76% with two doses per week.[6] Motivated by challenges of pill fatigue and PrEP accessibility, various biomedical developments have emerged aiming at improving therapeutic adherence and expanding HIV PrEP implementation.
Long-acting (LA) antiretroviral (ARV) formulations and delivery systems offer systemic delivery for prolonged periods, obviating the need for frequent dosing. Currently, LA ARV strategies for HIV PrEP are largely geared towards developing single-agent drugs for prevention instead of combinatorial formulations.[7–16] Focusing on a single drug allows for maximal drug loading, while minimizing injection volumes (for injectables). In the case of LA ARV implants, a single drug formulation affords smaller size dimensions for minimally-invasive and discreet implantation.[17, 18] Importantly, single-agent LA ARVs offer benefits of cost-effectiveness as well as reduced complexity in terms of development. Of relevance, a single-agent injectable LA ARV, cabotegravir, is currently in clinical trials for PrEP efficacy evaluation (NCT02076178, NCT02178800, NCT02720094, NCT03164564).[19, 20] Thus far, islatravir (MK-8591) remains the only single-agent ARV LA ARV implant to reach clinical testing for safety and pharmacokinetics assessment.[21]
Given the potency and safety advantages of TAF compared to TDF, numerous LA TAF strategies are under development involving biodegradable[9–11] or non-biodegradable[12, 13] polymeric implants, transcutaneously refillable devices[14], and an osmotic pump system.[15] While some LA TAF systems have achieved targeted preventive tenofovir diphosphate (TFV-DP) concentrations in peripheral blood mononuclear cells (PBMC) (40.0 fmol/106 cells)[9, 12, 14], none has undergone efficacy studies for protection from HIV transmission. Thus, considering the concentrated research efforts on developing LA TAF systems, it is of utmost importance to evaluate the efficacy of LA TAF as a single-agent drug for HIV prevention.
Here, we present the first efficacy study of LA TAF for HIV PrEP. We used a nonhuman primate (NHP) model of repeated low-dose rectal challenge with simian HIVSF162P3 (SHIVSF162P3), which recapitulates human HIV transmission. We assessed the efficacy of sustained subcutaneous delivery of TAF via a novel nanofluidic (nTAF) implant as a single-agent PrEP regimen for protection from SHIVSF162P3 infection. We investigated the pharmacokinetics and biodistribution of TAF, as well as safety and tolerability of the implant.
2. Results
2.1. Nanofluidic implant assembly
We leveraged a newly designed silicon nanofluidic membrane technology[22] for sustained drug elution independent of actuation or pumps. The nanofluidic membrane (6 mm × 6 mm with a height of 500 μm) is mounted within a medical-grade titanium drug reservoir (20 mm length × 13 mm width × 4.5 mm height) (Figure 1A). The nanofluidic membrane contains 199 circular microchannels, each measuring 200 μm in diameter and 490 μm in length. Hexagonally distributed in a circular configuration (Figure 1B), each microchannel leads to 1400 parallel slit-nanochannels (Figure 1C), for a total of 278,600 nanochannels per membrane. The nanochannels (length 10 μm, width 6 μm) are densely packed in square arrays organized in circular patterns. The whole membrane surface is coated by an innermost layer consisting of silicon dioxide (SiO2), and a surface layer of silicon carbide (SiC), which provides biochemical inertness for long term implantable applications (Figure 1D).[23, 24]
Figure 1.
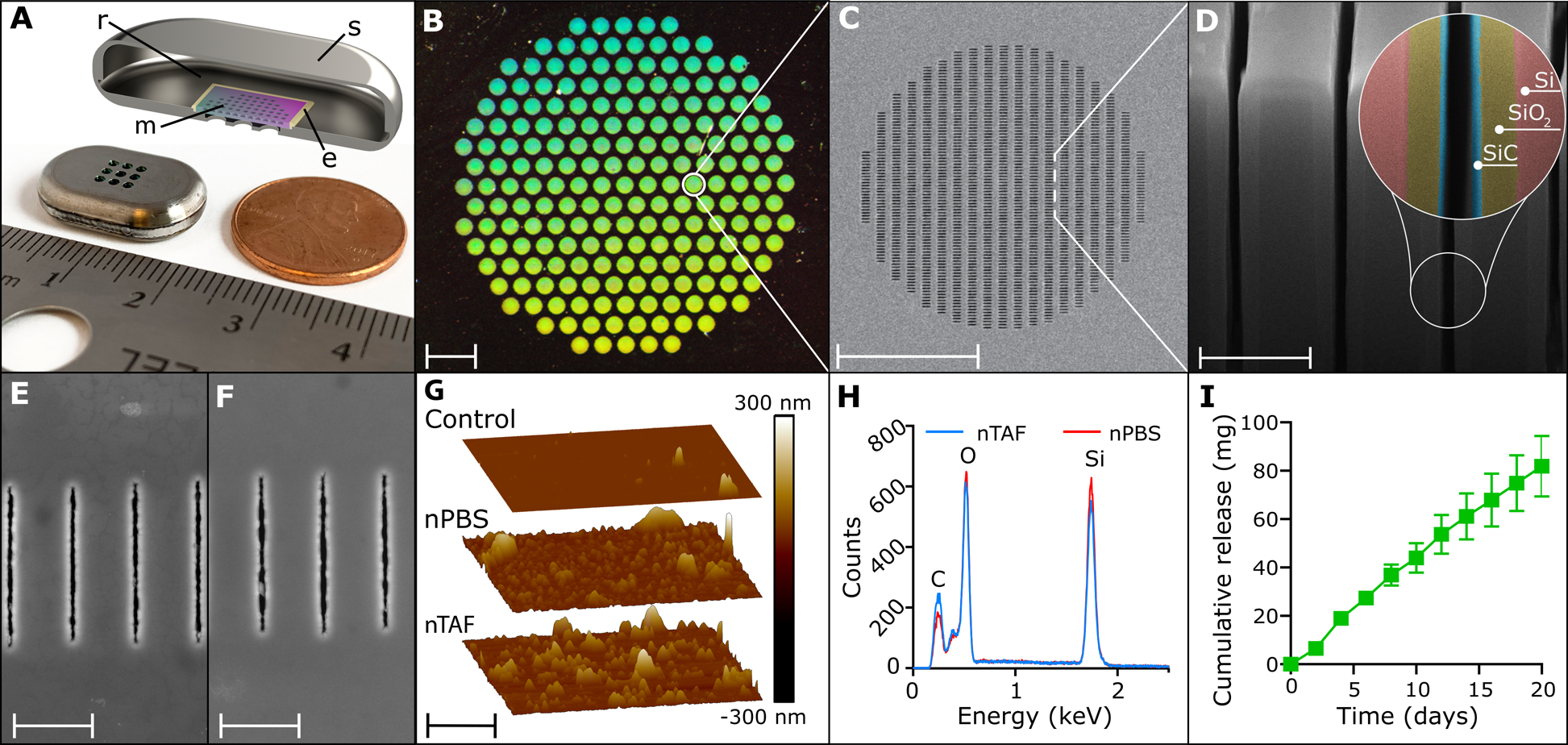
The nanofluidic implant for subcutaneous TAF HIV PrEP delivery. A) Rendered image of cross-section of titanium drug reservoir where r is the reservoir, s the implant shell, m is the nanochannel membrane, and e the epoxy. B) Assembled titanium TAF drug reservoir with 200 nm nanofluidic membrane. Image taken at 0.5 × magnification, scale bar is 1 mm. C) Top-view of SEM image of nanochannel membrane. Scale bar is 100 μm. D) FIB image of nanochannel membrane cross-section displaying perpendicular nanochannels. Zoom-in on nanochannel layers colored for identification. Scale bar is 2 μm (SiC, silicon carbide, SiO2, silicon oxide, Si, silicon). E) Representative top view SEM image of nanochannel membrane from nTAF after 4 months in vivo. Scale bar is 2.5 μm. F) Representative top view SEM image of nanochannel membrane from nPBS after 4 months in vivo. Scale bar is 2.5 μm. G) Comparison of representative AFM image of control membrane prior to implantation with nPBS and nTAF membranes after 4 months in vivo. Scale bar is 2.5 μm. H) EDX analysis of surface elements below SiC coating of membrane from nTAF compared to nPBS after 4 months in vivo. I) Cumulative release of drug in vitro (mean ± SEM) from nTAF into sink solution (n=5).
Drug diffusion across the membrane is driven by concentration difference between the drug reservoir and the subcutaneous space. The drug is loaded in the implant in powder form and is continuously solubilized in the interstitial fluids penetrated within the implant via capillary wetting of the membrane. Drug release is determined by both nanochannels and drug solubilization kinetics. Within the nanochannels, diffusivity of drug molecules is defined by steric and electrostatic interactions with channel walls. The size of nanochannels is selected to saturate drug transport, rendering it steady and independent from the concentration gradient.[25, 26] The release rate can be finely tuned by selecting the suitable number of nanochannels per membrane.[27] Therefore, the nanofluidic membrane passively achieves constant and sustained drug delivery obviating the need of mechanical components.[28–31]
In this study, we evaluated suitable nanochannel sizes through a molecular transport in silico model previously developed by Di Trani et al.[25], which is based on the molecular size and physicochemical properties of TAF. A nanochannel size of ~190 nm was ultimately selected. PrEP implants were loaded with solid powder TAF (nTAF), while control implants were loaded with phosphate buffered saline (nPBS) and welded shut. Membrane stability was evaluated after 4 months of subcutaneous implantation via scanning electron microscopy (SEM) (Figure 1E and F) along with atomic force microscopy (AFM) (Figure 1G) and energy dispersive x-ray spectroscopy (EDX) (Figure 1H). We compared surface morphology between a control non-implanted membrane and nTAF and nPBS membranes implanted after 4 months. We observed similar roughness surface morphology by AFM for the nPBS (Ra, 10.2 nm; Rq, 19.6 nm) membranes and nTAF (Ra, 14.9 nm; Rq, 25.4 nm), with a slight increase in roughness with respect to the control membrane (Ra, 1.23 nm; Rq, 6.15 nm). The EDX showed the same abundance of elements at the surface in both membranes, indicating that TAF does not alter the membrane composition. These results demonstrate that TAF does not affect membrane stability even after prolonged implantation.
Nanochannel size selection was confirmed via in vitro release testing. Short-term in vitro drug release from nTAF showed a linear cumulative release of 81.85 ± 12.55 mg (mean ± SEM) of TAF over 20 days (Figure 1I). However, an increase of TAF degradation products was observed throughout the study, attributable to decrease in TAF stability (Figure S1 and S2, Supporting Information).
2.2. nTAF pharmacokinetic profile in NHP
For in vivo evaluation of pharmacokinetic (PK) and PrEP efficacy, rhesus macaques were subcutaneously implanted with either nTAF (n=8) or control nPBS (n=6) in the dorsum for 4 months. We used TFV-DP concentration in PBMC of 100.00 fmol/106 cells as the benchmark prevention target, which exceeds the clinically protective level in the iPrEX trial.[6, 9] Preventive TFV-DP PBMC concentrations were surpassed one day post-implantation (median, 213.00 fmol/106 cells; IQR, 140.00 to 314.00 fmol/106 cells) and maintained at a median of 390.00 fmol/106 cells (IQR, 216.50 to 585.50 fmol/106 cells) for 4 months (Figure 2A). During the washout period, TFV-DP PBMC concentrations decreased to below the limit of quantitation (BLOQ) within 6 weeks of device retrieval.
Figure 2.
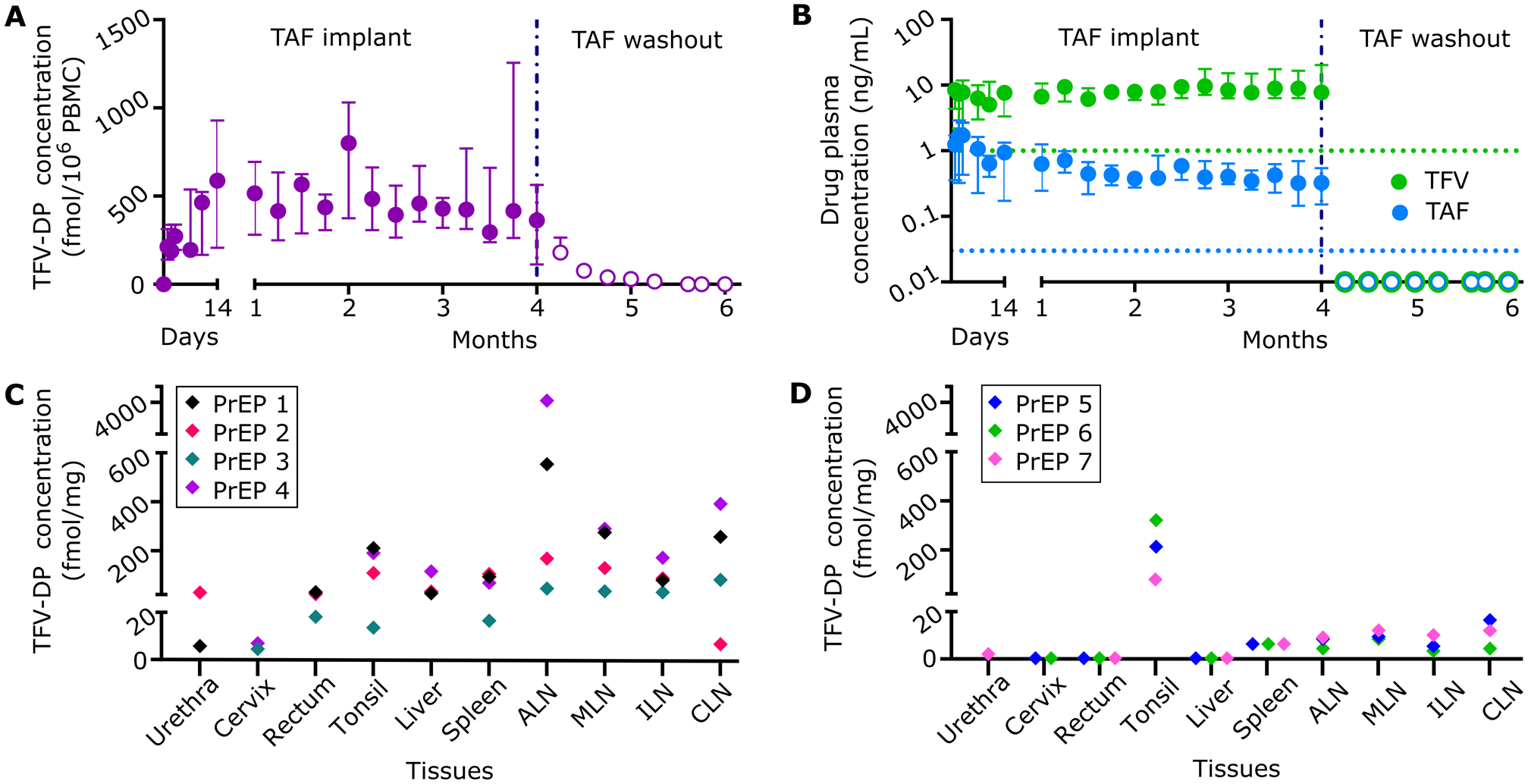
Pharmacokinetics and tissue distribution of TAF from PrEP group implanted with subcutaneous nTAF. nTAF implants (n=7) were retrieved after 4 months and washout concentrations (open circles) were followed in 3 animals. A) Intracellular TFV-DP PBMC concentrations of PrEP cohort throughout the study. B) TAF and TFV concentrations in the plasma of PrEP cohort throughout the study. Green and blue dotted horizontal lines represent lower LOQ TFV and TAF concentrations, 1.00 ng/mL and 0.03 ng/mL, respectively. C) Tissue TFV-DP concentrations upon nTAF removal after 4 months of implantation in a subset of animals (n=4). D) Tissue TFV-DP levels after the 2-month washout period in a subset of animals (n=3). Data are presented as median ± IQR in panels A and B.
Plasma TFV concentrations were consistently higher than plasma TAF for the duration of the PK study (Figure 2B). Notably, TFV concentrations increased as TAF concentrations decreased, beginning at the 3-month time point. This is attributable to the limited stability of TAF and degradation to TFV within the implant, as was observed in vitro (Figure S1 and S2, Supporting Information).[32] Plasma TAF and TFV levels (median, 0.51; IQR, 0.30 to 0.91 ng/mL; and median, 7.81; IQR, 6.17 to 9.97 ng/mL, respectively) were within range of that achieved with oral TAF dosing of NHP.[33] Within a week post-device retrieval, TAF and TFV concentrations were BLOQ.
Estimated half-life (t1/2) PK of TAF and TFV were below 1.87 ± 0.32 and 1.84 ± 0.63 days, respectively, as BLOQ was achieved in under a week (Table 1). Individual TFV-DP concentrations for each animal were fitted to an intravenous bolus injection two-compartment model (Figure S3A–D, Supporting Information). During the washout period, TFV-DP PBMC concentrations had an average first-order elimination rate constant of 0.14 ± 0.028 days−1.
Table 1.
Plasma TAF and TFV half-lives and PBMC TFV-DP elimination rate constant pharmacokinetics in nTAF washout NHPs.
| Analyte | NHP PrEP 5 | NHP PrEP 6 | NHP PrEP 7 | Average | Standard deviation |
|---|---|---|---|---|---|
| Plasma TAF t1/2 (days) | <2.24 | <1.71 | <1.67 | <1.87 | ±0.32 |
| Plasma TFV t1/2 (days) | <2.55 | <1.61 | <1.35 | <1.84 | ±0.63 |
| PBMC TFV-DP k10 (1/day) | 0.18 | 0.13 | 0.13 | 0.14 | ±0.028 |
We measured TFV-DP concentrations after device retrieval (n=4) (Figure 2C) and after the washout period (n=3) (Figure 2D) in tissues relevant to HIV-1 transmission or viral reservoirs. Specifically, we assessed cervix, urethra, rectum, tonsil, liver, spleen, axillary lymph nodes (ALN), mesenteric lymph nodes (MLN), inguinal lymph nodes (ILN), and cervical lymph nodes (CLN). Drug penetration from subcutaneous TAF delivery was observed at varying levels in all tissues after device retrieval (Figure 2C). After the two-month washout period, TFV-DP concentrations were quantifiable in the tonsil, spleen and lymph nodes (Figure 2D) and BLOQ in tissues highly associated with HIV-1 transmission, specifically the cervix and rectum. TFV-DP concentrations in the tonsil were above 75.00 fmol/mg, suggestive of longer clearance or better penetration.
2.3. nTAF efficacy protection against virus
We next assessed whether sustained nTAF delivery as a subcutaneously delivered monotherapy could protect the macaques against rectal SHIVSF162P3 infection. Prior to rectal challenge, the animals were subjected to a two-week “conditioning phase” (Figure 3A) to allow for reaching the benchmark target preventive intracellular TFV-DP PBMC concentrations of 100.00 fmol/106 cells (Figure 2A). Animals in both PrEP (n=8) and control (n=6) cohorts were rectally challenged weekly with low-dose SHIVSF162P3 for up to 10 inoculations and continually monitored for drug PK throughout the study (Figure 3A). The SHIV inoculation dosage used are similar to human semen HIV RNA levels during acute viremia, thus recapitulating high-risk or acute HIV infection in humans. Therefore, this animal model is considered more aggressive, as the risk of infection per exposure markedly exceeds the risk in clinical settings.[34]
Figure 3.
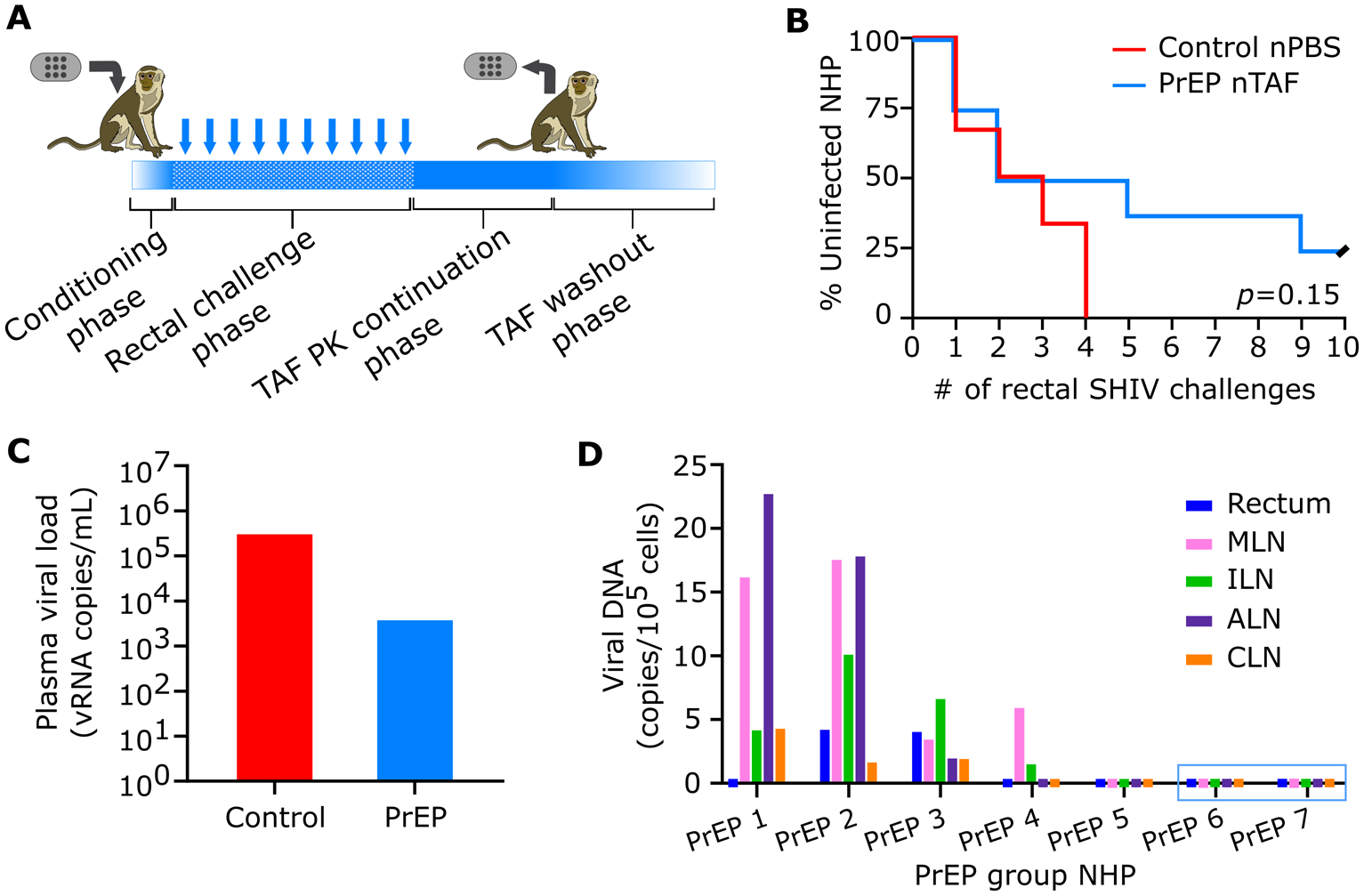
PrEP efficacy of nTAF. A) Schematic of study design. Conditioning phase to reach TFV-DP PBMC concentrations above 100 fmol/106 cells. Rectal challenge phase with up to 10 weekly low-dose SHIVSF162P3 exposures. TAF PK continuation phase followed by nTAF explantation from all animals and euthanasia of 4 animals. TAF washout was observed in the remaining 3 animals for 2 months prior to euthanasia. B) Kaplan-Meier curve representing the percentage of infected animals as a function of weekly SHIV exposure. PrEP (n=8) vs control (n=6) group; censored animals represented with black slash. Statistical analysis by Mantel-Cox test. C) Median peak viremia levels in breakthrough animals at initial viral load detection. D) Cell-associated viral DNA loads of tissues in PrEP group. Animals PrEP 1–5 were infected while PrEP 6 and 7 (blue box) remained uninfected throughout the study. MLN, mesenteric lymph nodes, ILN, inguinal lymph nodes, ALN, axillary lymph nodes, CLN, cervical lymph nodes.
To monitor for SHIVSF162P3 infection, we evaluated weekly cell-free viral RNA in the plasma. Rectal challenges were stopped upon initial detection of plasma viral RNA, which was confirmed after a consecutive positive assay. Two of eight macaques from the nTAF group (25.00%) were uninfected after 10 weekly rectal SHIVSF162P3 challenges (Figure 3B). Based on the number of infections per total number of challenges, the nTAF group had a reduced risk of infection per-exposure of 62.50% (95% CI, 1.72% to 85.69%; p=0.068), in comparison to the control group. However, because of the small sample size, the result is not very precise, as indicated by the lower bound of the confidence interval. Prophylaxis with nTAF increased the median time to infection to 5 challenges compared to 2 challenges in the control cohort (p=0.38). After device explantation, there was no spike in viremia, indicative of PrEP efficacy of nTAF monotherapy in the two uninfected animals. While Kaplan-Meier analysis demonstrated delayed and reduced infection in some animals, there was no statistical significance (p=0.15) between nTAF and nPBS groups.
TAF-treated infected NHPs had blunted SHIV RNA peak viremia (median; 3.80 × 104 vRNA copies/mL; IQR, 1.60 × 103 to 2.09 ×105 vRNA copies/mL) in comparison to control groups (median; 3.01 × 105 vRNA copies/mL; IQR, 9.00 × 103 to 7.25 × 106 vRNA copies/mL) (Figure 3C). However, differences in SHIV RNA levels at initial detection were not statistically significant between control and infected PrEP animals (p=0.18 by Mann-Whitney test).
At euthanasia, we assessed the residual SHIV infection in various tissues collected from the nTAF cohort by measuring cell-associated SHIVSF162P3 provirus DNA (Figure 3D). Tissues from PrEP 1–4 were assessed after 4 months of nTAF implantation, and after 2 months of drug washout for PrEP 5–7. SHIV DNA was detectable in the MLN in 4/5 of the infected PrEP NHPs. Animals PrEP 5 (infected) and PrEP 6 and 7 (uninfected), had no detectable SHIV DNA in any of the tissues analyzed.
2.4. Drug stability in vivo within nTAF
To evaluate drug stability in nTAF after 4 months of in vivo implantation, we extracted residual contents from the implant and analyzed for TAF and the sum of TAF with its hydrolysis products (TAF*) (Table 2). Residual drug within the implant ranged 30.75 – 71.12% of the initial loaded amount. Further, TAF* within the implant was predominantly composed of TAF hydrolysis products, including TFV, with TAF stability ranging 18.21 – 43.08% (Figure S4, Supporting Information). Therefore, augmented TAF hydrolysis to TFV within the implant most likely contributed to increased TFV levels observed in plasma towards the end of the study. The nTAF implants had a mean release rate of 1.40 ± 0.39 mg/day, which was sufficient to sustain intracellular TFV-DP concentrations above 100.00 fmol/106 PBMCs throughout the duration of the study.
Table 2.
Residual drug analysis from nTAF implants at explantation via high performance liquid chromatography (HPLC) and UV-Vis spectroscopy.
| NHP PrEP | TAF loaded (mg) | Residual TAF* (mg) | TAF stability (%) | TAF release rate (mg/day) |
|---|---|---|---|---|
| 1 | 341.50 | 161.87 | 30.76 | 1.60 |
| 2 | 330.10 | 217.65 | 12.28 | 1.00 |
| 3 | 337.10 | 215.57 | 18.21 | 1.09 |
| 4 | 382.10 | 241.01 | 31.78 | 1.26 |
| 5 | 457.60 | 325.43 | 43.08 | 1.18 |
| 6 | 449.30 | 279.46 | 18.70 | 1.52 |
| 7 | 342.60 | 105.34 | 22.26 | 2.12 |
2.5. nTAF safety and tolerability in NHP
To assess nTAF safety and tolerability, we histologically examined the tissue surrounding the implants after 4 months of implantation, through immunohistochemical analysis (Figure 4A) and semiquantitative histopathological assessment. Specifically, we evaluated the fibrotic capsule in contact with either the titanium reservoir (Figure 4B) or TAF-eluting nanofluidic membrane (Figures 4C–D, S5A–H, Supporting Information). Histological analysis via hematoxylin and eosin (H&E) demonstrated foreign-body response, which is typical of medical implants. While fibrotic capsules exhibited limited cellular infiltration (Figures S5A–H, Supporting Information), assessment of slides stained for the presence of macrophages (Figure 4E) and lymphocytes (Figure 4F) show a low-level influx of immune cells (Figures S6A and B, Supporting Information). DAPI staining demonstrated healthy nuclei in the areas with increased cellular infiltration. Also, the inflammatory response was localized as the surrounding subcutaneous tissue and underlying skeletal muscle was healthy (Figure 4G) Further, analysis of the fibrotic area in contact with TAF-releasing membrane via acid-fast bacteria (AFB) (Figure S7A, Supporting Information) and Grocott methenamine silver staining (Figure S7B, Supporting Information), which evaluates for presence of bacteria and fungi, respectively, were negative.
Figure 4.
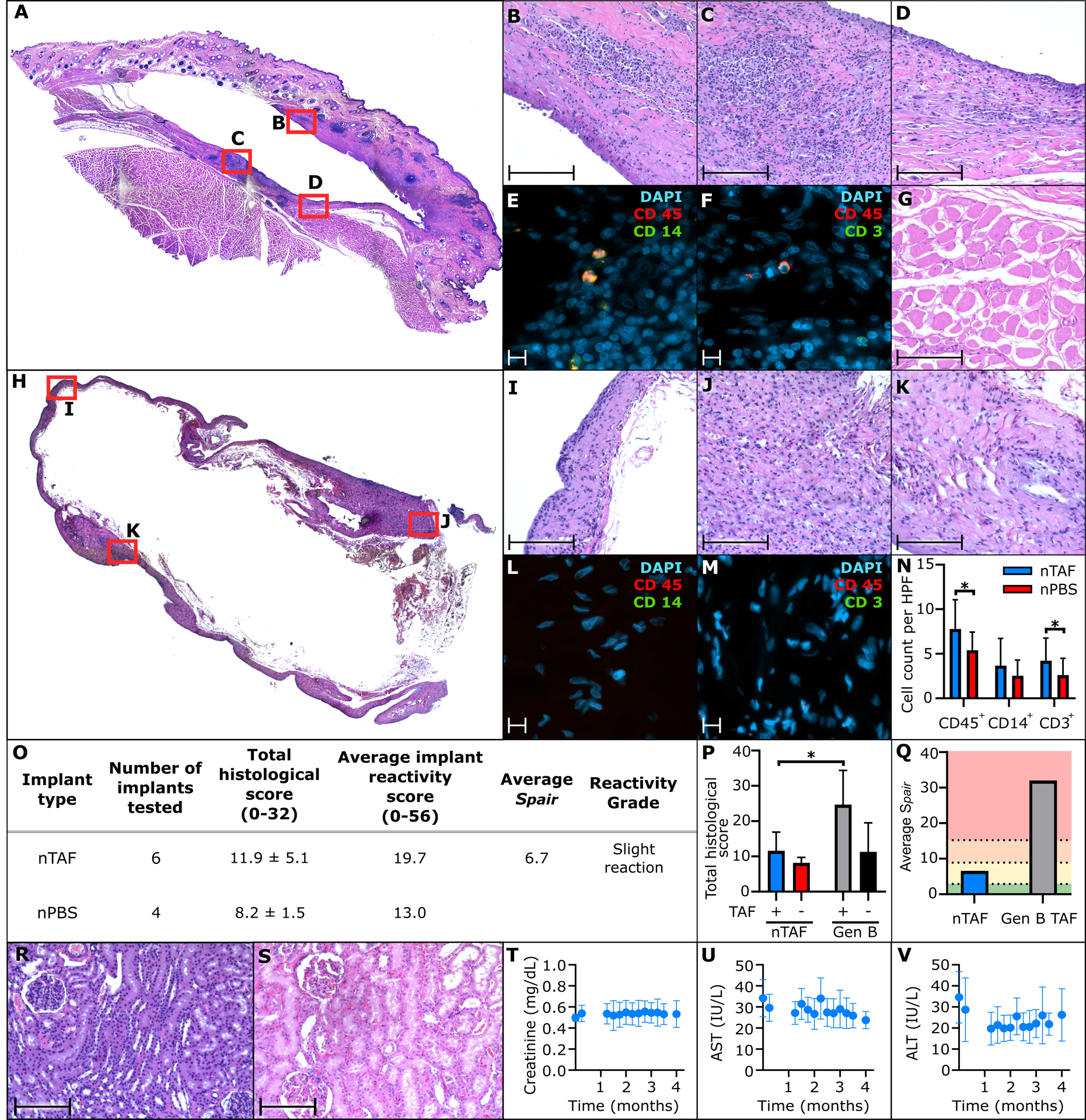
Histological inflammatory response to nTAF and control nPBS; and toxicology assessment of nTAF in the kidney and liver. A) Representative H&E stain of NHP skin surrounding PrEP nTAF, with B) fibrotic capsule in contact with titanium implant; 20 × magnification. Fibrotic capsule in contact with TAF-releasing membrane was assessed via C), D) H&E, 20 × magnification; E) immunofluorescence staining of CD45 (red), CD 14 (green) and DAPI nuclear stain (blue), 100 × magnification; F) immunofluorescence staining of CD45 (red), CD 3 (green) and DAPI nuclear stain (blue), 100 × magnification. G) Representative H&E stain of underlying skeletal muscle near implant site, 20 × magnification. H) Representative H&E stain of NHP skin surrounding control nPBS. Fibrotic capsule in contact with titanium implant was assessed via I), J), K) H&E, 20 × magnification, L) immunofluorescence staining of CD45 (red), CD 14 (green) and DAPI nuclear stain (blue), 100 × magnification; M) immunofluorescence staining of CD45 (red), CD 3 (green) and DAPI nuclear stain (blue), 100 × magnification. N) Quantification of CD45+, CD14+, and CD3+ cells in fibrotic capsule surrounding nTAF and nPBS implants. Data are presented as mean ± SD. Statistical analysis using two-tailed p<0.05 unpaired t test. O) Implant reactivity scores and placebo-adjusted implant reactivity scores (Spair). Data are presented as mean ± SD. P) Comparison of total histological characteristic scores between nTAF and nPBS implants with Generation B (Gen B) TAF and placebo implants. Data are presented as mean ± SD. Statistical analysis using two-tailed p<0.05 unpaired t test. Q) Comparison of average Spair reactivity grade between nTAF and Generation B (Gen B) implants from Su et al.[12] Spair values 0.0–2.9, 3.0–8.9, 9.0–15.0, and >15.1 colored as green, yellow, orange and red, respectively, represent no reaction, slight reaction, moderate reaction, and severe reaction, respectively. R) Representative H&E stain of kidney from PrEP nTAF group demonstrating normal histology, in comparison to S) representative H&E stain of kidney from control NHP similarly showing no nephrotoxicity; 20 × magnification. T) Creatinine activity measurements from nTAF cohort. Liver enzymes, U) aspartate aminotransferase (AST), and V) alanine aminotransferase (ALT) from nTAF cohort. Baseline levels (0 month) were measured before implantation of nTAF. All data are presented as mean ± SD (n=7). Images A and H taken at 4 × magnification and stitched together. Scale bar in 20 and 100 × magnification is 200 and 10 μm, respectively.
In parallel, as a control, the tissue surrounding nPBS implants were histologically assessed (Figure 4H), specifically the fibrotic capsule (Figure 4I–K), which was thinner and denser than the nTAF. Similarly, the tissue surrounding the control implant was negative for macrophages (Figure 4L), lymphocytes (Figure 4M), bacteria (Figure S7C, Supporting Information) or fungi (Figure S7D, Supporting Information). However, blinded quantification of CD45+, CD14+, and CD3+ cells in fibrotic capsules surrounding nTAF and nPBS implants (Figure 4N) revealed similar cellular findings in both groups. Although, the nTAF group exhibited a statistically significant increase in inflammatory cells (p=0.021) and lymphocytes (p=0.049).
Further, histopathological characteristics of tissue surrounding the implant site were scored by three board-certified pathologists from different institutions blinded to the groups. Briefly, the assessment of inflammatory response to a foreign-body was evaluated in accordance to the inflammation scoring system adopted from Su et al.[12, 35] The scoring system (scaled from 0 to 4) assessed the presence of characteristics relevant to inflammatory response to a foreign body: polymorphonuclear cells, lymphocytes, macrophages, giant cells, necrosis, capsule thickness and tissue infiltrate (Table S1, Supporting Information). After 4 months of implantation, the total histological score (scale 0 to 32) was 11.9 ± 5.1 and 8.2 ± 1.5 in the nTAF and nPBS groups, respectively (Figure 4O and Table S2, Supporting Information). Furthermore, the average implant reactivity score in the nTAF group was 19.7 (scale 0 to 56) and nPBS was 13.0 (Figure 4O). Notably, nTAF exhibited a statistically significant (p=0.025) lower total histological score than other TAF implants previously reported in the literature. As an example, TAF-releasing polymeric implants termed “Generation B TAF (Gen B TAF)” presented in Su et al.[12] showed significantly worse pathology scores (Figure 4P). Comparative analysis shows that nTAF implants were not statistically different than nPBS and placebo implant from Su et al. (Gen B placebo). Moreover, the average placebo-adjusted implant reactivity scores (Spair) for nTAF compared Gen B TAF were 6.7 and 32.0, respectively (Figure 4Q). In summary, tissue response to our nTAF implants (average release rate 1.40 ± 0.39 mg/day) was qualified as slight reaction. This is contrast with the results obtained with Gen B TAF implants, for which tissue response was determined as severe reaction despite the release rate was approximately 1/10 of nTAF.
As TFV is implicated in nephrotoxicity and hepatotoxicity, we evaluated the kidney and liver in the animals with nTAF implants. The kidney of an untreated NHP from a prior study was used as a historical control, because nPBS NHPs were transferred to another study after infection. Histological assessment of the kidney from nTAF cohort via H&E analysis (Figure 4R) did not demonstrate necrosis or signs of damage, in comparison to control (Figure 4S). Further, creatinine levels were within normal limits throughout the study, suggesting that there was no detectable kidney damage in the nTAF cohort (Figure 4T). Liver enzymes were monitored as surrogate markers for health; aspartate aminotransferase (AST) (Figure 4U), and alanine aminotransferase (ALT) (Figure 4V) measurements were within normal levels with respect to baseline values pre-nTAF implantation. Metabolic panel, complete blood count and urinalysis results were also within normal levels (Figures S8–V, S9A–N, Table S3, Supporting Information).
3. Discussion
This work represents the first ever preventive efficacy assessment of an implantable LA ARV platform and the foremost study of LA TAF as a single agent HIV PrEP regimen. Our finding that nTAF protected from SHIV infection with 62.50% reduction in risk of infection per exposure resembles that of TAF predecessor, tenofovir disoproxil fumarate (TDF). TDF monotherapy resulted in 60.00% protective efficacy in macaques[36], but clinically achieved 67% risk reduction and 86.00% preventive efficacy in individuals with detectable plasma tenofovir.[5, 37]
There is no benchmark preventive level of TFV-DP in PBMCs for sustained subcutaneous administration of TAF. We used as a reference the TFV-DP concentration in PBMCs of 100 fmol/106 cells, which conservatively exceeds the levels identified as protective in the iPrEX trial with Truvada® (cryopreserved PBMC, 16.00 fmol/106 cells; freshly lysed PBMC, 40.00 fmol/106 cells).[6] Other TAF-releasing implants are targeting 24–48 fmol/106, a target that takes into consideration the 66% TFV-DP loss during cryopreservation in the iPrEX trial.[9, 11, 12] While not directly comparable to oral Truvada administration, we used 100 fmol/106 cells as rational target to exceed prior to start the viral challenges. Nonetheless, this is the first efficacy study with continuous TAF administration via the subcutaneous route. Our results show that by maintaining a median TFV-DP concentration of 390 fmol/106 PBMC (IQR, 216.50 to 585.50 fmol/106 PBMC) we achieved partial protection with 62.50% efficacy (95% CI, 1.72% to 85.69%). In light of our studies, it remains unclear what the preventive benchmark could be to establish 100% efficacy in a rectal challenge model.
Most clinical studies evaluating PrEP adherence use plasma, PBMC or dried blood spots as surrogate markers to local tissue concentrations.[5, 6, 37, 38] However, breakthrough infection has occurred in individuals with high systemic drug concentrations, similar to the infected nTAF animals in our study. Therefore, it remains unclear if infection in some animals in our study could be attributable to inadequate TFV-DP concentrations in the site of viral transmission. In a study of weekly oral TAF as a single-agent PrEP against vaginal SHIV infection by the Center for Disease Control, TFV-DP PBMC levels were similar between the four infected and five uninfected animals.[33] However, only five out of nine animals had detectable vaginal TFV-DP concentrations (5 fmol/mg) prior to challenge.[33] It is also of interest to identify the turn-over rate of “TFV-DP positive” to “TFV-DP naïve” mononuclear cells systemically and locally at the site of transmission to improve dosing regimens. Garcia-Lerma et. al demonstrated that once weekly oral TAF dosing conferred low protection from HIV transmission, despite high systemic (>1000 fmol/106 PBMC) and rectal (median, 377 fmol/106 mononuclear cells) TFV-DP levels.[39] However, in the aforementioned study the animals were rectally challenged 3 days after the first weekly oral TAF dose. Thus, the long interval between drug dosing and virus exposure could have allowed for TFV-DP naïve mononuclear cells to repopulate at the site of transmission. Of relevance, on-demand local TFV delivery at HIV transmission sites, such as a TFV rectal douche, has shown to achieve high local tissue concentrations and favorable PK profiles in NHP with SHIV challenges.[40, 41] Therefore, we posit that PrEP efficacy could plausibly be improved if first-line target cells have sufficient TFV-DP concentrations prior to virus exposure.
The present study was limited by the number of animals and the use of both sexes for rectal SHIV prevention. Future studies could address this issue by increasing the sample size and conducting separate sexes studies to evaluate protection against rectal or vaginal exposure. Further, because Descovy® is clinically approved for oral administration, scientific rigor could be strengthened with an additional group with daily oral TAF dosing as opposed to weekly dosing as performed in literature, in comparison to sustained subcutaneous delivery. Moreover, our study was limited by the instability of TAF within our implant, potentially contributing to increased TFV levels in plasma at the end of the study. Although we did not observe nephrotoxicity and hepatotoxicity, future studies will use a more stable formulation of TAF with urocanic acid[32, 42] to maintain low TFV levels in plasma. Furthermore, assessing implant site concentrations of TAF, TFV, and TFV-DP could provide insight into tissue response to subcutaneous TAF administration.
In summary, our innovative strategy of continuous low-dose systemic delivery of TAF obviates adherence challenges and provides similar protective benefit to that observed with oral TDF. Taken together, this work provides optimism for implementing clinical studies to assess the safety and efficacy of LA TAF platforms for HIV PrEP.
Experimental Section
Nanofluidic implant assembly
Medical-grade 6AI4V titanium oval drug reservoirs were specifically designed and manufactured for this study. Briefly, a nanofluidic membrane possessing 278,600 nanochannels (mean; 194 nm) was mounted on the inside of the sterile drug reservoir as described previously.[16] Detailed information regarding the membrane structure and fabrication was described previously.[30, 31] Implants were welded together using Arc welding. PrEP implants were loaded with ~300 – 457 mg TAF fumarate in solid powder form (Table 2) using a funnel in the loading port, while control implants were left empty in view of subsequent loading of sterile 1 X Phosphate Buffered Saline (PBS). A titanium piece that resembled a small nail was inserted into the loading port and welded shut. Implants were primed for drug release through the nanofluidic membrane by placing implants in 1 X Phosphate Buffered Saline (PBS) under vacuum. This preparation method resulted in loading of control implants with PBS. Implants were maintained in sterile 1X PBS in a hermetically sealed container until implantation shortly after preparation. TAF was kindly provided by Gilead Sciences, Inc.
In vitro release from nanofluidic implant
In an effort to limit the amount of drug used, the in vitro release study was performed using nanochannel membranes with identical structure and channel size those adopted in vivo, but with a small number of nanochannels (n= 9,800 as compared to n=278,600 for the full-size membrane). In vitro release results were then linearly scaled to account for the difference in nanochannels number. Medical-grade 6AI4V titanium cylindrical drug reservoirs (n=5) were assembled as described above, loaded with ~20.00 mg TAF fumarate and placed in sink solution of 20 mL 1 × PBS with constant agitation at 37°C. For analysis, the entire sink solution was retrieved and replaced with fresh PBS every other day for 20 days. The maximum TAF concentration regarding TAF saturation in sink solution was <10%, therefore maintaining sink condition. High-performance liquid chromatography (HPLC) analysis was performed on an Agilent Infinity 1260 system equipped with a diode array and evaporative light scattering detectors using a 3.5-μm 4.6 × 100 mm Eclipse Plus C18 column and water/methanol as the eluent and 25 μL injection volume, as previously described.[32] Specifically, ammonia acetate buffered water (solvent A) and ammonia acetate buffered methanol (solvent B) at 2.00 mL/min flow rate gradient: 0 min (5% B), 0.8 min (5% B), 3.8 min (100% B), 4.6 min (100% B), 5.2 min (5% B). Peak areas were analyzed at 260 nm absorbance.[32]
Nanofluidic membrane assessment
Silicon nanofluidic membranes structure and composition was assessed using different imaging techniques at the Microscopy – SEM/AFM core of the Houston Methodist Research Institute (HMRI), Houston, TX, USA. Inspection of structural conformation was performed via scanning electron microscopy (SEM; Nova NanoSEM 230, FEI, Oregon, USA), nanochannel dimension was measured on membrane cross sections obtained using gallium ion milling (FIB, FEI 235). Surface roughness was measured by atomic force microscopy (AFM Catalyst), surface chemical composition was evaluated with Energy-dispersive X-ray spectroscopy (EDAX, Nova NanoSEM 230).
Animals and animal care
All animal procedures were conducted at the AAALAC-I accredited Michale E. Keeling Center for Comparative Medicine and Research, The University of Texas MD Anderson Cancer Center (UTMDACC), Bastrop, TX. All animal experiments were carried out according to the provisions of the Animal Welfare Act, PHS Animal Welfare Policy, and the principles of the NIH Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at UTMDACC, which has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare. IACUC #00001749-RN00. Indian rhesus macaques (Macaca mulatta; n=14; 6 males and 8 females) of 2–4 years and 2–5 kg bred at this facility were used in the study. All procedures were performed under anesthesia with ketamine (10 mg/kg, intramuscular) and phenytoin/pentobarbital (1 mL/10 lbs, intravenous [IV]).
All animals had access to clean, fresh water at all times and a standard laboratory diet. Prior to the initiation of virus inoculations, compatible macaques were pair-housed. Once inoculations were initiated, the macaques were separated into single housing (while permitting eye contact) to prevent the possibility of SHIV transmission between the macaques. Euthanasia of the macaques was accomplished in a humane manner (IV pentobarbital) by techniques recommended by the American Veterinary Medical Association Guidelines on Euthanasia. The senior medical veterinarian verified successful euthanasia by the lack of a heartbeat and respiration.
Minimally invasive implantation procedure
An approximately 1-cm dorsal skin incision was made on the right lateral side of the thoracic spine. Blunt dissection was used to make a subcutaneous pocket ventrally about 5 cm deep. The implant was placed into the pocket with the membrane facing the body. A simple interrupted tacking suture of 4–0 polydioxanone (PDS) was placed in the subcutaneous tissue to help close the dead space and continued intradermally to close the skin. All animals received a single 50,000 U/kg perioperative penicillin G benzathine/penicillin G procaine (Combi-Pen) injection and subcutaneous once-daily meloxicam (0.2 mg/kg on day 1 and 0.1 mg/kg on days 2 and 3) for postsurgical pain.
Blood collection and plasma and PBMC sample preparation
All animals had weekly blood draws to assess plasma TAF and TFV concentrations, intracellular TFV-DP PBMC concentrations, plasma viral RNA loads, and cell-associated SHIV DNA in PBMCs. Blood collection and sample preparation were performed as previously described.[14] Blood was collected in EDTA-coated vacutainer tubes before implantation; on days 1, 2, 3, 7, 10, and 14; and then once weekly until euthanasia. Plasma was separated from blood by centrifugation at 1200 × g for 10 min at 4 ⁰C and stored at −80 ⁰C until analysis. The remaining blood was used for PBMC separation by standard Ficoll-Hypaque centrifugation. Cell viability was > 95%. After cells were counted, they were pelleted by centrifugation at 400 × g for 10 min, resuspended in 500 μL of cold 70% methanol/30% water, and stored at −80 ⁰C until further use.
Pharmacokinetic analysis of TFV-DP in PBMC and TAF and TFV in plasma
The PK profiles of TFV-DP in PBMC and TAF and TFV in plasma were evaluated throughout the 4 months of nTAF implantation. Due to early implant removal in one animal on day 43, seven animals were evaluated for drug PK. After device explantation, drug washout was assessed for an additional 2 months (n=3).
Intracellular TFV-DP concentrations in PBMCs were quantified using previously described validated liquid chromatographic-tandem mass spectrometric (LC-MS/MS) analysis.[6, 43] The assay was linear from 5 to 6000 fmol/sample. Typically, 25 fmol/sample was used as the lower limit of quantitation (LLOQ). If additional sensitivity was needed, standards and quality controls were added down to 5 fmol/samples, as previously described.[43] Day 21 TFV-DP concentrations were omitted due to PBMC count below threshold.
Plasma TAF and TFV concentrations were quantified using a previously described LC-MS/MS assay.[44] Drugs were extracted from 0.1 mL plasma via solid phase extraction; assay lower limits of quantitation for TAF and TFV were 0.03 ng/mL and 1 ng/mL, respectively. The multiplexed assay was validated in accordance with FDA, Guidance for Industry: Bioanalytical Method Validation recommendations.[45]
Tissue TFV-DP quantification
Lymphoid tissues (mesenteric, axillary, and inguinal lymph nodes), rectum, urethra, cervix, tonsil, spleen, and liver were homogenized, and 50- to 75-mg aliquots were used for TFV-DP quantitation. Pharmacokinetic analysis of TFV-DP was conducted by the Clinical Pharmacology Analytical Laboratory at the Johns Hopkins University School of Medicine. TFV concentrations in aforementioned tissue biopsies were determined via LC-MS/MS analysis. TFV-DP was measured using a previously described indirect approach, in which TFV was quantitated following isolation of TFV-DP from homogenized tissue lysates and enzymatic conversion to the TFV molecule.[43] The assay LLOQ for TFV-DP in tissue was 5 fmol/sample, and drug concentrations were normalized to the amount of tissue analyzed.[46] The TFV-DP tissue was validated in luminal tissue (rectal and vaginal tissue) in accordance with FDA, Guidance for Industry: Bioanalytical Method Validation recommendations[45]; alternative tissue types were analyzed using this method.
PrEP nTAF efficacy against rectal SHIV challenge
To study the efficacy of the PrEP implant against SHIV transmission, animals were divided into two groups, PrEP nTAF-treated [n=8; 4 male (M) and 4 female (F)] or control nPBS (n=6; 3 M and 3 F), in a non-blinded study. The PrEP regimen consisted of subcutaneously implanted nTAF for sustained drug release over 112 days. The efficacy of nTAF in preventing rectal SHIV transmission was evaluated using a repeat low-dose exposure model described previously.[36, 39, 47] Animals were considered protected if they remained negative for SHIV RNA throughout the study. Briefly, after PrEP-treated macaques achieved intracellular TFV-DP concentrations above 100.00 fmol/106 PBMCs, both groups were rectally exposed to SHIVSF162P3 once a week for up to 10 weeks until infection was confirmed by two consecutive positive plasma viral RNA loads. The SHIVSF162P3 dose was in range of HIV-1 RNA levels found in human semen during acute viremia.[47]
Challenge stocks of SHIV162p3 were generously supplied by Dr. Nancy Miller, Division of AIDS, NIAID, through Quality Biological (QBI), under Contract No. HHSN272201100023C to the Vaccine Research Program, Division of AIDS, NIAID. The stock SHIV162p3 R922 derived harvest 4 dated 9/16/2016 (p27 content 173.33 ng/ml, viral RNA load >109 copies/ml, TCID50/ml in rhesus PBMC 1280) was diluted 1:300 and 1ml of virus was used for rectal challenge each time.
For the challenge, the animals were positioned in prone position and virus was inoculated approximately 4 cm into the rectum. Inoculated animals were maintained in the prone position with the perineum elevated for 20 minutes to ensure that virus did not leak out. Care was also taken to prevent any virus from contacting the vagina area and to not abrade the mucosal surface of the rectum.
Infection monitoring by SHIV RNA in plasma and SHIV DNA in tissues
Infection was monitored by the detection of SHIV RNA in plasma using previously described methods[48, 49] with modification. Viral RNA (vRNA) was isolated from blood plasma using the Qiagen QIAmp UltraSense Virus Kit (Qiagen #53704) in accordance with manufacturer’s instructions for 0.5 mL of plasma. vRNA levels were determined by quantitative real-time PCR (qRT-PCR) using Applied Biosystems™ TaqMan™ Fast Virus 1-Step Master Mix (Thermofisher #4444432) and a primer-probe combination recognizing a conserved region of gag (GAG5f: 5’-ACTTTCGGTCTTAGCTCCATTAGTG-3’; GAG3r: 5’-TTTTGCTTCCTCAGTGTGTTTCA-3’; and GAG1tq: FAM 5’- TTCTCTTCTGCGTGAATGCACCAGATGA-3’TAMRA). Each 20 μl reaction contained 900 nM of each primer and 250 nM of probe, and 1x Fast Virus 1-Step Master Mix, plasma-derived vRNA sample, SIV gag RNA transcript containing standard, or no template control.
qRT-PCR was performed in a ABI Step One Plus Cycler. PCR was performed with an initial step at 50°C for 5 min followed by a second step at 95°C for 20 sec, and then 40 cycles of 95°C for 15 sec and 60°C for 1 min. Ten-fold serial dilutions (1 to 1 × 106 copies per reaction) of an in vitro transcribed SIV gag RNA were used to generate standard curves. Each sample was tested in duplicate reactions. Plasma viral loads were calculated and shown as viral RNA copies/mL plasma. The limit of detection is 50 copies/ml. Infections were confirmed after a consecutive positive plasma viral load measurement.
To detect viral DNA in tissue samples, total DNA was isolated from PBMCs or tissue specimens using the Qiagen DNeasy Blood & Tissue Kit (Qiagen #69504) according to the manufacturer’s protocol. DNA was quantified using a nanodrop spectrophotometer. qRT-PCR was performed using the SIV gag primer probe set described above. Each 20 μl reaction contained 900 nM of each primer and 250 nM of probe, and 1x TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, CA), macaque-derived DNA sample, SIV gag DNA containing standard, or no template control. PCR was initiated in with an initial step of 50°C for 2 min and then 95°C for 10 min. This was followed by 40 cycles of 95°C for 15 sec, and 60°C for 1 min. Each sample was tested in triplicate reactions. Ten-fold serial dilutions of a SIV gag DNA template (1 to 1 × 105 per reaction) were used to generate standard curves. The limit of detection of this assay was determined to be 1 copy of SIV gag DNA.
Device retrieval and macaque euthanasia
A subset of PrEP-treated macaques (n=4), those with the highest viral load, were euthanized on day 112, while implants were retrieved on day 112 from the remaining PrEP-treated macaques (n=3) for continuation to a 2-month drug-washout period before euthanasia. SHIV-infected macaques in the control group (n=6) were transferred to another study (data not shown) and euthanized 28 days later. The implant was retrieved with a small incision in the skin and stored at −80 ⁰C until further analysis. Skin within a 2-cm margin surrounding the implant was excised from euthanized macaques and fixed in 10% buffered formalin for histological analysis. Macaques continuing in the washout period underwent a skin punch biopsy of the subcutaneous pocket, and the skin incision was sutured with a simple interrupted tacking suture of 4–0 PDS; the specimen was fixed in 10% buffered formalin for histological analysis. The following tissues were collected from all animals at euthanasia (n=13): lymphoid tissues (mesenteric, axillary, and inguinal lymph nodes), rectum, urethra, cervix, tonsil, spleen, and liver. Tissues were snap-frozen and stored at −80 ⁰C until further analysis of TAF concentrations, viral RNA loads, and cell-associated SHIV DNA.
Residual drug and nanofluidic membrane retrieval from explanted implants
Upon explantation, the implants were snap frozen with liquid nitrogen to preserve residual drug for stability analysis. For residual drug retrieval, the implants were thawed at 4°C overnight. A hole was drilled on the outermost corner on the back of the implant using a 3/64 titanium drill bit with a stopper. Drilling was performed on the back of the implant and distal to the membrane to avoid damage. Following drilling, 20 μL sample from the implant drug reservoir was aliquoted into respective 1.5 mL Eppendorf tubes with 0.5 mL 100% ethanol using a pipette. The implants were placed in 50 mL conical tubes with 40.0 g 70% ethanol. Each implant was flushed using a 19-gauge needle with 70% ethanol from the sink solution. For sterilization, the implants were incubated in 70% ethanol for 4 days and transferred to new conical tubes with fresh 70% ethanol for an additional 4 days. To ensure nanochannel membranes were dry, the implants were transferred to new conical tubes with 100% ethanol for a day and placed in 6-well plates to dry under vacuum. To protect the membrane during machining procedure, electrical tape was placed over the outlets. The implants were opened using a rotary tool with a diamond wheel. Titanium dust from machining procedure was gently cleaned from membrane with a cotton swab and 70% ethanol. To remove membrane from the implant, a drop of nitric acid (Trace Metal grade) was placed on the membrane overnight and rinsed with Millipore water the next day. Membranes were kept in hermetically sealed containers until analysis.
TAF stability analysis in drug reservoir
Liquid in the drug reservoir after explantation was collected with a pipette and diluted 25 times with 100% ethanol. The samples were transferred to 0.2 μm nylon centrifugal filters and centrifuged at 500 G for 8 minutes at room temperature. An aliquot of 50 μL from the filtered samples were further diluted in 100 μL 100% ethanol. HPLC analysis of TAF was performed as aforementioned in the in vitro release section.
Drug solids from within the implant were analyzed from the initial 40.0 g 70% ethanol sink solution. The samples were transferred to 0.2 μm nylon centrifugal filter and centrifuged at 500 G for 8 minutes at room temperature. An aliquot of 10 μL from the filtered samples was further diluted in 990 μL of deionized water. UV-vis spectroscopy was performed on a Beckman Coulter DU® 730 system. Peak areas were analyzed at 260 nm absorbance.
Assessment of PrEP nTAF safety and tolerability
Tissues were fixed in 10% buffered formalin and stored in 70% ethanol until analysis. Tissues were then embedded in paraffin, cut into 5 μm sections and stained with hematoxylin and eosin (H&E) staining at the Research Pathology Core HMRI, Houston, TX, USA. H&E staining was performed on tissue sections surrounding the implant site and kidney. For immunohistochemistry evaluation of tissue sections, slides were stained with anti-CD45 conjugated to Texas Red (CD45 Monoclonal Antibody (HI30), PE-Texas Red Thermo Fisher), anti-human CD14 and anti-human CD3 conjugated to fluorescein isothiocyanate (Pharmingen). For negative controls, corresponding immunoglobulin and species (IgG)-matched isotype control antibodies were used. Nonspecific binding in sections was blocked by a 1-hour treatment in tris-buffered saline (TBS) plus 0.1% w/v Tween containing defatted milk powder (30 mg ml–1). Stained sections were mounted in Slow Fade GOLD with 4’,6-diamidino-2-phenylindole (DAPI) (Molecular Probes, OR) and observed using a Nikon T300 Inverted Fluorescent microscope (Nikon Corp., Melville, NY). For verification of cell phenotype, each slide was scored by counting three replicate measurements by the same observer for each slide. All slides were counted at 20 × magnification without knowledge of the cell-specific marker being examined, and results were averaged with through a second reading by another observer.
Semiquantitative histopathological assessment of inflammatory response to a foreign body was evaluated in accordance to the inflammation scoring system presented in Su et al.[12], which was adopted from a published standard.[35] Briefly, cells were counted via high power field (HPF) and scored (0–4) based on histological characteristics: polymorphonuclear cells, lymphocytes, plasma cells, macrophages, giant cells, necrosis, capsule thickness, and tissue infiltrate (see Table S1, Supporting Information). The scores reported by each pathologist were averaged per implant (see Table S2, Supporting Information). Then, the total histological characteristic scores were reported per group as the average of the sum of all histological scores of all implants. The reactivity grade for each implant was computed using Equation 1 from Su et al.[12] and the average placebo-adjusted implant reactivity score (Spair) was calculated by subtracting the result obtained for nPBS from nTAF. The Spair classification used in Su et al.[12] and published standard[35] was adopted: minimal to no reaction (0.0 < Spair < 2.9), slight reaction (3.0 < Spair < 8.9), moderate reaction (9.0 < Spair < 15.0), and severe reaction (Spair > 15.1).
Assessment of TAF toxicity
To assess TAF toxicity, a comprehensive metabolic panel was analyzed for each animal weekly during the rectal challenge phase of the study and biweekly afterward. Urine and CBCs were analyzed monthly to assess kidney and liver function and monitor the well-being of the NHPs.
Statistical analysis
Plasma t1/2 PK analysis was performed in Microsoft Excel using 2 time points, days 112 and 119. Results were expressed as actual t1/2 is less than obtained t1/2 (because day 119 values were undetectable and were substituted with BLOQ values). PBMC PK analysis was performed using PKSolver add-in for Microsoft Excel developed by Zhang et al.[46] Data are represented as mean ± SD or median with interquartile range (IQR) between the first (25th percentile) and third (75th percentile) quartiles. The relative risk and relative risk reduction with 95% confidence intervals (95% CI) were estimated to examine the per-exposure effect of TAF, and the Fisher’s exact was used for the comparison. The Mann-Whitney test was used to compare the median survival time and the differences in SHIV RNA levels at initial detection. The Kaplan-Meier analysis was performed between the PrEP and control groups, with the use of the number of inoculations as the time variable. The exact log-rank test was used to test the survival between the two groups. Unpaired t test was used to compare quantification of inflammatory cells between nTAF and nPBS and total histological characteristic scores between nTAF and nPBS implants with Gen B TAF and placebo implants. Data are presented as mean ± SD. All statistical analysis for calculation of the efficacy of TAF were performed with GraphPad Prism 8 (version 8.2.0; GraphPad Software, Inc., La Jolla, CA). Statistical significance was defined as two-tailed p<0.05 for all tests.
Supplementary Material
Acknowledgements
We thank Dr. Andreana L. Rivera, Yuelan Ren, and Sandra Steptoe from the research pathology core of Houston Methodist Research Institute. Dr. Jianhua “James” Gu from the electron microscopy core. We thank Simone Capuani from the Houston Methodist Research Institute for implant design and rendering, Dixita Viswanath for help with tissue dissection, and Nicola Di Trani for obtaining FIB image. We thank Jesus Paez-Mayorga and Dr. Dorothy Lewis for the useful discussions. We thank Luke Segura, Elizabeth Lindemann and Dr. Greg Wilkerson from the Michale E. Keeling Center for Comparative medicine and Research at UTMDACC for support in animal studies and Bharti Nehete for plasma and PBMC isolation and virus challenge preparation. TAF fumarate was provided by Gilead Sciences, Inc. Funding: This work was supported by funding from the National Institutes of Health National Institute of Allergy and Infectious Diseases (R01AI120749; A.G.), the National Institutes of Health National Institute of General Medical Sciences (R01GM127558; A.G.) and Gilead Sciences (A.G.). F.P.P. received funding support from Tecnologico de Monterrey and Consejo Nacional de Ciencia y Tecnologia.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library and source data for figures and tables are available from the author upon request.
Competing interests
Study drugs were provided by Gilead Sciences. P.L.A. receives grants and contracts from Gilead Sciences paid to his institution and collects personal fees from Gilead Sciences. T.H. is an employee of Gilead Sciences. J.F.R. is an employee and stockholder of Gilead Sciences. All other authors declare that they have no competing interests.
Contributor Information
Fernanda P. Pons-Faudoa, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030, USA Tecnologico de Monterrey, School of Medicine and Health Sciences, Monterrey, NL, Mexico.
Antons Sizovs, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030, USA.
Kathryn A. Shelton, Department of Comparative Medicine, Michael E. Keeling Center for Comparative Medicine and Research, MD Anderson Cancer Center, Bastrop, TX 78602, USA
Zoha Momin, Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX 77030, USA.
Jean A. Niles, Division of Infectious Diseases, Department of Internal Medicine, University of Texas Medical Branch (UTMB), Galveston, TX 77555, USA
Lane R. Bushman, Deparment of Pharmaceutical Sciences, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado- Anschutz Medical Campus, Aurora, CO 80045, USA
Jiaqiong Xu, Center for Outcomes Research and DeBakey Heart and Vascular Center, Houston Methodist Research Institute, Houston, TX 77030, USA; Weill Medical College of Cornell University, New York, NY 10065, USA.
Corrine Ying Xuan Chua, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030, USA.
Joan E. Nichols, Division of Infectious Diseases, Department of Internal Medicine, University of Texas Medical Branch (UTMB), Galveston, TX 77555, USA
Sandra Demaria, Department of Radiation Oncology, Weill Cornell Medicine, New York, NY 10065, USA; Department of Pathology and Laboratory of Medicine, Weill Cornell Medicine, New York, NY 10065, USA.
Michael M. Ittmann, Department of Pathology and Immunology, Baylor College of Medicine, Houston, TX 77030, USA
James F. Rooney, Gilead Sciences, Inc., Foster City, CA 94404, USA
Mark A. Marzinke, Departments of Pathology and Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21224, USA
Jason T. Kimata, Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, TX 77030, USA
Peter L. Anderson, Deparment of Pharmaceutical Sciences, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado- Anschutz Medical Campus, Aurora, CO 80045, USA
Pramod N. Nehete, Department of Comparative Medicine, Michael E. Keeling Center for Comparative Medicine and Research, MD Anderson Cancer Center, Bastrop, TX 78602, USA The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, TX 77030, USA.
Roberto C. Arduino, Division of Infectious Diseases, Department of Internal Medicine, McGovern Medical School at The University of Texas Health Science Center, Houston, TX 77030, USA
Mauro Ferrari, School of Pharmacy, University of Washington, Seattle, WA 98195, USA.
K. Jagannadha Sastry, Department of Comparative Medicine, Michael E. Keeling Center for Comparative Medicine and Research, MD Anderson Cancer Center, Bastrop, TX 78602, USA; Department of Thoracic Head and Neck Medical Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Alessandro Grattoni, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030, USA; Department of Surgery, Houston Methodist Research Institute, Houston, TX 77030, USA; Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA.
References
- [1].Joint United Nations Programme on HIV/AIDS (UNAIDS), UNAIDS Information Production Unit, 2014, 40. [Google Scholar]
- [2].Ray AS, Fordyce MW, Hitchcock MJ, Antiviral research 2016, 125, 63. [DOI] [PubMed] [Google Scholar]
- [3].Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapía M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernández T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker L-G, Mayer KH, Kallás EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang F, McConnell JJ, Zheng J-H, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV, 2010, 363, 2587. [Google Scholar]
- [4].Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, Hakim JG, Kumwenda J, Grinsztejn B, Pilotto JHS, Godbole SV, Mehendale S, Chariyalertsak S, Santos BR, Mayer KH, Hoffman IF, Eshleman SH, Piwowar-Manning E, Wang L, Makhema J, Mills LA, de Bruyn G, Sanne I, Eron J, Gallant J, Havlir D, Swindells S, Ribaudo H, Elharrar V, Burns D, Taha TE, Nielsen-Saines K, Celentano D, Essex M, Fleming TR, New England Journal of Medicine 2011, 365, 493. [Google Scholar]
- [5].Baeten JM, Donnell D, Ndase P, Mugo NR, Campbell JD, Wangisi J, Tappero JW, Bukusi EA, Cohen CR, Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, Farquhar C, John-Stewart G, Kakia A, Odoyo J, Mucunguzi A, Nakku-Joloba E, Twesigye R, Ngure K, Apaka C, Tamooh H, Gabona F, Mujugira A, Panteleeff D, Thomas KK, Kidoguchi L, Krows M, Revall J, Morrison S, Haugen H, Emmanuel-Ogier M, Ondrejcek L, Coombs RW, Frenkel L, Hendrix C, Bumpus NN, Bangsberg D, Haberer JE, Stevens WS, Lingappa JR, Celum C, New England Journal of Medicine 2012, 367, 399. [Google Scholar]
- [6].Anderson PL, Glidden DV, Liu A, Buchbinder S, Lama JR, Guanira JV, McMahan V, Bushman LR, Casapia M, Montoya-Herrera O, Veloso VG, Mayer KH, Chariyalertsak S, Schechter M, Bekker LG, Kallas EG, Grant RM, iPrEx Study T, Sci Transl Med 2012, 4, 151ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boyd MA, Cooper DA, Lancet (London, England) 2017, 390, 1468. [DOI] [PubMed] [Google Scholar]
- [8].Clement ME, Kofron R, Landovitz RJ, Current opinion in HIV and AIDS 2020, 15, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gunawardana M, Remedios-Chan M, Miller CS, Fanter R, Yang F, Marzinke MA, Hendrix CW, Beliveau M, Moss JA, Smith TJ, Baum MM, 2015, 59, 3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schlesinger E, Johengen D, Luecke E, Rothrock G, McGowan I, van der Straten A, Desai T, Pharm Res 2016, 33, 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Johnson LM, Krovi SA, Li L, Girouard N, Demkovich ZR, Myers D, Creelman B, van der Straten A, Pharmaceutics 2019, 11, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Su JT, Simpson SM, Sung S, Tfaily EB, Veazey R, Marzinke M, Qiu J, Watrous D, Widanapathirana L, Pearson E, Peet MM, Karunakaran D, Grasperge B, Dobek G, Cain CM, Hope T, Kiser PF, 2020, 64, e01893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Simpson SM, Widanapathirana L, Su JT, Sung S, Watrous D, Qiu J, Pearson E, Evanoff A, Karunakaran D, Chacon JE, Kiser PF, Pharm Res 2020, 37, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chua CYX, Jain P, Ballerini A, Bruno G, Hood RL, Gupte M, Gao S, Di Trani N, Susnjar A, Shelton K, Bushman LR, Folci M, Filgueira CS, Marzinke MA, Anderson PL, Hu M, Nehete P, Arduino RC, Sastry JK, Grattoni A, Journal of controlled release : official journal of the Controlled Release Society 2018, 286, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Intarcia Therapeutics.
- [16].Pons-Faudoa FP, Sizovs A, Di Trani N, Paez-Mayorga J, Bruno G, Rhudy J, Manohar M, Gwenden K, Martini C, Chua CYX, Varchi G, Marzinke MA, Grattoni A, Journal of controlled release : official journal of the Controlled Release Society 2019, 306, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pons-Faudoa FP, Ballerini A, Sakamoto J, Grattoni A, Biomedical microdevices 2019, 21, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Flexner C, Current opinion in HIV and AIDS 2018, 13, 374. [DOI] [PubMed] [Google Scholar]
- [19].Markowitz M, Frank I, Grant RM, Mayer KH, Elion R, Goldstein D, Fisher C, Sobieszczyk ME, Gallant JE, Van Tieu H, Weinberg W, Margolis DA, Hudson KJ, Stancil BS, Ford SL, Patel P, Gould E, Rinehart AR, Smith KY, Spreen WR, The Lancet HIV 2017, 4, e331. [DOI] [PubMed] [Google Scholar]
- [20].Landovitz RJ, Li S, Grinsztejn B, Dawood H, Liu AY, Magnus M, Hosseinipour MC, Panchia R, Cottle L, Chau G, Richardson P, Marzinke MA, Hendrix CW, Eshleman SH, Zhang Y, Tolley E, Sugarman J, Kofron R, Adeyeye A, Burns D, Rinehart AR, Margolis D, Spreen WR, Cohen MS, McCauley M, Eron JJ, PLOS Medicine 2018, 15, e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Merck & Co., 2019.
- [22].Gated Nanofluidic Valve For Active And Passive Electrosteric Control Of Molecular Transport, And Methods Of Fabrication, U.S. Provisional Pat. Ser. No. 62/961,437, filed January 15. (2020).
- [23].Oliveros A, Guiseppi-Elie A, Saddow SE, Biomedical microdevices 2013, 15, 353. [DOI] [PubMed] [Google Scholar]
- [24].Zorman CA, Eldridge A, Du JG, Johnston M, Dubnisheva A, Manley S, Fissell W, Fleischman A, Roy S, “Amorphous silicon carbide as a non-biofouling structural material for biomedical microdevices”, presented at Materials Science Forum, 2012. [Google Scholar]
- [25].Di Trani N, Pimpinelli A, Grattoni A, ACS applied materials & interfaces 2020, 12, 12246. [DOI] [PubMed] [Google Scholar]
- [26].Bruno G, Di Trani N, Hood RL, Zabre E, Filgueira CS, Canavese G, Jain P, Smith Z, Demarchi D, Hosali S, Pimpinelli A, Ferrari M, Grattoni A, Nature Communications 2018, 9, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ferrati S, Fine D, You J, De Rosa E, Hudson L, Zabre E, Hosali S, Zhang L, Hickman C, Sunder Bansal S, Cordero-Reyes AM, Geninatti T, Sih J, Goodall R, Palapattu G, Kloc M, Ghobrial RM, Ferrari M, Grattoni A, Journal of controlled release : official journal of the Controlled Release Society 2013, 172, 1011. [DOI] [PubMed] [Google Scholar]
- [28].Di Trani N, Jain P, Chua CYX, Ho JS, Bruno G, Susnjar A, Pons-Faudoa FP, Sizovs A, Hood RL, Smith ZW, Ballerini A, Filgueira CS, Grattoni A, Nanomedicine : nanotechnology, biology, and medicine 2019, 16, 1. [DOI] [PubMed] [Google Scholar]
- [29].Di Trani N, Silvestri A, Bruno G, Geninatti T, Chua CYX, Gilbert A, Rizzo G, Filgueira CS, Demarchi D, Grattoni A, Lab on a Chip 2019, 19, 2192. [DOI] [PubMed] [Google Scholar]
- [30].Di Trani N, Silvestri A, Sizovs A, Wang Y, Erm DR, Demarchi D, Liu X, Grattoni A, Lab on a Chip 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Di Trani N, Silvestri A, Wang Y, Demarchi D, Liu X, Grattoni A, Pharmaceutics 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sizovs A, Pons-Faudoa FP, Malgir G, Shelton K, Bushman LR, Chua CYX, Anderson PL, Nehete P, Sastry JK, Grattoni A, International Journal Of Pharmaceutics 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Massud I, Cong M-E, Ruone S, Holder A, Dinh C, Nishiura K, Khalil G, Pan Y, Lipscomb J, Johnson R, Deyounks F, Rooney JF, Babusis D, Park Y, McCallister S, Callebaut C, Heneine W, García-Lerma JG, The Journal of Infectious Diseases 2019, 220, 1826. [DOI] [PubMed] [Google Scholar]
- [34].Anderson PL, García-Lerma JG, Heneine W, Current opinion in HIV and AIDS 2016, 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].W. H. O. E. C. o. B. Standardization, W. H. Organization, WHO Expert Committee on Biological Standardization: Sixty-sixth Report, Vol. 999, World Health Organization, 2016. [Google Scholar]
- [36].Subbarao S, Otten RA, Ramos A, Kim C, Jackson E, Monsour M, Adams DR, Bashirian S, Johnson J, Soriano V, Rendon A, Hudgens MG, Butera S, Janssen R, Paxton L, Greenberg AE, Folks TM, The Journal of Infectious Diseases 2006, 194, 904. [DOI] [PubMed] [Google Scholar]
- [37].Baeten JM, Donnell D, Mugo NR, Ndase P, Thomas KK, Campbell JD, Wangisi J, Tappero JW, Bukusi EA, Cohen CR, Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, Farquhar C, John-Stewart G, Kidoguchi L, Coombs RW, Hendrix C, Marzinke MA, Frenkel L, Haberer JE, Bangsberg D, Celum C, The Lancet Infectious Diseases 2014, 14, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Anderson PL, Liu AY, Castillo-Mancilla JR, Gardner EM, Seifert SM, McHugh C, Wagner T, Campbell K, Morrow M, Ibrahim M, Buchbinder S, Bushman LR, Kiser JJ, MaWhinney S, Antimicrobial Agents and Chemotherapy 2017, 62, 1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Garcia-Lerma JG, Aung W, Cong M. e., Zheng Q, Youngpairoj AS, Mitchell J, Holder A, Martin A, Kuklenyik S, Luo W, Lin CYC, Hanson DL, Kersh E, Pau CP, Ray AS, Rooney JF, Lee WA, Heneine W, Journal of Virology 2011, 85, 6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xiao P, Gumber S, Marzinke MA, Date AA, Hoang T, Hanes J, Ensign LM, Wang L, Rohan L, Fuchs EJ, Hendrix C, Villinger F, Antimicrobial Agents and Chemotherapy 2017, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Weld E, Fuchs E, Marzinke M, 2018.
- [42].Formulations for stabilizing phosphonamidate esters in long-term drug delivery systems, U.S. Provisional Pat. Ser. No. 62/961,294, filed January 15. (2020). [Google Scholar]
- [43].Bushman LR, Kiser JJ, Rower JE, Klein B, Zheng JH, Ray ML, Anderson PL, Journal of pharmaceutical and biomedical analysis 2011, 56, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hummert P, Parsons TL, Ensign LM, Hoang T, Marzinke MA, Journal of pharmaceutical and biomedical analysis 2018, 152, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research, C. f. V. Medicine, (Eds: U.S. Department of Health and Human Services, U.S. Food and Drug Administration; ), 2018, 44. [Google Scholar]
- [46].Shieh E, Marzinke MA, Fuchs EJ, Hamlin A, Bakshi R, Aung W, Breakey J, Poteat T, Brown T, Bumpus NN, Hendrix CW, Journal of the International AIDS Society 2019, 22, e25405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].García-Lerma JG, Otten RA, Qari SH, Jackson E, Cong M.-e., Masciotra S, Luo W, Kim C, Adams DR, Monsour M, Lipscomb J, Johnson JA, Delinsky D, Schinazi RF, Janssen R, Folks TM, Heneine W, PLoS Medicine 2008, 5, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Biesinger T, White R, Yu Kimata MT, Wilson BK, Allan JS, Kimata JT, Retrovirology 2010, 7, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Polacino P, Cleveland B, Zhu Y, Kimata JT, Overbaugh J, Anderson D, S. L. J. J. o. m. p. Hu, 2007, 36, 254. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.