

Abstract
Tissue engineered grafts show great potential as regenerative implants for diseased or injured tissues within the human body. However, these grafts suffer from poor nutrient perfusion and waste transport, thus decreasing their viability post-transplantation. Graft vascularization is therefore a major area of focus within tissue engineering because biologically relevant conduits for nutrient and oxygen perfusion can improve viability post-implantation. Many researchers utilize microphysiological systems as testing platforms for potential grafts due to an ability to integrate vascular networks as well as biological characteristics such as fluid perfusion, 3D architecture, compartmentalization of tissue-specific materials, and biophysical and biochemical cues. While many methods of vascularizing these systems exist, microvascular self-assembly has great potential for bench-to-clinic translation as it relies on naturally occurring physiological events. In this review, we highlight the past decade of literature and critically discuss the most important and tunable components yielding a self-assembled vascular network on chip: endothelial cell source, tissue-specific supporting cells, biomaterial scaffolds, biochemical cues, and biophysical forces. This article discusses the bioengineered systems of angiogenesis, vasculogenesis, and lymphangiogenesis, and includes a brief overview of multicellular systems. We conclude with future avenues of research to guide the next generation of vascularized microfluidic models and future tissue engineered grafts.
Keywords: organ-on-a-chip, angiogenesis, vasculogenesis, lymphangiogenesis, microvascular networks, self-assembly
Graphical Abstract
Microphysiological systems have recently seen increasing reports of on-chip microvascular network integration due to the endothelium’s critical role in natural physiology as well as tissue engineering. Herein, we review recent reports and identify that five tunable ingredients independently and interdependently contribute to microvascular network formation via angiogenesis, vasculogenesis, and lymphangiogenesis.
1. Introduction
The vascular endothelium is the largest organ in the body by surface area.[1] In fact, the vessel wall is five times the size of the heart in mass and six times the size of a tennis court in area.[2] Microvascular networks are dynamic structures composed of organized monolayers of endothelial cells (ECs), comprising a large percentage of this organ and serving as a key component of many tissues in the body. For example, the pancreatic islets are situated within a dense capillary bed to facilitate hormone delivery,[3–4] cardiac tissue features dense networks of vasculature for blood supply,[5] and the gastrointestinal tract is riddled with single-layered vessels to facilitate nutrient transport to the circulation.[6] Without a microvascular network surrounding and flowing throughout these and many other tissues, cells responsible for critical functions lose their source of oxygen and key nutrients while building up waste materials, all of which contribute to their death[7]. Additionally, the endothelium provides regulatory information contributing to the homeostasis of tissues through endothelium-tissue communication.[8–9] Indeed, the microvasculature is an essential component in maintaining healthy tissue.
Due to its apparent critical importance, incorporating a microvascular network into tissue engineered constructs has been a major focus in recent literature. Within this discipline, an exciting development has been the emergence of microphysiological systems (MPSs) for drug discovery, screening, and even biological investigation. These systems, often composed of cells and naturally derived or synthetic biomaterials in three-dimensional (3D) assemblies, are used to replicate the biochemical, electrical, and mechanical processes of natural tissues and organs. Unsurprisingly, some reports incorporate an endothelium due to its inherent importance in tissue-specific microenvironments (reviewed in [10–14]). In these reports, three methods of vascularizing MPSs are most common: 1) sacrificial molding of hollow lumens that are coated with ECs; 2) seeding of ECs on extracellular matrix (ECM) protein coated, rectangular polydimethylsiloxane (PDMS) microchannels, and; 3) self-assembly of microvessels within ECM-like biomaterials. Of these methods, we argue that microvascular self-assembly has the greatest potential for translation to tissue-specific models as well as microscale tissue engineered products (including cell replacement grafts such as islet and hepatocyte replacement therapy) because it relies on naturally occurring biological processes. Further, relying on microvascular self-assembly eliminates fabrication complexities when building MPSs or tissue engineered constructs with traditional methods such as 3D bioprinting.[15–17] However, for macroscale tissue engineering applications, microvascular self-assembly may not be an effective strategy due to the differential transport phenomena of oxygen and other solutes critical to cell survival.
Primary reports suggest five crucial components to be significantly important in fabricating a self-assembled, vascularized MPS: endothelial cell source, supporting cell source, biomaterials used to create the 3D vascular construct, activating growth factors, and mechanical stressors added into devices (Figure 1). Individually, these contribute to endothelial function, often influencing processes such as angiogenic sprouting and de novo vascular assembly, but in the past decade of literature, it appears that optimal combinations of these components yield vascularized MPSs and show potential for future devices. Therefore, as the tissue engineering and MPS fields progress, it is important to define what among these reports initiates vascular self-assembly. With this review, we present not only the impactful work accomplished in vascularizing in vitro systems, but also aim to categorize how each report falls into the 5-factor paradigm yielding physiological microvasculature in MPSs. A key significance of this article is that it will review angiogenesis (blood vessel sprouting), vasculogenesis (de novo vascular assembly), and lymphangiogenesis (lymphatic vessel sprouting) together, and concludes with a brief review of multicellular organ-systems that integrate these microvascular networks. Further, we will discuss the limitations of these devices and experimental protocols, as well as their benefits over other methods of vascularizing MPSs. Finally, we will offer perspectives on how these devices may be specifically designed to improve their utility in drug screening and disease modeling, as well as applications for translation into the healthcare pipeline.
Figure 1.
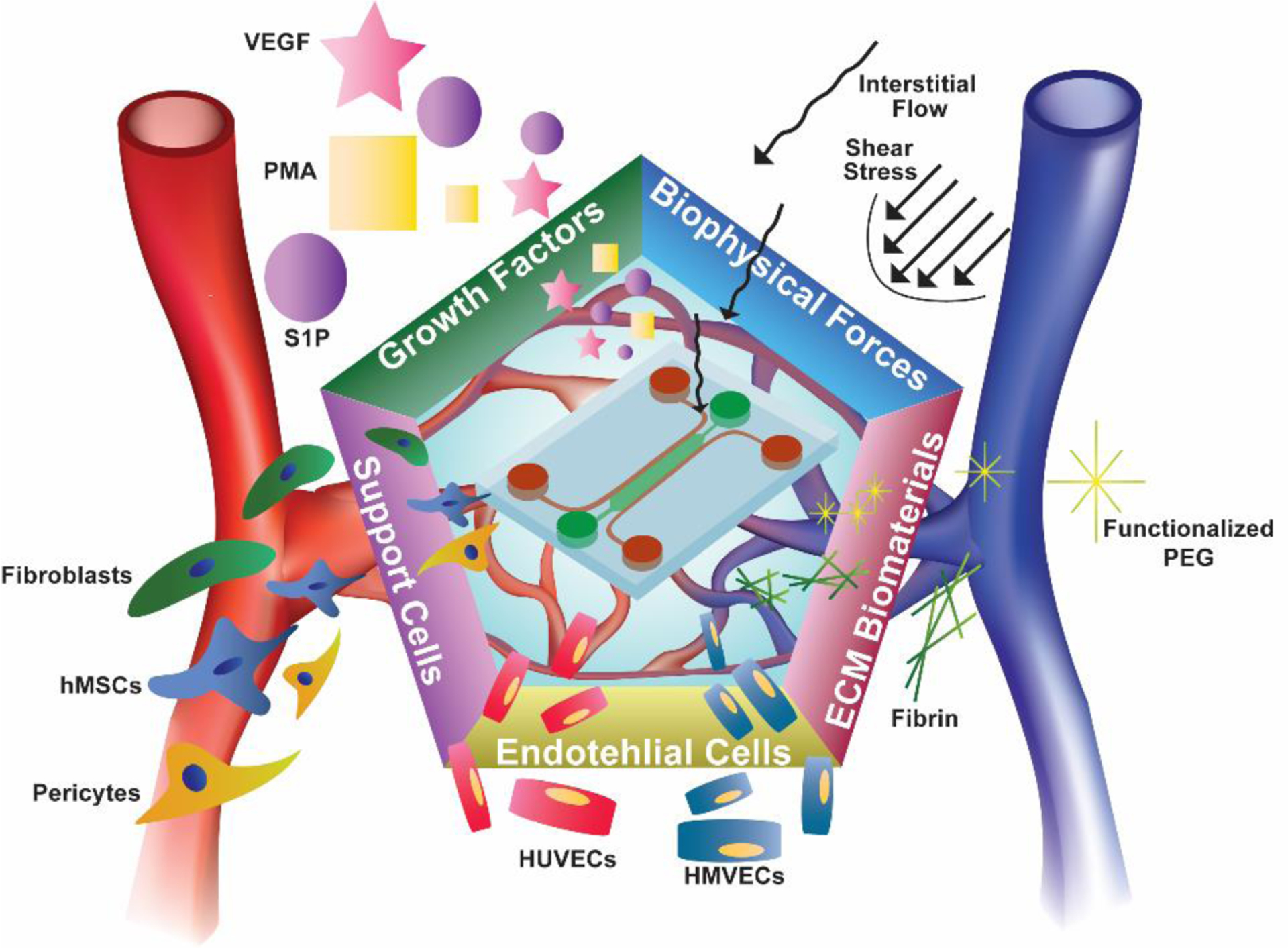
We argue that five critical components allow researchers to achieve vascularized MPSs: ECs, supporting cells, growth factors, ECM-mimicking biomaterials, and mechanical forces. As we will review, the combination of some or all of these components results in perfusable, self-assembled microvasculature for drug discovery and screening, healthy and diseased tissue modeling, and material testing for tissue engineering applications.
2. Angiogenesis-Chips: Microphysiological Systems of Angiogenesis
Angiogenesis – the formation of new vessels from pre-existing vessels – is a vital developmental process which contributes to the growth of vasculature in vivo. Although angiogenesis may occur via splitting of a parent vessel into two (intussusceptive angiogenesis), it is sprouting angiogenesis, characterized by the formation of sprouts of ECs, which is more understood and present throughout human life including embryonic development, wound healing, tissue regeneration, and in pathological conditions such as tumor growth.[18] Sprouting angiogenesis is typically characterized by three distinct endothelial cell processes: tip cell specification and migration, stalk cell proliferation, and tube maturation (Figure 2A).[19–20] Tip cell migration begins in response to a pro-angiogenic signal from a vasculature-deficient tissue. When receiving the signal, tip cells remodel tight junctions, degrade the basement membrane by secreting matrix metalloproteases (MMPs), and initiate pericyte detachment. Vascular endothelial growth factor (VEGF) is the most characterized pro-angiogenic signal and principally activates VEGF Receptor-2 (VEGFR-2). This signaling initiates a cascade within the tip cell that causes delta-like ligand 4 over-expression that laterally inhibits neighboring endothelial cells by Notch activation.[21–22] The Notch activated cells then follow tip cells and proliferate, specifying their stalk cell fate. Stalk cells continue to proliferate causing sprout elongation before laying down a new layer of laminin and secreting factors such as platelet-derived growth factor (PDGF) to attract pericytes (or smooth muscle cells for larger sprouts).[23] When pericytes begin forming cell-cell contacts with the proliferating stalk cells, proliferation halts and stalk cells assume a quiescent state.[20] Final tube formation through specification into phalanx cells occurs when fluid begins perfusing into the new lumen.[19] This leads to shear stress-activation of EC factors that upregulate tight junctions, oxygenation of surrounding tissue that halts pro-angiogenic signaling, and maturation of newly recruited pericytes. Several engineering approaches have led to microphysiological models of sprouting angiogenesis that replicate these physiological events. In this review, we specifically focus on models where capillary beds form in response to the critical components (Figure 1) that initiate vascular self-assembly and summarize these components in Table 1.
Figure 2.
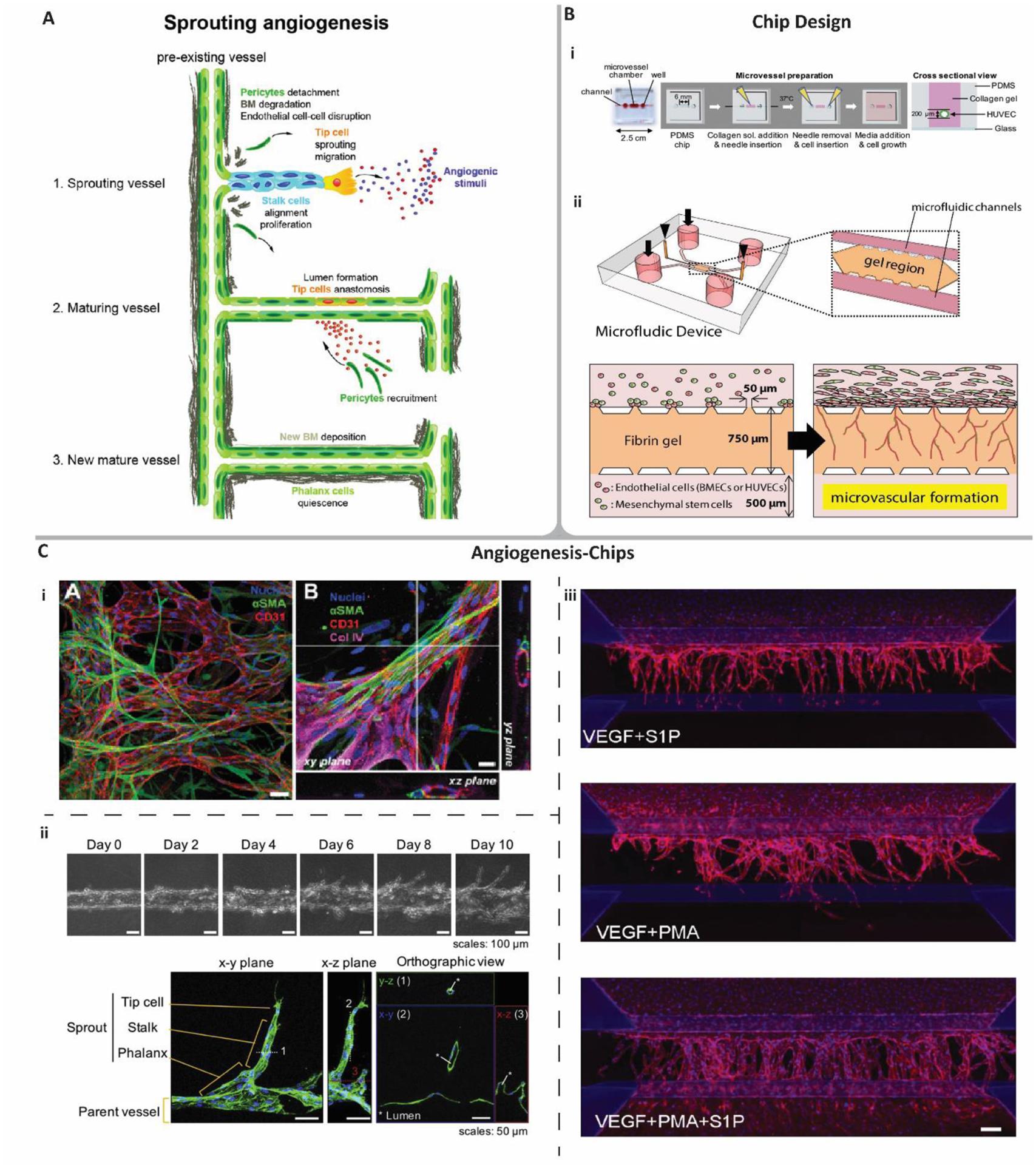
Angiogenesis and its MPS models. A) sprouting angiogenesis occurs first through tip cell migration in response to angiogenic stimuli. Stalk cell proliferation causes sprouts to elongate. Vessels mature via pericyte recruitment that forms cell-cell contacts with ECs. Angiogenesis ends when ECs and pericytes mature and lay down basement membrane (denoted BM) and enter a quiescent state. Reproduced with permission.[139] 2013, John Wiley and Sons. B) traditional microfluidic device designs used in angiogenic self-assembly. i) cylindrical lumen formation via gelation of hydrogels around a removable needle. Reproduced with permission.[35] 2018, Elsevier. ii) micropost device defined as multiple parallel channels boundaried by microposts forming perpendicular conduits through which cells, materials, and fluids can perfuse throughout a device. Angiogenesis-chips utilizing a micropost device typically coat a fluidic channel with ECs to mimic a parent vessel. Sprouts migrate into a hydrogel channel that remains within microposts due to the gel solution’s high viscosity and surface tension. Reproduced with permission.[46] 2019, Elsevier. C) Examples of angiogenesis-chips incorporating some or all of the five reviewed components initiating angiogenic self-assembly in MPSs. i) Left: microvascular networks formed via angiogenic sprouting supported by pericytes. Scale bar = 50 μm. Right: Higher magnification and confocal sections. Scale bar = 20 μm. Reproduced with permission.[27] 2013, Royal Society of Chemistry. ii) VEGF-induced angiogenic sprouting from a parent vessel recreated in a cylindrical lumen device. Reproduced with permission.[35] 2018, Elsevier. iii) Angiogenic sprouting assay to determine the optimal combination of different, previously reported pro-angiogenic growth factors. Scale bar = 100 μm. Reproduced with permission.[54] 2019, Springer Nature.
Table 1.
Reports of MPSs featuring angiogenic sprouting utilizing the five reviewed components of vascular self-assembly.
| EC type | Supporting Cells | Growth Factors | ECM Biomaterial | Mechanical Forces | Comments | References |
|---|---|---|---|---|---|---|
| HUVECs | N/A | VEGF | Collagen | N/A | Some models adapted for HTS[24] and drug studies[35] | [24, 31, 35] |
| N/A | VEGF | Collagen | IF | Studied the balance of VEGF and IF on sprout morphology | [60] | |
| NHLFs | VEGF | Fibrin | N/A | Found that adding exogenous VEGF hindered sprouting | [29] | |
| NHLFs | VEGF, S1P | Fibrin | IF | Model measured a de novo network’s angiogenic potential | [26] | |
| NHLFs and Pericytes | N/A | Fibrin | N/A | Platform variations used in cancer[27], inflammation[28] and bone tumor models[100–101] | [27–28, 100–101] | |
| hMSCs | N/A | Fibrin | N/A | Also studied BMECs to show translation to BBB models | [46] | |
| HS5 Bone Marrow Stromal Fibroblasts | N/A | Fibrin | N/A | Studied DLL4-Notch signaling with GNR-LNA biosensors | [53] | |
| Pericytes | VEGF, bFGF, PMA | Collagen | Laminar Flow | Utilized for BBB and cancer chips | [36] | |
| N/A | VEGF, TGF-β, PDGF, BMP | Collagen | N/A | Comparative study on single or combinatory GF gradients | [25] | |
| N/A | S1P, PMA, HGF, VEGF, MCP-1 | Collagen | N/A | Individual factors were unable to induce robust sprouting similar to the combination of the factors | [41] | |
| N/A | S1P, PMA | Collagen | N/A | Investigated the balance between tip cell migration and stalk cell proliferation | [40] | |
| N/A | VEGF, S1P, PMA | Collagen | IF | Utilized their device for anastomosis experiments | [54] | |
| 10T 1/2 Mouse SMCs | N/A | Collagen/Matrigel | N/A | Interestingly formed lumens via viscous fingering | [37] | |
| HUVECs, HAECs | N/A | VEGF | Collagen | N/A | Comparative study on EC source. Interestingly studied the impact of hydrogel stiffness on sprouting between the two EC types | [44] |
| HMVECs | N/A | VEGF, ANG-1 | Collagen | N/A | Utilized their device to perpendicularly diffuse VEGF against ANG-1 | [30] |
| HDF | VEGF | Collagen/Alginate | N/A | Also studied angiogenesis when fibroblasts were replaced with HT-1080 human fibrosarcoma cells | [43] |
2.1. Chip Designs
Most contemporary microfluidic models of angiogenesis rely on traditional soft lithography patterning using PDMS. While advantageous for its optical transparency, gas diffusivity, and rapid fabrication, cured PDMS is naturally hydrophobic and many reports of angiogenesis-chips utilize this property to compartmentalize tissue regions. For example, researchers have designed parallel and adjacent channels separated by microposts that run along the channel boundary at a given spacing (Figure 2B). The spacing of microposts in a hydrophobic channel enables injectable hydrogel pre-polymers with high viscosity and surface tension to flow through one channel without leaking into an adjacent channel. After gels are crosslinked and form semi-solid materials in the microchannel, fluid can be introduced into neighboring microchannels, thus creating a multi-compartment device with boundaries separated by fluid-hydrogel interfaces (micropost devices). Utilizing this general device design has enabled researchers to study diffusion of growth factors across hydrogel compartments while also providing a space for cell migration, proliferation, encapsulation, and ECM remodeling. This design also facilitates easy EC loading, where researchers can either form a parent vessel with a rectangular but rounded shape[24–25] or form a monolayer on the side of the hydrogel by tilting the device 90⁰ after injecting the EC suspension. [26–29] Both methods have been reported but more often researchers form an entire parent vessel as it allows for higher fidelity to in vivo angiogenesis. Other modifications include coating PDMS microchannels before loading hydrogels because some studies found that adjacent parent vessels sprout under the gel and form monolayers instead of sprouting three-dimensional tubular structures into the gel.[30–34] A study was presented specifically on preventing EC slippage, finding that coating channels with poly-D-lysine before injecting pre-hydrogel polymers created a better PDMS-hydrogel bond that prevented ECs from proliferating and migrating under the hydrogel.[31]
Later works focused on assembling parent vessels using the aforementioned sacrificially molded, cylindrical lumens that are later endothelialized. To accomplish this, researchers design single channels in PDMS devices and insert PDMS rods or medical-grade needles into the center of the channel (Figure 2B).[35–36] These components are considerably smaller than the PDMS channel, allowing injection of hydrogel pre-polymers into the remaining volume of the microchannel. The gel can then be solidified before removing the rod, leaving a cylindrical lumen that can be endothelialized. Other groups accomplish the same using a method termed “viscous fingering,” where the same PDMS microchannel is completely filled with a hydrogel pre-polymer and immediately injected with a less viscous fluid.[37] After crosslinking the hydrogel, a similar cylindrical lumen is left behind. Other engineers may utilize sacrificial molding of gels or other dissolvable materials (reviewed in [38]), but examples where this technology is used to model angiogenesis within MPSs are rare.[39] Studies have also utilized a dual-lumen setup where one lumen is endothelialized while the other serves as a support cell or pro-angiogenic factor source that can be endothelialized via sprouting angiogenesis of the parent vessel.[40–42]
2.2. Endothelial Cell Types
Angiogenesis-chips are made with ECs, but EC sources are widely varied between reports. Several groups use human umbilical vein endothelial cells (HUVECs) or human microvascular endothelial cells (HMVECs) (Table 1). Both cell types are commercially available and can come with optimized cell culture mediums (EGM-2 or EGM-2 MV). For researchers not experienced in cell culture, utilizing optimized but proprietary components helps decrease error. Other reports detail the use of red fluorescent protein (RFP)- or green fluorescent protein (GFP)-expressing HUVECs that are commercially available[24, 29] or transfected in-house[43] because they allow for live tracking of the device without fixing and staining the cells, thus saving time and materials. Interestingly, one report compared two commercially available EC lines, HUVECs and human aortic endothelial cells (HAECs), in an angiogenesis-chip and found that HAECs have a higher angiogenic potential defined by a higher fibroblast growth factor (FGF) secretion upon VEGF stimulation.[44]
Primary EC types usually depend on the platform application. For example, in models of the blood-brain barrier (BBB), researchers relied on angiogenesis to model the vascularized compartment between a parent vessel and brain cells such as astrocytes.[45–46] One BBB-on-a-chip study compared the angiogenic potential of HUVECs and human brain microvascular endothelial cells (HBMECs), finding that the two self-assembled into networks with similar network diameter but HUVECs had a more extensive network.[46] Diffusion assays also showed that HBMECs were more permeable, corroborated by their decreased tight junction expression. Another BBB-on-a-chip report utilized iPSC-derived ECs (iPSC-ECs), another cell type with tissue-specific differentiation potential.[45] The iPSC-ECs’ ability to also demonstrate angiogenic sprouting similar to HUVECs and HBMECs highlights an avenue for personalized medicine, since iPSCs can be derived from patient somatic cells. Clearly, the EC source is an important factor when testing functionality of a vascularized MPS, and the variability in angiogenic potential could be validated against the most extensive EC type, such as HUVEC, before conducting extensive studies.
2.3. Supporting Cell Types
A common design feature in angiogenesis chips is to situate stromal cells in hydrogels or in monolayers across from ECs (Table 1). The motivation for this is to recapitulate processes such as the proliferative phase of wound healing, where stromal cells migrate to inflammatory regions and secrete pro-angiogenic factors to initiate new blood vessel formation.[47–48] Early works detailed the use of normal human lung fibroblasts (NHLFs) encapsulated in separate hydrogel compartments that secreted factors across microfluidic devices towards microchannels housing monolayers of ECs.[27–28] In a relatively short time period (~5 days), sprouting and migration of tip cells, followed by proliferation of stalk cells, resulted in tubular network formation towards the fibroblasts. Further, networks matured and demonstrated perfusibility using traditional Dextran and fluorescent bead assays.
Other stromal cell types have been used in some angiogenesis-chip reports. In an earlier angiogenesis-chip, human placenta pericytes were included in the monolayer seeded on the side of the hydrogel wall (Figure 2C-i).[27] Pericytes migrated into the hydrogel following the sprouting EC branches, attached themselves to the newly forming vessels, and expressed α-smooth muscle actin (α-SMA). A follow-up experiment compared EC and EC-pericyte angiogenesis-chips.[28] They found that when pericytes wrapped themselves around the vessels, network diameter and permeability decreased while the expression of tight junctions increased, thus demonstrating a physiological relevance of their MPS.
Other reports detail alternative device organization. One study demonstrated angiogenic sprouting in the same exact micropost device mentioned earlier but with a modified cell compartmentalization: two EC monolayers coated the sides of a central hydrogel containing spheroids of fibroblasts or fibroblasts co-cultured with ECs.[29] Interestingly, the time for each EC monolayer to sprout toward the center of the hydrogel was double that of the monolayer of the original work, despite the monolayers in the newer work being much closer to the same pro-angiogenic source. This is indicative that the stromal network architecture, distribution within the chip, or proximity to the ECs affects the gradient of pro-angiogenic factors, thus altering the paracrine signaling inducing angiogenesis.
2.4. Synthetic and Biologically Derived Hydrogels to Support Angiogenesis
Biomaterials designed to support in vitro angiogenesis are plentiful (reviewed in [49–52]) However, on-chip examples of angiogenesis that can be integrated into other tissue-specific models utilize a limited range of hydrogels currently. Most chips using such biomaterials support compartmentalization of different components, diffusion across a device, and 3D assembly of supporting cell networks and vascular networks. These biomaterials aim to mimic the interstitial space or ECM of the vascular tissue. Hydrogels have been an attractive material in these chips because they support diffusion of pro-angiogenic factors and other cell signals, provide a scaffold for cells to assume 3D structure, and can be loaded in the aforementioned micropost devices. Further, hydrogels can be fabricated using synthetic or biologically derived materials, allowing researchers to engineer the angiogenic process to support rapid migration, proliferation, or desired microvasculature structure.
Early models relied on hydrogels sourced from collagen type I, usually from rat tail.[24, 30–31, 37, 41] This biomaterial has an easy fabrication process, usually involving neutralizing the stock material and diluting with cell culture medium before initiating crosslinking through heating. Further, the injectable hydrogel pre-polymer has been reported as having optimal rheological properties that allow loading of the solutions into micropost devices with a low failure rate. Interestingly, one report found that decreasing concentration of collagen I caused tip cells to migrate faster, sometimes breaking from the proliferating stalk cells.[40] Later reports utilized similar devices with fibrin hydrogels, sometimes supplemented with collagen type I, because the material is a more faithful representation of the proliferative phase of wound healing that the co-culture of NHLFs and ECs was replicating.[27–29, 53] Here, human or bovine fibrinogen was first prepared separately from thrombin. The two solutions were then mixed and immediately loaded into micropost devices before rapid solidification occurred. As with the collagen gels, injectable fibrin pre-polymers have suitable rheological properties that allow for repeated successful loading into devices. Some reports detail Matrigel mixed with collagen as a hydrogel source, but these are far less common than collagen and fibrin hydrogels.[37]
Synthetic materials are sometimes used in micropost devices as mimics of engineered grafts that can support angiogenesis. The motivation for this is to design optimal materials or decipher the minimum essential components needed in a tissue engineered graft that could support a vascularizable implant for in vivo studies. One study detailed the encapsulation of 150 μm alginate beads in 2.5 mg/mL collagen type I hydrogels before seeding flanking channels with HMVECs.[43] One motivation for this was to provide a scaffold where ECs could sprout through spaces made between microbeads. They later showed that fibrosarcoma cells encapsulated within the beads enhanced the angiogenic response, theoretically due to the addition of angiogenin-1 (ANG-1) and placental growth factor. Interestingly, this report presents a potential cell or biologic transplantation method, where cells needing to be vascularized or factors to induce vascularization can be housed within microbeads encapsulated in a hydrogel that supports host angiogenesis. Further, the encapsulating hydrogel can be readily engineered for additional properties such as immunoprotection.
2.5. Growth Factors
Another key area of research utilizing angiogenesis-chips is the addition of growth factors and biochemical cues that affect sprouting characteristics. Earlier reports detailed the use of VEGF, one of the most characterized pro-angiogenic factors (Figure 2C-ii).[30–31, 37, 43] Among these reports, a range of 20 – 100 ng/mL were enough to stimulate angiogenesis across devices. Later reports detailed combinations of factors in single culture systems in efforts to optimize vascular network formation. One study reported that four cocktails were tested in a dual lumen device featuring HUVECs and HMVECs and resulted in some angiogenic sprouting.[41] Two optimal cocktails were found utilizing monocyte chemotactic protein-1 (MCP-1), hepatocyte growth factor (HGF), basic fibroblast growth factor (bFGF), phorbol 12-myristate 13-acetate (PMA), VEGF, and sphingosine-1-phosphate (S1P). One cocktail, termed “MVPS,” featured MCP-1, VEGF, PMA, and S1P, while the other, termed “HMVPS,” contained the same factors and supplemented with HGF. Interestingly, when testing S1P or PMA alone, some tubular formation was observed but often tip cells broke away from stalk cells or stalk cells failed to proliferate at all. Similarly, another group tested the combination of VEGF, S1P, and PMA, finding these three factors were optimal in inducing angiogenic sprouting (Figure 2C-iii).[54] Recently, one report expanded upon these observations, finding that parent vessels formed with needle lumen scaffolds displayed angiogenic sprouting that depended on the balance between EC migration (S1P-induced) and proliferation (PMA-induced).[40] While it would be ideal that angiogenesis chips consist of growth factors in physiological concentrations, it appears there is no gold standard when used in engineered systems. This could be partly because the biological characterization of growth factors and its independent or collective influence on angiogenesis is lacking. Further, it is not fully known what the constitution of growth factors and their variability between organs is. Finally, device geometries differ between groups, so different initial growth factor concentrations will yield gradients of different magnitudes. Therefore, a consensus may be needed as to the formulation of growth factors induced within angiogenesis chips so that results can be reproduced faithfully amongst different laboratories and scientific groups.
2.6. Biophysical Forces
Microfluidic devices offer an attractive ability to add mechanical forces to an in vitro platform that traditional techniques cannot. Shear-stress induced by fluid flow is an important biophysical force to include in vascularized devices because many reports detail differential EC gene and protein expression and elongated morphology.[55–57] In in vitro studies of angiogenic sprouting outside of microfluidic devices, some reports found that shear stress activation coupled with S1P supplementation induced deeper and more frequent tip cell sprouts.[58–59] In recent literature, angiogenesis-chips more often dissect the effects of interstitial flow (IF), a creeping transport of fluid shown to affect cell migration and morphology.[26, 60] In studies adding IF to angiogenesis-chips, fluid flows across a microfluidic device perpendicular to the parent vessel, either originating from a microchannel opposite but parallel the parent vessel or originating from the parent vessel and flowing outwards. In each case, IF acts across the basal-apical or apical-basal plane of the ECs. Studies have shown that flow in the direction of the vessel induces sprouting in a magnitude-dependent manner, where varying basal-apical IF from an opposite microchannel to the parent vessel results in different microvascular network density, sprout length, and number of sprouts.[26, 60–62] The flow rate in these reports vary but are often induced by differential pressure heads at the fluidic channels’ inlets in the range of 2–25 mmH2O, resulting in flow rates of 0–10 μm/s across the compartment housing hydrogel scaffolds. One study showed that without IF, the concentration gradient of pro-angiogenic growth factors depended on simple diffusion, and thus the plot of concentration of VEGF across the hydrogel compartment had a sloping decrease from the source to the parent vessel.[62] However, the addition of IF from the growth factor channel disturbed the concentration gradient and saturated the hydrogel scaffold with VEGF, which in turn increased angiogenic sprouting. A later study further dissected the interaction of biophysical and biochemical events resulting from IF.[60] They showed that that different levels of IF with constant VEGF flux yielded differences in the resulting sprouts from a parent vessel, proving that ECs not only react to changing levels of pro-angiogenic stimuli, but also sprout in response to different magnitudes of fluid flow perpendicular to the vessel wall. Interestingly, one report swapped the parent vessel with a microvascular bed within a hydrogel and found that the angiogenic sprouting into an adjacent hydrogel compartment was flow direction dependent[26]. However, it is likely this resulted from a convection of VEGF and FGF that either saturated or deprived the angiogenesis compartment of the growth factors. In summary, it appears a balance between growth factor supplementation and biophysical cues contributes to robust angiogenic sprouting in vitro, highlighting that the addition of fluid forces is critical in recapitulating physiological characteristics of an angiogenic vessel.
3. Vasculogenesis-Chips: Microphysiological Systems of Vasculogenesis
While angiogenesis describes the sprouting of new vasculature from existing vessels, vasculogenesis has been characterized as endothelial precursor cells (EPCs) forming de novo vasculature through their self-assembly into tubular networks.[63] Because vasculogenesis is seen mostly during embryonic development, mouse or zebrafish models have been the standard in vivo models with which researchers dissect the mechanisms governing the physiological process. From these models, researchers have shown that mesoderm cells first differentiate into hemangioblasts, cells that have a multipotent phenotype and usually differentiate into ECs or hematopoietic cells (Figure 3A).[63–64] Hemangioblasts then migrate to form a blood island, a mass of cells containing an outer and inner population of cells. Blood islands have been described as the embryonic equivalent of bone marrow. The outer layer of the blood island differentiates into angioblasts and the inner layer obtains a hematopoietic cell fate. At this point, vasculogenesis begins and solely describes the maturation of the angioblasts on the outer side of the blood island. Angioblasts differentiate into ECs, forming tight junctions before laying down a basement membrane. Like angiogenesis, vasculogenesis proceeds with the final maturation of vessels before fluid flows through the newly established lumens. Some claim that de novo vascularization arising from angioblast self-assembly into EC networks occurs in adult humans due to the presence of circulating EPCs, but other disagree with the nomenclature, often citing the process as “neovascularization” to distinguish a difference.[65] Nevertheless, we review microfluidic models that successfully recapitulate this relatively less characterized vascular self-assembly process in the context of the critical components that make up these systems, as summarized in Table 2.
Figure 3.

Vasculogenesis and its MPS models. A) vasculogenesis in embryonic development begins as mesodermal cells aggregate to form a blood island. The outer cells specify to angioblasts, an EPC. As cells differentiate to ECs, tight junctions form and a basement membrane is laid down. Reproduced with permission.[64] 2010, UBC Press. B) Microfluidic models of vasculogenesis often follow two designs. i) 5-channel micropost device featuring compartmentalization of NHLFs and HUVECs in encapsulating fibrin hydrogels. Reproduced with permission.[27] 2013, Royal Society of Chemistry. ii) a variation of a micropost device featuring large diamond-shaped hydrogel compartments with small openings to fluidic microchannels. Reproduced with permission.[70] 2016, Springer Nature. C) Examples of vasculogenesis-chips formed using a combination of the five components of vascular self-assembly in MPSs. i) Vasculogenesis-chip formed by co-culturing encapsulated HUVECs in the same device as encapsulated NHLFs. Network functionality is confirmed via the perfusion of microbeads and fluorescent Dextran. Scale bar = 100 μm. Reproduced with permission.[27] 2013, Royal Society of Chemistry. ii) Vasculogenesis-chip displaying physiologically relevant tight junctions, von Willebrand Factor (vWF) expression, and collagen IV (basement membrane) deposition after co-culturing with senescent fibroblasts. Scale bar = 50 μm. Reproduced with permission.[72] 2019, John Wiley and Songs. iii) EC monocultures in vasculogenesis-chips setups are stable and selectively permeable when intraluminal flow is established. Scale bars = 100 μm. Reproduced with permission.[70] 2016, Springer Nature.
Table 2.
Summary of vasculogenesis-chips utilizing some or all of the five components of vascular self-assembly.
| EC Type | Supporting Cells | Growth Factors | ECM Biomaterial | Mechanical Forces | Comments | References |
|---|---|---|---|---|---|---|
| HUVECs | NHLFs | N/A | Fibrin | N/A | Common arrangement utilized in cancer studies[76–77] and particle coagulation in microvasculature[80] | [27, 76–77, 80] |
| Senescent NHLFs | N/A | Fibrin | N/A | Characterized secretome of proliferating and senescent fibroblasts | [72] | |
| NHLFs | VEGF, S1P | Fibrin | N/A | Tested different gel densities, GF concentrations, and cell densities and analyzed the resulting network characteristics | [73] | |
| hMSCs | N/A | Fibrin | N/A | Arrangement used with osteoblasts in bone tissue model | [78] | |
| hMSCs | VEGF, ANG-1, TGF-β1 | Fibrin | N/A | Found that hMSCs assume a mural phenotype and GF supplementation affects network characteristics | [79] | |
| N/A | VEGF | Fibrin, Collagen | N/A | Specifically tested ECM material for concentration and composition | [140] | |
| HUVECs, HMVEC-Ls | Lung Pericytes | N/A | Fibrin | N/A | Compartmentalized co-culture resulted in pericyte migration to microvasculature | [141] |
| ECFCs | NHLFs | N/A | Fibrin | IF | Component combination used in biological investigation[66–67] and drug testing[69–70] | [66–71] |
| iPSC-ECs | NHLFs | S1P | Fibrin | N/A | Study mainly focused on IPSC-EC characterization | [74] |
| Brain Pericytes | N/A | Fibrin | N/A | Studies often include brain astrocytes to mimic BBB | [32, 45] | |
| N/A | VEGF | Functionalized PEG | N/A | Optimized gel composition, VEGF concentration, and EC density | [75] |
3.1. Chip Designs
PDMS devices housing compartmentalized cultures of ECs with their pro-vasculogenic stimulants are closely related to angiogenesis-chips. Vasculogenesis-chips more often utilize micropost devices, as they easily allow any combination of the five components needed for microvascular self-assembly on chip (Figure 3B-i). One study even reported vasculogenesis- and angiogenesis-chips using the same device design but with different cell compartmentalization in each microchannel.[27] Generally, the experimental requirements (i.e. number of compartmentalized cell types needed to induce self-assembly) governs the number of parallel channels in micropost devices, but typical reports include 3–5 parallel channels with boundaries separated by microposts. Many other reports have emerged detailing devices featuring two parallel fluidic channels separated by large diamond compartments that house biomaterials supporting de novo vascular self-assembly (Figure 3B-ii).[66–71] Variations of this include situating multiple diamond-shaped compartments between more complex fluidic channels that are used to control fluid properties and forces across the vasculature chamber.[66]
3.2. Endothelial Cell Types
As with angiogenesis-chips, vasculogenesis-chips usually feature HUVECs and HMVECs because of their ease-of-use and commercial availability. Some studies have also utilized transfected HUVECs or HMVECs that produce RFP or GFP fluorescent signal.[70–72] Primary EC types are not as common, but have been used in a BBB model that combines angiogenic sprouting with de novo network formation via co-culture of HBMECs with brain astrocytes and pericytes.[45] Like angiogenesis-chips, the initial seeding density between vasculogenesis-chips reports varies, yet this has a greater impact on the characteristics of the microvascular network. No report indicates a difference in angiogenesis based on seeding density, but one report demonstrated that increasing the concentration of encapsulated HUVECs in a vasculogenesis-chip from 1 to 4 million cells/mL results in an increase in network branch length, diameter, and length, whereas a lower density results in thinner tubes encompassing the hydrogel compartment of a micropost device.[73]
It is suggested that vasculogenesis arises from angioblasts or EPCs, and thus vasculogenesis-chips featuring terminally differentiated HUVECs or HMVECs may not be a faithful representation of the in vivo process. Some precursor cells circulate in the blood and are thus a possible EC source for vasculogenesis chips. In one study, ECs from cord blood were isolated, referred to as “endothelial colony forming cells,” or ECFCs, with which de novo vascular networks on a chip were formed. While reports utilizing primary precursor cells are limited to one group,[66–71] many publications detail vasculogenesis-chips or multicellular MPSs with a de novo vascular network using commercially available iPSC-ECs.[32, 45, 74–75] This appears as a more physiologically relevant source for vasculogenesis-chips, yet iPSC cells are more stem-like than EPCs, thus highlighting potential misdifferentiation towards a different cell type. However, successful reports show vasculogenesis in microfluidic devices using iPSC-ECs cultured in proprietary but commercially available culturing reagents. Successful results here indicate potential for personalized, vascularized devices or tissue engineered constructs, similar to those shown with angiogenesis-chips. However, potential for this route requires advances in iPSC generation and their repeatable differentiation to EPCs or ECs.
3.3. Supporting Cell Types
Vasculogenesis-chips feature a range of supporting cells that induce the characteristic self-assembly of ECs into tubular structures. Like angiogenesis-chips, vasculogenesis-chips often feature stromal cell types as the supporting cell type. One investigation illustrated in its 5-channel device a central hydrogel compartment encapsulating HUVECs situated between two compartments of NHLF-encapsulating hydrogels (Figure 3C-i).[27] Like the angiogenesis-chip, this vasculogenesis-chip mimics the proliferative stage of wound healing where NHLFs secrete factors such as VEGF, thus activating the migration of ECs before inducing proliferation into immature tubular networks. Later, pericytes were added and demonstrated the characteristic vessel-wrapping phenotype indicative of a maturing vessel. Another study utilized a similar setup but induced senescence in their fibroblasts (“SFB”) because they found SFB conditioned medium had higher levels of VEGF, HGF, CXCL1, IL-6, IL-8, and MCP-1.[72] Because of this, microvascular networks with SFB were more mature and higher quality (Figure 3C-ii). Interestingly, they introduced a third cell type to model diabetic wound healing, where mouse islets were encapsulated with HUVECs and were subsequently vascularized. Finally, models with de novo assembly of vascular networks more commonly feature tumor cell co-culture with ECs, demonstrating the vasculogenic power of tumors.[70, 76–78] For example, cancer cell extravasation in vascularized constructs in micropost devices was shown in one study,[77] while another encapsulated a tri-culture of ECs, fibroblasts, and tumor cells and showed an increase in vascular self-assembly compared to a co-culture of ECs and fibroblasts alone.[70]
Human mesenchymal stem cells (hMSCs) are a common stromal cell type included in recent reports of vasculogenesis-chips. Similar to angiogenesis-chips, the inclusion of hMSCs yields their differentiation into pericytes that support the newly formed lumens.[78–79] A previous work found that the direct cell-cell contacts made between ECs and hMSCs caused hMSCs to adopt a mural-cell phenotype with increased α-SMA when stimulated with pro-vascular factors (discussed in Section 5).[79] Later, in a vascularized bone tissue model utilizing a tri-channel vasculogenesis-chip design, ECs were encapsulated with hMSCs and osteoblast-differentiated cells (OBs).[78] Like the earlier device, the hMSCs in this device differentiated into mural cells and supported the vasculature formed around OBs in fibrin hydrogel. These results showcase the ability of a supporting cell type to adopt a tissue-specific identity, therefore showing potential applications when designing multicellular MPSs that require self-assembled vasculature.
3.4. Synthetic and Biologically Derived Hydrogels to Support Vasculogenesis
Most vasculogenesis-chips consist of the same fibrin or collagen hydrogels featured in angiogenesis-chips, although fibrin gels appear more frequently. The concentration of fibrin in hydrogels is usually 2.5 mg/mL,[72, 74, 77, 80] but it was found that increasing fibrin concentration from 1.5 to 10 mg/mL resulted in increased number of branches within a vascular network and a decreased branch diameter and length.[73] Over the range, the area of coverage remained constant, therefore illustrating that higher concentrations of fibrin result in thinner but increased vascular branches, whereas a lower concentration results in a large-diameter network broadly encompassing the hydrogel compartment.
Synthetic hydrogels are less common. However, a micropost device was reported featuring poly(ethylene glycol) hydrogels containing MMP-degradable peptide crosslinks.[75] When forming iPSC-EC monolayers in adjacent compartments of their tri-channel device, de novo network formation was initiated in a cell density, MMP-crosslink percentage, and VEGF dependent gradient. They found that the optimal microvascular network formed when hydrogels contained a 50% MMP crosslink density, when ECs were encapsulated at a concentration of 10 million cells/mL, and when ECs received supplemented media containing 100 ng/mL VEGF. While other synthetic materials are common in other in vitro assays (reviewed in [81]), they often involve suspending ECs within gels and monitoring for tube formation, a method that prevents users from adding critical regulators of vasculogenesis including cell compartmentalization, addition of fluid forces through the hydrogels, and an inability to monitor barrier function of newly formed vasculature via Dextran perfusion.
3.5. Growth Factors
Due to the limited work on de novo vascularization in microfluidic devices, most reports rely on the traditional NHLF-ECs co-culture setup in micropost devices (Table 2). However, one group utilized VEGF and S1P in vasculogenesis-chips due to the positive results seen with other angiogenesis-chips.[73] They found that supplementing the EGM-2 MV medium with 50 ng/mL VEGF and 250 nM S1P did not increase vascular network coverage compared to a mono-culture of ECs encapsulated in fibrin hydrogel: vascular branch length, diameter, and number all decreased with the addition of the pro-angiogenic factors. When repeating the experiment with a co-culture device containing NHLFs, the addition of growth factors increased the number of branches but decreased the average branch length and diameter compared to devices containing NHLFs with unmodified EGM-2 MV media.
Later works focused on testing stimulating factors in mural cell supported vascular networks. For example, one report showcased a device featuring hMSC-derived mural cells that resulted in different vascular network characteristics when supplementing EGM-2 MV medium with VEGF, ANG-1, and transforming growth factor-β (TGF-β).[78] HUVECs encapsulated alone in tri-channel micropost devices with VEGF-supplemented medium resulted in robust networks with a large branch diameter. The addition of hMSCs decreased vessel diameter but increased the total length and number of branches. Co-cultured devices supplied with VEGF and ANG-1 supplemented EGM-2 MV resulted in a decrease in the branch diameter, length, and number but the microvasculature remained perfusable. VEGF combined with TGF-β exacerbated this decrease in network characteristics and formed a non-interconnected network. However, the combination of VEGF with ANG-1 or TGF-β resulted in an increase in the mural cell phenotype of hMSCs, which was also shown to be HUVEC dependent. Therefore, it appears that VEGF and ANG-1 can be utilized if thinner networks are desired in co-culture systems.
3.6. Biophysical Forces
Commonly reported vasculogenesis-chip setups involve a 3- or 5-channel device containing ECs with or without NHLFs encapsulated in hydrogels. Because cells are situated in hydrogels, shear stress cannot be applied against ECs until after the microvascular network forms. However, some reports from the same group illustrate the presence of IF across the hydrogel chamber containing encapsulated ECs.[66–71] In their diamond-chamber vasculogenesis-chip, flow through one fluidic channel resulted in a pressure drop across the EC encapsulating hydrogel. The pressure drop was modeled in the first report presenting the microfluidic device design, and later yielded vasculature development under this mechanical force.[67] Not too long after, the same group presented a related microfluidic device featuring long microchannels supplying the hydrogel compartment with oxygen and nutrients, but designed the channels such that they could control the Péclet number (Pe), or the ratio of convective to diffusive transport.[66] They found that low Pe, representing physiological hypoxia, and high Pe, representing IF, resulted in de novo vascularization, but intermediate Pe did not. These devices combined indicate the importance of including IF in regulating de novo vascularization. Interestingly, one group found that after networks formed, intraluminal flow induced by a pressure drop across the vascularization matrix caused stable network generation without a supporting cell source (Figure 3C-iii).[70] Such findings have physiological relevance because newly formed vasculature matures and exhibits selective permeability in response to intraluminal flow.
4. Lymphangiogenesis-Chips: Microphysiological Systems of Lymphangiogenesis
The lymphatic vasculature is closely related to the vascular endothelium. During embryonic development in mouse models, lymphatic vessels first appear through polarization of the anterior cardinal vein.[82–83] In response to VEGF-C stimulation, venous ECs begin expressing prospero homeobox protein-1 (PROX-1), VEGFR-3, and lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), initiating their specification as lymphatic endothelial cell (LEC) progenitors and subsequent lymphatic commitment (Figure 4A). Through continued VEGF-C signaling, the three aforementioned markers and podoplanin are continually expressed as LEC progenitors bud from the other vascular endothelial cells to form a lymph sac. The lymph sac, also known as a lymphatic vessel precursor, continues to mature into a lymphatic network through the sprouting of new vessels, or lymphangiogenesis.
Figure 4.

Lymphangiogenesis and its microfluidic models. A) Lymphangiogenesis in the developing embryo begins as ECs in the anterior cardinal vein begin expressing PROX-1, an event specifying their LEC fate. LECs migrate and form lymph sacs, or precursors to lymphatic vessels. As the lymph sacs mature, lymphangiogenic sprouts bud and form new lymphatic microvasculature. In the adult, lymphangiogenesis is uncommon except in pathological conditions, where sprouting follows similar EC events such as migration and proliferation in response to pro-lymphangiogenic factors such as VEGF-C. Reproduced with permission.[82] 2010, Company of Biologists. B) Common chip designs in lymphangiogenesis MPSs. i) Micropost device situated across from fibrin-encapsulated NHLFs, similar to early angiogenesis-chips. Reproduced with permission.[85] 2016, Elsevier. ii) Cylindrical lumen formed through sacrificial molding around solidified hydrogels, followed by endothelialization by LEC coating. Reproduced with permission.[87] 2020, Royal Society of Chemistry. C) Examples of lymphangiogenesis-chips. i) LECs sprout in response to IF toward a parent lymphatic vessel and a combination of growth factors that alone induce less sprouting. Reproduced with permission.[85] 2016, Elsevier. ii) Lymphatic vessels reported to exhibit higher lymphangiogenic sprouting in response to laminar flow (denoted LF) within the vessel. Scale bars = 100 μm. Reproduced with permission.[94] 2017, American Society for Clinical Investigation.
Unlike angiogenesis, lymphangiogenesis involves the sprouting of lymphatic endothelial cells (LECs). However, lymphangiogenesis and angiogenesis are similar in that lymphatic sprouts form from pre-existing lymphatic vessels in a coordinated process of cellular events. Indeed, lymphangiogenesis involves LEC migration, proliferation, and tubular maturation of LECs similar to angiogenesis of vascular ECs. These processes depend on VEGFR-2 and −3 signaling by VEGF-C and -D.[84] Lymphangiogenesis occurs post-development only during pathological conditions such as tumor growth, metastasis, and inflammation as well as during tissue repair.[83] As such, many MPSs demonstrating lymphangiognesis find their applications in tumor models, with which drugs can be tested. Few others examine other features of the five components of vascular self-assembly, such as pro-lymphangiogenic growth factor cocktails. In the following section we will review the different models in this growing field of research, which we also summarize in Table 3.
Table 3.
Cooperative effects of the five components of vascular self-assembly in MPS models incorporating lymphangiogenesis.
| EC type | Supporting Cells | Growth Factors | ECM Biomaterial | Mechanical Forces | Comments | References |
|---|---|---|---|---|---|---|
| LEC from Lymph Nodes | MCF7, MDA-MB-231 breast cancer cells | N/A | Collagen | N/A | Demonstrated cancer cell line dependent changes in lymphangiogenic activity | [86] |
| MDA-MB-231 breast cancer cells | IL-6 | Collagen | N/A | Studied different gel concentrations to mimic cancerous breast tissue | [87] | |
| N/A | VEGF-C, VEGF-D, IL-6 | Collagen | N/A | Studied gel concentration and inflammatory cytokine stimulation | [93] | |
| Primary LECs (unspecified) | N/A | N/A | Collagen | Laminar Flow | Found that Notch activity was downregulated causing increased sprouting | [94] |
| HMVEC-dLy-Neo | N/A | VEGF-C, PMA | Collagen | Laminar Flow | Co-cultured the lymphatic vessel across from a HUVEC vessel to induce angiogenic and lymphangiogenic sprouting to a central point | [88] |
| HMVEC-dLyAd | NHLFs | VEGF-A, VEGF-C, bFGF, S1P | Fibrin | IF | Demonstrated combinatory effect of the five components. Also tested VEGF-D, HGF, IGF-1, and PDGF-BB in supplementary experiments | [85] |
| MDA-MB-231 breast cancer cells | VEGF-C | GelMA | Laminar Flow | Utilized sacrificial molding by 3D printing agar rods | [95] |
4.1. Chip Designs
Microfluidic models of lymphangiogenesis have only emerged recently and reports are thus rare. In one of the first such reports, a 5-channel micropost device was utilized similar to angiogenesis-chip published earlier (Figure 4B-i).[85] The chip allowed for compartmentalization of hydrogels that served as lymphangiogenic remodeling and support matrices as well as stromal cell scaffolds. Later, another group published works with lymphatic vessels similar to other vascular lumen devices (Figure 4B-ii).[86–87] Here, the same chip featuring a straight channel with a wide hydrogel chamber housing a removable rod was used to form a hollow lumen after hydrogel crosslinking, on the inside walls of which they seeded LECs. Recent reports featuring lymphangiogenesis generally follow these design rules, sometimes varying the design to include multiple lumens or vessel microchannels in close proximity with the goal of providing a stimulating factor source or a vascular-lymphatic vessel co-culture on one chip.[88–89]
4.2. Endothelial Cell Types
As mentioned earlier, lymphatic vessels consist of LECs arranged in tubular structures. They are leaky, secrete basement membrane proteins such as laminin and collagen type IV, and are characterized by their surface expression of podoplanin, PROX-1, and LYVE-1.[90–92] One of the first lymphangiogenesis-chip reports utilized commercially available human dermal lymphatic microvascular endothelial cells (HMVEC-dLyAd), cultured in EGM-2 MV, and then seeded these as a monolayer on the hydrogel side wall in their micropost device.[85] A later report detailed the use of human neonatal dermal lymphatic microvascular endothelial cells (HMVEC-dLy-Neo) cultured in EGM-2 medium.[88] Other reports with cylindrical lumen devices detail the use of commercially available LECs from the lymph node in EGM-2 MV or EGM-2.[86–87, 93] To the best of our knowledge, only one study where lymphangiogenesis was reported utilized a non-commercial LEC source, where human primary LECs isolated from human neonatal foreskins were cultured in EGM-2.[94] As future models will be designed, it can be expected that differential effects of lymphatic endothelial cell sources and their culturing media will be characterized more rigorously.
4.3. Supporting Cell Types
Co-cultures of LECs with other cell types is common, especially in tumor models. In one device, a dual cylindrical lumen chip was presented where a lymphatic vessel was cultured in close proximity to different breast cancer cell lines.[86] Not only did sprouting of lymphatic vessels occur when co-cultured with cancer cells, but LECs exhibited a cancer cell line dependent differential expression of pro-lymphangiogenic genes. Co-culturing with MCF7 breast cancer cells led to overexpression of FGF2, VEGF-C, angiopoietin-2 (ANGPT2), and placental growth factor (PGF), factors reported to be promoters of lymphangiogenesis and neovascularization. Other chips featuring co-cultures of cancer cell lines and LECs demonstrate similar results, usually using cylindrical lumen chips formed by sacrificial molding.[95]
Few reports utilize fibroblasts in their lymphangiogenesis-chips.[85, 89, 93] One group designed a micropost device with similar cell compartmentalization as the earlier angiogenesis-chips where they encapsulated NHLFs across from a monolayer of LECs and observed sprouting.[85] However, broad sprouting was only seen when supplementing the cell culture medium with pro-lymphangiogenic factors such as VEGF-C. In another report, lymphangiogenic sprouting was reported when co-culturing lymphatic vessels with cancer associated fibroblasts that produced a pro-inflammatory lymphatic microenvironment.[93] It therefore seems that healthy stromal cells alone may not be enough to induce lymphangiogenesis, and that growth factor supplementation or co-culture with more potent cell types is a route for better success.
4.4. Synthetic and Biologically Derived Hydrogels to Support Lymphangiogenesis
One of the first lymphangiogenesis-chip reports featured the same collagen-supplemented fibrin hydrogel as their earlier angiogenesis- and vasculogenesis-chips.[85] In these micropost devices, low concentrations, typically 2.5 mg/mL fibrin, were sufficient to support lymphangiogenic sprouting and tube formation. One report detailed the bioprinting of a gelatin-methacrylate (GelMA) matrix containing breast cancer cells around a printed agar cylindrical mold within PDMS housing.[95] After the agar rod was removed, LECs were seeded on the inner walls of the hollow lumen, thus creating a bioprinted and perfusable lymphatic vessel. While the final device was not a PDMS-based microfluidic chip, the bioprinted construct shows potential for use in other micropost or cylindrical lumen devices.
Other reports of cylindrical lumens that eventually sprouted towards stimulants utilized fibrin hydrogels sourced purely from fibrinogen and thrombin. Interestingly, a collagen-fibrin hydrogel was used where each ECM protein had a concentration of 2.5 mg/mL.[89] While this is not the first ECM hydrogel to contain both fibrin and collagen, it is a rare report of a hydrogel containing equal concentrations of the two proteins. It shows potential for tuning ECM proteins in a hydrogel for tissue specific applications and ought to be a consideration in future tissue-specific lymphangiogenesis models.
As mentioned earlier, other reports usually utilize a cylindrical lumen formed by sacrificial molding. Because of this, higher ECM protein concentrations are used as they allow repeated hollow lumen formation without collapsing the hydrogel during rod removal. Some reports utilize low concentration collagen hydrogels (2.5–3 mg/mL),[86, 93–94] with one study comparing sprouting to a 6 mg/mL collagen hydrogel.[87] Here, a cylindrical lumen device was used to mimic healthy breast tissue (3 mg/mL collagen hydrogel) and cancerous breast tissue (6 mg/mL collagen hydrogel). When investigating lymphatic vessel morphology, sprouting, cytokine secretion, and barrier function, they saw that higher density matrices induce a higher proinflammatory cytokine secretion and leakier vessels, which were exacerbated when culturing their lumens in breast cancer cell-encapsulating hydrogels of the same concentrations. Relatedly, a recent study reported better cell viability when using 3 mg/mL collagen for culturing LECs from the same supplier.[93] Therefore, not only is ECM-protein source important for mimicking tissue-specific environments, but the concentration plays an important role in the response of cells of interest.
4.5. Growth Factors
Since most models of lymphatic vessels and their lymphangiogenic sprouting are used for cancer research, growth factors are not commonly supplemented into the cell culture medium as in angiogenesis- and vasculogenesis-chips. In these devices, co-culturing LECs in a lymphatic vessel architecture with cancer cells in the surrounding matrix or adjacent microchannels is most common. Therefore, an unknown number and concentration of factors are secreted to induce lymphangiogenesis unless proteomic data such as ELISA results are included in the report. A micropost device featuring LECs was the first lymphangiogenesis-chip using growth factor supplemented medium.[85] In this setup, the investigators supplemented EGM-2 MV medium with different pro-lymphangiogenic factors to decipher which may play the greatest role in lymphangiogenic sprouting. They found that VEGF-A, VEGF-C, bFGF, and S1P all induced some sprouting individually, but the combination of all four induced sprouts two times in length (Figure 4C-i). These factors are similar to those found in other reports of LECs co-cultured with cancer cells. For example, one study found VEGF-C and FGF-2, a relative of bFGF, was expressed by LECs exhibiting lymphangiogenic sprouting,[86] while another utilized VEGF-C, VEGF-D, and IL-6 to induce a pro-inflammatory microenvironment.[93] Other reports also indicate sprouting in response to proinflammatory cytokines but these involve co-cultures with cancer cell lines.
4.6. Biophysical Forces
In the limited number of reports of lymphangiogenesis-chips or lymphatic vessels-on-chips exhibiting lymphangiogenic sprouting, we could find only one work which integrated interstitial fluid forces into their devices.[85] They found that interstitial flow in a basal-apical direction perpendicular to an LEC monolayer induced greater lymphangiogenic sprouting compared to static conditions. IF-induced sprouting was highest when combined with all four growth factors utilized in their study, highlighting the interplay of the five ingredients necessary for microvascular self-assembly. Another study tested laminar flow within a lymphatic vessel-on-a-chip to determine whether shear stress against LECs could induce lymphangiogenic sprouting into the surrounding ECM hydrogel.[94] It was found that an intraluminal flow of 5 dyn/cm2 induced sprouts almost double in length as those in static conditions over 4 days (Figure 4C-ii). Later experiments in the same report used spheroid assays and showed that the shear stress from laminar flow downregulated Notch signaling, thus increasing the number of LECs migrating into the hydrogel.
4.7. Vascular/Lymphatic Co-cultures
Few MPSs feature a co-culture of ECs and LECs that sprout or form a de novo network. Some reasons include the recent emergence of lymphangiogenesis as an MPS research topic, as well as how difficult it is to induce two biological processes within the same device. Nevertheless, one report briefly presented a micropost device where NHLFs and tumor spheroids induced both angiogenic and lymphangiogenic sprouting toward the spheroid.[89] Later, another group presented a dual lumen system where HUVECs formed a parent vessel across from a LEC-coated lymphatic vessel.[88] Here, VEGF-C and VEGF-A were utilized, resulting in angiogenic and lymphangiogenic sprouting. Interestingly, they noticed that lymph microvasculature sprouted at a slower rate and formed vessels with a thinner diameter, which they claimed matched in vivo reports of tumor-induced lymphangiogenesis.
5. A Brief Review of Vascularized Multicellular Systems
The previously described devices are important for dissecting molecular regulators of vascularization. However, MPSs are also used as tissue specific models, disease models, and drug testing systems. Having established methods of vascularizing MPSs, a new research area has focused on vascularizing tissue specific models because the vasculature has been shown to regulate homeostasis of tissues and organs.[8–9] Further, tissue-specific diseases also affect the tissue vasculature, and thus neglecting an endothelium in a disease model could leave researchers with an incomplete picture of pathological characteristics.[96–97] Finally, in drug testing, the vasculature often reacts to drug candidates, sometimes resulting in drug-induced vascular injury and inflammation (thus releasing inflammatory signals to the surrounding tissues).[98] Therefore, some recent in vitro models for biological investigation, disease modeling, and drug evaluation have include a self-assembled vascular network to serve as better predictors of in vivo results.
Most multicellular, tissue-specific MPS designs utilize the micropost device in either an angiogenesis-chip or vasculogenesis-chip setup, situating ECs as parent vessels or allowing a de novo network to form within a tissue-mimicking matrix. One report utilized decellularized bone matrix to support a co-culture of HUVECs and hMSCs in a vascularized bone tissue model.[99] Further, they found that flow velocities, shear stresses, and even oxygen gradients affected the microvascular network in their bone tissue model. One similar report detailed that hMSC-derived osteoblasts expressed bone specific markers such as osteocalcin and bone alkaline phosphatase.[78] Interestingly, they compared the extravasation of breast cancer from the microvascular network in bone-mimicking and unconditioned matrices, finding that a bone-specific model induced higher extravasation rates, thus mimicking breast cancer’s tendency to metastasize to bone tissue. Reports from another group utilized an angiogenesis-chip in to form a microvascular network within a fibrin/hydroxyapatite hydrogel.[100–101] In their later report, the cancerous bone tissue model replicated the increased angiogenic sprouting characteristic of many tumors.[101]
Other tumor models have been presented with microvascular networks. For example, an early report of a vascularized tumor model replicated cancer cell extravasation, where human adenocarcinoma cells perfused through a microvascular network in collagen hydrogel extravasated into the matrix.[77] Later, a dual-lumen setup modeling pancreatic cancer situated HUVECs in one lumen and pancreatic cancer cells in the other.[42] Interestingly, pancreatic cancer cells sprouted to a greater degree in the presence of HUVECs, eventually reached the HUVEC lumen, and ablated the ECs. Some tumor models have even included de novo vascular or sprouting lymphatic networks.[70, 76–77, 89, 95, 102] One study showed that the lymphatic networks served as conduits for metastasis via enhanced lymphangiogenesis into the tumor mass.[95] Another showed that both vascular and lymphatic networks increase their sprouting and network formation into tumor masses.[89] Such works demonstrate the importance of including both EC lineages and their self-assembled networks in cancer models as both play a role in tumor cell extravasation and metastasis.
Recently, MPSs have been applied in drug discovery and development fields, with some blood brain barrier (BBB) models incorporating brain-specific cells with microvascular networks.[32, 45–46] One report encapsulated brain pericytes and astrocytes in fibrin hydrogel with iPSC-ECs.[45] While de novo vascularization took place, more ECs were seeded in adjacent channels forming two parent vessels. When the EC networks of the three channels anastomosed, the resulting BBB model was used to preclinically asses nanocarrier transport and uptake across the BBB. Other reports detail the use of similar cells in their BBB models but separate the vascular network from the neural cells into two adjacent hydrogel channels.[103] Another report ventured away from the BBB model but utilized microvascular networks in models of neuronal disorders.[104] Here, embryonic stem cell derived motor neuron (MN) spheroids were co-cultured with iPSC-ECs in a microfluidic device, resulting in the assembly of an intertwined microvascular around a neuronal network. They showed that the presence of the microvascular network improved neurite elongation and connectivity of the neurons in the chip. Further, delta-Notch signaling between the ECs and MNs promoted neuroprotection.
In summary, the incorporation of a microvascular network, and even lymphatic networks, recapitulates tissue and disease specific characteristics that might not be present in non-vascularized MPSs. Further, many of these examples utilize tissue-specific materials such as decellularized matrix as the vascular scaffold, thus narrowing the gap between in vitro results and possible in vivo outcomes. It is therefore important that future tissue and disease specific models incorporate vascular and/or lymphatic microvascular networks as they can help develop better drug screening platforms, tools for basic biological investigation, and effective disease modeling tools for drug development.
6. Perspectives and Future Directions
While there have been large advances made since the first reports of self-assembled vasculature-on-a-chip, some limitations in vascularizing MPSs still exist. In this section, we will discuss the current trends and limitations in vascularizing MPSs, the limitations in experimental protocols of current vascularized chips, and the future of vascularized chips in disease modeling and drug screening.
6.1. The State of Vascularized MPSs: Present and Future
In this review, we have discussed some vascularized systems featuring self-assembled, perfusable networks formed within tissue-specific microenvironments. However, another method of including an endothelium published regularly is to situate a tissue monolayer or 3D assembly across from a monolayer of ECs, separated by a thin membrane.[105–108] The membrane, usually made of PDMS but sometimes fibers of ECM proteins,[109–110] is typically less than 10 μm thick and has pores of diameters on the scale of single microns. Like the reviewed devices herein, these setups allow for shear-induced factor release by ECs, paracrine signaling between the two compartments, and a direct endothelium-cell or endothelium-tissue communication. However, these devices are not designed to produce an in vivo capillary-like structures of sub-100 μm perfusable tubes that supply the tissue of interest with nutrients and oxygen. Nevertheless, these devices have shown success for in vitro drug evaluation (some reports of which detail results that agree with in vivo findings), preliminary studies on personalizing therapy, and some disease models.[111–114]
While this method of endothelializing MPSs is common, improving the field requires the use of self-assembled networks in some cases for many reasons. First, many anti-cancer drug candidates target the angiogenic process or small capillary structures that feed into tumor masses.[115–117] Without recapitulating the enhanced angiogenic response and the resulting tubular structures, the efficacy of these candidates cannot be properly evaluated. Second, many tissues in the body are highly vascularized by thin, tubular structures, and thus the inability to spontaneously form tubular vessels around cells, tissues, or spheroids in these types of devices could become a limiting factor in some cases. Tissues within the liver, pancreas, heart, skin, eyes, and kidneys are either surrounded by or intermingled with capillaries and/or lymphatic vessels, and MPSs that include the functions of the tissues can have advantages.[10] As discussed in Section 5, this was important for modeling bone tissue and tumor systems that were used for biological investigation and therapy testing. Finally, engineering self-assembled vascularized MPSs can help isolate the necessary ingredients in tissue-engineered grafts and biomaterials. In cellular therapy, the lack of vasculature hinders success of cell transplantation procedures.[118] By building an MPSs containing the necessary components (i.e. ECs, tissue-specific cells, ECM components, growth factors, and mechanical forces), researchers can test different biomaterials, cell sources, and growth factors that not only support viability of the cells to be transplanted, but provide other features such as immunoprotection, directed differentiation of progenitors or stem cells to desired cell types, and factors to induce parent vessel angiogenesis into the mimicked implant. Successful combinations can then be combined in bioinks for 3D printing, from which microscale grafts can be assembled. Therefore, while some endothelialized MPSs show potential usefulness, vascularizing MPSs with capillary-like vessels via self-assembly offers better potential for drug discovery, targeting, and graft vascularization. Approaches to arrive at this higher level of biological complexity, but keeping the technology simple so that it can be adopted and reproduced beyond a single lab, remains a challenge. However, since these bioengineered systems are often reductionist, they offer an enormous promise to build complexity incrementally.
6.2. Other hydrogel materials in vascularized MPSs: A brief perspective
As reviewed, the MPS field rarely deviates from fibrin, collagen, or Matrigel hydrogels in vascularized models. However, there is a range of biomaterials that have been utilized in other fields such as bioprinting. Interestingly, many of these materials have mechanical or chemical properties that have been tuned to change material stiffness or increase biocompatibility and bioactivity, and thus might be easily tuned for injection into micropost devices or other hydrogel-based MPSs. Such materials can be classified as either biologically-derived or synthetic materials. Of the biologically derived materials, the bioprinting field largely employs gelatin, or its methacrylated version – GelMA. Gelatin alone is usually used as a sacrificial material that liquifies at 37 ⁰C, but GelMA is a stronger material crosslinked irreversibly by UV.[119] Within MPSs, it may be a promising material for lymphangiogenesis- or angiogenesis-based vascularization methods but encapsulating ECs within the material would require careful tuning of UV dosage to ensure that the encapsulated cells can still perform vasculogenesis following exposure. Interestingly, many reports detail the tuning of GelMA’s stiffness via addition of other polymers[120–121] or adjustment of the material handling[122–124], which shows potential translation to tissue-specific MPSs such as vascularized bone or tumor models, or soft-tissue MPSs replicating fat, lymphatic, or nerve tissues. Other works include the addition of drug-loadable nanoparticles which provide biological cues[125–127], which in turn can enhance vascularization within MPSs or even replace support cells to simplify the existing co-culture models.
Another interesting class of biological materials finding applications in macro-scale in vitro reports is tissue-derived decellularized extracellular matrix (dECM). dECM is especially attractive for tissue-specific MPSs as it helps strengthen the physiological relevance of the system. Further, dECM has been reported as promoting angiogenesis within constructs in vitro or post-implantation.[128–131] Interestingly, one report proved that the mechanical stiffness and viscosity of a dECM hydrogel can be adjusted by addition of vitamin B2.[132] Other reports detailed that dECM modification changed the resulting microvascular network phenotype, highlighting both successful use of dECM for vascularization as well as methods of tuning the material for a desired microvascular network quality.[133–134] These works highlight how pre-injection modification might help successfully integrate dECM into MPSs for the purposes of on-chip vascularization.
The second classification of hydrogel materials, synthetic polymers, offers engineers the ability to more specifically change mechanical, chemical, and biological properties to optimize vascular network formation. For tuning of mechanical properties, this can help in insuring successful injection into chips. For example, materials such as poly(ethylene glycol) diacrylate (PEGDA) or poly(ethylene glycol)-tetra-acrylate (PEGTA) have tunable rheological properties based on the molecular weight and/or concentration of the polymers.[135] Further, PEGDA has recently been combined with GelMA in a bioprinted vascularized bone tumor model where EC showed upregulated expression of angiogenesis-related genes.[136] While synthetic polymers can be argued as less physiological, MPSs looking to model microscale tissue engineered grafts might employ such materials in validating constructs before in vivo animal studies.
6.3. Standardization of Culturing Conditions in MPSs
It is apparent that vascular self-assembly in chips is an attractive field, but the current state of the literature is not without its limitations, primarily because the discipline is still being developed. First, the experimental setups for the reports of angiogenesis- and vasculogenesis-chips differ with regard to the basal culturing reagents and stimulating factors. The presence of NHLFs or other stromal cell types is a common method of inducing vascular self-assembly, however the secretions from the cells are rarely characterized in reports where they are utilized. Interestingly, a recent report even highlighted that donor source of a commercial fibroblast line induced different network characteristics, highlighting how easily different labs can achieve different microvascular networks even when using commercial materials.[137] Further, it remains to be seen whether stromal cells may also receive paracrine signals from ECs, which in turn might stimulate the release of pro-angiogenic or pro-vasculogenic factors. Finally, the variation in EC and stromal cell seeding density between reports highlights a changing signaling molecule density and gradient between devices from different labs utilizing the stromal cell induced vascularization method (possibly due to donor variability). Therefore, it is very plausible that the secretome within a device varies largely between different reports depending on their experimental conditions. Therefore, some consensus into standardizing cell sources and culture conditions is beneficial.
Another noticeable characteristic of many reports is the use of EGM-2 or EGM-2 cell-culture media as supplied from the manufacturer. While this medium may be important for maintaining the ECs, they do not provide a true baseline condition. First, the medium supports EC proliferation, yet in vivo parent vessels often contain phalanx ECs which are characteristically quiescent. Second, the medium often contains FBS or bovine brain extract, both of which contain undefined proteins that could vary depending upon the supplier. Similarly, the collagen, fibrin, and Matrigel proteins used to build the hydrogel scaffolds often come from rat or cow and could be variable in their composition depending upon the supplier.
One avenue to be explored is the use of pre-defined, serum-free, human sourced cell culture medium components. For example, a human collagen I hydrogel matrix was presented containing HUVECs and adipose-derived stromal cells (hAsCs) that enabled vascular network formation in a similar time span as the previously discussed angiogenesis- and vasculogenesis-chips.[138] Interestingly, M199 medium, a completely defined medium, supplemented with 10 ng/mL VEGF-A and 10 ng/mL FGF-2, along with various antibiotics, was sufficient to induce vascular network formation. However, there were still limitations: the species source of the proteins and antibiotics were not reported and the constructs were mounted on porcine small intestine submucosa. Therefore, while FBS was eliminated from the experiments, some reduction in xenogeneic cell culture supplements may be desirable to arrive at a standard basal condition, which may not replicate the exact in vivo serum-state in the near-term, but that can be practically used across a variety of such in vitro platforms.
7. Conclusion
EC source, supporting cell types, growth factors, ECM-incorporating or -mimicking biomaterials, and forces due to fluid flow have been presented as individually or combinatorially initiating vascular self-assembly in MPSs. We have shown successful reports where angiogenesis, de novo vascular assembly or vasculogenesis, and lymphangiogenesis were initiated separately or together, indicating major breakthroughs in incorporating not only an endothelium, but also a lymphatic system in tissue-specific models. Advances here show potential for translation to tissue engineering research and graft development because many components of an MPS can be bioprinted or molded into grafts. While limitations exist, perhaps due to lack of consensus and standardization of methods, the movement from monolayer endotehliums to perfusable capillary beds in 3D architectures represent a promising outlook for the future of tissue modeling, drug testing, and personalized therapy.
Acknowledgements
This material is supported by a National Science Foundation Graduate Research Fellowship under Grant No. 1650114 and a Texas A&M Biomedical Engineering Department National Excellence Fellowship to J.J.T.; and by the NIBIB of NIH under Award Number R21EB025945, NSF CAREER Award number 1944322, and Texas A&M University President’s Excellence in Research Award (T32/ X-Grant) to A.J.
Contributor Information
James J. Tronolone, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, TX 77843, USA
Abhishek Jain, Department of Medical Physiology, College of Medicine, Texas A&M Health Science Center, Bryan, TX 77808, USA.
References
- [1].Augustin HG, Koh GY, Science 2017, 357. [DOI] [PubMed] [Google Scholar]
- [2].Aird WC, Crit Care Med 2004, 32, S271. [DOI] [PubMed] [Google Scholar]
- [3].Ballian N, Brunicardi FC, World J Surg 2007, 31, 705. [DOI] [PubMed] [Google Scholar]
- [4].Brunicardi FC, Stagner J, Bonner-Weir S, Wayland H, Kleinman R, Livingston E, Guth P, Menger M, McCuskey R, Intaglietta M, Charles A, Ashley S, Cheung A, Ipp E, Gilman S, Howard T, Passaro E, Diabetes 1996, 45, 385. [DOI] [PubMed] [Google Scholar]
- [5].Factor SM, Okun EM, Minase T, Kirk ES, Circulation 1982, 66, 1241. [DOI] [PubMed] [Google Scholar]
- [6].Kvietys PR, in PanVascular Medicine, DOI: 10.1007/978-3-642-37393-0_141-1 2014, Ch. Chapter 141–1, p. 1. [DOI] [Google Scholar]
- [7].Carmeliet P, Jain RK, Nature 2000, 407, 249. [DOI] [PubMed] [Google Scholar]
- [8].Pi X, Xie L, Patterson C, Circ Res 2018, 123, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Witjas FMR, van den Berg BM, van den Berg CW, Engelse MA, Rabelink TJ, Stem Cells Transl Med 2019, 8, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Haase K, Kamm RD, Regenerative Medicine 2017, 12, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lee S, Ko J, Park D, Lee SR, Chung M, Lee Y, Jeon NL, Lab Chip 2018, 18, 2686. [DOI] [PubMed] [Google Scholar]
- [12].Osaki T, Sivathanu V, Kamm RD, Curr Opin Biotechnol 2018, 52, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tan SY, Leung Z, Wu AR, Small 2020, DOI: 10.1002/smll.201905055e1905055. [DOI] [PubMed] [Google Scholar]
- [14].Wang X, Sun Q, Pei J, Micromachines (Basel) 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yi HG, Jeong YH, Kim Y, Choi YJ, Moon HE, Park SH, Kang KS, Bae M, Jang J, Youn H, Paek SH, Cho DW, Nat Biomed Eng 2019, 3, 509. [DOI] [PubMed] [Google Scholar]
- [16].Park JY, Ryu H, Lee B, Ha DH, Ahn M, Kim S, Kim JY, Jeon NL, Cho DW, Biofabrication 2018, 11, 015002. [DOI] [PubMed] [Google Scholar]
- [17].Gao G, Park JY, Kim BS, Jang J, Cho DW, Adv Healthc Mater 2018, 7, e1801102. [DOI] [PubMed] [Google Scholar]
- [18].Otrock ZK, Mahfouz RA, Makarem JA, Shamseddine AI, Blood Cells Mol Dis 2007, 39, 212. [DOI] [PubMed] [Google Scholar]
- [19].Welti J, Loges S, Dimmeler S, Carmeliet P, J Clin Invest 2013, 123, 3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ribatti D, Crivellato E, Dev Biol 2012, 372, 157. [DOI] [PubMed] [Google Scholar]
- [21].Chen W, Xia P, Wang H, Tu J, Liang X, Zhang X, Li L, J Cell Commun Signal 2019, 13, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalen M, Gerhardt H, Betsholtz C, Nature 2007, 445, 776. [DOI] [PubMed] [Google Scholar]
- [23].Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C, Genes Dev 2003, 17, 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kim C, Kasuya J, Jeon J, Chung S, Kamm RD, Lab Chip 2015, 15, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Del Amo C, Borau C, Gutierrez R, Asin J, Garcia-Aznar JM, J Biomech 2016, 49, 1340. [DOI] [PubMed] [Google Scholar]
- [26].Kim S, Chung M, Ahn J, Lee S, Jeon NL, Lab Chip 2016, 16, 4189. [DOI] [PubMed] [Google Scholar]
- [27].Kim S, Lee H, Chung M, Jeon NL, Lab Chip 2013, 13, 1489. [DOI] [PubMed] [Google Scholar]
- [28].Kim J, Chung M, Kim S, Jo DH, Kim JH, Jeon NL, PLoS One 2015, 10, e0133880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nashimoto Y, Hayashi T, Kunita I, Nakamasu A, Torisawa YS, Nakayama M, Takigawa-Imamura H, Kotera H, Nishiyama K, Miura T, Yokokawa R, Integr Biol (Camb) 2017, 9, 506. [DOI] [PubMed] [Google Scholar]
- [30].Shin Y, Jeon JS, Han S, Jung GS, Shin S, Lee SH, Sudo R, Kamm RD, Chung S, Lab Chip 2011, 11, 2175. [DOI] [PubMed] [Google Scholar]
- [31].Chung S, Sudo R, Zervantonakis IK, Rimchala T, Kamm RD, Adv Mater 2009, 21, 4863. [DOI] [PubMed] [Google Scholar]
- [32].Campisi M, Shin Y, Osaki T, Hajal C, Chiono V, Kamm RD, Biomaterials 2018, 180, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Haase K, Gillrie MR, Hajal C, Kamm RD, Adv Sci (Weinh) 2019, 6, 1900878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shin Y, Han S, Jeon JS, Yamamoto K, Zervantonakis IK, Sudo R, Kamm RD, Chung S, Nat Protoc 2012, 7, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pauty J, Usuba R, Cheng IG, Hespel L, Takahashi H, Kato K, Kobayashi M, Nakajima H, Lee E, Yger F, Soncin F, Matsunaga YT, EBioMedicine 2018, 27, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tourovskaia A, Fauver M, Kramer G, Simonson S, Neumann T, Exp Biol Med (Maywood) 2014, 239, 1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bischel LL, Young EWK, Mader BR, Beebe DJ, Biomaterials 2013, 34, 1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li S, Wang K, Hu Q, Zhang C, Wang B, Mater Sci Eng C Mater Biol Appl 2019, 104, 109936. [DOI] [PubMed] [Google Scholar]
- [39].Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, Stroock AD, Proc Natl Acad Sci U S A 2012, 109, 9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang WY, Lin D, Jarman EH, Polacheck WJ, Baker BM, Lab Chip 2020, 20, 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nguyen DH, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA, Chen CS, Proc Natl Acad Sci U S A 2013, 110, 6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nguyen D-HT, Lee H, Alimperti S, Norgard RJ, Wong A, Lee JJ-K, Eyckmans J, Stanger BZ, Chen CS, Sci Adv 2019, 5, eaav6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chan JM, Zervantonakis IK, Rimchala T, Polacheck WJ, Whisler J, Kamm RD, PLoS One 2012, 7, e50582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Seo HR, Jeong HE, Joo HJ, Choi SC, Park CY, Kim JH, Choi JH, Cui LH, Hong SJ, Chung S, Lim DS, Sci Rep 2016, 6, 28832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee SWL, Campisi M, Osaki T, Possenti L, Mattu C, Adriani G, Kamm RD, Chiono V, Adv Healthc Mater 2020, DOI: 10.1002/adhm.201901486e1901486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Uwamori H, Ono Y, Yamashita T, Arai K, Sudo R, Microvasc Res 2019, 122, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Johnson KE, Wilgus TA, Adv Wound Care (New Rochelle) 2014, 3, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kumar P, Kumar S, Udupa EP, Kumar U, Rao P, Honnegowda T, Plastic and Aesthetic Research 2015, 2. [Google Scholar]
- [49].Mastrullo V, Cathery W, Velliou E, Madeddu P, Campagnolo P, Front Bioeng Biotechnol 2020, 8, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kant RJ, Coulombe KLK, Acta Biomater 2018, 69, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Karimi F, O’Connor AJ, Qiao GG, Heath DE, Adv Healthc Mater 2018, 7, e1701324. [DOI] [PubMed] [Google Scholar]
- [52].Crosby CO, Zoldan J, Regen Biomater 2019, 6, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zheng Y, Wang S, Xue X, Xu A, Liao W, Deng A, Dai G, Liu AP, Fu J, Lab Chip 2017, 17, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].van Duinen V, Zhu D, Ramakers C, van Zonneveld AJ, Vulto P, Hankemeier T, Angiogenesis 2019, 22, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gold K, Gaharwar AK, Jain A, Biomaterials 2019, 196, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhou J, Li YS, Chien S, Arterioscler Thromb Vasc Biol 2014, 34, 2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA, J Clin Invest 2016, 126, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kang H, Bayless KJ, Kaunas R, Am J Physiol Heart Circ Physiol 2008, 295, H2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kang H, Kwak HI, Kaunas R, Bayless KJ, J Biol Chem 2011, 286, 42017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Abe Y, Watanabe M, Chung S, Kamm RD, Tanishita K, Sudo R, APL Bioeng 2019, 3, 036102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Song JW, Munn LL, Proc Natl Acad Sci U S A 2011, 108, 15342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shirure VS, Lezia A, Tao A, Alonzo LF, George SC, Angiogenesis 2017, 20, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Risau W, Flamme I, Annu Rev Cell Dev Biol 1995, 11, 73. [DOI] [PubMed] [Google Scholar]
- [64].Patel-Hett S, D’Amore PA, Int J Dev Biol 2011, 55, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ribatti D, Vacca A, Nico B, Roncali L, Dammaco F, Mech Dev 2001, 100, 157. [DOI] [PubMed] [Google Scholar]
- [66].Hsu YH, Moya ML, Abiri P, Hughes CC, George SC, Lee AP, Lab Chip 2013, 13, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hsu YH, Moya ML, Hughes CC, George SC, Lee AP, Lab Chip 2013, 13, 2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Moya ML, Hsu Y-H, Lee AP, Hughes CCW, George SC, Tissue Engineering Part C: Methods 2013, 19, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Phan DTT, Wang X, Craver BM, Sobrino A, Zhao D, Chen JC, Lee LYN, George SC, Lee AP, Hughes CCW, Lab Chip 2017, 17, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sobrino A, Phan DT, Datta R, Wang X, Hachey SJ, Romero-Lopez M, Gratton E, Lee AP, George SC, Hughes CC, Sci Rep 2016, 6, 31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wang X, Phan DT, Sobrino A, George SC, Hughes CC, Lee AP, Lab Chip 2016, 16, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Xiao Y, Liu C, Chen Z, Blatchley MR, Kim D, Zhou J, Xu M, Gerecht S, Fan R, Adv Biosyst 2019, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Whisler JA, Chen MB, Kamm RD, Tissue Eng Part C Methods 2014, 20, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Belair DG, Whisler JA, Valdez J, Velazquez J, Molenda JA, Vickerman V, Lewis R, Daigh C, Hansen TD, Mann DA, Thomson JA, Griffith LG, Kamm RD, Schwartz MP, Murphy WL, Stem Cell Rev Rep 2015, 11, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zanotelli MR, Ardalani H, Zhang J, Hou Z, Nguyen EH, Swanson S, Nguyen BK, Bolin J, Elwell A, Bischel LL, Xie AW, Stewart R, Beebe DJ, Thomson JA, Schwartz MP, Murphy WL, Acta Biomater 2016, 35, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chen MB, Hajal C, Benjamin DC, Yu C, Azizgolshani H, Hynes RO, Kamm RD, Proc Natl Acad Sci U S A 2018, 115, 7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chen MB, Whisler JA, Jeon JS, Kamm RD, Integr Biol (Camb) 2013, 5, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD, Proc Natl Acad Sci U S A 2015, 112, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Jeon JS, Bersini S, Whisler JA, Chen MB, Dubini G, Charest JL, Moretti M, Kamm RD, Integr Biol (Camb) 2014, 6, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li Y, Pi Q-M, Wang P-C, Liu L-J, Han Z-G, Shao Y, Zhai Y, Zuo Z-Y, Gong Z-Y, Yang X, Wu Y, RSC Advances 2017, 7, 56108. [Google Scholar]
- [81].Chapla R, West J, Progress in Biomedical Engineering 2020, DOI: 10.1088/2516-1091/abc947. [DOI] [Google Scholar]
- [82].Oliver G, Srinivasan RS, Development 2010, 137, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wang Y, Oliver G, Genes Dev 2010, 24, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG, Nat Rev Cancer 2014, 14, 159. [DOI] [PubMed] [Google Scholar]
- [85].Kim S, Chung M, Jeon NL, Biomaterials 2016, 78, 115. [DOI] [PubMed] [Google Scholar]
- [86].Ayuso JM, Gong MM, Skala MC, Harari PM, Beebe DJ, Adv Healthc Mater 2020, 9, e1900925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lugo-Cintron KM, Ayuso JM, White BR, Harari PM, Ponik SM, Beebe DJ, Gong MM, Virumbrales-Munoz M, Lab Chip 2020, DOI: 10.1039/d0lc00099j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Osaki T, Serrano JC, Kamm RD, Regen Eng Transl Med 2018, 4, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Chung M, Ahn J, Son K, Kim S, Jeon NL, Adv Healthc Mater 2017, 6. [DOI] [PubMed] [Google Scholar]
- [90].Keuschnigg J, Karinen S, Auvinen K, Irjala H, Mpindi JP, Kallioniemi O, Hautaniemi S, Jalkanen S, Salmi M, PLoS One 2013, 8, e74293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Pepper MS, Skobe M, J Cell Biol 2003, 163, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Vainionpaa N, Butzow R, Hukkanen M, Jackson DG, Pihlajaniemi T, Sakai LY, Virtanen I, Cell Tissue Res 2007, 328, 317. [DOI] [PubMed] [Google Scholar]
- [93].Gong MM, Lugo-Cintron KM, White BR, Kerr SC, Harari PM, Beebe DJ, Biomaterials 2019, 214, 119225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Choi D, Park E, Jung E, Seong YJ, Yoo J, Lee E, Hong M, Lee S, Ishida H, Burford J, Peti-Peterdi J, Adams RH, Srikanth S, Gwack Y, Chen CS, Vogel HJ, Koh CJ, Wong AK, Hong YK, J Clin Invest 2017, 127, 1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Liu T, Liu Q, Anaya I, Huang D, Kong W, Mille LS, Zhang YS, Methods 2020, DOI: 10.1016/j.ymeth.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Monteiro JP, Bennett M, Rodor J, Caudrillier A, Ulitsky I, Baker AH, Cardiovasc Res 2019, 115, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rajendran P, Rengarajan T, Thangavel J, Nishigaki Y, Sakthisekaran D, Sethi G, Nishigaki I, Int J Biol Sci 2013, 9, 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gara E, Csiko KG, Ruzsa Z, Foldes G, Merkely B, Med Oncol 2019, 36, 72. [DOI] [PubMed] [Google Scholar]
- [99].Marturano-Kruik A, Nava MM, Yeager K, Chramiec A, Hao L, Robinson S, Guo E, Raimondi MT, Vunjak-Novakovic G, Proc Natl Acad Sci U S A 2018, 115, 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Jusoh N, Oh S, Kim S, Kim J, Jeon NL, Lab on a Chip 2015, 15, 3984. [DOI] [PubMed] [Google Scholar]
- [101].Ahn J, Lim J, Jusoh N, Lee J, Park TE, Kim Y, Kim J, Jeon NL, Front Bioeng Biotechnol 2019, 7, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Boussommier-Calleja A, Atiyas Y, Haase K, Headley M, Lewis C, Kamm RD, Biomaterials 2019, 198, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bang S, Lee SR, Ko J, Son K, Tahk D, Ahn J, Im C, Jeon NL, Sci Rep 2017, 7, 8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Osaki T, Sivathanu V, Kamm RD, Sci Rep 2018, 8, 5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chung HH, Mireles M, Kwarta BJ, Gaborski TR, Lab Chip 2018, 18, 1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Huh D, Hamilton GA, Ingber DE, Trends Cell Biol 2011, 21, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Huh D, Kim HJ, Fraser JP, Shea DE, Khan M, Bahinski A, Hamilton GA, Ingber DE, Nat Protoc 2013, 8, 2135. [DOI] [PubMed] [Google Scholar]
- [108].Quiros-Solano WF, Gaio N, Stassen O, Arik YB, Silvestri C, Van Engeland NCA, Van der Meer A, Passier R, Sahlgren CM, Bouten CVC, van den Berg A, Dekker R, Sarro PM, Sci Rep 2018, 8, 13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Mondrinos MJ, Yi YS, Wu NK, Ding X, Huh D, Lab Chip 2017, 17, 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wang C, Tanataweethum N, Karnik S, Bhushan A, ACS Biomaterials Science & Engineering 2018, 4, 1377. [DOI] [PubMed] [Google Scholar]
- [111].Herland A, Maoz BM, Das D, Somayaji MR, Prantil-Baun R, Novak R, Cronce M, Huffstater T, Jeanty SSF, Ingram M, Chalkiadaki A, Benson Chou D, Marquez S, Delahanty A, Jalili-Firoozinezhad S, Milton Y, Sontheimer-Phelps A, Swenor B, Levy O, Parker KK, Przekwas A, Ingber DE, Nature Biomedical Engineering 2020, 4, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee HH, Alves SE, Salmon M, Ferrante TC, Weaver JC, Bahinski A, Hamilton GA, Ingber DE, Nat Methods 2016, 13, 151. [DOI] [PubMed] [Google Scholar]
- [113].Park TE, Mustafaoglu N, Herland A, Hasselkus R, Mannix R, FitzGerald EA, Prantil-Baun R, Watters A, Henry O, Benz M, Sanchez H, McCrea HJ, Goumnerova LC, Song HW, Palecek SP, Shusta E, Ingber DE, Nat Commun 2019, 10, 2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Saha B, Mathur T, Handley KF, Hu W, Afshar-Kharghan V, Sood AK, Jain A, Blood Adv 2020, 4, 3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Huang J, Guo P, Moses MA, ACS Biomaterials Science & Engineering 2019, 6, 2563. [DOI] [PubMed] [Google Scholar]
- [116].Rajabi M, Mousa SA, Biomedicines 2017, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Al-Abd AM, Alamoudi AJ, Abdel-Naim AB, Neamatallah TA, Ashour OM, J Adv Res 2017, 8, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Mitrousis N, Fokina A, Shoichet MS, Nature Reviews Materials 2018, 3, 441. [Google Scholar]
- [119].Leucht A, Volz AC, Rogal J, Borchers K, Kluger PJ, Sci Rep 2020, 10, 5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kim P, Yuan A, Nam KH, Jiao A, Kim DH, Biofabrication 2014, 6, 024112. [DOI] [PubMed] [Google Scholar]
- [121].Wang Y, Ma M, Wang J, Zhang W, Lu W, Gao Y, Zhang B, Guo Y, Materials (Basel) 2018, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Lin CH, Su JJ, Lee SY, Lin YM, J Tissue Eng Regen Med 2018, 12, 2099. [DOI] [PubMed] [Google Scholar]
- [123].Puckert C, Tomaskovic-Crook E, Gambhir S, Wallace GG, Crook JM, Higgins MJ, Acta Biomater 2020, 106, 156. [DOI] [PubMed] [Google Scholar]
- [124].Rizwan M, Peh GSL, Ang HP, Lwin NC, Adnan K, Mehta JS, Tan WS, Yim EKF, Biomaterials 2017, 120, 139. [DOI] [PubMed] [Google Scholar]
- [125].Chimene D, Peak CW, Gentry JL, Carrow JK, Cross LM, Mondragon E, Cardoso GB, Kaunas R, Gaharwar AK, ACS Appl Mater Interfaces 2018, 10, 9957. [DOI] [PubMed] [Google Scholar]
- [126].Liu B, Li J, Lei X, Cheng P, Song Y, Gao Y, Hu J, Wang C, Zhang S, Li D, Wu H, Sang H, Bi L, Pei G, Mater Sci Eng C Mater Biol Appl 2020, 112, 110905. [DOI] [PubMed] [Google Scholar]
- [127].Modaresifar K, Hadjizadeh A, Niknejad H, Artif Cells Nanomed Biotechnol 2018, 46, 1799. [DOI] [PubMed] [Google Scholar]
- [128].Liguori GR, Liguori TTA, de Moraes SR, Sinkunas V, Terlizzi V, van Dongen JA, Sharma PK, Moreira LFP, Harmsen MC, Front Bioeng Biotechnol 2020, 8, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Giobbe GG, Crowley C, Luni C, Campinoti S, Khedr M, Kretzschmar K, De Santis MM, Zambaiti E, Michielin F, Meran L, Hu Q, van Son G, Urbani L, Manfredi A, Giomo M, Eaton S, Cacchiarelli D, Li VSW, Clevers H, Bonfanti P, Elvassore N, De Coppi P, Nat Commun 2019, 10, 5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Seo Y, Jung Y, Kim SH, Acta Biomater 2018, 67, 270. [DOI] [PubMed] [Google Scholar]
- [131].Jang J, Park HJ, Kim SW, Kim H, Park JY, Na SJ, Kim HJ, Park MN, Choi SH, Park SH, Kim SW, Kwon SM, Kim PJ, Cho DW, Biomaterials 2017, 112, 264. [DOI] [PubMed] [Google Scholar]
- [132].Jang J, Kim TG, Kim BS, Kim SW, Kwon SM, Cho DW, Acta Biomater 2016, 33, 88. [DOI] [PubMed] [Google Scholar]
- [133].Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, Mencia-Trinchant N, Negron Abril YL, Hassane DC, Bonassar LJ, Butcher JT, Weiss RS, Reinhart-King CA, Proc Natl Acad Sci U S A 2017, 114, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King CA, Acta Biomater 2013, 9, 4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Zhang Y, Kumar P, Lv S, Xiong D, Zhao H, Cai Z, Zhao X, Materials & Design 2020, DOI: 10.1016/j.matdes.2020.109398. [DOI] [Google Scholar]
- [136].Cui H, Esworthy T, Zhou X, Hann SY, Glazer RI, Li R, Zhang LG, Adv Healthc Mater 2020, 9, e1900924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Mejias JC, Nelson MR, Liseth O, Roy K, Lab Chip 2020, 20, 3601. [DOI] [PubMed] [Google Scholar]
- [138].Andree B, Ichanti H, Kalies S, Heisterkamp A, Strauss S, Vogt PM, Haverich A, Hilfiker A, Sci Rep 2019, 9, 5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Hernandez-Fernaud JR, Reid SE, Neilson LJ, Zanivan S, Proteomics Clin Appl 2013, 7, 464. [DOI] [PubMed] [Google Scholar]
- [140].Park YK, Tu TY, Lim SH, Clement IJ, Yang SY, Kamm RD, Cell Mol Bioeng 2014, 7, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Bichsel CA, Hall SR, Schmid RA, Guenat OT, Geiser T, Tissue Eng Part A 2015, 21, 2166. [DOI] [PubMed] [Google Scholar]