

Abstract
Mitochondrial complex I (NADH:ubiquinone oxidoreductase) is a crucial metabolic enzyme that couples the free energy released from NADH oxidation and ubiquinone reduction to the translocation of four protons across the inner mitochondrial membrane, creating the proton motive force for ATP synthesis. The mechanism by which the energy is captured, and the mechanism and pathways of proton pumping, remain elusive despite recent advances in structural knowledge. Progress has been limited by a lack of model systems able to combine functional and structural analyses with targeted mutagenic interrogation throughout the entire complex. Here, we develop and present the α-proteobacterium Paracoccus denitrificans as a suitable bacterial model system for mitochondrial complex I. First, we develop a robust purification protocol to isolate highly active complex I by introducing a His6-tag on the Nqo5 subunit. Then, we optimize the reconstitution of the enzyme into liposomes, demonstrating its proton pumping activity. Finally, we develop a strain of P. denitrificans that is amenable to complex I mutagenesis and create a catalytically inactive variant of the enzyme. Our model provides new opportunities to disentangle the mechanism of complex I by combining mutagenesis in every subunit with established interrogative biophysical measurements on both the soluble and membrane bound enzymes.
Subject terms: Bioenergetics, Enzymes, Oxidoreductases, Biochemistry, Membrane proteins, Metalloproteins, Mitochondrial proteins
Introduction
Complex I (NADH:ubiquinone oxidoreductase) is a large, multisubunit, membrane-bound enzyme that couples the oxidation of NADH and reduction of ubiquinone to the translocation of four protons across the inner mitochondrial membrane, or cytoplasmic membrane in prokaryotes, contributing to the proton motive force (∆p) that is used to power ATP synthesis1–3. Complex I is also a key site for cellular reactive oxygen species generation4,5, particularly during ischemia reperfusion injury6. Due to its essential role in metabolism and energy production, mutations in complex I and its associated proteins are the origin of a wide range of neuromuscular and metabolic disorders7,8.
In its most minimal form, as typically found in α-proteobacteria, complex I is composed of 14 conserved catalytic ‘core’ subunits; the seven core subunits of the hydrophilic domain catalyze electron transfer from NADH to ubiquinone, and the seven core subunits in the membrane domain contain the proton pumps9,10. The mammalian enzyme contains an additional 31 ‘supernumerary’ subunits that have been accumulated during evolution and are required for assembly, stability or regulation of the complex11,12. Despite the central importance of complex I in oxidative phosphorylation, both the coupling mechanism that captures the energy released from the electron transfer reaction and consumes it to drive proton translocation, and the mechanism and pathways of the proton pumps, remain elusive.
In the last decade, the ‘resolution revolution’ has resulted in a surge of complex I structural data, especially resulting from cryoEM analyses, including native enzyme structures from bacteria, yeast, plants and mammals10,11,13–16, and substrate and inhibitor bound states17–20. These structures, along with molecular dynamic simulations2,21,22, have identified intriguing structural elements and key residues that are likely involved in the coupling mechanism and proton transfer pathways. However, probing the catalytic roles of these elements and sites further is hindered by lack of model systems that first, are suitable for both functional and structural analyses, and second, allow genetic manipulation of the core membrane domain subunits, which are encoded on the mitochondrial DNA in eukaryotes. Robust methods for creating site-directed mutants in the mitochondrial encoded proteins in eukaryotes are not yet available, rendering the core membrane domain subunits inaccessible to mutagenesis in model systems such as Yarrowia lipolytica, the popular yeast model system for complex I, as well as in mammalian species. Development of an experimentally versatile and fully genetically tractable bacterial model system thus has a crucial role to play in addressing mechanistic questions.
The α-proteobacterium Paracoccus denitrificans is a strong candidate for a bacterial model of the mitochondrial enzyme. P. denitrificans is a close relative of the protomitochondrion23 and its respiratory chain possesses similarities to the contemporary mitochondrial chain that are uncommon among other bacterial species23,24. First, P. denitrificans uses ubiquinone-10 as its sole quinone in the cell, the same as used by the human respiratory chain and in many mammalian species23,25. Second, P. denitrificans complex I shares much higher sequence similarity with the mammalian complexes, particularly in the hydrophilic domain, than the other bacterial models currently in use (Escherichia coli and Thermus thermophilus)26. Third, P. denitrificans complex I possesses three supernumerary subunits with mitochondrial homologues26, which are not present in other bacterial models. Finally, P. denitrificans is known to form respiratory supercomplexes, including complex I-containing respirasomes27, and its electron transport chain composition matches that of the canonical mitochondrial chain; it contains both complex III (cytochrome bc1) and complex IV (cytochrome c oxidase). These features have led to P. denitrificans being usefully employed in the past as a model system for studies of complex III and IV28,29. In combination, these observations suggest that P. denitrificans presents exciting opportunities to develop a bacterial model system for detailed and comprehensive studies of mitochondrial complex I.
To date, only E. coli and T. thermophilus have been used as bacterial model systems for studying complex I. While the first published complex I structure came from T. thermophilus, it has not been utilized widely beyond structural work, likely due to the difficulty of growing cells at 70 °C and the challenge of biophysical experiments at elevated temperatures9. In contrast, the E. coli model system has been heavily employed in the functional interrogation of complex I. Extensive mutagenesis studies in E. coli have helped identify essential residues in the complex, such as conserved glutamate and lysine residues that span the central axis of the membrane domain, likely involved in charge propagation21,30–33. However, much of this work was carried out without the benefits of the detailed structural information now available. In addition, despite its wide use, the E. coli system has some drawbacks that limit its relevance as a model for the mitochondrial enzyme, particularly when compared to P. denitrificans: it uses menaquinone or ubiquinone-8 as a terminal electron acceptor25, has a lower sequence homology than P. denitrificans to the mitochondrial enzyme26 and E. coli lacks complex III and a cytochrome c oxidase and thus cannot form respirasome-like supercomplexes34.
Perhaps the most appealing aspect of using P. denitrificans complex I to study the enzyme’s catalytic mechanism is that it is possible to form well-coupled energy-transducing vesicles from P. denitrificans membranes, a feat that has not yet proved possible for other bacterial or yeast model systems. P. denitrificans sub-bacterial particles (SBPs, inverted cytoplasmic membrane vesicles) catalyze NADH-driven ATP synthesis both rapidly and efficiently, and they have been utilized to determine the number of protons translocated across the membrane by complex I for each NADH oxidized (the proton stoichiometry)35,36. Attempts to apply the same approach to E. coli vesicles did not succeed because the vesicles were not sufficiently well coupled and the data did not converge to provide a robust stoichiometry value. The only published study of the stoichiometry of the E. coli enzyme was only able to report a value of at least three37. The lack of a robust method to determine the proton pumping stoichiometry of E. coli complex I compromises its use, as the stoichiometry is a key biophysical property that underpins mechanistic assessment of site-directed mutants.
Here, we present P. denitrificans as a genetically tractable model system that can be used to generate and study mutations in every core subunit of complex I. Despite being considered unstable when not associated with its supercomplex27, a four-column purification strategy for purifying intact complex I from P. denitrificans (Pd-CI) has been described previously26. Here, we describe a robust, streamlined two-step purification to isolate catalytically active P. denitrificans complex I using a genetically engineered affinity tag. Following characterization of the pure enzyme, we describe its reconstitution into liposomes to demonstrate that it remains coupled and retains proton pumping activity. Finally, we describe a P. denitrificans strain engineered to be amenable to the creation of deleterious complex I variants, produced by introducing an alternative NADH dehydrogenase as an artificial bypass for complex I essentiality. A point mutation in a conserved charge residue of the Nqo13 (ND4) subunit was then created and confirmed to possess no catalytic activity, demonstrating the versatility and capability of our system.
Results
Insertion of a His6-tag into the complex I operon
To develop a robust protocol for isolating complex I from P. denitrificans we introduced an affinity purification tag onto the C-terminus of the Nqo5 (NDUFS3) subunit. The affinity tag contained six histidine residues attached to the Nqo5 subunit by six alanine linker residues. The same tag, on the homologous subunit, has been used to purify complex I from the yeast model Y. lipolytica17,38,39. Here, we introduced an unmarked insertion of the His6-tag into the chromosomal DNA of P. denitrificans strain Pd1222-∆Hy35 by suicide vector-mediated homologous recombination, an approach previously used to create unmarked deletions in the P. denitrificans genome (see “Materials and methods”)35,40,41. The genetic modification was supported by expression of an alternative NADH dehydrogenase (NDH-2), as discussed in detail below. The correct and specific insertion of the purification tag on nqo5 was confirmed by sequencing, and the sequences of the all known complex I subunits were checked in the final strain. We refer to this strain, which is formally termed Pd1222-∆Hy-Nqo5His6, as Pd-Nqo5His6. Expression of the tagged Nqo5 subunit was subsequently confirmed by Western blot analyses of SBPs following protein separation by SDS-PAGE (SI Fig. S1).
Purification of complex I from P. denitrificans
To purify P. denitrificans complex I, membranes were prepared from P. denitrificans cells grown aerobically on LB medium. The conditions for membrane solubilization with n-dodecyl β-D-maltoside (DDM) and the buffer compositions were based on those of Yip et al., with Bis–Tris replaced by MES and our affinity chromatography buffers supplemented with 0.02% asolectin26,42. Following solubilization, the proteins were loaded onto a Ni-affinity column and complex I eluted using 200 mM imidazole (see "Materials and methods" for details) (Fig. 1a). Complex IV coeluted with complex I, so the eluent was further separated by size exclusion chromatography; only the first peak exhibited complex I flavin site activity (NADH:APAD+ oxidoreduction) and it was thus assigned to complex I (Fig. 1b). The complex I peak eluted at the volume expected for intact Pd-CI, as predicted from the eluted volumes of standard proteins given in the manufacturer's guide (Cytiva). The purified enzyme contained 1.50 ± 0.13 Q10 molecules per complex I (S.E.M., n = 3 samples from three independent cultures and purifications), which is similar to the ubiquinone content measured for the purified E. coli enzyme (1.3 ± 0.1)43. The Pd-CI was analyzed by SDS-PAGE and prominent bands were excised, digested with trypsin and the proteins identified by mass spectrometry analyses (Fig. 1c). All the core complex I subunits were identified, apart from Nqo7 and Nqo11, which are likely missed from the analysis due to their small and hydrophobic nature (SI Table S1). Further to the core subunits, the three previously identified supernumerary subunits (PdNUMM, PdNUYM, PdN7BM) were also detected26. Our mass spectrometry analyses also identified several additional proteins with varying degrees of confidence (SI Table S1). Most of these are likely minor contaminants and unlikely to be associated with complex I, such as elongation factor Tu, transcription termination factor Rho and subunits from other respiratory complexes. A small number of other proteins were identified, notably a protein-L-isoaspartate O-methyltransferase (23.5 kDa), which cannot be either excluded from or assigned to complex I at the current time. Structural studies will be required to determine whether these are impurities or unique subunits associated with the P. denitrificans enzyme.
Figure 1.

Purification and subunit composition of P. denitrificans complex I. (a) Typical Ni-affinity chromatography trace produced using a HisTrap HP column. The column was washed with 80 mM imidazole and protein eluted with 200 mM imidazole. (b) Typical size exclusion chromatography trace using a Superdex 200 increase 5/150 GL column. Complex I was identified by measuring the relative NADH:APAD+ activity in eluted fractions (black trace). (c) SDS-PAGE analysis of purified P. denitrificans complex I. Two full-length lanes are shown from a single gel; the image has been cut and they have been moved to be adjacent to each other. The original image is shown in SI Fig. S6. Individual complex I subunits were identified and assigned by excising each band, treating the sample with trypsin and analyzing the resultant peptides by mass spectrometry. Peptides were assigned to a subunit/protein by peptide mass fingerprinting.
EPR characterization of purified complex I
In order to compare the electron transfer pathways in Pd-CI and its mammalian counterpart, electron paramagnetic resonance (EPR) spectroscopy was performed. Previous EPR studies of Pd-CI have been limited to membrane particles44–46, which make deconvolution of the CI specific signals difficult. EPR spectra of the NADH-reduced purified Pd-CI were measured at different temperatures (Fig. 2a). The spectra clearly show the presence of four reduced FeS clusters (N1b, N2, N3 and N4), confirming the integrity of the hydrophilic domain in the purified enzyme. At 40 K, a single axial FeS cluster corresponding to the slow-relaxing [2Fe-2S] N1b is observed. As the temperature is decreased, additional [4Fe-4S] FeS signals appear in accordance with the established relaxation properties for mammalian complex I (N1b < N2 < N3 < N4)47,48. At 7 K, the FeS signals from N2 and N4 are largely saturated. The fast-relaxing N5 cluster was not observed during these experiments as high power and low temperature are typically required to resolve its signal; however, overexpression of the P. denitrificans Nqo3 (NDUFS1) subunit in E. coli has previously given rise to a signal attributable to N549.
Figure 2.

EPR spectra of NADH-reduced purified complex I from P. denitrificans. (a) Complex I (5.8 mg mL–1) was reduced anaerobically with 15 mM NADH. EPR spectra were recorded at 100 kHz modulation frequency with a modulation amplitude of 7 G and a microwave power of 2.02 mW at the temperatures indicated. Vertical lines correspond to the g factors for the individual FeS clusters. (b) Simulation of the EPR spectrum of purified Pd-CI at 16 K. The total simulation of the combined FeS cluster signals is shown in red, with the individual simulations labelled according to the established nomenclature. FeS clusters N1b-N4 were simulated at a 1:1 ratio. Simulation parameters are given in SI Table S2.
A simulation of the Pd-CI EPR spectrum recorded at 16 K, using an equal stoichiometry of the four observed FeS clusters, is shown (Fig. 2b). This ‘signature’ spectrum for Pd-CI shows remarkable similarity to that observed for mitochondrial complex I from Y. lipolytica, which could also be simulated with an equal stoichiometry for the same four reduced clusters50. In contrast, mammalian complex I exhibits sub-stoichiometric reduction of N1b when NADH is used as a reductant, owing to its low reduction potential48. As in both of these mitochondrial complexes, but unlike in complex I from E. coli, the [2Fe-2S] cluster in Nqo2 (NDUFV2), referred to as N1a, is not reduced in NADH treated Pd-CI. In E. coli complex I, N1a is a high potential cluster that is reduced during turnover whereas N3, which is fully reduced by NADH in both mitochondrial and Pd-CI, appears to be reduced sub-stoichiometrically51,52. The EPR signal for N1a has thus far only been observed in overexpressed Pd-Nqo2 reduced with sodium dithionite, with rhombic geometry and characteristic g values of 2.00, 1.94 and 1.9253,54 that are clearly not present in Fig. 2a. The absence of the reduced N1a signal is consistent with its reduction potential being below that of NADH54,55, and with data on mammalian complex I that show reduction of N1a only in the dithionite-reduced flavoprotein subcomplex, not in the intact enzyme48,55,56. The properties of the [2Fe-2S] N1a cluster and pattern of FeS cluster reduction thus further establish the close similarity between Pd-CI and mitochondrial complex I.
Catalysis by isolated (detergent-solubilized) P. denitrificans complex I
The integrity and catalytic competence of the complex I isolated from P. denitrificans were determined by measuring its catalytic activity. The optimized assay conditions previously described for Pd-CI by Yip et al. were confirmed here26: the activity was highest in buffer at pH 6.5 and in the presence of divalent cations, Mg2+ or Ca2+ (SI Fig. S2). The addition of sucrose (to make the buffer more hypertonic) had no effect on activity. Under optimal assay conditions, our preparations of Pd-CI were able to catalyze the reduction of 200 µM decylubiquinone (DQ) by NADH at typically 21.9 ± 4.2 µmol min–1 mg–1 (~ 204 s–1, S.D. from 11 independent purifications). For comparison, the purified bovine, ovine and Y. lipolytica enzymes have been reported to catalyze the reduction of 200 µM DQ at 22.2–24.7 µmol min–1 mg–1 (~ 390 s–1)57, 5–6 µmol min–1 mg–1 (~ 90 s–1)20 and 13.9 µmol min–1 mg–1 (~ 208 s–1)17, respectively, and the bacterial enzyme from E. coli to catalyze reduction of 100 µM DQ at 20–25 µmol min–1 mg–1 (~ 206 s–1)43,58. The activities of two soluble quinone analogues, Q1 and DQ, were then compared on the same enzyme preparation (Fig. 3a). Pd-CI catalyzed DQ reduction to higher rates than Q1 reduction, however, the high KM values observed for both DQ (169 ± 15 µM) and Q1 (124 ± 8 µM) suggest that neither is a good substrate for the P. denitrificans enzyme. It is worth noting that DQ and Q1 have limited solubility in aqueous solution and so activity data recorded at high substrate concentrations, where they tend to aggregate or form micelles, may not provide an accurate picture. Unless otherwise stated, activities reported here were measured using 200 µM DQ, to match standard data on the bovine enzyme57.
Figure 3.
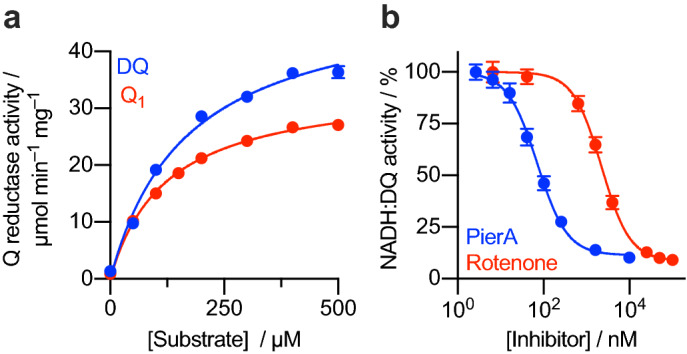
Characterization of purified complex I from P. denitrificans. (a) KM curve for DQ and Q1 substrates for a typical sample of soluble complex I. The KM values for DQ and Q1 are 169 ± 15 and 124 ± 8 µM, respectively (± S.E. of the fit). (b) Piericidin A and rotenone IC50 titration curves for soluble complex I, using DQ as the substrate. The IC50 values of piericidin A and rotenone are 72 ± 7 nM and 2226 ± 185 nM, respectively (± S.E. of the fit).
To estimate the Q-reductase activity of native Pd-CI in the membrane, and thus the highest possible activity we may achieve for the purified enzyme, we compared activities in the membranes with those of the soluble form. The membranes and isolated enzyme were able to catalyze flavin-catalyzed NADH:APAD+ oxidoreduction at 0.497 ± 0.015 µmol min–1 mg–1 and 9.98 ± 0.05 µmol min–1 mg–1, respectively (S.E.M. of three technical replicates). By assuming that the flavin site activity is fully retained during purification, and that the only NADH:APAD+ activity in the membrane is from complex I, the complex I content of the membranes was estimated at 5.0 ± 0.2% (by protein mass). The membranes were able to catalyze NADH:O2 oxidoreduction at 2.02 ± 0.09 µmol min–1 mg–1, which translates to a Q-reductase activity of 40 ± 2 µmol min–1 mg–1, based on the estimated complex I content. Therefore, our rate of DQ reduction suggests we have retained 54 ± 11% of the Q-reductase activity in our preparation – although greater activities may be observed at higher concentrations of DQ, or by using the native Q10 as the electron acceptor. Overall, these data provide clear evidence that our preparation of Pd-CI is highly active for NADH:ubiquinone oxidoreduction.
The sensitivity of the purified enzyme to the canonical complex I inhibitors piericidin A and rotenone was then determined (Fig. 3b). The NADH:DQ activity was inhibited ≥ 90% by piericidin A and rotenone. However, the enzyme was more sensitive to piericidin A (IC50: 72 ± 7 nM) than rotenone (IC50: 2226 ± 185 nM). A previous study has also shown that, unlike the mitochondrial enzyme, Pd-CI is poorly inhibited by rotenone59. Surprisingly, Pd-CI purified by Yip et al. was reported to exhibit similarly high sensitivities to both rotenone (IC50: 170 nM) and piericidin A (IC50: 100 nM)26. These values do not agree with our data or with previous data, despite our similar buffer and assay conditions. The enzyme preparation of Yip et al. was reported to have a lower activity (15.0 µmol min–1 mg–1) than ours (21.9 µmol min–1 mg–1), although it was assayed with a lower concentration of DQ. In addition, we note our preparation time was much shorter (hours vs. days), so it is possible we have maintained greater structural integrity at known rotenone binding sites in the Q-channel and near the transverse helix20.
Finally, we assessed the rate of H2O2 generation by our purified enzyme using the horseradish peroxidase-dependent oxidation of Amplex Red to resorufin (see "Materials and methods"). The Amplex Red system detects the H2O2 formed, either directly or by the dismutation of superoxide, assisted by the presence of SOD. H2O2 was generated at 138.6 ± 1.1 nmol min–1 mg–1 (78 min–1) using 30 µM NADH to activate superoxide/H2O2 production at the flavin site, approximately four times faster than generated by the purified complex I from bovine heart mitochondria (21.1 ± 2.9 nmol min–1 mg–1, 21 min–1)60. However, the different pH values at which the two assays were performed (pH 6.5 vs. pH 7.5) may also influence the rate of H2O2 production60.
Optimization of the reconstitution of Pd-CI into liposomes
To study proton pumping of wild-type and mutated Pd-CI in a controlled minimal membrane environment, the purified enzyme needs to be reconstituted into a compartmentalized membrane system that supports formation of a ∆p. We have established protocols for the assembly of proteoliposome (PL) systems comprising mitochondrial complex I, ubiquinone Q10, and Trypanosoma brucei brucei alternative oxidase (AOX), which re-oxidizes ubiquinol Q10H2 to ubiquinone Q10 and reduces oxygen to water61,62. The system was later expanded to couple ∆p formation by complex I to ATP synthesis by further incorporating E. coli ATP synthase into the proteoliposomes63. AOX is a monotopic membrane protein that associates spontaneously to the liposomal membrane and thus it was added after reconstitution (to the mature proteoliposomes) in all experiments. Here, we adapted a protocol optimized for mammalian CI63 for the efficient incorporation of Pd-CI. As soluble Pd-CI was found to be most active at pH 6.5 (SI Fig. S2a), the pH of the reconstitution buffer was adjusted to pH 6.5 with MES replacing MOPS as the buffering component. Reconstitution of Pd-CI into our standard liposomes that mimic the mitochondrial inner membrane lipid composition (80:10:10 dioleoyl phosphocholine (DOPC):dioleoyl phosphoethanolamine (DOPE):tetraoleoyl cardiolipin (TOCL), in % (w/w)) for the reconstitution of mitochondrial CI61,63,64, resulted in only low activities of 7.08 ± 0.63 μmol NADH min-1 mg-1 (n = 8) compared to NADH:Q10 oxidoreduction activities of up to 40 ± 2 µmol min–1 mg–1 estimated for Pd-CI in membranes. Thus, we amended the lipid mixture to more closely mimic the reported lipid composition of exponentially growing P. denitrificans65, to 52 dioleoyl phosphoglycerol (DOPG):37 DOPC:8 DOPE:3 TOCL. Although this did not affect the activity of reconstituted Pd-CI (SI Fig. S3a) we adopted it for all further experiments. Significantly increased activities were subsequently observed (SI Fig. S3a) when 250 mM sucrose was included in the reconstitution and catalytic assay buffers (see below). Next, the amount of AOX was varied to achieve optimal turnover (SI Fig. S3b). The optimal amount of AOX was found to be 40–80 µg mL–1 AOX per 0.5 µg mL–1 outward facing Pd-CI, which translates to 1200–2400 molecules of AOX added (to the cuvette) per Pd-CI. These optimized conditions were used for all further experiments.
Characterization of Pd-CI proteoliposomes
The reconstitution protocol optimized for Pd-CI activity led to a random orientation of Pd-CI in the membrane, with 46.7 ± 16.8% (n = 9, S.D., nine reconstitutions using Pd-CI from four different preparations) of the NADH:APAD+ oxidoreductase activity on the outside of the proteoliposomes, reflecting around half of the Pd-CI being inserted in an outward facing orientation. As NADH is membrane impermeable, the inward facing CI is catalytically silent in kinetic assays. The average protein retention (relative to the amount added at the start of the preparation) was determined as 36.9 ± 12.7% (n = 10, S.D.). The average NADH:O2 activity (for outward facing Pd-CI) for a standard preparation was 24.5 µmol min–1 mg–1 ± 8.4 µmol min-1 mg-1 (n = 11, S.D.). However, the NADH:O2 activity varied substantially, from, 13.8 to 41.5 µmol min–1 mg–1, with more than 50% of the preparations giving activities between 22.6 and 26.6 µmol min–1 mg–1. Only small respiratory control ratios (RCRs) of 1.2 ± 0.2 (n = 8, S.D.), similar to our observations for bovine CI PLs61, were measured. Comparing the average NADH:O2 activity to our estimate for the Q-reductase activity of native Pd-CI in the membranes, 61 ± 15% of the total activity is retained during purification and reconstitution of Pd-CI.
The KM for the reduction of native ubiquinone Q10 was measured in Pd-CI PLs (Fig. 4a, in which all the data points are from individual reconstitutions and determinations). The KM value of 1.1 ± 0.2 mM for ubiquinone Q10 is within the range (0.48–3.94 mM)61–63 of experimentally determined Q10 KM values for bovine CI, indicating similar affinities for Q10 for both enzymes. The inhibitor titrations for piericidin A and rotenone carried out with the soluble enzyme were repeated in Pd-CI proteoliposomes to investigate how the membrane affects the inhibition (Fig. 4b). The IC50 values for both inhibitors were decreased in PLs from 2226 ± 185 nM to 452 ± 48 nM and from 72 ± 7 nM to 24 ± 3 nM for rotenone and piericidin A, respectively. This is likely due to the smaller hydrophobic phase volume in the PL measurements, which concentrates the inhibitor in the vicinity of the enzyme. However, the same trend of weaker inhibition by rotenone than piericidin A was confirmed. Pd-CI appears to not bind canonical inhibitors of complex I, particularly rotenone, as strongly as the mitochondrial enzymes do. Understanding the fundamental differences in inhibitor binding will require structural work and comparison to known piericidin A and rotenone-bound complex I structures18–20.
Figure 4.

Characterization of Pd-CI in liposomes. (a) KM curve for Q10 in proteoliposomes. The KM and Vmax values were determined to be 1.1 ± 0.2 mM and 27.9 ± 1.2 µmol min–1 (mg CI)–1, respectively (± S.E.M. of the fit). (b) Inhibition by piericidin A and rotenone. The measured IC50 values for piericidin A and rotenone were 24 ± 3 nM and 452 ± 48 nM, respectively (± S.E. of the fit). (c) Proton pumping measured by an ACMA fluorescence quench assay. Proton pumping was initiated by addition of 1 mM NADH and proteoliposomes uncoupled by addition of 25 µg mL–1 alamethicin (AlaM). Liposomes without Pd-CI were added as a control. (d) Coupling Pd-CI catalysis to ATP synthesis. Pd-CI was co-reconstituted in liposomes with E. coli ATP synthase (CI-AOX-F1FO) and NADH-coupled ATP production was measured as luminescence using a luciferase-based assay. No ATP was generated by proteoliposomes containing only ATP synthase (F1FO only) while addition of 2 μM piericidin A (pierA) or 20 μg mL–1 gramicidin A (gramA) inhibited ATP synthesis. Alternative oxidase (AOX) was directly added to the assay mixture at 20 µg mL–1 in panels (a–c) and at 5 µg mL–1 in panel (d). See “Materials and methods” for the assembly of proteoliposomes and other assay conditions.
Next, it was tested whether NADH oxidation is coupled to proton pumping in liposome-reconstituted Pd-CI by using the standard fluorescence quench assay, ACMA (9-amino-6-chloro-2-methoxyacridine) quenching (Fig. 4c)61. Addition of NADH to Pd-CI-AOX PLs led to a rapid quench of the fluorescence signal indicating formation of a ΔpH. Addition of the uncoupler alamethicin dissipated the ΔpH, while empty liposomes did not show any fluorescence quench upon NADH addition. The addition of piericidin A to the liposomes also prevented fluorescence quenching after NADH addition by inhibiting catalysis and proton pumping (SI Fig. S4). In a separate experiment, Pd-CI was co-reconstituted with E. coli ATP synthase into liposomes to test if a substantial ∆p is being produced by the Pd-CI (Fig. 4d). Indeed, addition of NADH to CI-AOX-F1FO PLs coupled NADH oxidation (and ubiquinone reduction) to proton pumping, establishing a ∆p high enough to drive ATP synthesis. No ATP was produced when NADH was added to proteoliposomes containing only E. coli ATP synthase, and ATP synthesis in CI-AOX-F1FO PLs was inhibited by piericidin A, or when the ∆p was collapsed by the uncoupler gramicidin A. Together these results indicate specific reconstitution of intact Pd-CI into sealed liposomes, which couple NADH oxidation to ∆p formation, and demonstrate its proton pumping capability. Therefore, our reconstitution protocol can be applied to aid study of the proton pumping mechanism of mutant and wild-type CI in future experiments.
A genetic system to study complex I variants
Complex I is essential for the survival of P. denitrificans cells, preventing the creation of severely deleterious complex I variants. To overcome this issue, Finel and coworkers inserted the alternative NADH dehydrogenase (NDH-2) from E. coli directly into the P. denitrificans genome to compensate for loss of activity in variants generated in the Nqo8 (ND1) subunit66,67. Here, we introduced ndh2 from E. coli into P. denitrificans on the inducible expression vector pLMB50968. Using this approach, we could control whether or not NDH-2 was expressed by including the inducer, taurine, in the growth medium, allowing us to generate deleterious complex I mutants. The tunability of the system means that we are also able to turn off NDH-2 expression to prevent it interfering with activity measurements on non-deleterious mutants. The complex I specific substrate deaminoNADH (dNADH), which is not oxidized by NDH-2, can be used in those cases where NDH-2 is required for cell growth. To show that expression of NDH-2 allows cells to grow when complex I catalysis is severely compromised, we grew P. denitrificans cells in succinate media and inhibited complex I with piericidin A in both cells expressing or not expressing NDH-2 (Fig. 5a). While the addition of piericidin A severely limited cell growth, the expression of NDH-2 rescued it to near untreated levels: the cells remain viable despite a severely compromised complex I.
Figure 5.

Creation of a catalytically inactive complex I variant. (a) Growth of wild-type P. denitrificans in succinate minimal media40,41. Cells were grown with/without 10 mM taurine to induce NDH-2 expression and with/without 5 µM piericidin A to inhibit complex I and cell growth. (b) Comparison of the complex I flavin site activity (NADH:APAD+) of wild-type and the K232QNqo13 variant measured in three different contexts. Membrane activities were measured using dNADH due to the expression of NDH-2 during cell growth. The specific activities have been normalized to the wild type activities; in membranes (0.527 ± 0.006 µmol min–1 mg–1, S.E.M. n = 3), purified CI (12.13 ± 0.14 µmol min–1 mg–1, S.E.M. n = 4), and proteoliposomes (10.71 ± 0.12 µmol min–1 mg–1, S.E.M. n = 3). (c) Comparison of the quinone reductase activity (Q10 or DQ) of wild-type and the K232QNqo13 variant measured in three different contexts. The specific activities have been normalized to the wild type activities (minus the average piericidin A insensitive rates) in membranes (2.41 ± 0.02 µmol min–1 mg–1, S.E.M. n = 3), purified enzyme (19.73 ± 0.058 µmol min–1 mg–1, S.E.M. n = 3), and proteoliposomes (41.45 ± 0.17 µmol min–1 mg–1, S.E.M. n = 3).
To demonstrate the application of our system, we generated a complex I variant that we expected would abolish catalytic activity. We chose to remove the charge from the conserved charged residue Lys232 in the Nqo13 (ND4) subunit by mutation to a Gln residue (K232QNqo13). Mutation of the equivalent residue in E. coli to Ala rendered complex I inactive69. The mutation was generated using a method similar to that used to insert the affinity purification tag. Crucially, NDH-2 was expressed throughout the growth protocol, essential for successful mutagenesis. The K232QNqo13 mutant was confirmed by sequencing and the cells were only able to grow when expressing NDH-2. Membranes were prepared for the K232QNqo13 mutant from cells expressing NDH-2 and the activities of the membranes were measured using dNADH. While the dNADH:APAD+ activity was similar between the wild-type (i.e. Pd-Nqo5His6) and K232QNqo13, the quinone reductase activity was abolished in the mutant (Fig. 5b,c). We further characterized the mutant by first purifying K232QNqo13 and then reconstituting it into liposomes. The K232QNqo13 mutant purified as normal, showed essentially identical elution profiles to wild-type on the size exclusion column, and a matching band pattern to wild-type in SDS-PAGE analyses (SI Fig. S5). These observations suggest that the structural integrity of the mutant was maintained throughout purification and its loss of activity has a mechanistic basis rather than being due to structural instability. Both the soluble enzyme and the proteoliposomes showed similar relative activities to those measured in membranes (Fig. 5b,c) and consequently proteoliposomes of the K232QNqo13 mutant showed no proton pumping activity as measured by ACMA (SI Fig. S4).
Discussion
In order to answer long-standing questions about the energy coupling and proton translocation mechanisms of respiratory complex I, functional and structural analyses should be combined with targeted mutagenic interrogation of specific residues and protein features. The α-proteobacterium P. denitrificans, a close relative of the protomitochondrion, is thus a prime candidate for a model system that could be exploited to unlock the catalytic mechanism of mitochondrial complex I, as it enables all three approaches. In addition to the similarities Pd-CI possesses with the mitochondrial enzyme that are uncommon in other bacterial models (Q10 as an electron acceptor; higher subunit sequence homology; supernumerary subunits and complex I-containing supercomplexes), it is possible to form well-coupled SBPs as a powerful tool for insightful biophysical and biochemical characterization. In particular, using SBPs, we have already established a method that can be used to determine the number of protons translocated across the membrane by complex I for every NADH oxidized (the proton stoichiometry)35,36. Thus P. denitrificans provides an exciting platform to combine site-directed mutagenesis with biochemical and biophysical measurements essential for gaining mechanistic insight into complex I.
Here, we establish the potential of P. denitrificans as a suitable bacterial model for mitochondrial complex I. First, an affinity purification tag was engineered onto the Nqo5 subunit, allowing reproducible and straightforward purification of intact, catalytically active complex I. The reconstitution of the detergent-solubilized complex into liposomes was then optimized, demonstrating that the complex I retained proton pumping activity. The two protocols allow Pd-CI to be studied in both its soluble form and when reconstituted in a controllable minimal membrane system, providing a complementary perspective to that which can be gained from well-established measurements in SBPs. Second, we created a strain of P. denitrificans that is fully genetically tractable to complex I mutagenesis. As complex I is essential to growth of P. denitrificans, its mutagenesis was otherwise limited by the inability to create catalytically inactive or compromised variants that would kill the cell. By introducing NDH-2 from E. coli into P. denitrificans on a taurine-inducible expression plasmid, we were able to show cell survival and growth when complex I was compromised, by expressing NDH-2. By creating a catalytically inactive complex I variant by a mutation in the Nqo13 subunit we demonstrate the power of our approach, both in being able to generate mutations in the membrane domain, not currently routinely possible in mitochondrial models, and being able to target even essential residues, regardless of their loss of activity.
Despite the many advantages of using P. denitrificans as a model for mitochondrial complex I, some current technical limitations should be noted. Our approach generates unmarked mutations in the P. denitrificans genome which, while beneficial as it ensures complex I variants will be constitutively expressed, is more technically challenging/slower than introducing mutations on a gene expressed on a plasmid, as employed in Y. lipolytica39 or E. coli21,30. Further to this, the high GC content of the P. denitrificans genome requires careful consideration during even basic cloning procedures. Perhaps the most obvious limitation is that there is no P. denitrificans complex I structure yet available, a crucial element that is required to fully realize its potential as a model for combining both functional and structural data with mutagenesis. Although we have demonstrated we can isolate intact and highly active complex I from P. denitrificans, the lack of stabilizing supernumerary subunits in the bacterial enzyme likely decreases its stability compared to its mitochondrial counterparts. This possibility may explain the relative lack of bacterial complex I structures solved by cryo-EM to date. It is known that P. denitrificans complex I dissociates under Blue-Native PAGE conditions27 despite remaining intact during solubilization and in our two-step purification, and the freezing methods used in cryo-EM grid preparation may also be detrimental. A potential alternative route to the P. denitrificans complex I structure may be isolation of the respiratory supercomplex, which is known to stabilize Pd-CI and can be visualized on Blue-Native PAGE27. Although we are confident in expecting the enzyme to have a similar overall architecture to current models, and the positions of key conserved residues to be in similar configurations, pursuit of the P. denitrificans complex I structure will be both beneficial and complementary to the work described here.
As described above, complex I from P. denitrificans possesses many similarities to the mitochondrial enzyme, making it an attractive model system. However, there are also fundamental differences between the two that can be considered both beneficial and disadvantageous. An important property of mitochondrial complex I is that it can undergo an active/deactive (A/D) transition13,57,70 in which the deactive state cannot catalyze quinone reduction and the enzyme is considered to be in an off-pathway resting state. The deactivate state is identified biochemically by the binding of N-ethylmaleimide (NEM) to a conserved cysteine residue in Nqo7 (ND3) that is only exposed in the deactive state, locking it in this state. P. denitrificans has not been observed to undergo an A/D transition46,70 and here we also tested whether Pd-CI could be deactivated in membranes by assaying the membrane activity in control and heat-treated membranes (conditions which deactivate the mouse and bovine enzymes13,57) with and without NEM. We observed no differences in activity to suggest the enzyme is able to form an (NEM-sensitive) deactive state (SI Table S3). Hence, Pd-CI cannot be used as a model to assess or investigate the A/D transition that occurs in mitochondria, which may be a unique feature of the mitochondrial enzyme. However, Pd-CI can instead be considered a model for the ‘active’ mitochondrial enzyme, which is an advantage for studies of the mechanism of catalysis as the effects of mutations in catalytically relevant residues can be deconvoluted from possible effects on the A/D transition in the mitochondrial enzyme. In this regard, as well as in many others, P. denitrificans is a powerful system for probing the mechanism of complex I catalysis.
Materials and methods
Chemicals
All chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Creation of bacterial strains
Insertion of NDH-2 into P. denitrificans
The NDH-2-encoding gene from E. coli (ndh2) was optimized for the codon usage in P. denitrificans and the construct synthesized by GENEWIZ®. Two restriction sites were added to the 5ʹ (NsiI) and 3ʹ (SacI) ends of the construct, which was then digested and inserted into the expression vector pLMB509 kindly provided by Dr Andrew Gates (University of East Anglia)68. The expression vector used here was a modified version that has had the GFP marker removed. The His6-tag present in the vector was removed during the ndh2 insertion. The vector was transformed into the MFDpir E. coli donor strain and subsequently conjugated into the Pd1222-∆Hy parental strain35 of P. denitrificans at a 10:1 donor to recipient ratio. After two days of incubation at 30 °C on LB plates containing 0.5 mM diaminopimelic acid, successful conjugation was selected for by plating serial dilutions onto LB agar containing rifampicin (50 µg mL–1) and gentamicin (20 µg mL–1).
Generation of the affinity-tagged complex I strain
The location and design for the affinity tag replicated the purification tag previously engineered onto Y. lipolytica complex I38,39. The affinity tag was designed to contain six histidine residues attached to the C-terminus of the Nqo5 subunit by six alanine linker residues. The insertion of the affinity tag onto the C-terminus of the Nqo5 subunit of P. denitrificans complex I was achieved using a similar strategy as described previously for creating deletions in the P. denitrificans genome35,40,41. Insertion cassettes were designed containing two sequences homologous to regions on either side of nqo5 (Pden 2248, nt 2,250,063–689). The first homologous flanking region of 1292 bp began at nucleotide position 2,248,772 and extended to position 2,250,065 to include the STOP codon of the nqo5 gene. The sequence for the His6Ala6-tag (5ʹ-GTG ATG GTG ATG ATG ATG CGC GGC TGC CGC GGC GGC-3ʹ) was then inserted to become the new C-terminal sequence of nqo5. The next flanking region contained the entire nqo5 gene (minus the STOP codon) and extended for a further 1071 bp to nucleotide position 2,251,760. This second flanking region was followed by a kanamycin (kanR) selection marker of 815 bp length. EcoRI restriction sites were added to the end of the construct and any EcoRI sites within the construct itself were removed by silent mutagenesis. The construct was assembled by GENEWIZ® and then inserted into the lacZ-containing pRVS1 suicide plasmid via an EcoRI restriction site. The plasmid was transformed into the MFDpir E. coli donor strain and conjugated into the Pd1222-∆Hy parental strain containing the ndh2 gene on pLMB509. NDH-2 was expressed throughout conjugation and homologous recombination by including taurine (10 mM) in all media. Successful conjugation and first recombination events were selected by using LB-agar plates containing 50 µg mL–1 kanamycin. Successful colonies were then plated on X-gal (200 µg mL–1), and white colonies were selected as positive for the second recombination event, and screened by PCR amplification of the DNA sequence across the nqo5 gene to confirm the expected insertion of base pairs. The integrity of the strain was determined by full sequencing of all complex I subunits. Here, we refer to the His6-tag strain as Pd-Nqo5His6, or as the ‘wild-type’ strain in comparisons with the K232QNqo13 variant.
Generation of the K232QNqo13 complex I variant
The K232QNqo13 complex I variant was created in our Pd-Nqo5His6 strain that also contained the ndh2 gene on pLMB509. The same procedure as above was followed to generate the mutation, ensuring NDH-2 was expressed continuously throughout the protocol by the inclusion of 10 mM taurine in all media. The point mutation cassettes were designed to include homologous flanking regions 1000 bp upstream and downstream of nqo13 (Pden_2232). The kanamycin (kanR) selection marker followed the second flanking region. The lysine residue in Nqo13 at amino acid position 232 was mutated to a glutamine (AAG to CAG). The creation of the mutant was confirmed by sequencing.
Bacterial growth and membrane preparations
Succinate media growth curves
Colonies from the Pd-Nqo5His6 strain were picked and grown for 24 h in 40 mL succinate minimal media with/without 10 mM taurine (30 °C, 225 rpm); the culture was then used to inoculate 200 µL aliquots of succinate minimal medium in a 96-well plate to a starting optical density of 0.02 (at 600 nm). Media were supplemented with/without 10 mM taurine and with/without 5 µM piericidin A and each experiment was carried out in quadruplicate. Plates were incubated at 30 °C with 200 rpm orbital shaking in a CLARIOstar Plus microplate reader (BMG Labtech) and the optical density was recorded every 30 min. The minimal medium used was adjusted to pH 7.2 and contained 50 mM succinate, 9.35 mM NH4Cl, 2 mM MgSO4, 0.07 mM CaCl2, 0.29 mM KH2PO4, 0.69 mM K2HPO4, 25.2 mM Na-Hepes, 19.6 μM Na2-EDTA, 9 μM FeSO4, 0.1 μM MnCl2, 0.8 μM CuCl2, 1 μM Na2MoO4 and 2.5 μM ZnCl240,41.
Membrane preparations
Colonies from the Pd-Nqo5His6 strain were picked and grown for 24 h in 50 mL LB (30 °C, 225 rpm) then 12 × 500 mL flasks of LB medium were each inoculated with 500 µL of preculture. Flasks were grown for 16–20 h (30 °C, 225 rpm) and harvested at late log phase when the OD600 was 3.5–4.5. Cells were collected by centrifugation in a Sorvall RC 12BP centrifuge and resuspended in 2–2.5 mL of buffer per gram of cells. The resuspension buffer consisted of 50 mM MES pH 6.5 at 4 °C, 0.002% (w/v) phenylmethanesulfonyl fluoride (PMSF) and one cOmplete™ EDTA-free protease inhibitor cocktail (Roche) per 50 mL. After homogenizing the cells, the resuspension was passed through a Z-plus 2.2 kW cell disruptor (Constant Systems Limited) once at 15,000 and twice at 30,000 psi. Cell debris was removed by centrifugation at 31,900×g in an SLA-1500 rotor (Sorvall) for 1 h and the membrane fraction was collected by ultracentrifugation at 234,800×g in a Ti45 rotor (Beckman) for 2 h. Membranes were suspended in 50 mM MES pH 6.5 at 4 °C and flash frozen in liquid N2.
Enzyme purifications
P. denitrificans membranes (200–600 mg) were diluted to a concentration of 9.5 mg mL–1 in buffer with a composition of 20 mM MES pH 6.5 at 4 °C, 100 mM NaCl, 5 mM CaCl2, 10% glycerol, 0.002% PMSF, and the cOmplete™ EDTA-free protease inhibitor cocktail. Membranes were solubilized for 30 min at a 3:1 detergent to protein ratio by addition of 2.85% DDM (Anatrace) to membranes continuously stirred at 4 °C. Non-solubilized material was removed by centrifugation (172,000×g, 45 min). Imidazole was added to the supernatant to a final concentration of 20 mM, then the supernatant was filtered through a 0.45 µm filter. The supernatant was loaded onto either a 2 or 5 mL HisTrap HP column (Cytiva) and washed with buffer A (20 mM MES pH 6.5 at 4 °C, 400 mM NaCl, 5 mM CaCl2, 10% glycerol, 0.1% DDM, 40–80 mM imidazole) before complex I was eluted with buffer B (buffer A plus 200 mM imidazole). Fractions from the single eluted peak were combined, concentrated to 1 mL, and loaded onto either a Superdex 200 increase 10/300 GL or Superdex 200 5/150 GL column (Cytiva) equilibrated in buffer C (20 mM MES pH 6.5 at 4 °C, 150 mM NaCl, 10 mM CaCl2, 10% glycerol, 0.05% DDM). Fractions from the first peak corresponding to complex I were collected and concentrated to 10–20 mg mL–1 before glycerol was added at 20% and the protein flash frozen in liquid N2.
ATP synthase of E. coli71 and AOX of Trypanosoma brucei brucei61 were purified as described previously.
Membrane and soluble Pd-CI kinetic assays
All kinetic assays were carried out at 32 °C in a Molecular Devices SpectraMax 348 96-well plate reader in buffer containing 10 mM MES (pH 6.5 at 32 °C), 25 mM NaCl and 2 mM CaCl2. NADH:O2 oxidoreduction was measured at 340–380 nm (ε = 4.81 mM–1 cm–1) and NADH:APAD+ (3-acetylpyridine adenine dinucleotide) oxidoreduction was measured at 400–450 nm (ε = 3.16 mM–1 cm–1). For NADH:O2 measurements, typically 5–10 µg mL–1 of membranes were assayed with 200 µM NADH and 12.5 µg mL–1 alamethicin. Complex I was inhibited with 5 µM piericidin A. Detergent-solubilized complex I (0.5 µg mL–1) was typically assayed for NADH:DQ activity in buffer containing 200 µM NADH, 200 µM DQ, 0.15% asolectin, 0.15% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate). The NADH:APAD+ activity of membranes and Pd-CI were assayed in buffer containing 100 µM NADH, 500 µM APAD+ and 1 µM piericidin A. For membranes, 12.5 µg mL–1 alamethicin was also added to the buffer and for Pd-CI, 0.15% asolectin and 0.15% CHAPS were included. dNADH was used for comparison of membrane activities for the wild-type and NDH-2 containing variants. H2O2 production from Pd-CI was measured at 557–620 nm (ε = 51.6 mM–1 cm–1) using the horseradish peroxidase dependent oxidation of Amplex red to resorufin72. The reaction contained 30 µM NADH, 2 units mL–1 horseradish peroxidase, 10 µM Amplex red and 10 units mL–1 superoxide dismutase (SOD).
Reconstitution of complex I into liposomes
For a standard reconstitution, 5 mg of synthetic lipids (all from Avanti Polar Lipids as 25 mg mL−1 stocks in chloroform) at a ratio of 52:37:8:3 DOPG:DOPC:DOPE:TOCL (% (w/w)), mimicking the natural lipid composition of P. denitrificans membranes65, and 50 nmol ubiquinone Q10 (from a 6.8 mM stock in chloroform) were mixed in a 25 mL round bottomed flask. For optimization of the reconstitution protocol, the total and relative amounts of lipids were varied as described in the figure legends. The chloroform was removed by rotating the flask under a nitrogen stream, then further for at least 1 h under vacuum in a desiccator. Liposomes were formed by the addition of 1 mL reconstitution buffer (10 mM MES, pH 6.5, 50 mM KCl, 250 mM sucrose), to achieve 5 mg lipid mL−1, and by vigorous mixing (Vortex Genie 2, Scientific Instruments) followed by 11 extrusions through a 100 nm Nucleopore polycarbonate membrane (Whatman). For reconstitution of Pd-CI, 200 µL liposomes (5 mg lipid mL−1) were partially solubilized by addition of 6 μL 20% (w/v) sodium cholate (0.5% final concentration) and the solution was incubated for 10 min on ice after mixing briefly by inversion. Then, 100 µg purified Pd-CI (13.4 mg mL−1) (and 50 µg E. coli ATP synthase for co-reconstitutions) and reconstitution buffer were added to yield a final volume of 250 μL. The solution was mixed by inversion, incubated on ice for 15 min, and cholate was removed using a PD10 desalting column (Cytiva) at 4 °C. PLs were collected by centrifugation at 100,000×g (4 °C for 45 min), resuspended in 100 μL ice-cold reconstitution buffer, and stored on ice until required.
Characterization and kinetic assays of proteoliposomes
The amount of Pd-CI in PLs was calculated using a flavin-site NADH:APAD+ oxidoreduction assay73. The specific NADH:APAD+ activity of purified Pd-CI was measured in reconstitution buffer containing 1 µg mL–1 Pd-CI, 100 µM NADH, 500 µM APAD+, 1 µM piericidin A and 0.2% (w/v) DDM. To calculate the amount of outward facing Pd-CI in PLs, the specific activity of purified Pd-CI was compared to the activity of PLs in reconstitution buffer containing 100 µM NADH, 500 µM APAD+ and 1 µM piericidin A. Orientation and retention of Pd-CI were determined by adding 20 µg mL–1 alamethicin61 or 0.2% DDM, respectively, to the assay mixture. The total phospholipid and Q10 contents were determined as described previously61,63. Rates of NADH:O2 oxidoreduction were measured with 0.5 μg outward-facing CI mL−1 in the reconstitution buffer containing 200 μM NADH and 20 μg mL−1 AOX. For determination of the respiratory control ratio, an additional 10 µg mL-1 gramicidin A were used in the assay buffer61. Fluorescence quench assays were conducted in a Shimadzu RF-5301 PC spectrofluorometer as described previously63 using reconstitution buffer containing 1 μg outward facing CI mL−1, 20 μg AOX mL−1, 500 nM ACMA and 5 µM valinomycin. Fluorescence quenches were initiated by addition of 1 mM NADH and the ΔpH formed was released by the addition of 25 μg mL−1 alamethicin. ATP synthesis was measured at ambient temperature for 1 μg outward-facing CI mL−1 as described63.
EPR spectroscopy
EPR samples were prepared anaerobically by transferring 100 µL of purified complex I (5.8 mg mL–1) to a 4 mm (O.D.) EPR tube (Wilmad), then the complex I was reduced by the addition of 15 mM NADH. The sample was immediately frozen in dry-ice/acetone before transfer to liquid nitrogen for storage. EPR measurements were performed using an X/Q-band Bruker Elexsys E580 Spectrometer (Bruker BioSpin) operating in X-band mode and equipped with a closed-cycle cryostat (Cryogenic Ltd) and X-band split-ring resonator module (ER 4118X-MD5). EPR measurement conditions were 100 kHz modulation frequency, 7 G modulation amplitude, 2 mW microwave power; other relevant measurement conditions are given in the figure legend. All spectra presented are baseline corrected using a buffer-only sample. EPR simulations were performed using the EasySpin package for MATLAB74.
SDS-PAGE and mass spectrometry
Purified complex I was incubated in loading buffer (0.125 M Tris–HCl (pH 6.8), 20% (w/v) glycerol, 4% (w/v) SDS, 0.005% (w/v) bromophenol blue and 0.1 M DTT) for 10 min at room temperature and 10 µg loaded on a Novex WedgeWell 10–20% tris–glycine gel. The gel was run as described previously and bands visualized using Coomassie R25075. Bands excised from SDS-PAGE gels were digested with trypsin and analyzed by matrix-assisted laser-desorption ionization (MALDI) using an Applied Biosystems spectrometer. Spectra were assigned to peptide sequences and their originating proteins using the Mascot 2.4 application (Matrix Science Ltd.) with a peptide precursor mass tolerance of 360 ppm and fragment mass tolerance of 0.8 Da. One missed cleavage, plus methionine oxidation and cysteine propionamide formation as variable modifications were allowed.
Supplementary Information
Acknowledgements
We thank Ian Fearnley and Michael Harbour (MBU) for carrying out mass spectrometry analyses. EPR measurements were conducted at Queen Mary University of London, and Katherine Richardson and Dr. Guy Hanke are gratefully acknowledged for providing access to the EPR spectrometer. We thank Dr. Andrew Gates (University of East Anglia) for providing us with the pLMB509 plasmid.
Author contributions
O.D.J. designed, created and characterized all P. denitrificans strains and variants. O.D.J. optimized the purification of complex I and carried out its kinetic characterization and the full characterization of the K232QNqo13 mutant. J.J.W. performed EPR experiments and analysis. O.B. optimized and characterized the reconstitution of complex I into liposomes with help from O.D.J.. O.D.J. and J.H. wrote the manuscript with contributions from all authors.
Funding
This work was supported by the Medical Research Council (MC_UU_00015/2 to J.H.) and by the Swiss National Science Foundation (P400PB_191096 to O.B.).
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files) and/or are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-89575-9.
References
- 1.Hirst J. Mitochondrial complex I. Annu. Rev. Biochem. 2013;82:551–575. doi: 10.1146/annurev-biochem-070511-103700. [DOI] [PubMed] [Google Scholar]
- 2.Kaila VRI. Long-range proton-coupled electron transfer in biological energy conversion: Towards mechanistic understanding of respiratory complex I. J. R. Soc. Interface. 2018;15:20170916. doi: 10.1098/rsif.2017.0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parey K, Wirth C, Vonck J, Zickermann V. Respiratory complex I: Structure, mechanism and evolution. Curr. Opin. Struct. Biol. 2020;63:1–9. doi: 10.1016/j.sbi.2020.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Murphy MP. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chouchani ET, et al. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016;23:254–263. doi: 10.1016/j.cmet.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Fassone E, Rahman S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012;49:578–590. doi: 10.1136/jmedgenet-2012-101159. [DOI] [PubMed] [Google Scholar]
- 8.Fiedorczuk K, Sazanov LA. Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. 2018;28:835–867. doi: 10.1016/j.tcb.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–445. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- 10.Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu J, Vinothkumar KR, Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536:354–358. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stroud DA, et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature. 2016;538:123–126. doi: 10.1038/nature19754. [DOI] [PubMed] [Google Scholar]
- 13.Agip A-NA, et al. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018;25:548–556. doi: 10.1038/s41594-018-0073-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parey K, et al. High-resolution cryo-EM structures of respiratory complex I: Mechanism, assembly, and disease. Sci. Adv. 2019;5:eaax9484. doi: 10.1126/sciadv.aax9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soufari H, Parrot C, Kuhn L, Waltz F, Hashem Y. Specific features and assembly of the plant mitochondrial complex I revealed by cryo-EM. Nat. Commun. 2020;11:5195. doi: 10.1038/s41467-020-18814-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grba DN, Hirst J. High-resolution cryo-EM structure of mitochondrial complex I reveals ordered water molecules for catalysis and proton translocation. Nat. Struct. Mol. Biol. 2020;27:892–900. doi: 10.1038/s41594-020-0473-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parey K, et al. Cryo-EM structure of respiratory complex I at work. Elife. 2018;7:e39213. doi: 10.7554/eLife.39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutiérrez-fernández J, et al. Key role of quinone in the mechanism of respiratory complex I. Nat. Commun. 2020;11:4135. doi: 10.1038/s41467-020-17957-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bridges HR, et al. Structure of inhibitor-bound mammalian complex I. Nat. Commun. 2020;11:5261. doi: 10.1038/s41467-020-18950-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kampjut D, Sazanov LA. The coupling mechanism of mammalian respiratory complex I. Science. 2020;370:eabc4209. doi: 10.1126/science.abc4209. [DOI] [PubMed] [Google Scholar]
- 21.Mühlbauer ME, et al. Water-gated proton transfer dynamics in respiratory complex I. J. Am. Chem. Soc. 2020;142:13718–13728. doi: 10.1021/jacs.0c02789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Röpke M, Saura P, Riepl D, Pöverlein MC, Kaila VRI. Functional water wires catalyze long-range proton pumping in the mammalian respiratory complex I. J. Am. Chem. Soc. 2020;142:21758–21766. doi: 10.1021/jacs.0c09209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John P, Whatley FR. Paracoccus denitrificans and the evolutionary origin of the mitochondrion. Nature. 1975;254:495–498. doi: 10.1038/254495a0. [DOI] [PubMed] [Google Scholar]
- 24.John P, Whatley FR. The bioenergetics of Paracoccus denitrificans. Biochem. Biophys. Acta. 1977;463:129–153. doi: 10.1016/0304-4173(77)90006-4. [DOI] [PubMed] [Google Scholar]
- 25.Castro PJ, Silva AF, Marreiros BC, Batista AP, Pereira MM. Respiratory complex I: A dual relation with H+ and Na+? Biochim. Biophys. Acta. 2016;1857:928–937. doi: 10.1016/j.bbabio.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Yip C-Y, Harbour ME, Jayawardena K, Fearnley IM, Sazanov LA. Evolution of respiratory complex I ‘supernumerary’ subunits are present in the α-proteobacterial enzyme. J. Biol. Chem. 2011;286:5023–5033. doi: 10.1074/jbc.M110.194993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stroh A, et al. Assembly of respiratory complexes I, III, and IV into NADH oxidase supercomplex stabilizes complex I in Paracoccus denitrificans. J. Biol. Chem. 2004;279:5000–5007. doi: 10.1074/jbc.M309505200. [DOI] [PubMed] [Google Scholar]
- 28.Trumpower BL. The three-subunit cytochrome bc1 complex of Paracoccus denitrificans. Its physiological function, structure, and mechanism of electron transfer and energy transduction. J. Bioenerg. Biomembr. 1991;23:241–255. doi: 10.1007/BF00762220. [DOI] [PubMed] [Google Scholar]
- 29.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 30.Michel J, DeLeon-Rangel J, Zhu S, Van Ree K, Vik SB. Mutagenesis of the L, M, and N subunits of complex I from Escherichia coli indicates a common role in function. PLoS ONE. 2011;6:e17420. doi: 10.1371/journal.pone.0017420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Euro L, Belevich G, Verkhovsky MI, Wikström M, Verkhovskaya M. Conserved lysine residues of the membrane subunit NuoM are involved in energy conversion by the proton-pumping NADH:ubiquinone oxidoreductase (complex I) Biochim. Biophys. Acta. 2008;1777:1166–1172. doi: 10.1016/j.bbabio.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Steimle S, et al. Asp563 of the horizontal helix of subunit NuoL is involved in proton translocation by the respiratory complex I. FEBS Lett. 2012;586:699–704. doi: 10.1016/j.febslet.2012.01.056. [DOI] [PubMed] [Google Scholar]
- 33.Sato M, Torres-Bacete J, Sinha PK, Matsuno-Yagi A, Yagi T. Essential regions in the membrane domain of bacterial complex I (NDH-1): The machinery for proton translocation. J. Bioenerg. Biomembr. 2014;46:279–287. doi: 10.1007/s10863-014-9558-8. [DOI] [PubMed] [Google Scholar]
- 34.Anraku Y, Gennis RB. The aerobic respiratory chain of Escherichia coli. Trends Biochem. Sci. 1987;12:262–266. doi: 10.1016/0968-0004(87)90131-9. [DOI] [Google Scholar]
- 35.Jones AJY, Blaza JN, Varghese F, Hirst J. Respiratory complex I in Bos taurus and Paracoccus denitrificans pumps four protons across the membrane for every NADH oxidized. J. Biol. Chem. 2017;292:4987–4995. doi: 10.1074/jbc.M116.771899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fedor JG, Hirst J. Mitochondrial supercomplexes do not enhance catalysis by quinone channeling. Cell Metab. 2018;28:525–531.e4. doi: 10.1016/j.cmet.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bogachev AV, Murtazina RA, Skulachev VP. H+/e– stoichiometry for NADH dehydrogenase I and dimethyl sulfoxide reductase in anaerobically grown Escherichia coli cells. J. Bacteriol. 1996;178:6233–6237. doi: 10.1128/JB.178.21.6233-6237.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kashani-Poor N, Kerscher S, Zickermann V, Brandt U. Efficient large scale purification of his-tagged proton translocating NADH:ubiquinone oxidoreductase (complex I) from the strictly aerobic yeast Yarrowia lipolytica. Biochim. Biophys. Acta. 2001;1504:363–370. doi: 10.1016/S0005-2728(00)00266-8. [DOI] [PubMed] [Google Scholar]
- 39.Kerscher S, Dröse S, Zwicker K, Zickermann V, Brandt U. Yarrowia lipolytica, a yeast genetic system to study mitochondrial complex I. Biochim. Biophys. Acta. 2002;1555:83–91. doi: 10.1016/S0005-2728(02)00259-1. [DOI] [PubMed] [Google Scholar]
- 40.Varghese F, Blaza JN, Jones AJY, Jarman OD, Hirst J. Deleting the IF1-like ζ subunit from Paracoccus denitrificans ATP synthase is not sufficient to activate ATP hydrolysis. Open Biol. 2018;8:170206–170215. doi: 10.1098/rsob.170206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarman OD, Biner O, Hirst J. Regulation of ATP hydrolysis by the ε subunit, ζ subunit and Mg-ADP in the ATP synthase of Paracoccus denitrificans. Biochim. Biophys. Acta. 2021;1862:148355. doi: 10.1016/j.bbabio.2020.148355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharpley MS, Shannon RJ, Draghi F, Hirst J. Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondrial. Biochemistry. 2006;45:241–248. doi: 10.1021/bi051809x. [DOI] [PubMed] [Google Scholar]
- 43.Verkhovsky M, Bloch DA, Verkhovskaya M. Tightly-bound ubiquinone in the Escherichia coli respiratory complex I. Biochim. Biophys. Acta. 2012;1817:1550–1556. doi: 10.1016/j.bbabio.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 44.Albracht SPJ, Van Verseveld HW, Hagen WR, Kalkman ML. A comparison of the respiratory chain in particles from Paracoccus denitrificans and bovine heart mitochondria by EPR spectroscopy. Biochem. Biophys. Acta. 1980;593:173–186. doi: 10.1016/0005-2728(80)90055-9. [DOI] [PubMed] [Google Scholar]
- 45.Meinhardt SW, Kula T, Yagi T, Lillich T, Ohnishi T. EPR characterization of the iron-sulfur clusters in the NADH:ubiquinone oxidoreductase segment of the respiratory chain in Paracoccus denitrificans. J. Biol. Chem. 1987;262:9147–9153. doi: 10.1016/S0021-9258(18)48060-X. [DOI] [PubMed] [Google Scholar]
- 46.Kotlyar AB, Albracht SPJ, van Spanning RJM. Comparison of energization of complex I in membrane particles from Paracoccus denitrificans and bovine heart mitochondria. J. Bioenerg. Biomembr. 1998;1365:53–59. doi: 10.1016/s0005-2728(98)00042-5. [DOI] [PubMed] [Google Scholar]
- 47.Ohnishi T. Iron-sulfer clusters/semiquinones in complex I. Biochem. Biophys. Acta. 1998;1364:186–206. doi: 10.1016/s0005-2728(98)00027-9. [DOI] [PubMed] [Google Scholar]
- 48.Reda T, Barker CD, Hirst J. Reduction of the iron-sulfur clusters in mitochondrial NADH:ubiquinone oxidoreductase (complex I) by EuII-DTPA, a very low potential reductant. Biochemistry. 2008;47:8885–8893. doi: 10.1021/bi800437g. [DOI] [PubMed] [Google Scholar]
- 49.Yano T, et al. Characterization of cluster N5 as a fast-relaxing [4Fe-4S] cluster in the Nqo3 subunit of the proton-translocating NADH-ubiquinone oxidoreductase from Paracoccus denitrificans. J. Biol. Chem. 2003;278:15514–15522. doi: 10.1074/jbc.M212275200. [DOI] [PubMed] [Google Scholar]
- 50.Djafarzadeh R, et al. Biophysical and structural characterization of proton-translocating NADH-dehydrogenase (complex I) from the strictly aerobic yeast Yarrowia lipolytica. Biochim. Biophys. Acta. 2000;1459:230–238. doi: 10.1016/S0005-2728(00)00154-7. [DOI] [PubMed] [Google Scholar]
- 51.Verkhovskaya ML, Belevich N, Euro L, Wikström M, Verkhovsky MI. Real-time electron transfer in respiratory complex I. Proc. Natl Acad. Sci. USA. 2008;105:3763–3767. doi: 10.1073/pnas.0711249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Vries S, Dörner K, Strampraad MJF, Friedrich T. Electron tunneling rates in respiratory complex I are tuned for efficient energy conversion. Angew. Chem. Int. Ed. 2015;54:2844–2848. doi: 10.1002/anie.201410967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crouse BR, Yano T, Finnegan MG, Yagi T, Johnson MK. Properties of the iron-sulfur center in the 25-kilodalton subunit of the proton-translocating NADH-quinone oxidoreductase of Paracoccus denitrificans. J. Biol. Chem. 1994;269:21030–21036. doi: 10.1016/S0021-9258(17)31925-7. [DOI] [PubMed] [Google Scholar]
- 54.Zu Y, Di Bernardo S, Yagi T, Hirst J. Redox properties of the [2Fe-2S] center in the 24 kDa (NQO2) subunit of NADH: Ubiquinone oxidoreductase (complex I) Biochemistry. 2002;41:10056–10069. doi: 10.1021/bi026026f. [DOI] [PubMed] [Google Scholar]
- 55.Birrell JA, Morina K, Bridges HR, Friedrich T, Hirst J. Investigating the function of [2Fe–2S] cluster N1a, the off-pathway cluster in complex I, by manipulating its reduction potential. Biochem. J. 2013;456:139–146. doi: 10.1042/BJ20130606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barker CD, Reda T, Hirst J. The flavoprotein subcomplex of complex I (NADH:ubiquinone oxidoreductase) from bovine heart mitochondria: Insights into the mechanisms of NADH oxidation and NAD+ reduction from protein film voltammetry. Biochemistry. 2007;46:3454–3464. doi: 10.1021/bi061988y. [DOI] [PubMed] [Google Scholar]
- 57.Blaza JN, Vinothkumar KR, Hirst J. Structure of the deactive state of mammalian respiratory complex I. Structure. 2018;26:312–319.e3. doi: 10.1016/j.str.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sazanov LA, Carroll J, Holt P, Toime L, Fearnley IM. A role for native lipids in the stabilization and two-dimensional crystallization of the Escherichia coli NADH-ubiquinone oxidoreductase (complex I) J. Biol. Chem. 2003;278:19483–19491. doi: 10.1074/jbc.M208959200. [DOI] [PubMed] [Google Scholar]
- 59.Kotlyar AB, Borovok N. NADH oxidation and NAD+ reduction catalysed by tightly coupled inside-out vesicles from Paracoccus denitrificans. Eur. J. Biochem. 2002;269:4020–4024. doi: 10.1046/j.1432-1033.2002.03091.x. [DOI] [PubMed] [Google Scholar]
- 60.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl Acad. Sci. USA. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones AJY, et al. A self-assembled respiratory chain that catalyzes NADH oxidation by ubiquinone-10 cycling between complex I and the alternative oxidase. Angew. Chem. Int. Ed. 2016;55:728–731. doi: 10.1002/anie.201507332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fedor JG, Jones AJY, Di Luca A, Kaila VRI, Hirst J. Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl Acad. Sci. USA. 2017;114:12737–12742. doi: 10.1073/pnas.1714074114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Biner O, Fedor JG, Yin Z, Hirst J. Bottom-up construction of a minimal system for cellular respiration and energy regeneration. ACS Synth. Biol. 2020;9:1450–1459. doi: 10.1021/acssynbio.0c00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horvath SE, Daum G. Lipids of mitochondria. Prog. Lipid Res. 2013;52:590–614. doi: 10.1016/j.plipres.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 65.Wilkinson BJ, Morman MR, White DC. Phospholipid composition and metabolism of Micrococcus denitrificans. J. Bacteriol. 1972;112:1288–1294. doi: 10.1128/JB.112.3.1288-1294.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Finel M. Genetic inactivation of the H+-translocating NADH:ubiquinone oxidoreductase of Paracoccus denitrificans is facilitated by insertion of the ndh gene from Escherichia coli. FEBS Lett. 1996;393:81–85. doi: 10.1016/0014-5793(96)00831-9. [DOI] [PubMed] [Google Scholar]
- 67.Zickermann V, Barquera B, Wikström M, Finel M. Analysis of the pathogenic human mitochondrial mutation ND1/3460, and mutations of strictly conserved residues in its vicinity, using the bacterium Paracoccus denitrificans. Biochemistry. 1998;37:11792–11796. doi: 10.1021/bi9810555. [DOI] [PubMed] [Google Scholar]
- 68.Tett AJ, Rudder SJ, Bourdès A, Karunakaran R, Poole PS. Regulatable vectors for environmental gene expression in Alphaproteobacteria. Appl. Environ. Microbiol. 2012;78:7137–7140. doi: 10.1128/AEM.01188-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torres-Bacete J, Nakamaru-Ogiso E, Matsuno-Yagi A, Yagi T. Characterization of the NuoM (ND4) subunit in Escherichia coli NDH-1: Conserved charged residues essential for energy-coupled activities. J. Biol. Chem. 2007;282:36914–36922. doi: 10.1074/jbc.M707855200. [DOI] [PubMed] [Google Scholar]
- 70.Grivennikova VG, Serebryanaya DV, Isakova EP, Belozerskaya TA, Vinogradov AD. The transition between active and de-activated forms of NADH:ubiquinone oxidoreductase (complex I) in the mitochondrial membrane of Neurospora crassa. Biochem. J. 2003;369:619–626. doi: 10.1042/bj20021165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishmukhametov RR, Galkin MA, Vik SB. Ultrafast purification and reconstitution of His-tagged cysteine-less Escherichia coli F1FO ATP synthase. Biochim. Biophys. Acta. 2005;1706:110–116. doi: 10.1016/j.bbabio.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 72.Pryde KR, Hirst J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011;286:18056–18065. doi: 10.1074/jbc.M110.186841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Birrell JA, Yakovlev G, Hirst J. Reactions of the flavin mononucleotide in complex I: A combined mechanism describes NADH oxidation coupled to the reduction of APAD+, ferricyanide, or molecular oxygen. Biochemistry. 2009;48:12005–12013. doi: 10.1021/bi901706w. [DOI] [PubMed] [Google Scholar]
- 74.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 75.Bridges HR, Mohammed K, Harbour ME, Hirst J. Subunit NDUFV3 is present in two distinct isoforms in mammalian complex I. Biochem. Biophys. Acta. 2017;1858:197–207. doi: 10.1016/j.bbabio.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files) and/or are available from the corresponding author on reasonable request.