

Abstract
Glioblastoma multiforme (GBM) is the most common and deadliest form of brain tumor and remains amongst the most difficult cancers to treat. Brevican (Bcan), a central nervous system (CNS)-specific extracellular matrix protein, is upregulated in high-grade glioma cells, including GBM. A Bcan isoform lacking most glycosylation, dg-Bcan, is found only in GBM tissues. Here, dg-Bcan is explored as a molecular target for GBM. In this study, we screened a d-peptide library to identify a small 8-amino acid dg-Bcan-Targeting Peptide (BTP) candidate, called BTP-7 that binds dg-Bcan with high affinity and specificity. BTP-7 is preferentially internalized by dg-Bcan-expressing patient-derived GBM cells. To demonstrate GBM targeting, we radiolabeled BTP-7 with 18F, a radioisotope of fluorine, and found increased radiotracer accumulation in intracranial GBM established in mice using positron emission tomography (PET) imaging. dg-Bcan is an attractive molecular target for GBM, and BTP-7 represents a promising lead candidate for further development into novel imaging agents and targeted therapeutics.
Keywords: peptide, molecular targeting, glioblastoma, PET imaging
Graphical Abstract
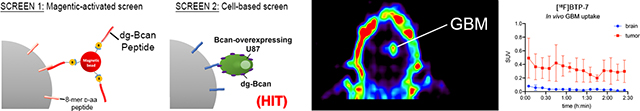
Here, we describe the discovery of a novel peptide, called dg-Bcan-Targeting Peptide (BTP)-7, that can selectively home to GBM cells. Extensive characterization of the peptide through binding kinetic analyses and cell uptake tests demonstrate its potential for targeting dg-Bcan and GBM cells. Radiolabeling of BTP-7 with 18F enables PET imaging of intracranial xenograft GBM in mice.
1. Introduction
GBM is the most malignant of all brain tumors and is incurable with dismal patient prognosis even after surgery and chemo-radiotherapy.[1] Invasive tumor cells are protected from most systemic chemotherapeutics by the blood-brain-barrier (BBB).[2] Moreover, potent chemotherapeutics can also have off-target effects leading to toxicity in healthy brain tissues.[3] For these reasons, there is a critical need for the development of novel biomolecular agents with improved GBM-recognition capability that can effectively target the tumor for delivery of imaging agents or therapeutics specifically to malignant but not healthy brain tissues.
GBM are also well-known for their cellular and genetic heterogeneity. The tumor extracellular matrix (ECM) however, has less spatial variability. Therefore, strategies to target the tumor ECM have advantages over conventional targeted-therapies that are specific for membrane receptors on select tumor cell populations, such as epidermal growth factor receptor variant III (EGFRvIII).[4,5] Brevican (Bcan), a major ECM glycoprotein expressed exclusively in the central nervous system (CNS),[6] is upregulated in GBM,[7,8] and is associated with increased tumor invasion and aggressiveness.[9] Although Bcan has multiple isoforms in normal brain, a unique under-glycosylated isoform (dg-Bcan) has only been found in human high-grade gliomas (grade III and GBM).[7] Importantly, dg-Bcan is absent in the normal CNS and other neuropathologies.[7] Thus, dg-Bcan could serve as a promising tumor-specific marker for the development of novel therapeutic targeting strategies for GBM.
Peptides are attractive tools for rationally-designed therapeutics as they are small, cost-effective, scalable, and can be easily modified and tailored to further optimize binding specificity and BBB penetration.[10] They have massive therapeutic potential and have been described as a “coming peptide tidal wave”.[11] Imaging agents or therapeutic cargoes can be covalently linked in a defined and reversible manner, and non-natural amino acids including d-amino acids, as well as a variety of functional groups can be readily incorporated to increase proteolytic stability, reduce immunogenicity and fine-tune their properties.[12,13] To this end, we have chosen to develop d-amino acid peptides that can bind dg-Bcan with high specificity and affinity for targeting GBM.
To do so, we have screened a one-bead-one-compound (OBOC) combinatorial library built on micro-sized polystyrene beads,[14] with each bead displaying only one unique peptide species composed of 8 d-amino acids. Through a combination of high-throughput screening strategies including a magnetic-capture technique and cell-bead binding approach[15–18] we have identified a novel dg-Bcan-Targeting Peptide (BTP), called BTP-7 that specifically binds recombinant dg-Bcan protein. Fluorescently labeled BTP-7 (BTP-7-Cy5.5) is taken up by patient derived GBM cells (PDGCs) expressing dg-Bcan and targets human GBM in vivo in an intracranial xenograft mouse model. Furthermore, we show that BTP-7-Cy5.5 can cross the BBB in mice and in human BBB organoids. Finally, we have radiolabeled BTP-7 with the 18F radionuclide ([18F]BTP-7) and utilized PET imaging to validate BTP-7 as an in vivo GBM targeting platform and study its biodistribution.
2. Results
2.1. dg-Bcan expression in primary and recurrent glioma cells and tissues
We used the BG1 polyclonal antibody (which detects a 14-amino acids epitope specifically exposed in the dg-Bcan protein) to verify that dg-Bcan was expressed exclusively in GBM specimen, as previously reported.[7,19] Western blot analysis of dg-Bcan using BG1 as well as a “pan-brevican” antibody confirmed that dg-Bcan lacked normal Bcan glycosylation (Figure S1a). We also confirmed that normally glycosylated Bcan was found preferentially in the media of cultured GBM cells, while the dg-Bcan isoform was found associated with the cell membrane (Figure S1a), as previously reported.[7] Immunofluorescence staining and western blot analysis showed that dg-Bcan was expressed in human GBM surgical specimens but not in normal brain tissues (Figure 1a,b), consistent with previous findings[7] and strengthening the evidence of dg-Bcan as a marker for GBM. We observed that dg-Bcan expression was detected in both primary and recurrent GBM specimens (Figure 1b). Furthermore, immunofluorescence staining of GBM biopsy samples isolated from each patient’s tumor core and border revealed expression of dg-Bcan throughout the tissue specimen (Figure S1b).
Figure 1. dg-Bcan is expressed in primary and recurrent glioma tissues and cells.

a) Immunofluorescence images of frozen GBM specimens (using BG1 antibody) showing dg-Bcan (red) and nuclei (blue). Scale bar: 500 μm. b) Western blot of high-grade glioma tissues (grade III or GBM) and control samples (using pan-brevican antibody) with corresponding clinical information (tables). PMI: Post-mortem interval for samples that were recovered after autopsy. c) Immunofluorescence images of patient-derived glioma cells (PDGCs) (using BG1 antibody) showing dg-Bcan (red) and nuclei (blue). (20x scale bar 100μm, 40x scale bar 50μm) d) Brevican RNA level in PDGCs analyzed by qRT-PCR (n = 3).
Immunofluorescence staining of PDGCs revealed that dg-Bcan expression is retained in undifferentiated primary cells (cultured in serum-free media supplemented with epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF)[20]), though expression levels varied with BT188 cells showing the highest and BT145 cells the lowest dg-Bcan expression (Figure 1c). qRT-PCR analysis showed similar levels of Bcan mRNA in BT188 and BT145 despite their different levels of dg-Bcan, suggesting that dg-Bcan levels do not necessarily correlate with Bcan expression (Figure 1d). We further confirmed the expression of dg-Bcan by western blot analysis in other PDGCs. (Figure S1c). Collectively, these data suggest that dg-Bcan could serve as a promising marker for the specific detection or targeting of GBM. From our observations and consistent with previous findings, most established GBM cell lines, including U87 do not express endogenous Bcan.[8]
2.2. OBOC peptide library screen for dg-Bcan targeting peptides
The fully glycosylated Bcan contains chondroitin sulfate glycosaminoglycan (GAG) chains, as well as N-linked and O-linked oligosaccharides,[19] which are not present in dg-Bcan (schematic shown in Figure 2a). We reasoned that dg-Bcan contains exposed epitopes lacking normal glycosylation, which are masked by carbohydrates in fully-glycosylated Bcan. Accordingly, we chose one of those epitopes in dg-Bcan for the development of dg-Bcan-specific binding peptides. Here, we employed a high-throughput two-stage screening strategy[15,18] to search for high-affinity dg-Bcan ligands with stringency and low false positives.
Figure 2. OBOC peptide library screen for dg-Bcan-binding peptides yields 7 candidates.

a) Schematic of the brevican protein, Bcan (top) and its deglycosylated isoform, dg-Bcan (bottom). The dg-Bcan isoform lacks the majority of N- and O-linked carbohydrates (including chondroitin sulfate chains), thus exposing specific epitopes that are masked in the glycosylated isoform. Red box denotes dg-Bcan epitope targeted for screening. b) Schematic of magnetic-activated OBOC library screen with a microfluidic sorter. c,d) Western blot analysis and immunofluorescence images showing astrocytes, U87 (empty vector) and Bcan-overexpressing U87 cells. e) Schematic of the secondary cell-based screen.
Screen 1:
A high-throughput magnetic capture approach was utilized, where OBOC library beads with high affinity for the dg-Bcan epitope would be labeled with magnetic “prey” particles (Figure 2b) (see SI for details). First, library beads coated with magnetic particles conjugated to the scramble(dg-Bcan)-peptide were discarded to minimize the incidence of false positives.[17] The remaining beads that associated with magnetic particles displaying the dg-Bcan epitope peptide were subjected to screen 2.
Screen 2:
To ensure the highest chance of identifying peptides that can recognize dg-Bcan in its native conformation on the cell surface, a secondary cell-based screen was conducted using dg-Bcan-overexpressing U87,[21] as well as dg-Bcan-negative astrocytes and U87 cells (Figure 2c,d,e). Beads that did not associate with live astrocytes and U87 cells but had high interaction with dg-Bcan-overexpressing U87 cells were isolated. Seven hit beads were isolated and the peptides sequenced by Edman degradation.
2.3. Characterization of dg-Bcan-targeting peptides (BTP)
The identified peptides all had a high isoelectric point (pI > 10) (Figure 3a). We observed that BTP-2, 5 and 10 were insoluble and precipitated in aqueous solution. To characterize dg-Bcan specificity, each peptide was conjugated to fluorescein (FITC) on the C-terminus according to the peptide orientation displayed on the library bead (Figure 3b, Figure S2a).
Figure 3. Cell uptake assessments reveal three promising dg-Bcan-targeting peptides (BTP).

a) Table summarizing the properties of the seven peptides from the OBOC library screen. Each letter abbreviates a d-amino acid. MW = molecular weight. b) Chemical structure of the peptide labeled with fluorescein dye. c) Western blot of dg-Bcan and Bcan in human embryo kidney (HEK) cells, and Bcan-overexpressing HEK cells (HEK-Bcan). d) Flow cytometry analysis showing cell uptake of fluorescein-labeled peptides with or without blocking (50x excess native peptide) in HEK-Bcan cells (nevents = 20,000, two-way ANOVA). e) Cell uptake analysis of fluorescein-labeled peptides in BT188 (dg-Bcan-high) or BT145 (dg-Bcan-low) patient derived glioma cells (PDGCs) as measured by fluorescence microscopy. (nPDGCs = 10, two-way ANOVA). f) Chemical structure of the peptide labeled with Cy5.5 dye. g) Western blot analysis showing dg-Bcan expression in GBM-6 cells, astrocytes and neural progenitor cells (NPCs). h) Flow cytometry analysis showing GBM-6 cell uptake of the top 3 candidates, BTP-7-Cy5.5, BTP-8-Cy5.5 and BTP-9-Cy5.5 (each group normalized to a control scramble peptide (Scr-8-Cy5.5) (nevents = 20,000, two-way ANOVA) significance measured in comparison to Scr-8-Cy5.5). i) Flow cytometry analysis showing cell uptake of BTP-7-Cy5.5, BTP-8-Cy5.5 and BTP-9-Cy5.5 in GBM-6 cells and NPCs (nevents = 20,000 cells, two-way ANOVA).
The peptides were characterized using HEK293 cells that do not normally express endogenous brevican and engineered HEK293 cells with stable brevican overexpression (HEK-Bcan) (Figure 3c). Similar to primary PDGCs (Figure S1a), western blot analysis showed that dg-Bcan was found in the cell lysates while full-length glycosylated brevican was secreted into the culture medium (Figure 3c). In a competitive binding study, FITC-labeled BTP candidates, BTP-2, 7, 8, 9 and 10 demonstrated reduced uptake in HEK-Bcan cells in the presence of 50x molar excess of unlabeled (blocking) peptide as analyzed by flow cytometry (Figure 3d), suggesting specific peptide interaction. Flow cytometry analyses confirmed that these five peptides showed increased uptake in HEK-Bcan cells compared to regular HEK cells (Figure S3a). Similarly, these peptides exhibited increased cell uptake in endogenous dg-Bcan-expressing BT-188 PDGCs compared to dg-Bcan-null astrocytes when analyzed by fluorescence microscopy (Figure S3b). To further narrow down the top candidates, we investigated whether these peptides had differential uptake in dg-Bcan-high BT188 cells compared with dg-Bcan-low BT145 cells (Figure 1c and 3e). Three peptides, BTP-7, 8 and 9, exhibited the highest uptake in BT188 compared with BT145 cells and were brought forward for further evaluation in vivo (Figure 3e).
To facilitate in vivo characterization, we labeled BTP-7, 8 and 9 with a Cy5.5 dye (Figure 3f, Figure S2c) that emits light in the near-infrared (NIR) region to enable deeper tissue penetration. The well-characterized GBM-6 patient-derived xenoline (PDX)[22] was used due to its stable expression of dg-Bcan (Figure 3g) and its ability to form aggressive intracranial tumors in nude mice. Flow cytometry analysis showed that Cy5.5-labeled BTP-7 displayed the highest uptake in GBM-6 cells (Figure 3h). The Cy5.5-labeled peptides exhibited greater uptake in GBM-6 cells compared with dg-Bcan-null astrocytes (Figure S3c,d,e). Additionally, GBM-6 cells showed significantly higher peptide uptake than neural progenitor cells (NPC), which do not express dg-Bcan (Figure 3g,i). Flow cytometry analysis also confirmed increased uptake of Cy5.5-labeled peptides in HEK-Bcan cells compared to regular HEK cells (Figure S3f). Incubation of GBM-6 cells with each peptide at 4°C resulted in a dramatic reduction in peptide uptake compared to 37°C (Figure S3g), suggesting ligand-receptor internalization. This was confirmed by confocal microscopy (Figure S3h).
2.4. Affinity of BTP-7 for dg-Bcan protein
Next, we sought to assess the binding of BTP-7, 8 and 9 to purified recombinant dg-Bcan protein by performing binding kinetics analyses using the Octet RED platform. This revealed that native BTP-7 bound dg-Bcan protein with the highest affinity (KD = 0.26 μM) compared to BTP-8 and BTP-9 (KD = 1.57 μM and 1.50 μM, respectively) (Figure 4a,b and c), thus making BTP-7 the top candidate for dg-Bcan targeting. These peptides did not bind the normally glycosylated form of brevican (Figure 4a,b and c), highlighting their specificity for the dg-Bcan isoform. A scrambled sequence of the BTP-7 peptide (Scr-7) did not exhibit dg-Bcan protein binding (Figure 4a). Fluorescein-conjugated BTP-7 retained its specificity for dg-Bcan and did not bind glycosylated brevican, although the binding affinity (KD = 3.80 μM) was more than 10x weaker than the native unlabeled peptide (Figure S4a). Conversely, BTP-7 conjugated to a Cy5.5 dye was found to have a slightly higher affinity for dg-Bcan (KD = 0.19 μM) than the native peptide (Figure S4b).
Figure 4. Binding affinity and specificity of top 3 peptides to recombinant dg-Bcan protein.

a,b,c) Steady state binding kinetics analysis of BTP-7, Scr-7 (scramble BTP-7; green), BTP-8 and BTP-9 to recombinant dg-Bcan (blue) or the fully glycosylated Bcan (red) protein using the Octet (ForteBio) platform. KD = dissociation constant, Rmax = maximal response in response unit. KD, Rmax and curves were calculated using a non-linear regression (one site specific binding) fit in Graphpad Prism. d) Competitive binding assay using biotinylated BTP-7 functionalized with a UV-crosslinker. Peptide binding/crosslinking to recombinant dg-Bcan (in the presence of native BTP-7 blocking peptide) is detected by western blot using streptavidin-HRP (n = 3).
The specificity of BTP-7 binding to dg-Bcan was further confirmed using BTP-7 functionalized with an ultraviolet (UV)-sensitive crosslinker and biotin (Figure S2b). Upon interaction, the peptide can be crosslinked to dg-Bcan by exposure to UV light, and detected by western blot using streptavidin-HRP. Competitive binding studies showed that the presence of native BTP-7 (blocking) peptide decreased the level of crosslinking in a dose-dependent manner (Figure 4d). This effect was not observed when blocking was performed with the scramble peptide, Scr-7 (Figure S5a).
Next, we investigated whether BTP-7 would interact specifically with the dg-Bcan epitope which was mentioned previously. Analysis by circular dichroism (CD) spectrometry indicated that the BTP-7 structure was altered in the presence of the dg-Bcan-peptide, and this effect was negligible in the presence of the scramble(dg-Bcan) peptide (Figure S5b). Meanwhile, BTP-8 structure appeared to be unaffected in the presence of the dg-Bcan or scramble(dg-Bcan) peptide (Figure S5c). Increasing dg-Bcan-peptide concentration resulted in alteration of BTP-7 structure in a dose-dependent manner (Figure S5d). This was not observed when BTP-7 was mixed with the scramble(dg-Bcan) peptide (Figure S5d), suggesting that BTP-7 interacted with the dg-Bcan epitope specifically.
2.5. BTP-7 targets intracranial GBM xenografts
We showed that BTP-7 is relatively stable in human serum, with approximately 50% of peptide detected after 12 hours at 37°C (Figure S6a). In contrast, when each d-residue in the BTP-7 peptide was substituted with the corresponding L-amino acid, the peptide was rapidly degraded (half-life = 0.44 hours) (Figure S6b).
For in vivo studies, GBM-6 PDGCs were implanted intracranially into the right frontal lobe of nude mice and tumor formation was confirmed by MRI after two weeks. (Figure S6c). Mice were then injected with either Cy5.5-labeled BTP-7, 8, 9 or Cy5.5 dye (without peptide) as a control via the tail vein. Fluorescence imaging of brain cryo-sections revealed that BTP-7-Cy5.5 had significantly higher tumor uptake (more than 10x) compared to other peptides 7 hours post-injection (Figure 5a,b), reaffirming BTP-7 as the prime GBM-targeting peptide. We also found that tumor tissues in the BTP-7-Cy5.5 group exhibited 4-fold higher fluorescence intensity compared to normal brain, underlining tumor specificity (Figure 5c). IVIS fluorescence imaging showed that BTP-7-Cy5.5 had the highest accumulation in the liver and kidneys four hours post-intravenous administration (Figure S6d,e), suggesting that the major routes of BTP-7-Cy5.5 clearance occurred through these organs. The brain showed low peptide uptake compared to other organs (Figure S6e).
Figure 5. Functional characterization of BTP-7 in vitro and in vivo.

a) Fluorescence images showing brain cryo-sections 7 hrs post intravenous injection of Cy5.5-labeled BTP-7, BTP-8 or BTP-9 (100 μL of 500 μM peptide solution, n = 3) at Day 15 post-tumor implantation. Cy5.5 dye (no peptide) was injected as a negative control. Tumor regions (right (R) brain hemisphere) are marked with yellow dotted lines. b) Quantification of the mean Cy5.5 intensity in the tumor (nsections = 15, one-way ANOVA). c) Quantification of tumor:non-tumor ratio (from a); non-tumor regions (left hemisphere) are marked with red dotted lines. Significance was compared with the Cy5.5 dye control group. d) Permeability of BTP-7-Cy5.5 and Scr-7-Cy5.5 in BBB organoids (BH3-Cy5.5 peptide was used as a non-permeable control) (nspheres = 4–6, cpeptide = 10 μM, t = 3 hrs, one-way ANOVA). e) Fluorescence images of brain cryo-sections (30 μm slices) showing the distribution of BTP-7-Cy5.5 and Scr-7-Cy5.5 (white) in the frontal lobe of non-tumor bearing mice upon intravenous administration. Dextran rhodamine (155 kDa; red) was injected 4 hrs later intravenously, and the mice were sacrificed 30 min later. Red areas indicate regions of high perfusion (blood vessels). Scale bar: 100 microns. f) BBB permeability was quantified by measuring the mean Cy5.5 intensity within the brain parenchyma outside of highly perfused areas (cyan circles in panel e) (ntissues = 2; nareas = 10, one- way ANOVA).
2.6. BBB penetration
Next, we examined whether BTP-7 could cross the BBB using an in vitro human BBB organoid model.[23,24] BTP-7-Cy5.5 exhibited influx into the organoids, demonstrating some potential for BBB-penetration (Figure 5d). Influx was also observed in the organoids with the scramble variant, Scr-7-Cy5.5 (compared to the control non-penetrating BH3-Cy5.5 peptide[23]), though the level of penetration was significantly lower than that observed with BTP-7-Cy5.5 (Figure 5d). In the presence of BTP-7-Cy5.5, the organoids remained impermeable to high molecular weight dextran molecules (4.4 kDa) (Figure S7), showing that BTP-7-Cy5.5 and Scr-7-Cy5.5 did not damage the BBB spheroid barrier. To confirm this observation in vivo, BTP-7-Cy5.5 or Scr-7-Cy5.5 was administered intravenously into healthy non-tumor bearing mice. After 4 hours, dextran-rhodamine (155 kDa) was administered intravenously so that perfused blood vessels could be visualized. Confocal microscopy revealed that both BTP-7-Cy5.5 and Scr-7-Cy5.5 was detected within the brain parenchyma (outside of regions labeled by dextran-rhodamine), with the level of BTP-7-Cy5.5 being slightly, though significantly higher than Scr-7-Cy5.5 (Figure 5e,f).
Radiolabeling of BTP-7 and PET imaging
To further demonstrate dg-Bcan dependent GBM targeting, we labeled BTP-7 with a 18F radionuclide (Figure 6a). In vitro cell uptake studies showed increased [18F]BTP-7 uptake in dg-Bcan overexpressing cells (HEK-Bcan) compared to HEK cells, similar to our observations with fluorescently labeled BTP-7 (Figure 6b, Figure S3a). In addition, [18F]BTP-7 uptake in GBM-6-cells was significantly reduced when blocking with “cold” [19F]BTP-7, confirming its specificity (Figure 6b).
Figure 6. Radiolabeling of BTP-7 and PET imaging.

a) [18F]BTP-7 was prepared in a 2-step synthesis. The labeling synthon 1-[18F]fluoro-4-pentyne was prepared via nucleophilic fluorination of 1-tosyloxy-4-pentyne, isolated and then trapped in a receiving vial containing BTP-7-azide precursor with prepared Copper-catalyzed Azide-Alkyne Cycloaddition (CuAAC) solvent. b) Cell uptake studies showing (left graph) increased [18F]BTP-7 uptake in dg-Bcan-overexpressing HEK cells in comparison to non-expressing HEK cells (n = 8, added activity/well = 2.06 mCi, t = 60 min, Mann-Whitney-U test); and (right graph) [18F]BTP-7 uptake is blocked by “cold” [19F]BTP-7 in GBM-6 cells (n = 4, added activity/well = 0.7 mCi, cblock = 10mM, t = 90 min, Mann-Whitney-U test). Y-axis displays percentage of administered dose (%AD). c) Representative image (axial) showing in vivo biodistribution by PET/CT imaging (left: CT, right: merge PET/CT). Organ marked with * = kidney and + = liver. d) Quantification of [18F]BTP-7 organ activity from images obtained from (c). Graph shows biodistribution (activity in standard uptake value (SUV)) in nude mice over 2.5 hours (n = 3, i.v. administered activity = 600 – 800 mCi, t = 150 min). e) Representative images showing PET imaging in nude mice bearing intracranial GBM-6 tumor with the following panels; (I) MRI (axial view) confirming tumor formation in right brain hemisphere (delineated with red dashed line), scale bar: 2mm. (II, III, IV) axial, coronal and sagittal view of PET images show high contrast between tumor and brain. f) Quantification of [18F]BTP-7 activity from images from (e). Graph showing activity in SUV in the GBM tumor and normal brain tissues in nude mice over 2.5 hours (p<0.0001) (n = 3, i.v. administered activity = 600 – 800 μCi, t = 150min).
Next, we evaluated the biodistribution of [18F]BTP-7 in GBM-bearing mice. Intracranial GBM-6 tumor was established in nude mice and tumor formation and volume were confirmed via MRI after 35 days (ranging from 6 – 60mm3). Tumor-bearing mice were then administered with 600–800 μCi of [18F]BTP-7 intravenously and PET imaging was performed over 150 minutes. Quantitative image analysis revealed that the highest tracer uptake was in the kidneys, followed by the heart (Figure 6c,d). High kidney uptake suggests renal excretion as the major route of clearance for the peptide tracer in the animals. Low tracer uptake was observed in the liver, bone and brain (Figure 6c,d). Ex vivo biodistribution analyzed by gamma-counting revealed similar results, with the brain showing the lowest tracer uptake (Figure S9a). Importantly, significantly higher tracer uptake was observed in intracranial GBM-6 tumors compared to the healthy brain tissues, allowing clear visualization of the tumor by PET imaging (Figure 6e,f), with the tumor mostly retaining an average standard uptake value (SUV) that is more than 10 times higher than normal brain tissues between 45 and 150 mins post-administration (Figure S9b).
3. Discussion
Tumor-specific targeting agents represent an attractive approach to developing targeted imaging agents and precision medicines. In this work, to overcome tumor genetic heterogeneity,[25] we have focused our efforts on an ECM protein, dg-Bcan that is expressed in high-grade gliomas but not found in other tissues within or outside the CNS. The tumor specificity and consistency of dg-Bcan expression throughout the tumor tissues underscore its potential as a novel GBM-specific marker for the development of new targeting agents.
Here, we have undertaken a stringent peptide library screen against the dg-Bcan protein as well as dg-Bcan-expressing cells to discover BTP-7, a novel d-peptide that is stable in serum, binds recombinant human dg-Bcan with little to no cross-reactivity with the major full-length glycosylated Bcan protein, and labels cultured cells in a dg-Bcan-dependent manner. We have also demonstrated that BTP-7 labeled with a NIR dye or 18F radionuclide homes to human GBM xenografts established intracranially in mice, highlighting its potential as a targeted delivery system for imaging agents or anti-tumor therapies. While the mechanism of BTP-7 penetration across the BBB is yet to be elucidated, we postulate that transport may be occurring via absorption-mediated transcytosis (AMT), as many short positively charged and hydrophilic BBB-penetrating peptides typically cross the BBB through this mechanism.[26–28]
Bcan is a ECM proteoglycan that is highly upregulated in high-grade gliomas and implicated in tumor invasion and progression.[21,29] It has multiple isoforms produced by glycosylation, cleavage, and alternative splicing but only one of these isoforms, dg-Bcan is uniquely expressed in human high-grade gliomas.[30] Primary PDGCs in their undifferentiated state retain dg-Bcan expression, even after being passaged in mice, and can be used for both in vitro and in vivo GBM modeling and drug targeting analyses. Bcan activates EGFR/mitogen-activated protein kinase (MAPK) signaling and fibronectin production in glioma cells, increasing their migratory and invasive properties.[21] The specific function of dg-Bcan as well as its correlation with clinical parameters remains largely unknown, due to our limited ability to manipulate dg-Bcan expression and detect it by any means other than western blotting of tumor samples. Nevertheless, its unique and widespread expression in high-grade gliomas provides an excellent and accessible target that is tumor-specific.
BTP-7 is the first dg-Bcan-binding peptide described, which gives this peptide a unique advantage to label the ECM of GBM cells. Accordingly, imaging agents derived from BTP-7 for specific GBM detection will likely be useful for neuroimaging to accurately distinguish real tumor progression from pseudoprogression,[31,32] monitor treatment response, and overall efficacy of GBM therapies. This peptide can therefore serve as a companion diagnostic with standard of care. Additionally, this peptide tracer could potentially be useful in patients requiring longitudinal follow up from immunotherapy or viral therapy.
PET imaging is a sensitive imaging modality that can provide additional biochemical data to complement structural MRI for non-invasive diagnosis, staging and monitoring treatment for GBM.[33] Several radioligands have been used for the evaluation of patients with GBM in the clinic. Fluorine-18 glucose analogue ([18F]FDG) is a commonly used radiotracer that has been shown to have high uptake in tumor cells due to high glucose consumption in the tissue.[34,35] However, [18F]FDG has been widely criticized for its lack of specificity as a contrast agent for imaging brain tumors as physiological high FDG uptake in the healthy brain parenchyma hampers the delineation of brain tumors, thereby diminishing its diagnostic performance.[33,36] Higher diagnostic accuracy in GBM patients has been reported with amino acid PET tracers, such as [18F]fluoro-ethyl-L-tyrosine) ([18F]FET), and deoxynucleoside bases ([18F]fluoro-thymidine ([18F]FLT) and [18F]clofarabine ([18F]CFA)) that depend on the metabolism of highly proliferative GBM cells.[33,35,37] Here, we have demonstrated that the tumor-to-brain uptake of [18F]BTP-7 is higher in comparison to values reported from similar studies evaluating [18F]FDG and [18F]FET tracers.[35,36] Newer radiotracers targeting specific GBM biomarkers such as sigma-1 and 2 receptors, programmed death ligand 1 (PD-L1), poly (ADP-ribose) polymerases (PARP) and isocitrate dehydrogenase (IDH) 1 have also been explored for imaging GBM lesions.[37] However, these biomarkers are not unique to GBM. [18F]BTP-7 represents a first-in-class dg-Bcan-specific radioligand that demonstrates potential for targeted imaging of GBM, and warrants further development for clinical translation in the future. The overall low brain uptake also makes it ideal for tumor detection. Our findings show tumor uptake in less than an hour, allowing for rapid tumor identification non-invasively by PET imaging. We have also demonstrated that [18F]BTP-7 is sensitive enough to identify tumors (as small as 6 mm3 volume) in our orthotopic tumor model studies.
Our next aims are to perform rational peptide modifications to develop second-generation peptides with increased dg-Bcan affinity and BBB permeability, investigate stable cleavable linkers such as acid or enzyme-cleavable linkers[38] and explore BTP-7 conjugation to a wide array of other anti-cancer therapeutics to further promote therapeutic efficacy. Additionally, BTP-7 could provide a robust platform for the development of novel cytotoxic radionuclides for targeted radiotherapy, as well as other targeted non-invasive molecular imaging agents, such as gadolinium or iron oxide nanoparticles (for MRI).
4. Conclusion
Taken together, this work shows that targeting dg-Bcan is a novel approach towards combating GBM. This study opens the door for further development of BTP-7 into clinically useful imaging agents and targeted therapeutics in the near future.
5. Experimental Section/Methods
OBOC library screening
An OBOC combinatorial library was synthesized on 1 g of 90 μm TentaGel resin (Tentagel S –NH2; 0.33 mmol/g loading) using a “split and pool” strategy so that each bead carried multiple copies of a unique peptide ligand.[14,15,18] See SI for detailed screening approach.
Cell uptake analysis
Cells were washed once with PBS and dissociated by treatment with 1 mL of 2.5 mM ethylenediaminetetraacetic acid (EDTA). After centrifugation, PBS supplemented with 10% FBS was added to create a single-cell suspension. All peptide stocks were dissolved in dimethyl sulfoxide (DMSO) and stored at −20 °C in the dark. Cells were incubated with each peptide (1–10 μM) for 3 hrs at 37 °C in the dark under constant rotation. Cells were then washed 3x with PBS/10% FBS (7000 rpm centrifugation for 3 min). Cells were resuspended in 3.7% formaldehyde and washed once with PBS. Peptide uptake (mean fluorescence intensity) was determined by flow cytometry (10,000–20,000 events) or fluorescence microscopy (nPDGCs=10). For blocking studies, cells were incubated with a 50x molar excess of unlabeled peptide for 10 min on ice, before adding the corresponding fluorescently labeled peptide at a final concentration of 1 μM and incubated for 1.5 hrs on ice. To measure peptide internalization, cells were incubated at 37 °C or 4 °C (to inhibit endocytosis) for 3.5 hrs with Cy5.5-labeled peptide (5 μM). Cells were then washed with cold PBS/10% FBS, fixed and analyzed by flow cytometry or confocal microscopy.
Octet binding kinetic analyses
The FortéBio OctetRed384 was used to study the binding kinetics of each peptide to recombinant human brevican (in PBS and 0.1 mM EDTA, pH 6.8). The brevican protein was deglycosylated prior to experimental use (see SI for details). All binding kinetics assays were performed using the OctetRed instrument under agitation at 1000 rpm in 0.9% NaCl irrigation with 0.05% Tween (working buffer). Assays were performed at 30°C in solid black 384-well plates (Geiger Bio-One). See SI for details.
Synthesis of [18F]BTP-7
BTP-7 was functionalized with an azide to facilitate fast and reliable labeling with a 5-[18F]fluoro-1-pentyne by copper-catalyzed alkyne-azide cycloaddition (CuAAC). See SI for detailed synthesis.
Animal studies
All animal protocols were reviewed and approved by the in-house Institutional Animal Care and Use Committee (IACUC)
Intracranial GBM implantation and tumor uptake analysis
100,000 GBM-6 PDGCs resuspended in 2μl PBS were inoculated into the striatum of female 6–8-week-old athymic mice using a stereotactic frame as described.[39] After two weeks, tumor formation was confirmed via T2-weighted MRI. The following day, mice were injected with either Cy5.5-labeled BTP-7, BTP-8 or BTP-9 (n = 3; 100 μL of 500 μM peptide solution) via the tail vein. A ‘no peptide’ control group (Cy5.5 dye) was also included. After 8 hrs, mice were sacrificed and their brains excised, cryo-sectioned, stained with Hoechst dye and imaged by fluorescence microscopy. The mean Cy5.5 fluorescence intensity of the tumor was quantified using ImageJ and compared with the mean fluorescence intensity of the non-tumor bearing contralateral (left) side of the brain.
PET imaging for biodistribution and tumor targeting studies in GBM-bearing mice
GBM-6 intracranial tumors were established as above. At Day 35 post-implantation, intracranial tumor formation was confirmed using T2-weighted MRI. Animals were anesthetized with 1–3% isoflurane-oxygen and then injected with 500–800 μCi of [18F]BTP-7 via the tail vein. Whole body PET imaging was performed for each animal using a microPET/CT scanner (SuperArgus, Sedecal, Madrid) for 150 minutes; CT images were acquired immediately afterward. The time-activity curves for all organs were generated by manually placing volumes of interest over the organs / tumor using AMIDE-bin 1.0.5. (See SI for details)
Statistical analysis
Data are presented as means ± SD. Significance was determined via one-way (two-way when applicable) ANOVA and Tukey’s Multiple Comparison test or Mann-Whitney-U and Komogorov-Smirnov test. Details are provided in the figure legends. Significance is indicated as follows: (* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001). Statistical analysis was performed using GraphPad Prism v8.4.0
Use of human specimens
All human subjects had given informed consent and signed consent forms inspected, approved by the Institutional Review Board (IRB), and provided to the PI in de-identified manner for research.
Supplementary Material
Fig S1. Expression of dg-Bcan in high-grade glioma tumor samples and patient derived glioma cell lines
Figure S2. Workflow for the synthesis of labeled peptides
Figure S3. Characterization of peptides for dg-Bcan specificity using cell uptake studies
Figure S4. Binding affinity of functionalized BTP-7 to dg-Bcan protein
Figure S5. Binding of BTP-7 to dg-Bcan and the dg-Bcan-derived peptide
Figure S6. In vivo analysis of BTP-7
Figure S7. BBB permeability of BTP-7 in vitro
Figure S8. No effect of BTP-7 on GBM cell proliferation and invasion
Figure S9. Ex vivo biodistribution and in vivo uptake of [18F]BTP-7
Supplementary Notes: Sequences and chromatograms
Acknowledgements
Funding: N.v.S. is supported by the Deutsche Forschungsgemeinschaft (DFG) (research fellowship grant number 400975596). C.-F.C. is supported by the Sperling Family Foundation, Harvard Neuro-Discovery Center, BWH Women’s Brain Initiative, BWH Connors Center, Brigham Research Institute and DoD (grant number W81XWH1910791). J.M.W. is supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374. S.E.L. is supported by (NCI R01CA237063) the BWH Innovator Award and B*CURED Foundation. B.L.P. is supported by (NCI R01CA237063). E.A.C. is supported by (RO1CA166172). L.G.L. is support by NSERC.
Conflict of Interest
An invention disclosure describing BTP-7 entitled ‘Brevican binding peptides for brain tumor targeting’ has been filed within BWH. Partners Healthcare Innovation has supported filing of a provisional patent application (Serial # 62/739845) on Oct 1st, 2018, and a full PCT application has been filed on Oct 1st, 2019.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Stupp R, Mason WP, Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, New England Journal of Medicine 2005, 352, 987. [DOI] [PubMed] [Google Scholar]
- [2].Kemper EM, Boogerd W, Thuis I, Beijnen JH, Tellingen O, Cancer Treat. Rev 2004, 30, 415. [DOI] [PubMed] [Google Scholar]
- [3].Niewald M, Berdel C, Fleckenstein J, Licht N, Ketter R, Rube C, Radiat Oncol 2011, 6, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Raavé R, Kuppevelt TH, Daamen WF, J Control Release 2018, 274, 1. [DOI] [PubMed] [Google Scholar]
- [5].An Z, Aksoy O, Zheng T, Fan Q-W, Weiss WA, Oncogene 2018, 37, 1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jaworski DM, Kelly GM, Hockfield S, Cell Biol J. 1994, 125, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Viapiano MS, Bi WL, Piepmeier J, Hockfield S, Matthews RT, Cancer Res 2005, 65, 6726. [DOI] [PubMed] [Google Scholar]
- [8].Jaworski DM, Kelly GM, Piepmeier JM, Hockfield S, Cancer Res 1996, 56, 2293. [PubMed] [Google Scholar]
- [9].Lu R, Wu C, Guo L, Liu Y, Mo W, Wang H, Ding J, Wong ET, Yu M, BMC Cancer 2012, 12, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ladner RC, Sato AK, Gorzelany J, Souza M, Drug Discov. Today 2004, 9, 525. [DOI] [PubMed] [Google Scholar]
- [11].Kruger RP, Cell 2017, 171, 497. [DOI] [PubMed] [Google Scholar]
- [12].Milton R, Milton S, Kent S, Science 1992, 256, 1445. [DOI] [PubMed] [Google Scholar]
- [13].Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS, Proceedings of the National Academy of Sciences 2007, 104, 16828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ, Nature 1991, 354, 82. [DOI] [PubMed] [Google Scholar]
- [15].Cho C-F, Amadei GA, Breadner D, Luyt LG, Lewis JD, Nano Lett 2012, 12, 5957. [DOI] [PubMed] [Google Scholar]
- [16].Cho C-F, Behnam Azad B, Luyt LG, Lewis JD, ACS Comb Sci 2013, 15, 393. [DOI] [PubMed] [Google Scholar]
- [17].Cho C-F, Lee K, Speranza M-C, Bononi FC, Viapiano MS, Luyt LG, Weissleder R, Chiocca EA, Lee H, Lawler SE, ACS Comb Sci 2016, 18, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cho C-F, Yu L, Nsiama TK, Kadam AN, Raturi A, Shukla S, Amadei GA, Steinmetz NF, Luyt LG, Lewis JD, Nanoscale 2017, 9, 12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Viapiano MS, Matthews RT, Hockfield S, J. Biol. Chem 2003, 278, 33239. [DOI] [PubMed] [Google Scholar]
- [20].Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA, Cancer Cell 2006, 9, 391. [DOI] [PubMed] [Google Scholar]
- [21].Hu B, Kong LL, Matthews RT, Viapiano MS, J. Biol. Chem 2008, 283, 24848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, James CD, Neuro-oncology 2005, 7, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cho C-F, Wolfe JM, Fadzen CM, Calligaris D, Hornburg K, Chiocca EA, Agar NYR, Pentelute BL, Lawler SE, Nat Commun 2017, 8, 15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bergmann S, Lawler SE, Qu Y, Fadzen CM, Wolfe JM, Regan MS, Pentelute BL, Agar NY, Cho C-F, Nature protocols 2018, 13, 2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, Weiss WA, Smirnov IV, Song JS, Olshen AB, Cha S, Zhao Y, Moore RA, Mungall AJ, Jones SJM, Hirst M, Marra MA, Saito N, Aburatani H, Mukasa A, Berger MS, Chang SM, Taylor BS, Costello JF, Science 2014, 343, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hervé F, Ghinea N, Scherrmann J-M, The AAPS Journal 2008, 10, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oller-Salvia B, Sánchez-Navarro M, Giralt E, Teixidó M, Chem. Soc. Rev 2016, 45, 4690. [DOI] [PubMed] [Google Scholar]
- [28].Rousselle C, Smirnova M, Clair P, Lefauconnier J-M, Chavanieu A, Calas B, Scherrmann J-M, Temsamani J, Journal of Pharmacology and Experimental Therapeutics 2001, 296, 124. [PubMed] [Google Scholar]
- [29].Dwyer CA, Bi WL, Viapiano MS, Matthews RT, J. Neurooncol 2014, 120, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Viapiano MS, Matthews RT, Trends in molecular medicine 2006, 12, 488. [DOI] [PubMed] [Google Scholar]
- [31].Dhermain FG, Hau P, Lanfermann H, Jacobs AH, Bent MJ, Lancet Neurol 2010, 9, 906. [DOI] [PubMed] [Google Scholar]
- [32].Stuplich M, Hadizadeh DR, Kuchelmeister K, Scorzin J, Filss C, Langen K-J, Schäfer N, Mack F, Schüller H, Simon M, Glas M, Pietsch T, Urbach H, Herrlinger U, JCO 2012, 30, e180. [DOI] [PubMed] [Google Scholar]
- [33].Galldiks N, Lohmann P, Albert NL, Tonn JC, Langen K-J, Neuro-Oncology Advances 2019, 1, DOI 10.1093/noajnl/vdz010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Toyama H, Ichise M, Liow J-S, Vines DC, Seneca NM, Modell KJ, Seidel J, Green MV, Innis RB, Nuclear medicine and biology 2004, 31, 251. [DOI] [PubMed] [Google Scholar]
- [35].Wang H-E, Wu S-Y, Chang C-W, Liu R-S, Hwang L-C, Lee T-W, Chen J-C, Hwang J-J, Nuclear medicine and biology 2005, 32, 367. [DOI] [PubMed] [Google Scholar]
- [36].Sai KKS, Huang C, Yuan L, Zhou D, Piwnica-Worms D, Garbow JR, Engelbach JA, Mach RH, Rich KM, McConathy J, Journal of nuclear medicine 2013, 54, 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Drake LR, Hillmer AT, Cai Z, Molecules 2020, 25, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Leriche G, Chisholm L, Wagner A, Bioorg. Med. Chem 2012, 20, 571. [DOI] [PubMed] [Google Scholar]
- [39].Williams SP, Nowicki MO, Liu F, Press R, Godlewski J, Abdel-Rasoul M, Kaur B, Fernandez SA, Chiocca EA, Lawler SE, Cancer research 2011, 71, 5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Expression of dg-Bcan in high-grade glioma tumor samples and patient derived glioma cell lines
Figure S2. Workflow for the synthesis of labeled peptides
Figure S3. Characterization of peptides for dg-Bcan specificity using cell uptake studies
Figure S4. Binding affinity of functionalized BTP-7 to dg-Bcan protein
Figure S5. Binding of BTP-7 to dg-Bcan and the dg-Bcan-derived peptide
Figure S6. In vivo analysis of BTP-7
Figure S7. BBB permeability of BTP-7 in vitro
Figure S8. No effect of BTP-7 on GBM cell proliferation and invasion
Figure S9. Ex vivo biodistribution and in vivo uptake of [18F]BTP-7
Supplementary Notes: Sequences and chromatograms