

Abstract
Cerebral small vessel disease (CSVD) is the most important cause of vascular cognitive impairment (VCI). Most CSVD cases are sporadic but familial monogenic forms of the disorder have also been described. Despite the variants identified, many CSVD cases remain unexplained genetically. We used whole-exome sequencing in an attempt to identify novel gene variants underlying CSVD. A cohort of 35 Finnish patients with suspected CSVD was analyzed. Patients were screened negative for the most common variants affecting function in NOTCH3 in Finland (p.Arg133Cys and p.Arg182Cys). Whole-exome sequencing was performed to search for a genetic cause of CSVD. Our study resulted in the detection of possibly pathogenic variants or variants of unknown significance in genes known to associate with CSVD in six patients, accounting for 17% of cases. Those genes included NOTCH3, HTRA1, COL4A1, and COL4A2. We also identified variants with predicted pathogenic effect in genes associated with other neurological or stroke-related conditions in seven patients, accounting for 20% of cases. This study supports pathogenic roles of variants in COL4A1, COL4A2, and HTRA1 in CSVD and VCI. Our results also suggest that vascular pathogenic mechanisms are linked to neurodegenerative conditions and provide novel insights into the molecular basis of VCI.
Subject terms: Stroke, Sequencing, Genetics research, Dementia
Introduction
Vascular cognitive impairment (VCI) is a term used for cognitive impairment associated with cerebrovascular disease [1]. Vascular dementia (VaD) is the most severe form of VCI and it is the second most common cause of dementia after Alzheimer’s disease (AD) [2]. An important cause of VCI is cerebral small vessel disease (CSVD) which consists of a heterogeneous group of pathological processes that affect the small vessels of the brain [3]. Most CSVD patients suffer from a sporadic disorder but familial monogenic forms of the disorder have also been described [4]. CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) is the most frequent subtype of familial CSVD and is caused by variants affecting function in the NOTCH3 gene [5]. CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy) is an autosomal recessive CSVD caused by pathogenic HTRA1 gene variants [6], although an autosomal dominant form of the disease has been identified [7]. Autosomal dominant COL4A1-related CSVD is usually caused by pathogenic glycine missense variants within the triple-helical domain of COL4A1/COL4A2 collagen genes [8]. Multi-infarct dementia of Swedish type and PADMAL (pontine autosomal dominant microangiopathy and leukoencephalopathy) were recently found to be caused by variants of a predicted binding site for miR-29 microRNA located within the 3′UTR of COL4A1 gene [9, 10]. These diseases differ from other COL4A1-related CSVD, and variants found both in Swedish multi-infarct dementia family and PADMAL cases disrupt the same miR-29 binding site leading to upregulation of COL4A1 [9, 10].
Despite the variants identified, many CSVD cases remain unexplained genetically even when they appear familial. In this study, we used whole-exome sequencing (WES) to study the genetic background of a cohort of 35 Finnish CSVD patients. We also investigated the prevalence of variants in miR-29 binding site of COL4A1 in a cohort of 60 Finnish CSVD patients.
Subjects and methods
The study was approved by the Ethical Committee of the Hospital District of Southwest Finland. The approval for the use of patient DNA samples was obtained from the National Supervisory Authority for Welfare and Health (Valvira) and Hospital District of Southwest Finland. Permit for the access to medical records was obtained from the National Institute for Health and Welfare.
Patients
A cohort of Finnish patients with suspected CADASIL was selected from 365 patients referred for diagnostic testing for NOTCH3 in the Department of Medical Genetics of Turku University Hospital between years 1998 and 2004. All patients were screened negative for the most common variants affecting function in NOTCH3 (p.Arg133Cys and p.Arg182Cys). Two of the patients were also screened negative for variants in NOTCH3 exons 3–8, 11, and 18–20 and one patient was screened negative for variants in NOTCH3 exons 3, 4, and 8 (NOTCH3 exons numbered consecutively from 1 to 33 according to NM_000435.2). Medical records of the cohort of 365 patients were reviewed to confirm the diagnosis or clinical phenotype. Characteristics of the whole Finnish cohort are summarized in Supplementary Table I. After examining the medical records, 60 patients from the cohort of 365 patients were confirmed to have a diagnosis of VCI and were selected for sequence analysis of the miR-29 microRNA binding site in the 3′UTR of COL4A1 (Fig. 1). Of these 60 VCI patients, 35 patients were selected for whole-exome sequencing (Fig. 1). The inclusion criteria included the presence of VCI with white matter changes in magnetic resonance imaging, age at onset up to 75 years and/or family history of dementia or stroke. Family history was defined from the medical notes and was considered positive if patient had at least one relative suffering from dementia or stroke. The inclusion criteria were used to select the best candidates with adequate clinical information from the cohort of 60 patients to investigate familial forms of VCI.
Fig. 1. Schematic presentation of the study describing the workflow of selection of patients and genetic examinations.
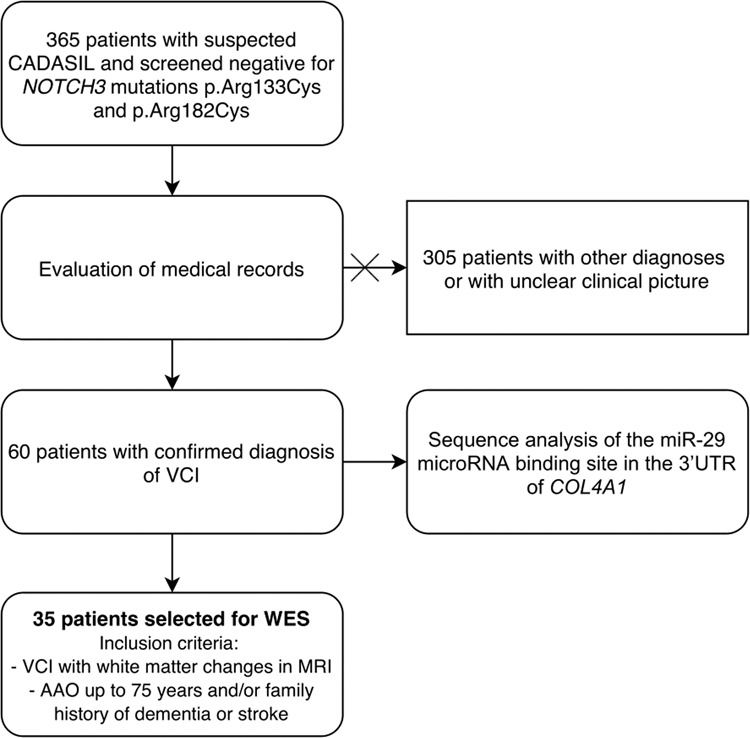
AAO age-at-onset, MRI magnetic resonance imaging, VCI vascular cognitive impairment, WES whole-exome sequencing.
Sanger sequencing of the miR-29 microRNA binding site in the 3′UTR of COL4A1
The miR-29 microRNA binding site in the 3′UTR of COL4A1 was sequenced in 60 of the samples studied. Sequencing was performed after PCR amplification with Applied Biosystems BigDye terminator version 3.1 sequencing chemistry in an ABI3730xl DNA analyzer (region sequenced: NG_011544.2(NM_001845.5):c.5001_*145). Primers are available upon request. Sequences were analysed using SeqScape Software (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA).
Whole-exome sequencing (WES)
Details of library preparation and data processing are shown in Supplemental Materials. The stroke-gene panels SGP1 and SGP2 compiled by Ilinca et al. [11] was utilized in the variant analysis. Variants located in 168 genes/loci known to be associated with monogenic causes of stroke [11] were extracted from the whole-exome data. Mitochondrial genes were excluded from this analysis. Variants were filtered out if they were located in a known genomic duplication region and if they did not pass the VQSR score. Variants included in subsequent analyses had a high or moderate impact annotation score, which excluded synonymous and intronic variants that were not located within splice sites. Variants reported at this stage had an allele frequency <1% in gnomAD (v2.1.1) and passed the QC filters described by Patel et al. [12]. In addition, applying the same QC steps, we searched for rare variants by evaluating all non-synonymous and splice site variants that were absent from gnomAD. In addition, we used the Exomiser software (v11.0.0) to prioritize variants related to CADASIL (ORPHA:136). Exomiser aids finding disease-causing variants from WES data by annotating, filtering, and prioritising variants according to user-defined criteria. With Exomiser, autosomal dominant and recessive inheritance models were analyzed to compile a list of the three to four top ranked candidate variants. Only variants that had allele frequency <1% in gnomAD were considered in Exomiser analysis. The workflow of the WES data analysis is presented in Supplementary Fig. 1. In silico prediction tools SIFT, PolyPhen2, MutationTaster, LRT, MutationAssessor and CADD were used to predict variant pathogenicity. Only variants with CADD score ≥10 were considered as potentially pathogenic. Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) criteria [13]. Possibly causative variants were submitted to ClinVar (submission ID: SUB7388577, accession numbers SCV001250686, SCV001250687, SCV001250688, SCV001250689, SCV001250690, SCV001250691, SCV001250692, SCV001250693, SCV001250694, SCV001250695, SCV001250696, SCV001250697, SCV001250698, SCV001250699, SCV001250700).
Results
WES results
We used WES to identify the variants underlying CSVD in 35 Finnish patients. A positive family history was identified from the patient records for 46% (16/35) of the patients. Of the subjects, 54% (19/35) were women. Clinical characteristics of the patients studied by WES are summarized in Table 1.
Table 1.
Characteristics of the 35 patients selected for WES.
| Sample | Gender | AAO | Diagnosis/clinical features | Family history | Affected family members | Migraine | Hypertension | Other risk factors | Other conditions |
|---|---|---|---|---|---|---|---|---|---|
| 9 | F | 74 | VaD | Yes | Father, eight siblings (dementia and multiple cerebral strokes) | No | Yes | Diabetes | |
| 43 | M | 64 | VaD/dementia NAS | n/a | No | Yes | |||
| 48 | M | 55 | VaD, depression, psychosis | Yes | Sibling with same clinical features. Also uncle with moton neuron disease | No | No | ||
| 57 | F | 61 | VaD, schizoaffective psychosis | No | No | No | Obesity, myocardial infarction | ||
| 102 | M | 57 | VaD | n/a | No | Yes | |||
| 108 | M | 58 | VaD | Yes | Father (dementia, AAO 60 years) | No | No | ||
| 110 | M | 58 | VaD | n/a | No | Yes | |||
| 125 | F | 65 | VaD | Yes | Uncle (dementia, before age 60), sibling (died of cerebral hemorrage at age 59 years) | Yes | Yes | Hypercholesterolemia | |
| 137 | F | 65 | VaD, parkinsonismus secundaris | No | Yes | Yes | Coronary artery disease | ||
| 140 | F | 43 | VaD | Yes | Father (died of stroke at age 57), several siblings (strokes), one sibling (epilepsy) | No | Yes | Hypercholesterolemia, diabetes, myocardial infarction, coronary artery disease | |
| 147 | F | 74 | VaD | Yes | Sibling (progressive dementing disorder, AAO 60 years) | No | Yes | Coronary artery disease | |
| 156 | M | 17 | VaD, epilepsy, psychiatric features | n/a | Yes | No | |||
| 160 | F | 56 | VCI | n/a | Yes | Yes | Hypercholesterolemia | ||
| 161 | M | 65 | VaD | n/a | No | No | |||
| 184 | F | 71 | VaD | n/a | No | Yes | Angina pectoris | ||
| 185 | F | 62 | VaD | Yes | Father (stroke), mother (cognitive impairment). Also child and several relatives suffering from hearing loss. | No | Yes | ||
| 204 | M | 69 | VaD | n/a | No | No | Myocardial infarction | ||
| 207 | F | 74 | VaD | Yes | Father (cerebral hemorrage), sibling (aphasia and hemiplegia, AAO 66 years) | Yes | Yes | Hypercholesterolemia | |
| 233 | F | 73 | VaD | Yes | Mother, identical twin (dementia, before age 70) | No | Yes | Diabetes, coronary artery disease | |
| 235 | M | 56 | VaD | n/a | No | Yes | Atherosclerosis, cardiomyopathy, heart failure, diabetes, obesity, smoking | ||
| 236 | M | 64 | VaD | n/a | No | Yes | Diabetes, hypercholesterolemia | ||
| 255 | F | 31 | VaD | Yes | Grandmother (hemiplegia, several strokes, dementia) | Yes | No | Smoking | Chronic obstructive pulmonary disease |
| 260 | F | 68 | VaD | n/a | No | Yes | Hypercholesterolemia | ||
| 266 | F | 61 | VaD, depression | Yes | Sibling (dementia, before age 50) | No | No | ||
| 269 | M | 54 | VaD | No | Yes | No | Heart failure, atrial fibrillation, previous heavy alcohol consumption | Colitis ulcerosa | |
| 273 | M | 61 | VCI | Yes | Mother (multiple strokes, first at age 42), sibling (stroke at age 50), sibling (multiple strokes, first at age 55), two aunts (stroke at young age), uncle (stroke at young age) | No | No | Hypercholesterolemia | |
| 283 | F | 60 | VaD | Yes | Father (multiple strokes, first at age 46), maternal aunt (stroke, migraine and dementia). Several relatives suffering from cardiovascular disease and migraine | No | Yes | Hypercholesterolemia | |
| 289 | M | 48 | VaD | n/a | No | Yes | Heart arrhythmia, smoking | ||
| 290 | F | 56 | VaD | n/a | No | No | Hypercholesterolemia | ||
| 293 | M | 56 | VaD | n/a | Yes | Yes | |||
| 343 | M | 68 | VaD | No | No | No | Hypercholesterolemia | ||
| 379 | F | 70 | VaD | Yes | Sibling with same clinical features (dementia, hearing loss). Child suffering from migraine. | Yes | Yes | Hypercholesterolemia | Ménière’s disease |
| 380 | F | 60 | VaD, epilepsy | Yes | Mother (multiple strokes) | No | Yes | Obesity | |
| 383 | M | 68 | VaD | Yes | Mother and sibling (strokes before age 70) | No | Yes | Coronary artery disease, hypercholesterolemia | |
| 387 | F | 73 | VaD | n/a | No | Yes | Smoking |
AAO age at onset, F female, M male, NAS Non Aliter Specificatus (Not Further Specified), n/a no information available, VaD vascular dementia, VCI vascular cognitive impairment.
Six of the patients (17%) carried variants possibly affecting function in NOTCH3, HTRA1, COL4A1, or COL4A2, which are genes known to be associated with CSVD (Table 2). In addition, seven of the patients (20%) carried variants possibly affecting function in genes associated with other neurological or stroke-related conditions (Table 2). All results of the analyses are presented in Supplementary Tables II–IV. Heterozygous NOTCH3 variants were identified in two patients: c.323 G > A, p.(Cys108Tyr) in exon 3 and c.2149 C > T, p.(Arg717Cys) in exon 14. Both variants are missense variants resulting either in the gain or loss of a cysteine residue in the EGF-like repeats of NOTCH3 protein that is the most common type of variant causing CADASIL. NOTCH3 variant c.323 G > A, p.(Cys108Tyr) has been reported earlier in the literature in a CADASIL patient [14]. The patient carrying the c.323 G > A variant had a phenotype consistent with CADASIL and positive family history. The other NOTCH3 variant c.2149 C > T, p.(Arg717Cys) has not been reported before. It was detected in a VaD patient whose phenotype included multiple strokes, atherosclerosis, cardiomyopathy, and heart failure. The patient also had multiple vascular risk factors; diabetes, obesity, and smoking.
Table 2.
Possibly causative variants identified by WES.
| Patient | AAO | Family history | Gene | Nucleotide change | Amino acid change | Zygocity | Reference sequence | dbSNP ID | Allele frequency (gnomAD total) | Allele frequency (gnomAD Finnish) | CADD phred | PolyPhen2 | Mutation Taster | SIFT | ACMG classification | Condition associated with the gene | Condition associated with the variant, reference | Detected in stroke-panel analysis | Detected in Exomiser analysis | Detected in rare variant analysis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 102 | 57 | n/a | CCM1 (KRIT1) | c.1565 T > C | p.(Ile522Thr) | Het | NM_194456.1 | rs758188972 | 0.000007118 | 0.00001558 | 22,2 | P | D | T | 3 | Cerebral cavernous malformations [24, 25] | Novel | Yes | No | No |
| 102 | ITM2B | c.193 C > T | p.(Leu65Phe) | Het | NM_021999.4 | 0 | 0 | 32 | D | D | D | 3 | Cerebral amyloid angiopathy, retinal dystrophy [21–23] | Novel | Yes | Yes | Yes | |||
| 137 | 65 | No | CACNA1A | c.1348 T > C | p.(Ser450Pro) | Het | NM_001127222.1 | rs1308599413 | 0.000007 | 0.00008 | 26,4 | P | D | D | 3 | Episodic ataxia, familial hemiplegic migraine, spinocerebellar ataxia [27] | Novel | Yes | Yes | No |
| 140 | 43 | Yes | C1R | c.336 G > C | p.(Met112Ile) | Het | NM_001733.7 | rs139531404 | 0.002823 | 0.002157 | 22,2 | D | D | D | 3 | Ehlers–Danlos syndrome, periodontal type 1 [30] | Novel | Yes | No | No |
| 140 | CCM2 | c.1346 T > G | p.(Ile449Ser) | Het | NM_001029835.2 | 0 | 0 | 22 | D | D | D | 3 | Cerebral cavernous malformations [25, 26] | Novel | Yes | Yes | Yes | |||
| 156 | 17 | n/a | COL4A1 | c.2440 G > A | p.(Gly814Arg) | Het | NM_001845.5 | 0 | 0 | 26,5 | D | D | D | 3 | SVD [8] | Novel | Yes | Yes | Yes | |
| 156 | PSEN2 | c.53 C > T | p. (Thr18Met) | Het | NM_000447.2 | rs143061887 | 0.00002 | 0 | 29 | D | D | D | 3 | AD [17] | Novel | No | Yes | No | ||
| 160 | 56 | n/a | COL4A2 | c.4291 C > T | p.(Arg1431Cys) | Het | NM_001846.3 | rs139124960 | 0.000007 | 0 | 14,11 | P | D | D | 3 | SVD [8] | Novel | Yes | Yes | No |
| 184 | 71 | n/a | NPPA | c.377 G > A | p.(Arg126Gln) | Het | NM_006172.4 | rs1803268 | 0.00007592 | 0.0006427 | 19,69 | D | N | D | 3 | Atrial fibrillation familial [32, 33] | Novel | Yes | No | No |
| 185 | 62 | Yes | COL4A1 | c.401 C > T | p.(Pro134Leu) | Het | NM_001845.5 | rs140517831 | 0.00042 | 0.002867 | 22,8 | D | D | T | 3 | SVD [8] | Novel | Yes | No | No |
| 204 | 69 | n/a | APP | c.1795G > A | p.(Glu599Lys) | Het | NM_000484.3 | rs140304729 | 0.00149 | 0.00836 | 20,5 | D | D | D | 3 | AD [17] | PD, LBD [18–20] | Yes | No | No |
| 235 | 56 | n/a | NOTCH3 | c.2149 C > T | p.(Arg717Cys) | Het | NM_000435.2 | rs144163298 | 0.000036 | 0 | 29,3 | D | D | T | 3 | CADASIL1 [5] | Novel | Yes | Yes | No |
| 273 | 61 | Yes | NOTCH3 | c.323 G > A | p.(Cys108Tyr) | Het | NM_000435.2 | 0 | 0 | 33 | D | D | D | 4 | CADASIL1 [14] | CADASIL [14] | Yes | Yes | Yes | |
| 290 | 56 | n/a | TMEM106B | c.115 G > C | p.(Asp39His) | Het | NM_018374.3 | 0 | 0 | 25,3 | D | D | D | 3 | FTLD, hypomyelinating leukodystrophy [28, 29] | Novel | No | Yes | Yes | |
| 379 | 70 | Yes | HTRA1 | c.961 G > A | p.(Ala321Thr) | Het | NM_002775.4 | rs587776449 | 0.000081 | 0.00089 | 34 | D | D | D | 3 | CARASIL [6], CADASIL2 [7, 16] | CARASIL [15] | Yes | No | No |
| 379 | COL4A1 | c.401 C > T | p.(Pro134Leu) | Het | NM_001845.5 | rs140517831 | 0.00042 | 0.002867 | 22,8 | D | D | T | 3 | SVD [8] | Novel | Yes | No | No |
ACMG variant classification [13] (5 = pathogenic, 4 = likely pathogenic, 3 = variant of unknown significance, 2 = likely benign, 1 = benign).
CADD Combined Annotation Dependent Depletion [40], algorithm for scoring the deleteriousness of variants (≥10 = belongs to 10% most deleterious variants in the human genome, ≥20 = belongs to 1% most deleterious variants in the human genome).
Polyphen2, MutationTaster, SIFT pathogenicity prediction tools (Polyphen2, D = damaging, P = possibly damaging, B = benign; MutationTaster, D = disease causing, N = polymorphism; SIFT, D = damaging, T = tolerated).
AAO age at onset, AD Alzheimer’s disease, FTLD frontotemporal lobar degeneration, het heterozygous, LBD Lewy Body Dementia, n/a no information available, PD Parkinson’s disease.
Furthermore, we identified a heterozygous HTRA1 variant c.961 G > A, p.(Ala321Thr), which has been reported in a CARASIL patient compound heterozygous with another HTRA1 variant [15]. Homozygous or compound heterozygous variants affecting function in HTRA1 are known to cause CARASIL, rare autosomal recessive CSVD [6], whereas heterozygous HTRA1 variants have been identified in autosomal dominant CSVD which is characterized by delayed onset and absence of extra-neurological features typical for CARASIL [7, 16]. The VaD patient carrying the HTRA1 c.961 G > A variant had a phenotype consistent with HTRA1-CSVD. The age at onset of the patient was 70 years and her phenotype included cerebral microangiopathy, lacunar infarcts, migraine with aura, hypertension and she also suffered from Ménière’s disease. Her sibling had a similar phenotype. The patient was not recorded to have extra-neurological features. In addition to the HTRA1 variant, the patient carried the COL4A1 variant c.401 C > T, p.(Pro134Leu) which was also identified in another patient in our study. We also detected two other collagen variants in two patients, COL4A1 c.2440 G > A, p.(Gly814Arg) and COL4A2 c.4291 C > T, p.(Arg1431Cys), both occurring on the triple-helical domain of the protein. The COL4A1 c.2440 G > A, p.(Gly814Arg) variant was identified in a patient who also carried the PSEN2 variant c.53 C > T, p.(Thr18Met). The patient had the youngest age of onset in the study cohort (17 years) and his phenotype included vascular leukoencephalopathy, multiple strokes, epilepsy, and psychiatric features. Variants affecting function in PSEN2 have been found in patients with early-onset AD [17]. The COL4A2 variant c.4291 C > T, p.(Arg1431Cys) was identified in a CSVD patient whose phenotype included VCI, migraine, mild hearing impairment, and balance impairment.
One of the patients carried the APP missense variant c.1795G > A, p.(Glu599Lys), which has previously been reported in patients with Parkinson’s disease or dementia with Lewy bodies [18–20]. Variants affecting function in APP are a well-known cause of early-onset AD and cerebral amyloid angiopathy (CAA). Heterozygous variants CCM1 (KRIT1) c.1565 T > C, p.(Ile522Thr) and ITM2B c.193 C > T, p.(Leu65Phe) were identified in a VaD patient whose phenotype also included behavioral changes and hearing impairment. ITM2B loss-of-function variants resulting lengthened protein products cause autosomal dominant CAA (Familial British and Danish dementia) [21, 22], but ITM2B gene has also been linked to retinal dystrophy [23]. Variants in the KRIT1 (CCM1) and CCM2 genes cause autosomal dominant cerebral cavernous malformations, which are vascular anomalies in the brain [24–26]. We also identified a novel heterozygous CACNA1A variant c.1348 T > C, p.(Ser450Pro), CACNA1A is a gene associated with familial hemiplegic migraine, episodic ataxia type 2 and spinocerebellar ataxia type 6 [27]. The patient carrying the CACNA1A variant c.1348 T > C, p.(Ser450Pro) suffered from migraine with aura and her phenotype also included secondary parkinsonism and dysphagia. In addition, we detected a novel heterozygous variant c.115 G > C, p.(Asp39His) in the TMEM106B gene. TMEM106B gene is identified as a risk factor for frontotemporal dementia (FTD), but the gene is also linked to hypomyelinating leukodystrophy [28, 29].
Furthermore, we detected variants in C1R and NPPA. These genes are linked to stroke-related conditions. Pathogenic variants in the C1R gene are associated with autosomal dominant periodontal Ehlers–Danlos syndrome [30], which is a syndrome that may include vascular anomalies [31]. However, the heterozygous C1R variant c.336 G > C, p.(Met112Ile) detected in our study is present in 0.2% of the Finnish population according to the gnomAD database and the clinical significance of the variant is interpreted both as uncertain and likely benign in ClinVar database. The patient carrying the C1R variant c.336 G > C, p.(Met112Ile) also carried the CCM2 variant c.1346 T > G, p.(Ile449Ser) and her phenotype included walking and balance impairment, hypercholesterolemia, diabetes, myocardial infarction, and coronary artery disease, and she had family history positive for strokes. The NPPA gene is linked to familial atrial fibrillation [32, 33], which may cause cardioembolic stroke. In our study, the heterozygous NPPA variant c.377 G > A, p.(Arg126Gln) was identified in a patient who suffered from angina pectoris.
Sanger sequencing of the miR-29 microRNA binding site in 3′UTR of COL4A1
A total of 60 Finnish CSVD patients were screened for variants in the miR-29 microRNA binding site in 3′UTR of COL4A1. Sanger sequencing did not reveal any variants in the miR-29 microRNA binding site in 3′UTR of COL4A1.
Discussion
Although VCI is very commonly found in subjects with dementia, research of the disease lags behind other dementing conditions. There are no common standards in the studies of VCI or universally accepted diagnostic criteria for the disease, which complicates reproducibility of research in this area. Research of VCI has also lacked large, well-characterized patient cohorts. Even though monogenic forms of VCI are considered rare, the identification and characterizations of these forms of disease may considerably contribute to the understanding of the molecular pathogenesis of dementing diseases. With this in mind, we investigated the genetics of VCI by studying a homogenous Finnish cohort with well-defined clinical features, ascertained by the individual revision of medical records. Our study resulted in the detection of several variants possibly affecting function both in known CSVD genes and in genes linked to other neurological disorders or stroke-related conditions.
Six patients carried variants possibly affecting function in the known CSVD genes: NOTCH3, COL4A1, COL4A2, and HTRA1, accounting for as high as 17% of all the patients. The relatively high proportion of these variants probably reflects the original selection of patients for CADASIL (NOTCH3) testing, and our selection criteria for exome sequencing might have further favored a CSVD type phenotype. Even so, these results support pathogenic roles of variants in COL4A1, COL4A2, and HTRA1 in CSVD and VCI. This is in line with the recent study by Ilinca et al., where variants in NOTCH3, COL4A1, and COL4A2 were found in a WES study in patients with suspected monogenic form of stroke [34].
Interestingly, we also detected several variants in genes associated with other dementing or neurodegenerative disorders, which may indicate the overlapping pathologies between these disorders. Detection of variants in the AD-linked genes APP and PSEN2 may represent a genetic connection of CSVD with AD pathology. Several studies have shown a relationship between CSVD and AD [35]. AD very often occurs concomitantly with vascular or other neurodegenerative pathology [36], but it is still unknown how pathologies of AD and CSVD interact with each other [37]. One of the study subjects carried variants both in CSVD-linked gene COL4A1 and AD-linked gene PSEN2, so it is possible that both variants had a role in his disease, which started at an exceptionally early age (17 years). In this study, three patients carried more than one variant that possibly affect function and may have roles in patients’ disease, indicating possible oligogenic cause of VCI. In addition to AD-linked genes, we observed variants possibly affecting function in genes linked to FTD and migraine. Although there are not many studies on the relationship between vascular impairment and FTD, an effect of vascular lesions in the pathogenesis of FTD has been suggested [38]. It is also possible that phenotypic similarities may have been the cause for detection of variants in genes linked to FTD and migraine in our study.
Distinguishing VCI from other forms of dementia and neurodegenerative diseases may be challenging, highlighting the importance of the evaluation of the clinical phenotype of the study subjects when studying a particular disease entity. In our study, the clinical information of the patients was obtained from the medical records, but the amount of the available information varied between patients. A large proportion of the subjects were later diagnosed with another disease than VCI, although CADASIL testing was originally performed (Supplementary Table I). Furthermore, less than half (46%) of the patients showed a positive family history, the rest of the subjects possibly representing sporadic cases. Samples from the relatives of the patients were not available and therefore we could not analyse the segregation of the detected variants. In addition, the cohort did not include any cases confirmed by neuropathological examination, which could have facilitated the diagnosing and characterization of patients.
Previous studies have shown that PADMAL and multi-infarct dementia of Swedish type are caused by variants in an untranslated region of COL4A1 [9, 10], but there is limited knowledge on the prevalence of these variants among CSVD patients in different populations. Here we screened the miR-29 microRNA binding site in 3′UTR of COL4A1 in 60 CSVD patients of Finnish origin, but found no variants to be present in our cohort. The small sample size and possible clinical heterogeneity of the cohort included in this analysis can be possible reasons for the negative results obtained. Despite these, this analysis suggests that COL4A1 3′UTR variants are a very rare cause of CSVD and they may be restricted to certain populations and/or clinical phenotypes. Further studies including larger sample sizes from different ethnicities are needed to fully reveal the role of COL4A1 3′UTR variants in the whole spectrum of CSVD.
Patients that remained negative may represent disorders that are inherited in a polygenic rather than a Mendelian manner. Two patients carried variants in genes associated with atrial fibrillation or Ehlers–Danlos syndrome, which are distinct from other variants detected in genes linked to CSVD or other neurological disorders, but which could also have roles in the vascular phenotypes of the patients. Vascular risk factors, such as hypertension and type 2 diabetes, and environmental risk factors, such as smoking and alcohol consumption, have also a role in the pathogenesis of VCI [39]. Some of the patients may carry pathogenic intronic variants, copy number variants, repeat expansions, structural variants, or methylation changes that were not possible to detect with WES. In addition, some of the patients may carry variants in novel genes that have not yet been found to be associated with VCI or other forms of neurodegeneration.
It should also be noted, that the stroke-gene panel used in the variant analysis needs to be updated in future studies, as more data on the genetic background of cerebrovascular phenotypes will accumulate. Pathogenicity of the identified variants with uncertain significance should be confirmed with functional studies and larger data sets.
These data provide evidence for improved information and guidance in genetic testing of familial VCI. Although there are no curative treatments available for VCI, identifying disease-causing variants may aid making a precise diagnosis and provide information on the prognosis. Genetic diagnosis provides the opportunity for diagnostic testing of other affected family members and predictive screening of the unaffected relatives.
In conclusion, our results support pathogenic roles of variants in COL4A1, COL4A2, and HTRA1 in CSVD and VCI. The variants identified in genes linked with neurodegenerative diseases suggest that vascular pathogenic mechanisms are linked to neurodegenerative conditions. Although more research needs to be done to reveal how these variants cause disease, our study provides novel insights into the molecular basis of VCI.
Supplementary information
Acknowledgements
We thank Prof. (emeritus) Hannu Kalimo and the CADASIL group for discussions when planning this study.
Funding
This study was supported by Academy of Finland (294817), Helsinki University Hospital Competetive Fund, and The Finnish Medical Society Duodecim.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Rita Guerreiro, Liisa Myllykangas
Supplementary information
The online version of this article (10.1038/s41431-020-00775-9) contains supplementary material, which is available to authorized users.
References
- 1.Dichgans M, Leys D. Vascular cognitive impairment. Circ Res. 2017;120:573–91. doi: 10.1161/CIRCRESAHA.116.308426. [DOI] [PubMed] [Google Scholar]
- 2.O’Brien JT, Thomas A. Vascular dementia. Lancet. 2015;386:1698–706. doi: 10.1016/S0140-6736(15)00463-8. [DOI] [PubMed] [Google Scholar]
- 3.Pasi M, Cordonnier C. Clinical relevance of cerebral small vessel diseases. Stroke. 2020;51:47–53. doi: 10.1161/STROKEAHA.119.024148. [DOI] [PubMed] [Google Scholar]
- 4.Marini S, Anderson CD, Rosand J. Genetics of cerebral small vessel disease. Stroke. 2020;51:12–20. doi: 10.1161/STROKEAHA.119.024151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–10. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 6.Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360:1729–39. doi: 10.1056/NEJMoa0801560. [DOI] [PubMed] [Google Scholar]
- 7.Verdura E, Herve D, Scharrer E, Amador Mdel M, Guyant-Marechal L, Philippi A, et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain. 2015;138:2347–58. doi: 10.1093/brain/awv155. [DOI] [PubMed] [Google Scholar]
- 8.Labelle-Dumais C, Kuo DS, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. hmg. 2012;21:R97–R110. doi: 10.1093/hmg/dds346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siitonen M, Borjesson-Hanson A, Poyhonen M, Ora A, Pasanen P, Bras J, et al. Multi-infarct dementia of Swedish type is caused by a 3’UTR mutation of COL4A1. Brain. 2017;140:e29. doi: 10.1093/brain/awx062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verdura E, Herve D, Bergametti F, Jacquet C, Morvan T, Prieto-Morin C, et al. Disruption of a miR-29 binding site leading to COL4A1 upregulation causes pontine autosomal dominant microangiopathy with leukoencephalopathy. Ann Neurol. 2016;80:741–53. doi: 10.1002/ana.24782. [DOI] [PubMed] [Google Scholar]
- 11.Ilinca A, Samuelsson S, Piccinelli P, Soller M, Kristoffersson U, Lindgren AG. A stroke gene panel for whole-exome sequencing. Eur J Hum Genet. 2019;27:317–24. doi: 10.1038/s41431-018-0274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel ZH, Kottyan LC, Lazaro S, Williams MS, Ledbetter DH, Tromp H, et al. The struggle to find reliable results in exome sequencing data: filtering out Mendelian errors. Front Genet. 2014;5:16. doi: 10.3389/fgene.2014.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opherk C, Herzog J, Dichgans M, Peters N, Luedtke R. Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain. 2004;127:2533–9. doi: 10.1093/brain/awh282. [DOI] [PubMed] [Google Scholar]
- 15.Bianchi S, Di Palma C, Gallus GN, Taglia I, Poggiani A, Rosini F, et al. Two novel HTRA1 mutations in a European CARASIL patient. Neurology. 2014;82:898–900. doi: 10.1212/WNL.0000000000000202. [DOI] [PubMed] [Google Scholar]
- 16.Nozaki H, Kato T, Nihonmatsu M, Saito Y, Mizuta I, Noda T, et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology. 2016;86:1964–74. doi: 10.1212/WNL.0000000000002694. [DOI] [PubMed] [Google Scholar]
- 17.Guerreiro RJ, Gustafson DR, Hardy J. The genetic architecture of Alzheimer’s disease: beyond APP, PSENs and APOE. Neurobiol Aging. 2012;33:437–56. doi: 10.1016/j.neurobiolaging.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghani M, Lang AE, Zinman L, Nacmias B, Sorbi S, Bessi V, et al. Mutation analysis of patients with neurodegenerative disorders using NeuroX array. Neurobiol Aging. 2015;36:545.e9–545.14. doi: 10.1016/j.neurobiolaging.2014.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulte EC, Fukumori A, Mollenhauer B, Hor H, Arzberger T, Perneczky R, et al. Rare variants in beta-Amyloid precursor protein (APP) and Parkinson’s disease. Eur J Hum Genet. 2015;23:1328–33. doi: 10.1038/ejhg.2014.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geiger JT, Ding J, Crain B, Pletnikova O, Letson C, Dawson TM, et al. Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol Dis. 2016;94:55–62. doi: 10.1016/j.nbd.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, et al. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–81. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 22.Vidal R, Révész T, Rostagno A, Kim E, Holton JL, Bek T, et al. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci USA. 2000;97:4920–5. doi: 10.1073/pnas.080076097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Audo I, Bujakowska K, Orhan E, El Shamieh S, Sennlaub F, Guillonneau X, et al. The familial dementia gene revisited: a missense mutation revealed by whole-exome sequencing identifies ITM2B as a candidate gene underlying a novel autosomal dominant retinal dystrophy in a large family. Hum Mol Genet. 2014;23:491–501. doi: 10.1093/hmg/ddt439. [DOI] [PubMed] [Google Scholar]
- 24.Notelet L, Chapon F, Khoury S, Vahedi K, Chodkiewicz JP, Courtheoux P, et al. Familial cavernous malformations in a large French kindred: mapping of the gene to the CCM1 locus on chromosome 7q. J Neurol Neurosurg Psychiatry. 1997;63:40–45. doi: 10.1136/jnnp.63.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Craig HD, Gunel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851–8. doi: 10.1093/hmg/7.12.1851. [DOI] [PubMed] [Google Scholar]
- 26.Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, et al. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–64. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajakulendran S, Kaski D, Hanna MG. Neuronal P/Q-type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol. 2012;8:86–96. doi: 10.1038/nrneurol.2011.228. [DOI] [PubMed] [Google Scholar]
- 28.Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–9. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simons C, Dyment D, Bent SJ, Crawford J, D’Hooghe M, Kohlschutter A, et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain. 2017;140:3105–11. doi: 10.1093/brain/awx314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapferer-Seebacher I, Pepin M, Werner R, Aitman TJ, Nordgren A, Stoiber H, et al. Periodontal Ehlers-Danlos syndrome is caused by mutations in C1R and C1S, which encode subcomponents C1r and C1s of complement. Am J Hum Genet. 2016;99:1005–14. doi: 10.1016/j.ajhg.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cikla U, Sadighi A, Bauer A, Baskaya MK. Fatal ruptured blood blister-like aneurysm of middle cerebral artery associated with Ehlers-Danlos syndrome type VIII (Periodontitis Type) J Neurol Surg Rep. 2014;75:210. doi: 10.1055/s-0034-1387185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, et al. Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N Engl J Med. 2008;359:158–65. doi: 10.1056/NEJMoa0706300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham RL, Yang T, Blair M, Roden DM, Darbar D. Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J Mol Cell Cardiol. 2010;48:181–90. doi: 10.1016/j.yjmcc.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ilinca A, Martinez-Majander N, Samuelsson S, Piccinelli P, Truve K, Cole J, et al. Whole-Exome Sequencing in 22 Young Ischemic Stroke Patients With Familial Clustering of Stroke. Stroke. 2020;51:1056–63. doi: 10.1161/STROKEAHA.119.027474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai Z, Wang C, He W, Tu H, Tang Z, Xiao M, et al. Cerebral small vessel disease and Alzheimer’s disease. Clin Interv Aging. 2015;10:1695–704. doi: 10.2147/CIA.S90871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol. 2018;83:74–83. doi: 10.1002/ana.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Braidy N, Poljak A, Chan DKY, Sachdev P. Cerebral small vessel disease and the risk of Alzheimer’s disease: A systematic review. Ageing Res Rev. 2018;47:41–48. doi: 10.1016/j.arr.2018.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Thal DR, von Arnim CA, Griffin WS, Mrak RE, Walker L, Attems J, et al. Frontotemporal lobar degeneration FTLD-tau: preclinical lesions, vascular, and Alzheimer-related co-pathologies. J Neural Transm (Vienna) 2015;122:1007–18. doi: 10.1007/s00702-014-1360-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dichgans M, Zietemann V. Prevention of vascular cognitive impairment. Stroke. 2012;43:3137–46. doi: 10.1161/STROKEAHA.112.651778. [DOI] [PubMed] [Google Scholar]
- 40.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.