

Abstract
Insulin resistance (IR) is a major metabolic risk factor even before the onset of hyperglycemia. Recently, berberine (BBR) is found to improve hyperglycemia and IR. In this study, we investigated whether BBR could improve IR independent of hyperglycemia. Acute insulin-resistant state was induced in rats by systemic infusion of intralipid (6.6%). BBR was administered via different delivery routes before or after the beginning of a 2-h euglycemic-hyperinsulinemic clamp. At the end of experiment, rats were sacrificed, gastrocnemius muscle was collected for detecting mitochondrial swelling, phosphorylation of Akt and AMPK, as well as the mitochondrial permeability regulator cyclophilin D (CypD) protein expression. We showed that BBR administration markedly ameliorated intralipid-induced IR without affecting blood glucose, which was accompanied by alleviated mitochondrial swelling in skeletal muscle. We used human skeletal muscle cells (HSMCs), AML12 hepatocytes, human umbilical vein endothelial cells, and CypD knockout mice to investigate metabolic and molecular alternations. In either HSMCs or AML12 hepatocytes, BBR (5 μM) abolished palmitate acid (PA)-induced increase of CypD protein levels. In CypD-deficient mice, intralipid-induced IR was greatly attenuated and the beneficial effect of BBR was diminished. Furthermore, we demonstrated that the inhibitory effect of BBR on intralipid-induced IR was mainly mediated by skeletal muscle, but not by intestine, liver, or microvasculature; BBR administration suppressed intralipid-induced upregulation of CypD expression in skeletal muscle. These results suggest that BBR alleviates intralipid-induced IR, which is related to the inhibition of CypD protein expression in skeletal muscle.
Keywords: berberine, intralipid, insulin resistance, skeletal muscle, mitochondrial swelling, cyclophilin D, euglycemic-hyperinsulinemic clamp
Introduction
Insulin resistance (IR) plays a crucial role in the development of type 2 diabetes mellitus and is associated with hypertension, obesity, and cardiovascular diseases [1]. Studies have indicated that IR precedes the clinical diagnosis of type 2 diabetes and is a major metabolic risk factor even in normoglycemic states [2, 3]. Several clinical trials, such as the IR atherosclerosis study [4] and the San Antonio Heart Study [3], have shown that IR is associated with atherosclerosis even in nondiabetic patients. These studies suggest that current clinical interventions for IR occur relatively late considering diabetes-related complications. Therefore, earlier pharmaceutical intervention for IR before the onset of diabetes is a very promising strategy for the prevention of diabetes-related complications.
Although the pathogenesis of IR has not yet been elucidated, elevated free fatty acid (FFA) levels are a well-established risk factor for IR [5]. Acute exposure to FFAs leads to IR without affecting blood glucose in healthy humans and rodents [6–10]. In the process of FFA induction, mitochondrial dysfunction is considered a major cause of IR formation [11–18]. Mitochondrial function is regulated by cyclophilin D (CypD), the key regulator of the mitochondrial permeability transition pore (mPTP), which is closely associated with IR [19, 20]. Studies have revealed that CypD-deficient mice are protected from high-fat diet (HFD)-induced IR, and the increased glucose uptake is observed in only skeletal muscle but not in liver or adipose tissue [20]. The skeletal muscle-specific regulation of insulin sensitivity has been demonstrated by variations in the composition and function of the mPTP [21–23]. These studies suggest that the inhibition of CypD might have potential therapeutic value in IR by regulating skeletal muscle mitochondrial dysfunction.
Berberine (BBR), an isoquinoline alkaloid, has been used to treat intestinal infections for thousands of years. Recently, several studies have shown its chronic beneficial effects on weight loss, hyperglycemia and IR in humans and rodents with diabetes [24–26]. One study in db/db mice indicated that BBR reduces body weight and causes an improvement in glucose tolerance [24]. Another study in type 2 diabetic patients showed that three months of BBR treatment reduces fasting blood glucose and was accompanied by improved insulin sensitivity and decreased body weight [25]. However, it is hard to rule out the effect of weight loss on IR in the chronic treatment model because weight loss is sufficient to improve IR [27]. Meanwhile, these studies focused on the hypoglycemic effect of BBR in the diabetic state. Whether BBR has a therapeutic role in the treatment of IR before the onset of hyperglycemia has not yet been elucidated.
In the current study, we demonstrated that BBR alleviated FFA-induced IR independent of glucose homeostasis by inhibiting skeletal muscle CypD, suggesting a therapeutic role of BBR in the early intervention of IR.
Materials and methods
Animal experimental protocols
Before the study, male Sprague-Dawley rats (Vital River Laboratory Animal Technology Co., Ltd, Beijing, China) were fed standard laboratory chow and water ad libitum and housed at a constant temperature on a 12:12-h light-dark cycle in the SPF Experimental Animal Center of Shandong Provincial Hospital Affiliated to Shandong University. Rats weighing 250–350 g (aged 7–9 weeks) were fasted overnight before the surgery. After being anesthetized (pentobarbital sodium, 50 mg/kg, i.p.), rats were placed in a supine position and intubated to maintain a patent airway. Polyethylene cannulae (PE-50, Fisher Scientific, Newark, DE, USA) were inserted into the carotid artery, portal vein, and jugular vein for arterial blood sampling, arterial blood pressure monitoring, and various infusions, as previously described [28]. After a 30-min baseline period to assure stable anesthesia and hemodynamic stability, rats were studied with the following three protocols.
In each protocol, euglycemic-hyperinsulinemic clamps were performed as follows. Rats received systemic infusions with regular insulin (3 mU · kg−1 · min−1) for 2 h. The arterial blood glucose level was determined every 10 min using an Accu-Chek Advantage glucometer (Roche Diagnostics, Indianapolis, IN, USA), and 30% dextrose (30% wt/vol) was infused into the jugular vein at a variable rate to maintain blood glucose levels within 10% of basal levels. The time course and steady-state whole-body glucose infusion rate (GIR) were calculated [29, 30]. The clamp quality was assessed by the coefficient of variation of blood glucose concentrations (CVBG), which was calculated as 100× (SD device/mean device). At the end of the study, rats were sacrificed, and the gastrocnemius muscle was obtained to evaluate mitochondrial swelling, Akt, AMP-activated protein kinase (AMPK) phosphorylation, and CypD protein expression was determined using Western blotting.
Protocol 1. Rats received an intragastric (i.g.) administration of BBR (10 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) 1 h before or after the insulin clamp in the presence or absence of an intralipid (6.6%, Sigma-Aldrich) plus heparin (60 U/mL) infusion (5 μL/min).
Protocol 4. Rats received BBR via three routes [i.g., 10 mg/kg, intraportal vein (p.v., 20 μg/kg), or intravenous (i.v., 15 μg/kg)], 1 h before the insulin clamp was performed in the presence of intralipid infusion. The dose we used for the p.v. delivery (20 μg/kg) was calculated according to BBR pharmacokinetics [31], which could make the intraportal BBR concentration similar to that after BBR (10 mg/kg) i.g. delivery. The dose we used for the i.v. delivery (15 μg/kg) was calculated according to BBR pharmacokinetics [31], which could make the plasma BBR concentration similar to that after BBR (20 μg/kg) p.v. delivery.
Protocol 5. Rats received an intralipid + heparin infusion with or without BBR (10 mg/kg) i.g. delivery 1 h before the insulin clamp in the presence or absence of NG-nitro-L-arginine methyl ester (L-NAME; Sigma-Aldrich; 50 μg · kg−1 · min−1) via i.v. infusion [32]. The dose we used for L-NAME was sufficient to inhibit NO-dependent vasodilation, as previously reported [33].
Ppif−/− (the gene encoding CypD) mice on a C57BL/6 background were provided by Dr. Heng Du (The University of Texas, Dallas, USA). These mice were backcrossed to the C57BL/6 strain for ten generations to obtain stable Ppif−/− heterozygosis. Mice were housed in colony cages with a 12-h light/dark cycle in a temperature-controlled SPF environment and were maintained on a regular chow diet (Beijing Keao Xieli Co., Ltd, China). Male wild-type mice aged 19–20 weeks were divided into three groups. Each group received saline or intralipid injection through the caudal vein with or without BBR i.g. delivery 60 min before the insulin tolerance test (ITT; protocol 2). In protocol 3, mice were divided into three Ppif+/+ (wild-type) groups and three Ppif−/− (CypD deficiency) groups. Each group received saline or intralipid injection through the caudal vein with or without BBR i.g. delivery 60 min before the ITT.
The investigation conformed to the guidelines for the ethical review of animal welfare in China. The animal experiments were approved by the Animal Ethics Committee of Shandong Provincial Hospital.
Insulin tolerance test
After fasting for 8 h, mice received an i.p. injection of insulin (1.0 U/kg, human insulin; Lily, Indianapolis, IN, USA). The blood glucose levels were determined at 0, 15, 30, 60, 90, and 120 min after the injection of insulin. The insulin sensitivity index (Kitt) was calculated as (0.693/t1/2) × 100 [34].
Cell culture
AML12 hepatocytes were maintained in DMEM/F12 supplemented with 1% ITS Liquid Media Supplement (Sigma-Aldrich), 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), and 40 ng/mL dexamethasone. Human skeletal muscle cells (HSMCs; Otwo Biotech Inc., Shenzhen, Guangdong, China) were cultured in 89% H-DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS (Gibco, Grand Island, NY, USA). Human umbilical vein endothelial cells (HUVECs; ATCC, Manassas, VA, USA) were cultured in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS, 0.5% antibiotics, and 2 mM L-glutamine. All of these cells were cultured in a humidified atmosphere with 5% CO2 at 37 °C. Cells received the indicated treatment and were then collected for further analysis.
Assessment of mPTP opening
Mitochondria were prepared as previously described [35] with several modifications. Rats were anesthetized, and the gastrocnemius muscles were dissected from the surrounding connective tissue. Samples were immersed in ice-cold PBS with 10 mM EDTA supplementation. The tissues were cut into small pieces with scissors and resuspended in 5 mL of PBS supplemented with 10 mM EDTA and 0.05% trypsin for 30 min. Samples were centrifuged at 200 × g for 5 min, and the supernatant was discarded. The pellet was resuspended in the isolation medium (67 mM sucrose, 50 mM KCl, 50 mM Tris-HCl, 10 mM EDTA, 0.2% BSA, pH 7.4). The samples were homogenized with a Plexiglas pestle and centrifuged at 700 × g for 10 min. The supernatant was decanted and then centrifuged at 8000 × g for 10 min. The resulting pellet was resuspended in medium (250 mM sucrose, 3 mM EGTA-Tris, and 10 mM Tris-HCl, pH 7.4) and centrifuged at 8000 × g for 10 min. The mitochondrial pellet was resuspended in the same medium. All procedures were carried out at 4 °C. Protein concentrations were measured at 540 nm by a NanoDrop 2000c (Thermo Scientific). The mPTP opening was determined according to a previous method [36] with slight modifications. Two hundred micrograms of freshly isolated mitochondria were suspended in 200 μL of swelling assay buffer (mM: 150 KCl, 5 HEPES, 2 K2HPO4, 5 glutamate, and 5 malate, pH 7.2). Mitochondrial swelling was triggered by adding calcium (1 μmol/mg protein) and then recorded at 540 nm on a microplate reader (Molecular Devices, LLC) after 12 min. The vehicle was measured without adding calcium.
Western blotting
Skeletal muscle tissue and cells were lysed in lysis buffer containing protease inhibitors and phosphatase inhibitors (Shenergy Biocolor Bioscience & Technology Company, Shanghai, China). The following antibodies were used in the study: anti-p-Ser1177 eNOS (Cell Signaling Technology, Boston, MA, USA), anti-p-Thr172 AMPK (Cell Signaling Technology), anti-p-Ser473 Akt (Cell Signaling Technology), anti-AMPK (Abcam, Cambridge, UK), anti-eNOS (Abcam), anti-β-actin (Abcam), anti-CypD (Abcam), and anti-GAPDH (Proteintech, Wuhan, China).
Statistical analysis
The results are displayed as the mean ± SD. The comparison of two groups was performed using two-tailed unpaired Student’s t tests. Multiple groups were compared using one-way ANOVA. P < 0.05 was considered statistically significant.
Results
BBR improves intralipid-induced IR
Previous studies have shown the beneficial effects of chronic BBR use on hyperglycemia and IR. To examine whether BBR improves intralipid-induced acute IR before the onset of hyperglycemia in rats, we performed insulin clamp experiments for 2 h with or without BBR i.g. administration at −60 min in rats receiving a 3-h systemic intralipid infusion (Fig. 1a). Intralipid infusion inhibited insulin-stimulated glucose disposal by ~70%, demonstrating whole-body IR. BBR i.g. delivery partially improved IR, as indicated by an ~2.2-fold increase in the GIR at steady state (Fig. 1b, c). To further verify whether BBR still plays its role after the onset of IR, BBR was i.g. delivered 2 h after intralipid infusion. The GIR was acutely increased 30 min after BBR i.g. administration, and the steady-state GIR was similar to that after BBR i.g. delivery at −60 min (Fig. 1b, c). The CVBG, which was indicative of the quality of the clamp, was 4.9%. The basal fasting blood glucose levels of rats were within the normal range and were not affected by either intralipid or BBR treatment (Fig. 1d). The arterial blood pressures did not change during the experiments (data not shown). Similar experiments were also performed in C56BL/6 mice, and insulin sensitivity was measured by the ITT (Fig. 1e, f). The Kitt decreased in the intralipid delivery group but was restored by BBR supplementation (Fig. 1g).
Fig. 1. Berberine (BBR) improves intralipid-induced IR in vivo.

a Animal study protocol for SD rats. Each rat received a systemic infusion of either saline or intralipid 60 min before a 2-h insulin clamp (3 mU · kg−1 · min−1). Berberine (10 mg/kg) was i.g. administered 60 min before or after the initiation of the insulin clamp. b Time courses of the glucose infusion rate (GIR) during the euglycemic-hyperinsulinemic clamps. The data are shown as the mean ± SD. ***P < 0.001 vs. saline group; ###P < 0.001 vs. intralipid group, n = 3–4. c Steady-state GIR. The data are shown as the mean ± SD. ***P < 0.001 vs. saline group, ###P < 0.001 vs. intralipid group, n = 3–4. d Fasting blood glucose. e Animal study protocol for C57BL/6 mice. f ITT. g Kitt. The data are shown as the mean ± SD. *P < 0.05 vs. saline group, #P < 0.05 vs. intralipid group, n = 4. h Ca2+-overload induced skeletal muscle mitochondrial swelling in the indicated groups in protocol 1. The data are shown as the mean ± SD. ###P < 0.001 vs. intralipid group, n = 3.
It was reported that BBR accumulated in mitochondria [37]. To further investigate whether the insulin-sensitizing effects of BBR were related to improved mitochondrial function, we examined Ca2+-overload-induced mitochondrial swelling in the skeletal muscle of SD rats. Mitochondria from intralipid-treated skeletal muscle were more sensitive to Ca2+ overload, indicating that more mPTP opened in skeletal muscle mitochondria after intralipid treatment than saline treatment (P < 0.05, ANOVA). BBR i.g. administration before intralipid infusion alleviated intralipid-induced mitochondrial swelling (Fig. 1h). These results indicated that BBR improved intralipid-induced IR independent of glucose homeostasis and was accompanied by attenuated mitochondrial swelling.
BBR inhibits intralipid-induced CypD expression
Since CypD is a key regulator of mitochondrial permeability and is closely associated with IR [20], we next investigated the effects of BBR on CypD protein expression in AML12 hepatocytes and HSMCs in vitro.
BBR treatment strongly inhibited palmitate acid (PA)-induced CypD protein expression in AML12 hepatocytes and HSMCs (Fig. 2a, b). BBR treatment decreased CypD protein expression and increased AMPK phosphorylation levels in a dose-dependent manner without influencing Akt phosphorylation in HSMCs (Fig. 2c).
Fig. 2. Berberine regulates CypD protein expression in vitro.
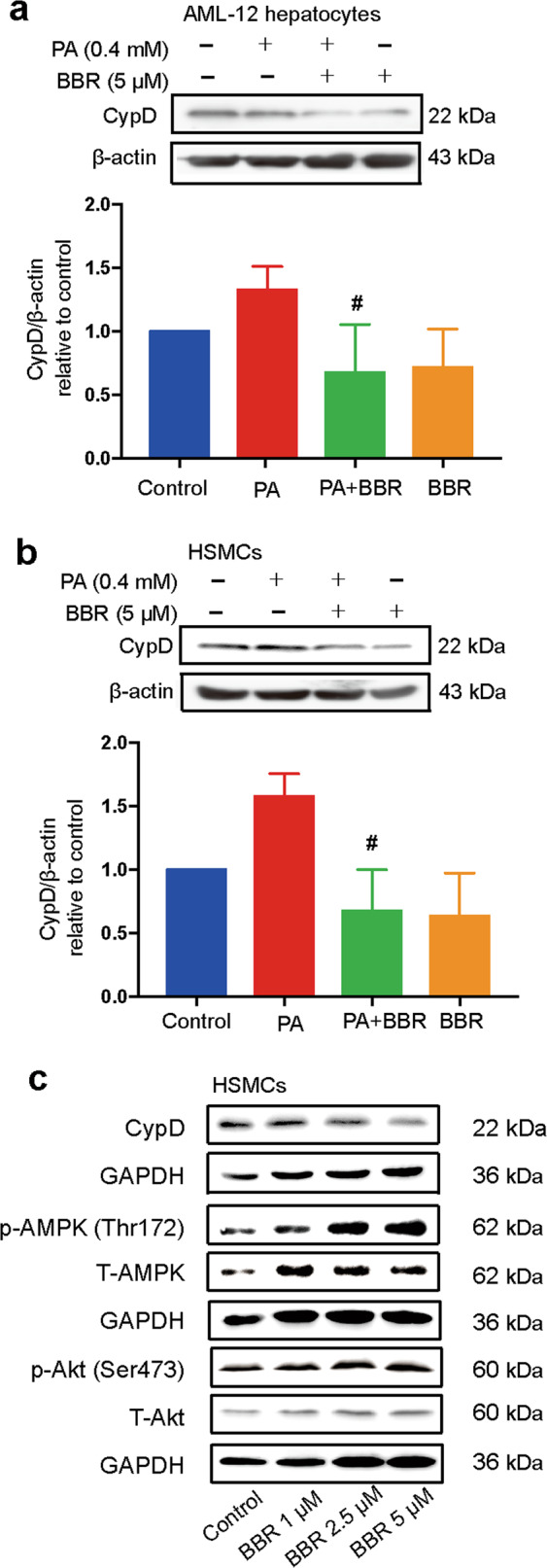
AML12 hepatocytes (a) and HSMCs (b) were pretreated with 0.4 M palmitate acid (PA) for 2 h before coincubation with 5 μM BBR for 24 h. CypD protein expression was detected through Western blot analysis and quantified by densitometry. The data are shown as the mean ± SD. #P < 0.05 vs. PA. c HSMCs were exposed to BBR at different concentrations (1, 2.5, and 5 μM) for 24 h. Western blot analysis of CypD, phospho-AMPK (Thr172), total AMPK, phospho-Akt (Ser473), and total Akt protein expression normalized to GAPDH.
The improvement of IR in response to BBR is associated with CypD inhibition
Since BBR inhibited CypD protein expression in vitro and CypD ablation protected mice from HFD-induced mitochondrial swelling [38, 39] and IR [20], we next determined whether the improvement of intralipid-induced IR in response to BBR was dependent on CypD inhibition.
All mice were divided into wild-type (Ppif+/+) and CypD-deficient (Ppif−/−) groups. Each group was divided into three subgroups that received saline i.v., intralipid i.v., or intralipid i.v. + BBR i.g. 60 min before the ITT (Fig. 3a). The absence of CypD protein expression in skeletal muscle validated the CypD deficiency model (Fig. 3b). The Kitt was lower in the intralipid-treated group than in the saline-treated group (P < 0.01, ANOVA), while Kitt was not different in the two subgroups of CypD-deficient mice. These results suggest that intralipid-induced IR was alleviated due to CypD deficiency. BBR cotreatment with intralipid partially restored Kitt in wild-type mice (compared with the intralipid-treated subgroup, P < 0.05, ANOVA) but not in CypD-deficient mice (P > 0.05, ANOVA; Fig. 3c, d). Collectively, these results revealed that CypD participated in the protective effect of BBR on intralipid-induced IR.
Fig. 3. CypD is involved in the effect of berberine on insulin resistance.

a Animal study protocol. Mice were divided into three Ppif+/+ (wild-type) groups and three Ppif−/− (CypD deficiency) groups, and each group received saline or intralipid i.v. administration with or without BBR i.g. administration 60 min before the insulin tolerance test (ITT). b Immunoblotting analysis of CypD protein expression in the skeletal muscle of Ppif+/+ and Ppif−/− mice normalized to GAPDH. The data are shown as the mean ± SD. ###P < 0.0001 vs. Ppif−/−. c ITT and d Kitt. The data are shown as the mean ± SD. **P < 0.01 vs. Ppif+/+-saline group, #P < 0.05 vs. Ppif+/+-intralipid group; ns not significant; n = 4.
The insulin-sensitizing effect of BBR is independent of the gut and liver
As above, the beneficial effect of BBR was dependent on CypD inhibition, but the main target organ has not yet been clarified. Extensive studies have revealed that BBR affects many tissues, such as the intestine, liver, skeletal muscle, adipose tissue, and vasculature [24, 40–43]. Notably, the concentration of BBR in the intestine and liver was much higher than that in other organs, as the first-pass (intestinal and hepatic) elimination of BBR after oral administration is strong.
To examine the effect on the intestine, we injected a much lower dose of BBR directly into the portal veins of rats, which could make the intraportal BBR concentration similar to that after BBR i.g. administration while avoiding high intestinal concentrations. Then, we performed insulin clamp experiments on the rats (Fig. 4a). Compared with i.g. delivery, the p.v. delivery of BBR did not weaken the effect of BBR on IR (P > 0.05, ANOVA), indicating that the insulin-sensitizing effect of BBR was independent of the intestine. Similarly, we injected a lower dose of BBR directly into the jugular veins of the rats, which could make the plasma BBR concentration similar to that after BBR p.v. administration while avoiding high liver concentrations. Compared with p.v. delivery, the i.v. delivery of BBR did not weaken the effect of BBR on IR (P > 0.05, ANOVA; Fig. 4b, c), indicating that the insulin-sensitizing effect of BBR was independent of the liver.
Fig. 4. The effect of berberine is independent of the liver and intestine.

a Animal study protocols. Each rat received a systemic infusion of intralipid 60 min before a 2 h euglycemic insulin clamp was performed (3 mU · kg−1 · min−1). BBR was intragastrically (i.g., 10 mg/kg), intravenously (i.v., 15 μg/kg) or intraportally (p.v., 20 μg/kg) administered 60 min before the insulin clamp. b Time courses of the glucose infusion rate (GIR) during the euglycemic-hyperinsulinemic clamp. The data are shown as the mean ± SD. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. intralipid group; ns not significant; n = 3–4. c Steady-state GIR. The data are shown as the mean ± SD. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. intralipid group; ns not significant; n = 3–4.
Endothelial-dependent vasodilation is not associated with BBR’s insulin-sensitizing effect
As impaired endothelium-dependent vasodilation plays a crucial role in the pathogenesis of IR [8, 44], we next examined whether the increased insulin sensitivity after BBR administration was caused by improved NO-dependent vasodilation.
BBR treatment increased eNOS phosphorylation at Ser1177 in HUVECs in a dose-dependent manner (Fig. 5a). To examine the role of NO-dependent vasodilation on the insulin-sensitizing effect of BBR, L-NAME was used to inhibit NO-dependent vasodilation in rats receiving BBR and intralipid administration (Fig. 5b). However, L-NAME coinfusion with BBR failed to abolish the effect of BBR on IR (Fig. 5c, d), indicating that endothelial-dependent vasodilation was not involved in the insulin-sensitizing effect of BBR.
Fig. 5. The effect of berberine is independent of endothelial-dependent vasodilation.

a Western blot analysis of p-eNOS (Ser1177) and total eNOS in HUVECs. b Animal study protocol. Rats received an intralipid + heparin (6.6%, 30 U/mL) infusion with or without BBR (10 mg/kg) i.g. delivery 1 h before the insulin clamp in the presence or absence of NO inhibitor NG-nitro-L-arginine (L-NAME, 50 μg · kg−1 · min−1) infusion. c Time courses of the glucose infusion rate (GIR) during euglycemic-hyperinsulinemic clamps. The data are shown as the mean ± SD. ***P < 0.001 vs. intralipid group, n = 3–4. d Steady-state GIR. The data are shown as the mean ± SD. ***P < 0.001 vs. intralipid group; ns not significant; n = 3–4.
BBR inhibits skeletal muscle CypD expression
Since the insulin-sensitizing effect of BBR was independent of the gut-, liver-, and endothelial-dependent vasodilation, we hypothesized that skeletal muscle is the main target organ of BBR in mediating intralipid-induced IR. To further examine this issue, we measured the protein level of CypD and the phosphorylation level of AMPK and Akt in skeletal muscle of SD rats, as described in protocol 1.
CypD protein expression was upregulated in the intralipid-treated group and was dramatically inhibited by BBR cotreatment in skeletal muscle (Fig. 6a, b). AMPK phosphorylation levels were increased by BBR treatment in skeletal muscle. However, this effect was strongly inhibited by intralipid administration (Fig. 6c). The phosphorylation of Akt was not influenced by BBR in the skeletal muscle of SD rats (Fig. 6d). These data indicated that BBR improved intralipid-induced IR associated with the inhibition of CypD expression in skeletal muscle.
Fig. 6. Berberine affects CypD protein expression in the skeletal muscle.
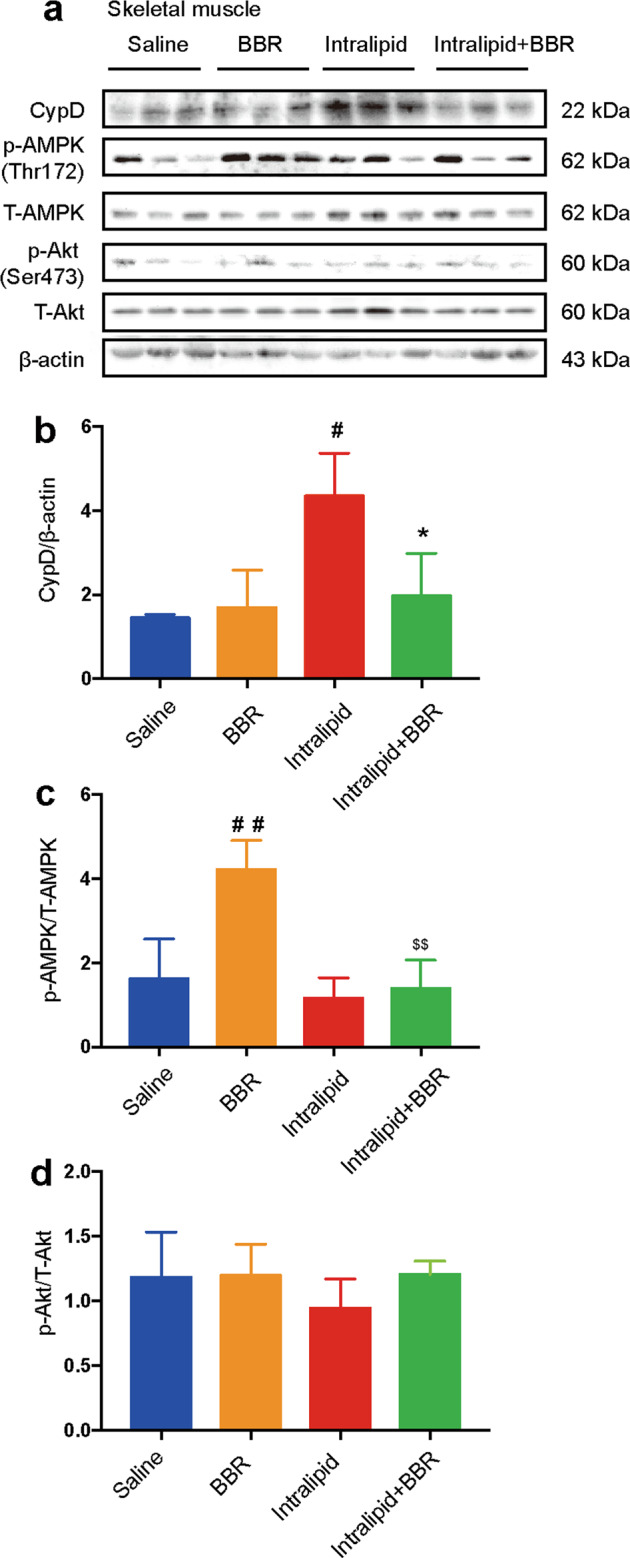
a Changes in CypD, phospho-AMPK (Thr172), total AMPK, phospho-Akt (Ser473), and total Akt protein expression in the skeletal muscle of rats in protocol 1. β-actin served as a loading control. b–d Quantification of CypD immunoblots, phospho/total AMPK, and phospho/total Akt. The data are shown as the mean ± SD. #P < 0.05 and ##P < 0.01 vs. saline group, *P < 0.05 vs. intralipid group, $$P < 0.01 vs. BBR group; n = 3.
Discussion
In the current study, we found that BBR treatment acutely rescued intralipid-induced IR before the onset of hyperglycemia. These findings provide evidence for the use of BBR in the early intervention of insulin-resistant patients, such as those with prediabetes, polycystic ovary syndrome, and total parenteral nutrition. Moreover, BBR i.g. administration reversed IR both before and after intralipid infusion, indicating the preventive and therapeutic role of BBR in mediating IR.
We demonstrated that BBR inhibited FFA-induced mitochondrial swelling. Our findings that BBR improved mitochondrial swelling were in agreement with those of several other studies. For example, Gomes et al. showed that the beneficial effects of BBR were accompanied by increased mitochondrial function in skeletal muscle in vivo [45], and Qin et al. demonstrated that BBR improved FFA-induced mitochondrial dysfunction in glomerular podocytes [46]. However, some in vitro studies reported that BBR inhibited ETC complex I and attenuated mitochondrial function at concentrations thousands of times higher than its plasma concentration [47]. We did not rule out the possibility of toxic effects of BBR on mitochondria at higher concentrations. We observed that BBR exerted a beneficial effect on IR at a much lower dose (10 mg/kg) than previously described (100–380 mg · kg−1 per day) [24, 48, 49]. The low effective dose suggests that BBR might be a good pharmacologic agent to treat FFA-induced IR.
Our study demonstrated for the first time that the insulin-sensitizing effect of BBR was derived from its inhibition of mitochondrial CypD. These results were in line with a study that showed that CypD deficiency protected mice from HFD-induced IR [20]. We also measured the phosphorylation of AMPK and Akt, which play crucial roles in two distinct pathways in glucose uptake. AMPK is the target of many insulin-sensitizing agents, such as metformin and thiazolidinediones [50, 51]. BBR has been reported to activate the AMPK pathway, which was proposed as a mechanism for the insulin-sensitizing effect of BBR [24, 52]. However, another group demonstrated that AMPK activation was not necessary for the glucose-lowering effect of BBR [53]. They found that the effect of BBR on glucose utilization could not be blocked by compound C, siRNA, or adenoviruses encoding dominant-negative forms of AMPKα1/α2 [53]. In the current study, we observed that AMPK phosphorylation was increased by BBR oral administration and inhibited by intralipid infusion in the skeletal muscle of SD rats. However, BBR was still effective in ameliorating IR in the presence of intralipid, as the GIR was not different between the intralipid + BBR group and the BBR group. These results suggest that the activation of AMPK might be a concomitant effect in the insulin-sensitizing effect of BBR. Moreover, we observed that the phosphorylation of Akt, a key molecule in the insulin signaling pathway, was not influenced by BBR in vivo and in vitro. Our results were in agreement with those of study showing that BBR-stimulated GLUT4 translocation was not inhibited by the phosphatidylinositol 3 kinase inhibitor wortmannin in L6 myotubes [24], suggesting that Akt may not be involved in the glucose-lowering effect of BBR.
The target organs of insulin-sensitizing agents include the intestine, liver, skeletal muscle, adipose tissue, and microvasculature [29, 54–56]. Similar to some other herbal products, the first-pass (gut and liver) elimination effect of BBR was very strong. BBR that entered the portal vein was present at 0.2% of the total oral dose, while the hepatic elimination of BBR was 28.2% [31]. Thus, the concentrations of BBR in the intestine and liver were much higher than those in peripheral tissues after oral administration. To rule out the effect of the gut and liver, we used two different (i.v., p.v.) delivery routes to make plasma BBR concentrations similar to those after BBR i.g. administration. Although these three injection routes differed greatly in BBR concentrations in the gut and liver, the insulin-sensitizing effects of BBR were not different among those three groups. Our results implied that the insulin-sensitizing effect of BBR was independent of the intestine and liver. In addition, the microvasculature offers endothelial exchange surface area to regulate nutrient exchanges between the plasma and muscle interstitium. Recent evidence has shown that intralipid infusion leads to microvascular IR by inhibiting endothelial-dependent vasodilation [6]. Here, we used the NO inhibitor L-NAME and found that the improved IR in response to BBR was independent of endothelial-dependent vasodilation. Furthermore, adipose tissue was also ruled out in the acute effect of BBR because lipolysis in adipose tissue was suppressed during intralipid infusion [57]. Therefore, we propose that skeletal muscle is the main target organ of BBR in mediating intralipid-induced IR. We also observed that CypD protein expression is inhibited by BBR in skeletal muscle. Indeed, skeletal muscle accounted for 80% of insulin-stimulated glucose uptake and was the main organ influenced by IR [58]. Although we did not use skeletal muscle-specific CypD knockout mice, it is reasonable to propose that skeletal muscle CypD is associated with the insulin-sensitizing effect of BBR. This tissue-specific phenomenon resulted from the structural variation of the mPTP complex in different tissues [59]. mPTP opening is regulated by the CypD-ANT interaction [60]. However, different isoforms of ANT are expressed in a tissue-specific manner. ANT-1 is mainly expressed in skeletal muscle, kidney, heart, and brain, while ANT-2 is expressed in other tissues [21, 23]. CypD had a higher binding affinity for ANT-1 than ANT-2 [22], which led to more effective CypD inhibition in the skeletal muscle than other glucose metabolic tissues [20]. However, there were still some limitations of this study. The influences of holistic factors could not be ruled out in the Ppif conventional knockout mouse model. The skeletal muscle Ppif conditional knockout mouse model and Ppif overexpression mouse model may provide more support for the mechanism of the effect of BBR.
In summary, this study indicated that low concentrations of BBR acutely improved FFA-induced IR, which was related to the inhibition of CypD protein expression in the skeletal muscle. These findings provide new evidence for the use of BBR in the early clinical intervention of IR.
Acknowledgements
We would like to acknowledge Dr. Heng Du (The University of Texas, Dallas, USA) for kindly providing Ppif mice. This work was supported by the National Key Research and Development Program of China (grant number: 2017YFC1309800), National Natural Science Foundation of China (grant number: 81400788), Medical and Health Technology Development Project of Shandong province (grant number: 2019WS076), Scientific Research Project of Ji-nan (201909039), and Scientific Research Project of Weifang Health Committee (wfwsjk_2019_121).
Author contributions
ZHD, ZH, LG, and JJZ designed the experiments. ZHD and FLC performed the experiments, testing, and data analyses. ZHD and ZH wrote the manuscript. ZHD, HYL, WKB, XQC, JW, FLC, and SLS cared for the mice. All authors edited the manuscript and provided comments.
Competing interests
The authors declare no competing interests.
Contributor Information
Ling Gao, Email: linggao@sdu.edu.cn.
Zhao He, Email: zhaohe7711@qq.com.
Jia-jun Zhao, Email: jjzhao@sdu.edu.cn.
References
- 1.Eriksson J, Franssila-Kallunki A, Ekstrand A, Saloranta C, Widen E, Schalin C, et al. Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. N Engl J Med. 1989;321:337–43. doi: 10.1056/NEJM198908103210601. [DOI] [PubMed] [Google Scholar]
- 2.UK Prospective Diabetes Study (UKPDS) Group Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–65. [PubMed] [Google Scholar]
- 3.Hanley AJ, Williams K, Stern MP, Haffner SM. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: the San Antonio Heart Study. Diabetes Care. 2002;25:1177–84. doi: 10.2337/diacare.25.7.1177. [DOI] [PubMed] [Google Scholar]
- 4.Howard G, O’Leary DH, Zaccaro D, Haffner S, Rewers M, Hamman R, et al. Insulin sensitivity and atherosclerosis. The Insulin Resistance Atherosclerosis Study (IRAS) Investigators. Circulation. 1996;93:1809–17. doi: 10.1161/01.cir.93.10.1809. [DOI] [PubMed] [Google Scholar]
- 5.Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care. 2007;10:142–8. doi: 10.1097/MCO.0b013e328042ba90. [DOI] [PubMed] [Google Scholar]
- 6.Liu Z, Liu J, Jahn LA, Fowler DE, Barrett EJ. Infusing lipid raises plasma free fatty acids and induces insulin resistance in muscle microvasculature. J Clin Endocrinol Metab. 2009;94:3543–9. doi: 10.1210/jc.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA. Effect of fatty acids on glucose production and utilization in man. J Clin Invest. 1983;72:1737–47. doi: 10.1172/JCI111133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clerk LH, Rattigan S, Clark MG. Lipid infusion impairs physiologic insulin-mediated capillary recruitment and muscle glucose uptake in vivo. Diabetes. 2002;51:1138–45. doi: 10.2337/diabetes.51.4.1138. [DOI] [PubMed] [Google Scholar]
- 9.Liang H, Lum H, Alvarez A, Garduno-Garcia JJ, Daniel BJ, Musi N. A low dose lipid infusion is sufficient to induce insulin resistance and a pro-inflammatory response in human subjects. PLoS One. 2018;13:e0195810. doi: 10.1371/journal.pone.0195810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rachek LI. Free fatty acids and skeletal muscle insulin resistance. Prog Mol Biol Transl Sci. 2014;121:267–92. doi: 10.1016/B978-0-12-800101-1.00008-9. [DOI] [PubMed] [Google Scholar]
- 11.Tripathy D, Mohanty P, Dhindsa S, Syed T, Ghanim H, Aljada A, et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003;52:2882–7. doi: 10.2337/diabetes.52.12.2882. [DOI] [PubMed] [Google Scholar]
- 12.de Jongh RT, Serne EH, Ijzerman RG, de Vries G, Stehouwer CD. Free fatty acid levels modulate microvascular function: relevance for obesity-associated insulin resistance, hypertension, and microangiopathy. Diabetes. 2004;53:2873–82. doi: 10.2337/diabetes.53.11.2873. [DOI] [PubMed] [Google Scholar]
- 13.Martins AR, Nachbar RT, Gorjao R, Vinolo MA, Festuccia WT, Lambertucci RH, et al. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis. 2012;11:30. doi: 10.1186/1476-511X-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–79. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–11. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 16.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murrow BA, Hoehn KL. Mitochondrial regulation of insulin action. Int J Biochem Cell Biol. 2010;42:1936–9. doi: 10.1016/j.biocel.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 18.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–81. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tavecchio M, Lisanti S, Bennett MJ, Languino LR, Altieri DC. Deletion of cyclophilin D impairs beta-oxidation and promotes glucose metabolism. Sci Rep. 2015;5:15981. doi: 10.1038/srep15981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taddeo EP, Laker RC, Breen DS, Akhtar YN, Kenwood BM, Liao JA, et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol Metab. 2014;3:124–34. doi: 10.1016/j.molmet.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem. 1992;267:14592–7. [PubMed] [Google Scholar]
- 22.Vyssokikh MY, Katz A, Rueck A, Wuensch C, Dorner A, Zorov DB, et al. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem J. 2001;358:349–58. doi: 10.1042/0264-6021:3580349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doerner A, Pauschinger M, Badorff A, Noutsias M, Giessen S, Schulze K, et al. Tissue-specific transcription pattern of the adenine nucleotide translocase isoforms in humans. FEBS Lett. 1997;414:258–62. doi: 10.1016/s0014-5793(97)01000-4. [DOI] [PubMed] [Google Scholar]
- 24.Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55:2256–64. doi: 10.2337/db06-0006. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Li X, Zou D, Liu W, Yang J, Zhu N, et al. Treatment of type 2 diabetes and dyslipidemia with the natural plant alkaloid berberine. J Clin Endocrinol Metab. 2008;93:2559–65. doi: 10.1210/jc.2007-2404. [DOI] [PubMed] [Google Scholar]
- 26.Mi J, He W, Lv J, Zhuang K, Huang H, Quan S. Effect of berberine on the HPA-axis pathway and skeletal muscle GLUT4 in type 2 diabetes mellitus rats. Diabetes Metab Syndr Obes. 2019;12:1717–25.. doi: 10.2147/DMSO.S211188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niskanen L, Uusitupa M, Sarlund H, Siitonen O, Paljarvi L, Laakso M. The effects of weight loss on insulin sensitivity, skeletal muscle composition and capillary density in obese non-diabetic subjects. Int J Obes Relat Metab Disord. 1996;20:154–60. [PubMed] [Google Scholar]
- 28.Dong Z, Chai W, Wang W, Zhao L, Fu Z, Cao W, et al. Protein kinase A mediates glucagon-like peptide 1-induced nitric oxide production and muscle microvascular recruitment. Am J Physiol Endocrinol Metab. 2013;304:E222–8. doi: 10.1152/ajpendo.00473.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chai W, Wang W, Dong Z, Cao W, Liu Z. Angiotensin II receptors modulate muscle microvascular and metabolic responses to insulin in vivo. Diabetes. 2011;60:2939–46. doi: 10.2337/db10-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chai W, Zhang X, Barrett EJ, Liu Z. Glucagon-like peptide 1 recruits muscle microvasculature and improves insulin’s metabolic action in the presence of insulin resistance. Diabetes. 2014;63:2788–99. doi: 10.2337/db13-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YT, Hao HP, Xie HG, Lai L, Wang Q, Liu CX, et al. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab Dispos. 2010;38:1779–84. doi: 10.1124/dmd.110.033936. [DOI] [PubMed] [Google Scholar]
- 32.Wang N, Chai W, Zhao L, Tao L, Cao W, Liu Z. Losartan increases muscle insulin delivery and rescues insulin’s metabolic action during lipid infusion via microvascular recruitment. Am J Physiol Endocrinol Metab. 2013;304:E538–45. doi: 10.1152/ajpendo.00537.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chai W, Dong Z, Wang N, Wang W, Tao L, Cao W, et al. Glucagon-like peptide 1 recruits microvasculature and increases glucose use in muscle via a nitric oxide-dependent mechanism. Diabetes. 2012;61:888–96. doi: 10.2337/db11-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi X, Guo Y, Song Y, Yu C, Zhao L, Fang L, et al. Follicle-stimulating hormone enhances hepatic gluconeogenesis by GRK2-mediated AMPK hyperphosphorylation at Ser485 in mice. Diabetologia. 2018;61:1180–92.. doi: 10.1007/s00125-018-4562-x. [DOI] [PubMed] [Google Scholar]
- 35.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–95. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 36.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Q, Zhou T, Liu C, Wang XY, Zhang JQ, Wu F, et al. Mitochondrial membrane potential played crucial roles in the accumulation of berberine in HepG2 cells. Biosci Rep. 2019. 10.1042/BSR20190477. [DOI] [PMC free article] [PubMed]
- 38.Liu X, Du H, Chen D, Yuan H, Chen W, Jia W, et al. Cyclophilin D deficiency protects against the development of mitochondrial ROS and cellular inflammation in aorta. Biochem Biophys Res Commun. 2019;508:1202–8. doi: 10.1016/j.bbrc.2018.12.064. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Du H, Shao S, Bo T, Yu C, Chen W, et al. Cyclophilin D deficiency attenuates mitochondrial perturbation and ameliorates hepatic steatosis. Hepatology. 2018;68:62–77. doi: 10.1002/hep.29788. [DOI] [PubMed] [Google Scholar]
- 40.Xia X, Yan J, Shen Y, Tang K, Yin J, Zhang Y, et al. Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic gluconeogenesis. PLoS One. 2011;6:e16556. doi: 10.1371/journal.pone.0016556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geng FH, Li GH, Zhang X, Zhang P, Dong MQ, Zhao ZJ, et al. Berberine improves mesenteric artery insulin sensitivity through up-regulating insulin receptor-mediated signalling in diabetic rats. Br J Pharmacol. 2016;173:1569–79. doi: 10.1111/bph.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yi P, Lu FE, Xu LJ, Chen G, Dong H, Wang KF. Berberine reverses free-fatty-acid-induced insulin resistance in 3T3-L1 adipocytes through targeting IKKbeta. World J Gastroenterol. 2008;14:876–83. doi: 10.3748/wjg.14.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng Z, Pang T, Gu M, Gao AH, Xie CM, Li JY, et al. Berberine-stimulated glucose uptake in L6 myotubes involves both AMPK and p38 MAPK. Biochim Biophys Acta. 2006;1760:1682–9. doi: 10.1016/j.bbagen.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Steinberg HO, Tarshoby M, Monestel R, Hook G, Cronin J, Johnson A, et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J Clin Invest. 1997;100:1230–9. doi: 10.1172/JCI119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomes AP, Duarte FV, Nunes P, Hubbard BP, Teodoro JS, Varela AT, et al. Berberine protects against high fat diet-induced dysfunction in muscle mitochondria by inducing SIRT1-dependent mitochondrial biogenesis. Biochim Biophys Acta. 2012;1822:185–95. doi: 10.1016/j.bbadis.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics. 2019;9:1698–713.. doi: 10.7150/thno.30640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes. 2008;57:1414–8. doi: 10.2337/db07-1552. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Campbell T, Perry B, Beaurepaire C, Qin L. Hypoglycemic and insulin-sensitizing effects of berberine in high-fat diet- and streptozotocin-induced diabetic rats. Metabolism. 2011;60:298–305. doi: 10.1016/j.metabol.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 49.Wu YY, Zha Y, Liu J, Wang F, Xu J, Chen ZP, et al. Effect of berberine on the ratio of high-molecular weight adiponectin to total adiponectin and adiponectin receptors expressions in high-fat diet fed rats. Chin J Integr Med. 2016. 10.1007/s11655-016-2518-x. [DOI] [PubMed]
- 50.Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–32. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 51.Riddle MC. Oral pharmacologic management of type 2 diabetes. Am Fam Phys. 1999;60:2613–20. [PubMed] [Google Scholar]
- 52.Zhao HL, Sui Y, Qiao CF, Yip KY, Leung RK, Tsui SK, et al. Sustained antidiabetic effects of a berberine-containing Chinese herbal medicine through regulation of hepatic gene expression. Diabetes. 2012;61:933–43. doi: 10.2337/db11-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu M, Xiao Y, Yin J, Hou W, Yu X, Shen L, et al. Berberine promotes glucose consumption independently of AMP-activated protein kinase activation. PLoS One. 2014;9:e103702. doi: 10.1371/journal.pone.0103702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okada K, Hosooka T, Shinohara M, Ogawa W. Modulation of lipid mediator profile may contribute to amelioration of chronic inflammation in adipose tissue of obese mice by pioglitazone. Biochem Biophys Res Commun. 2018;505:29–35. doi: 10.1016/j.bbrc.2018.09.081. [DOI] [PubMed] [Google Scholar]
- 55.Duca FA, Cote CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, et al. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat Med. 2015;21:506–11. doi: 10.1038/nm.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Gao Y, Duan L, Wei S, Liu J, Tian L, et al. Metformin ameliorates skeletal muscle insulin resistance by inhibiting miR-21 expression in a high-fat dietary rat model. Oncotarget. 2017;8:98029–39.. doi: 10.18632/oncotarget.20442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samra JS, Giles SL, Summers LK, Evans RD, Arner P, Humphreys SM, et al. Peripheral fat metabolism during infusion of an exogenous triacylglycerol emulsion. Int J Obes Relat Metab Disord. 1998;22:806–12. doi: 10.1038/sj.ijo.0800664. [DOI] [PubMed] [Google Scholar]
- 58.DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest. 1985;76:149–55. doi: 10.1172/JCI111938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83:213–25. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. 2013;77:1111–22. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]