

Abstract
Inflammatory markers like C-reactive protein (CRP) have been associated with post-traumatic stress disorder (PTSD) and traumatic experiences, but the underlying mechanisms are unclear. We investigated the relationship among serum CRP, PTSD, and traits related to traumatic events and social support using genetic association data from the Psychiatric Genomics Consortium (23,185 PTSD cases and 151,309 controls), the UK Biobank (UKB; up to 117,900 individuals), and the CHARGE study (Cohorts for Heart and Aging Research in Genomic Epidemiology, 148,164 individual). Linkage disequilibrium score regression, polygenic risk scoring, and two-sample Mendelian randomization (MR) analyses were used to investigate genetic overlap and causal relationships. Genetic correlations of CRP were observed with PTSD (rg = 0.16, p = 0.026) and traits related to traumatic events, and the presence of social support (−0.28 < rg < 0.20; p < 0.008). We observed a bidirectional association between CRP and PTSD (CRP → PTSD: β = 0.065, p = 0.015; PTSD → CRP: β = 0.008, p = 0.009). CRP also showed a negative association with the “felt loved as a child” trait (UKB, β = −0.017, p = 0.008). Owing to the known association of socioeconomic status (SES) on PTSD, a multivariable MR was performed to investigate SES as potential mediator. We found that household income (univariate MR: β = −0.22, p = 1.57 × 10−7; multivariate MR: β = −0.17, p = 0.005) and deprivation index (univariate MR: β = 0.38, p = 1.63 × 10−9; multivariate MR: β = 0.27, p = 0.016) were driving the causal estimates of “felt loved as a child” and CRP on PTSD. The present findings highlight a bidirectional genetic association between PTSD and CRP, also suggesting a potential role of SES in the interplay between childhood support and inflammatory processes with respect to PTSD risk.
Subject terms: Post-traumatic stress disorder, Trauma, Genetics
Introduction
Post-traumatic stress disorder (PTSD) is a common psychiatric condition that can occur after experiencing or witnessing traumatic events [1]. Inflammatory processes, reflected by blood-based markers like C-reactive protein (CRP), could be involved in the biology of the trauma response and PTSD [2]. CRP is a protein that responds to inflammatory stimuli by triggering cellular reactions such as those related to macrophage recruitment during inflammation initiation [3]. Elevated levels of pro-inflammatory cytokines (e.g., interleukins; a downstream product of CRP signaling) and acute-phase proteins (e.g., CRP) have been observed in the blood of individuals with PTSD [4, 5]. Meta-analyses of cross-sectional studies have confirmed the association of inflammation with traumatic events and PTSD [6, 7]. Longitudinal studies reported evidence supporting a bidirectional association: elevated inflammation may contribute to PTSD and that PTSD contributes to elevated inflammation [8–13]. Additionally, PTSD is well known to be associated with many chronic diseases (e.g., cardiovascular diseases, type-2 diabetes, and rheumatoid arthritis) with an established inflammatory component [14–16].
Understanding the mechanisms underlying the PTSD-inflammation association can potentially lead to important translational implications [17]. Leveraging the role of CRP as an inflammatory marker, we will test the putative causal mechanisms at the basis of the PTSD-inflammation association. This analysis could reveal three different translation scenarios. First, if increased inflammation is causal for PTSD, inflammatory markers may be useful in identifying persons at risk, and anti-inflammatory treatments may be therapeutic. Second, if PTSD causes increased inflammation, evidence for one pathway via which PTSD increases risk of chronic disease would support the inflammation screening in persons with PTSD. The third scenario is that the relationship between inflammation and PTSD is not causal in either direction but accounted for by a third factor.
Analytic approaches based on datasets generated from large-scale genome-wide association studies (GWAS) may inform how inflammatory processes and CRP levels are related to etiology or consequences of PTSD. Mendelian randomization (MR) can be used to investigate causal relationships between phenotypes using genetic variants (e.g., single-nucleotide polymorphisms, SNPs) as instrumental variables in genetic epidemiologic experiments analogous to randomized control trials [18–20].
In this context, we investigated the genetic overlap and the putative causal relationships linking CRP and PTSD, leveraging large-scale genome-wide datasets from the Psychiatric Genomic Consortium PTSD Workgroup (PGC-PTSD) [21], the UK Biobank (UKB) [22], and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) study [23]. To investigate more broadly the relationship between trauma and inflammation, we also tested traits related to traumatic events, and the presence of social support. Additionally, since socioeconomic status (SES) is associated with the traits investigated (i.e., PTSD, CRP, traumatic events, and social support) [24–26] and has been shown to mediate the effect of other PTSD risk factors in a previous MR analysis [27], we tested whether the associations observed are independent from the effects that are mediated by SES.
Materials and methods
Study design
The present study was designed to leverage multiple methods based on large-scale genome-wide datasets to investigate genetic overlap and causal relationships involved in the PTSD-inflammation association (Fig. 1). Accordingly, CRP GWAS data [23] were used as an indicator of inflammatory response. To conduct a comprehensive analysis, we also investigated GWAS data generated from traits related to exposure and response to traumatic events and to social support, which may mediate the PTSD-inflammation association. Another potential mediator is SES. Thus, we included genetic information related to SES-related traits in the analysis as well. The linkage disequilibrium score regression (LDSC) method [28] was used to estimate SNP heritability and genetic correlation among the phenotypes tested (i.e., PTSD, CRP, traits related to traumatic events and social support, and SES traits). As explained later (see Heritability and Genetic Correlation section), this initial step permitted us to identify informative datasets and trait pairs with significant genetic overlap. Based on the significant LDSC results, we conducted a polygenic risk scoring (PRS) to define the genetic instruments to be used in MR. Finally, multiple MR approaches were applied to informative genetic datasets to investigate causal relationship and mediation effect among the traits potentially involved in the PTSD-inflammation association. Each analytic step is described in detail in the corresponding method section.
Fig. 1. Study Design.
Flowchart of the analytic steps and methods included in the present study.
Data sources
GWAS data for PTSD were obtained from PGC-PTSD, which is an international collaborative group that investigates the genetic basis of PTSD [29]. We used two versions (i.e., Freeze 2 and Freeze 1.5) of the latest PGC-PTSD GWAS [21]. PGC-PTSD Freeze 2 includes 23,185 cases and 151,309 controls of European descent, including both PGC and UKB samples. PGC-PTSD Freeze 1.5 includes only 12,823 cases and 35,648 controls available from PGC samples (i.e., UKB samples were not included in the Freeze 1.5 meta-analysis). We used the PGC-PTSD Freeze 1.5 dataset to exclude the potential bias induced by the overlap between PGC-PTSD Freeze 2 and UKB samples in the PRS and MR analyses, both of which are sensitive to such sample overlap. PTSD diagnosis across PGC-PTSD cohorts was obtained considering different version of Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria (DSM-III-R, DSM IV, DSM-5) that was based on current or lifetime PTSD. The control group was composed of a majority of trauma-exposed participants. A detailed description of the quality control criteria and GWAS methods was reported previously [21].
To investigate inflammatory processes, we used the genome-wide data generated by the CHARGE Inflammation Working Group, which investigated the genetics of circulating CRP in up to 204,402 subjects of European ancestry (including both sexes). Serum CRP were measured via standard laboratory techniques and transformed using a natural log transformation. This GWAS identified 58 distinct genetic loci (p < 5 × 10−8) associated with serum CRP. The lead variants in these loci explained up to 7% of the variance in circulating amounts of CRP [23]. In our analysis, we used the subset (148,164 individuals) of the GWAS data based on 1000 Genomes Project imputed data, which provide information about 10,019,203 variants. CRP GWAS data from this subset showed 12% (standard error, SE = 0.03) SNP heritability.
We leveraged data from the UKB to test traits related to traumatic events and social support [22]. These include 21 phenotypes grouped in the category “traumatic events” (UKB Field ID: 145). Among them, we can distinguish three different phenotypic classes: (i) information regarding the exposure to traumatic events; (ii) behavioral and emotional response to traumatic events, and (iii) information regarding social support (Supplemental Table 1). These traits were assessed via the UKB online mental health questionnaire in more than 150,000 UKB participants (both sexes; aged from 40- to 69-years-old) [30]. Information regarding SES were also derived from UKB. Specifically, we investigated two SES traits: “Average total household income before tax” (UKB Field ID: 738, household income) and “Townsend deprivation index at recruitment” (UKB Field ID: 189, deprivation index) [31, 32]. With respect to these UKB traits, we used previously generated GWAS data. Details regarding quality control criteria and methods of this previous analysis are available at https://github.com/Nealelab/UK_Biobank_GWAS/tree/master/imputed-v2-gwas. Briefly, the association analyses for all phenotypes were conducted using regression models available in Hail (available at https://github.com/hail-is/hail) including the first 20 ancestry principal components, sex, age, age2, sex × age, and sex × age2 as covariates. Details regarding the UKB traits investigated are reported in Supplemental Table 1.
Heritability and genetic correlation
To estimate observed-scale SNP heritability and genetic correlation among the phenotypes interrogated in this study, we performed Linkage Disequilibrium Score Regression (LDSC) analyses according to the pipeline described previously (available at https://github.com/bulik/ldsc) [28]. We calculated the heritability z-score, which is defined as the LDSC heritability estimate divided by its standard error. The heritability z-score is able to capture information about the genetic architecture of a trait and the reliability of each heritability estimate [28]. SNP heritability reflects the proportion of phenotypic variance attributable to the additive effects of a given set of SNPs. SNP heritability was calculated using GWAS data generated from PGC-PTSD (Freeze 2), CHARGE CRP, UKB traumatic events (exposure to traumatic events; behavioral and emotional response to traumatic events; social support), and UKB SES (household income; deprivation index). Then, we estimate the genetic correlation among the traits investigated. The genetic correlation represents the proportion of SNP heritability shared between two traits. As recommended by LDSC developers [28], we conducted genetic correlation analysis only using traits with heritability z-score≥4. Genetic correlations were adjusted for multiple testing using false discovery rate (FDR), considering Q < 0.05 as the significance threshold.
Polygenic risk score and definition of the genetic instruments
PRSs were calculated and tested to define genetic instruments sufficiently powered to be used in a MR. The PRS analysis was performed using the gtx R package embedded in PRSice v1.25 software [33]. This approach permitted us to calculate an approximate estimate of the variance explained from the PRS tested from a multivariate regression model [34]. In the present study, we conducted a bidirectional PRS analysis considering the trait pairs that showed significant genetic correlation in the LDSC analysis (i.e., we tested trait 1 predicting trait 2 and trait 2 predicting trait 1). The PRSs were calculated considering the following parameters: clumping with a linkage disequilibrium (LD) cutoff of R2 = 0.001, 10,000-kb as clumping window, and exclusion of major histocompatibility complex (MHC) region of the genome because of its complex LD structure. European samples from the 1000 Genomes Project were used as the LD reference panel. In the PRS analysis, we used PGC-PTSD freeze 1.5 to avoid sample overlap between PGC-PTSD Freeze 2, and UKB. We applied FDR multiple testing correction (Q < 0.05) to account for the number of PRS tested within each analysis.
Mendelian randomization (MR)
Mendelian randomization (MR) permits to infer putative causal relationships between a modifiable exposure and the health outcome of interest using genetic variants (SNPs) as instrumental variables [35]. MR is based on three assumptions about the genetic instrument: (i) the instrumental variables (genetic variants) are associated with the exposure, (ii) the genetic variants are not associated with confounders related to exposure and outcome, and (iii) the genetic variants affect the outcome only through the exposure factor [36]. In the present study, we used the R package TwoSampleMR [37] to test the causality among the phenotype combinations that showed genetic instruments surviving the PRS FDR correction. In our analysis, we compared five MR methods: inverse variance weighted (IVW), MR-Egger, weighted median, simple mode, and weighted mode [37]. These permitted us to verify the stability of the causal effects of MR results considering the sensitivities unique to each of the stated methods. Unless otherwise noted, the IVW approach was reported as the main results because of the higher statistical power of this method [38]. MR was conducted in accordance with the STROBE (STrenghtening the Reporting of Observational studies in Epidemiology)—MR reporting guidelines [39]. The genetic instruments used were based on the best-fit PRS threshold to identify the most predictive inclusion threshold. As this relaxed genetic instrument selection threshold potentially violates the above MR assumptions, we verified the IVW estimates using the MR–Robust Adjusted Profile Score (MR-RAPS) approach, which is a method designed to identify and estimate confounded associations using weak genetic instrument variables [40]. Additionally, multiple MR sensitivity tests were performed to investigate the presence of confounders (e.g., horizontal pleiotropy and heterogeneity) within the genetic instruments tested: MR-Egger regression intercept [41], MR-PRESSO (Pleiotropy RESidual Sum and Outlier) global test [42], and MR heterogeneity test [43]. Outliers contributing to the heterogeneity and horizontal pleiotropy within the genetic instruments were identified via a leave-one-out analysis and considering MR-RAPS [40] standardized residuals that fall outside the 95% confidence level. The significant MR results not biased by horizontal pleiotropy and heterogeneity were entered in the multivariable MR (MVMR) to estimate the independent effect of two or more exposures on a specific outcome. This analysis was conducted using the MendelianRandomization R package [44]. The MVMR method permits evaluation of the independent association of each risk exposure with the outcome, similar to the simultaneous assessment of several treatments in a factorial randomized trial [44].
Results
Genetic correlation and polygenic risk scores analyses
SNP heritability was calculated for the 25 GWAS datasets related to the traits investigated in the present study. We found that five GWAS datasets were not informative for the subsequent analyses due to a lack of statistical power (SNP heritability z-score < 4). Twenty GWAS datasets were investigated further. These include PGC-PTSD, CHARGE-CRP, four UKB GWAS of traits related to trauma response (i.e., behavioral and emotional response to traumatic events), nine UKB GWAS of traits related to trauma exposure, three UKB GWAS of traits related to social support, and two UKB GWAS of SES traits (Supplemental Table 2). The significant heritability observed supports the informativeness of the datasets investigated.
We found a positive genetic correlation between CRP and PTSD diagnosis (PGC-PTSD Freeze 2: rg = 0.155, p = 0.026). After multiple testing correction (FDR 5%), we identified eight significant genetic correlates of CRP: one trait related to trauma response (i.e., UKB Data-Field 20498: Felt very upset when reminded of stressful experience in past month), three traits related to exposure to traumatic events, and four traits related to social support (Fig. 2). As previously shown [27], there is a high degree of genetic correlation among PTSD, trauma response, trauma exposure, social support, and SES. Positive genetic correlations also were observed between CRP and SES: deprivation index, rg = 0.20, p = 3.38 × 10−5; household income, rg = -0.25, p = 4.08 × 10−10. The direction of genetic correlations involving CRP were in line with expectations: positive genetic correlation (increased inflammation) with PTSD, traumatic events, and deprivation index and negative genetic correlation (reduced inflammation) with social support and increased household income.
Fig. 2. Genetic correlation (x-axis: genetic correlation, rg; y-axis: –log10p-value) of serum C-reactive protein (CRP) with post-traumatic stress disorder (PTSD), trauma response, trauma exposure, social support, and socioeconomic status.
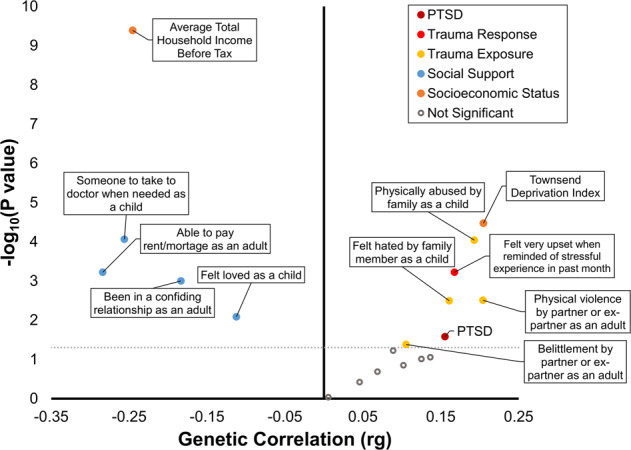
Full circles represent nominally significant genetic correlations (p < 0.05). As expected, CRP showed positive genetic correlation with PTSD, trauma response, trauma exposure and deprivation index, and negative genetic correlation with social support and household income.
Considering these genetic correlations, we conducted bidirectional PRS to identify the direction of association between: (i) CRP and PTSD (PGC-PTSD Freeze 1.5 case-control cohort), (ii) CRP and eight traits related to traumatic events (i.e., trauma response, trauma exposure, and social support), (iii) CRP and SES traits. After FDR 5% correction for the number of PRS thresholds tested, we found 12 significant associations (Table 1 and Supplemental Fig. 1). The most significant PRS association was observed between household income and CRP (R2 = 0.08%; FDR Q = 7.45 × 10–25). In addition, we observed that only 3 traits have bidirectional prediction (1: “Felt loved as a child” and CRP, 2: household income and CRP, and 3: PTSD and household income; Table 1), suggesting that these traits may share genetic etiology. PRS analyses also confirmed that the datasets investigated can be used to derive informative instrumental variables.
Table 1.
Significant PRS results for serum CRP, PTSD, traits related to traumatic events and SES traits.
| PRS | Target phenotype | FDR Q-value |
|---|---|---|
| CRP | PTSD | 3.70 × 10−2 |
| Physically abused by family as a child | 1.80 × 10−4 | |
| Able to pay rent/mortgage as an adult | 8.10 × 10−4 | |
| Felt loved as a child | 4.20 × 10−3 | |
| Household income | 3.10 × 10−2 | |
| PTSD | CRP | 2.70 × 10−2 |
| Household income | 5.52 × 10−4 | |
| Felt loved as a child | CRP | 1.60 × 10−2 |
| Deprivation index | CRP | 2.70 × 10−4 |
| PTSD | 6.00 × 10−5 | |
| Household income | CRP | 7.45 × 10−25 |
| PTSD | 1.36 × 10−7 |
Mendelian randomization (MR)
Based on the PRS findings, we performed two-sample MR to assess causality leveraging by genetic associations related to the combinations of phenotypes reported in Table 1. When heterogeneity or pleiotropy was identified within the genetic instruments, we removed the outlier variants to verify that the causal estimates were not generated by potential biases within the genetic instruments. The effects reported in Fig. 3 are those estimated in the absence of genetic instruments affected by heterogeneity and/or horizontal pleiotropy. We found evidence supporting several associations among the traits investigated (Fig. 3 and Supplemental Table 3). Our results suggest a bidirectional association between PTSD and CRP, where genetically determined CRP is positively associated with increased PTSD risk (β = 0.065, 95% CI: 0.012 to 0.119, p = 0.015), and genetic liability to PTSD has a positive association with CRP (β = 0.008, 95% CI: 0.001 to 0.015, p = 0.024). We observed the presence of possible heterogeneity in SNPs evaluated in the PTSD→CRP association (heterogeneity test p = 0.001). However, after removal of the outlier variants from the genetic instrument, the PTSD→CRP genetic association remained significant (β = 0.008, 95% CI: 0.002 to 0.015, p = 0.009).
Fig. 3. Significant Mendelian randomization (MR) tests based on the inverse variance weighted method (IVW; p < 0.05).

Effect size (beta) and 95% confidence interval are reported for each MR test. We observed a bidirectional effect between serum C-reactive protein and post-traumatic stress disorder.
Among the traits related to traumatic events and social support, we observed that genetically determined CRP also has a negative effect on the “Felt loved as a child” trait from UKB (β = –0.02, 95% CI: –0.034 to –0.007, p = 0.003). However, strong evidence of heterogeneity was present among the variants within the genetic instrument (heterogeneity test p = 4.26 × 10−5). After removing these variants, we confirmed the CRP→“Felt loved as a child” association (β = –0.017, 95% CI: –0.031 to –0.004, p = 0.008). SNPs associated with “Felt loved as a child” trait showed a protective effect on genetic liability to PTSD (β = –0.139, 95% CI: –0.206 to –0.071, p = 5.64 × 10−6). However, the genetic instrument was affected by heterogeneity (heterogeneity test p = 0.002). Removing these variants confirmed the protective effect of “Felt loved as a child” trait on PTSD was robust (β = –0.19, 95% CI = –0.124 to –0.257, p = 2.1 × 10−8).
SNPs associated with indicators of SES affect both genetically determined CRP and genetic liability to PTSD. Household income showed a negative bidirectional association with CRP (household income→CRP β = –0.131, 95% CI = –0.161 to –0.102, p = 1.06 × 10−18; CRP→household income β = –0.016, 95% CI = –0.027 to –0.005, p = 0.004). Both household income and deprivation index had a causal effect on PTSD (Household income: β = –0.277, 95% CI: –0.312 to –0.142, p = 1.57 × 10−7; Deprivation index: β = 0.389, 95% CI: 0.262 to 0.515, p = 1.63 × 10−9). The effects were confirmed after removing outlier variants when heterogeneity or horizontal pleiotropy were observed within the genetic instruments (Supplemental Table 3).
As we found that CRP, “Felt loved as a child”, and SES (deprivation index and household income) have causal effects on genetic liability to PTSD, we performed a MVMR analysis to verify whether the effects observed among PTSD, CRP, social support, and SES are independent from each other (Fig. 4). We confirmed that genetically determined CRP has an effect on PTSD independently of the “Felt loved as a child” association (β = 0.066, 95% CI: 0.007 to 0.126, p = 0.029; Fig. 4a). However, the CRP→PTSD genetic association (CRP→PTSD univariate MR β = 0.066, 95% CI = 0.013–0.119, p = 0.015; multivariate MR β = 0.043, 95% CI = −0.027–0.113, p = 0.232) was not independent from the effect of deprivation index (deprivation index→PTSD: univariate MR: β = 0.38, p = 1.63 × 10–9, 95% CI = 0.263–0.516; multivariate MR β = 0.27, p = 0.016; 95% CI = 0.050–0.498; Fig. 4b) and household income (Household income→PTSD: univariate MR β = –0.22, 95% CI = –0.312 to –0.142, p = 1.57 × 10−7; multivariate MR β = –0.17, 95% CI = –0.294 to –0.051, p = 0.005; Fig. 4c).
Fig. 4. Multivariable Mendelian randomization (MVMR) results considering the following models.
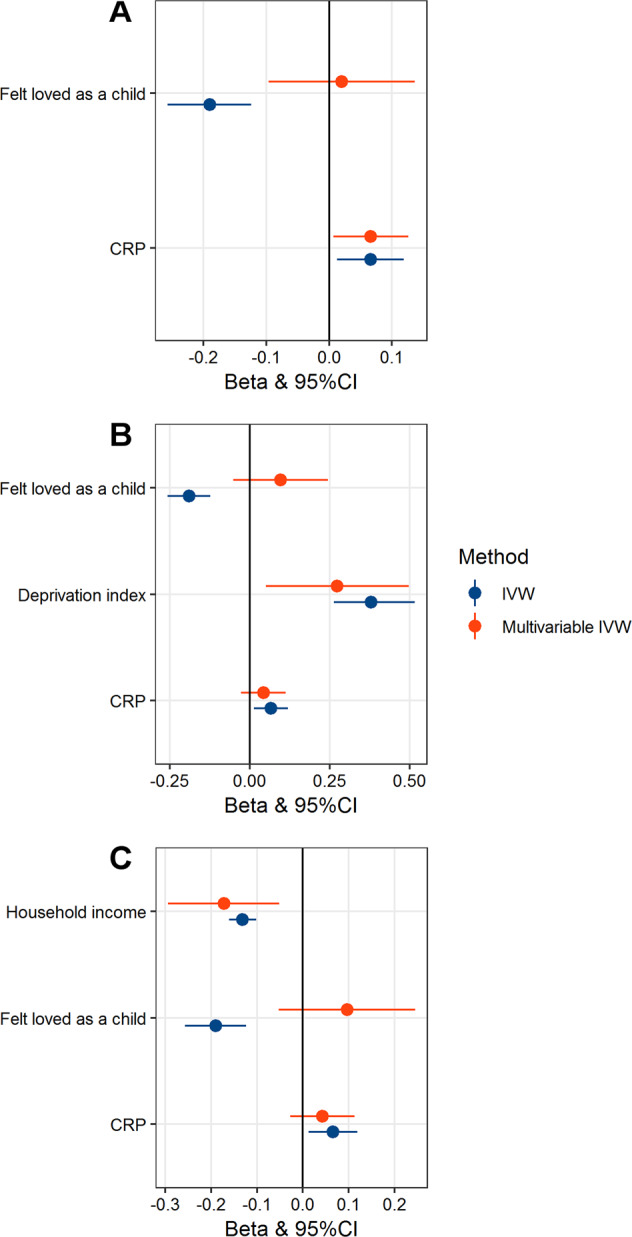
a Association of genetically determined serum C-reactive protein (CRP) with post-traumatic stress disorder (PTSD) independently of “Felt love as a child”; b Association of deprivation-index genetic instrument with PTSD independent of “Felt loved as a child” and serum CRP; c Association of household-income genetic instrument with PTSD independent of “Felt loved as a child” and CRP. The analysis was conducted using the MVMR inverse variance weighted method (IVW) and compared with the univariate MR result. For each test, effect size (beta) and 95% confidence interval are reported. The effect of CRP and childhood social support (i.e., “Felt love as a child”) appears to be not independent from socioeconomic status (i.e., household income and deprivation-index).
Discussion
Increased levels of inflammation markers, such as serum CRP, are consistently associated with several adverse events, including PTSD, traumatic experiences, and low SES [45–47]. However, the mechanisms underlying these associations are unclear. In the present study, we used genome-wide data to investigate the genetic overlap and the causal mechanisms between CRP and PTSD, also considering the effects of genetic variants associated with SES, traumatic experiences, and social support. Our findings support a bidirectional genetic association between PTSD and CRP, which appears to be affected by SES. This suggests that, among possible scenarios hypothesized, the relationship between CRP and PTSD is not causal in either direction but accounted for by a third factor (i.e., SES).
CRP is a protein that responds to inflammatory stimuli by triggering cellular reactions and it is normally induced through pro-inflammatory cytokines, mainly by interleukin-6 secretion [48, 49]. CRP may be produced in the microvessel endothelial cells that form the blood-brain-barrier, and peripheral CRP can affect the central nervous system via mechanisms of blood-brain barrier disruption associated with elevated levels of peripheral inflammatory cytokines [50, 51]. Higher levels of pro-inflammatory cytokines may influence neurotransmission, leading to altered production of serotonin, norepinephrine, dopamine, and brain-derived neurotrophic factor. This effect on neurotransmitter concentration may be associated with specific psychiatric symptoms, such as anhedonia [52]. Furthermore, these cytokines may influence neurocircuitry, causing alterations in motivation status, anxiety, arousal, and alarm response [53]. Twin and family studies estimated that heritability of CRP is around 35%–40% [23]. Several environmental factors, such as sociodemographic variables, dietary habits, behavioral and lifestyle factors, are also strongly associated with CRP level [54]. Previous cross-sectional and longitudinal studies highlighted the presence of a bidirectional association between PTSD and CRP levels. Previous genetic studies confirmed that CRP genetic variation was associated with PTSD [6–13]. Our findings confirm this scenario. However, we also observed that the CRP-PTSD genetic association is not independent from SNPs associated with SES. The effect of SES on serum CRP was previously observed in observational studies that highlighted the association of increased CRP levels with low SES and childhood traumatic experiences [55]. In addition, low SES has been associated with DNA methylation of genes involved in inflammation, suggesting that alterations in epigenetics of specific genes might influence the inflammation response, leading to increased inflammatory markers [56, 57].
A recent GWAS of social stratification in Great Britain showed that genetic factors related to SES (educational, income, employment) are associated with a wide range of physical and mental health outcomes [32]. An independent analysis also showed how genetic effects contribute towards some of the observed socioeconomic inequalities [31]. Lower economic status might be a mediator of the negative association between educational attainment and PTSD [27]. In line with these previous findings, our analysis based on genetic information found that SES might affect the interplay among inflammatory processes and consequently cause alterations in serum CRP and possibly increase the risk of PTSD. This indicates that SES may affect several of the comorbid traits associated with PTSD.
Furthermore, we found that SNPs associated with “Felt loved as a child” were collectively a protective factor with respect to genetic liability to PTSD. Social support systems (e.g., positive social interaction, love, care, respect, affection, acceptance, and financial assistance) are important protective factors for mental health and a driving force for individual well-being [58, 59]. Social support might influence trauma perception and cognitive processes and consequently reduce the risk to develop psychiatric disorders. Individuals with low social support had worse prognosis of PTSD [60, 61]. Additionally, high levels of social support were associated with reduced CRP levels [62, 63]. Our analysis showed that the protective effect of childhood social support (i.e., “Felt loved as a child”) on PTSD is not independent from CRP association. Accordingly, we hypothesized that environments in early life may influence inflammatory response in adulthood. Also, we found that SNPs associated with serum CRP affect the information reported by UKB participants regarding childhood social support (i.e., “Felt loved as a child”). This suggests that CRP and potentially other inflammatory biomarkers may be informative of the individual response to adverse environments.
Although we present novel results contributing to understand the genetic association among PTSD, CRP, traumatic events, and social support, our study has several limitations. Our analyses focused on CRP only, because no large-scale GWAS has investigated other inflammatory biomarkers. Further studies will be needed to verify whether the same effects are present across inflammation-related molecules such as IL-6. The analyses were conducted using GWAS in subjects of European ancestry, thus we cannot generalize these results to other populations. The estimated magnitude of the effect of serum CRP on PTSD is relatively small, which may indicate a minor contribution of the associations reported in the biology of PTSD. The genetic information about PTSD and trauma traits were obtained across different cohorts, which may cause heterogeneity in the outcomes related to stress exposition. The datasets investigated were generated from participants assessed using different instruments, also including online questionnaires. Although this approach was needed to collect information from a large cohort and the online instruments were validated [30], this may have affected the quality of the data collected. Finally, although we used appropriate statistical methods and conducted the analyses across multiple independent cohorts, findings related to genetic data related to these phenotypes need to be interpreted cautiously [64, 65]. Although we conducted multiple sensitivity analyses and the significant MR results appear to be not affected by biases, our findings could be still affected by unaccounted confounders, which may cause a violation of MR assumptions II and III. Finally, it is not possible to exclude different mechanisms implying the non-genetic components of these conditions and traits (e.g., the non-genetic component of CRP could be associated with PTSD also independently from SES).
In conclusion, this study is the first analysis leveraging large-scale genome-wide datasets to investigate the underlying mechanisms linking serum CRP, PTSD, traumatic experiences, and social support. Our findings support a bidirectional association between PTSD and CRP, which appears to be strongly affected by SES. Additionally, SES traits seem to also mediate the interplay among serum CRP, PTSD, and childhood emotional support.
Funding and disclosure
This research was supported by the Veterans Affairs National Center for Post-traumatic Stress Disorder Research, the Simons Foundation Autism Research Initiative (SFARI Explorer Award: 534858), the American Foundation for Suicide Prevention (YIG-1-109-16), and the National Institute of Mental Health (R01MH106595). CMC and SIB were supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2018/05995-4) international fellowship and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES Code 001). M.B.S. is paid for his editorial work on the journals Biological Psychiatry and Depression and Anxiety, and the health professional reference Up-To-Date. D.J.S. received personal fees from Lundbeck and Sun Pharmaceutical Industries. R.P. and J.G. are paid for their editorial work on the journal Complex Psychiatry. The other authors declare no competing interests.
Supplementary information
Author contributions
Conception and design: CMC, FRW, RP; acquisition of data: CMC, RP; analysis and interpretation of data: CMC, FRW, AXM, DJS, MBS, JAS, SMJH, CMN, KCK, JG, SIB, and RP; drafting the article: CMC, RP; revising the article critically for important intellectual content: CMC, FRW, AXM, DJS, MBS, JAS, SMJH, CMN, KCK, JG, SIB, and RP; final approval of the version to be published: CMC, FRW, AXM, DJS, MBS, JAS, SMJH, CMN, KCK, JG, SIB, and RP.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41386-020-0655-6).
References
- 1.North CS, Suris AM, Smith RP, King RV. The evolution of PTSD criteria across editions of DSM. Ann Clin Psychiatry. 2016;28:197–208. [PubMed] [Google Scholar]
- 2.Michopoulos V, Rothbaum AO, Jovanovic T, Almli LM, Bradley B, Rothbaum BO, et al. Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am J Psychiatry. 2015;172:353–62. doi: 10.1176/appi.ajp.2014.14020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vashist SK, Venkatesh AG, Marion Schneider E, Beaudoin C, Luppa PB, Luong JH. Bioanalytical advances in assays for C-reactive protein. Biotechnol Adv. 2016;34:272–90. doi: 10.1016/j.biotechadv.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 4.Rosen RL, Levy-Carrick N, Reibman J, Xu N, Shao Y, Liu M, et al. Elevated C-reactive protein and posttraumatic stress pathology among survivors of the 9/11 World Trade Center attacks. J Psychiatr Res. 2017;89:14–21. doi: 10.1016/j.jpsychires.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Heath NM, Chesney SA, Gerhart JI, Goldsmith RE, Luborsky JL, Stevens NR, et al. Interpersonal violence, PTSD, and inflammation: potential psychogenic pathways to higher C-reactive protein levels. Cytokine. 2013;63:172–8. doi: 10.1016/j.cyto.2013.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tursich M, Neufeld RW, Frewen PA, Harricharan S, Kibler JL, Rhind SG, et al. Association of trauma exposure with proinflammatory activity: a transdiagnostic meta-analysis. Transl Psychiatry. 2014;4:e413. doi: 10.1038/tp.2014.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2015;2:1002–12. doi: 10.1016/S2215-0366(15)00309-0. [DOI] [PubMed] [Google Scholar]
- 8.Eraly SA, Nievergelt CM, Maihofer AX, Barkauskas DA, Biswas N, Agorastos A, et al. Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry. 2014;71:423–31. doi: 10.1001/jamapsychiatry.2013.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen M, Meir T, Klein E, Volpin G, Assaf M, Pollack S. Cytokine levels as potential biomarkers for predicting the development of posttraumatic stress symptoms in casualties of accidents. Int J Psychiatry Med. 2011;42:117–31. doi: 10.2190/PM.42.2.b. [DOI] [PubMed] [Google Scholar]
- 10.Glaus J, von Kanel R, Lasserre AM, Strippoli MF, Vandeleur CL, Castelao E, et al. The bidirectional relationship between anxiety disorders and circulating levels of inflammatory markers: results from a large longitudinal population-based study. Depress Anxiety. 2018;35:360–71.. doi: 10.1002/da.22710. [DOI] [PubMed] [Google Scholar]
- 11.Michopoulos V, Beurel E, Gould F, Dhabhar FS, Schultebraucks K, Galatzer-Levy I, et al. Association of prospective risk for chronic PTSD symptoms with low TNFalpha and IFNgamma concentrations in the immediate aftermath of trauma exposure. Am J Psychiatry. 2019. 10.1176/appi.ajp.2019.19010039. [DOI] [PubMed]
- 12.Sumner JA, Chen Q, Roberts AL, Winning A, Rimm EB, Gilsanz P, et al. Posttraumatic stress disorder onset and inflammatory and endothelial function biomarkers in women. Brain Behav Immun. 2018;69:203–09.. doi: 10.1016/j.bbi.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sumner JA, Chen Q, Roberts AL, Winning A, Rimm EB, Gilsanz P, et al. Cross-sectional and longitudinal associations of chronic posttraumatic stress disorder with inflammatory and endothelial function markers in women. Biol Psychiatry. 2017;82:875–84.. doi: 10.1016/j.biopsych.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bielas H, Meister-Langraf RE, Schmid JP, Barth J, Znoj H, Schnyder U, et al. C-reactive protein as a predictor of posttraumatic stress induced by acute myocardial infarction. Gen Hosp Psychiatry. 2018;53:125–30.. doi: 10.1016/j.genhosppsych.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Farr OM, Ko BJ, Joung KE, Zaichenko L, Usher N, Tsoukas M, et al. Posttraumatic stress disorder, alone or additively with early life adversity, is associated with obesity and cardiometabolic risk. Nutr Metab Cardiovasc Dis. 2015;25:479–88. doi: 10.1016/j.numecd.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Donovan A, Cohen BE, Seal KH, Bertenthal D, Margaretten M, Nishimi K, et al. Elevated risk for autoimmune disorders in iraq and afghanistan veterans with posttraumatic stress disorder. Biol Psychiatry. 2015;77:365–74. doi: 10.1016/j.biopsych.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sumner JA, Nishimi KM, Koenen KC, Roberts AL, Kubzansky LD. Posttraumatic stress disorder and inflammation: untangling issues of bidirectionality. Biol Psychiatry. 2019. 10.1016/j.biopsych.2019.11.005. [DOI] [PMC free article] [PubMed]
- 18.Evans DM, Davey Smith G. Mendelian randomization: new applications in the coming age of hypothesis-free causality. Annu Rev Genomics Hum Genet. 2015;16:327–50. doi: 10.1146/annurev-genom-090314-050016. [DOI] [PubMed] [Google Scholar]
- 19.Ravera S, Carrasco N, Gelernter J, Polimanti R. Phenomic impact of genetically-determined euthyroid function and molecular differences between thyroid disorders. J Clin Med. 2018;7:E296.. doi: 10.3390/jcm7100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polimanti R, Peterson RE, Ong JS, MacGregor S, Edwards AC, Clarke TK, et al. Evidence of causal effect of major depression on alcohol dependence: findings from the psychiatric genomics consortium. Psychol Med. 2019;49:1218–26. doi: 10.1017/S0033291719000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nievergelt CM, Maihofer AX, Klengel T, Atkinson E, Chen CY, Choi K, et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun. 2019;10:4558. doi: 10.1038/s41467-019-12576-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9.. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ligthart S, Vaez A, Vosa U, Stathopoulou MG, de Vries PS, Prins BP, et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet. 2018;103:691–706. doi: 10.1016/j.ajhg.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prag P, Mills MC, Wittek R. Subjective socioeconomic status and health in cross-national comparison. Soc Sci Med. 2016;149:84–92. doi: 10.1016/j.socscimed.2015.11.044. [DOI] [PubMed] [Google Scholar]
- 25.Stepanikova I, Bateman LB, Oates GR. Systemic inflammation in midlife: race, socioeconomic status, and perceived discrimination. Am J Prev Med. 2017;52(1S1):S63–76. doi: 10.1016/j.amepre.2016.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams DR, Priest N, Anderson NB. Understanding associations among race, socioeconomic status, and health: Patterns and prospects. Health Psychol. 2016;35:407–11.. doi: 10.1037/hea0000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polimanti R, Ratanatharathorn A, Maihofer AX, Choi KW, Stein MB, Morey RA, et al. Association of economic status and educational attainment with posttraumatic stress disorder: a Mendelian randomization study. JAMA Netw Open. 2019;2:e193447. doi: 10.1001/jamanetworkopen.2019.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Logue MW, Amstadter AB, Baker DG, Duncan L, Koenen KC, Liberzon I, et al. The psychiatric genomics consortium posttraumatic stress disorder workgroup: posttraumatic stress disorder enters the age of large-scale genomic collaboration. Neuropsychopharmacology. 2015;40:2287–97. doi: 10.1038/npp.2015.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis KAS, Coleman JRI, Adams M, Allen N, Breen G, Cullen B, et al. Mental health in UK Biobank Revised. medRxiv. 2019:19001214.
- 31.Hill WD, Davies NM, Ritchie SJ, Skene NG, Bryois J, Bell S, et al. Genome-wide analysis identifies molecular systems and 149 genetic loci associated with income. Nat Commun. 2019;10:5741. doi: 10.1038/s41467-019-13585-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdellaoui A, Hugh-Jones D, Yengo L, Kemper KE, Nivard MG, Veul L, et al. Genetic correlates of social stratification in Great Britain. Nat Hum Behav. 2019;3:1332–42. doi: 10.1038/s41562-019-0757-5. [DOI] [PubMed] [Google Scholar]
- 33.Euesden J, Lewis CM, O’Reilly PF. PRSice: polygenic risk score software. Bioinformatics. 2015;31:1466–8. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dastani Z, Hivert MF, Timpson N, Perry JR, Yuan X, Scott RA, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012;8:e1002607. doi: 10.1371/journal.pgen.1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng J, Baird D, Borges MC, Bowden J, Hemani G, Haycock P, et al. Recent developments in Mendelian randomization studies. Curr Epidemiol Rep. 2017;4:330–45. doi: 10.1007/s40471-017-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi: 10.1136/bmj.k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. 2017;36:1783–802. doi: 10.1002/sim.7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davey Smith G, Davies NM, Dimou N, Egger M, Gallo V, Golub R, et al. STROBE-MR: guidelines for strengthening the reporting of Mendelian randomization studies. PeerJ Prepr. 2019;7:e27857v1. [Google Scholar]
- 40.Zhao Q, Wang J, Hemani G, Bowden J, Small DS. Statistical inference in two-sample summary-data Mendelian randomization using robust adjusted profile score. arXiv. 2019:1801.09652.
- 41.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–98. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28:30–42. doi: 10.1097/EDE.0000000000000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181:251–60. doi: 10.1093/aje/kwu283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michopoulos V, Powers A, Gillespie CF, Ressler KJ, Jovanovic T. Inflammation in fear- and anxiety-based disorders: PTSD, GAD, and beyond. Neuropsychopharmacology. 2017;42:254–70. doi: 10.1038/npp.2016.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Z, Young MR. PTSD, a disorder with an immunological component. Front Immunol. 2016;7:219. doi: 10.3389/fimmu.2016.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang YC, Boen C, Gerken K, Li T, Schorpp K, Harris KM. Social relationships and physiological determinants of longevity across the human life span. Proc Natl Acad Sci USA. 2016;113:578–83. doi: 10.1073/pnas.1511085112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fernandes BS, Steiner J, Molendijk ML, Dodd S, Nardin P, Goncalves CA, et al. C-reactive protein concentrations across the mood spectrum in bipolar disorder: a systematic review and meta-analysis. Lancet Psychiatry. 2016;3:1147–56. doi: 10.1016/S2215-0366(16)30370-4. [DOI] [PubMed] [Google Scholar]
- 50.Hsuchou H, Kastin AJ, Mishra PK, Pan W. C-reactive protein increases BBB permeability: implications for obesity and neuroinflammation. Cell Physiol Biochem. 2012;30:1109–19. doi: 10.1159/000343302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol. 2018;9:754. doi: 10.3389/fimmu.2018.00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety. 2013;30:297–306. doi: 10.1002/da.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felger JC, Haroon E, Miller AH. What’s CRP got to do with it? Tackling the complexities of the relationship between CRP and depression. Brain Behav Immun. 2018;73:163–64. doi: 10.1016/j.bbi.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 54.Shen J, Ordovas JM. Impact of genetic and environmental factors on hsCRP concentrations and response to therapeutic agents. Clin Chem. 2009;55:256–64. doi: 10.1373/clinchem.2008.117754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.O’Donovan A, Neylan TC, Metzler T, Cohen BE. Lifetime exposure to traumatic psychological stress is associated with elevated inflammation in the Heart and Soul Study. Brain Behav Immun. 2012;26:642–9. doi: 10.1016/j.bbi.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stringhini S, Polidoro S, Sacerdote C, Kelly RS, van Veldhoven K, Agnoli C, et al. Life-course socioeconomic status and DNA methylation of genes regulating inflammation. Int J Epidemiol. 2015;44:1320–30. doi: 10.1093/ije/dyv060. [DOI] [PubMed] [Google Scholar]
- 57.Lin YH, Jen MH, Chien KL. Association between life-course socioeconomic position and inflammatory biomarkers in older age: a nationally representative cohort study in Taiwan. BMC Geriatr. 2017;17:201. doi: 10.1186/s12877-017-0598-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lakey B, Orehek E. Relational regulation theory: a new approach to explain the link between perceived social support and mental health. Psychol Rev. 2011;118:482–95. doi: 10.1037/a0023477. [DOI] [PubMed] [Google Scholar]
- 59.Hakulinen C, Pulkki-Raback L, Jokela M, E Ferrie J, Aalto AM, Virtanen M, et al. Structural and functional aspects of social support as predictors of mental and physical health trajectories: Whitehall II cohort study. J Epidemiol Community Health. 2016;70:710–5. doi: 10.1136/jech-2015-206165. [DOI] [PubMed] [Google Scholar]
- 60.Lee JS. Perceived social support functions as a resilience in buffering the impact of trauma exposure on PTSD symptoms via intrusive rumination and entrapment in firefighters. PLoS ONE. 2019;14:e0220454. doi: 10.1371/journal.pone.0220454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Nutte L, Okello J, Derluyn I. Social relationships and social support among post-war youth in Northern Uganda. Int J Psychol. 2017;52:291–9. doi: 10.1002/ijop.12221. [DOI] [PubMed] [Google Scholar]
- 62.Shimanoe C, Hara M, Nishida Y, Nanri H, Otsuka Y, Horita M, et al. Coping strategy and social support modify the association between perceived stress and C-reactive protein: a longitudinal study of healthy men and women. Stress. 2018;21:237–46.. doi: 10.1080/10253890.2018.1435638. [DOI] [PubMed] [Google Scholar]
- 63.Elliot AJ, Heffner KL, Mooney CJ, Moynihan JA, Chapman BP. Social relationships and inflammatory markers in the MIDUS cohort: the role of age and gender differences. J Aging Health. 2018;30:904–23. doi: 10.1177/0898264317698551. [DOI] [PubMed] [Google Scholar]
- 64.Martschenko D, Trejo S, Domingue BW. Genetics and education: recent developments in the context of an ugly history and an uncertain future. AERA Open. 2019;5:2332858418810516. [Google Scholar]
- 65.Trejo S, Domingue BW. Genetic nature or genetic nurture? Introducing social genetic parameters to quantify bias in polygenic score analyses. Biodemography Soc Biol. 2018;64:187–215. doi: 10.1080/19485565.2019.1681257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.