

Abstract
Nitrogen-centered radicals are powerful reaction intermediates owing in part to their ability to guide position-selective C(sp3)–H functionalization reactions. Typically, these reactive species dictate the site of functionalization by preferentially engaging in 1,5-hydrogen-atom transfer (1,5-HAT) processes. Broadly relevant approaches to alter the site-selectivity of HAT pathways would be valuable because they could be paired with a variety of tactics to install diverse functional groups. Yet, until recently, there have been no generalizable strategies to modify the position-selectivity observed in these HAT processes. This Synpacts article reviews transformations in which nitrogen-centered radicals preferentially react through 1,6-HAT pathways. Specific attention will be focused on strategies that employ alcohol- and amine-anchored sulfamate esters and sulfamides as templates to achieve otherwise rare γ-selective functionalization reactions.
Keywords: hydrogen-atom transfer, remote C–H functionalization, radicals, nitrogen-centered radicals, sulfamate ester, sulfamide
Graphical Abstract
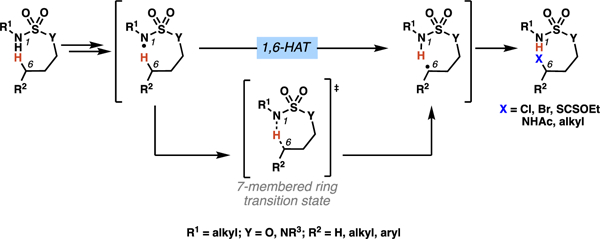
1. Introduction
Position-selective C(sp3)–H functionalization reactions have emerged as formidable strategies to manipulate complex molecular scaffolds.1 Recently, innovations in methods to access and employ radical intermediates have contributed to a dramatic expansion in C(sp3)–H functionalization technologies.2 Among reactive radical species, nitrogen-centered radicals are particularly useful,3,4 as they can facilitate position-selective hydrogen-atom transfer (HAT) processes to furnish carbon-centered radicals.5 These carbon-centered radical intermediates are poised to be trapped by a range of reagents to furnish diverse molecular architectures. Consequently, intramolecular HAT pathways mediated by nitrogen-centered radicals have become powerful and practical approaches to enable selective C(sp3)–H functionalization reactions.6
In this context, nitrogen-centered radicals predominantly participate in 1,5-HAT processes (Scheme 1).7 These 1,5-HAT pathways engage six-membered ring transition states and are generally kinetically favored over transition states of other ring sizes. Smaller ring transition states are enthalpically disfavored, as they cannot easily accommodate the necessary geometry for HAT to proceed. Specifically, HAT processes require a nearly colinear arrangement of the nitrogen-centered radical, the abstracted hydrogen atom and the pendant carbon atom.8 For larger rings, transition state preorganization incurs an entropic penalty due to the increased number of endocyclic atoms. As a result of these energetic factors, nitrogen-centered radicals engage in predictable and selective intramolecular HAT processes, which make precursors to nitrogen-centered radicals attractive directing moieties for C(sp3)–H functionalization reactions.
Scheme 1.
Traditionally, nitrogen-centered radicals react through kinetically favored 6-membered ring transition states resulting in 1,5-HAT processes.
The Hofmann-Löffler-Freytag Reaction
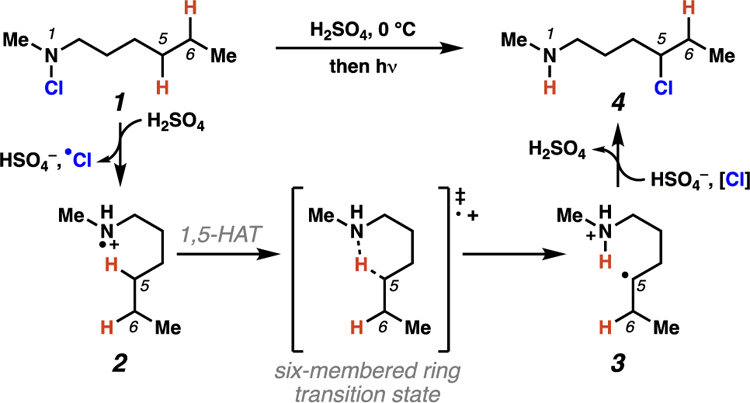
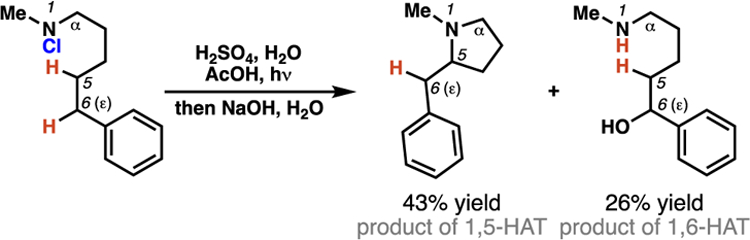
The Hofmann-Löffler-Freytag (HLF) reaction is the earliest example of a directed C–H functionalization process.9 To achieve site-selectivity, HLF reactions make use of the 1,5-HAT processes mediated by nitrogen-centered radicals. In traditional HLF transformations, N-haloamines (i.e. 1) serve as precursors to reactive nitrogen-centered radical intermediates. The earliest examples of HLF reactions are conducted in strongly acidic media (i.e. neat sulfuric acid, c.f. Schemes 1,2), and rely on direct thermal or photochemical homolysis of a weak nitrogen-halogen bond to access the critical nitrogen-centered radical (i.e. 2). Selective, intramolecular 1,5-HAT then ensues to furnish carbon-centered radical intermediates. Traditionally, these carbon-centered radicals trap a halogen atom to yield alkyl halide intermediates (i.e. 4), which can be treated with base to promote cyclization to pyrrolidine compounds (i.e. 5).
Scheme 2.
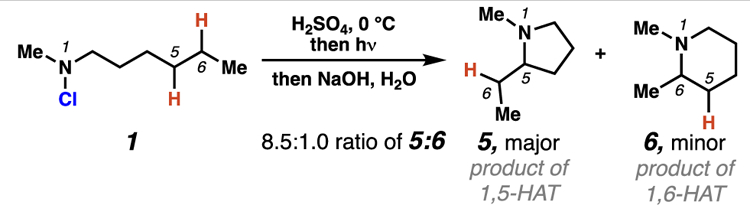
In 1960, E. J. Corey and Hertler determined that in some traditional HLF reactions, where position-selectivity arises predominantly from 1,5-HAT processes, minor products can form through 1,6-HAT processes.
To improve on the harsh conditions associated with early examples of the HLF reaction, subsequent investigations demonstrated that these transformations can proceed in neutral or basic media. In the 1980s, Suárez and co-workers identified a protocol for in situ oxidation, expanding the range of viable substrates.10 More recently, tactical advances in photo- and electrochemical approaches have resulted in further expansion of the HLF reaction.11 With the emergence of these versatile strategies to access nitrogen-centered radicals, opportunities to employ the HLF reaction have increased substantially. Despite the renewed focus on the HLF reaction, strategies to alter the site-selectivity of HAT have remained limited.
Evolution of 1,6-HAT Processes in HLF Reactions
While pyrrolidines are the predominant products of most HLF transformations, piperidines (i.e. 6) have been observed as minor byproducts in some HLF reactions (Scheme 2).12 These piperidines are generated through 1,6-HAT processes, which rely on higher energy seven-membered transition states. Despite early observations indicating the feasibility of 1,6-HAT processes, HLF reactions that exploit this complementary site-selectivity have been underexplored.13 Indeed, until recently, examples of nitrogen-centered radicals that participate in selective 1,6-HAT processes were limited to substrates that (1) lacked abstractable hydrogen atoms six positions away from the reactive nitrogen atom, (2) featured rigid architectures precluding the necessary six-membered ring transition state, or (3) relied on proximate functional groups to weaken a C–H bond so as to favor a 1,6-HAT pathway. Owing to these restrictions, there were no strategies to accomplish 1,6-HAT processes that were broadly applicable to unactivated C–H bonds in structurally unbiased substrates. Furthermore, in many transformations that are proposed to engage 1,6-HAT processes, the obtained products would similarly be expected to arise from unguided, intermolecular hydrogen atom abstraction. This mechanistic ambiguity complicates our understanding of substrate-guided 1,6-HAT reactions.
With these factors in mind, our laboratory sought to identify a template for nitrogen-centered radicals that would preferentially engage in 1,6-HAT processes. Our research has been directed toward the development of generalizable 1,6-HAT technologies that achieve site-selectivity when functionalizing otherwise unactivated C(sp3)–H bonds. We envisioned that such a platform would deliver products functionalized at positions that do not tend to react using traditional templates for HLF transformations. Importantly, throughout our investigations, substrates have been judiciously chosen to confirm that reactivity proceeds through 1,6-HAT pathways, rather than through intermolecular hydrogen atom abstraction processes. This approach enables previously infeasible synthetic disconnections and facilitates late-stage diversification of complex molecular scaffolds.
To this end, alcohol-anchored sulfamate esters and amine-anchored sulfamides have emerged as suitable precursors to nitrogen-centered radicals that favor reactivity through 1,6-HAT processes. This Synpacts article will summarize key advances in 1,6-HAT technologies, with a specific focus on radical-mediated sulfamate ester- and sulfamide-guided C(sp3)–H functionalization reactions. For the purposes of this article, Arabic numerals will be used in the traditional manner to number the atoms involved in HAT processes. Conversely, Greek letters will be used describe positional selectivity of C(sp3)–H functionalization, relative to the anchoring position of the directing group.
2. Transformations that Rely on Structural Constraints or Weakened C–H Bonds to Favor 1,6-HAT Processes
Absent Viable 1,5-HAT Pathways, 1,6-HAT Processes May Dominate
Early investigations by the Ban laboratory demonstrated the ability to achieve selective 1,6-HAT in substrates which lack available C–H bonds for 1,5-HAT pathways (Scheme 3).14a In this report, 1,5-diamine compounds (i.e. 7) are employed in a traditional HLF manifold. However, in such substrates, there are no C–H bonds at the δ-position relative to the nitrogen-centered radical, precluding the standard 1,5-HAT site-selectivity. Instead, the 1,6-HAT pathway occurs exclusively.
Scheme 3.
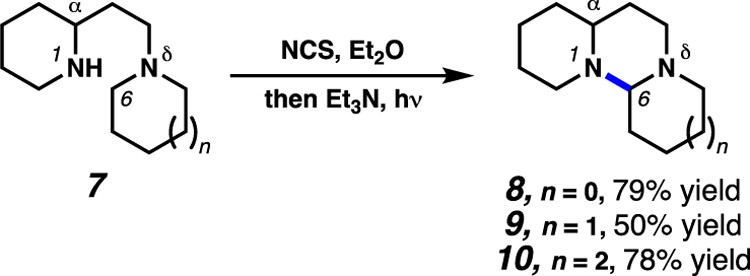
Ban and co-workers demonstrate that substrates lacking δ-C(sp3)–H bonds react through exclusive 1,6-HAT pathways.
This strategy continues to be employed in radical-mediated processes. As an example, in cutting-edge copper-catalyzed, intramolecular amination reactions, Chiba and co-workers disclose that select amidinyl radicals engage 1,6-HAT pathways in the course of C–H oxygenation reactions.15 Specifically, in seven reported examples, the reaction proceeds through 1,6-HAT processes when there are no β-C(sp3)–H bonds available for abstraction (Scheme 4).
Scheme 4.
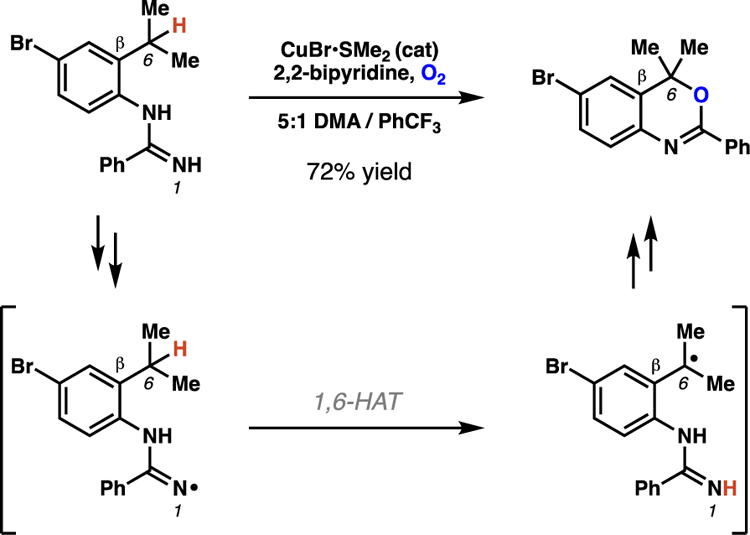
Chiba and co-workers describe amidinyl radicals reacting through 1,6-HAT pathways when no β-C(sp3)–H bonds are present for abstraction through 1,5-HAT processes.
Geometric Constraints Can Be Used to Favor 1,6-HAT Processes
With some traditional HLF substrates, infeasible transition state geometries disfavor or preclude 1,5-HAT pathways even when δ-C–H bonds are present. Early examples of this phenomenon were described by Wawzonek and co-workers when employing N-halocycloalkylamines for the synthesis of bicyclic amine compounds (Scheme 5).16 Specifically, upon irradiation with UV light, N-chloro-N-methylcyclooctylamine (11) generates only N-methylgranatanine (12) and none of the 9-azabicylco-[4.2.1]-nonane derivative (13) expected from a 1,5-HAT process (Scheme 5).16a The selectivity of this transformation is presumed to result from the geometric constraint imposed by the cycloalkane core. Due to this structural restriction, the energy barrier is raised for abstraction of a hydrogen atom five positions away from the reactive nitrogen-centered radical. Similar geometric constraints have been invoked to explain the transformation of N-halo-4-alkylpiperidine substrates into quinuclidine derivatives by way of 1,6-HAT pathways.16b
Scheme 5.
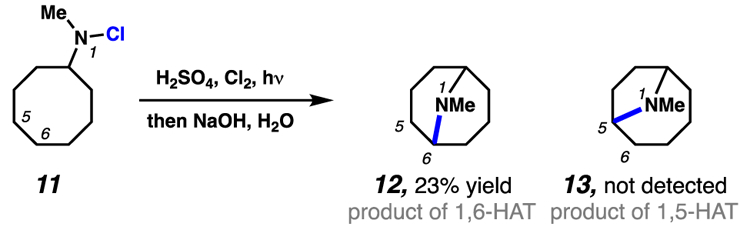
Wawzonek and Thelen demonstrate that 1,6-HAT pathways are operative when employing cycloalkylamine substrates where geometric constraints preclude 1,5-HAT processes.
Heteroatoms Can Weaken Adjacent C–H Bonds and Favor 1,6-HAT Processes
Ban and co-workers have also demonstrated that lone-pair bearing heteroatoms can be used to induce 1,6-HAT processes through the weakening of adjacent C(sp3)–H bonds (Scheme 6).14b In such examples, 1,5-diamino substrates (i.e. 14) are employed which feature abstractable C(sp3)–H bonds at both δ- and ε-positions, relative to the reactive nitrogen-centered radical. Despite the availability of canonical 1,5-HAT pathways, the authors report exclusive formation of products resultant from the less common 1,6-HAT processes (i.e. 15). This change in site-selectivity has been attributed to the ability of heteroatoms to weaken adjacent C–H bonds, which renders the 1,6-HAT pathway sufficiently facile to predominate.
Scheme 6.
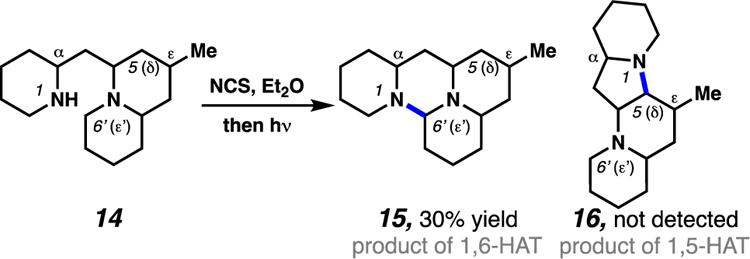
Heteroatoms can serve to weaken adjacent C(sp3)–H bonds as a strategy to enable exclusive 1,6-HAT reactivity.
Suárez and co-workers extended this insight to enable the functionalization of complex small molecules under more mild conditions. Specifically, they report that the combination of an appropriate oxidant in the presence of iodine and light can provide nitrogen-centered radicals from a variety of precursors.17 While these intermediates generally favored functionalization following 1,5-HAT processes, the authors described several examples of products arising from 1,6-HAT when employing sufficiently biased substrates (Scheme 7). In particular, 1,6-HAT pathways predominate for substrates where geometric restrictions necessitate seven-membered ring transition states for C–H abstraction, or when the abstracted hydrogen atom is weakened by a vicinal heteroatom. Notably, this approach to selective 1,6-HAT processes has been employed as a key step in the total syntheses of complex molecular scaffolds.
Scheme 7.
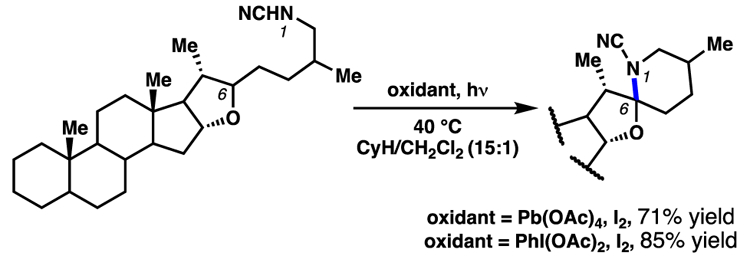
Suárez and co-workers employ oxidants in the presence of iodine to generate nitrogen-centered radicals. These transformations rely on low C–H bond dissociation energies to achieve site-selective 1,6-HAT processes.
Allylic, Benzylic, and Tertiary Centers Present Weakened C–H Bonds Which Can Facilitate 1,6-HAT Processes
For many decades, qualitative observations have suggested that HLF processes are often more efficient when the engaged C–H bond is weaker (i.e. primary < secondary < tertiary < benzylic).18 In an investigation to further probe this phenomenon, Neale, et al., examined the selectivity of 1,5- versus 1,6-HAT processes as a function of ε-C–H bond strength (Scheme 8).19 Notably, when the ε-carbon center presented a relatively weak benzylic C–H bond, only a slight preference for 1,5-HAT was observed. Until recently, similar exploitations of bond dissociation energy differences have proven to be the most common strategy to achieve selectivity for 1,6-HAT processes when 1,5-HAT pathways are feasible.
Scheme 8.
Neale et al. observed only a slight preference for 1,5-HAT pathways when weak, benzylic C–H bonds can be transformed through 1,6-HAT pathways.
Pioneering research by Baran and co-workers has documented that a series of carbamyl radicals can engage 1,6-HAT processes to access 1,3-diol products. Within the disclosed multi-step reaction sequence, an HLF reaction is employed to access γ-brominated carbamate intermediates (Scheme 9).20 The authors discovered that the involved carbamyl radical intermediates preferentially abstract a tertiary or benzylic hydrogen-atom six positions away (i.e. 1,6-HAT), when a 1,5-HAT pathway would result in functionalization of an unactivated methylene center. When a substrate contains tertiary centers that could react through either a 1,5- or 1,6-HAT process, similar amounts of 1,5- and 1,6-HAT-derived products are observed (Scheme 10). This suggests that these carbamyl radical intermediates are selective for functionalization of tertiary or benzylic C–H bonds, a subtle restriction that presents predictable constraints on the breadth of substrates that productively engage 1,6-HAT processes.
Scheme 9.
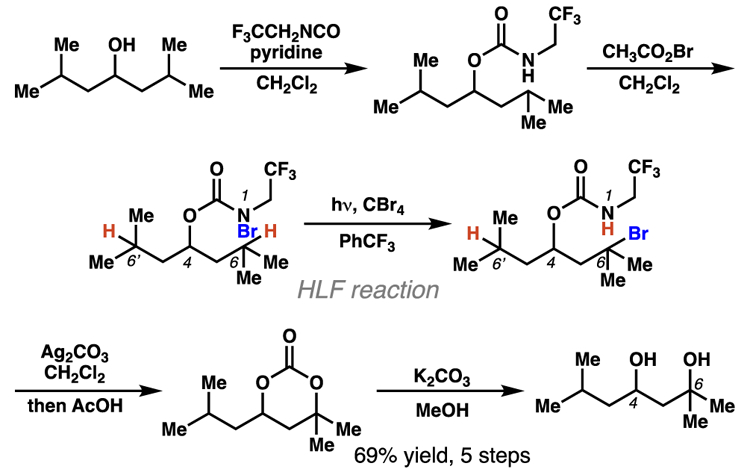
Baran and co-workers describe a multi-step sequence to prepare 1,3-diols. The key step relies on an HLF-type 1,6-HAT process to achieve the desired positional selectivity.
Scheme 10.
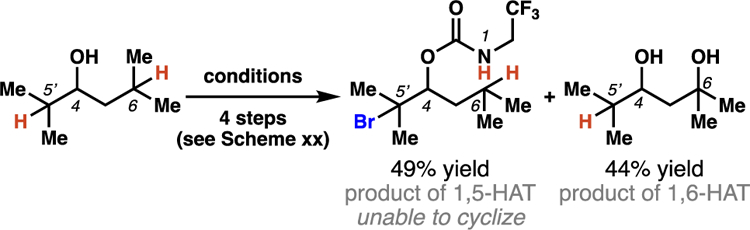
When Baran, et al. employ a substrate in which either 1,5- or 1,6-HAT processes can engage tertiary C–H bonds, both pathways proceed with nearly equivalent selectivity.
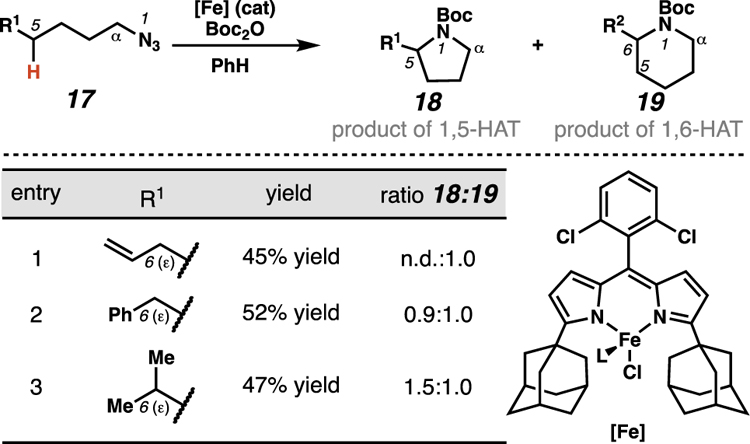
The use of small differences in C–H bond dissociation energies remains a relevant strategy to influence the site-selectivity of directed transformations in modern approaches to the generation of radical intermediates. Betley and co-workers have described formation of product mixtures that arise from competitive 1,5- and 1,6-HAT processes in the course of iron-mediated C–H amination reactions.21 In these transformations, alkyl azides serve as precursors to Fe(III) imido radicals that engage in 1,6-HAT pathways when the ε-C(sp3)–H bond is sufficiently weakened by neighboring groups (Table 1). For instance, when 1,6-HAT processes result in abstraction of an allylic C–H bond, this pathway is favored such that only piperidine product 19 forms (entry 1). By contrast, when 1,6-HAT processes require abstraction of benzylic or tertiary C–H bonds, mixtures of pyrrolidine (18) and piperidine (19) products are generated (entries 2–3). Accordingly, under these conditions, the abstracted C–H bonds must be significantly weakened to selectively engage 1,6-HAT pathways. This energetic requirement limits the generalizability of this approach to facilitate site-selective 1,6-HAT processes.
Table 1.
When employing substrates with sufficiently weakened ε-C(sp3)–H bonds, Betley and Hennessy show that Fe(III) imido radicals engage competitive 1,5- and 1,6-HAT pathways.
 |
Similarly, Nagib and co-workers have illustrated that 1,6-HAT pathways can predominate in select remote C–H arylation processes. In these HLF-type reactions, copper-mediated N–F bond homolysis generates a sulfonamidyl radical that preferentially abstracts a hydrogen atom from a benzylic center through a 1,6-HAT event over transformation of a stronger, unactivated secondary C–H bond (22 → 23, Scheme 11).22
Scheme 11.
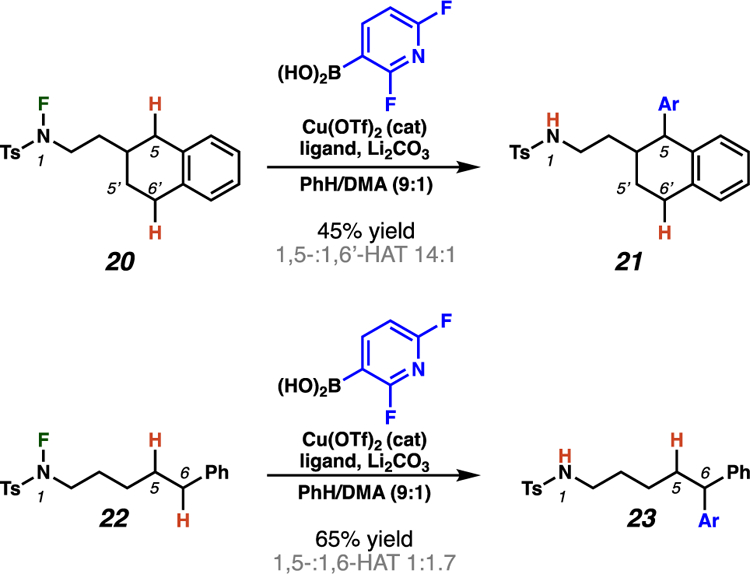
The system developed by Nagib and co-workers can preferentially engage 1,6-HAT pathways based on abstraction of weak, benzylic hydrogen atoms.
Recent tactical advances to affect the generation of nitrogen-centered free radicals have provided occasional examples of products generated based on 1,6-HAT pathways. In particular, these position-selective C–H functionalization reactions also favor 1,6-HAT processes when the abstracted C–H bond is weakened relative to vicinal C–H bonds. Leonori and co-workers23 have disclosed examples of photochemically-driven radical cascade reactions for remote C–H fluorination. In these transformations, select carbamyl (from 24) and amidyl (from 25) radical intermediates preferentially abstract hydrogen atoms at tertiary centers through putative 1,6-HAT pathways (Scheme 12). To put this into context, related substrate 28 reacts to generate mixtures of δ- and ε-functionalized products. The erosion of selectivity when using these substrates could be a result of their increased alkyl chain flexibility, or relatively diminished inductive deactivation of the hydrogen atoms, which are more distal to electron withdrawing groups.
Scheme 12.
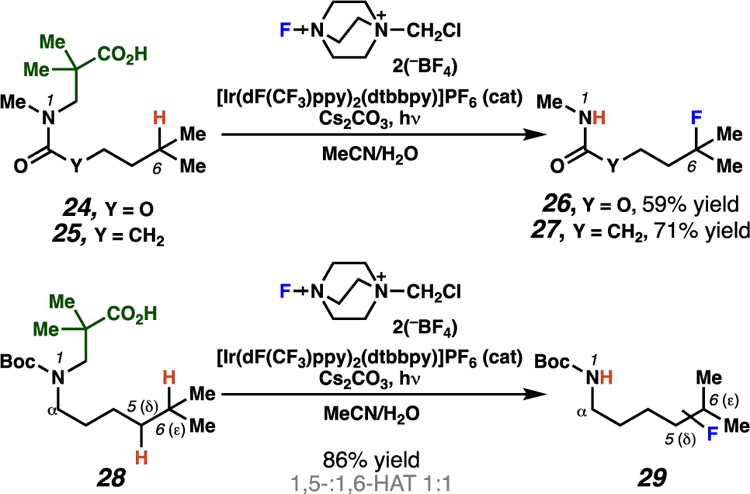
Leonori and co-workers provide examples of radical-mediated remote C–H fluorination reactions.
There Is An Unmet Need for Broadly Relevant 1,6-HAT Processes
While these examples demonstrate the capability of nitrogen-centered radicals to react through 1,6-HAT processes, the current methods suffer from limitations. Notably, each of these transformations requires significant substrate bias to enable preferential 1,6-HAT, indicating that such technologies are confined to very select classes of substrates. Thus, there is a significant unmet need to develop generalizable manifolds that engage 1,6-HAT pathways to provide a broad array of selectively functionalized products.
3. Sulfamate Esters Engage Selective 1,6-HAT Processes
Metal-Catalyzed Amination Technologies Provide Inspiration
To overcome some of the current limitations in position-selective C–H functionalization reactions with nitrogen-centered radical intermediates, our laboratory sought to identify directing groups that would preferentially react through 1,6-HAT processes. To this end, we were inspired by research into metal-mediated, γ-selective amination technologies employing sulfamate ester substrates.24,25 Investigations by Du Bois and co-workers suggest that the site-selectivity observed with sulfamate esters arises from elongated N–S and S–O bond lengths.24 Building upon this knowledge, we anticipated that nitrogen-centered radicals generated from sulfamate esters (i.e. sulfamyl radicals) may geometrically favor 7-membered transition states for C–H abstraction, a prerequisite for 1,6-HAT processes.26
Owing to this vision, sulfamate esters have now emerged as powerful precursors to sulfamyl radicals that engage selective 1,6-HAT pathways. Recent research from our laboratory, as well as others, has established that sulfamate esters guide position-selective, radical-mediated C(sp3)–H functionalization reactions. Importantly, these technologies do not require otherwise geometrically or electronically biased substrates. Indeed, HLF reactions exploiting sulfamyl radicals have been found to proceed with good to exquisite selectivity even when equally or more reactive C–H bonds are present at adjacent atoms. The unique site-selectivity afforded by these sulfamate ester directing groups has provided a diverse array of γ-functionalized alcohol derivatives, classes of products that would be challenging to prepare using more conventional methods.27 To access the key sulfamyl radicals, the traditional strategy of N–X bond homolysis has proven to be broadly effective. Alternatively, more modern approaches to generate the sulfamyl radicals have been disclosed, most notably through photoredox-promoted single electron oxidation pathways.
Reactions Employing N-Functionalized Sulfamate Esters
Our initial research into sulfamate ester-guided 1,6-HAT processes began with a transformation that most closely mimics a traditional HLF reaction.28 We chose to evaluate N-chlorosulfamate esters as a means to limit mechanistic uncertainty and establish the feasibility of sulfamate esters as templates for 1,6-HAT pathways. To our delight, photoirradiation of N-chlorosulfamate 30 delivered alkyl chloride 33 with exquisite positional selectivity (Scheme 14). Notably, this protocol maintains exceptional site-selectivity even when weaker C–H bonds are present and provides the first unambiguous evidence that sulfamate esters selectively participate in intramolecular 1,6-HAT pathways.
Scheme 14.
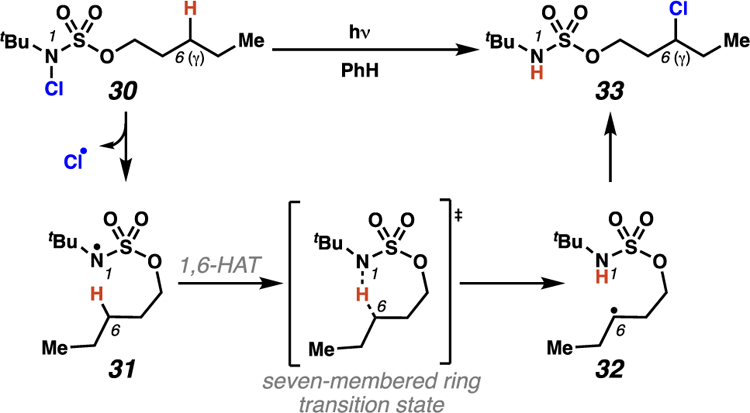
Sulfamate esters selectively engage in 1,6-HAT processes to chlorinate alkanes by a radical chain propagation mechanism.
To probe the mechanism of this chlorine-transfer reaction, a quantum yield experiment was performed.29 This experiment revealed that a radical chain propagation process is operative (ϕ = 77), indicating that carbon-centered radical 32 is capable of abstracting a chlorine atom from another equivalent of N-chlorosulfamate 30. This step proceeds with release of the desired alkyl chloride 33 and generates an additional molecule of sulfamyl radical 31 to propagate the chain reaction. These findings are consistent with the prevailing mechanism of traditional amine-directed HLF reactions, in which radical chain propagation processes have been confirmed through efficient reactions initiated by radical initiators (catalytic quantities).12 Our chlorine-transfer process constitutes the first example of sulfamate ester-guided, remote C–H functionalization that proceeds through exogenous atom transfer.
Overall, this sulfamate ester-directed method for C(sp3)–H chlorination offers complementary site-selectivity to many other radical-mediated chlorination procedures4b,11b,30 and yields only mono-chlorinated products (Scheme 15). A variety of N-alkyl substituents, which span broad electronic and steric profiles,31 are well-tolerated in the reaction. Furthermore, the transformation successfully chlorinates benzylic, tertiary, secondary, and even primary centers, while tolerating an array of functional groups.
Scheme 15.
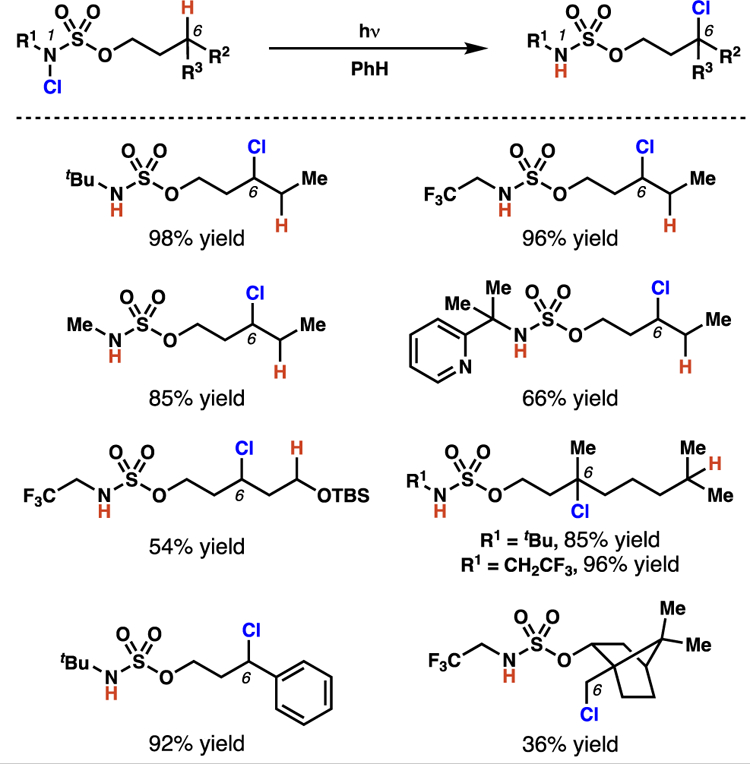
Sulfamate esters direct chlorine-transfer at primary, secondary, tertiary, and benzylic centers with site-selectivity that is complementary to that available based on innate selectivity.
Concurrent to our research, Du Bois, Burns, Zare and co-workers discovered that sulfamyl radicals could engage in 1,6-HAT processes to induce site-selective alkane bromination (Schemes 16–17).32a Using N-methyl sulfamate ester substrates, N-bromosulfamate esters 35 are generated in situ, and are proposed to undergo rhodium-initiated N–Br bond homolysis to furnish reactive sulfamyl radicals.32b As in the chlorination process, 1,6-HAT provides exquisite position-selectivity, further documenting sulfamate esters as general tools for site-selective exogenous atom-transfer reactions. Mechanistically, this transformation may also proceed via a radical-chain propagation process. To support this claim, a cross-over experiment was carried out between two N-halosulfamate esters: N-bromosulfamate 39 and N-chlorosulfamate 40. The identity of the halogen in products 41–44 did not depend on the halogen in the substrate. Accordingly, this result provides evidence that these reactions proceed through radical-chain propagation mechanisms and exogenous atom transfer processes (Scheme 16B).
Scheme 16.
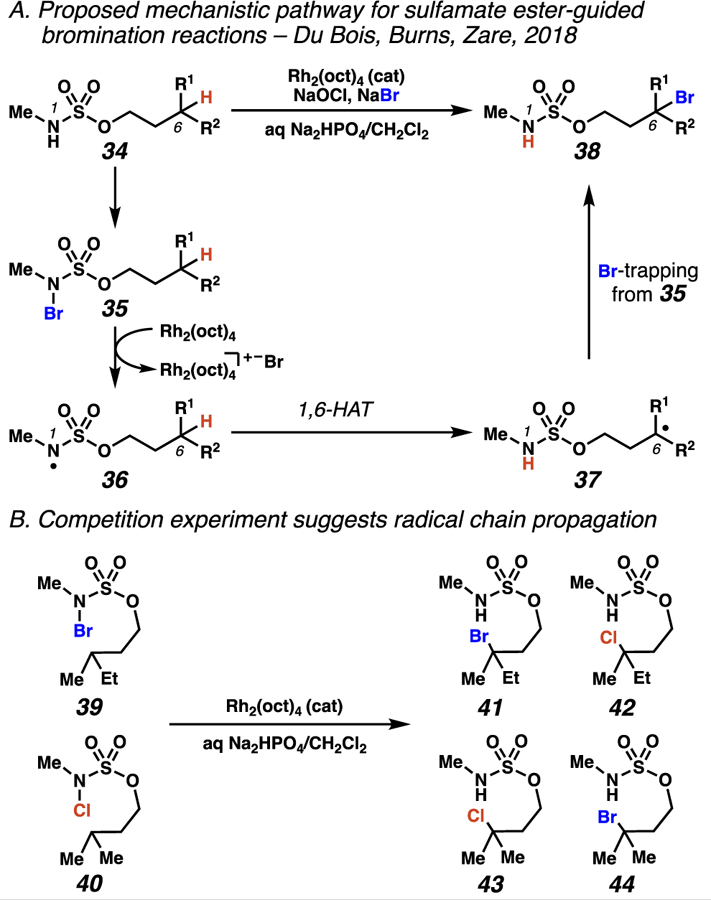
Du Bois, Burns, and Zare propose that sulfamate esters direct bromination based on a radical chain propagation mechanism (A), and use mass spectrometry and a crossover experiment (B) to support this claim.
Scheme 17.
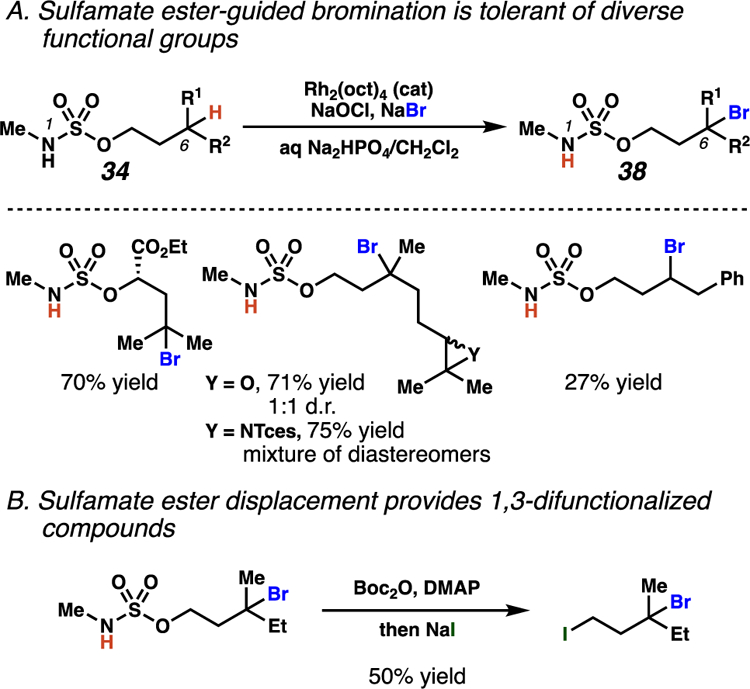
A. Sulfamate ester-directed, rhodium-mediated bromination is well-documented at tertiary centers and tolerates esters, epoxides, and aziridines. B. Subsequent sulfamate ester displacement furnishes 1,3-difunctionalized compounds.
Using these bromination conditions, efficient HAT from benzylic and tertiary centers occurs in the presence of diverse functional groups, including esters, epoxides, and aziridines (Scheme 17A). Yet, only a single example transforming an unactivated secondary C–H bond is reported. Finally, the authors also disclose two methods for sulfamate displacement, demonstrating the utility of the halogenation method to access 1,3-difunctinalized products (Scheme 17B).
Building on these advances, Muñiz and co-workers have determined that sulfamate esters can guide dihalogenation processes based on sequential 1,6-HAT events (Scheme 18).33 By employing hydantoin derivatives as halogenating reagents, N-halosulfamate esters are generated in situ, and halogen-transfer occurs upon photoirradiation. Interestingly, this method can be translated to a multi-step protocol to install two different halogen atoms (50 and 51) in sequential HLF reactions.
Scheme 18.
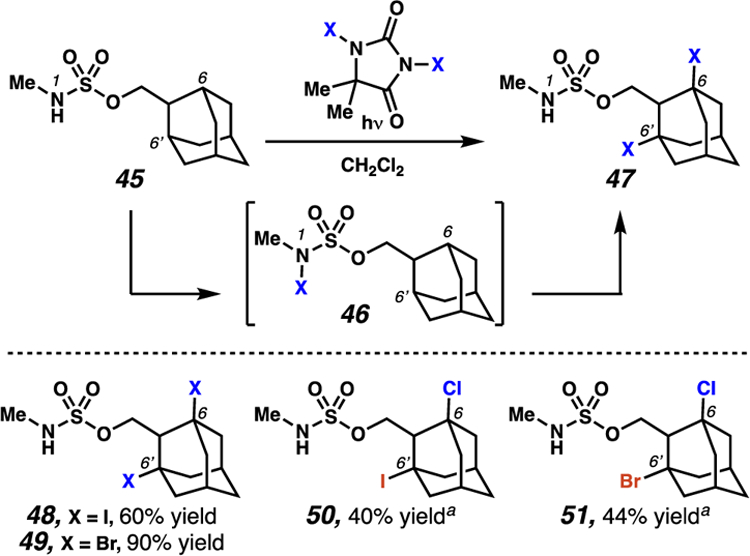
Muñiz and co-workers confirm that sulfamate esters guide 1,6-HAT processes to achieve C–H halogenation, and invent protocol to dihalogenate alkanes. a Yields reported over 3 steps.
In a subsequent disclosure, the Minakata group demonstrated that primary sulfamate esters were also effective substrates when used in conjunction with electrophilic iodine oxidants (Scheme 19).34 Following C–H functionalization, cyclization to the γ-aminated products ensues in situ, closely mirroring the standard outcome of traditional HLF technologies. The process was found to be most effective for the transformation of benzylic C–H bonds, with diminished yields observed for the functionalization of unactivated secondary and tertiary centers. Overall, this reaction constitutes a metal-free alternative to previously reported sulfamate ester-guided amination protocols.24,25
Scheme 19.
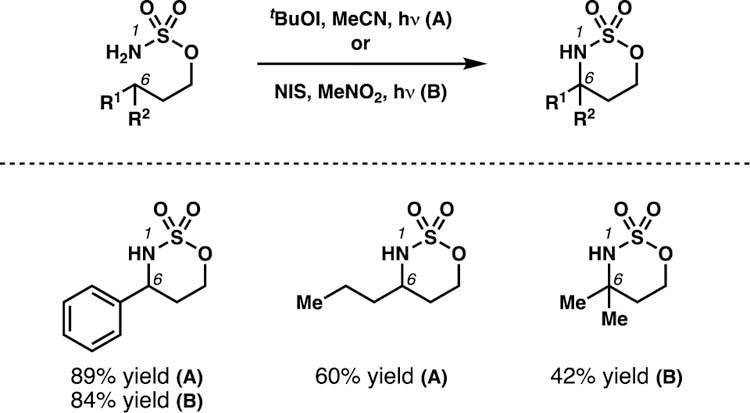
As an alternative to metal-mediated amination technologies, Minakata et al. have disclosed a protocol for sulfamate ester-guided amination promoted by electrophilic iodine oxidants.
Beyond atom-transfer processes, our laboratory has demonstrated that sulfamate esters direct site- and diastereoselective group-transfer processes, including xanthate-transfer reactions (Scheme 20).35,4c Pre-generation of N-xanthyl sulfamate esters with subsequent photoirradiation yields a diverse array of alkyl xanthate products with predictable position selectivity. The transformation is applied to guide the functionalization of structurally complex scaffolds, and is robust to N- and O-alkyl substitution. Similar to the analogous chlorination technology,28 quantum yield experiments are used to demonstrate that the reaction proceeds through a radical chain propagation mechanism (ϕ = 3).
Scheme 20.
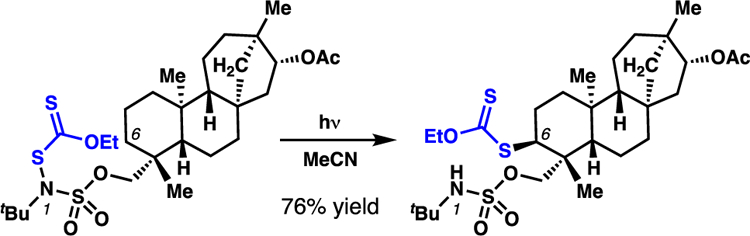
Sulfamate esters guide position-selective xanthate-transfer processes, even on structurally complex scaffolds.
Given the ability of alkyl xanthates to serve as latent alkyl radicals, this xanthylation platform provides valuable γ-functionalized products poised for further elaboration.36 To demonstrate the utility of the alkyl xanthate products, a variety of transformations of the xanthate moiety were performed to access a range of γ-functionalized sulfamate ester derivatives (Scheme 21).
Scheme 21.
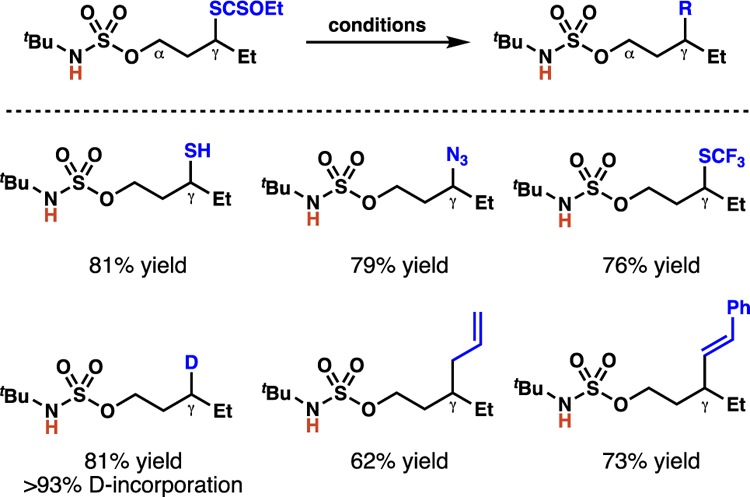
Alkyl xanthate products can undergo facile conversions to other functional motifs.
These initial investigations into atom- and group-transfer based on N-functionalized sulfamate esters have been instrumental in establishing the fundamental basis for a broadly relevant strategy to enable position-selective 1,6-HAT processes. Furthermore, these methods provide valuable γ-functionalized products, which would be challenging to access using previously known technologies. Unfortunately, these methods rely on a discrete synthetic step to oxidize the sulfamate esters, or in situ use of strong chemical oxidants, both of which potentially limit the substrate tolerance of the associated process. For instance, pre-oxidation of the sulfamate ester substrates has precluded the use of substrates containing electron-rich aromatic moieties as well pendant electron-rich olefins. To overcome some of these limitations, methods that generate the requisite sulfamyl radical under mild conditions, ideally from a native N–H bond, will further the synthetic utility of sulfamate ester-guided technologies.
Reactions Exploiting Photoredox Catalysis to Access Sulfamyl Radicals
Photoredox catalysis provides a mild, environmentally benign approach to generate radical intermediates, and has transformed the way organic radicals are employed in chemical synthesis.37 As such, methods that exploit photocatalysis to form nitrogen-centered radical intermediates are advantageous, as they often avoid the use of strong chemical/two-electron oxidants and toxic radical initiators. Previously, photoredox catalysis has been utilized to access amidyl, carbamyl, and sulfonamidyl radicals for use in remote C(sp3)–H functionalization reactions.11c,d,f As a complement to these 1,5-HAT processes, sulfamate ester-guided transformations have emerged, and proceed with complementary site-selectivity through 1,6-HAT pathways.38
The first reports to utilize photocatalytic strategies to access sulfamyl radicals have been described in contemporaneous research by the Duan, Roizen, and Huang laboratories (Scheme 22).38 These technologies each employ sulfamate ester substrates and electron-deficient olefins under photoredox catalysis to facilitate position-selective C(sp3)–C(sp3) bond-forming processes.39 The three reports detail broad sulfamate ester and electrophile functional group tolerance when transforming tertiary C(sp3)–H bonds; however, each protocol is distinctive, and provides unique insights into sulfamate ester-guided alkylation processes.
Scheme 22.
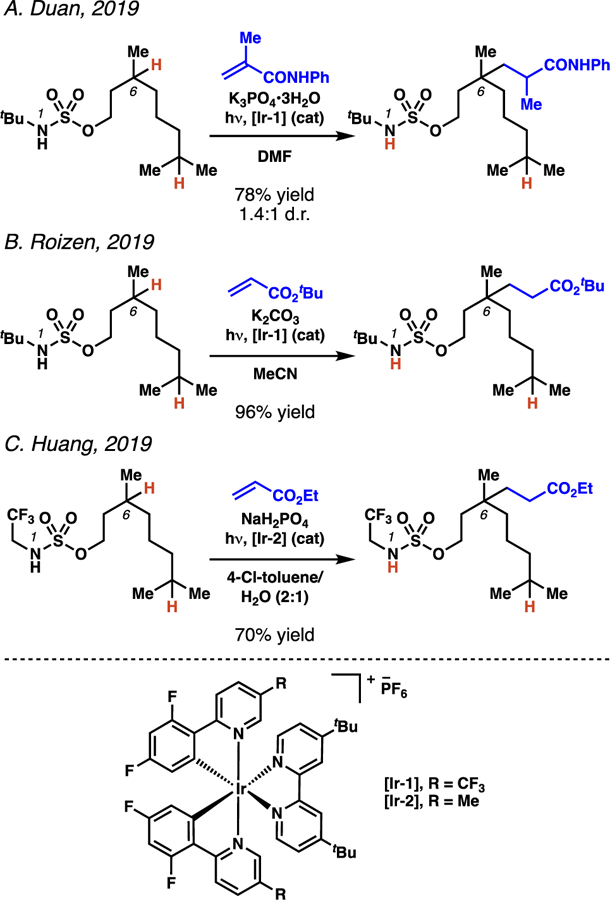
Representative examples of photocatalytic alkylation reactions guided by sulfamate esters.
Duan and co-workers employ N-tert-butylsulfamate esters to efficiently transform tertiary C–H bonds into new C–C bonds (Scheme 23).38a The disclosed protocol is also used to functionalize secondary centers, albeit in diminished yields. Additionally, the authors discover that not only are electron-deficient olefins competent electrophiles, but styrene derivatives can also serve as radical trapping agents in the alkylation process. This change in electrophilic partner does require modification of the standard reaction conditions, specifically alteration of the photocatalyst. This reaction demonstrates that this radical-mediated C(sp3)–H functionalization can be extended to enable the hydroalkylation of styrenes.40
Scheme 23.
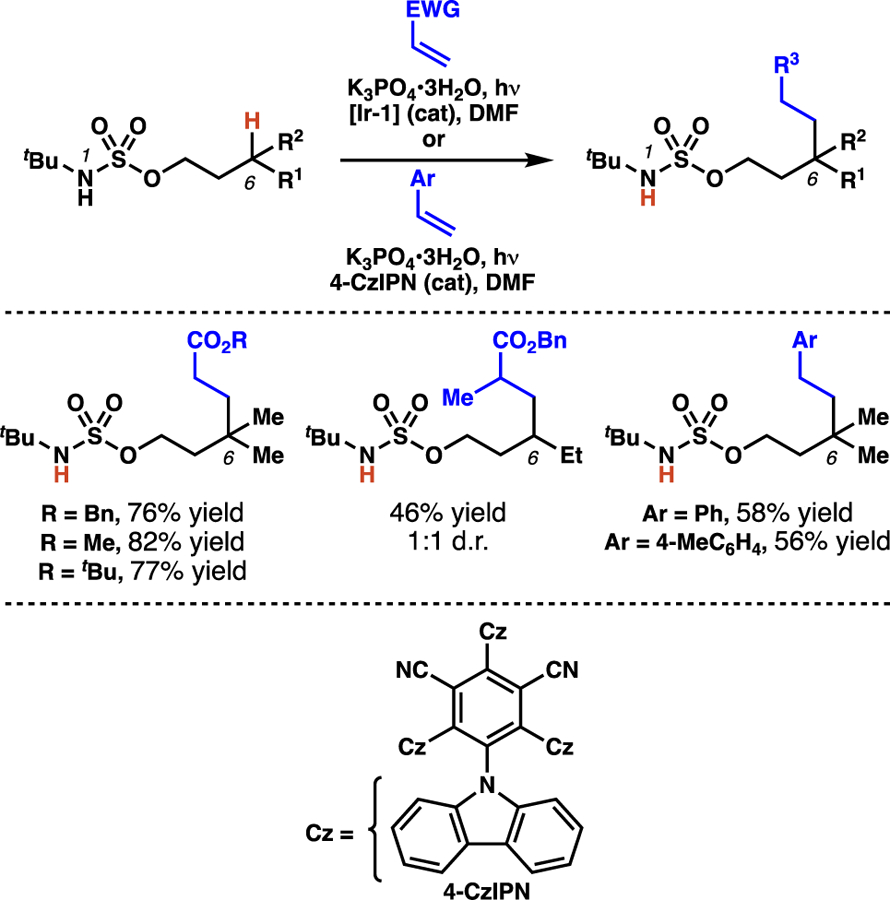
Duan and co-workers show that, in addition to Michael acceptors, styrene derivatives are competent radical trapping agents in photoredox-mediated alkylation processes.
Concurrently, our laboratory investigated the use of N-tert-butylsulfamate esters to dictate the site of functionalization in photoredox alkylation reactions.38b While the C–C bond-forming procedure was most effective at tertiary sites, secondary centers could also be transformed. Our research established that bisalkylation processes are feasible (53) and that monoalkylation of methylene centers is favored by increasing steric encumberance proximal to the site of alkylation (54). (Scheme 24). Furthermore, some enantioenriched Michael acceptors were found to engage in diastereoselective alkylation processes (55). Finally, structurally complex substrates could be alkylated with exquisite site-selectivity and modest diastereoselectivity, despite the presence of additional, weaker C(sp3)–H bonds elsewhere in the molecule (56–57).
Scheme 24.
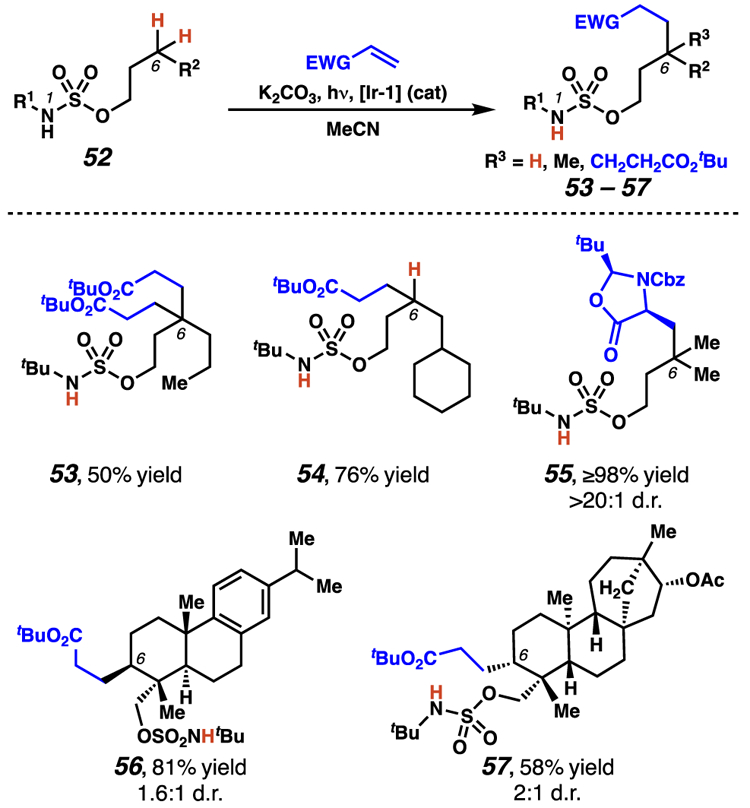
Roizen and co-workers interrogate the influence of steric encumbrance on alkylation at secondary centers.
A third alkylation protocol guided by sulfamate esters was also disclosed by Huang and co-workers around the same time.38c In their report, the authors describe the use of N-(2,2,2-trifluoro)ethylsulfamate esters in alkylation reactions with electron-deficient olefins and styrene derivatives (Scheme 25). Again, the procedure is most effective for the transformation of tertiary C(sp3)–H bonds, but several substrates were functionalized at secondary C(sp3) centers. By employing the N-(2,2,2-trifluoro)ethylsulfamate esters, a weaker base promotes the desired reaction, and more facile sulfamate displacement may be feasible.
Scheme 25.
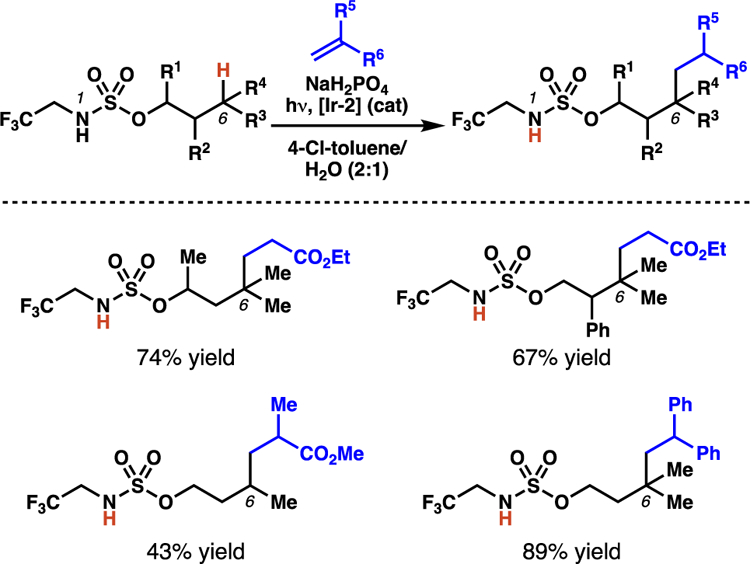
Huang and co-workers employ trifluoromethyl sulfamate esters as templates for photoredox-mediated Giese reactions.
Mechanistically, there are multiple reaction pathways that may be operative to convert sulfamate ester 58 to the neutral sulfamyl radical 60.41 In one reaction pathway to generate neutral sulfamyl radical 60, deprotonation with an appropriate base (58 → 59) followed by single electron transfer (SET) with an excited-state iridium photocatalyst provides a sulfamyl radical (60) poised for 1,6-HAT. Alternatively, a proton-coupled electron transfer (PCET) pathway could be viable, in which the proton- and electron-transfer events occur simultaneously.42 Our laboratory has performed Stern-Volmer quenching studies43 to interrogate the method by which radical 60 is generated. These experiments provide evidence that an SET process is feasible between sulfamate ester anions and the [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 photocatalyst; however, as the anionic substrate is not directly employed in the reaction protocol, a concerted PCET process cannot be ruled out. Subsequently, sulfamyl radical-mediated translocation of the hydrogen atom (60 → 61) occurs to generate the carbon-centered radical 61. Our laboratory has used an enantioenriched substrate to demonstrate that this carbon-centered radical is generated irreversibly.44 Next, radical addition to the alkene acceptor furnishes stabilized carbon-centered radical 63, which can be reduced to the corresponding anion by single-electron transfer with the iridium catalyst. Finally, protonation of anion 64 generates the final alkylated product.
By obviating the necessity to pre-install the atom or group to be transferred, these sulfamate ester-guided alkylation processes lay the foundation for a rich future in modular, position-selective C(sp3)–H functionalization protocols directed by sulfamate ester-masked alcohols.
4. Expansion to 1,6-HAT Processes with Masked Amine Substrates
As a complement to alcohol-derived sulfamate esters, sulfamoylated amine derivatives facilitate selective 1,6-HAT processes. Seminal investigations employing sulfamides45 in rhodium-catalyzed intramolecular amination reactions have provided inspiring precedent for more recent research into free radical-mediated processes guided by these masked amine substrates. Similar to their investigations into sulfamate ester-guided intramolecular amination, Du Bois and co-workers have suggested that sulfamide substrates induce γ-position-selective functionalization due to their elongated S–N bond lengths. Taking advantage of these geometric constraints, sulfamoyl azides and sulfamides are valuable counterparts to sulfamate esters as general directing groups for γ-selective functionalization processes.46
Following the research by Du Bois, Zhang and co-workers implicated 1,6-HAT processes in intramolecular amination technologies employing sulfamoyl azide substrates (Scheme 27).47 Specifically, sulfamoyl azides serve as precursors to metalloradical species (73) in the presence cobalt porphyrin catalysts. Once formed, metalloradical 73 engages an intramolecular 1,6-HAT process to generate carbon-centered radical 74. Subsequent intramolecular homolytic substitution rapidly delivers the aminated products with concomitant regeneration the cobalt catalyst. These conditions facilitate the intramolecular functionalization of a variety of C(sp3)–H bonds,47a–d and demonstrate the generalizability of these metalloradical-mediated C–H amination procedures. Building on these pioneering reactions, recent investigations have revealed that a chiral cobalt porphyrin catalyst can render the process enantioselective, providing chiral cyclic sulfamides poised for ready transformation into enantioenriched 1,3-diamine derivatives.47e These metalloradical-mediated protocols have served as the foundation for C–H functionalization mediated by sulfamidyl free radicals proceeding through 1,6-HAT pathways.
Scheme 27.
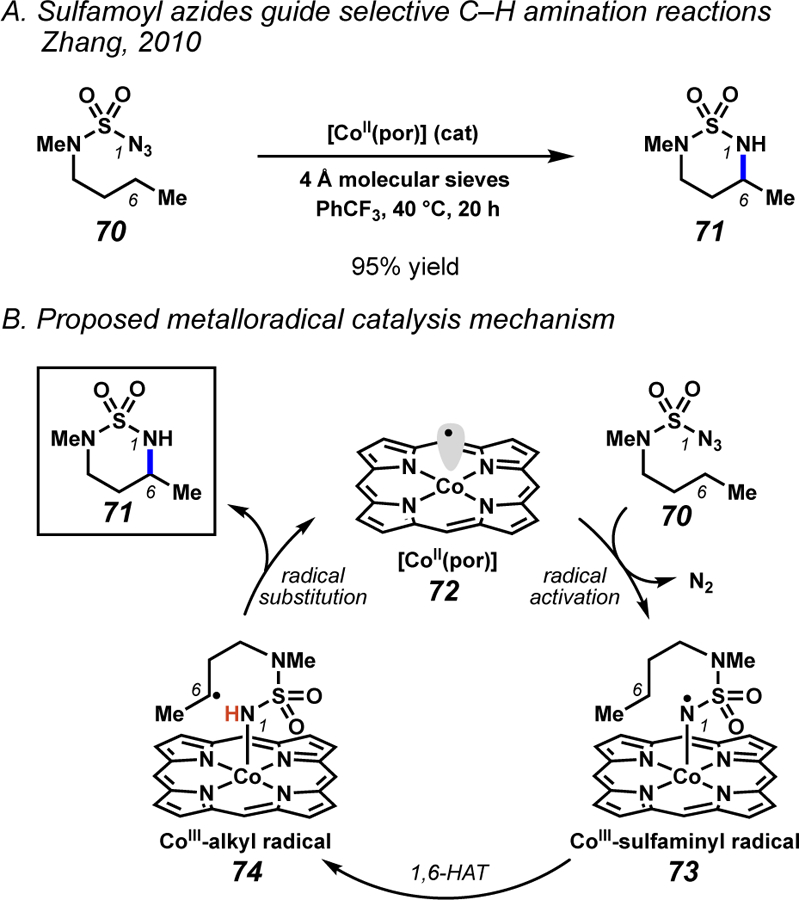
Zhang and co-workers have achieved position-selective C(sp3)–H amination reactions mediated by metalloradical 1,6-HAT processes.
Muñiz and co-workers have disclosed an “interrupted” HLF-type process, guided by sulfamides (Schemes 28–29).48 This transformation is proposed to proceed via in situ N-iodination with subsequent light-promoted N–I bond homolysis to generate the pivotal sulfamidyl radical (78). By analogy to sulfamate ester-derived sulfamyl radicals, sulfamidyl radicals geometrically favor 7-membered transition states for C–H abstraction, resulting in a selective 1,6-HAT process. Carbon-centered radical 79 can then be trapped via iodine-abstraction from an additional molecule of 77 through a radical-chain pathway that was confirmed by a quantum yield experiment (ϕ = 120). Next, oxidation followed by a Ritter-type amination provides the aminated product (Scheme 29). While this amidation reaction has thus far only been detailed for tertiary centers, the developed transformation provides a manifold for position-selective intermolecular amination facilitated by an iodine-catalyzed 1,6-HAT process.
Scheme 28.
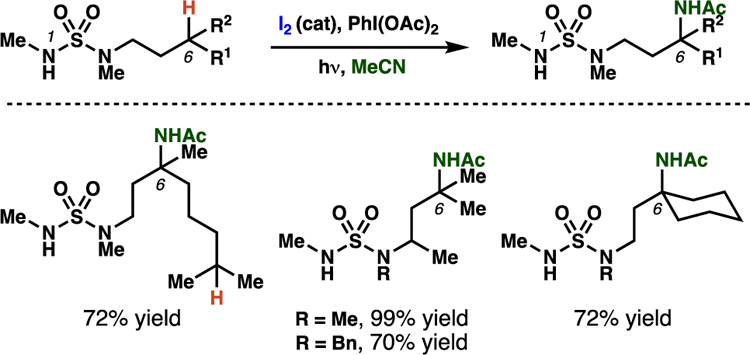
Select examples for the sulfamide-guided Ritter-type amination through an interrupted iodine-catalyzed HLF process reported by Muñiz and co-workers.
Scheme 29.
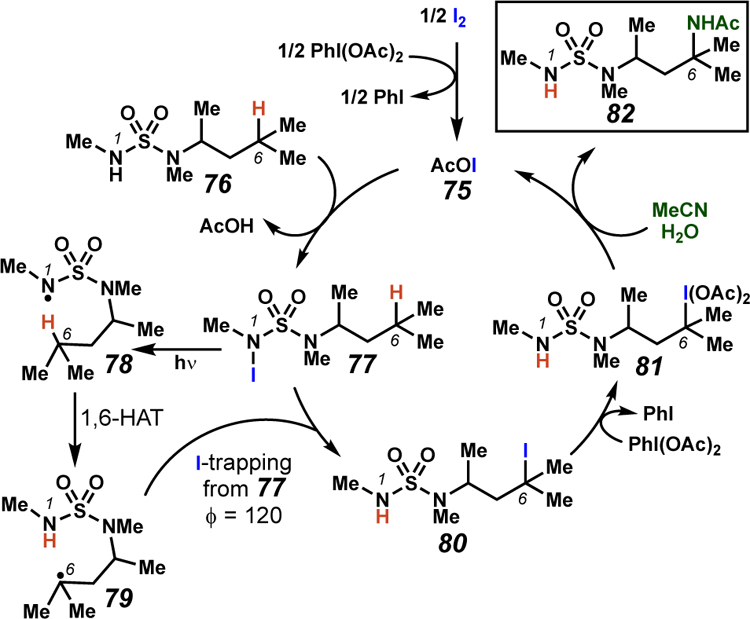
Plausible reaction mechanism for the interrupted HLF reaction employing sulfamide templates.
Within their same report on metal-free sulfamate ester-directed amination protocol, the Minakata group demonstrates that primary sulfamides (83) can engage in 1,6-HAT pathways in the presence of electrophilic iodine oxidants (Scheme 30).34 Similar to the investigations by the Muñiz group, this transformation also furnishes 1,3-diamine derivatives. The products of this method, however, are cyclic sulfamides, as the reaction proceeds with intramolecular cyclization of putative alkyl iodide intermediates. The authors demonstrate that the protocol is effective for the transformation of a variety of benzylic C–H bonds (including one β-C–H bond), but examples of unactivated C(sp3)–H bonds are not reported.
Scheme 30.
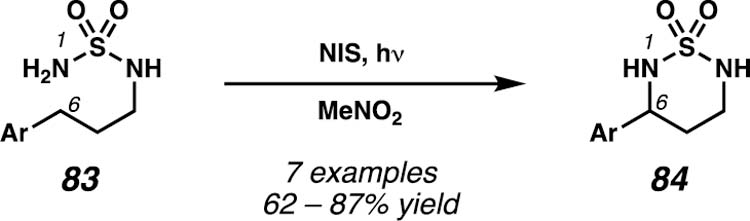
Minakata and co-workers demonstrate access to cyclic sulfamides through a metal-free approach with an electrophilic iodine oxidant.
Contemporaneous to the research disclosed by the groups of Muñiz and Minakata, our laboratory has revealed the generality of sulfamide substrates to guide 1,6-HAT processes by developing a mechanistically straightforward chlorine-transfer reaction (Schemes 31–32).49 Similar to sulfamate esters, we have found that sulfamide-masked amines and amides are effective directing groups for functionalization of remote C(sp3)–H bonds.50 In these investigations, we have observed that sulfamidyl radicals are capable of abstracting C(sp3)–H bonds from primary, secondary, and tertiary centers to provide the corresponding alkyl chlorides in remarkable yields. Importantly, this method is the first broadly demonstrated extension of sulfamide directing groups beyond γ-selective C–N bond-forming technologies.
Scheme 31.
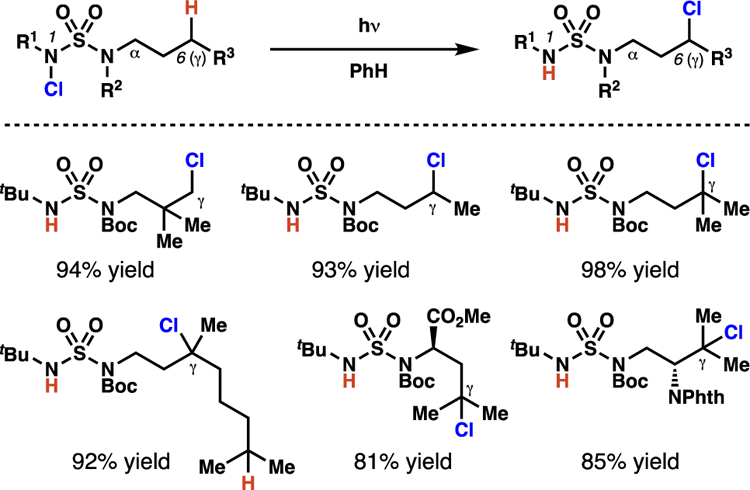
Sulfamides direct γ-selective chlorination of C(sp3)–H bonds.
Scheme 32.
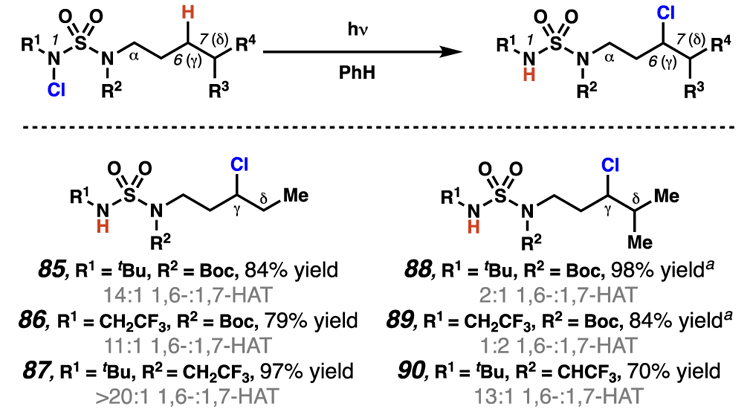
Selectivity can be modulated by the steric and electronic properties of sulfamide nitrogen substituents. Isolated yields of products from 1,6-HAT. a Isolated yields of mixture of 6- and 7-chlorinated products.
These investigations have demonstrated that position-selectivity can be modulated based on variations in the sulfamide nitrogen substituents (Scheme 32). Specifically, sulfamides that contain vicinal secondary centers appear to engage competing 1,6- and 1,7-HAT pathways (85 – 87). By manipulating the N- and N’-substituents of the sulfamide, the 1,7-HAT process can be suppressed, yielding only γ-functionalized products (87). In addition, the same reactivity trends hold when comparing substrates for which 1,6-HAT pathways require abstraction of a hydrogen atom at a secondary center, whereas 1,7-HAT results in abstraction of a weaker, δ-tertiary hydrogen atom (88 – 90). These investigations are the first to exploit modifications in sulfamide substituents to modulate the site-selectivity of C–H functionalization processes.
Collectively, these reports provide a foundation for future applications of masked amine substrates as templates for position-selective C(sp3)–H functionalization processes. Additionally, facile sulfamide cleavage is known,45 and affords rapid access to 1,3-difunctionalized products, further demonstrating the utility of these methods.
5. Conclusions and Outlook
These selective 1,6-HAT technologies unlock previously infeasible synthetic disconnections, and expand synthetic access to uncharted chemical space. Accordingly, the ongoing development of 1,6-HAT processes is of great importance for diversification of complex molecular scaffolds. To this end, sulfamate ester- and sulfamide-guided methods provide complementary site-selectivity to most radical-mediated protocols. These generalizable transformations exploit the unique physicochemical parameters of sulfamate esters and sulfamides to overcome traditional 1,5-HAT processes with nitrogen-centered radical intermediates. To date, a range of carbon–carbon and carbon–heteroatom bond-forming processes have been disclosed, which enable installation of high-value functional groups at C–H bonds that are challenging to transform using traditional approaches. Already, the research into these C(sp3)–H functionalization reactions has opened the door to previously infeasible synthetic disconnections. Nevertheless, the landscape remains rich for future innovation, both in terms of methods to access the powerful sulfamyl and sulfamidyl radical intermediates, as well as strategies to functionalize the resultant carbon-centered radical intermediates. Given the promise offered by sulfamate ester and sulfamide directing groups, we are hopeful that 1,6-HAT processes will become prominent strategies in the construction of intricate molecular architectures.
Scheme 13.
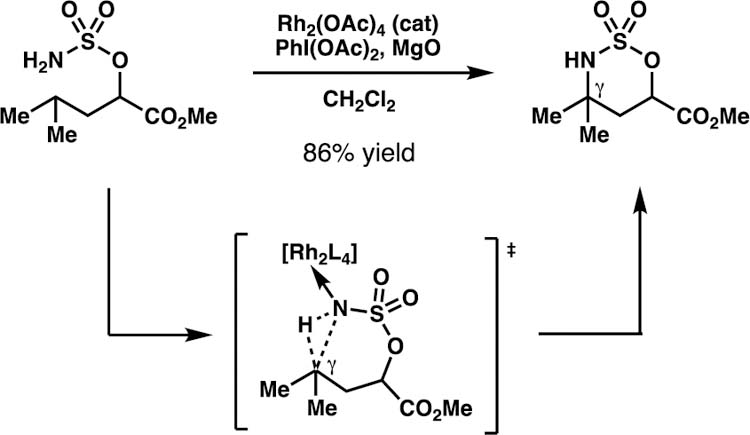
Rhodium-catalyzed intramolecular amination reactions inspired our idea that sulfamate esters might direct site-selective 1,6-HAT processes.
Scheme 26.
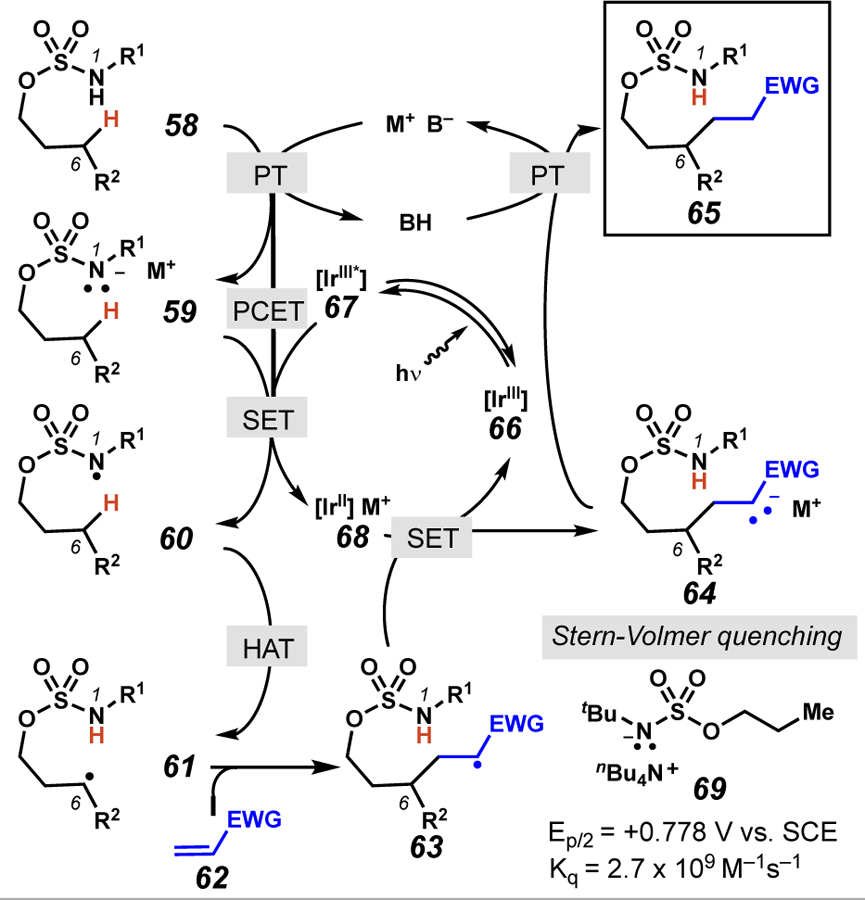
Plausible mechanistic pathways for photoredox-mediated alkylation reactions employing sulfamate ester substrates.
Acknowledgment
We gratefully acknowledge Mina F. Shehata, Anastasia L. G. Kanegusuku, Suraj K. Ayer, and Thomas Castanheiro – our colleagues who have made important contributions to our investigations of sulfamate ester- and sulfamide-guided 1,6-HAT processes.
Funding Information
The National Institutes of Health (R35GM128741–01) funds research documenting sulfamate ester reactivity, and has supported the development of this manuscript.
Biosketches

Jennifer L. Roizen (center) is an Assistant Professor at Duke University and a 2017 Thieme Chemistry Journals Award recipient. She had her first taste of synthetic research with J. Hodge Markgraf and Tom Smith as a Williams College undergraduate, where she advanced syntheses of benzoisocanthenones and contributed to publications on the total synthesis of hennoxazole A (a marine natural product). She moved to the California Institute of Technology to earn a Ph.D. with Brian Stoltz, researching approaches to access the ineleganolide core. These Cope-centric approaches remain the only published strategies to access the all carbon framework of ineleganolide, a small molecule that continues to elude synthetic campaigns. Upon graduation, Dr. Roizen became an NIH postdoctoral researcher and CMAD fellow with Justin Du Bois at Stanford University, where they extended intermolecular amination technologies. Dr. Roizen’s laboratory researches total synthesis and the development of cross-coupling and C–H functionalization processes. Drs. Melanie A. Short and J. Miles Blackburn are founding members of the PI’s independent research laboratory and of the program described in this Synpacts article.
Melanie A. Short (left) grew up in Birmingham, Alabama and received her bachelor’s degree from Birmingham-Southern College (BSC) in 2013. As an undergraduate, she performed mathematics research with Prof. Douglas Riley and organic chemistry research with Profs. Laura Stultz and David Schedler. Melanie chose to pursue graduate research at Duke University with Prof. Roizen as an NSF Graduate Research Fellow. Melanie earned her Ph.D. in 2019 for her investigations into the development of sulfamate ester- and sulfamide-guided C–H halogenation processes.
J. Miles Blackburn (right) is alumnus of Hamilton College, where he studied chemistry and mathematics. While at Hamilton, he performed research with Robin Kinnel, contributing to the total syntheses of recently isolated marine natural products, Ian Rosenstein, studying ring-opening reactions of cyclopropylcarbinyl radicals, and Martin Brechbiel (at the National Cancer Institute), investigating the development of new PET imaging agents. In 2013, Miles began his Ph.D. in chemistry, joining the laboratory of Prof. Roizen at Duke University. His doctoral research has focused Suzuki-Miyaura reactions to access alkylpyridines, new preparations of N-substituted sulfamate esters, and uses of these sulfamate esters in radical-mediated C–H functionalization processes.
Footnotes
Dedicated to Prof. Brian M. Stoltz as an early celebration of his fiftieth birthday
References
- (1).(a) For select reviews on C–H functionalization, see: Bergman RG Nature, 2007, 446, 391. [DOI] [PubMed] [Google Scholar]; (b) Davies HML; Manning JR Nature, 2008, 451, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lyons TW; Sanford MS Chem. Rev 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) White MC Science, 2012, 335, 807. [DOI] [PubMed] [Google Scholar]; (e) He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Chem. Rev 2017, 117, 8754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chu JCK; Rovis T Angew. Chem. Int. Ed 2018, 57, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Karimov RR; Hartwig JF Angew. Chem. Int. Ed 2018, 57, 4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For a recent perspective, see: Yan M; Lo JC; Edwards JT; Baran PS J. Am. Chem. Soc 2016, 138, 12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) For reviews focusing on the generation and use of nitrogen-centered radicals, see: Zard SZ Chem. Rev 2008, 108, 1603. [DOI] [PubMed] [Google Scholar]; (b) Kärkäs MD ACS Catal 2017, 7, 4999. [Google Scholar]; (c) Kärkäs MD Chem. Soc. Rev 2018, 47, 5786. [DOI] [PubMed] [Google Scholar]
- (4).(a) For recent, elegant innovations surrounding nitrogen-centered radicals in intermolecular C–H functionalization, see: Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ J. Am. Chem. Soc 2014, 136, 14389. [DOI] [PubMed] [Google Scholar]; (b) Quinn R; Könst Z; Michalak S; Schmidt Y; Szklarski A; Flores A; Nam S; Horne D; Vanderwal C; Alexanian EJ J. Am. Chem. Soc 2016. 138, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Czaplyski WL; Na CG; Alexanian EJ J. Am. Chem. Soc 2016, 138, 13854. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Carestia AM; Ravelli D; Alexanian EJ Chem. Sci 2018, 9, 5360. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tierney MM; Crespi S; Ravelli D; Alexanian EJ J. Org. Chem 2019, 84, 12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).a) For a discussion of the fundamentals of HAT, see: Mayer JM. Acc. Chem. Res 2011, 44, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) For a recent review elaborating on the utility of HAT, see Capaldo L; Ravelli D Eur. J. Org. Chem, 2017, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For a review of selective hydrogen-atom transfer, see: Stateman LM; Nakafuku KM; Nagib DA Synthesis, 2018, 50, 1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) For computational analyses of intramolecular HAT mediated by alkoxyl radicals, see: Dorigo AE; Houk KN J. Am. Chem. Soc 1987, 109, 2195. [Google Scholar]; (b) Dorigo AE; Houk KN J. Org. Chem 1988, 53, 1650. [Google Scholar]; (c) Dorigo AE; McCarrick MA; Loncharich RJ; Houk KN J. Am Chem. Soc 1990, 112, 7508. [Google Scholar]; (d) Zou Y; Xue X-S; Deng Y; Smith AB; Houk KN Org. Lett 2019, 21, 5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Beckwith ALJ; Ingold KU In Rearrangements in Ground and Excited States, Vol. 1; DeMayo P, Ed.; Academic: New York, 1980, 251. [Google Scholar]
- (9).(a) Hofmann AW Ber. Dtsch. Chem. Ges 1883, 16, 558. [Google Scholar]; (b) ffler K; Freytag C Ber. Dtsch. Chem. Ges 1909, 42, 3427. [Google Scholar]
- (10).(a) For the seminal disclosure, see: De Armas P; Carrau R; Concepción JI; Francisco CG; Hernández R; Suárez E Tetrahedron Lett 1985, 26, 2493. [Google Scholar]; (b) For further investigations and applications: see: Carrau R; Hernández R; Suárez E; Betancour CJ Chem Soc., Perkin Trans 1, 1987, 937. [Google Scholar]; (c) Dorta RL; Francisco CG; Suárez EJ Chem. Soc.; Chem. Commun 1989, 1168. [Google Scholar]; (d) Francisco CG; Herrera AJ; Suárez EJ Org. Chem 2003, 68, 1012. [DOI] [PubMed] [Google Scholar]
- (11).(a) Matínez C; Muñiz K Angew. Chem. Int. Ed 2015, 54, 8287. [DOI] [PubMed] [Google Scholar]; (b) Qin Q; Yu S; Org. Lett 2015, 17, 1894. [DOI] [PubMed] [Google Scholar]; (c) Choi GJ; Zhu Q; Miller DX; Gu CJ; Knowles RR Nature, 2016, 539, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chu JCK; Rovis T Nature, 2016, 539, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wappes EA; Fosu SC; Chopko TC; Nagib DA Angew. Chem. Int. Ed 2016, 55, 9974. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen D-F; Chu JCK; Rovis TJ Am. Chem. Soc 2017, 139, 14897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hu X; Zhang G; Bu X; Nie L; Lei A ACS Catal 2018, 8, 9370. [Google Scholar]; (h) Wang F; Stahl SS Angew. Chem. Int. Ed 2019, 58, 6385. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Nikolaienko P; Jentsch M; Kale AP; Cai Y; Rueping M; Chem. Eur. J 2019, 25, 7177. [DOI] [PubMed] [Google Scholar]
- (12).Corey EJ; Hertler WR J. Am. Chem. Soc 1960, 82, 1657. [Google Scholar]
- (13).For a review of 1,n-HAT processes where n≠5, see: Nechab M; Mondal S; Bertrand MP Chem. Eur. J 2014, 20, 16034. [DOI] [PubMed] [Google Scholar]
- (14).(a) Kimura M; Ban Y Synthesis, 1976, 201. [Google Scholar]; (b) Ban Y; Kimura M; Oishi T Heterocycles, 1974, 2, 323. [Google Scholar]
- (15).(a) Wang Y-F; Chen H; Zhu X; Chiba SJ Am. Chem. Soc 2012, 134, 11980. [DOI] [PubMed] [Google Scholar]; (b) Chen H; Sanjaya S; Wang Y-F; Chiba S Org. Lett 2013, 15, 212. [DOI] [PubMed] [Google Scholar]
- (16).(a) Wawzonek S; Thelen PJ J. Am. Chem. Soc 1950, 72, 2118. [Google Scholar]; (b) Wawzonek S; Nelson MF Jr.; Thelen JP J. Am. Chem. Soc 1951, 73, 2806. [Google Scholar]
- (17).(a) Hernández R; Medina MC; Salazar JA; Suárez E Tetrahedron Lett 1987, 28, 2533. [Google Scholar]; (b) Carrau R; Hernández, Suárez E J. Chem. Soc. Perkin Trans 1, 1987, 937. [Google Scholar]; (c) Freire R; Martín A; Pérez-Martín I; Suárez E Tetrahedron Lett 2002, 43, 5113. [Google Scholar]; (d) Francisco CG; Herrera AJ; Martín Á; Pérez-Martín I; Suárez E Tetrahedron Lett 2007, 48, 6384. [Google Scholar]
- (18).Wolff ME Chem. Rev 1963, 63, 55. [Google Scholar]
- (19).Neale RS; Walsh MR; Marcus NL J. Org. Chem 1965, 30, 3683. [Google Scholar]
- (20).Chen K; Richter JM; Baran PS J. Am. Chem. Soc 2008, 130, 7247. [DOI] [PubMed] [Google Scholar]
- (21).Hennessy ET; Betley TA Science, 2013, 340, 591. [DOI] [PubMed] [Google Scholar]
- (22).Zhang Z; Stateman LM; Nagib DA Chem. Sci 2019, 10, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Morcillo SP; Dauncey EM; Kim JH; Douglas JJ; Sheikh NS; Leonori D Angew. Chem. Int. Ed 2018, 57, 12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) For a seminal report, see: Espino CG; When PM; Chow J; Du Bois JJ Am. Chem. Soc 2001, 123, 6935. [Google Scholar]; (b) For mechanistic insights, see: Fiori KW; Espino CG; Brodsky BH; Du Bois J Tetrahedron 2009, 65, 3042. [Google Scholar]
- (25).(a) For select innovations of C−H amination reactions using sulfamate esters, see: Milczek E; Boudet N; Blakey S Angew. Chem. Int. Ed 2008, 47, 6825. [DOI] [PubMed] [Google Scholar]; (b) Liu Y; Guan X; Lai-Ming Wong E; Liu P; Huang J-S; Che C-MJ Am. Chem. Soc 2013, 135, 7194. [DOI] [PubMed] [Google Scholar]; (c) Alderson JM; Phelps AM; Scamp RJ; Dolan NS; Schomaker JM J. Am. Chem. Soc 2014, 136, 16720. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Paradine SM; Griffin JR; Zhao J; Petronico AL; Miller SM; White MC Nat. Chem 2015, 7, 987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) For reviews discussing the use of sulfamate esters in position-selective amination technologies, see: Collett F; Dodd RH; Dauban P Chem. Commun 2009, 5061. [DOI] [PubMed] [Google Scholar]; (f) Roizen JL; Harvey ME; Du Bois J Acc. Chem. Res 2012, 45, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).For an early example of metal-mediated, HLF-type reaction with sulfamate esters, see: Zalatan DN; Du Bois J Synlett, 2009, 1, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).(a) Fore rare examples of directed γ-C–H functionalization reactions of alcohol derivatives, see: Kasuya S; Kamijo S; Inoue M Org. Lett 2009, 11, 3630. [DOI] [PubMed] [Google Scholar]; (b) Voica A-F; Medoza A; Gutekunst WR; Fraga JO; Baran PS Nat. Chem 2012, 4, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Simmons EM; Hartwig JF; Nature 2012, 483, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Parasram M; Cheuntragool P; Shi Y; Gevorgyan VJ Am. Chem. Soc 2017, 139, 12857. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tanaka K; Ewing WR; Yu J-QJ Am. Chem. Soc 2019, 141, 15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Short MA; Blackburn JM; Roizen JL Angew. Chem. Int. Ed 2018, 57, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).(a) Cismesia MA; Yoon TP Chem. Sci 2015, 6, 5426. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hatchard CG; Parker CA; Proc. R. Soc. London Ser. A 1956, 235, 518. [Google Scholar]
- (30).(a) For some pioneering examples, see: Barton DHR; Beaton JM; Geller LE; Pechet MM J. Am. Chem. Soc 1960, 82, 2640. [Google Scholar]; (b) Minisci F; Galli R; Galli A; Bernardi A Tetrahedron Lett 1967, 8, 2207. [Google Scholar]; (c) Deno NC; Fishbein JR; Wyckoff JC; J. Am. Chem. Soc 1971, 93, 2065. [Google Scholar]; (d) Breslow R Heyer D; J. Am. Chem. Soc 1982, 104, 2045. [Google Scholar]; (e) For select modern approaches, see: Liu W; Groves JT J. Am. Chem. Soc 2010, 132, 12847. [DOI] [PubMed] [Google Scholar]; (f) Liu W; Groves JT Acc. Chem. Res 2015, 48, 1727. [DOI] [PubMed] [Google Scholar]; (g) Ozawa J; Kanai M Org. Lett 2017, 19, 1430. [DOI] [PubMed] [Google Scholar]
- (31).Blackburn JM; Short MA; Castanheiro T; Ayer SK; Muellers TD; Roizen JL Org. Lett 2017, 19, 6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) Sathyamoorthi S; Banerjee S; Du Bois J; Burns NZ; Zare RN Chem. Sci 2018, 9, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Control reactions excluding catalyst in the presence of light gave modest amounts of product, suggesting that light-mediated N–Br homolysis may be operative. [Google Scholar]
- (33).Del Castillo E; Martínez MD; Bosnidou AE; Duhamel T; O’Broin CQ; Zhang H; Escudero-Adán EC; Martínez-Belmonte M; Muñiz K Chem. Eur. J 2018, 24, 17225. [DOI] [PubMed] [Google Scholar]
- (34).Kiyokawa K; Nakamura S; Jou K; Iwaida K; Minakata S Chem. Commun 2019, 55, 11782. [DOI] [PubMed] [Google Scholar]
- (35).(a) For sulfamate ester-directed C–H xanthylation reactions, see: Ayer SK; Roizen JL J. Org. Chem 2019, 84, 3508. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) For prior art featuring xanthate transfer processes, see: Na CG; Alexanian EJ Angew. Chem. Int. Ed 2018, 57, 13106; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sato A; Yorimitsu H; Oshima K Chem. Asian J 2007, 2, 1568. [DOI] [PubMed] [Google Scholar]
- (36).(a) Ollivier C; Renaud PJ Am. Chem. Soc 2000, 122, 6496. [Google Scholar]; (b) Ollivier C; Renaud PJ Am. Chem. Soc 2001, 123, 4717. [DOI] [PubMed] [Google Scholar]; (c) Bertrand F; Quiclet-Sire B; Zard SZ Angew. Chem. Int. Ed 1999, 38, 1943. [DOI] [PubMed] [Google Scholar]; (d) Quiclet-Sire B; Seguin S; Zard SZ Angew. Chem. Int. Ed 1998, 37, 2864. [DOI] [PubMed] [Google Scholar]; (e) Spiegel DA; Wilberg KB; Schacherer LN; Medeiros MR; Wood JL J. Am. Chem. Soc 2005, 127, 12513. [DOI] [PubMed] [Google Scholar]; (f) Soulard V; Villa G; Vollmar DP; Renaud PJ Am. Chem. Soc 2018, 140, 155. [DOI] [PubMed] [Google Scholar]
- (37).(a) For select reviews on photoredox catalysis, see: McAtee RC; McClain EJ; Stephenson CR J. Trends in Chemistry, 2019, 1, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romer NA; Nicewicz DA Chem. Rev 2016, 116, 10075. [DOI] [PubMed] [Google Scholar]; (c) Schultz DM; Yoon TP Science, 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Prier CK; Rankic DA; MacMillan DW C. Chem. Rev 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xuan J; Xiao W-J Angew. Chem. Int. Ed 2012, 51, 6828. [DOI] [PubMed] [Google Scholar]; (f) Narayanam JMR; Stephenson CR J. Chem. Soc. Rev 2011, 40, 102. [DOI] [PubMed] [Google Scholar]
- (38).(a) Ma Z-Y; Guo L-N; You Y; Yang F; Hu M; Duan X-H Org. Lett 2019, 21, 5500. [DOI] [PubMed] [Google Scholar]; (b) Kanegusuku ALG; Castanheiro T; Ayer SK; Roizen JL Org. Lett 2019, 21, 6089. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shu W; Zhang H; Huang Y Org. Lett 2019, 21, 6107. [DOI] [PubMed] [Google Scholar]
- (39).(a) For the seminal disclosures of the Giese reaction, see: Giese B; Meixner J Chem. Ber 1981, 114, 2138. [Google Scholar]; (b) Minisci F; Pallini U Gazz. Chim. Ital 1961, 91, 1030. [Google Scholar]; (c) For leading reviews regarding the addition of carbon-centered radicals to alkenes, see: Giese B; Angew. Chem. Int. Ed 1980, 22, 753. [Google Scholar]; (d) Zhang W Tetrahedron 2001, 57, 7237. [Google Scholar]; (e) Srikanth GSC; Castle SL Tetrahedron 2005, 61, 10377. [Google Scholar]
- (40).For a previous example, see: Capacci AG; Malinowski JT; McAlpine NJ; Kuhne J; MacMillan DW C. Nat. Chem 2017, 9, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41). Slight variations on the discussed pathways could be envisioned if radical cation intermediates are formed in lieu of the proposed neutral nitrogen-radical species.
- (42).For a review on PCET, see: Yayla HG; Knowles RR Synlett, 2014, 25, 2819. [Google Scholar]
- (43).(a) Fraiji LK; Hayes DM; Werner TC J. Chem. Ed 1992, 69, 424. [Google Scholar]; (b) Hopkinson MN; Gómez-Suárez A; Teders M; Sahoo B; Glorius F Angew. Chem. Int. Ed 2016, 55, 4361. [DOI] [PubMed] [Google Scholar]
- (44). Use of an enantioenriched sulfamate ester substrate was found to deliver racemic C(3) alkylated product. Recovered starting material, however, was isolated with no detectable erosion in enantioenrichment. For specific details, see ref. 38b.
- (45).Kurokawa T; Kim M; Du Bois J Angew. Chem. Int. Ed 2009, 48, 2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).(a) For a review which discusses γ-C–H functionalization reactions of amine derivatives, see: Xu Y; Dong G Chem. Sci 2018, 9, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) For additional γ-C–H functionalization reactions of amine derivatives, see: Zaitsev VG; Shabashov D, Daugulis OJ Am. Chem. Soc 2005, 27, 3154. [DOI] [PubMed] [Google Scholar]; (c) Callela J; Pla D; Gorma TW; Domingo V; Haffemayer B; Gaunt MJ Nat. Chem 2015, 7, 1009. [DOI] [PubMed] [Google Scholar]; (d) Xu Y; Young MC; Wang C; Magness DM; Dong G Angew. Chem. Int. Ed 2016, 55, 9084. [DOI] [PubMed] [Google Scholar]; (e) Wu Y; Chen Y-Q; Liu T; Eastgate MD; Yu J-QJ Am. Chem. Soc 2016, 138, 14554. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liu Y; Ge H Nat. Chem 2017, 9, 26. [Google Scholar]; (g) Yada A; Liao W; Sato Y; Murakami M; Angew. Chem. Int. Ed 2017, 56, 1073. [DOI] [PubMed] [Google Scholar]
- (47).(a) For the seminal report, see: Lu H; Jiang H; Wojtas L; Zhang XP Angew. Chem. Int. Ed 2010, 49, 10192. [DOI] [PubMed] [Google Scholar]; (b) For extensions transforming other types of C–H bonds, see: Lu H; Jiang H; Hu Y; Wojtas L; Zhang XP Chem. Sci 2011, 2, 2361. [Google Scholar]; (c) Lu H; Hu Y; Jiang H; Wojtas L; Zhang XP Org. Lett 2012, 14, 5158. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu H; Li C; Jiang H; Lizardi CL; Zhang XP Angew. Chem. Int. Ed 2014, 53, 7028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) For an enantioselective variant, see: Li C; Lang K; Lu H; Hu Y; Cui Z; Wojtas L; Zhang XP Angew. Chem. Int. Ed 2018, 57, 16837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Duhamel T; Martínez MD; Sideri IK; Muñiz K ACS Catal 2019, 9, 7741. [Google Scholar]
- (49).Short MA; Shehata MF; Sanders MA; Roizen JL Chem. Sci 2019, accepted article. DOI: 10.1039/C9SC03428E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shehata MF; Short MA; Sanders MA; Roizen JL Tetrahedron 2019, 75, 3186. [Google Scholar]