

Abstract
Identifying genetic factors that contribute to the evolution of adaptive phenotypes in pathogenic bacteria is key to understanding the establishment of infectious diseases. In this study, we performed mutation accumulation experiments to record the frequency of mutations and their effect on fitness in hypermutator strains of the environmental bacterium Pseudomonas aeruginosa in comparison to the host-niche-adapted Salmonella enterica. We demonstrate that P. aeruginosa, but not S. enterica, hypermutators evolve toward higher fitness under planktonic conditions. Adaptation to increased growth performance was accompanied by a reversible perturbing of the local genetic context of membrane and cell wall biosynthesis genes. Furthermore, we observed a fine-tuning of complex regulatory circuits involving multiple di-guanylate modulating enzymes that regulate the transition between fast growing planktonic and sessile biofilm-associated lifestyles. The redundancy and local specificity of the di-guanylate signaling pathways seem to allow a convergent shift toward increased growth performance across niche-adapted clonal P. aeruginosa lineages, which is accompanied by a pronounced heterogeneity of their motility, virulence, and biofilm phenotypes.
Subject terms: Molecular evolution, Bacterial genetics, Functional genomics, Transcriptomics, Biofilms
Introduction
Pseudomonas aeruginosa is an environmental generalist and an opportunistic pathogen capable of infecting a wide variety of hosts ranging from plants to humans [1, 2]. Morphological, physiological, and behavioral changes that allow an organism to adjust to changing conditions arise as the result of two key mechanisms: phenotypic plasticity and natural selection [3]. Phenotypic plasticity is conferred by regulatory circuits that coordinate the perception of different signals, integrate them, and respond by altering gene transcription, protein translation, and protein activity. Natural selection acts upon a genetically diverse population and promotes the maintenance of alleles that are beneficial and the loss of alleles that are deleterious in a given environment. In addition to the acquisition of mutations and new genes via horizontal gene transfer, phase variation can produce genetic diversity in changing environments. This reversible process involves the variation of gene expression via the stochastic on–off switching of gene activity [4], mediated by insertions and deletions in simple sequence repeats [5–8], rearrangements in gene or promoter regions [9–11] or by epigenetic mechanisms [12].
The genomes of P. aeruginosa strains are large (5–7 Mbp) and encode a variety of regulatory circuits. These provide a remarkable phenotypic plasticity and a high adaptation potential to the opportunistic pathogen. For the successful colonizing of new habitats, P. aeruginosa must maintain this adaptation potential even under constant conditions. However, as it has been observed for various bacteria and yeasts, constant conditions favor the genetic selection of niche specialists, which have a higher fitness, but show fitness decay in other environments [13]. The trade-offs between higher fitness in one, but lower fitness in other environments result from the accumulation of mutations, which are beneficial or neutral under the selective conditions, but deleterious in other environments [13–18].
In this study, we performed mutation accumulation (MA) experiments in the environmental bacterium P. aeruginosa and recorded the type and frequency of mutations as well as their effect on bacterial fitness as compared to S. enterica. We used DNA repair-deficient bacterial strains characterized with ~100 times higher mutation rates in order to accelerate MA. Each of the species was continuously cultivated in the exponential phase under weak and strong bottlenecking conditions. In the context of experimental evolution, strong bottlenecking and a lower size of the effective population are thought to promote fixation of random mutations and, since most mutations are deleterious, lead to a loss of fitness. Conversely, weak bottlenecking enables natural selection and is more favorable to the fixation of beneficial and elimination of harmful mutations [19, 20]. Of note, the use of hypermutators in this study can undermine adaptive evolution, because a hypermutator with a beneficial mutation has a higher chance to acquire a detrimental second mutation, so that the evolutional behavior will differ to non-mutator populations. We demonstrate that under weak bottlenecking, P. aeruginosa, but not S. enterica hypermutator strains adapt to planktonic rich medium conditions by evolving increased growth performance. We show that the increase in fitness in P. aeruginosa was associated with an increased rate of phase variation events in the promoter regions of genes involved in membrane and cell wall biogenesis. Furthermore, the fine-tuning of cyclic di-guanylate (c-di-GMP) signaling-governed pathways was observed, indicating that P. aeruginosa adapts to lifestyles between planktonic and biofilm growth modes.
Materials and methods
Strains and culture conditions
Mismatch-repair-deficient mutants of P. aeruginosa and S. enterica were taken from a PA14 transposon insertion library [21] and provided by Prof. M. Erhardt (the mutS gene was replaced with an Frt-Kanamycin-Frt cassette in the S. enterica serovar Typhimurium LT2 background), respectively. Bacterial cultures were grown in 96-well plates with continuous shaking at 180 rpm. To synchronize the growth of the studied species, P. aeruginosa and S. enterica cultures were grown at 37 °C in LB medium (1% tryptone, 0.5% yeast extract, 0.75% NaCl) and at 30 °C in LB medium supplemented with 0.2% glucose, respectively (Fig. S1).
Experimental evolution
Starting cell lines of P. aeruginosa PA14 mutS::Tn and S. enterica LT2 ∆mutS were streaked out on LB plates, and six randomly selected colonies were used to inoculate starting cultures (Fig. 1). After 10.5 h of growth, P. aeruginosa strong bottleneck (SBN) and weak bottleneck (WBN) cultures were diluted to 1 and 100 cells per 100 μl, respectively, and S. enterica cultures were diluted to 1 (SBN) and 250 (WBN) cells per 100 μl (see Supplementary Note S1). New WBN and SBN cultures were prepared every 12 h. All the cultures were propagated for 24 days. At the end of the MA experiment, SBN cultures were used to prepare 4 ml overnight cultures in LB medium or LB + 0.2% glucose, which were subsequently aliquoted and stored at −70 °C. The WBN cultures were streaked on LB agar plates to isolate single colonies. Nine colonies were randomly selected per plate and subsequently used to prepare 4 ml overnight cultures in LB medium or LB + 0.2% glucose. Following overnight growth, aliquots from the WBN clonal cultures were stored at −70 °C.
Fig. 1. Schematic overview of the mutation accumulation experiment.

P. aeruginosa mutS::Tn (the starting cell line) was streaked on LB agar. Six colonies were picked and used to inoculate the overnight cultures (the starting cultures). Each of the starting cultures was used to inoculate a weak bottleneck (WBN) cell line with 100 cells and a strong bottleneck (SBN) cell line with one cell. WBN and SBN cell lines were propagated for 24 days. Six single-colony-derived clones for each WBN cell line were saved in liquid nitrogen. Subsequently, whole-genome sequencing of cryo-stocked starting cultures, last passage of the SBN cell lines and the six WBN clones per each WBN cell line was performed. We also performed an analogous mutation accumulation experiment with S. enterica LT2 ∆mutS using 250 cells for passaging WBN cell lines.
Genome sequencing and mutation analysis
For whole-genome sequencing, genomic DNA was isolated from, in total, 83 planktonic overnight cultures using the DNeasy Blood & Tissue kit (Qiagen, Hilden, Germany) and fragmented using the S2/E210 Focused-ultrasonicator (Covaris, Woburn, MA) to achieve 400-nt-long fragments. Libraries were prepared using the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, Ipswich, MA) according to the manufacturer’s instructions. The libraries were sequenced using Illumina MiSeq (2 × 250-bp paired-ended reads). Reads were mapped to PA14 reference genome using Burrow–Wheeler Aligner [22], re-mapped with Stampy [23], and subsequently processed using SAMtools [24] and custom scripts written in Python (Python Software Foundation, https://www.python.org/). The sequencing coverage was analyzed using SAMtools and was determined to be on average 32×. After quality filtering of specific nucleotide variations, mutations were checked using Integrative Genomics Viewer (v.2.3.98) [25]. The analysis of the types and topology of mutations was performed using custom R scripts (for details concerning indel analysis, see Supplementary Note S2).
Transcriptome sequencing and analysis
RNA sequencing was performed for three randomly selected clonal isolates from each WBN cell line. RNA was extracted from the logarithmic stage cultures of the selected clones and further processed as described in the protocol in Supplementary Note S3. The cDNA libraries yielded by the protocol were subsequently sequenced with an Illumina HiSeq 2500 in single end mode. Reads were mapped to PA14 reference genome with Stampy, and the data were processed using custom Perl (The Perl Foundation, https://www.perl.org/) and C shell scripts. Differential gene expression was estimated with the help of custom R scripts utilizing DESeq2 [26], tidyverse, pheatmap, and RColorBrewer packages.
Phenotypic assays
Fitness was analyzed by determining the optical density of the cultures throughout the growth phase. Swimming assays were performed as previously described [27]. Virulence was determined using a Galleria mellonella (greater wax moth) infection model [28]. Biofilm thickness and volume were evaluated using automated confocal microscopy [29, 30]. In addition, biofilm-forming capacity of the evolved strains was assessed based on their ability to adhere to a polyvinyl chloride surface, as measured by the crystal violet assay [31]. The detailed protocols are described in Supplementary Notes S4–S8.
Results
MA experiment
We performed serial passaging of a mismatch-repair-deficient P. aeruginosa mutant through defined weak bottlenecks (WBNs) in standard laboratory conditions (Fig. 1). Five independent WBN cell lines were continuously propagated in the exponential growth phase in LB medium for 24 days. WBN cell lines were passed through 45 bottlenecks by transferring 100 cells into fresh culture medium every 12 h. We estimate that WBN cell lines were grown for ~870 generations with 19 generations between each bottleneck. In order to evaluate the spontaneous mutation rate, and to analyze the influence of randomly accumulated mutations on bacterial fitness, we included a control experiment. In this experiment, selection and adaptation were prevented by passaging P. aeruginosa through SBNs. Six independent SBN cell lines were passaged through 44 bottlenecks by transferring a single cell into fresh medium over the course of 24 days. The SBN cell lines were grown for around 950 generations, resulting in ~21 generations between passages.
P. aeruginosa adapts to exponential LB growth conditions and gains fitness
To elucidate whether the five evolved P. aeruginosa WBN cell lines adapted to the cultivation conditions of the MA experiment, each WBN cell line was streaked out on LB agar, six colonies per cell line were isolated and their growth behavior in LB was compared with the growth behavior of the starting cultures (Fig. 1). Optical density (OD600) measurements demonstrated that evolved P. aeruginosa WBN clones developed a shortened lag phase (Fig. 2A, B), grew faster in the exponential phase (Fig. 2C), and reached higher maximal OD600 values (Fig. 2D). We confirmed for three randomly picked WBN clones that they evolved superior growth behavior by determining colony-forming units/ml (Fig. S2). In addition, we showed that the cells of the starting line and one of the randomly selected WBN clone do not differ in size (Fig. S3 and Supplementary Note S9), corroborating that higher OD600 values in evolved clones reflect higher cell numbers. Regression analysis demonstrated a positive correlation between growth rate and maximal cell density in the clones from four out of five WBN cell lines (Fig. S4). Taken together, P. aeruginosa WBN cell lines adapted to the conditions of the MA experiment by evolving shorter lag phase, faster growth rates, and increased growth yield.
Fig. 2. Growth characteristics of the evolved P. aeruginosa clones.

The growth of six P. aeruginosa starting cultures, six SBN cell lines, and the average growth of 30 clones from overall five WBN cell lines was recorded in standard LB medium (three independent experiments, technical triplicates) (A). The lag phase (B) and the growth rate (C) were calculated from growth curves using linear regression. D depicts the maximal ODs. Graph A shows average values and standard deviation, graphs B–D show individual values, average values, and standard deviation. The differences between growth parameters were calculated using the Mann–Whitney test. * corresponds to p < 0.05, ** corresponds to p < 0.01, and *** corresponds to p < 0.001.
In a control experiment, we analyzed the fitness of P. aeruginosa SBN cell lines. Under extreme bottlenecking conditions, genetic drift dominates evolution as the influence of natural selection is reduced and MA occurs mainly by chance [13]. In accordance with previous observations that random MA in SBN cultures results in a decrease in fitness [32], the analysis of growth behavior of the evolved P. aeruginosa SBN cell lines revealed no signs of adaptation to the LB cultivation conditions. Instead, the evolved SBN cell lines had a growth disadvantage. This was characterized by a longer lag phase and a slower growth during the first hours of cultivation (Fig. 2A, B).
We then investigated whether the fitness advantage of the P. aeruginosa WBN clones in LB produced trade-offs between growth in LB and in chemically defined nutrient-rich BM2 medium. We observed that the phenotypes of the studied clones ranged from the complete loss of ability to grow in BM2 to even increased growth rates, shortened lag phase, and higher maximal density in this medium (Fig. S5). Thus, the adaptation of the WBN clones to growth in LB did not necessarily produce fitness trade-offs in BM2 medium. Furthermore, the variability in BM2 growth parameters among the different clones indicates that they reached higher fitness in LB via different adaptive mechanisms.
S. enterica did not gain fitness upon sustained growth in LB
In an analogous MA experiment, WBN and SBN cell lines of mismatch-repair-deficient S. enterica were cultivated for 24 days in logarithmic phase. During this time, S. enterica cultures were passed through 45 bottlenecks. WBN cultures of S. enterica were grown for ~840 generations, and SBN cultures of S. enterica were grown for ~1000 generations, equivalent to 18 and 22 generations between bottlenecks, respectively. The subsequent evaluation of the growth behavior of the evolved S. enterica WBN and SBN cell lines revealed considerable differences to that of P. aeruginosa. While we observed a consistent increase in fitness of the P. aeruginosa WBN clones, the S. enterica WBN clones showed no significant increase in fitness (Fig. 3A, B). In addition, unlike the P. aeruginosa SBN cell lines, the S. enterica SBN cell lines exhibited no significant fitness loss.
Fig. 3. Growth characteristics of the evolved S. enterica clones.

The growth of six S. enterica starting cultures, six SBN cell lines, and the average growth of 30 clones from overall five WBN cell lines was recorded in LB medium + 0.2% glucose (three independent experiments, technical triplicates) (A). The lag phase of S. enterica was calculated from their growth curves using linear regression (B). Graph A shows average values and standard deviation and graph B shows individual values, average values, and standard deviation. No statistically significant differences between lag phases were identified as calculated using the Mann–Whitney test.
Intergenic indels show strong selection patterns in P. aeruginosa but not in S. enterica
For the identification of accumulated mutations, the WBN and SBN cell lines were subjected to whole-genome sequencing at the end of the MA experiments (Fig. 1). In addition, we sequenced the genomic DNA of the six starting cultures used to inoculate our WBN and SBN cell lines. The evolved P. aeruginosa WBN and SBN cell lines accumulated 123.2 ± 13.3 and 146.0 ± 15.7 mutations, respectively, which corresponded to 22 ± 2 and 23 ± 2 mutations/Mbp/103 generations. The evolved S. enterica WBN and SBN cell lines acquired 69.3 ± 8.4 and 80.7 ± 9.3 mutations, respectively, which corresponded to 17 ± 1 and 17 ± 2 mutations/Mbp/103 generations and thus closely resembled the mutation rates in P. aeruginosa. The rates were comparable to those reported previously in P. aeruginosa and Escherichia coli MA experiments using DNA repair-deficient strains [32, 33]. The most prevalent type of mutations were nucleotide substitutions (Fig. S6). As was to be expected in the hypermutator strains [32], the increase in mutation rates resulted primarily from the increase in the rate of transitions (Fig. S7). These accounted for more than 80% of all accumulated mutations and more than 95% of accumulated nucleotide substitutions in the WBN and SBN lines of P. aeruginosa and S. enterica.
To identify mutation patterns that reflect the P. aeruginosa fitness increase in the WBN and the fitness decay in the SBN cell lines, we performed a more in-depth mutation analysis. The rate of intragenic as well as intergenic nucleotide substitutions did not differ between WBN and SBN cell lines of P. aeruginosa or S. enterica. However, the intragenic mutation rate was ~25% higher in P. aeruginosa as compared to S. enterica (p < 0.001, Mann–Whitney test) (Fig. 4). Among the genes that harbored intragenic nucleotide substitutions, we did not identify an enrichment of any Clusters of Orthologous Groups (COG) functional category in the WBN or SBN cell lines, in both P. aeruginosa and S. enterica. Taken together, despite the overall higher single nucleotide polymorphism (SNP) rates in P. aeruginosa, no differences in nucleotide substitution rates between WBN and SBN cell lines in either of the species were detected, indicating that there was no clear selection pattern for SNPs in the various bottlenecking conditions of our MA experiment.
Fig. 4. The rates of nucleotide substitutions and indels according to location in coding and intergenic regions.
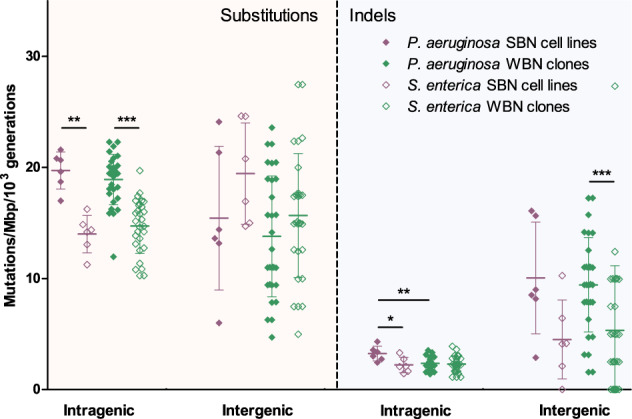
The individual values, the mean values, and the standard deviations for each class of mutations and each experimental group are shown. All the mutation rates are normalized to the number of generations in the corresponding experimental group. The rates of nucleotide substitutions and indels in the coding regions are normalized to the total length of coding regions in the corresponding species. The rates of nucleotide substitutions and indels in the intergenic regions are normalized to the total length of intergenic regions in the corresponding species. The differences between growth parameters were calculated using the Mann–Whitney test. * corresponds to p < 0.05, ** corresponds to p < 0.01, and *** corresponds to p < 0.001.
We then focused our analysis on indels. We found indels mostly in DNA homopolymeric tracts, which are the simplest of simple sequence repeats, and which are considered to be mutation hotspots [34]. The rate of intragenic indels did not differ between S. enterica WBN and SBN cell lines, and the rate of intragenic indels in P. aeruginosa WBN cell lines was comparable to that of the S. enterica WBN cell lines. However, the rate of intragenic indels in P. aeruginosa SBN cell lines was ~50% higher as compared to the P. aeruginosa WBN cell lines (p < 0.01, Mann–Whitney test) (Fig. 4). Intragenic indels frequently cause frameshifts, and frameshift variants tend to be more deleterious. The fact that we found less indels in the intragenic regions of the WBN cell lines indicates that they were subject to purifying selection in P. aeruginosa. At the same time, the intragenic indels in P. aeruginosa WBN cell lines showed no enrichment in any functional gene category. Thus, the indels that were found, did not seem to be under positive selection.
In the intergenic regions, the rate of indels did not differ between WBN and SBN cell lines of the same species (Fig. 4). However, the rate of intergenic indels was twice as high in P. aeruginosa as compared to S. enterica (p < 0.001 for WBN cell lines and p = 0.09 (nonsignificant) in SBN cell lines, Mann–Whitney test). Interestingly, we found an enrichment in intergenic indels that preceded genes of the functional COG category “cell wall/membrane/envelope biogenesis” (p < 0.01, chi-square test) exclusively in the P. aeruginosa WBN cell lines, indicating that there was a positive selection for the respective indels.
To further confirm that the indels in the intergenic homopolymeric tracts are under positive selection in the P. aeruginosa WBN clones, we re-analyzed the previously recorded genome sequences of 414 P. aeruginosa clinical isolates [35]. Thirty out of 45 P. aeruginosa indels that were exclusively found in WBN cell lines were also found in at least one and up to 213 clinical isolates. At the same time only 9 out of 22 indels that recorded exclusively in upstream gene regions in P. aeruginosa SBN cell lines were found in at least one clinical isolate. The significantly higher rate of intragenic WBN indels that were found in the clinical isolates (p < 0.05, chi-square test) implies that the intergenic indels in the P. aeruginosa WBN cell lines are subject to positive selection also in P. aeruginosa clinical isolates, probably, due to their effect on the transcription and/or translation of downstream genes. Since a majority of the indels are within homopolymeric tracts, phase variation might facilitate adaptation to rapidly varying environments without the requirement for random mutations.
Gene expression analysis in P. aeruginosa WBN cell lines showed convergent and divergent gene expression changes
To obtain a deeper understanding of how the genetic changes in the P. aeruginosa WBN clones are translated into fitness changes, we randomly selected three isolates from each of the five WBN cell lines and compared the gene expression profile of these 15 evolved WBN clones with that of the starting cell line (Table S1). The expression profiles of the three clones derived from each of the five WBN cell lines typically clustered together, reflecting their common origin (Fig. 5). Each clone also exhibited a number of unique gene expression changes. However, there were also transcriptional changes that were shared by the clonal isolates from different WBN cell lines. We found a functional enrichment of differentially expressed genes, which were assigned to the COG categories “signal transduction,” “cell motility,” and “intracellular trafficking, secretion, and vesicular transport” and to the PseudoCAP categories “cell wall/lipopolysaccharides/capsule,” “chemotaxis,” “motility and attachment,” “protein secretion/export apparatus” and “secreted factors (toxins, enzymes, alginate).” It was shown that the majority of WBN clones (12 of 15) exhibited downregulation of rsmY, which regulates the transition from planktonic lifestyle to biofilm lifestyle, and seven exhibited an additional downregulation of rsmZ (Fig. 5), which acts in synergy with rsmY. Six of the seven rsmZ/rsmY-low expressing clones, and one of the rsmY-low expressing clones, showed a significant upregulation of type-III secretion system (T3SS) genes, including PA14_14330 (spcS ortholog), exoY, pscBCDE, exsABC, PA14_42380 (exsD ortholog), PA14_42410 (exsE ortholog), popDBN, pcrDGHRV, pcr1-4 orthologs, pscNOPQRTU, and PA14_42630. The rsmZ/rsmY-downregulating clones also showed lower expression of type-VI secretion system (T6SS) genes (tagQ1R1S1T1, ppkA, pppA, tagF1J1, icmF1, tssA1, tssE1F1G1, tssJ1K1L1, PA14_00970 (fha ortholog), fha1, hsiB1, hsiC1, hcp1, clpV1) (Fig. 5). Four of the clones, which did not downregulate rsmZ, downregulated at least seven genes involved in chemotaxis signaling (pctA, PA14_20750 (cheW ortholog), PA14_20760 (cheR ortholog), and chemotaxis transducers PA14_28050, PA14_58650, PA14_61300, and PA14_67010) (Fig. 5). Interestingly, only one of four clones with downregulated expression of chemotaxis-related genes showed a minor upregulation of the T3SS, while the other three clones showed upregulation of the T6SS. Last, the downregulation of four genes known to encode di-guanylate cyclases (morA, PA14_42220 (mucR ortholog), PA14_64050 (pleD ortholog), and PA14_56280 (sadC ortholog)) were found in two thirds of the clones (9 out of 15). Changes in expression of rsmZ, rsmY, and genes encoding T3SS, T6SS, and di-guanylate cyclases are well known to be linked to the switch between motile and sessile lifestyles [36–43]. This prompted us to investigate if the aforementioned alterations in gene expression result in coherent changes in motility, virulence, and biofilm formation.
Fig. 5. Heat map depicting the selected gene expression changes of the evolved clones.

RNA sequencing was performed for three randomly selected clonal isolates from each WBN cell line. Gene expression changes in 15 WBN clones as compared to the starting cell line are indicated as a color code. Selected genes that were found to be differentially regulated in at least four WBN clones and that belonged to the enriched COG categories (“signal transduction,” “cell motility,” and “intracellular trafficking, secretion, and vesicular transport”) are shown.
Swimming motility, virulence, and biofilm formation were highly variable between individual evolved clones
We observed a high variation in swimming motility between individual WBN clones (Fig. 6). Their phenotypes ranged from the complete absence of swimming motility to much higher motility than observed for the starting cell line. The differences were high even between WBN clones that belonged to the same cell line. As expected, all four WBN clones with low expression of chemotaxis genes showed low swimming motility.
Fig. 6. Motility phenotypes of the evolved clones.

Swimming motility of the starting cell line and of the 15 WBN clones with recorded transcriptional profiles was estimated based on the diameter of the swimming zones on BM2 agar plates. The graph shows individual values, average values, and standard deviation (three independent experiments with three technical replicates). To facilitate the comparison of phenotypic data and transcriptional profiles, the WBN clones are arranged along x axis in the same order as they are arranged by DESeq2 clustering analysis.
Similarly to swimming motility, virulence of the evolved WBN clones, as determined by the use of the G. mellonella model, showed extreme variability with phenotypes ranging from almost avirulent to more virulent than the starting cell line (Fig. 7). Moreover, virulence also differed between clones derived from the same WBN cell line. Of note, virulence of WBN clones did not correlate with the expression of the T3SS. The estimation of biofilm production using confocal microscopy and the crystal violet assay demonstrated that the WBN clones had variable phenotypes (Figs. 8A–C and S8) but did not surpass the starting cell line according to biofilm thickness and volume.
Fig. 7. Pathogenicity phenotypes of the evolved clones in the Galleria mellonella assay.

Survival rates of G. mellonella after 24 h (A) and 30 h (B) following infection of the larvae with the starting cell line and the 15 WBN clones with recorded transcriptional profiles are shown. PBS was used as negative control. The graph shows individual values, average values, and standard deviation (in at least two independent experiments with two technical replicates, minimally 40 larvae per clone were infected.). To facilitate the comparison of phenotypic data and transcriptional profiles, the WBN clones are arranged along x axis in the same order as they are arranged by DESeq2 clustering analysis.
Fig. 8. Biofilm phenotypes of the evolved clones.

Biofilm thickness (A) and biofilm volume (B) was recorded in two independent experiments, two technical replicates using automated confocal microscopy. Biofilm-forming capacity according to the crystal violet assay (C) was recorded in two independent experiments with eight technical replicates. All the graphs show individual and average values and standard deviation. To facilitate the comparison of phenotypic data and transcriptional profiles, the WBN clones are arranged along x axis in the same order as they are arranged by DESeq2 clustering analysis.
Discussion
P. aeruginosa has become a model organism to study experimental evolution and adaptation to diverse and challenging conditions, including adaptation to various carbon sources [44], antibiotic pressure [45], biofilms [46], and conditions encountered during an infection process [47–49]. In this study, we subjected hypermutator strains of P. aeruginosa to continuous exponential phase cultivation in rich LB medium under SBN and WBN regimens. While the fitness of P. aeruginosa SBN cell lines consistently decayed, competition, enabled by passaging P. aeruginosa WBN cell lines through 100-cell bottlenecks, led to the selection of P. aeruginosa clones with increased fitness in all five independently propagated WBN cell lines. Growth parameters that were under selective pressure showed improvement in almost all evolved P. aeruginosa WBN clones. Moreover, roughly half of the P. aeruginosa WBN clones developed higher cell densities in stationary phase, despite a lack of selection for this trait. In the majority of WBN clones, we observed a positive correlation between the growth rate and yield, which distinguished our results from the previous observations of a trade-off between growth rate and growth yield in bacteria characterized by faster growth [50–56]. Nevertheless, in one of the lines (WBN2 cell line) there was evidence for a growth rate/yield trade-off as predicted previously [57].
In BM2, an alternative medium, approximately half of WBN clones retained their full growth advantage. Thus, though adaptation of P. aeruginosa to one condition produced fitness trade-offs in other conditions, adaptation of P. aeruginosa to one condition also led to simultaneous improvement of fitness in selected alternative conditions as it was previously shown in a different range of bacterial species [13, 15, 58].
Furthermore, our finding of varying growth curve changes in LB and in BM2 medium indicates that P. aeruginosa WBN clones acquired fitness via different and independent mechanisms. Of note, passaging S. enterica in LB did not result in a change of fitness under any of the cultivation regimens even though the numbers of mutations accumulated by S. enterica and P. aeruginosa were comparable. Further research would be needed to establish which traits account for different fitness changes in the studied species.
Our finding, and that of a previous study, which reported no increase of growth rates of S. enterica following serial propagation in LB medium [59], seems to be contrasting the results of a long-term evolution experiment, in which E. coli evolved a shorter lag phase and higher growth rates [60]. However, the experiment was performed in minimal medium with glucose as the sole carbon source, and E. coli adapted by increasing glucose uptake [61].
Closer inspection of the mutation profiles of the evolved P. aeruginosa and S. enterica hypermutator strains revealed that transitions accounted for more than 95% of the nucleotide substitutions and more than 80% of total accumulated mutations. This is in line with previous reports on hypermutator mutation profiles [32, 62]. Despite the fact that most mutations are weakly deleterious [63], SNPs did not seem to be subject of purifying selection in our experimental conditions. According to the drift-barrier hypothesis, weak purifying selection may indicate that the disadvantage of most nucleotide substitutions is so small that the selection against them is insufficient to prevent their fixation [64]. Furthermore, we did not observe an enrichment of SNPs in any functional gene category in P. aeruginosa WBN cell lines, which exhibited increased fitness. Thus, on the genome scale, SNPs did not show evidence of positive selection. However, one cannot exclude that the fitness increase in each of WBN clones is at least partially driven by very few strongly beneficial SNPs in genes from different functional categories.
In contrast to SNPs, the indels, the majority of which were found in DNA homopolymeric tracts, displayed clear patterns of natural selection. We found a lower frequency of indels within the coding sequences in P. aeruginosa WBN cell lines. Under the assumption that those indels frequently cause frameshifts and thus negatively affect the gene function, their higher frequency in the SBN cell lines might explain the loss of fitness, while their lower frequency in the WBN cell lines might be the result of purifying selection. Unlike the intragenic indels, the intergenic indels were not counter-selected and became fixed in P. aeruginosa WBN cell lines. Homopolymeric tract-mediated phase variation has been identified as an adaptive mechanism in a wide range of bacteria [5, 34]. Interestingly, intergenic indels that were exclusively found in P. aeruginosa WBN cell lines were enriched in the upstream regions of the genes involved in membrane and cell wall biogenesis, implicating that they were positively selected. Most notably, a large part of intergenic indels encountered in the adapted WBN cell lines, but not in maladapted SBN cell lines, could also be found in a collection of more than 400 clinical P. aeruginosa isolates. Our results suggest that, in P. aeruginosa, indels in intergenic homopolymeric tracts promote adaptation both, to our experimental conditions and to the environment of the human host. The data are also supported by a recent study, which highlighted the importance of mutations in the intergenic regions for the adaptation of P. aeruginosa to the conditions of cystic fibrosis lungs [65]. We suggest that changes in the length of intergenic homopolymeric tracts mediate phase variation, giving rise to a plethora of phenotypically distinct individuals, which may have advantages in colonizing new niches or in coping with adverse conditions [4]. Since efficient adaptation seemed to be based on the positive selection of intergenic indels in our experimental setup, and since indels are not known to be enriched in hypermutator strains, our data also imply that indels in intergenic homopolymeric tracts might not only promote adaptation of P. aeruginosa hypermutator but also wild-type populations.
In order to elucidate whether there is another common theme associated with P. aeruginosa adaptation to fast growth, we performed transcriptional profiling. Indeed, we found transcriptional changes that were shared between P. aeruginosa WBN clones even if they originated from independently evolved cell lines. These results indicate that independent groups of adapting clones followed partly convergent evolutional paths, which has been previously shown both for P. aeruginosa [47, 49] and closely related P. fluorescens [66]. Common transcriptional changes mainly affected signaling, motility, secretion, and virulence factor production. More specifically, we found a group of clones that exhibited a decreased expression of rsmZ and rsmY, associated with an increased expression of the T3SS and decreased expression of the T6SS, and another group of clones with a decreased expression of chemotaxis transducers and an induced T6SS. Notably, the trade-off between chemotaxis, motility, and the rapid growth has been described as typical in various bacterial species [67–70]. Also, two thirds of all the clones showed a decreased expression of cyclases that produce the intracellular signaling molecule c-di-GMP.
Low levels of c-di-GMP and low transcription of rsmZ and rsmY are associated with planktonic lifestyles, characterized by fast growth, high swimming motility, and high virulence [36–43], whereas high levels of c-di-GMP contribute to the switch from planktonic to biofilm lifestyles [40, 71, 72]. Many cyclases increase cellular pools of c-di-GMP locally, thereby impacting selected downstream effector molecules. As a result, the impact of the activity of different cyclases on motility, biofilm, and virulence phenotypes can vary extensively [40, 73–77] e.g., whereas gcbA, a PA14_64050 homolog, was shown to impact motility, surface attachment, and biofilm dispersal, but not biofilm formation [78, 79], morA showed strain-dependent effects on motility [80, 81] and biofilm formation [77, 81].
The results of the transcriptional analysis and phenotypic assays imply that, in P. aeruginosa, adapted clones occupy a large space between the two extreme modes of growth—rapid planktonic growth associated with high virulence and slow biofilm-associated growth with reduced motility and virulence. It seems that the phenotypes of the individual evolved clones are impacted by the variable expression of small regulatory rRNAs, such as rsmY and rsmZ. In addition, the evolved phenotypes could be the result of an adaptation of the activity of different c-di-GMP modifying enzymes (>45 can be found in P. aeruginosa PA14 [77]), which modulate local c-di-GMP pools with different outcomes for motility and virulence. The overall redundancy and local specificity of these regulatory pathways of P. aeruginosa allow a convergent shift from the biofilm to planktonic lifestyle, beneficial under the experimental conditions, whereas individual evolved clones still display divergent biofilm, virulence, and motility phenotypes. It must, however, be considered that in our experimental setup an extensive diversification might have resulted from using a hypermutator strain of P. aeruginosa. Nevertheless, rapid diversification in nutritionally or structurally complex environments has been demonstrated before for mutator as well as non-mutator P. aeruginosa populations [46, 48]. A bet-hedging strategy could ensure that, on the population level, P. aeruginosa maintains its versatility and preserves high adaptive potential.
In conclusion, we demonstrated that the WBN clones of the hypermutator P. aeruginosa strain evolved higher fitness in LB through a multitude of different pathways related to membrane biogenesis and fine-tuning of the balance between fast growing planktonic and sessile biofilm-associated lifestyles. We speculate that phase variation due to indels in intergenic homopolymeric tracts and adaptive changes engaging complex regulatory circuits with highly redundant c-di-GMP metabolic enzymes act in synergy, allowing P. aeruginosa to produce phenotypic diversity, maintain high versatility, and efficiently colonize new habitats.
Supplementary information
Acknowledgements
S.H. was funded by the EU (ERC Consolidator Grant COMBAT 724290) and received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC 2155 “RESIST”—Project ID 39087428. We thank M. Erhardt for providing S. enterica LT2 ∆mutS, M. Preusse and J. Tomasch for assistance with the bioinformatic analysis, M. Müsken for performing electron microscopy experiments and assistance with the biofilm image analysis, and A. Arce-Rodríguez for fruitful discussions. IG designed and performed the experiments and analyzed the data. JGT performed confocal microscopy experiments. AK performed virulence experiments. IG and SH wrote the paper. SH acquired funding.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
The DNA sequencing data are deposited in the National Center for Biotechnology Information sequence read archive (http://www.ncbi.nlm.nih.gov/sra) under the BioProject accession no. PRJNA612188. The RNA sequencing data are deposited in the GEO repository (https://www.ncbi.nlm.nih.gov/geo) with the accession no. GSE146906. Custom scripts used to facilitate the processing of sequencing data can be provided upon request.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
2/11/2021
The following funding note was missing: Open Access funding enabled and organized by Projekt DEAL.
Supplementary information
The online version of this article (10.1038/s41396-020-00841-6) contains supplementary material, which is available to authorized users.
References
- 1.Walker TS, Bais HP, Deziel E, Schweizer HP, Rahme LG, Fall R, et al. Pseudomonas aeruginosa-plant root interactions. Pathogenicity, biofilm formation, and root exudation. Plant Physiol. 2004;134:320–31. doi: 10.1104/pp.103.027888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rashid MH, Kornberg A. Inorganic polyphosphate is needed for swimming, swarming, and twitching motilities of Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 2000;97:4885–90. doi: 10.1073/pnas.060030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grenier S, Barre P, Litrico I. Phenotypic plasticity and selection: nonexclusive mechanisms of adaptation. Scientifica. 2016;2016:7021701. doi: 10.1155/2016/7021701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phillips ZN, Tram G, Seib KL, Atack JM. Phase-variable bacterial loci: how bacteria gamble to maximise fitness in changing environments. Biochem Soc Trans. 2019;47:1131–41. doi: 10.1042/BST20180633. [DOI] [PubMed] [Google Scholar]
- 5.Orsi RH, Bowen BM, Wiedmann M. Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genom. 2010;11:102. doi: 10.1186/1471-2164-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy GL, Connell TD, Barritt DS, Koomey M, Cannon JG. Phase variation of gonococcal protein II: regulation of gene expression by slipped-strand mispairing of a repetitive DNA sequence. Cell. 1989;56:539–47. doi: 10.1016/0092-8674(89)90577-1. [DOI] [PubMed] [Google Scholar]
- 7.Stibitz S, Aaronson W, Monack D, Falkow S. Phase variation in Bordetella pertussis by frameshift mutation in a gene for a novel two-component system. Nature. 1989;338:266–9. doi: 10.1038/338266a0. [DOI] [PubMed] [Google Scholar]
- 8.Palmer ME, Lipsitch M, Moxon ER, Bayliss CD. Broad conditions favor the evolution of phase-variable loci. mBio. 2013;4:e00430–12. doi: 10.1128/mBio.00430-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zieg J, Silverman M, Hilmen M, Simon M. Recombinational switch for gene expression. Science. 1977;196:170–2. doi: 10.1126/science.322276. [DOI] [PubMed] [Google Scholar]
- 10.De Ste Croix M, Chen KY, Vacca I, Manso AS, Johnston C, Polard P, et al. Recombination of the phase-variable spnIII locus is independent of all known pneumococcal site-specific recombinases. J Bacteriol. 2019;201:e00233–19. [DOI] [PMC free article] [PubMed]
- 11.Koomey M, Gotschlich EC, Robbins K, Bergstrom S, Swanson J. Effects of recA mutations on pilus antigenic variation and phase transitions in Neisseria gonorrhoeae. Genetics. 1987;117:391–8. doi: 10.1093/genetics/117.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez-Romero MA, Casadesus J. The bacterial epigenome. Nat Rev Microbiol. 2020;18:7–20. doi: 10.1038/s41579-019-0286-2. [DOI] [PubMed] [Google Scholar]
- 13.Van den Bergh B, Swings T, Fauvart M, Michiels J. Experimental design, population dynamics, and diversity in microbial experimental evolution. Microbiol Mol Biol Rev. 2018;82. [DOI] [PMC free article] [PubMed]
- 14.Philippe N, Pelosi L, Lenski RE, Schneider D. Evolution of penicillin-binding protein 2 concentration and cell shape during a long-term experiment with Escherichia coli. J Bacteriol. 2009;191:909–21. doi: 10.1128/JB.01419-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leiby N, Marx CJ. Metabolic erosion primarily through mutation accumulation, and not tradeoffs, drives limited evolution of substrate specificity in Escherichia coli. PLoS Biol. 2014;12:e1001789. doi: 10.1371/journal.pbio.1001789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kvitek DJ, Sherlock G. Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet. 2013;9:e1003972. doi: 10.1371/journal.pgen.1003972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Brien EJ, Utrilla J, Palsson BO. Quantification and classification of E. coli proteome utilization and unused protein costs across environments. PLoS Comput Biol. 2016;12:e1004998. doi: 10.1371/journal.pcbi.1004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper VS, Lenski RE. The population genetics of ecological specialization in evolving Escherichia coli populations. Nature. 2000;407:736–9. doi: 10.1038/35037572. [DOI] [PubMed] [Google Scholar]
- 19.Wahl LM, Gerrish PJ. The probability that beneficial mutations are lost in populations with periodic bottlenecks. Evol; Int J Org Evol. 2001;55:2606–10. doi: 10.1111/j.0014-3820.2001.tb00772.x. [DOI] [PubMed] [Google Scholar]
- 20.de Visser JA, Rozen DE. Limits to adaptation in asexual populations. J Evol Biol. 2005;18:779–88. doi: 10.1111/j.1420-9101.2005.00879.x. [DOI] [PubMed] [Google Scholar]
- 21.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A. 2006;103:2833–8. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–9. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Overhage J, Bains M, Brazas MD, Hancock RE. Swarming of Pseudomonas aeruginosa is a complex adaptation leading to increased production of virulence factors and antibiotic resistance. J Bacteriol. 2008;190:2671–9. doi: 10.1128/JB.01659-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyata S, Casey M, Frank DW, Ausubel FM, Drenkard E. Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect Immun. 2003;71:2404–13. doi: 10.1128/IAI.71.5.2404-2413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Müsken M, Di Fiore S, Römling U, Häussler S. A 96-well-plateg-based optical method for the quantitative and qualitative evaluation of Pseudomonas aeruginosa biofilm formation and its application to susceptibility testing. Nat Protoc. 2010;5:1460–9. doi: 10.1038/nprot.2010.110. [DOI] [PubMed] [Google Scholar]
- 30.Thoming JG, Tomasch J, Preusse M, Koska M, Grahl N, Pohl S, et al. Parallel evolutionary paths to produce more than one Pseudomonas aeruginosa biofilm phenotype. NPJ Biofilms Microbiomes. 2020;6:2. doi: 10.1038/s41522-019-0113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Toole GA. Microtiter dish biofilm formation assay. J Vis Exp. 2011;47:2437. [DOI] [PMC free article] [PubMed]
- 32.Heilbron K, Toll-Riera M, Kojadinovic M, MacLean RC. Fitness is strongly influenced by rare mutations of large effect in a microbial mutation accumulation experiment. Genetics. 2014;197:981–90. doi: 10.1534/genetics.114.163147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wielgoss S, Barrick JE, Tenaillon O, Wiser MJ, Dittmar WJ, Cruveiller S, et al. Mutation rate dynamics in a bacterial population reflect tension between adaptation and genetic load. Proc Natl Acad Sci U S A. 2013;110:222–7. doi: 10.1073/pnas.1219574110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin WH, Kussell E. Evolutionary pressures on simple sequence repeats in prokaryotic coding regions. Nucleic Acids Res. 2012;40:2399–413. doi: 10.1093/nar/gkr1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khaledi A, Weimann A, Schniederjans M, Asgari E, Kuo TH, Oliver A, et al. Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol Med. 2020;12:e10264. doi: 10.15252/emmm.201910264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valentini M, Gonzalez D, Mavridou DA, Filloux A. Lifestyle transitions and adaptive pathogenesis of Pseudomonas aeruginosa. Curr Opin Microbiol. 2018;41:15–20. doi: 10.1016/j.mib.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Dotsch A, Eckweiler D, Schniederjans M, Zimmermann A, Jensen V, Scharfe M, et al. The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS ONE. 2012;7:e31092. doi: 10.1371/journal.pone.0031092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lapouge K, Schubert M, Allain FH, Haas D. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol. 2008;67:241–53. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- 39.Moscoso JA, Jaeger T, Valentini M, Hui K, Jenal U, Filloux A. The diguanylate cyclase SadC is a central player in Gac/Rsm-mediated biofilm formation in Pseudomonas aeruginosa. J Bacteriol. 2014;196:4081–8. doi: 10.1128/JB.01850-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valentini M, Filloux A. Multiple roles of c-di-GMP signaling in bacterial pathogenesis. Annu Rev Microbiol. 2019;73:387–406. doi: 10.1146/annurev-micro-020518-115555. [DOI] [PubMed] [Google Scholar]
- 41.Sisti F, Ha DG, O’Toole GA, Hozbor D, Fernandez J. Cyclic-di-GMP signalling regulates motility and biofilm formation in Bordetella bronchiseptica. Microbiology. 2013;159:869–79. doi: 10.1099/mic.0.064345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–73. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 43.Romling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toll-Riera M, San Millan A, Wagner A, MacLean RC. The genomic basis of evolutionary innovation in Pseudomonas aeruginosa. PLoS Genet. 2016;12:e1006005. doi: 10.1371/journal.pgen.1006005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gifford DR, Furio V, Papkou A, Vogwill T, Oliver A, MacLean RC. Identifying and exploiting genes that potentiate the evolution of antibiotic resistance. Nat Ecol Evol. 2018;2:1033–9. doi: 10.1038/s41559-018-0547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flynn KM, Dowell G, Johnson TM, Koestler BJ, Waters CM, Cooper VS. Evolution of ecological diversity in biofilms of Pseudomonas aeruginosa by altered cyclic diguanylate signaling. J Bacteriol. 2016;198:2608–18. doi: 10.1128/JB.00048-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Racey D, Inglis RF, Harrison F, Oliver A, Buckling A. The effect of elevated mutation rates on the evolution of cooperation and virulence of Pseudomonas aeruginosa. Evol; Int J Org Evol. 2010;64:515–21. doi: 10.1111/j.1558-5646.2009.00821.x. [DOI] [PubMed] [Google Scholar]
- 48.Schick A, Kassen R. Rapid diversification of Pseudomonas aeruginosa in cystic fibrosis lung-like conditions. Proc Natl Acad Sci USA. 2018;115:10714–9. doi: 10.1073/pnas.1721270115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Granato ET, Ziegenhain C, Marvig RL, Kummerli R. Low spatial structure and selection against secreted virulence factors attenuates pathogenicity in Pseudomonas aeruginosa. ISME J. 2018;12:2907–18. doi: 10.1038/s41396-018-0231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lipson DA. The complex relationship between microbial growth rate and yield and its implications for ecosystem processes. Front Microbiol. 2015;6:615. doi: 10.3389/fmicb.2015.00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu Y, Beck DA, Lidstrom ME. Difference in C3-C4 metabolism underlies tradeoff between growth rate and biomass yield in Methylobacterium extorquens AM1. BMC Microbiol. 2016;16:156. doi: 10.1186/s12866-016-0778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bachmann H, Fischlechner M, Rabbers I, Barfa N, Branco dos Santos F, Molenaar D, et al. Availability of public goods shapes the evolution of competing metabolic strategies. Proc Natl Acad Sci U S A. 2013;110:14302–7. doi: 10.1073/pnas.1308523110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Novak M, Pfeiffer T, Lenski RE, Sauer U, Bonhoeffer S. Experimental tests for an evolutionary trade-off between growth rate and yield in E. coli. Am Nat. 2006;168:242–51. doi: 10.1086/506527. [DOI] [PubMed] [Google Scholar]
- 54.Cheng C, O’Brien EJ, McCloskey D, Utrilla J, Olson C, LaCroix RA, et al. Laboratory evolution reveals a two-dimensional rate-yield tradeoff in microbial metabolism. PLoS Comput Biol. 2019;15:e1007066. doi: 10.1371/journal.pcbi.1007066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basan M, Hui S, Okano H, Zhang Z, Shen Y, Williamson JR, et al. Overflow metabolism in Escherichia coli results from efficient proteome allocation. Nature. 2015;528:99–104. doi: 10.1038/nature15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mori M, Hwa T, Martin OC, De Martino A, Marinari E. Constrained allocation flux balance analysis. PLoS Comput Biol. 2016;12:e1004913. doi: 10.1371/journal.pcbi.1004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504–7. doi: 10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez-Verdugo A, Gaut BS, Tenaillon O. Evolution of Escherichia coli rifampicin resistance in an antibiotic-free environment during thermal stress. BMC Evol Biol. 2013;13:50. doi: 10.1186/1471-2148-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knoppel A, Knopp M, Albrecht LM, Lundin E, Lustig U, Nasvall J, et al. Genetic adaptation to growth under laboratory conditions in Escherichia coli and Salmonella enterica. Front Microbiol. 2018;9:756. doi: 10.3389/fmicb.2018.00756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farida V, Travisano M, Lenski RE. Long-term experimental evolution in Escherichi coli. II. Changes in life-history traits during adaptation to a seasonal environment. Am Nat. 1994;144:432–56. doi: 10.1086/285685. [DOI] [Google Scholar]
- 61.Travisano M, Vasi F, Lenski RE. Long-term experimental evolution in Escherichia Coli. III. Variation among replicate populations in correlated responses to novel environments. Evol; Int J Org Evol. 1995;49:189–200. doi: 10.1111/j.1558-5646.1995.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 62.Couce A, Caudwell LV, Feinauer C, Hindre T, Feugeas JP, Weigt M, et al. Mutator genomes decay, despite sustained fitness gains, in a long-term experiment with bacteria. Proc Natl Acad Sci USA. 2017;114:E9026–E35. doi: 10.1073/pnas.1705887114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Trindade S, Perfeito L, Gordo I. Rate and effects of spontaneous mutations that affect fitness in mutator Escherichia coli. Philos Trans R Soc Lond B Biol Sci. 2010;365:1177–86. doi: 10.1098/rstb.2009.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lynch M, Ackerman MS, Gout JF, Long H, Sung W, Thomas WK, et al. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. 2016;17:704–14. doi: 10.1038/nrg.2016.104. [DOI] [PubMed] [Google Scholar]
- 65.Khademi SMH, Sazinas P, Jelsbak L. Within-host adaptation mediated by intergenic evolution in Pseudomonas aeruginosa. Genome Biol Evol. 2019;11:1385–97. doi: 10.1093/gbe/evz083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vogwill T, Phillips RL, Gifford DR, MacLean RC. Divergent evolution peaks under intermediate population bottlenecks during bacterial experimental evolution. Proc R Soc B Biol Sci. 2016;283:20160749. [DOI] [PMC free article] [PubMed]
- 67.Martinez-Garcia E, Nikel PI, Chavarria M, de Lorenzo V. The metabolic cost of flagellar motion in Pseudomonas putida KT2440. Environ Microbiol. 2014;16:291–303. doi: 10.1111/1462-2920.12309. [DOI] [PubMed] [Google Scholar]
- 68.Wang J, Ma W, Wang Y, Lin L, Wang T, Wang Y, et al. Deletion of 76 genes relevant to flagella and pili formation to facilitate polyhydroxyalkanoate production in Pseudomonas putida. Appl Microbiol Biotechnol. 2018;102:10523–39. doi: 10.1007/s00253-018-9439-x. [DOI] [PubMed] [Google Scholar]
- 69.Ni B, Ghosh B, Paldy FS, Colin R, Heimerl T, Sourjik V. Evolutionary remodeling of bacterial motility checkpoint control. Cell Rep. 2017;18:866–77. doi: 10.1016/j.celrep.2016.12.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cui Z, Yang CH, Kharadi RR, Yuan X, Sundin GW, Triplett LR, et al. Cell-length heterogeneity: a population-level solution to growth/virulence trade-offs in the plant pathogen Dickeya dadantii. PLoS Pathog. 2019;15:e1007703. doi: 10.1371/journal.ppat.1007703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Conner JG, Zamorano-Sanchez D, Park JH, Sondermann H, Yildiz FH. The ins and outs of cyclic di-GMP signaling in Vibrio cholerae. Curr Opin Microbiol. 2017;36:20–9. doi: 10.1016/j.mib.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Teschler JK, Zamorano-Sanchez D, Utada AS, Warner CJ, Wong GC, Linington RG, et al. Living in the matrix: assembly and control of Vibrio cholerae biofilms. Nat Rev Microbiol. 2015;13:255–68. doi: 10.1038/nrmicro3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valentini M, Laventie BJ, Moscoso J, Jenal U, Filloux A. The diguanylate cyclase HsbD intersects with the HptB regulatory cascade to control Pseudomonas aeruginosa biofilm and motility. PLoS Genet. 2016;12:e1006354. doi: 10.1371/journal.pgen.1006354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dahlstrom KM, O’Toole GA. A symphony of cyclases: specificity in diguanylate cyclase signaling. Annu Rev Microbiol. 2017;71:179–95. doi: 10.1146/annurev-micro-090816-093325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sarenko O, Klauck G, Wilke FM, Pfiffer V, Richter AM, Herbst S, et al. More than enzymes that make or break cyclic Di-GMP-local signaling in the interactome of GGDEF/EAL domain proteins of Escherichia coli. mBio. 2017;8. 10.1128/mBio.01639-17. [DOI] [PMC free article] [PubMed]
- 76.Zamorano-Sanchez D, Xian W, Lee CK, Salinas M, Thongsomboon W, Cegelski L, et al. Functional specialization in vibrio cholerae diguanylate cyclases: distinct modes of motility suppression and c-di-GMP production. mBio. 2019;10. [DOI] [PMC free article] [PubMed]
- 77.Kulasakara H, Lee V, Brencic A, Liberati N, Urbach J, Miyata S, et al. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3’-5’)-cyclic-GMP in virulence. Proc Natl Acad Sci USA. 2006;103:2839–44. doi: 10.1073/pnas.0511090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Petrova OE, Cherny KE, Sauer K. The Pseudomonas aeruginosa diguanylate cyclase GcbA, a homolog of P. fluorescens GcbA, promotes initial attachment to surfaces, but not biofilm formation, via regulation of motility. J Bacteriol. 2014;196:2827–41. doi: 10.1128/JB.01628-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Petrova OE, Cherny KE, Sauer K. The diguanylate cyclase GcbA facilitates Pseudomonas aeruginosa biofilm dispersion by activating BdlA. J Bacteriol. 2015;197:174–87. doi: 10.1128/JB.02244-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meissner A, Wild V, Simm R, Rohde M, Erck C, Bredenbruch F, et al. Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ Microbiol. 2007;9:2475–85. doi: 10.1111/j.1462-2920.2007.01366.x. [DOI] [PubMed] [Google Scholar]
- 81.Choy WK, Zhou L, Syn CK, Zhang LH, Swarup S. MorA defines a new class of regulators affecting flagellar development and biofilm formation in diverse Pseudomonas species. J Bacteriol. 2004;186:7221–8. doi: 10.1128/JB.186.21.7221-7228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DNA sequencing data are deposited in the National Center for Biotechnology Information sequence read archive (http://www.ncbi.nlm.nih.gov/sra) under the BioProject accession no. PRJNA612188. The RNA sequencing data are deposited in the GEO repository (https://www.ncbi.nlm.nih.gov/geo) with the accession no. GSE146906. Custom scripts used to facilitate the processing of sequencing data can be provided upon request.