

Abstract
The clinical success of cancer immune checkpoint blockade (ICB) has refocused attention on tumor-infiltrating lymphocytes (TILs) across cancer types. The outcome of immune checkpoint inhibitor therapy in cancer patients has been linked to the quality and magnitude of T cell, NK cell, and more recently, B cell responses within the tumor microenvironment. State-of-the-art single-cell analysis of TIL gene expression profiles and clonality has revealed a remarkable degree of cellular heterogeneity and distinct patterns of immune activation and exhaustion. Many of these states are conserved across tumor types, in line with the broad responses observed clinically. Despite this homology, not all cancer types with similar TIL landscapes respond similarly to immunotherapy, highlighting the complexity of the underlying tumor-immune interactions. This observation is further confounded by the strong prognostic benefit of TILs observed for tumor types that have so far respond poorly to immunotherapy. Thus, while a holistic view of lymphocyte infiltration and dysfunction on a single-cell level is emerging, the search for response and prognostic biomarkers is just beginning. Within this review, we discuss recent advances in the understanding of TIL biology, their prognostic benefit, and their predictive value for therapy.
Keywords: Tumor infiltrating lymphocytes, B cells, T cells, Tertiary lymphoid structures, immunotherapy
Subject terms: Immunosurveillance, Immunology
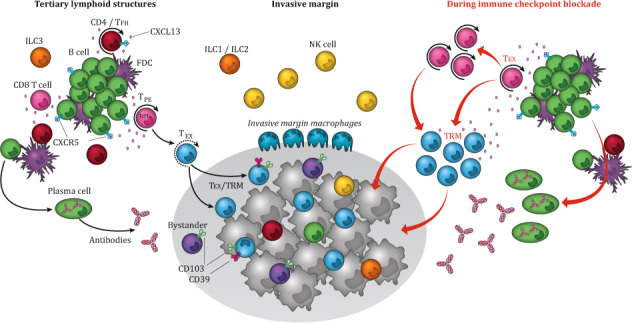
The schematic representation of the tumor immune environment shows the composition and function of a tertiary lymphoid structures (TLS), who are usually found peritumorally in the stroma and/or in the invasive margin. The chemokine CXCL13, produced by CD8+ T cells, induces chemotaxis by binding to the receptor CXCR5, mainly expressed by B cells and TFH cells, and regulates the organization of B cells inside the follicles of lymphoid tissues. The TLS consists out of a T cell-rich zone containing mature dendritic cells (DCs), in close proximity to GC containing follicle like-B cells, intermingled with follicular dendritic cells (FDCs) and surrounded by plasma cells and helper-innate lymphoid cell group 3 (ILC3) at the edge of the TLS. In the optimally organized TLS immune structure, DCs, FDCs, T cells and B cells interact and activate each other, promoting a local sustained immune response including the induction of T cell effector function, antibody generation, and clonal expansion. The stroma surrounding the tumor epithelium and the invasive margin further harbors cellular immune components including NK cells, macrophages, ILC1s and ILC2s, and a nonimmune cellular component, including fibroblasts. Within the tumor epithelium ILCs, NK cells, B cells, and different T cell subsets are present, including TEX cells,-tumor-specific CD103+CD39+ TRM CTLs and CD103+CD39- bystander TRM cells. Upon ICB, both T and B cell signaling increases. TCF1-expressing TPE cells expand and differentiate into TRM cells migrating to the tumor, where they can exert their cytolytic potential. The ICB response also increases B cell receptor diversity by means of SMH and CSR and induces their clonal expansion and differentiation into advanced antibody-producing plasma cells. TLS: tertiary lymphoid structure, TFH cells: follicular helper T cells, DCs: dendritic cells, GC: germinal center, FDCs: follicular dendritic cells, ILC3: helper-innate lymphoid cell group 3, NK cells: natural killer cells, ILC1: helper-innate lymphoid cell group 1, ILC2: helper-innate lymphoid cell group 2, TEX: terminally exhausted T cells, CTLs: cytotoxic lymphocytes, TRM: tissue resident memory, ICB: immune checkpoint blockade, TCF1: transcription factor 1, TPE: progenitor STEM-like exhausted cells, SMH: somatic hypermutation, RCS: recombinant class switch
Introduction
It has become abundantly clear that a successful antitumor immune response requires the presence, activation, and costimulation of all lymphoid components of the immune system, including CD8+ T cells, CD4+ T cells, B cells, and innate lymphoid cells. This is especially demonstrated by the discovery of tertiary lymphoid structures (TLSs), which represent well-organized clusters of TILs and give rise to an advanced immune response. Interestingly, not only TIL presence but also TIL differentiation and localization have been shown to determine clinical outcome. To translate these relationships into a usable diagnostic tool to predict prognosis and determine treatment strategy, state-of-the-art advanced computational techniques are making their way into the clinic. Within this review, we discuss recent advances in the understanding TIL of biology, their prognostic benefit, and their predictive value for therapy. Herein, we particularly address the recently identified role of tumor-resident memory cells and T cell exhaustion as key cellular effectors of immune surveillance and therapy. In addition, we elaborate on the role of TLSs and B cells as crucial supportive regulators in immune tumor control.
Search strategy
Studies relevant to the subject were searched for via PubMed. Several high-impact journals were searched specifically for the literature of interest, including Cell, Nature, Nature Communications, Nature Medicine, Clinical Cancer Research, and Cancer Immunology Research. Diverse search terms were used, including “tumor-infiltrating lymphocytes”, “T cells”, “B cells”, “natural killer cells”, “innate lymphoid cells”, “TCRαβ+”, “TCRγδ+”, “T helper cells”, “CD4 T cells”, “follicular helper T cells”, “tissue-resident memory cells”, “bystander cells”, “effector memory T cells”, “T cell exhaustion”, “progenitor stem-like exhausted cells”, “CD103”, “survival”, “cancer”, “tertiary lymphoid structures”, “stromal-infiltrating lymphocytes”, “digital immune scores”, “immunotherapy”, “checkpoint inhibition”, “microsatellite instability”, and “adoptive T cell transfer”.
When possible, studies published from 2018 to the 1st of June 2020 were used, when no studies were available, older literature was used. Searches were updated until the 1st of June 2020.
Tumor-infiltrating lymphocytes
T cells
T cells are broadly classified according to their T cell receptor (TCR) subunits, as well as the core lineage markers CD8 and CD4. The αβ TCR complex endows T cells with the capacity for recognition of peptides presented on the cell surface in the context of major histocompatibility complex (MHC) class I (CD8 T cells) or class II (CD4 T cells). By contrast, the γδ TCR subunit is thought to function largely independent of MHC class I and II. In general, CD8+ and CD4+ TCRαβ T cells are the most abundant T cell subsets in tissues, including tumor tissues (Table 1).
Table. 1.
Phenotype and functional properties of tumor infiltrating lymphocytes.
| Phenotype | Functional properties | |
|---|---|---|
| T cells | ||
| TCRγδ+ |
Express NK-cell markers such as NKG2D. Two main subsets; Vδ1 γδ T cells and Vγ9Vδ2 T cells. |
Display both innate and adaptive immune features and are described to exhibit both effector like and regulatory like functions. |
| TCRαβ+ CD4+ CD8+ Double positive T cells |
Effector memory-like phenotype Four main subpopulations: CD4highCD8low, CD4highCD8high, CD4medCD8high, CD4lowCD8high. |
Cytokine production, expression of inhibitory receptors (PD1, TIM3, TIGIT) and activation markers (HLA-DR, CD38, 4-1BB, Ki67). |
| TCRαβ+ CD4− CD8−Double negative T cells | Regulatory-like and/or effector memory-like phenotype. | Different functional properties which might reflect differences between circulating double-negative T cells from healthy donors versus tumor infiltrated double-negative T cells. |
| CD3+ T cell Memory subsets | ||
| Stem cell-like memory (TSCM) |
CD45RO−CD45RA+CCR7+CD62L+CD27+CD28+ IL7Rα+CD95+IL2Rβ+ |
Self-renewal, high proliferative capacity, circulation through lymphoid organs, cytokine production. |
| Central memory (TCM) |
CD45RA−CD45RO+CCR7+CD62L+ Diverse CD27CD28 expression. |
Reduced self-renewal and multipotentcy compared to TSCM. Circulation through lymphoid organs. Limited effector functions. |
| Effector memory (TEM) |
CD45RA−CD45RO+CCR7−CD62L-. Diverse CD27CD28 expression. |
Exhibit proinflammatory effector functions. Preferentially traffic through peripheral tissue. |
| effector memory RA+ (TEMRA) | CD45RA+CCR7−CD27−CD28− | Terminally differentiated effector T cell. Exhibit cytolytic capacity. |
| CD8+ TCRαβ subsets—Cytotoxic T lymphocytes (CTL) | ||
| Tissue-resident memory (TRM) | CD103+CD39+ | Cancer-specific CTL that reside in the tumor epithelium. Often coexpress inhibitory receptors such as PD-1. |
| Bystander | CD103+CD39− | Non-cancer-specific CTL that reside in the tumor epithelium. Capable of inducing antitumor immune response. |
| Progenitor stem-like exhausted (TPE) |
TCF1+Slamf6+CXCR5+PD1+ CD39− CX3CR1− |
Maintain antigen-specific immune response, persist long-term, self-renewal, differentiation into TEX. |
| Exhausted (TEX) |
CX3CR1+ CD39+PD1+Tim3+ TCF1−CXCR5− |
Exhibit high cytolytic and cytotoxic function. |
| CD4+ TCRαβ+ | ||
| Helper (THC) | STAT activated | Direct lysis of tumor cells, inducing CD8+ T cells activation and expansion. Improvement the antigen-presenting capacity of dendritic cells |
| Follicular helper (TFH) | CXCR5, BCL-6 expression | Promoting B cell activation, expansion, and differentiation into plasma cells. CXCL13 production. |
| Regulatory (Treg) | FOXP3+CD25+ | Production of suppressive cytokines, modification of antigen-presenting cells, nutrient deprivation, IL-2 exhaustion, and cytolysis. |
| B cells | CD19+CD20+ | |
| Antigen-presenting | MHC-mediated | Antigen presentation to T cells |
| Plasma cells (PC) | CD20−CD38+CD138+CD79a+ | Production of antibodies |
| Regulatory (Breg) | Lack of phenotypic markers | Production of suppressive cytokines IL-10, IL-35, and TGFβ. |
| Germinal center | Bcl-6+, activation-induced deaminase (AID+), Ki67 expression | Enables recombinant class switching of the constant region from IgM/IgD to IgG, IgE, or IgA, and somatic hypermutation of the BCR resulting in increased antigen affinity. |
| Class-switched | IgG, IgA, or IgE | Contain affinity-matured antibodies; give rise to a highly advanced immune response. |
| Innate lymphoid cells | ||
| Natural killer cells (NK) | CD16+NKp30+NK46+NKp44+NKG2D+NKG2A+ | Proinflammatory; high cytolytic capacity; release of granzymes, perforins, and IFNγ production. |
| Helper-like innate lymphoid cells (ILC) | ||
| ILC group 1 (ILC1) | NK1.1+ and NKp46+ | NK-like cells. Production of IFNγ. |
| ILC group 2 (ILC2) | IL33 receptor ST and CD127+ | GATA3-dependent function. Proinflammatory. |
| ILC group 3 (ILC3) | RORγt+CD127+ | Controversial role; both proinflammatory and immune regulatory. |
TCRαβ+ T cells
TCRαβ+ T cells of all states of differentiation have been observed in tumors, including nonclassical TCRαβ+CD4−CD8− and TCRαβ+CD4+CD8+ T cells. ‘Double-positive’ (CD4+CD8+) TCRαβ+ T cells have been identified in multiple tumors, including melanoma and lung, colon, and renal cancer. These double-positive T cells can be broadly subdivided into four major subpopulations, CD4highCD8low, CD4highCD8high, CD4medCD8high, and CD4lowCD8high, although many studies assess them as a single subset.1 To date, most work has focused on CD4lowCD8high T cells, the subset that is thought to develop from peripheral CD8 T cells, which coexpress low levels of CD4 after activation.2 In renal cell carcinoma, CD4lowCD8positive T cells have a CD8-like effector memory phenotype (CD45RO+CCR7−) with expression of the inhibitory receptors PD1, TIM-3, and TIGIT and the activation markers HLA-DR, CD38, 4-1BB, and Ki67.3 In melanoma, transcriptome analysis revealed a gene signature closer to that of CD8 single-positive T cells than that of CD4 single-positive T cells for CD4lowCD8positive T cells. However, the cells shared functional similarities with CD4 single-positive cells, including reduced cytolytic potential.2 These findings were confirmed in urological cancers, in which both CD4highCD8low and CD4lowCD8high T cells showed an effector memory-like phenotype, along with the production of the classical Th2 cytokines IL-4, IL5, and IL-13.4–6
‘Double-negative’ (CD4−CD8−) T cells are the subject of conflicting reports. Some studies in healthy donors have ascribed a regulatory-like phenotype to these cells,7,8 consisting of both CD45RA+CCR7+ and CD45RA+CCR7− cells, with an intermediate maturation stage (CD27+CD28−), high expression of CD95 and lack of activation markers such as CD25 and CD69.7 Other work argues specifically for the use of double-negative T cells from healthy donors as a source for adoptive cellular therapy due to their observed phenotype which is more consistent with that of effector memory cells: expression of CD45RA, CD44, and CD49d and low expression of CCR7, CD62L, CD127 and the inhibitory molecules ICOS, CTLA-4, and PD1.9 These differences may reflect changes in double-negative T cells infiltrating tumors. Indeed, a study comparing the reported phenotypes of double-negative cells across tumors found a comparable phenotype across human melanoma, renal cell carcinoma and glioblastoma, and the TILs in cancer tissues were phenotypically distinct from the double-negative T cells found in nonmalignant tissues. Interestingly, the double-negative population seemed to expand shortly after initiation of BRAF inhibitor treatment.10
CD8+ TCRαβ T cells
CD8+ TCRαβ T cells, referred to as CD8+ T cells, are mostly known for their exquisite antiviral and antitumor functions and are often referred to as cytotoxic T lymphocytes (CTLs). CTLs have the ability to produce high levels of antitumor cytokines and cytotoxic molecules, such as interferon-γ (IFNγ), tumor necrosis factor-α (TNFα), perforin, and granzymes.11 Accordingly, CD8+ CTLs are associated with improved prognosis in almost all types of cancer. Under physiological conditions and following elimination of their targets, CTLs generally form a number of memory subsets that provide long-term protection against reinfection after the resolution of the immune response. These memory T-cell subsets form a heterogeneous compartment and range from cells exhibiting a more naive-like phenotype to cells presenting an effector-like phenotype, they roughly follow along the line of stem cell-like memory T (TSCM), central memory T (TCM), effector memory T (TEM), and effector memory RA+ T (TEMRA) cells.12,13 TSCM cells are arguably the most naive cells (CD45RA+CCR7+CD27+) and have consistent recirculation patterns in vivo, mostly localized in the lymph nodes and to a lesser extent in the spleen and bone marrow but rarely found in peripheral mucosae. TSCM cells maintain their own pool through self-renewal. In addition, TSCM cells retain the ability to proliferate rapidly and release inflammatory cytokines. TCM cells differ from TSCM cells in their CD45RA−CD45RO+ phenotype and a reduced capacity for self-renewal and multipotency.14 However, TCM cells do possess naive-like functions and express lymph node-homing molecules such as C–C chemokine receptor type 7 (CCR7) and CD62L. TCM cells are also thought to have limited direct effector functions.12 TEM cells are characterized by cell surface expression of CD45RO+CCR7−CD62L−, and although these cells can (re)circulate through the blood, they preferentially traffic to peripheral tissues. In addition, these cells exhibit proinflammatory effector functions upon secondary antigen encounter with a cognate antigen and have diverse expression of CD27 and CD28.15 In line with their role in long-term protection, early work in colorectal cancer (CRC) demonstrated that the presence of a TEM cell immune infiltrate correlated with less advanced tumor stage and no signs of metastatic disease or lymph node involvement. Accordingly, the presence of TEM cells in the tumor was an independent prognostic factor for overall survival.16 Other studies around the same time highlighted the role of TCM cells in tumor control, as they possess high proliferative capacity and are suitable for adoptive T cell transfer, especially when combined with a tumor-antigen vaccination.17 Accordingly, a recent study highlights the correlation of CD45RO+ TILs with overall and disease-free survival in breast cancer.18
More recently, the role of peripheral tissue-resident memory (TRM) CTLs in tumor immunity has come into focus. After resolution of an immune response, TRM cells normally stay in the peripheral tissues without recirculating, providing a first line of defense against reinfection. TRM cells in peripheral tissues expresses canonical markers CD103, also known as integrin αE; CXCR6, which is involved in TRM development; CD49a, which is needed for retention and cytotoxic function; and CD69, an inhibitor of S1PR1 that mediates T cell recirculation.19–21 In tumors, TRM cells are also characterized by the expression of CD103. CD103 complexes exclusively with integrin β7, forming the αEβ7 complex; this complex interacts with E-cadherin, which is often expressed on tumor cells. Accordingly, CD103+ CTLs have been correlated with improved survival in a multitude of solid tumors, including several gynecological malignancies, lung cancer, breast cancer, melanoma, CRC and several head and cancers.22–26 In cancer mouse models, loss of E-cadherin or CD103+ CTLs was associated with loss of tumor control.27 Importantly, a recent study identified coexpression of CD103 and the immunosuppressive molecule CD39 as definitive markers of cancer-specific CTLs in tumors, further supporting the key role of the TRM cell subset.28
Finally, bystander TRM cells, which are not specific for tumor antigens but for epitopes unrelated to cancer, have also been identified in multiple solid tumors. These cells have diverse phenotypes but lack CD39 expression, which distinguishes them from the tumor-specific TRM cell population.29 Interestingly, it has been demonstrated that although unspecific for tumor antigens, these bystander cells are capable of contributing to the antitumor response. For instance, intratumoral viral-specific CTLs can be activated via the delivery of adjuvant-free viral peptides, which induce a broad immune response evidenced by accumulation and activation of CD8+ T cells and natural killer (NK) cells, increased expression of markers associated with dendritic cell (DC) activation and upregulation of PDL1. Consequently, tumor-bearing mice are more susceptible to PDL1 blockade when it is combined with viral peptide therapy than when it is used as a monotherapy.30
CD4+ TCRαβ+ T cells
CD8+ T cells do not function in isolation; there is also a well-established role for conventional CD4+ helper TCRαβ T (THC) cells in the antitumor immune response.31–34 THC cells promote CD8+ T cell priming through stimulation of CD40 on DCs via the expression of CD40 ligand (CD40L), resulting in the release of cytokines, such as IL-12, IL-15, and IFNγ, the upregulation of costimulatory ligands such as CD70, recruitment of B cells and naive CD8+ T cells and increased antigen presentation. In this two-step process, CD4+ and CD8+ T cells first independently interact with DCs in different areas of the lymphoid organs, whereas in the second-step of priming, both CD4+ and CD8+ T cells recognize their cognate antigens on the same DCs.34 In addition to a helper role in priming, THC cells can also possess cytolytic mechanisms that enable them to directly lyse tumor cells.32,34,35
In addition to these conventional THC cells, recent work has also identified T follicular helper (TFH) cells as crucial cells supporting B cell activation, expansion, and differentiation into plasma cells (PCs) and memory B cells in multiple human tumors.36 The presence of these TFH cells has been associated with improved prognosis in breast cancer and CRC. The CD4+ TFH cells, are characterized by CXCR5 expression, which is indispensable for T cell migration from T zones towards CXC chemokine ligand 13 (CXCL13)-rich B cell follicles, where they activate B cells through interactions with CD40 ligand (CD40L) and the production of interleukin (IL) 21. In addition, they are characterized by high expression of B cell lymphoma 6 (BCL-6). They also possess the capacity to produce CXCL13 and seem to be involved in the formation of TLSs, via which they shape intratumoral CD8+ T cell and B cell responses.37,38 Also capable of CXCL13 production are CD4+ TRM cells, which have a phenotype comparable to that of CD8+ TRM cells, including the capacity for the production of cytokines such as IFNγ and TNFα.21
In contrast to THC and TFH cells, CD4+ regulatory T (Treg) cells are known as tumor-promoting CD4+CD25+FoxP3+ T cells and have been shown to counteract tumor-specific immune responses by suppressing CD8+ cells, amongst other cell types.39 Consequently, Treg cells have been associated with poorer survival in multiple solid tumors, including pancreatic, ovarian, gastric, cervical, breast, and colon cancers.40–45 Several mechanisms exist by which Treg cells limit an effective antitumor response. Treg cells are known to produce immune-suppressive cytokines, including IL-10, IL-35, and TGFβ, but can also suppress productive immunity through nutrient deprivation, IL-2 exhaustion, and cytolysis.39 Complementary research specifically implicated the role of IL-10 and IL-35 in promoting BLIMP1-dependent inhibition of CD8+ TILs.46 Cell–cell-mediated suppression can also occur by CD28 costimulatory competition. Treg cells constitutively express CTLA4, which has a high affinity for CD80 and CD86 expressed on antigen-presenting cells (APCs). As CD80/CD86 also interacts, at a lower affinity, with the costimulatory receptor CD28 on T cells, Treg cells inhibit T cell activation by competitive CTLA4-CD80/CD86 binding.39,47 In addition, Tim3-positive Treg cells have displayed a superior capacity to inhibit naive T cell proliferation compared to Tim3-negative Treg cells, which is partially reversed by IFNγ production.39,48 Moreover, IFNγ was shown to drive the fragility of Treg cells, which in turn boosts antitumor immunity.49 Perhaps counterintuitively, two studies in gastric cancer and four in CRC demonstrate a good prognosis for patients with tumors with high densities of Treg cells (summarized by Fridman et al. in ref. 50). These results might be explained by the technical difficulties surrounding Treg cell quantification, the inability to detect multiple relevant markers at the same time, and the concomitant infiltration of other immune cells such as CD8+ TILs.50
TCRγδ+ T cells
TCRγδ+ T cells are mostly negative for CD4 and CD8, but these cells coexpress NK cell markers such as NKG2D. Consequently, TCRγδ+ cells have been proposed as a link between the innate and adaptive immune systems. Two main subsets have been described, the Vδ1γδ T cells and the Vγ9Vδ2 T cells, both displaying innate and adaptive immune features to differing extents.51,52 γδ T cells are described with different phenotypes, including CD4 T cell-like effector-like and regulatory phenotypes.53 In mice with lupus, it was demonstrated that a subset of γδ T cells express CXCR5 after activation. These TCRγδ+ CXCR5+ cells can then present antigens to naive CD4+ T cells and can induce follicular helper T cell differentiation, which in turn can induce a B cell response.54,55 Effector-like functions such as cytokine production have also been attributed to γδ T cells. Interestingly, TGF-β signaling upregulated the expression of CD54, CD103, IFNγ, and granzyme-B in Vγ9Vδ2 T cells, augmenting their cytotoxic effector activity.56
T cell exhaustion
Upon persistent antigen stimulation, T cells show a gradual decrease in various effector functions known as T cell exhaustion, which is characterized by a decrease in proliferative and cytolytic capacity and upregulation of multiple inhibitory signals, including PD1, LAG-3, CD160, 2B4, TIM-3, and TIGIT.13 Although characterized as an exhausted phenotype, these T cells can retain their cytolytic and proliferative capacity. The identification of this T cell phenotype led to the theory that exhaustion is a gradually developing state with various functional and phenotypic substates. Exhausted CD8+ T cells are thought to comprise both progenitor stem-like exhausted (TPE) cells and terminally exhausted T (TEX) cells, in a scheme that is similar to the classical T cell differentiation described above13,57,58 (Fig. 1). The classic view of T cell differentiation using TSCM, TCM, TEM, and TEMRA phenotypes is thus giving way to a TPE/TEX-based classification.
Fig. 1.

CD8+ T cell exhaustion states. CD8+ T cell exhaustion is thought of as a gradual process, with various functional and phenotypic states, including TPE and TEX states. The TPE phenotype is characterized by the expression of TCF1, which is lost upon differentiation into TEX cells. Cell surface markers identified on TPE cells include Slamf6, PD1, and CXCR5, and their functional capacity comprises the ability to produce an antigen-specific immune response, persist long-term, self-renew, and eventually differentiate into TEX cells. In contrast, TEX cells expresses mostly coinhibitory cell surface receptors and transcription factors associated with effector and exhausted cells, including CX3CR1, PD1, CD39, and TIM3, which reflects the functional capacity of TEX cells, that is, mostly cytotoxic functions. TEX cells: terminally exhausted T cells, TPE cells: progenitor STEM-like exhausted cells, TCF1: transcription factor 1
TPE cells are known to maintain antigen-specific immune responses, persist long-term, be capable of self-renewal and eventually differentiate into TEX cells.57,59 The TPE cell phenotype is characterized by the expression of the transcription factor T cell factor 1 (TCF1), encoded by the gene Tcf7, which is lost upon differentiation into TEX cells and is essential for the stem-like functions of TPE cells.57,58,60 Differential gene expression analysis has recently identified a CD39−Tim3−Slamf6+Tcf1+PD1+CD8+ cell phenotype to identify precursor states of exhaustion.57,59 In addition, CD127 and killer cell lectin-like receptor subfamily G member 1, a protein critical for T cell homeostasis and involved in the lysis of tumor cells, are found to be nearly absent on PD1+CD8+ TEX cells in breast and melanoma tumors.13 A recent report has also suggested CXCR5 as a marker for TPE cells that is coexpressed with Tcf1 in the absence of Tim3, and cells with CXCR5 expression showed similar functionality and persistence to cells with Tim3 expression.61 Interestingly, CXCR5 expression on CD8+ T cells has also been used to define follicular CD8+ T cells, which are able to migrate into B cell follicles and promote B cell differentiation. These cells also express lower levels of inhibitory receptors and exhibit more potent cytotoxicity than CXCR5−CD8+ cells, similar to the TPE cells phenotype.62 Considering the recent insights into ectopic B cell follicles in human tumors, the role and localization of CXCR5+ TPE cells might be of particular interest (see also corresponding sections below).
TPE cells have relatively high transcript levels of genes encoding cytokines, costimulatory molecules, and survival/memory molecules compared to TEX cells. TEX cells mostly expresses coinhibitory cell surface receptors and transcription factors associated with effector and exhausted cells. These differences are reflected in the functional capacity of both subsets: TPE cells contain the ability to proliferate, generating Tcf1+PD1+ and differentiated Tcf1−PD1+ cells, and TEX cells exhibit mostly cytolytic functions. Together, these data suggest that a delicate balance of both TPE cells and TEX cells is required for an effective antitumor immune response.57,58 Accordingly, data from mouse models of chronic viral infection have demonstrated that both TPE and TEX cell subsets are required for long-term viral control.59
Complementary studies used cell surface expression of CX3C chemokine receptor 1 (CX3CR1) as a marker for T cell differentiation and exhaustion.63,64 A recent in vivo study divided CX3CR1+CD8+ T cells into three subsets ranging from less to more terminally differentiated: CX3CR1−, CX3CR1int and CX3CR1high. Indeed, the CX3CR1− cells were characterized by high Tcf1 expression and possessed high proliferative capability upon activation. Moreover, PD1, LAG-3, and TIGIT expression decreased when CX3CR1 expression increased. Conversely, the CXC3C1high population exhibited the highest cytotoxicity. In addition, CX3CR1− cells were found to delay tumor growth and increase survival.64 The nuclear factor TOX has also been identified as a crucial regulator of T cell exhaustion. TOX expression was increased upon chronic TCR stimulation and was low during acute infection. In the absence of TOX, TEX cells do not form; T cells no longer upregulate inhibitory receptors, chromatin remains largely inaccessible, and Tcf1 expression is maintained. Although these cells are phenotypically “nonexhausted”, they are still dysfunctional.65–67 Interestingly, the aforementioned studies on TOX indicate that T cell exhaustion may be a beneficial process because it protects T cells from tumor and/or activation-induced cell death.
The T cell exhaustion phenotype appears to largely overlap with that observed for the TRM cell population. Indeed, the tumor-reactive TRM marker CD39 is a marker of persistent TCR stimulation, as demonstrated in both mice and human models.28,68 RNA sequencing of CD39+CD8+ cells revealed an exhausted transcriptome with PD1, Tim-3, Lag-3, TIGIT, and 2BA highly coexpressed. In addition, these cells demonstrated impaired production of IL-2, IFNγ, and TNF.68 Gene expression profiles of double-positive CD103+CD39+CD8+ cells (DP CTLs) versus double-negative CD103−CD39−CD8+ cells (DN CTLs) also identified a gene signature of DP CTLs consistent with that of cells with an exhausted, tissue-resident phenotype. This included high expression of PDCD1 (PD1), CTLA4 (CTLA-4), and HAVCR2 (Tim3) and decreased expression of T cell recirculation genes such as KLF2, SELL, and S1PR1 as well as lower expression of CCR7, CD127, and CD28 indicative of an effector memory phenotype. However, contrary to the findings of Canale et al., these DP CTLs exhibited more cytotoxic potential than DN CTLs, as more cells were granzyme-B positive, although this was not reflected in the production of IFNγ and TNFα.28 CD4+ TRM cells are also characterized by high expression of CD103, CD69, and CD49a and inhibitory molecules such as PD1, CTLA-4, and B24. Altogether, these findings support earlier observations that suggested CD103+ CTLs comprise tumor-reactive CD8 T cells in ovarian and lung cancer, characterized by the expression of exhaustion markers but without complete loss of functional competence.69–71
B lymphocytes
Signatures for patient stratification and response evaluation in clinical immunotherapy have focused predominantly on T cell responses. However, recent work has also identified a key role for B lymphocytes in immunotherapy, and their presence has been associated with an improved prognosis across different cancer types, including breast cancer, melanoma, renal cell carcinoma, CRC, hepatocellular carcinoma, and head and neck squamous cell carcinoma.72–76 However, the tumor-promoting effects of B cells have also been extensively described.77–80
Functionally, B cells may act as APCs for T cells, promoting local tumor-associated T cell responses.81,82 The observation of B cell clonal expansion and immunoglobulin phenotype switching across human cancers further indicates a possible role for antibody-dependent cell-mediated cytotoxicity (ADCC) in the antitumor humoral immune response, facilitated by antibody-secreting plasma B cells.83 Tumor-infiltrating B (TIL B) cells can also kill tumor cells directly by secreting toxic cytokines such as IFNγ and granzyme B or indirectly by promoting tumor-specific T cell secretion of immunostimulatory cytokines76,84 (Fig. 2, Table 1).
Fig. 2.

Antitumor- and protumor-related functional properties of B cells. B cells and plasma cells have several ways to promote local tumor-associated T cell responses. Functionally, B cells may act as antigen-presenting cells and facilitate tumor antigen-derived presentation to T cells. B cells also promote antitumor immunity by secreting immunostimulatory cytokines, such as IFNγ, that drive cytotoxic immune responses. In addition, B cells can directly kill tumor cells by secreting toxic cytokines, such as granzyme B. Plasma cells promote the antitumor immune response by secreting tumor-specific antibodies that can mediate ADCC, resulting in phagocytosis of tumor cells. In contrast, Breg cells suppress the antitumor immune response indirectly by secreting the immunoregulatory cytokines IL-10, IL-35, and TGFβ and directly by inhibiting effector cells, such as cytotoxic CD8+ T cells. Furthermore, Breg cells suppress antitumor immunity by converting CD4+ T cells into Treg cells via TGFβ. ADCC: antibody-dependent cell-mediated cytotoxicity, Breg cells: regulatory B cells, Treg cells: regulatory T cells
Antigen-presenting B cells
Professional APCs are characterized by their ability to take up antigens and load the processed antigen product onto MHC class molecules for presentation to T cells.85 Decades ago, B cells were found to be able to act as APCs, although they seem to function less efficiently than DCs, probably due to their reduced, nonspecific antigen uptake. When B cells encounter antigens, the binding affinity is relatively high (multivalency), resulting in B cells that are more sensitive to antigens at lower concentrations than DCs.86
Before immunization, antigen-specific B cells are very rare compared to DCs. Therefore, it was long assumed that B cells only minimally contributed as APCs to activate naive CD4+ T cells. However, by using RNA phage Qβ-derived virus-like particles as a nanoparticle antigen model, Hong et al. demonstrated that B cells, and not DCs, were responsible for the initial activation of CD4+ T cells and promoted CD4+ T cell differentiation into CD4+ TFH cells. Additionally, a germinal center (GC) response could be induced in this model in the absence of DCs.87 Similar results were observed when another type of immunization, a soluble protein, was used. Again, B cells acted herein as professional APCs upon immunization with inactivated influenza virus and initiated activation of naive CD4+ T cells. These results suggest an important role for B cells in initiating CD4+ T cell responses, with an emphasis on viral infections. However, it has also been shown in murine and human models that B cells efficiently present tumor-associated antigens (TAAs) to T cells.88,89 For instance, TIL B cells efficiently presented TAAs to CD4+ T cells in non-small-cell lung cancer patients and influenced the CD4+ phenotype. Specifically, activated TIL B cells (CD69+HLA-DR+CD27+CD21+) were associated with a CD4+ effector T cell response (CD4+IFNγ+), demonstrating the plausible role of B cells as professional APCs in promoting the antitumor immune response.88
Antibody-producing (plasma) B cells
PCs are characterized by the absence of CD20 and the coexpression of CD38, CD138, and cytosolic CD79a and are the dominant antibody-producing B cell subset. Recently, it was shown that PCs seem to have an important role in promoting antitumor immunity.
Kroeger et al. found that the prognostically favorable effects of CD8+ TILs accompanied by CD20+ B cells were even further enhanced by the presence of stromal PCs.90 In high-grade serious ovarian cancer patients, tumors infiltrated with CD20+ B cells and CD4+ and CD8+ T cells together with PCs were associated with increased survival, with ~65% of the patients being alive at 10 years after diagnosis. Interestingly, tumors containing CD8+ TILs accompanied by solely CD4+ TILs, CD20+ TILs, or PCs were associated with minor insignificant survival increases, suggesting the importance of interplay between these different immune subsets in promoting antitumor immunity.90 Several studies further analyzed the association between class-switched B cells with an increased B cell receptor (BCR) diversity and clonal fraction resulting from tumor-related GC activity. Hu et al. identified widespread clonal B expansion and Ig subclass switch events in various human cancers by observing the same complementarity-determining region 3, containing both IgG1 and IgG3 isotypes (IgG3-1 sCSR).83 These results were comparable to Kroeger et al., who detected clonally expanded PCs as well as the presence of somatic hypermutation (SMH) within VDJ families. Additionally, IgG transcripts, specifically IgG1, IgG2, and IgG3, represented the majority of immunoglobulin subtypes.90 Increased BCR diversity and clonal expansion were also observed in tumors of melanoma patients.75
Of note, some autoantibodies have also been found to be tumor promoting. Coussens et al. showed that antibodies that are deposited at tumor sites in the form of immune complexes recruit myeloid cells and macrophages to become tumor promoting by binding to the immune complexes via Fcγ-activating receptors. These myeloid cells and macrophages were found to secrete proangiogenic factors and immunoregulatory cytokines, enabling tumor progression.91
Regulatory B cells
Regulatory B (Breg) cells are a subpopulation of B cells characterized by their unique immunoregulatory and immunosuppressive qualities, possessing an important role in peripheral tolerance.92 Accordingly, Breg cells have been associated with worse clinical outcome in cancer.93,94 Phenotypic markers to characterize Breg cells, other than IL-10 production, are not yet definitive, complicating in-depth analysis.
Nevertheless, it is clear that Breg cells suppress the immune response by secreting IL-10, thereby inhibiting DC differentiation, suppressing helper T1 (TH1) and helper T17 (TH17) cell proliferation, and inducing the differentiation of Treg cells.94 Accordingly, Breg suppressive immune functions are favorable in autoimmune diseases, as the absence of Breg cells results in the exacerbation of rheumatoid arthritis (RA) and systemic lupus erythematosus.95,96
The antitumor immune response is likely indirectly suppressed by Breg cells secreting immunoregulatory cytokines (IL-10, IL-35, and TGFβ) but also directly suppressed by inhibition of effector cells such as cytotoxic CD8+ T cells. In ovarian cancer, IL-10 secretion by Breg cells significantly suppressed the production of cytotoxic effectors, such as IFNγ, by CD8+ T cells.78 Additionally, in human hepatoma, IL-10 secretion by Breg cells supported tumor growth and suppressed tumor-specific T cells.79 In glioblastoma, Breg cells were characterized by the immunosuppressive molecules PDL1 and CD155 and the production of IL-10 and TGF-β and were found to suppress CD8+ T cell activation, proliferation and production of IFNγ and granzyme B. Furthermore, local B cell depletion in mice using CD20 immunotherapy significantly improved OS, which correlated with increased tumor-infiltrating CD8+ T cells and production of granzyme B and IFNγ. Interestingly, this survival benefit was not observed in mice receiving systemic anti-CD20 immunotherapy. This suggests that B cells have different functions depending on their location and that naive B cells might differentiate into a Breg phenotype when localized in the immunosuppressive tumor microenvironment (TME).97
Finally, Breg cells were shown to suppress antitumor immunity by influencing the conversion of CD4+ T cells into Treg cells via TGFβ, which was observed in a 4T1 breast cancer mouse model of human gastric and tongue squamous cell carcinoma.80,98,99
Innate lymphoid cells
Innate lymphoid cells are a more recently appreciated subset of tumor-infiltrating lymphocytes with key roles under physiological immune homeostasis. In general, these cells are characterized as NK cells, type 1 innate lymphoid cells (ILC1s), ILC2s, or ILC3s (Table 1).
Natural killer cells
Natural killer cells (NK cells) are defined by the absence of antigen-specific B or TCRs due to their lack of recombination activating genes. The majority of peripheral NK cells are CD56dimCD16+ and characterized by the ability to rapidly mediate cytotoxicity. In addition, the CD56bright CD16− NK cell population accounts for ~10% of peripheral NK cells and is characterized by low perforin production but normal production of IFN‐γ and TNF‐α.100,101 NK cell activity is dependent on a repertoire of costimulatory and inhibitory signals that bind to their respective ligands on the cell surface. The dominant activation receptors are CD16, NKp30, NK46, NKp44, and NK group 2, member D (NKG2D). Inhibitory receptors include killer Ig-like receptors and CD94/NKG2A-B, which recognizes HLA-E molecules. When activated, NK cells exhibit antitumor activity via the release of granzymes and perforins, the induction of TNF-related apoptosis and the production of IFNγ.100,102 In mice, indirect antitumor activity of NK cells has also been demonstrated; NK cells were recruited in lymph nodes undergoing an immune response and produced IFNγ, which was necessary for the priming of T-helper cells.103 In addition, more recent research has demonstrated cancer immune control by NK cells through the accumulation of conventional type 1 dendritic cells (cDC1s) via the production of the chemoattractants CCL5, XCL1, and XCL2. The tumor cells were able to counteract this axis by the production of prostaglandin E2, which caused impaired chemokine production by NK cells, consequently leading to reduced intratumoral cDC1 recruitment.104
The activity and presence of both circulating and intratumoral NK cells have been associated with disease progression, metastatic disease development, and survival.100,102,105 In gastric cancer, a low percentage of NK cells in the tumor was associated with poor survival and disease progression. Ex vivo studies showed impairment of NK cells through TGF-β signaling by monocytes, which resulted in decreased IFNγ, TNFα, and Ki-67 expression in NK cells.106 Interestingly, surgical stress impairs peripheral NK cell function. In patients undergoing surgery for CRC, IFNγ production by NK cells was significantly suppressed for up to 2 months.101 Taking into account the cytolytic potential of NK cells, there is an increased interest in the use of NK cells for immunotherapy, either in adoptive transfer therapies or reactivation strategies affecting their activation and inhibitory ligands.
Helper-like innate lymphoid cells
Based on function, cell surface markers and transcription factors, ILCs have been categorized into three groups: group 1 (ILC1s), group 2 (ILC2s), and group 3 (ILC3s). Overall, the role of helper-like lymphoid cells in cancer remains poorly understood, with these cells having high plasticity and seemingly occupying controversial roles.107
The ILC1s are most comparable to NK cells, as both require the transcription factor Tbet to function; both express NK1.1 and NKp46, and they mostly produce IFNγ. Unlike NK cells, ILC1s are not dependent on Eomes expression.108 In mice, it has been demonstrated that ILC1s can arise from NK cells as a result of TGFβ signaling. NK cells are known to limit tumor growth and metastatic outgrowth. However, their conversion into ILC1s leads to inferior tumor control.109 Indeed, complementary research in mice suggested that SMAD4 impeded the conversion of NK cells into ILC1s via TGFβ signaling.110 This was recently confirmed in in vitro human cell cultures.111 In melanoma patients, ILC1s were found to be an enriched subset, although dysfunctional, as demonstrated by impaired IFNγ production. Follow-up experiments in mice identified the production of adenosine (ADO) and kynurenines by melanoma cells as possible causes of ILC1 disruption and impaired IFNγ production. These data suggest that the exploration of targeting IDO and the adenosinergic immunosuppressive axis in melanoma patients is warranted.112 Overall, these data suggest that at least part of the ILC1 subset emerges from NK cells. In addition, the function of ILC1s should be further explored to investigate their role in tumor immunology and therapy.109,110,112
ILC2s are mostly described as proinflammatory, although some studies highlight tumor-promoting characteristics. Their function and development are GATA3-dependent, and the cells are characterized by the expression of the IL-33 receptors ST2 and CD127.113 ILC2s have been detected in multiple tumor types, including breast, pancreatic, gastric, bladder, and prostate cancer.114 As ST2 is highly expressed on ILC2s, it was demonstrated that they are dependent on IL-33 for their expansion and cytokine production. Furthermore, IL-33-activated ILC2s are implicated in the priming of tissue-specific CD8+ T cells, as ILC2 expansion is accompanied by increased cytokine capacity and PD1 upregulation in CD8+ T cells, implicating a possible role of ILC2s in the antitumor response to PD1 blockade.113,115 In contrast, in acute leukemia, ILC2s have been shown to promote myeloid-derived suppressor cells through the production of IL-13.116 In mice, ILC2s were shown to activate Treg cells through IL-9 production, although this was in the context of chronic inflammation where treatment with IL-9 induced resolution of the inflammation.117
The overall role of ILC3s seems controversial, and they have been described as both proinflammatory and immune regulatory. They are characterized by the expression of RORγt and CD127. In non-small-cell squamous lung cancer, ILC3s were found to accumulate and produce the proinflammatory cytokines TNFα and IL-22. Moreover, ILC3s were specifically found at the edge of TLSs, suggesting that they may contribute to the formation of protective tumor-associated TLSs.118 In contrast, another study in squamous cell lung carcinoma demonstrated tumor immune evasion by the conversion of ILC1s into ILC3s via IL-23 production by tumor cells. The converted ILC3s were capable of IL-17 production, which promoted tumor growth and was associated with poorer prognosis.119 In addition, in breast cancer, increased numbers of ILC3s were correlated with the likelihood of developing lymph node metastasis, and in consecutive mouse experiments, depletion of ILC3s was sufficient to decrease lymph node metastasis.120
Organization of TILs in tumors
The presence of TILs in tumors has been associated with improved clinical outcomes. However, the type and function of TILs (e.g., CTL versus Treg cell) as well as the TME localization of different TILs are key with respect to eventual tumor control or tumor progression.121 Therefore, a deeper analysis of the spatial organization of TILs in the TME, e.g., marginal zone versus tumor stroma, is needed to provide a better understanding of antitumor immunity and to discover potentially new biomarkers.122
Early histopathological analyses of tumor samples already demonstrated varying TIL distribution across tumor types and showed that different types of immune cells are found in different locations, around and inside the tumor. Specifically, the distribution of TILs was found to be not random but well organized in specific areas. B cells, for instance, are mainly found in the invasive margin and clustered inside TLSs, close to the tumor, with NK cells mainly found in the stroma.121
TIL infiltration of tumors
The initial step in the formation of TILs from circulating lymphocytes requires the migration of immune cells from the blood to the tumor across the tumor endothelial barrier. The tumor endothelium is often disturbed and able to directly suppress T cell function, thereby preventing tumor infiltration. For instance, proangiogenic growth factors such as VEGF-A impair lymphocyte adhesion due to an associated defect in vascular cell adhesion molecule (VCAM-1) and intracellular adhesion molecule (ICAM-1).123 Proangiogenic factors can also induce overexpression of the endothelin B receptor (ETBR), which is associated with a lack of TILs in ovarian cancer patients.124 These changes are therapeutically targetable, as in vitro inhibition of VEGF-A and ETBR resulted in a restored amount of TILs and an improved response to immunotherapy.124,125 Similarly, FasL (CD95L or CD178), a pro-apoptotic cell surface protein, might also be targeted, as it is frequently overexpressed on endothelial tumor cells of humans and mice.126,127 To address this, Motz et al. studied FasL expression in tissue microarrays (TMAs) of human breast, renal, bladder, colon, prostate and ovarian adenocarcinomas and control TMAs derived from healthy tissues.128 Normal vasculature tissue did not express FasL, whereas the blood vessels of primary and metastatic tumors did, which was associated with reduced CD8+ T cell infiltration. VEGF-A, IL-10 and prostaglandin E2, three tumor-derived factors, together induced FasL expression, resulting in the elimination of CD8+ CTLs. Treg cells were resistant to FasL-mediated apoptosis due to their higher levels of the anti-apoptotic gene c-FLIP, which resulted in decreased levels of intratumoral CD8+ T cells and accumulation of intratumoral Treg cells. Conversely, FasL suppression resulted in increased infiltration of CD8+ T cells in tumors, improving the CD8+ T cell/Treg cell equilibrium, leading to reduced tumor volumes in mice. Of note, vessels carrying circulating lymphocytes were largely absent from the tumor core, localizing in the surrounding stroma and/or invasive margin. This suggests a direction of travel from vessels to the stroma by cancer cells and highlights a key role for the stroma in tumors.128
Tumor stroma
The stroma surrounding the tumor cells is an important component of the TME and harbors a cellular immune component including various innate and adaptive immune cells (B cells, T cells, macrophages, DCs, myeloid-derived suppressor cells, and NK cells) and a nonimmune cellular component (fibroblasts, endothelial cells, pericytes, and mesenchymal cells). Stromal cells in the TME can be either tumor promoting or tumor suppressing. Physiologically, in most nonmalignant tissues, stromal cells are suppressive, regulating the proliferation and migration of differentiated epithelial cells, as well as maintaining the structure and size of organs.129 Immunologically active cytokines, comprising growth factors, chemokines, angiogenic factors, and interferons, are major driving forces in tumor-stroma interactions.130
Stromal TILs, such as B and T cells, serve as key immune organizers in the TME through the secretion of cytokines. One of the most relevant and well-characterized chemokines in the structural organization of the immune cell cluster is CXCL13, which induces chemotaxis of CXCR5-expressing B cells and T cells towards the invasive margin, where they cluster together in well-organized structures, referred to as TLSs. This invasive margin represents the first line of defense against cancer metastasis. In CRC, the immune cell density is even higher at the tumor boundary than in the tumor core. As in other solid malignancies, CRC patients who exhibit TLSs in the invasive margin, also known as a ‘Crohn’s-Like reaction (CLR)’, have better OS than CRC patients who exhibit only diffuse inflammatory infiltration (DII).131,132 Accordingly, the existence of immune infiltrates in TLSs at the invasive margin was associated with a decreased presence of early metastatic processes such as vascular, lymphatic, and perineural invasion in CRC.133 In endometrial cancer, the number of TLSs is directly correlated with specific tumor mutations, such as the ultramutated POLE exonuclease domain or hypermutated microsatellite unstable (MSI) mutations.
Tertiary lymphoid structures
Many tumors are associated with TLSs, de novo lymphoid tissue resembling secondary lymphoid organs (SLOs). TLSs have been observed near zones of infection and tumors and less frequently near transplanted organs and autoimmune syndromes, where there is continued need for lymphocyte extravasation.82,134–137 In tumors, TLSs are associated with favorable prognosis and responses to immune checkpoint inhibitors.73,138 TLSs are mostly found peritumorally in the stroma and/or in the invasive margin, creating an optimally organized immune structure where DCs, T cells, and B cells interact and activate each other, promoting a local sustained immune response, e.g., induction of effector function, antibody generation, SMH, class switch recombination (CSR), and clonal expansion. As is the case for SLOs, the chemokine CXCL13, secreted by activated T cells, plays a crucial role in the formation of TLSs.82
Neogenesis of tertiary lymphoid structures
The neogenesis of TLSs starts with local production of IL-7 and CXCL13 by stromal cells or lymphocytes, which leads to the recruitment of IL-17-secreting CD4+ lymphoid-tissue inducer (LTI) cells.139 LTI cells express membrane-bound lymphotoxin α1β2 (LTα1β2), which can interact with stromal cells via the lymphotoxin β (LTβ) receptor, initiating NFκB signaling.140 Of note, it has been shown that TLS neogenesis can occur independent of CD4+ LTI cells, as B cells, T-helper 17 cells, and M1 macrophages were also found to be able to initiate TLS neogenesis.141–144
Together with IL-17 secretion, NFκB signaling in CD4+ LTI cells results in the production of homeostatic chemokines, notably CXCL12, CXCL13, CCL19, and CCL21.82 Additionally, in stromal cells, LTα1β2-LTβ signaling leads to the secretion of adhesion molecules (VCAM1, ICAM1, and MADCAM1) and vascular endothelial growth factor C, thereby stimulating the formation of high endothelial venules (HEVs).145 HEVs, MECA-79-expressing specialized postcapillary venules, enable lymphocyte migration and extravasation into TLSs.82,146,147 Finally, the organization of the recruited lymphocytes results in the formation of a nodular TLS consisting of a CD3+ T cell-rich zone with mature DCs in close proximity to CD20+ GC-like follicle B cells intermingled with follicular dendritic cells (FDCs), and surrounded by CD8+CD138+ PCs CD38+CD138+ PCs82 (Fig. 3).
Fig. 3.

TLS maturation state and CXCL13. TLSs are optimally organized nodular immune structures consisting of a CD3+ T cell-rich zone with mature DCs in close proximity to CD20+ GC-like follicle B cells intermingled with FDCs and surrounded by plasma cells. The CXCL13–CXCR5 axis regulates the organization of B cells inside the follicles. CXCL13-secreting CD8+ T cells induce chemotaxis by binding to the receptor CXCR5, which is mainly expressed by B cells and TFH cells. Inside the TLS, B cells, T cells, DCs, and FDCs interact and activate each other, promoting a local, sustained, organized immune response. TLS maturation varies from dense lymphocyte aggregates to primary TLSs and secondary follicle-like mature TLSs. The difference between primary and secondary TLSs is the presence of germinal center activity, which is dependent on B cells expressing AID, facilitating SHM and CSR and resulting in high-affinity antibody production by class-switched plasma cells. In addition, mature TLSs are surrounded by HEVs, facilitating lymphocyte migration and extravasation. TLSs: tertiary lymphoid structures, DCs: dendritic cells, FDCs: follicular dendritic cells, TFH cells follicular helper T cells, AID: activation-induced deaminase, SMH: somatic hypermutation, CSR: class switch recombination, HEVs: high endothelial venules
Cellular components, locations, and maturation of tertiary lymphoid structures
Two important subsets of the represented T cells in TLSs are TFH cells and FDCs. Differentiation of conventional CD4+ T cells into TFH cells is stimulated by TGF-β, IL-12, IL-23, and Activin A signaling, followed by upregulation of the TFH cell-associated genes Bcl6, PD1, ICOS, and CXCR5.148–153 In SLOs, TFH cells are specialized in helping B cells in helping B cells and are essential for GC formation, affinity maturation, SMH of immunoglobulin light chains and CSR of and immunoglobulin heavy chains.
FDCs facilitate long-term retention of the intact antigen in the form of immune complexes, enabling the positive selection of the SMH-mutated BCR by testing its antigen affinity.154 Furthermore, FDCs contribute to GC B cell survival and GC affinity maturation, as demonstrated by the inactivation of FDCs by Toll-like receptor 4, which is normally upregulated during GC responses, resulting in smaller GCs and decreased antibody titers in response to immunization.155 Finally, FDCs secrete increased levels of transforming growth factor β1 and express increased levels of the chemokine CXCL13.156 Although the described functional capacities of TFH cells and FDCs are mainly applicable in SLOs, the presence of these cells in TLSs has been identified, and similar functioning is assumed.82,157–159
TLSs are mostly located peritumorally in the stroma and/or in the invasive margin where the TLS maturation varies from dense lymphocytic aggregates (early TLSs) to primary and secondary follicle-like TLSs, depending on the presence of follicular dendritic cells (FDCs) and a GC reaction.158,160 Mature TLSs contain GC activity, defined by B cells expressing activation-induced deaminase (AID) and the proliferation marker Ki67, and are surrounded by HEVs.161 Interestingly, it seems that not only the presence of TLSs but also the TLS cellular components, such as T cells, B cells, FDCs, TFH cells, Treg cells, macrophages, HEVs, and chemokines, representing the TLS maturation state, are important for functionality in terms of a prosperous antitumor response. This was demonstrated in colorectal cancerCRC (CRC) stage II and III where not TLS density, but TLS maturation was associated with disease recurrence. Tumors with mature GC-harboring TLSs (secondary TLSs) had significantly decreased risk of recurrence compared to tumors without GC-harboring TLSs (early/primary TLSs).158 Similar results were found in chemotherapy-naive lung squamous cell carcinoma patients; only secondary TLSs, but not early or primary TLSs, were correlated with improved survival.160
These results are further supported by Yamaguchi et al., who demonstrated that TLSs can be categorized based on the different cellular component densities.159 CRC samples were stained for CD3, CD8, CD20, FDC, CD68, and Bcl-6 and counterstained with DAPI, and TLSs were defined as those structures that included specific T cells (THC cells: CD3+CD8−Bcl-6−; CTLs: CD3+CD8+; TFH cells: CD3+CD8−Bcl-6+), B cells (B cells: CD20+Bcl-6−; GC B cells: CD20+Bcl-6+) and FDCs (FDC+). TLS densities of CD4+ THC cells and macrophages were significantly higher in patients with disease recurrence than in patients without disease recurrence. Interestingly, on multivariate analysis, there was a significant correlation between CRC recurrence and the proportion of CD4+ T-helper cells (CD3+CD8−Bcl-6−), suggesting that a high CD4+ T-helper cell density hampers the antitumor immune reaction in TLSs and might be an independent predictor for CRC recurrence.159 On the other hand, the expression of TFH cell-related genes, such as CXCL13 and IL-21, was found to predict improved survival in CRC. Indeed, loss of CXCL13 was associated with a higher risk of relapse and lower densities of B and TFH cells in the invasive tumor margin.162
Because TLSs are only present in the invasive margin, Schürch et al. analyzed this region in CRC TMAs of CLR and DII patients.163 When further exploring the spatial organization of the invasive margin, they identified “nine coordinated cellular neighborhoods (CNs)”, specific areas of tissue within which every cell has a comparable surrounding neighborhood defined by the relative frequencies of cell types inside a defined radius. Similar sets of CNs were observed in both patient groups (CLR and DII), except for the follicle-enriched CN, representing TLSs, which was significantly more abundant in CLR patients.163
Strikingly, in CLR patients, the tumor immune compartments were isolated from the tumor compartments, but in DII patients, the immune compartments were increasingly interspersed with tumor compartments, suggesting that in DII patients, the tumor might interfere with proper development of the immune response and prevent efficient communication between CNs, which otherwise might result in the formation of follicular structures (TLSs). Furthermore, while T cells and macrophages were among the most common immune cells in the invasive margin, in DII patients, the CN1 (T cell-enriched) and CN4 (macrophage-enriched) areas were highly intertwined, having close physical contact and communication. Additionally, the CN functional states were different: in CLR patients, the CN1 (T cell-enriched) areas were more cytotoxic, and in DII patients, the CN4 (macrophage-enriched) areas were more immunosuppressive. Thus, the immune escape resulting in poor survival in DII patients might be due to factors released by the tumor, resulting in the coupling of CN1 (T cell-enriched) and CN4 (macrophage-enriched) areas and thus causing a shift towards an immunosuppressive macrophage phenotype and suppressed cytotoxic activity of the T cell-enriched CN, resulting in poor tumor immune control.163 These results highlight the importance of understanding the underlying immune architecture in the TME. Whether this CN spatial organization is applicable across tumor types needs to be further explored.
The role of CXCL13 in tertiary lymphoid structure formation
The chemokine CXCL13 induces chemotaxis by binding to the receptor CXCR5, which is mainly expressed by B cells and TFH cells. The CXCL13–CXCR5 axis regulates the organization of B cells inside the follicles of lymphoid tissues.164
Interestingly, Thommen et al. showed a potential link between CXCL13-secreting exhausted CD8+ T cells (high expression of PD1) and the formation of TLSs.165 They analyzed and compared the functional, metabolic, and transcriptional signatures of CD8+ TIL populations with PD1-high (PD1hi), PD1-intermediate, and no PD1 expression (PD1−) from tumor samples of non-small-cell lung cancer patients. Indeed, PD1hi CD8+ T cells were highly dysfunctional concerning classic cytotoxic functions such as IFNγ production compared to the other subsets, but strikingly, PD1hi CD8+ T cells highly expressed and constitutively secreted CXCL13. To study the function of CXCL13 in recruiting CXCR5-expressing cells towards the TME, colocalization of PD1hi CD8+ T cells with CD4+ TFH and B cells within the TME was analyzed. PD1hi CD8+ T cells were most represented in peritumoral and intratumoral TLSs, in close proximity to B cell infiltrates and CD4+ TFH cells. In the majority of the tumors, PD1hi CD8+ T cells were localized at the tumor-host interface, surrounding the central B cells, suggesting an active role of PD1hi CD8+ T cells in recruiting immune cells and forming TLSs. Additionally, the presence of PD1hi CD8+ T cells was predictive of the response to PD1 blockade treatment in non-small-cell lung cancer patients and correlated with OS and durable responses, demonstrating the reinvigoration capacity of PD1hi CD8+ T cells upon PD1 blockade treatment.165
A similar relationship between exhausted CXCL13-secreting tissue-resident CD8+ T cells (CXCL13+CD103+CD8+) and TLS formation was found by Workel et al.166 They analyzed pretreatment tumorous tissue of stage IIIC high-grade serous ovarian cancer patients (one patient received three cycles of chemotherapy prior to interval debulking surgery) and found that the phenotype of exhausted CD8+ tissue-resident T cells was similar to the exhausted CD8+ subpopulation of the study of Thommens et al, with both populations expressing equal PDCD1 (PD1) levels. Indeed, CXCL13 expression and secretion were observed in exhausted CD103+CD8+ TILs. Interestingly, as demonstrated by its ability to reactivate CD8+ T cells isolated from peripheral blood from healthy donors in vitro, TGFβ turned out to be a specific inducer of CXCL13 and CD103 in CD8+ T cells. Furthermore, the association between CXCL13-secreting tissue-resident CD8+ T cells and TLS formation was assessed by analyzing TCGA mRNA expression across different tumor types, including ovarian, uterine, lung, and breast cancers. The TLS-related genes of all four tumor types correlated with the CXCL13+CD103+CD8+ cell-related genes, suggesting that exhausted tissue-resident CD8+ T cells recruit lymphocytes towards the tumor and promote the formation of TLSs across tumor types.166
Similar results were found by Duhen et al., who identified CD39 and CD103 double-positive intratumoral CD8 T cells (CD103+CD39+CD8+), which displayed an exhausted TRM phenotype (expression of PD1, CTLA-4, and TIM-3), as tumor-reactive T cells in human solid tumors.28 Accordingly, TGFβ presence was needed for the maximum coexpression of CD39 and CD103 on CD8+ T cells, and indeed, this CD8+ TIL subset highly expressed CXCL13. In addition, CD103+CD39+CD8+ TILs were associated with increased survival in head and neck squamous cell carcinoma, lung adenocarcinoma and lung squamous cell carcinoma patients.28
TILs in clinical practice
In clinical practice, TILs have been suggested as potential prognostic and therapeutic biomarkers, most notably in the context of immune checkpoint blockade (ICB) therapy. Interestingly, the established prognostic benefit of TILs in ovarian and breast cancer does not directly translate to therapeutic benefit for ICB treatment in these malignancies, suggesting differences in the quality of the TIL response. Nevertheless, TIL quantification is steadily making a clinical impact in combination with the traditional parameters of disease staging.
Prognostic benefit of TILs
As discussed above, intraepithelial CD8+ T cells are associated with improved survival; however, some studies have also highlighted the importance and prognostic relevance of stromal TILs.167 In epithelial ovarian cancer, stromal TILs were associated with improved OS, specifically in high-grade serous ovarian cancer.168 In contrast, another study with similar research techniques found that increased levels of both intratumoral and stromal TILs were associated with a better prognosis, but statistical significance was only found for intratumoral TILs.169 In HER2-positive breast cancer patients, higher levels of stromal TILs are associated with improved prognosis.170–172 In one of the largest retrospective studies, Kim et al. assessed 1581 eligible B-31 cases for TILs in the NSABP trial and analyzed the association between infiltration of stromal TILs and clinical outcome in early-stage HER2-positive breast cancer patients receiving combined adjuvant trastuzumab plus chemotherapy or adjuvant chemotherapy alone. They found that higher levels of stromal TILs were associated with improved DFS in both groups. However, there was no association between stromal TILs and trastuzumab benefit. The authors concluded that “stromal TILs may have utility as a prognostic biomarker identifying HER2-positive early BC at low recurrence risk”.173
Stromal TILs were also found to have increased prognostic value in CRC compared to intraepithelial TILs.174 The importance of stromal TILs is reflected in the existence of a standardized methodology for evaluating TILs, designed by the International TILs Working Group (ITWG) in 2014. This methodology was initially designed to assess TILs in breast cancer, but subsequently, the ITWG also proposed a model for other solid malignancies. In short, stromal TILs residing in the stromal areas, in-between carcinoma cell islets, are scored as a percentage. The surface areas occupied by the carcinoma cell islets are not included in the total valued surface area.175,176 Fuchs et al. assessed the efficacy of the methodology in all-stage CRC patients (n = 1034). They used the ITWG method to estimate the stromal TIL density and found that the assessed stromal TILs had a superior predictive value compared to intraepithelial TILs using a traditional system proposed in the Royal College of Pathologists of Australia protocol (using the criterion of ≥5 intraepithelial lymphocytes directly in contact with tumor cells per high-power field).177 This study demonstrated that estimating stromal TILs, which are not in direct contact with carcinoma cells, seems to be a more adequate parameter than estimating intraepithelial TILs. This does not imply that intraepithelial TILs are not important but rather reflects the difficulties in determining intraepithelial TILs on H&E staining due to the small numbers and heterogeneous appearance of TILs. Another advantage of solely assessing stromal TILs is that the density or growth pattern of carcinoma cell islets does not affect the stromal TIL score.174 Recent advances in machine-learning approaches may help provide new insight into the utility of stromal versus intraepithelial TILs.
Clinical quantification of TILs
Traditionally, TIL infiltration has been manually quantified by pathological assessment. However, with the ever-increasing complexity in the understanding of TIL composition and localization, novel quantification approaches are under active development. The current development of digital immune scores, digital prognostic scores integrating multiple immune features into a single model, provides the opportunity to translate the prognostic benefit of TILs into a clinically usable diagnostic tool to aid clinical decisions and to improve personalized therapy. Digital pathology is earning more attention due to the advent of whole-slide imaging.178
In colon cancer, an internationally validated immune score is predictive of time to disease recurrence independent of existing prognostic factors, such as age, sex, tumor stage, node stage, and MSI. Of all clinical parameters, the immune score had the highest relative contribution to the risk of recurrence. This immune scoring system represents the first standardized immune-based assay for the classification of cancer.179 In the metastatic setting, response to treatment and prolonged survival were both significantly associated with high immune infiltration.180 In addition, a prognostic score for oral squamous cell carcinoma incorporating four immune markers, including the levels of immune cells present in both the invasive margin and center of the tumor, was recently published. This seven-immune-feature-based prognostic score is significantly correlated with disease-free survival.181 An independent study in oral squamous cell carcinoma identified the abundance, location, and spatial patterns of TILs as strong predictors of survival.182 While both scoring methods were tested in small cohorts and need to be cross-validated in larger patient groups, these studies indicate oral squamous cell carcinoma as a promising next candidate for the implementation of digital immune scoring in the clinic. The use of immune scores has also been suggested for gastric and bladder cancer.183,184 The accuracy of a digital immune score is dependent on the markers, regions of interest, procedures, measurements, and strategies used for quantification.179,181 Therefore, immune scores should be evaluated per cancer, and the method should be validated in multiple independent cohorts.
It is worth noting that manual quantification of TILs may have limited application as a diagnostic tool due to interobserver variability and the lack of diagnostic reproducibility. With automatic machine learning, these limitations may be overcome by the quantification and classification of digitized tissue samples by supervised deep learning.185 Studies presenting deep learning-based models for nuclei segmentation have been published.178,186,187 In addition, a deep learning model to differentiate between adenocarcinoma and squamous cell carcinoma and predict commonly mutated genes was validated in lung cancer, and the classification is expected to be extended to other less common lung cancers.188 In colon cancer, the accuracy of a deep learning classifier to predict 5-year disease-specific survival was compared with that of visual assessment. Patients were categorized into low-risk or high-risk, and the machine learning-based method demonstrated superior accuracy in patient stratification compared to human observers.185 The application of deep learning algorithms was also suggested for the detection of lymph node metastases in breast cancer.189
Overall, recent developments support the implementation of immune scores as a new component in the classification of cancer and advocate for the development and use of automatic machine learning. The use of immune scores will likely be extended to a wide variety of tumors, and the application could be extended to predict the development of metastatic disease and even the response to immunotherapy.179,181,190,191
TILs and response to immunotherapy
Immunotherapy, and in particular ICB, is targeted towards the reactivation of TEX cells in tumors. Accordingly, gene profiling of responders includes assessment of (exhausted) T cell signatures, IFN-related genes, enrichment of both immunosuppressive checkpoints and immune signaling of T, B, and NK cells and increased cytokine and chemokine signaling. To elucidate determinants of response, one recent study compared pretreatment biopsies of metastatic melanoma patients responding and not responding to ICB. The gene profile of responders included IFN-related genes and genes related to enrichment of both immunosuppressive checkpoints and immune signaling of T, B, and NK cells and increased cytokine and chemokine signaling. In addition, there was an abundance of TRM cells in ICB responders.192 Complementary research has demonstrated an increased prevalence of TRM cells in treatment-naive tumors versus healthy adjacent tissue, as well as demonstrating that ICB responders are characterized by CD8+ TCM cell accumulation.193 During ICB, these TCM cells develop an effector-like phenotype with a cytolytic gene signature. Characterization of nonresponders revealed increased coexpression of LAG-3, BTLA4, and PD1 during treatment.194
Interestingly, several recent studies have suggested that treatment with anti-PD1 therapies does not necessarily reverse the state of already TEX cells but rather acts on the activation of TPE cells. In human lung cancer, transcriptional analysis of PD1high CD8+ T cells identified that these cells had a low cytolytic capacity but high proliferative function compared to PD1low/negative CD8+ T cells, corresponding with a TPE cell phenotype. PD1high CD8+ T cells were predictive for both survival and response to PD1 blockade and were shown to secrete CXCL13, indicating their involvement in the formation of TLSs (see also corresponding section above).195 This observation is supported by several studies describing that ICB induces a proliferative response of less differentiated CD8+ T cells, whereas TEX cells cannot respond to anti-PD1 therapy.57,58,64 This is supported by a high-dimensional single-cell RNA analysis of melanoma tumors treated with checkpoint inhibition, which identified TCF7+CD8+ T cells in particular to be associated with better tumor regression and overall response in checkpoint-treated patients.60 Taken together, these findings suggest that signature genes consistent with TPE cells may serve as a potential biomarker for ICB response.57,58,60,63,64
In addition to the role of CD8+ T cells and MHC class I, Elspach et al. recently reported on a series of elegant experiments demonstrating the importance of MHC-II during ICB. In their work, mice were challenged with a sarcoma tumor cell line expressing MHC-I and/or MHC-II neoantigens in the absence or presence of administration of ICB. Interestingly, only the mice with functioning MHC-I and MHC-II were able to slow down tumor growth in the absence of ICB and were able to completely reject the tumor with the administration of ICB, thus demonstrating the requirement of MHC-II-mediated THC cell responses for optimal priming of MHC-I-restricted CD8+ cells and their maturation into CTLs. Unresponsiveness to ICB in the presence of a favorable mutational burden could therefore be explained by the lack of MHC-II expression.32 In human tumors, MHC-II expression was also associated with the response to ICB. In addition, low MHC-II expression was associated with primary resistance to ICB.31
ICB does not only affect beneficial antitumor immune cells. In particular, Treg cells are known to express both PD1 and CTLA-4, in addition to GITR, ICOS, and OX40.196,197 In some cases, patients treated with an anti-PD1 antibody develop hyperprogressive disease (HPD). A recent study revealed a markedly increased proliferation of Treg cells in HPD patients, while there was a reduction in Treg cells in patients with no HPD. This suggests that Treg cell depletion before anti-PD1 therapy may help prevent the induction of HPD.197 Accordingly, the success of anti-CTLA-4 treatment seems to be at least partially attributable to the depletion of intratumoral Treg cells.196
Mutational load, immune infiltrates, and immune checkpoint blockade
As mentioned, both the quantity and quality of TILs are likely factors in determining prognostic and therapeutic benefits. In general, the quality of T cell responses is determined by the antigen recognized through their cognate TCR. In a recent study of ICB nonresponders and responders, TCR sequencing from the tumor, normal adjacent tissue and peripheral blood revealed expansion of T cell clones in the periphery, normal adjacent tissue and tumor. Moreover, the expansion of both peripheral and intratumoral T cells was correlated with the response to ICB. This suggests a relationship between peripheral clonal expansion and tumor infiltration. Interestingly, peripherally expanded T cells infiltrating the tumor acquired a TRM-like phenotype during successful ICB responses, reconfirming the observed link between T cell exhaustion and TRM phenotypes. Considering this link, liquid biopsies to identify peripheral expanded T cell clones may help predict ICB response.193 Accordingly, single-cell RNA and TCR sequencing from site-matched tumors after anti-PD1 treatment revealed expansion of CD8+CD39+ T cells, yet these T cell clones did not derive from pre-existing intratumoral T cells, suggesting that they were derived from peripheral T cells.198 Similarly, high TCR clonality but lack of TCR diversity in pretreatment liquid biopsies was associated with longer PFS and good response to PD1 blockade but a poor response to CTLA4 inhibition. Multivariate regression models confirmed both TCR clonality and diversity as independent predictive factors for response.199 Altogether, these studies suggest that TCR specificity and cognate antigens are key determinants of the quality of the TILs and the corresponding ICB response.
During successful ICB, TCR specificity is mainly directed against neoantigens and mutation-induced changes in (generally nondriver) cancer cell proteins. Accordingly, tumor mutational burden is directly related to immune infiltration and is associated with response to ICB. A subset of cancers is characterized by mismatch repair deficiency (dMMR), which leads to the accumulation of mutation-associated neoantigens (MANAs) and TAAs that stimulate the activation, differentiation, and infiltration of TILs and are associated with a better prognosis.200,201 In endometrial cancer, increased immune infiltration and improved clinical outcome are seen in molecular subtypes harboring more mutations, such as POLE-mutant, MSI, and p53-mutant tumors.202–204 Similar results were shown in MSI CRC, which showed increased immune infiltration and a superior prognosis compared with microsatellite-stable (MSS) CRC.205,206 Interestingly, the results of the study presenting a validated colorectal immune score showed better prognostic benefits in highly infiltrated MSI and MSS tumors than poorly infiltrated MSI and MSS tumors. In line with the literature, relatively more MSI tumors than MSS tumors are infiltrated with a high number of TILs, indicating that mutational load does not directly contribute to improved survival but rather that high immune infiltration occurs as a consequence of high mutational load.179
Tumors with a high mutational load and thus increased immune infiltration have a favorable response to ICB. A recent meta-analysis including 939 patients with MSI advanced cancer from 14 studies demonstrated a response rate of 41.5%, a disease control rate of 62.8% and a 1- and 2-year overall survival of 75.6% and 56.5%, respectively.207 Earlier work showed a response rate of 53% and a complete response rate of 21% in ICB-treated dMMR tumors. Moreover, the expansion of MANA-specific T cells in the peripheral blood was documented as early as 2 weeks after the start of treatment.208 The application of neoadjuvant ICB has also been successfully demonstrated in multiple tumors characterized by high mutational burden, such as melanoma and small-cell lung cancer, demonstrating impressive response rates of 78% and 45%, respectively.209,210 In addition, neoadjuvant administration of ICB produces a superior response in MSI CRC, as demonstrated by the 100% pathological response rate in dMMR tumors and only 27% response rate in mismatch repair-proficient tumors.211 Taken together, these data demonstrate that mutational burden is highly predictive of the pathological response to ICB and highlight a role for neoadjuvant ICB administration.209–212
In tumors with low mutational burden, treatment with ICB has relatively poor response rates.213–215 A recent phase II trial compared anti-CTLA4-treated prostate cancer patients with a favorable outcome to those with an unfavorable outcome, demonstrating a gene signature enriched for the IFNγ response and CTL pathways, which was confirmed by the increased immunohistochemistry staining of CD3, CD8, granzyme-B, and PD1 in the favorable cohort. Interestingly, although there was no difference in total mutational burden between the cohorts, 8/9 patients in the favorable cohort showed peripheral T cell expansion in response to TAAs/MANAs, whereas this was only demonstrated in 4/10 patients in the unfavorable cohort. These data demonstrate that despite a low mutational burden, some mutations are capable of inducing antigen-specific T cell responses that facilitate the ICB response.216 To improve the ICB response in tumors with a low mutational burden, strategies to elicit immune induction are being explored, including vaccination, chemotherapy, and radiation strategies that target either T cells alone or a combination of TIL subsets.217–219
Reactivation of T cells coactivates NK cells
Some cosignaling receptors are expressed on both NK cells and T cells and have been of interest due to their therapeutic potential of simultaneously activating both cell types.220–222 For example, TIGIT is expressed on both tumor-infiltrating T cells and NK cells and has been associated with tumor progression. Interestingly, TIGIT expression was found to be significantly higher on intratumoral NK cells than on peritumoral NK cells. In mice, TIGIT blockade was capable of reversing NK cell exhaustion and delaying tumor growth. Moreover, NK cells were proven critical for therapeutic effects with TIGIT blockade, PD1 blockade and combination therapy.220
Another receptor expressed on ~50% of peripheral NK cells and on a subset of activated CD8+ T cells is NKG2A. NKG2A is an inhibitory receptor that binds to HLA-E; upon engagement, NK cells transmit intracellular signals, preventing its activation. In tumor-bearing mice, treatment with the anti-NKG2A antibody monalizumab resulted in both T and NK cell effector functions.221,223 A first-in-human trial of MSS CRC treated with monalizumab and durvalumab demonstrated preliminary activity and manageable toxicity.221 In head and neck cancer, overall response rates to cetuximab improved when it was combined with monalizumab, and the combination resulted in manageable side effects.223 Finally, monotherapy with monalizumab in advanced-stage gynecological malignancies demonstrated no dose-limiting toxicities and manageable adverse events. However, no clinical effects were elicited.222 This could suggest that NKG2A inhibition is more suitable for combination strategies. Overall, these studies indicate an important role of NK cells in the successful application of immunotherapy, advocating the dual targeting of both CD8+ T cells and cytotoxic NK cells.220–222
Adoptive T cell transfer
In addition to immune checkpoint inhibition, adoptive cell therapy (ACT) has emerged as a promising cancer immunotherapy. ACT involves three different strategies using either TILs or genetically modified T cells with novel TCRs or chimeric antigen receptors (CAR-T).224 For the first approach, naturally occurring TILs are harvested, expanded ex vivo, and subsequently transfused back into the patient to induce a robust immune-mediated antitumor response. In addition to TILs, peripherally obtained T cells can be genetically modified in vitro to T cells expressing TCRs that recognize specific TAAs expressed by tumor cells. However, recognition of the TAA by the TCR requires antigen presentation via MHC, which is often downregulated in cancer cells.225 ACT with CAR-T cells circumvents this problem because the CAR molecules, containing an extracellular antigen-binding domain and intracellular signaling and cosignaling domains, possess the properties to facilitate binding of CAR-T cells to their target and subsequently activate CAR-T cells independent of MHC.226 The overall application of ACT with TILs has been mainly investigated in solid tumors, especially in the context of metastatic melanoma, and multiple independent studies have demonstrated durable responses, including in patients resistant to ICB.227,228 In addition, adoptive transfer of autologous lymphocytes specifically targeting somatic mutations has elicited objective responses in gastrointestinal, colon, and breast cancer.229–231 To optimize ACT using TILs, selection of the TIL subtype most suitable for expansion and reinfusion with the highest antitumor response is crucial. A phase II clinical trial in melanoma showed a correlation between the total number of CD8+ T cells and clinical response, whereas nonresponders were characterized by higher percentages of CD4+ TILs. During more in depth analysis into CD8 differentiation memory status, especially the more differentiated effector memory T cells compared to TCM were found in responding patients. effector memory T cells is toch TEM? en TEM is TEM?.232 However, other studies suggested TCM cells as a suitable candidate for ACT as they possess high proliferative capacity.17 More recently, γδ T cells have been of interests because they have been reported to possess effector-like functions.53,233 One study successfully expanded γδ T cells from PBMCs and demonstrated that adoptive cell transfer of these cells in an ovarian carcinoma mouse model was capable of suppressing tumor growth via specific cytotoxic activities.234 However, although generally well tolerated, no impressive therapeutic response was seen in the initial clinical trials.233,235,236 In the context of CAR-T cells, high effectivity has been demonstrated in B cell malignancies targeting CD19. In addition, CAR-T cells have been investigated in multiple myeloma and leukemia. However, the treatment of solid tumors with TAA-specific CAR-T cells has achieved limited success, mostly due to restricted antitumor activity or severe toxicity.237
B cells in the response to immunotherapy
In addition to T cells and NK cells, a subset of B cells, ICOSL+ B cells, emerges after chemotherapy treatment and boosts antitumor immunity. Pretreatment breast cancer samples were enriched with high IL-10 and low complement-receptor 2 (CR2) expression, while postchemotherapy samples showed an increase in ICOSL- and CR2-expressing B cells, but IL-10+CD19+ B cells were dramatically decreased.238 In a mouse model, ICOSL blockade significantly inhibited chemotherapy efficacy and was accompanied by increased Treg cells and decreased levels of cytotoxic CD8+ T and TH1 cells in the tumors.238 Interestingly, Griss et al. observed similar results in melanoma patients, in which tumor-infiltrating CD8+ and CD4+ T cells diminished after anti-CD20 treatment.239 Moreover, ICOSL+ B cells were associated with improved therapeutic efficacy and improved DFS and OS in breast cancer patients receiving neoadjuvant chemotherapy, except the HR+Her2− subtype.238
Pretreatment TIL B cells by themselves are also associated with improved immunotherapy responses and better survival.75,240,241 Helmink et al. observed significantly higher expression levels of B cell-related genes in tumor samples from melanoma and renal cell carcinoma patients who responded to ICB treatment than in those from nonresponding patients. Moreover, increased BCR diversity, clonal expansion, and switched memory B cell signatures were more frequently observed in tumor samples from ICB responders than in those from nonresponders.75 Accordingly, Petitprez et al. found that a B cell lineage signature correlated with improved survival in soft tissue sarcoma patients, regardless of the amount of tumor-infiltrating CD8+ T cells. Furthermore, tumors enriched with B cell lineage signatures exhibited the highest response rate to ICB treatment.241 Comparable results were found in tumor samples of metastatic melanoma patients.240
In short, TIL B cells can suppress antitumor immunity, although accumulating evidence shows that TIL B cells support antitumor immunity and promote immunotherapy responses by acting as APCs, producing high-affinity antibodies and secreting antitumor cytokines. These conflicting observations might be caused by the heterogeneity of B cell subsets as well as the various responses of these B cells to different anticancer treatments.242 Future research and new immunotherapy strategies should focus on TIL B cells and how to exploit plasma B cells to promote lymphocyte infiltration and stimulate cytotoxic T cell activation to increase the antitumor immune response.
Of note, while corticosteroids are often used to treat the side effects of chemotherapy, radiotherapy, and immunotherapy, in addition to other cancer patient comorbidities, in lung squamous cell carcinoma patients, it was recently suggested that GC formation inside TLSs, reflecting a mature (secondary) TLS, could be impaired by corticosteroid treatment.160 Tumors of chemotherapy-naive lung squamous cell carcinoma patients treated with corticosteroids before surgery, either systemically or locally, showed a significantly lower TLS density and size of GCs than those of noncorticosteroid-treated patients. Using a mouse model, the causal role of corticosteroids in TLS impairment was studied. TLSs were induced in mice by intranasal administration of alum, a chemical compound, with or without Ova as an antigen, followed by systemic low-dose dexamethasone treatment. Interestingly, TLS development was observed in both models, but high TLS density and GC formation were only found in mice challenged with Ova antigen, suggesting that GC formation requires antigen-dependent interactions between TLS-residing lymphocytes.160 Supporting their hypothesis, dexamethasone treatment did not affect TLS density but significantly reduced the development of mature GC-bearing TLSs. Altogether, these data suggest that corticosteroids negatively influence TLS maturation in the lungs. The use of corticosteroid treatment in cancer patients deserves further exploration.
Perspectives
It is evident that CD8+ TILs are crucial for an effective antitumor immune response. With the rise of single-cell sequencing, it has become clear that CD8+ T cells are divided into a wide variety of subsets ranging from more naive-like and proliferative to more differentiated and cytolytic immune cells. In particular, CD103+CD39+ TRM cells seem capable of tumor control. Interestingly, CD8+CD103+CD39− bystander T cells, although unspecific for tumor antigens, seem capable of contributing to the antitumor response. In our review, we further emphasize the importance of B cells that promote immune surveillance and improve the ICB response as a single-cell type. But even more when clustered together with T cells in well-organized TLSs, initiated by chemokine CXCL13. TLSs give rise to a highly advanced immune response, reflected by the production of affinity-matured antibodies secreted by plasma B cells and created in GC-harboring TLSs near tumor areas. Interestingly, the presence of a TLS itself is associated with improved clinical outcome and response to ICB; however, when TLSs harbor GC activity, reflecting a mature TLS, this survival benefit is even further increased. Accordingly, proper identification of the presence and location of TILs and their spatial organization and identification of highly advanced immune structures such as TLSs is important to accurately predict prognosis, including the development of metastatic disease or recurrence, and response to ICB. A valuable tool to realize this could be found in automatic digital machine learning. Automatic machine learning enables the assessment of complex TIL composition and localization using multiple defining markers simultaneously, without interobserver variability. The success of ICB, especially anti-PD1 therapy, is undeniable, especially in a subset of solid tumors harboring a high mutational load. This clinical efficacy has been mostly attributed to the reactivation of TEX cells. However, the current literature suggests that anti-PD1 therapy does not necessarily reverse the exhausted state of T cells but rather acts on the activation of TPE cells, as T cell exhaustion seems to be a gradual developing state. Tcf7-expressing TPE cells persist long term, are capable of self-renewal and can eventually differentiate into TEX cells exhibiting high cytolytic capacity. In this overview, we show that TPE-like cells are especially associated with better tumor regression and overall response in ICB-treated patients. We believe that combinatorial immunotherapy regimes are the key to successful optimization of response rates and clinical outcomes of immunotherapy-treated cancer patients. Such strategies include the immune checkpoint inhibitors simultaneously targeting NK cells and T cells that are now entering the clinic, including anti-NKG2A and anti-TIGIT agents. In addition, strategies priming patients prior or during ICB treatment to induce an immune response via vaccination, chemotherapy, or radiotherapy also exist. More research into the recently discovered helper-like innate lymphoid cells might reveal new opportunities for the application of immunotherapy. Finally, we believe that future research should focus on the development of new immunotherapy strategies that can induce and exploit TLS formation.
In conclusion, tumor-infiltrating lymphocytes play a significant role in the tumor immune environment. No individual lymphocyte subset is responsible for tumor immune control; rather, the location, clustering, interplay, and costimulation of all lymphocyte subsets are required for a successful antitumor immune response.
Declarative statement on findings
Tumor-infiltrating lymphocytes play a significant role in the tumor immune environment. Individual lymphocyte subsets are not solely responsible for tumor immune control. Rather, the proper location, clustering, interplay and costimulation of all lymphocyte subsets are required for a successful antitumor immune response.
Author contributions
A.V. and S.T.P. contributed equally to the design and writing of the manuscript. M.B. and H.W.N. designed and supervised the writing of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Sterre T. Paijens, Annegé Vledder
These authors jointly supervised this work: Marco de Bruyn, Hans W. Nijman
References
- 1.Zloza A, Al-Harthi L. Multiple populations of T lymphocytes are distinguished by the level of CD4 and CD8 coexpression and require individual consideration. J. Leukoc. Biol. 2006;79:4–6. doi: 10.1189/jlb.0805455. [DOI] [PubMed] [Google Scholar]
- 2.Parrot T, et al. Transcriptomic features of tumour-infiltrating CD4lowCD8high double positive αβ T cells in melanoma. Sci Rep. 2020;10:5900. doi: 10.1038/s41598-020-62664-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menard LC, et al. Renal cell carcinoma (RCC) tumors display large expansion of double positive (DP) CD4+CD8+ T cells with expression of exhaustion markers. Front Immunol. 2018;9:2728. doi: 10.3389/fimmu.2018.02728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohner P, et al. Double positive CD4+CD8+ T cells are enriched in urological cancers and favor T helper-2 polarization. Front Immunol. 2019;10:622. doi: 10.3389/fimmu.2019.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desfrançois J, et al. Double positive CD4CD8 αβ T cells: a new tumor-reactive population in human melanomas. PLoS One. 2010;5:e8437. doi: 10.1371/journal.pone.0008437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desfrançois J, et al. Increased frequency of nonconventional double positive CD4CD8 αβ T cells in human breast pleural effusions. Int J. Cancer. 2009;125:374–380. doi: 10.1002/ijc.24366. [DOI] [PubMed] [Google Scholar]
- 7.Fischer K, et al. Isolation and characterization of human antigen-specific TCRαβ+ CD4-CD8- double-negative regulatory T cells. Blood. 2005;105:2828–2835. doi: 10.1182/blood-2004-07-2583. [DOI] [PubMed] [Google Scholar]
- 8.Priatel JJ, Utting O, Teh H-S. TCR/self-antigen interactions drive double-negative T cell peripheral expansion and differentiation into suppressor cells. J. Immunol. 2001;167:6188–6194. doi: 10.4049/jimmunol.167.11.6188. [DOI] [PubMed] [Google Scholar]
- 9.Lee JB, et al. Developing allogeneic double-negative T cells as a novel off-the-shelf adoptive cellular therapy for cancer. Clin. Cancer Res. 2019;25:2241–2253. doi: 10.1158/1078-0432.CCR-18-2291. [DOI] [PubMed] [Google Scholar]
- 10.Greenplate AR, et al. Computational immune monitoring reveals abnormal double-negative T cells present across human tumor types. Cancer Immunol. Res. 2019;7:86–99. doi: 10.1158/2326-6066.CIR-17-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamann D, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 1997;186:1407–CD18. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gattinoni L, et al. T memory stem cells in health and disease. Nat Med. 2017;23:18–27. doi: 10.1038/nm.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egelston CA, et al. Human breast tumor-infiltrating CD8+ T cells retain polyfunctionality despite PD-1 expression. Nat Commun. 2018;9:4297. doi: 10.1038/s41467-018-06653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gattinoni L, et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romero P, et al. Four functionally distinct populations of human effector-memory CD8+ T lymphocytes. J. Immunol. 2007;178:4112–4119. doi: 10.4049/jimmunol.178.7.4112. [DOI] [PubMed] [Google Scholar]
- 16.Pagès F, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005;353:2654–2666. doi: 10.1056/NEJMoa051424. [DOI] [PubMed] [Google Scholar]
- 17.Klebanoff CA, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl Acad. Sci. USA. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahmadvand S, et al. Importance of CD45RO+ tumor-infiltrating lymphocytes in post-operative survival of breast cancer patients. Cell Oncol. 2019;42:343–356. doi: 10.1007/s13402-019-00430-6. [DOI] [PubMed] [Google Scholar]
- 19.Cheuk S, et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity. 2017;46:287–300. doi: 10.1016/j.immuni.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zaid A, et al. Chemokine receptor–dependent control of skin tissue–resident memory T cell formation. J. Immunol. 2017;199:2451–2459. doi: 10.4049/jimmunol.1700571. [DOI] [PubMed] [Google Scholar]
- 21.Oja AE, et al. Functional heterogeneity of CD4+ tumor-infiltrating lymphocytes with a resident memory phenotype in NSCLC. Front Immunol. 2018;9:2654. doi: 10.3389/fimmu.2018.02654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y, Shin Y, Kang GH. Prognostic significance of CD103+ immune cells in solid tumor: a systemic review and meta-analysis. Sci. Rep. 2019;9:1–7. doi: 10.1038/s41598-019-40527-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solomon B, et al. Identification of an excellent prognosis subset of human papillomavirus-associated oropharyngeal cancer patients by quantification of intratumoral CD103+ immune cell abundance. Ann. Oncol. Off J. Eur. Soc. Med. Oncol. 2019;30:1638–1646. doi: 10.1093/annonc/mdz271. [DOI] [PubMed] [Google Scholar]
- 24.Mann JE, et al. Analysis of tumor-infiltrating CD103 resident memory T-cell content in recurrent laryngeal squamous cell carcinoma. Cancer Immunol. Immunother. 2019;68:213–20.. doi: 10.1007/s00262-018-2256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu W, Sun R, Chen L, Zheng X, Jiang J. Prognostic significance of resident CD103+CD8+T cells in human colorectal cancer tissues. Acta Histochem. 2019;121:657–663. doi: 10.1016/j.acthis.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Edwards J, et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy-naïve melanoma patients and expand significantly during anti-PD-1 treatment. Clin. Cancer Res. 2018;24:3036–3045. doi: 10.1158/1078-0432.CCR-17-2257. [DOI] [PubMed] [Google Scholar]
- 27.Shields BD, et al. Loss of E-cadherin inhibits CD103 antitumor activity and reduces checkpoint blockade responsiveness in melanoma. Cancer Res. 2019;79:1113–1123. doi: 10.1158/0008-5472.CAN-18-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duhen T, et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018;9:1–13. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simoni Y, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 30.Rosato P. C., et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. 10.1038/s41467-019-08534-1. [DOI] [PMC free article] [PubMed]
- 31.Liu D, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019;25:1916–1927. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alspach E, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marty R, Thompson WK, Salem RM, Zanetti M, Carter H. Evolutionary pressure against MHC class II binding cancer mutations. Cell. 2018;175:416–428.e13. doi: 10.1016/j.cell.2018.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018;18:635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 35.Bevan MJ. Helping the CD8+ T-cell response. Nat. Rev. Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]
- 36.Qiu L, et al. Functionally impaired follicular helper T cells induce regulatory B cells and CD14+ human leukocyte antigen-DR− cell differentiation in non-small cell lung cancer. Cancer Sci. 2018;109:3751–3761. doi: 10.1111/cas.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crotty ST. Follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50:1132–1148. doi: 10.1016/j.immuni.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin L., et al. Insights into the molecular mechanisms of T follicular helper-mediated immunity and pathology. Front Immunol. 9. 10.3389/fimmu.2018.01884/full (2018). [DOI] [PMC free article] [PubMed]
- 39.Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J. Autoimmun. 2018;95:77–99. doi: 10.1016/j.jaut.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 41.Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006;12:5423–5434. doi: 10.1158/1078-0432.CCR-06-0369. [DOI] [PubMed] [Google Scholar]
- 42.Perrone G, et al. Intratumoural FOXP3-positive regulatory T cells are associated with adverse prognosis in radically resected gastric cancer. Eur. J. Cancer. 2008;44:1875–1882. doi: 10.1016/j.ejca.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 43.Jordanova ES, et al. Human leukocyte antigen class I, MHC class I chain-related molecule A, and CD8+/regulatory T-cell ratio: Which variable determines survival of cervical cancer patients? Clin. Cancer Res. 2008;14:2028–2035. doi: 10.1158/1078-0432.CCR-07-4554. [DOI] [PubMed] [Google Scholar]
- 44.Bates GJ, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006;24:5373–5380. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- 45.Sinicrope FA, et al. Intraepithelial effector (CD3+)/regulatory (FoxP3+) T-cell ratio predicts a clinical outcome of human colon carcinoma. Gastroenterology. 2009;137:1270–1279. doi: 10.1053/j.gastro.2009.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sawant DV, et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019;20:724–735. doi: 10.1038/s41590-019-0346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Z, et al. Novel effector phenotype of TIM-3þ Regulatory T cells leads to enhanced suppressive function in head and neck cancer patients. Clin. Cancer Res. 2018;24:4529–4538. doi: 10.1158/1078-0432.CCR-17-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Overacre-Delgoffe AE, et al. Interferon-γ drives treg fragility to promote anti-tumor immunity. Cell. 2017;169:1130–1141.e11. doi: 10.1016/j.cell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017;14:717–734. doi: 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- 51.Morrow E. S., Roseweir A., Edwards J. The role of gamma delta T lymphocytes in breast cancer: a review. Transl. Res. 203, 88–96 (2019). [DOI] [PubMed]
- 52.Xiang Z., Tu W. Dual face of Vγ9Vdδ2-T cells in tumor immunology: Anti- versus pro-tumoral activities. Front. Immunol. 8, 1041 (2017). [DOI] [PMC free article] [PubMed]
- 53.Künkele K-P, et al. Vγ9Vδ2 T cells: can we re-purpose a potent anti-infection mechanism for cancer therapy? Cells. 2020;9:829. doi: 10.3390/cells9040829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rezende RM, et al. γδ T cells control humoral immune response by inducing T follicular helper cell differentiation. Nat Commun. 2018;9:3151. doi: 10.1038/s41467-018-05487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caccamo, N. et al. IL-21 regulates the differentiation of a human γδ T cell subset equipped with B cell helper activity. PLoS One. 7, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0223172 (2012). [DOI] [PMC free article] [PubMed] [Retracted]
- 56.Peters C, et al. TGF-β enhances the cytotoxic activity of Vδ2 T cells. Oncoimmunology. 2019;8:e1522471. doi: 10.1080/2162402X.2018.1522471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller BC, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siddiqui I, et al. Intratumoral Tcf1 + PD-1 + CD8 + T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Usatorre A, et al. Enhanced phenotype definition for precision isolation of precursor exhausted tumor-infiltrating CD8 T cells. Front Immunol. 2020;11:340. doi: 10.3389/fimmu.2020.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sade-Feldman M, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brummelman J, et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med. 2018;215:2520–2535. doi: 10.1084/jem.20180684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He R, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature. 2016;537:412–416. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 63.Guo X, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 64.Yamauchi T, et al. CX3CR1–CD8+ T cells are critical in antitumor efficacy but functionally suppressed in the tumor microenvironment. JCI Insight. 2020;5:e133920. doi: 10.1172/jci.insight.133920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khan O, et al. TOX transcriptionally and epigenetically programs CD8 + T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scott AC, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mann TH, Kaech SM. Tick-TOX, it’s time for T cell exhaustion. Nat. Immunol. 2019;20:1092–1094. doi: 10.1038/s41590-019-0478-y. [DOI] [PubMed] [Google Scholar]
- 68.Canale FP, et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8+ T cells. Cancer Res. 2018;78:115–128. doi: 10.1158/0008-5472.CAN-16-2684. [DOI] [PubMed] [Google Scholar]
- 69.Webb JR, Milne K, Nelson BH. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol. Res. 2015;3:926–935. doi: 10.1158/2326-6066.CIR-14-0239. [DOI] [PubMed] [Google Scholar]
- 70.Djenidi F, et al. CD8 + CD103 + tumor–infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015;194:3475–3486. doi: 10.4049/jimmunol.1402711. [DOI] [PubMed] [Google Scholar]
- 71.Ganesan AP, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017;18:940–950. doi: 10.1038/ni.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berntsson J, Nodin B, Eberhard J, Micke P, Jirström K. Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int J. Cancer. 2016;139:1129–1139. doi: 10.1002/ijc.30138. [DOI] [PubMed] [Google Scholar]
- 73.Cillo AR, et al. Immune landscape of viral- and carcinogen-driven head and neck cancer. Immunity. 2020;52:183–199.e9. doi: 10.1016/j.immuni.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garaud S, et al. Tumor-infiltrating B cells signal functional humoral immune responses in breast cancer. JCI Insight. 2019;4:1–20. doi: 10.1172/jci.insight.129641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Helmink BA, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi JY, et al. Margin-infiltrating CD20+ B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin. Cancer Res. 2013;19:5994–6005. doi: 10.1158/1078-0432.CCR-12-3497. [DOI] [PubMed] [Google Scholar]
- 77.Shalapour S, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature. 2015;521:94–98. doi: 10.1038/nature14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei X, et al. Regulatory B cells contribute to the impaired antitumor immunity in ovarian cancer patients. Tumor Biol. 2016;37:6581–6588. doi: 10.1007/s13277-015-4538-0. [DOI] [PubMed] [Google Scholar]
- 79.Xiao X, et al. PD-1hi identifies a novel regulatory b-cell population in human hepatoma that promotes disease progression. Cancer Discov. 2016;6:546–559. doi: 10.1158/2159-8290.CD-15-1408. [DOI] [PubMed] [Google Scholar]
- 80.Zhou X, Su YX, Lao XM, Liang YJ, Liao GQ. CD19+IL-10+ regulatory B cells affect survival of tongue squamous cell carcinoma patients and induce resting CD4+ T cells to CD4+Foxp3+ regulatory T cells. Oral. Oncol. 2016;53:27–35. doi: 10.1016/j.oraloncology.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 81.Dieu-Nosjean M-C, et al. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol. Rev. 2016;271:260–275. doi: 10.1111/imr.12405. [DOI] [PubMed] [Google Scholar]
- 82.Sautès-Fridman C., Petitprez F., Calderaro J., Fridman W. H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer19, 307–325 (2019). [DOI] [PubMed]
- 83.Hu X, et al. Landscape of B cell immunity and related immune evasion in human cancers. Nat. Genet. 2019;51:1068. doi: 10.1038/s41588-019-0437-4. [DOI] [PubMed] [Google Scholar]
- 84.Wouters M. C. A., Nelson B. H. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin. Cancer Res. 24, 6125–6135. http://www.ncbi.nlm.nih.gov/pubmed/30049748 (2018). [DOI] [PubMed]
- 85.Ana M, Avalos HLP. Early BCR events and antigen capture, processing, and loading on MHC class II on B Cells. Front Immunol. 2014;5:92. doi: 10.3389/fimmu.2014.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hua Z, Hou B. The role of B cell antigen presentation in the initiation of CD4 + T cell response. Immunol. Rev. 2020;296:1–12. doi: 10.1111/imr.12859. [DOI] [PubMed] [Google Scholar]
- 87.Hong S, et al. B cells are the dominant antigen-presenting cells that activate naive CD4+ T cells upon immunization with a virus-derived nanoparticle antigen. Immunity. 2018;49:695–708.e4. doi: 10.1016/j.immuni.2018.08.012. [DOI] [PubMed] [Google Scholar]
- 88.Bruno TC, et al. Antigen-presenting intratumoral B cells affect CD4+ TIL phenotypes in non–small cell lung cancer patients. Cancer Immunol. Res. 2017;5:898–907. doi: 10.1158/2326-6066.CIR-17-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jones H., Wang Y., Aldridge B., Archive J. W.-C. I. Lung and splenic B cells facilitate diverse effects on in vitro measures of antitumor immune responses. AACR. https://cancerimmunolres.aacrjournals.org/content/canimmarch/8/1/4.short (2008). [PMC free article] [PubMed]
- 90.Kroeger DR, Milne K, Nelson BH. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res. 2016;22:3005–3015. doi: 10.1158/1078-0432.CCR-15-2762. [DOI] [PubMed] [Google Scholar]
- 91.Andreu P, et al. FcRγ activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17:121–134. doi: 10.1016/j.ccr.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev. Immunol. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 93.Cai X., Zhang L., Wei W. Regulatory B cells in inflammatory diseases and tumor. Int. Immunopharmacol. 67, 281–286 (2019). [DOI] [PubMed]
- 94.Madan R, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J. Immunol. 2009;183:2312–2320. doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang X., et al. T follicular helper cells and regulatory B cells dynamics in systemic lupus erythematosus. PLoS One. 9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3925141/ (2014) [DOI] [PMC free article] [PubMed]
- 96.Daien CI, et al. Regulatory B10 cells are decreased in patients with rheumatoid arthritis and are inversely correlated with disease activity. Arthritis Rheumatol. 2014;66:2037–2046. doi: 10.1002/art.38666. [DOI] [PubMed] [Google Scholar]
- 97.Lee-Chang C, et al. Myeloid-derived suppressive cells promote B cell-mediated immunosuppression via transfer of PD-L1 in glioblastoma. Cancer. Immunol. Res. 2019;7:1928–1943. doi: 10.1158/2326-6066.CIR-19-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Olkhanud PB, et al. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4+ T cells to T-regulatory cells. Cancer Res. 2011;71:3505–3515. doi: 10.1158/0008-5472.CAN-10-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang W, et al. CD19+CD24hiCD38hi Bregs involved in downregulate helper T cells and upregulate regulatory T cells in gastric cancer. Oncotarget. 2015;6:33486–33499. doi: 10.18632/oncotarget.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Du Y., Wei Y. Therapeutic potential of natural killer cells in gastric cancer. Front. Immunol. 10, 3095 (2019). [DOI] [PMC free article] [PubMed]
- 101.Angka L, et al. Natural killer cell IFNγ secretion is profoundly suppressed following colorectal cancer surgery. Ann. Surg. Oncol. 2018;25:3747–3754. doi: 10.1245/s10434-018-6691-3. [DOI] [PubMed] [Google Scholar]
- 102.Hoogstad-Van Evert J. S., et al. Harnessing natural killer cells for the treatment of ovarian cancer. 10.1016/j.ygyno.2020.03.020 (2020). [DOI] [PubMed]
- 103.Martín-Fontecha A, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nat. Immunol. 2004;5:1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 104.Böttcher JP, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172:1022–1037.e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cui F, Qu D, Sun R, Nan K. Circulating CD16+CD56+ nature killer cells indicate the prognosis of colorectal cancer after initial chemotherapy. Med Oncol. 2019;36:84. doi: 10.1007/s12032-019-1307-8. [DOI] [PubMed] [Google Scholar]
- 106.Peng LS, et al. Tumor-associated monocytes/macrophages impair NK-cell function via TGFβ1 in human gastric cancer. Cancer Immunol. Res. 2017;5:248–256. doi: 10.1158/2326-6066.CIR-16-0152. [DOI] [PubMed] [Google Scholar]
- 107.Vivier, E. et al. Innate Lymphoid Cells: 10 Years On. Cell174, 1054–1066 (2018). [DOI] [PubMed]
- 108.Cortez, V. S., Colonna, M. Diversity and function of group 1 innate lymphoid cells. Immunol. Lett. 179, 19–24 (2016). [DOI] [PMC free article] [PubMed]
- 109.Gao Y, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017;18:1004–1015. doi: 10.1038/ni.3800. [DOI] [PubMed] [Google Scholar]
- 110.Cortez VS, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat. Immunol. 2017;18:995–1003. doi: 10.1038/ni.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hawke LG, Mitchell BZ, Ormiston ML. TGF-β and IL-15 synergize through MAPK pathways to drive the conversion of human NK cells to an innate lymphoid cell 1–like phenotype. J. Immunol. 2020;24:ji1900866. doi: 10.4049/jimmunol.1900866. [DOI] [PubMed] [Google Scholar]
- 112.Ercolano, G. et al. Immunosuppressive mediators impair proinflammatory innate lymphoid cell function in human malignant melanoma. Cancer Immunol Res. http://www.ncbi.nlm.nih.gov/pubmed/32019778 (2020). [DOI] [PubMed]
- 113.Moral JA, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature. 2020;579:130–135. doi: 10.1038/s41586-020-2015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Demaria O, Vivier E. Immuno-oncology beyond TILs: unleashing TILCs. Cancer Cell. 2020;37:428–430. doi: 10.1016/j.ccell.2020.03.021. [DOI] [PubMed] [Google Scholar]
- 115.Saranchova I, et al. Type 2 innate lymphocytes actuate immunity against tumours and limit cancer metastasis. Sci. Rep. 2018;8:1–17. doi: 10.1038/s41598-018-20608-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Trabanelli S, et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat. Commun. 2017;8:1–14. doi: 10.1038/s41467-017-00678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rauber S, et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 2017;23:938–944. doi: 10.1038/nm.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Carrega P, et al. NCR + ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015;6:8280. doi: 10.1038/ncomms9280. [DOI] [PubMed] [Google Scholar]
- 119.Koh J, et al. IL23-producing human lung cancer cells promote tumor growth via conversion of innate lymphoid cell 1 (ILC1) into ILC3. Clin. Cancer Res. 2019;25:4026–4037. doi: 10.1158/1078-0432.CCR-18-3458. [DOI] [PubMed] [Google Scholar]
- 120.Irshad S, et al. RORγt+ innate lymphoid cells promote lymph node metastasis of breast cancers. Cancer Res. 2017;77:1083–1096. doi: 10.1158/0008-5472.CAN-16-0598. [DOI] [PubMed] [Google Scholar]
- 121.Fridman, W. H., Pagès, F., Saut̀s-Fridman, C., Galon, J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 12, 298–306 (2012). [DOI] [PubMed]
- 122.Tsakiroglou AM, et al. Spatial proximity between T and PD-L1 expressing cells as a prognostic biomarker for oropharyngeal squamous cell carcinoma. Br. J. Cancer. 2020;122:539–544. doi: 10.1038/s41416-019-0634-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bouzin C, Brouet A, De Vriese J, DeWever J, Feron O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J. Immunol. 2007;178:1505–1511. doi: 10.4049/jimmunol.178.3.1505. [DOI] [PubMed] [Google Scholar]
- 124.Buckanovich RJ, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008;14:28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- 125.Shrimali RK, et al. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–6180. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yu JS, et al. Intratumoral T cell subset ratios and Fas ligand expression on brain tumor endothelium. J. Neurooncol. 2003;64:55–61. doi: 10.1007/BF02700020. [DOI] [PubMed] [Google Scholar]
- 127.Bajou K, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14:324–334. doi: 10.1016/j.ccr.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Motz GT, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014;20:607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Valkenburg, K. C., De Groot, A. E., Pienta, K. J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol.15, 366–381 (2018). [DOI] [PMC free article] [PubMed]
- 130.Bazzichetto, C. et al. Advances in tumor-stroma interactions: emerging role of cytokine network in colorectal and pancreatic cancer. J. Oncol. 2019. https://www.ncbi.nlm.nih.gov/pubmed/31191652 (2019). [DOI] [PMC free article] [PubMed]
- 131.Di Caro G, et al. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. 2014;20:2147–2158. doi: 10.1158/1078-0432.CCR-13-2590. [DOI] [PubMed] [Google Scholar]
- 132.Graham DM, Appelman HD. Crohn’s-like lymphoid reaction and colorectal carcinoma: a potential histologic prognosticator. Mod. Pathol. 1990;3:332–335. [PubMed] [Google Scholar]
- 133.Pagès F, Galon J, Fridman WH. The essential role of the in situ immune reaction in human colorectal cancer. J. Leukoc. Biol. 2008;84:981–987. doi: 10.1189/jlb.1107773. [DOI] [PubMed] [Google Scholar]
- 134.Thaunat O, et al. Chronic rejection triggers the development of an aggressive intragraft immune response through recapitulation of lymphoid organogenesis. J. Immunol. 2010;185:717–728. doi: 10.4049/jimmunol.0903589. [DOI] [PubMed] [Google Scholar]
- 135.Lee RS, et al. Indirect recognition of allopeptides promotes the development of cardiac allograft vasculopathy. Proc. Natl Acad. Sci. USA. 2001;98:3276–3281. doi: 10.1073/pnas.051584498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Neyt K., Perros F., GeurtsvanKessel C. H., Hammad H., Lambrecht B. N. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol.33, 297–305. 10.1016/j.it.2012.04.006 (2012). [DOI] [PMC free article] [PubMed]
- 137.Lee Y, et al. Recruitment and activation of naive T cells in the islets by lymphotoxin β receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 138.Cabrita R, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 139.Meier D, et al. Ectopic lymphoid-organ development occurs through interleukin 7-mediated enhanced survival of lymphoid-tissue-inducer cells. Immunity. 2007;26:643–654. doi: 10.1016/j.immuni.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 140.Furtado GC, et al. Lymphotoxin β receptor signaling is required for inflammatory lymphangiogenesis in the thyroid. Proc. Natl. Acad. Sci. USA. 2007;104:5026–5031. doi: 10.1073/pnas.0606697104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peters A, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011;35:986–996. doi: 10.1016/j.immuni.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Deteix C, et al. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J. Immunol. 2010;184:5344–5351. doi: 10.4049/jimmunol.0902999. [DOI] [PubMed] [Google Scholar]
- 143.Guedj K, et al. M1 macrophages act as LTβR-independent lymphoid tissue inducer cells during atherosclerosis-related lymphoid neogenesis. Cardiovasc Res. 2014;101:434–443. doi: 10.1093/cvr/cvt263. [DOI] [PubMed] [Google Scholar]
- 144.Lochner M, et al. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORγt and LTi cells. J. Exp. Med. 2011;208:125–134. doi: 10.1084/jem.20100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vondenhoff MF, et al. LTβR signaling induces cytokine expression and up-regulates lymphangiogenic factors in lymph node anlagen. J. Immunol. 2009;182:5439–5445. doi: 10.4049/jimmunol.0801165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Martinet L, et al. High endothelial venules (HEVs) in human melanoma lesions: major gateways for tumor-infiltrating lymphocytes. Oncoimmunology. 2012;1:829–839. doi: 10.4161/onci.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Streeter PR, Rouse BT, Butcher EC. Immunohistologic and functional characterization of a vascular addressin involved in lymphocyte homing into peripheral lymph nodes. J. Cell Biol. 1988;107:1853–1862. doi: 10.1083/jcb.107.5.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nurieva RI, et al. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schmitt N, et al. The cytokine TGF-β 2 co-opts signaling via STAT3-STAT4 to promote the differentiation of human T FH cells. Nat. Immunol. 2014;15:856–865. doi: 10.1038/ni.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Locci M, et al. Activin A programs the differentiation of human T FH cells. Nat. Immunol. 2016;17:976–984. doi: 10.1038/ni.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nurieva RI, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Karnowski A, et al. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J. Exp. Med. 2012;209:2049–2064. doi: 10.1084/jem.20111504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rasheed AU, Rahn HP, Sallusto F, Lipp M, Müller G. Follicular B helper T cell activity is confined to CXCR5hiICOShi CD4 T cells and is independent of CD57 expression. Eur. J. Immunol. 2006;36:1892–1903. doi: 10.1002/eji.200636136. [DOI] [PubMed] [Google Scholar]
- 154.Heesters BA, et al. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity. 2013;38:1164–1175. doi: 10.1016/j.immuni.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Garin A, et al. Toll-like receptor 4 signaling by follicular dendritic cells is pivotal for germinal center onset and affinity maturation. Immunity. 2010;33:84–95. doi: 10.1016/j.immuni.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 156.Cazac BB, Roes J. TGF-β receptor controls B cell responsiveness and induction of IgA in vivo. Immunity. 2000;13:443–451. doi: 10.1016/s1074-7613(00)00044-3. [DOI] [PubMed] [Google Scholar]
- 157.Dieu-Nosjean MC, Goc J, Giraldo NA, Sautès-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014;35:571–580. doi: 10.1016/j.it.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 158.Posch F, et al. Maturation of tertiary lymphoid structures and recurrence of stage II and III colorectal cancer. Oncoimmunology. 2018;7:e1378844. doi: 10.1080/2162402X.2017.1378844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Yamaguchi K, et al. Helper T cell-dominant tertiary lymphoid structures are associated with disease relapse of advanced colorectal cancer. Oncoimmunology. 2020;9:1724763. doi: 10.1080/2162402X.2020.1724763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Silina K, et al. Germinal centers determine the prognostic relevance of tertiary lymphoid structures and are impaired by corticosteroids in lung squamous cell carcinoma. Cancer Res. 2018;78:1308–1320. doi: 10.1158/0008-5472.CAN-17-1987. [DOI] [PubMed] [Google Scholar]
- 161.Germain C, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 2014;189:832–844. doi: 10.1164/rccm.201309-1611OC. [DOI] [PubMed] [Google Scholar]
- 162.Bindea G, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 163.Schürch, C. M. et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. bioRxiv. 743989 (2019). [DOI] [PMC free article] [PubMed]
- 164.Ansel KM, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406:309–314. doi: 10.1038/35018581. [DOI] [PubMed] [Google Scholar]
- 165.Thommen DS, et al. A transcriptionally and functionally distinct pd-1 + cd8 + t cell pool with predictive potential in non-small-cell lung cancer treated with pd-1 blockade. Nat. Med. 2018;24:994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Workel HH, et al. A transcriptionally distinct CXCL13þCD103þCD8þ T-cell population is associated with b-cell recruitment and neoantigen load in human cancer. Cancer Immunol. Res. 2019;7:784–796. doi: 10.1158/2326-6066.CIR-18-0517. [DOI] [PubMed] [Google Scholar]
- 167.Barnes, T. A., Amir, E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br. J. Cancer. 117, 451–460 (2017). [DOI] [PMC free article] [PubMed]
- 168.Hwang C, et al. Stromal tumor-infiltrating lymphocytes evaluated on H&E-stained slides are an independent prognostic factor in epithelial ovarian cancer and ovarian serous carcinoma. Oncol. Lett. 2019;17:4557–4565. doi: 10.3892/ol.2019.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.James FR, et al. Association between tumour infiltrating lymphocytes, histotype and clinical outcome in epithelial ovarian cancer. BMC Cancer. 2017;17:657. doi: 10.1186/s12885-017-3585-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Salgado R, et al. Tumor-infiltrating lymphocytes and associations with pathological complete response and event-free survival in HER2-positive early-stage breast cancer treated with lapatinib and trastuzumab: a secondary analysis of the NeoALTTO trial. JAMA Oncol. 2015;1:448–455. doi: 10.1001/jamaoncol.2015.0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Luen SJ, et al. Tumour-infiltrating lymphocytes in advanced HER2-positive breast cancer treated with pertuzumab or placebo in addition to trastuzumab and docetaxel: a retrospective analysis of the CLEOPATRA study. Lancet Oncol. 2017;18:52–62. doi: 10.1016/S1470-2045(16)30631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Denkert C, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015;33:983–991. doi: 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 173.Rim SKim, Stromal, et al. Tumor-infiltrating lymphocytes in NRG oncology/NSABP B-31 adjuvant trial for early-stage HER2-positive breast cancer. J. Natl Cancer Inst. 2019;111:867–871. doi: 10.1093/jnci/djz032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fuchs TL, et al. Assessment of tumor-infiltrating lymphocytes using international TILs working group (ITWG) system is a strong predictor of overall survival in colorectal carcinoma. Am. J. Surg. Pathol. 2020;44:536–544. doi: 10.1097/PAS.0000000000001409. [DOI] [PubMed] [Google Scholar]
- 175.Hendry, S. et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immunooncology biomarkers working group: Part 1: assessing the host immune response, TILs in invasive breast carcinoma and ductal carcinoma in situ, metastatic tumor deposits and areas for further research. Adv Anatomic Pathol. 24, 235–251 (2017). [DOI] [PMC free article] [PubMed]
- 176.Hendry, S. et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immuno-oncology biomarkers working group: Part 2: TILs in melanoma, gastrointestinal tract carcinom. Adv. Anatomic Pathol. 24, 311–335 (2017). [DOI] [PMC free article] [PubMed]
- 177.The Royal College of Pathologists of Australasia. Colorectal Cancer, Structured Reporting Protocol. (3rd ed). 1–77. (The Royal College of Pathologists of Australasia, 2016).
- 178.Cui Y, Zhang G, Liu Z, Xiong Z, Hu J. A deep learning algorithm for one-step contour aware nuclei segmentation of histopathology images. Med Biol. Eng. Comput. 2019;57:2027–2043. doi: 10.1007/s11517-019-02008-8. [DOI] [PubMed] [Google Scholar]
- 179.Pagès F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391:2128–2139. doi: 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 180.Mlecnik B, et al. Comprehensive intrametastatic immune quantification and major impact of immunoscore on survival. J. Natl Cancer Inst. 2018;110:97–108. doi: 10.1093/jnci/djx123. [DOI] [PubMed] [Google Scholar]
- 181.Zhou C, et al. Development and validation of a seven-immune-feature-based prognostic score for oral squamous cell carcinoma after curative resection. Int. J. Cancer. 2020;146:1152–1163. doi: 10.1002/ijc.32571. [DOI] [PubMed] [Google Scholar]
- 182.Shaban M, et al. A novel digital score for abundance of tumour infiltrating lymphocytes predicts disease free survival in oral squamous cell carcinoma. Sci. Rep. 2019;9:1–13. doi: 10.1038/s41598-019-49710-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zeng D, et al. Gene expression profiles for a prognostic immunoscore in gastric cancer. Br. J. Surg. 2018;105:1338–1348. doi: 10.1002/bjs.10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Li XD, et al. Prognostic role of the immunoscore for patients with urothelial carcinoma of the bladder who underwent radical cystectomy. Ann. Surg. Oncol. 2019;26:4148–4156. doi: 10.1245/s10434-019-07529-y. [DOI] [PubMed] [Google Scholar]
- 185.Bychkov D, et al. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci. Rep. 2018;8:1–11. doi: 10.1038/s41598-018-21758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ronneberger, O., Fischer, P., Brox, T. U-Net: Convolutional Networks for Biomedical Image Segmentation. http://lmb.informatik.uni-freiburg.de/.
- 187.Kurc T, et al. Segmentation and classification in digital pathology for glioma research: challenges and deep learning approaches. Front Neurosci. 2020;14:27. doi: 10.3389/fnins.2020.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Coudray, N., Santiago Ocampo, P. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat Med. 10.1038/s41591-018-0177-5. [DOI] [PMC free article] [PubMed]
- 189.Bejnordi BE, et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA. 2017;318:2199–2210. doi: 10.1001/jama.2017.14585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jiang Y, et al. ImmunoScore Signature. Ann. Surg. 2018;267:504–513. doi: 10.1097/SLA.0000000000002116. [DOI] [PubMed] [Google Scholar]
- 191.Tahkola K, et al. High immune cell score predicts improved survival in pancreatic cancer. Virchows Arch. 2018;472:653–665. doi: 10.1007/s00428-018-2297-1. [DOI] [PubMed] [Google Scholar]
- 192.Gide TN, et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell. 2019;35:238–255.e6. doi: 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 193.Wu TD, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579:274–278. doi: 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 194.Araujo B, et al. Common phenotypic dynamics of tumor-infiltrating lymphocytes across different histologies upon checkpoint inhibition: impact on clinical outcome. Cytotherapy. 2020;22:204–213. doi: 10.1016/j.jcyt.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 195.Thommen DS, et al. A transcriptionally and functionally distinct pd-1 + cd8 + t cell pool with predictive potential in non-small-cell lung cancer treated with pd-1 blockade. Nat. Med. 2018;24:994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Arce Vargas F, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. 2018;33:649–663.e4. doi: 10.1016/j.ccell.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kamada T, et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl Acad. Sci. USA. 2019;116:9999–10008. doi: 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yost KE, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019;25:1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hogan, S. A. et al. Peripheral blood TCR repertoire profiling may facilitate patient stratification for immunotherapy against melanoma. http://www.imgt.org (2019). [DOI] [PubMed]
- 200.Rizvi NA, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.McGranahan N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Eggink FA, et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Oncoimmunology. 2017;6:e1264565. doi: 10.1080/2162402X.2016.1264565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yamashita H, et al. Microsatellite instability is a biomarker for immune checkpoint inhibitors in endometrial cancer. Oncotarget. 2018;9:5652–5664. doi: 10.18632/oncotarget.23790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Jones NL, Xiu J, Rocconi RP, Herzog TJ, Winer IS. Immune checkpoint expression, microsatellite instability, and mutational burden: Identifying immune biomarker phenotypes in uterine cancer. Gynecol. Oncol. 2020;156:393–399. doi: 10.1016/j.ygyno.2019.11.035. [DOI] [PubMed] [Google Scholar]
- 205.Maby P, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: A rationale for personalized immunotherapy. Cancer Res. 2015;75:3446–3455. doi: 10.1158/0008-5472.CAN-14-3051. [DOI] [PubMed] [Google Scholar]
- 206.Gryfe R, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000;342:69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- 207.Petrelli F., Ghidini M., Ghidini A., Tomasello G. Outcomes following immune checkpoint inhibitor treatment of patients with microsatellite instability-high cancers. JAMA Oncol. https://jamanetwork.com/journals/jamaoncology/fullarticle/2765752 (2020). [DOI] [PMC free article] [PubMed]
- 208.Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Forde PM, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018;378:1976–1986. doi: 10.1056/NEJMoa1716078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Blank CU, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018;24:1655–1661. doi: 10.1038/s41591-018-0198-0. [DOI] [PubMed] [Google Scholar]
- 211.Chalabi M, et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020;26:566–576. doi: 10.1038/s41591-020-0805-8. [DOI] [PubMed] [Google Scholar]
- 212.Hellmann MD, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. 2018;33:843–852.e4. doi: 10.1016/j.ccell.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Boland JL, et al. Early disease progression and treatment discontinuation in patients with advanced ovarian cancer receiving immune checkpoint blockade. Gynecol. Oncol. 2019;152:251–258. doi: 10.1016/j.ygyno.2018.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Adams S, et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019;30:397–404. doi: 10.1093/annonc/mdy517. [DOI] [PubMed] [Google Scholar]
- 215.Beer TM, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J. Clin. Oncol. 2017;35:40–47. doi: 10.1200/JCO.2016.69.1584. [DOI] [PubMed] [Google Scholar]
- 216.Subudhi SK, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci. Transl. Med. 2020;12:eaaz3577. doi: 10.1126/scitranslmed.aaz3577. [DOI] [PubMed] [Google Scholar]
- 217.Adams S, et al. Atezolizumab plus nab-paclitaxel in the treatment of metastatic triple-negative breast cancer with 2-year survival follow-up. JAMA Oncol. 2019;5:334. doi: 10.1001/jamaoncol.2018.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Voorwerk L, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat. Med. 2019;25:920–928. doi: 10.1038/s41591-019-0432-4. [DOI] [PubMed] [Google Scholar]
- 219.Keskin DB, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–239. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhang Q, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018;19:723–732. doi: 10.1038/s41590-018-0132-0. [DOI] [PubMed] [Google Scholar]
- 221.Van Hall, T. et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J. ImmunoTher. Cancer. 7, 263 (2019). [DOI] [PMC free article] [PubMed]
- 222.Tinker AV, et al. Dose-ranging and cohort-expansion study of monalizumab (IPH2201) in patients with advanced gynecologic malignancies: a trial of the canadian cancer trials group (CCTG): IND221. Clin. Cancer Res. 2019;25:6052–6060. doi: 10.1158/1078-0432.CCR-19-0298. [DOI] [PubMed] [Google Scholar]
- 223.André P, et al. Anti-NKG2A mAb Is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175:1731–1743.e13. doi: 10.1016/j.cell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rohaan, M. W., Wilgenhof, S., Haanen, J. B. A. G. Adoptive cellular therapies: the current landscape. Virchows Archiv. 474, 449–461 /pmc/articles/PMC6447513/?report=abstract (2019). [DOI] [PMC free article] [PubMed]
- 225.Garrido, F., Ruiz-Cabello, F., Aptsiauri, N. Rejection versus escape: the tumor MHC dilemma. Cancer Immunol. Immunother. 66, 259–271. https://pubmed.ncbi.nlm.nih.gov/28040849/ (2017). [DOI] [PMC free article] [PubMed]
- 226.Guedan, S., Calderon, H., Posey, A. D., Maus, M. V. Engineering and design of chimeric antigen receptors. Mol. Ther.12, 145–156. 10.1016/j.omtm.2018.12.009 (2019). [DOI] [PMC free article] [PubMed]
- 227.Met, Ö., Jensen, K. M., Chamberlain, C. A., Donia, M., Svane, I. M. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 41, 49–58. 10.1007/s00281-018-0703-z (2019). [DOI] [PubMed]
- 228.Chandran SS, Klebanoff CA. T cell receptor‐based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol. Rev. 2019;290:127–147. doi: 10.1111/imr.12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tran E, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tran E, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016;3752:255–262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zacharakis N, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018;24:724–730. doi: 10.1038/s41591-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Radvanyi LG, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012;18:6758–6770. doi: 10.1158/1078-0432.CCR-12-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Joalland, N., Scotet, E. Emerging challenges of preclinical models of anti-tumor immunotherapeutic strategies utilizing Vγ9Vδ2 T Cells. Front Immunol. 11, 992 https://pubmed.ncbi.nlm.nih.gov/32528477/ (2020). [DOI] [PMC free article] [PubMed]
- 234.Mao TL, et al. Ex vivo expanded human Vγ9vδ2 T-cells can suppress epithelial ovarian cancer cell growth. Int J Mol Sci. 2019;20:1139. doi: 10.3390/ijms20051139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Dieli F, et al. Targeting human γδ T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007;67:7450–7457. doi: 10.1158/0008-5472.CAN-07-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Meraviglia S, et al. In vivo manipulation of V9V2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin. Exp. Immunol. 2010;161:290–297. doi: 10.1111/j.1365-2249.2010.04167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S., Milone, M. C. CAR T cell immunotherapy for human cancer. Science. 359, 1361–1365. http://science.sciencemag.org/ (2018). [DOI] [PubMed]
- 238.Lu Y, et al. Complement signals determine opposite effects of B cells in chemotherapy-induced immunity. Cell. 2020;180:1081–1097.e24. doi: 10.1016/j.cell.2020.02.015. [DOI] [PubMed] [Google Scholar]
- 239.Griss J, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019;10:1–14. doi: 10.1038/s41467-019-12160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Cabrita R, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 241.Petitprez F, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–60.. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 242.Liu Z, Fu YX. Chemotherapy induces cancer-fighting B cells. Cell. 2020;180:1037–1039. doi: 10.1016/j.cell.2020.02.046. [DOI] [PubMed] [Google Scholar]