

Abstract
Impaired glutamate homeostasis is a key characteristic of the neurobiology of drug addiction in rodent models and contributes to the vulnerability to relapse to drug seeking. Although disrupted astrocytic and presynaptic regulation of glutamate release has been considered to constitute with impaired glutamate homeostasis in rodent model of drug relapse, the involvement of endocannabinoids (eCBs) in this neurobiological process has remained largely unknown. Here, using cocaine self-administration in rats, we investigated the role of endocannabinoids in impaired glutamate homeostasis in the core of nucleus accumbens (NAcore), which was indicated by augmentation of spontaneous synaptic glutamate release, downregulation of metabotropic glutamate receptor 2/3 (mGluR2/3), and mGluR5-mediated astrocytic glutamate release. We found that the endocannabinoid, anandamide (AEA), rather than 2-arachidonoylglycerol elicited glutamate release through presynaptic transient receptor potential vanilloid 1 (TRPV1) and astrocytic cannabinoid type-1 receptors (CB1Rs) in the NAcore of saline-yoked rats. In rats with a history of cocaine self-administration and extinction training, AEA failed to alter synaptic glutamate release in the NAcore, whereas CB1R-mediated astrocytic glutamate release by AEA remained functional. In order to induce increased astrocytic glutamate release via exogenous AEA, (R)-methanandamide (methAEA, a metabolically stable form of AEA) was chronically infused in the NAcore via osmotic pumps during extinction training. Restoration of mGluR2/3 function and mGluR5-mediated astrocytic glutamate release was observed after chronic methAEA infusion. Additionally, priming or cue-induced reinstatement of cocaine seeking was inhibited in methAEA-infused rats. These results demonstrate that enhancing endocannabinoid signaling is a potential pathway to restore glutamate homeostasis and may represent a promising therapeutic strategy for preventing cocaine relapse.
Subject terms: Addiction, Neurotransmitters
Introduction
Glutamate homeostasis represents the balance between glutamate release and elimination and depends on neuron-glia interactions. Impaired glutamate homeostasis in the core of nucleus accumbens (NAcore) contributes to neurobiological processes in drug self-administration and underlies biological vulnerability to drug relapse in rodent models [1, 2]. Depletion of extrasynaptic glutamate and downregulated group-II metabotropic glutamate receptor 2/3 (mGluR2/3) have been observed after chronic cocaine administration [3]. Also, heroin self-administration produces long-lasting downregulation of glutamate reuptake and surface expression of glutamate transporter 1 (GLT-1) [4], which is associated with enhancing spillover of synaptic glutamate to stimulate extrasynaptic N-methyl-D-aspartate receptors (NMDARs) within the NAcore [4]. Alteration in prefrontal-cortex (PFc) glutamatergic top-down projections to the NAc has been demonstrated to be crucial for reinstatement of drug-seeking behavior [5, 6]. Withdrawal from cocaine downregulates presynaptic mGluR2/3 autoreceptors [1], which is followed by a concomitant increase in PFc-to-NAc glutamatergic synapse release probability [7]. Depletion of extrasynaptic glutamate in the NAc may further increase the signal-to-noise ratio of spillover synaptic glutamate. In consequence, robust transient synaptic glutamate release during reinstatement may be amplified by insufficient glutamate elimination and dysfunctional mGluR2/3 autoreceptors in the NAc after drug exposure [1, 8, 9].
The principal endocannabinoids (eCBs) are 2-arachidonoylglycerol (2-AG) and anandamide (N-arachidonoylethanolamine, AEA), which primarily modulate synaptic transmission and plasticity via retrograde signaling to presynaptic cannabinoid type-1 receptor (CB1R) [10, 11]. In addition to activating CB1Rs, AEA also activates transient receptor potential vanilloid 1 (TRPV1), which serves as ionotropic cannabinoid signaling and modulates presynaptic release in the CNS [12, 13]. Both 2-AG and AEA are produced in both neurons and astrocytes [14] and are hydrolyzed by monoacylglycerol lipase (MAGL) and fatty acid amide hydrolase (FAAH), respectively [15]. The endocannabinoid system is considered to be involved in brain reward signaling, although some of the results supporting this hypothesis remain controversial [16, 17]. AEA-induced activation of CB1R increases extracellular dopaminergic levels in the NAc [18], whereas CB1R agonists or eCBs inhibit optogenetic-activation-induced dopaminergic release [19]. CB1R antagonist AM251 inhibited cocaine self-administration [20] and reinstatement of cocaine seeking [21], while CB1R agonist WIN 55,212-2 also decreased cocaine self-administration [22].
Recent evidence suggests that endocannabinoid signaling may impact neurotransmission via densely expressed CB1R on astrocytes. In the hippocampus, activation of CB1R promotes astrocytic glutamate release and induces astrocytic calcium elevation through Gq/11 protein activation of phospholipase C, and then promotes astrocytic glutamate release that can be monitored via measuring slow inward currents (SICs) in adjacent neurons [23]. In human brain tissue, CB1R have been observed to trigger astrocytic calcium surges [24]. Astrocytic glutamate release, as a consequence of astrocytic activity, has been shown to actively modulate neuronal excitability, synaptic transmission and plasticity. These findings suggest that eCBs may exert both presynaptic and astrocytic influences to contribute to impaired glutamate homeostasis in rodent models of drug addiction. However, the involvement of eCBs in this process has remained unknown. In the present study, using cocaine self-administration in rats, we investigated the role of eCBs in presynaptic and astrocytic glutamate release in the NAcore, and tested the ability of CB1R activation via (R)-methanandamide (methAEA, a metabolic stable form of AEA) in inhibiting the reinstatement of cocaine seeking.
Materials and methods
Animals
Sprague-Dawley rats (3–6 months old) were group-housed and allowed to acclimate to their new environment for 1 week before any experimental procedures were performed. Total 299 rats were used in this study. 139 rats were used for Saline group. And 136 were trained for cocaine self-administration, in which 19 rats were excluded latterly due to lost catheters or cannulas. All animal care and experimental procedures were performed strictly following standard procedures approved by the Institutional Animal Care and Use committee at Peking University Health Science Center or Ningbo University (Supplementary material accompanies this paper).
Cocaine self-administration
Figure 1A shows the training procedure that we used for cocaine self-administration in rats. A single press on the active lever resulted in one delivery of cocaine (0.6 mg/kg/infusion, 0.6 mg/ml delivered over 3–5 s; Fixed ratio 1, FR1). The cue light and a discrete tone were each simultaneously presented for 5 s during cocaine infusion. Each cocaine delivery was followed by a 20 s timeout period. Extinction training started after at least 10 training days. Successfully trained rats were operationally defined as those that averaged more than 20 infusions per day in the last three days of training. During extinction training, lever presses did not result in cocaine infusion or any activation of tone or light cues. Extinction training continued for at least 14 days until rats performed <20 active lever presses in three consecutive days, and then was followed by reinstatement testing. Three days after cue-induced reinstatement, cocaine-primed reinstatement testing was conducted in the same groups of animals. During cue- or priming-induced reinstatement trial, rats did not receive intravenous drug infusions. Training sessions for self-administration, extinction, and reinstatement each lasted for 2 h.
Fig. 1. Decreased mGluR2/3-mediated feedback inhibition at NAcore glutamatergic synapses after cocaine self-administration and extinction training.

A Training procedure of cocaine self-administration and saline control. Recovery on the left-hand side denotes the number of days of recovery provided after catheter implantation. All measurements in all figures were performed after two weeks of extinction training and consisted of patch-clamp recordings from medium spiny neurons (MSNs) in NAc slices. B Active lever pressings and infusions of all cocaine and saline groups used in all data. C Upper: Representative traces of sEPSC. Lower: Cumulative distributions of inter-event intervals (left) or event amplitudes (right). Inset, mean event frequencies (left) or amplitudes (right). sEPSC frequency and amplitude in cocaine group were significantly higher than those in the saline group (***p < 0.001, unpaired t test). This convention is used in all other figures as well. Upper: Cumulative distributions of inter-event intervals pre- and post-LY341495 (or LY354740). Lower: The event frequencies from individual neurons. Synaptic glutamate release probability was less sensitive to LY341495 (0.03 μM & 0.3 μM) in the cocaine group (D, E). LY354740 (0.2 μM) had a significant impact on sEPSC frequency regardless of training condition (F). **p < 0.01, ***p < 0.001 comparing cocaine with saline group using two-way ANOVA with repeated measures followed by a Bonferroni’s post hoc test. The first and second N numbers in parentheses correspond to the numbers of recorded neurons and rats, respectively. Dots in B presented the mean values ± SEMs. Each dot in C–F represents an individual cell. Data with the lines connecting indicate within-subjects designs.
Mini-osmotic pumps for drug delivery to the NAcore
Mini-osmotic pumps (model 2001, infuse rate 0.1 ul/h, Alzet DURECT Corporation) were applied to chronically infuse the catalytically stable AEA analog, R(+)-methanandamide (methAEA, 100 μM, Tocris Bioscience) into the NAcore. Two cannulas connecting with mini-osmotic pumps (one for each hemisphere) were implanted into the NAcore at 14 days after extinction training, and the infusion would last 1 week. Total 24 (cocaine group) rats were used for mini-osmotic pumps implantation.
Whole-cell recordings in acute brain slices
Recording chambers were continuously perfused with aCSF at a flow rate of 2 ml/min at 32 °C. Patch electrodes (1.5–2 MΩ) were filled with a cesium-based internal solution (in mM: 120 cesium methanesulfonate, 0.5 EGTA, 10 HEPES, 5 QX-314 Br, 2 MgCl2, 10 NaCl, 2 Mg-ATP, and 0.25 Na-GTP). Recording solutions used for sEPSCs contained 1.2 mM of Mg2+. Picrotoxin (100 μM) was bath-applied during all patch-clamp recordings to block GABAA receptors. Slow inward current was recorded under low-Mg2+ aCSF (0.1 mM). Each cell was recorded for at least 40 min to avoid any floor effect caused by the inherently low frequency of slow inward current. Inward currents with 20–80% rising times no shorter than 10 ms, decaying times longer than 50 ms, and amplitudes larger than 10 ρA were defined as slow inward currents. The numbers of cells and rats used for whole-cell recordings were shown in figures.
Statistics
Statistical analyses between only two groups were assessed using two-tailed unpaired t-tests or two-tailed paired t-tests. When comparing a single factor or two factors across more than two groups, one-way or two-way ANOVAs, respectively, were used followed by Bonferroni’s post-hoc multiple pairwise comparisons. Statistical analyses were performed using GraphPad Prism 8.01 software.
Results
Impaired mGluR2/3 function in the NAcore after cocaine self-administration and extinction training
After cocaine self-administration and extinction training, rats were used for experiments involving either patch-clamp electrophysiology in NAc slices ex vivo or reinstatement testing in vivo (Fig. 1A&B). The averaged numbers of active lever presses and infusions was 73 ± 8.906 (±SEM) and 35 ± 2.341 on the last day of self-administration. The average number of active lever press on the last day of extinction is 13 ± 1.639. In the cocaine group, the average sEPSC frequency (unpaired t test, t(366) = 4.252, p < 0.001; Fig. 1C, left) and sEPSC amplitude were significantly higher (unpaired t test, t(366) = 4.196, p < 0.001; Fig. 1C, right) compared to these corresponding parameters in the saline-yoked group. These results are consistent with prior studies [7, 25, 26]. We next investigated the function of glutamatergic mGluR2/3 autoreceptors via perfusion of the mGluR2/3 antagonist LY341495 or agonist LY354740 into the recording chamber. A repeated-measures two-way ANOVA revealed that LY341495 with 0.03 or 0.3 μM significantly increased sEPSC frequency in the saline group but not in the cocaine group (Fig. 1D,E lower; factor of drug: F(1, 20) = 19.03, p < 0.001; factor of training condition: F(1, 20) = 0.309, p = 0.584; interaction: F(1, 20) = 14.9, p < 0.01 for 0.03 μM; factor of drug: F(1, 17) = 1.645, p = 0.2168; factor of training condition: F(1, 17) = 16.98, p < 0.001; interaction: F(1, 17) = 9.421, p < 0.01 for 0.3 μM). The mGluR2/3 agonist LY354740 (0.2 μM) significantly reduced sEPSC frequency in both saline group and cocaine group (Fig. 1F lower; factor of drug: F(1, 18)=2.088, p = 0.1656; factor of training condition: F(1, 18) = 10.11, p = 0.0052; interaction: F(1, 18) = 0.6988, p = 0.4142).
In contrast, no change in sEPSC amplitude was observed after LY341495 or LY354740 administration in either the saline or cocaine group (Fig. S1), confirming that mGluR2/3 antagonist did not influence postsynaptic-based sEPSC amplitudes.
The different role of CB1Rs and TRPV1 in presynaptic modulation of glutamate release in the NAcore of saline-yoked rats
In the saline-yoked group, CP55,940, an agonist for both CB1Rs and CB2Rs, significantly decreased sEPSC frequency (paired t test, t(14) = 4.524, p < 0.001; Fig. 2A), but did not change sEPSC peak amplitude (Supplementary Fig. S2A). These findings suggest that CB1Rs/CB2Rs activation presynaptically reduced glutamatergic drive to MSNs within the NAcore without affecting postsynaptic-based sEPSC amplitudes, which is consistent with the known retrograde presynaptic mechanism of cannabinoid receptors (CBRs) mediated negative feedback of glutamate release.
Fig. 2. AEA increases sEPSC frequency in the NAcore of saline group via TRPV1 rather than CB1Rs.

Cumulative distributions of inter-event intervals and mean event frequencies (insets) pre- and post-drug perfusion. The CB1Rs agonist CP55,940 (0.3 μM) or 2-AG hydrolase inhibitor JZL184 (10 μM) significantly reduced sEPSC frequency (A, C), while methAEA (30 μM), AEA hydrolase inhibitor URB597 (10 μM), methAEA (30 μM) or TRPV1 agonist capsaicin (10 μM) increased sEPSC frequency (B, D, H). MethAEA increased sEPSC frequency in the presence of the CB1R antagonist, AM251 (5 μM), but decreased sEPSC frequency in the presence of the TRPV1 antagonist, SB366791 (1 μM) (E, F). Hence, the action of AEA on increasing synaptic glutamate release probability may be mediated by TRPV1. *p < 0.05, **p < 0.01, ***p < 0.001, compared with vehicle perfusion using paired t test. The first and second N numbers in parentheses correspond to the numbers of recorded neurons and rats, respectively.
We next investigated the particular roles of AEA and 2-AG on sEPSCs of NAc MSNs. We elevated the extracellular concentrations of both endogenous 2-AG and AEA within our NAc slices via perfusion of hydrolytic enzyme inhibitors, JZL184 (10 μM) and URB597 (10 μM), respectively. Surprisingly, either URB597 (Fig. 2B) or methAEA (metabolic stable form of AEA; 30 μM, Fig. 2D) significantly increased sEPSC frequency (paired t test, URB597: t(10) = 4.786, p < 0.001; methAEA: t(12) = 7.537, p < 0.001), whereas JZL184 (Fig. 2C) attenuated sEPSC frequency (paired t test, t(9) = 3.471, p = 0.007). These findings suggest that boosting endogenous AEA, but not 2-AG, presynaptically increased glutamatergic drive to MSNs within the NAcore, which is in contrast to our findings of CBR agonist CP55,940 and suggests that our experimentally boosted endogenous AEA may have acted through non-CB receptors to increase MSN glutamatergic drive. The amplitudes of sEPSCs weren’t influenced by URB597, JZL184 or methAEA (Supplementary Fig. S2B–D).
Then we examined which receptor mediated the endogenous upregulation of synaptic glutamate release by AEA in the saline-yoked group. The selective TRPV1 antagonist SB366791 (paired t test: t(17) = 3.216, p = 0.0051; Fig. 2F), but not the CB1R antagonist AM251 (t(9) = 3.668, p = 0.0052; Fig. 2E), blocked the methAEA-induced increase in sEPSC frequencies. SB366791 also blocked the URB597-induced increase in sEPSC frequencies (paired t test: t(8) = 5.131, p = 0.0009; Fig. 2G). Moreover, capsaicin, a TRPV1 agonist, significantly increased sEPSC (paired t test: t(7) = 3.267, p = 0.0137; Fig. 2H) but not amplitude (Supplementary Fig. S2H). These results indicated that the AEA-induced enhancement of glutamate release to NAcore MSNs was potentially mediated by TRPV1 in the NAcore.
Impaired CB1R and TRPV1 signaling in the NAcore of rats after cocaine self-administration and extinction training
Strikingly, in the cocaine group, there was no significant effect of CP55,940, URB597, or methAEA on sEPSC frequencies (Fig. 3, paired t test, CP55,940: t(13)=1.004, p = 0.3338; URB597: t(8) = 2.054, p = 0.0741; methAEA: t(12) = 1.396, p = 0.1881). These results revealed the endocannabinoid-mediated presynaptic modulation of glutamate release was impaired in the NAcore from cocaine group. The amplitudes of sEPSCs were not altered by these drugs (Supplementary Fig. S2I–K).
Fig. 3. AEA fails to alter synaptic glutamate release probability in the NAcore of rats after cocaine self-administration and extinction training.
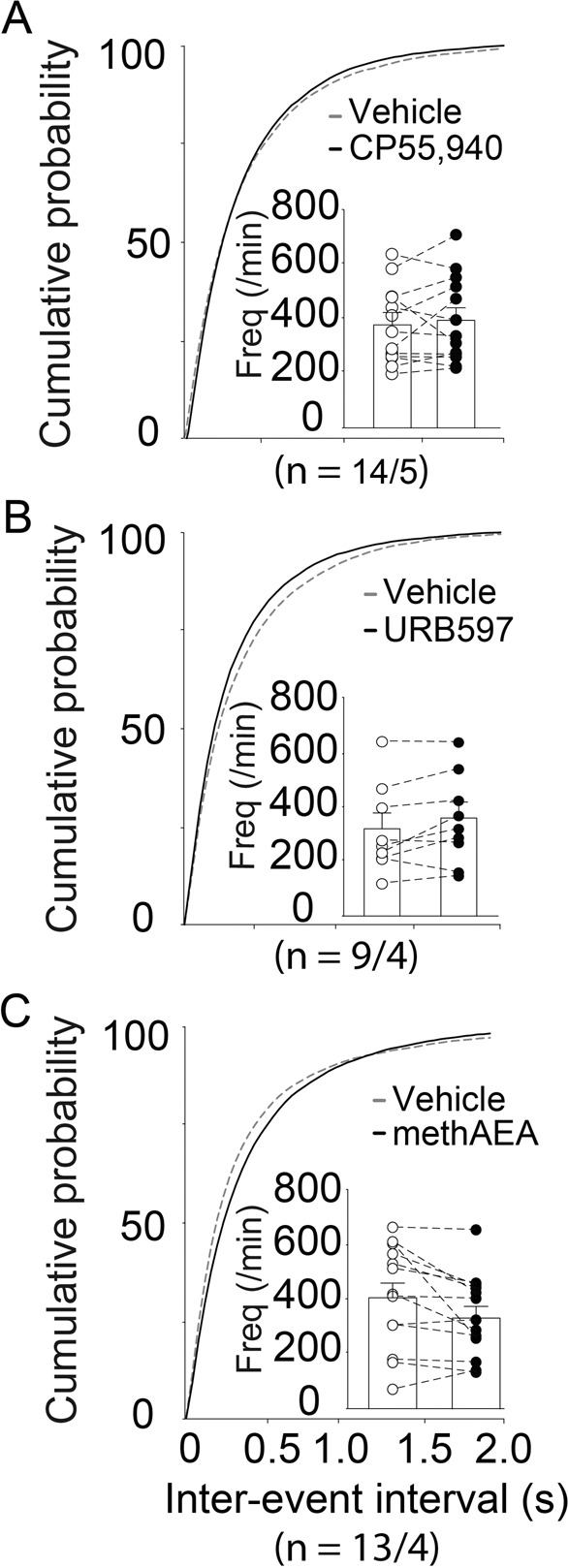
A–C Cumulative distributions of inter-event intervals and mean event frequencies (insets) pre- and post-drugs perfusion. The concentrations and perfusion times of CP55,940, URB597, or methAEA used in the NAc slices from the cocaine group were the same as those used in the saline group. None of these drugs had an influence on sEPSC frequencies in the NAc of the cocaine group (paired t test). The first and second N numbers in parentheses correspond to the numbers of recorded neurons and rats, respectively.
Differential mGluR5- and CB1R-mediated astrocytic glutamate release in the NAcore of rats after cocaine self-administration and extinction training
Astrocytic glutamate release triggers extrasynaptic NMDAR-mediated SICs that exhibit slower kinetics and longer rising times [27] (Fig. 4A). Because slow inward current occurs spontaneously and can be enhanced by astrocytic GPCR activation via mGluR5 and CB1Rs in the NAcore [23, 28], we compared the basic and evoked slow inward currents by GPCR agonists between saline and cocaine groups. While there was no difference in frequencies of slow inward currents at the baseline level between the two groups, activating mGluR1/5 with DHPG significantly increased frequencies of slow inward currents in the saline group but not in the cocaine group (two-way ANOVA; DHPG: F(1, 48) = 11.09, p = 0.0017; training condition: F(1, 48) = 8.811, p = 0.0047; interaction: F(1, 48) = 9.345, p = 0.0036; Fig. 4C left). In contrast, neither training condition nor DHPG application had an impact on slow inward currents amplitudes (Two-way ANOVA; DHPG: F(1, 48)=0.3865, p = 0.5371; training condition: F(1, 48) = 0.1677, p = 0.6839; interaction: F(1, 48) = 0.9105, p = 0.3448; Fig. 4C right).
Fig. 4. The effects of mGluR5 and CB1 activation on astrocytic glutamate release in the NAcore of the saline group or cocaine group.

A Upper: Recordings from a medium spiny neuron showing SICs and sEPSCs. Lower: Overlay of scaled sEPSC and SICs shown in the upper lane to compare their kinetics. B Sample traces of SICs from saline and cocaine groups with or without DHPG (10 μM) perfusion. C The mGluR5 agonist, DHPG, remarkably increased SICs frequency in the saline group but not in the cocaine group. DHPG did not change the peak amplitude of SICs in the two groups. ** p < 0.01 compared to vehicle-treated neurons using one-way ANOVA with repeated measures followed by a Bonferroni’s post hoc test. D CB1R agonist CP55,940 (0.3 μM) or methAEA (30 μM) increased SICs frequency in both saline and cocaine groups. CB1R antagonist AM251 (10 μM) blocked the effect of methAEA on SICs frequency. However, AEA hydrolase inhibitor URB597 (1 μM) increased SICs frequency in the saline group but not in the cocaine group. E In the absence of LY341495, sEPSC frequency before the SIC is significantly higher than sEPSC frequency after SIC in saline group, but not in cocaine group. *p < 0.05, **p < 0.01 compared to vehicle-treated neurons using one-way or two-way ANOVA followed by a Bonferroni’s post hoc test. Data are represented as the mean values ± SEMs. N numbers shown in parentheses correspond to the number of recorded neurons and the number of recorded rats per group, respectively. SIC: slow inward current. Data with the lines connecting indicates within-subjects designs. Data without the lines connecting indicates between-subjects designs.
MTEP (20 μM; mGluR5 antagonist) and MK801 (1 mM) blocked DHPG- and methAEA-induced increase of slow inward currents frequencies in saline group respectively (one-way ANOVA, F(4, 51)=23.88, p < 0.0001; Fig. S3C), while tetrodotoxin (0.5 μM) had no effect on those altered frequencies. Our findings are consistent with previous reports that slow inward currents are triggered by mGluR5 and mediated by NMDA receptor, and don’t rely on action potential [28].
Next, we compared the response of astrocytic glutamate release to CB1Rs activation in saline and cocaine groups. In the NAcore from the saline group, CP55,940 (0.3 μM), JZL184 (10 μM), URB597 (1 μM), and methAEA (30 μM) each significantly increased slow inward currents frequencies, while pre-incubation with the CB1R antagonist AM251 blocked methAEA-induced increases (one-way ANOVA, F(5, 76) = 6.864, p < 0.0001; Fig. 4D left). In the NAcore of cocaine group, CP55,940 or methAEA significantly increased frequencies of slow inward currents, while URB597 had no effect (one-way ANOVA, F(3, 50) = 7.972, p = 0.0002; Fig. 4D right). The amplitudes of slow inward currents weren’t affected by those drugs in either the saline or cocaine group (Supplementary Fig. S3A, B). These results suggest that CB1R-mediated astrocytic glutamate release was left intact in the cocaine group. In contrast, unlike the efficacy in the saline group, the inability of URB597 to induce slow inward currents in the cocaine group suggested that there was insufficient endogenous tonic AEA release in the NAcore of cocaine group.
We further calculated the sEPSC frequencies within 30 s duration immediately before and after the event of slow inward current. Repeated-measures two-way ANOVA analysis revealed that the occurrence of slow inward current had significantly impact on sEPSC frequencies in the absence of LY341495 (0.03 μM) in saline group (factor of LY341495: F(1, 18) = 2.479, p = 0.1328; factor of slow inward current: F(1, 18) = 10.63, p = 0.0043; interaction: F(1, 18) = 12.35, p < 0.01; Fig. 4E left). The effect of slow inward current on sEPSC frequency wasn’t observed in the cocaine group regardless of LY341495 existing (factor of drug: F(1, 22)=0.1831, p = 0.6729; factor of SICs: F(1, 22) = 1.688, p = 0.2074; interaction: F(1, 22) = 0.91, p = 0.3505; Fig. 4E right). Alterations in sEPSC amplitude were not observed in either group (Supplementary Fig. S3D). These results imply that the astrocytic glutamate release may be due to inhibition of synaptic glutamate release via mGluR2/3 activation in saline group, but not in cocaine group. It is possible that although the mGluR2/3 in the NAcore of cocaine group could be activated by agonist LY354740 (Fig. 1F), it was still less sensitive to endogenous extracellular glutamate.
Chronic methAEA infusions induce sustained CB1R activation that restores mGluR2/3-mediated presynaptic inhibition and mGluR5-mediated astrocytic glutamate release in the NAcore of rats after cocaine self-administration and extinction training
The preserved function of CB1R-mediated astrocytic glutamate release in the cocaine group provides the possibility that sustained stimulation of astrocytic CB1Rs may restore extracellular glutamate levels and thereby revive presynaptic modulation of glutamate release. To test this supposition, methAEA were bilaterally infused into the NAcore of cocaine group for one week, then the function of presynaptic modulation by mGluR2/3 and CB1Rs was tested via monitoring sEPSC frequencies in NAcore MSNs from cocaine group. A two-way ANOVA revealed that the mGluR2/3 antagonist LY341495 significantly increased sEPSC frequencies in the methAEA-infused group, but not in the PBS-infused group (factor of LY341495, F(1, 17) = 0.7854, p = 0.3879; infusion, F(1, 17) = 7.39, p = 0.0146; interaction: F(1, 17) = 6.294, p = 0.0225; Fig. 5A). LY341495 increases sEPSCs frequency by 17.1% in cocaine group with chronic methAEA infusion, which is in the same range as that in saline group (34.6% increasing with 0.03uM; 19.7% with 0.3 μM; Fig. 1D, E). Notably, methAEA infusion did not restore the failed responses of MSN sEPSC frequencies to CP55,940 in the NAcore from cocaine group (paired t test, t(12) = 1.455, p = 0.1714; Fig. 5B).
Fig. 5. methAEA infusion in the NAcore restores the glutamate homeostasis in the cocaine group, and inhibits reinstatement to cocaine seeking.

A LY341495 (0.03 μM) increased sEPSC frequency of MSNs in the methAEA -infused group. **p < 0.01 compared to PBS-infused group using two-way ANOVA with repeated measures followed by a Bonferroni’s post hoc test. B CP55,940 (0.3 μM) failed to alter sEPSC frequency of MSNs in the methAEA-infused group. C DHPG (10 μM) increased SICs frequency of MSNs in the methAEA-infused group. ***p < 0.001 compared to PBS-infused group using two-way ANOVA with repeated measures followed by a Bonferroni’s post hoc test. N numbers shown in bars and parentheses corresponds to the number of recorded neurons and the number of recorded rats per group, respectively. D Cocaine priming- or cue-induced reinstatement of active lever pressing was reduced by pretreatment via methAEA infusions (100 μM). E No difference in locomotor activity was observed between methAEA- and PBS-infused groups. F Illustration of the location of cannula tips where infusions were made into the NAcore according to the atlas of Paxinos and Watson (1986). N numbers shown in parentheses corresponds to the number of rats assigned in behavioral experiments. **p < 0.01, ***p < 0.001 compared to PBS-infused group using two-way ANOVA with repeated measures followed by a Bonferroni’s post hoc test. Data are represented as the mean values ± SEMs. Data with the lines connecting indicates within-subjects designs. Data without the lines connecting indicates between-subjects designs.
Next, we investigated whether methAEA infusion repaired the failed response of astrocytic glutamate release to mGluR5 activation in the cocaine group. A two-way ANOVA revealed that DHPG significantly impacted slow inward currents frequencies in the methAEA-infused group (factor of DHPG, F(1, 37) = 7.846, p = 0.0081; infusion, F(1, 37) = 7.831, p = 0.0081; interaction F(1, 37) = 13.88, p = 0.0006; Fig. 5C). There are no alterations of sEPSC or slow inward currents amplitudes in both PBS- and methAEA-infused groups (Supplementary Fig. S4).
Chronic methAEA infusions in the NAcore inhibit either cue- or priming-induced reinstatement to cocaine seeking
As demonstrated in previous studies, deficient functionalities of GLT-1 and mGluR2/3 in cocaine or heroin self-administered rats are unable to homeostatically control spillover of synaptic glutamate during reinstatement of drug seeking [3, 4]. Therefore, preventing excessive spillover of glutamate may represent a strategy to reduce reinstatement of drug seeking. Given that mGluR2/3 function were restored via methAEA infusions in the NAcore of cocaine group, we next tested the ability of methAEA to inhibit reinstatement of cocaine seeking. The data during extinction and reinstatement were analyzed using two-way ANOVAs with repeated measures (factor of infusion, F(1, 23) = 13.2, p = 0.0014; factor of session F(2, 46) = 49.89, p < 0.001; interaction, F(2, 46) = 4.698, p = 0.0139; Fig. 5D). For PBS-infused rats, prominent reinstatement of cocaine seeking was induced by either cocaine priming (10 mg/kg) or the presentation of a light/tone cue. In contrast, chronic infusion of methAEA significantly reduced the reinstatement triggered by either priming or the conditional cue (Bonferroni’s post-hoc test, t = 3.711, DF = 69, p = 0.0012 for cue; t = 3.301, DF = 69, p = 0.0046 for priming). There was no difference in the total traveled distance in the open field between the two groups (Fig. 5E). Infusion sites were verified during the tissue harvesting process for patch-clamp recordings (Fig. 5F).
Discussion
The present study demonstrated that in terms of endocannabinoid signaling in the NAcore from saline rats, AEA, but not 2-AG, elicited presynaptic and astrocytic glutamate release via TRPV1 and CB1Rs, respectively. After cocaine self-administration and extinction training, although AEA no longer changed synaptic glutamate release, its effect on astrocytic glutamate release was preserved. The restorations of mGluR2/3 function and mGluR5-activated astrocytic glutamate release were obtained after chronic methAEA infusion in the NAcore of cocaine group, which inhibited either priming or cue-induced reinstatement of cocaine seeking. The multiple roles of AEA in glutamate homeostasis and cocaine seeking behavior elucidated in the present study suggest that the endocannabinoid system represents a promising therapeutic target for cocaine addiction.
Impaired glutamate homeostasis and astrocytic glutamate release after cocaine self-administration and extinction training
The increase of presynaptic glutamate release and failed presynaptic mGluR2/3 regulation during withdrawal or extinction after chronic cocaine treatment have been reported by previous studies [5, 29–33]. Insufficient extrasynaptic glutamate in cocaine-treated rats plays a key role in their disturbed glutamate homeostasis [21, 34–36], which has been linked to dysregulated mGluR2/3 function and glutamate uptake [37]. However, it has not been fully understood as to how extrasynaptic glutamate is regulated by astrocytes in the NAcore of rats trained to self-administer cocaine. Accumulated evidence has demonstrated that reactive astrocytes release neuroactive molecules in a Ca2+-dependent manner [28, 38, 39]. Astrocytes express Gq/11-coupled receptors such as mGluR5, the activities of which drive inositol-1,4,5-trisphosphate (IP3)-induced Ca2+ release from the endoplasmic reticulum and activates extracellular glutamate release [28, 40]. At the synaptic level, glutamate released by astrocytes acts on extrasynaptic NMDARs to generate slow inward currents [40] and simultaneously activates presynaptic mGluR2/3 to inhibit synaptic glutamate release [37]. With these signaling mechanisms, astrocytes detect excitatory synaptic activities and act as a processor and coordinator for activation of adjacent neurons [41]. Hence, we monitored astrocytic glutamate release via quantifying slow inward currents. Activating mGluR5 with DHPG significantly increased frequencies of slow inward currents in the saline group but not the cocaine group, indicating long-term disruption of mGluR5-mediated astrocytic activity in cocaine group. While impaired postsynaptic mGluR5 function in animal models of cocaine self-administration has been reported by previous studies [31, 42–46], our present findings are the first to validate that astrocytic mGluR5 is involved. We hypothesize that this reduction in mGluR5-mediated astrocytic glutamate release potentially contributed to a decrease of extrasynaptic glutamate in the NAcore, and consequently resulted in decreased glutamatergic tone on presynaptic mGluR2/3 [37, 47]. It is noteworthy that the present study could not distinguish whether the impaired glutamate homeostasis resulted from cocaine self-administration or from extinction training.
Whether the mGluR5 distributes on mature astrocytes in the rodent brain are still debated. Several studies revealed the mRNA expression of mGluR5s in isolated astrocytes from cortex and hippocampus declined throughout development [48–50], and mGluR5-immunostaining was detected mainly in striatal neurons, not in GFAP-labeled astrocytes [51]. However, other researches showed that mGluR5 expressed on processes of astrocytes of the adult or aged brain [52–55], and remained sufficient function to process astrocytic Ca2+ signaling [56, 57]. From our own data, we found that DHPG, a mGluR1/5 agonist significantly increased slow inward currents frequency in adult control, implying functional activity of astrocytic mGluR5 in the NAcore of those adult rats.
Slow inward currents exhibited prominent influence on neuronal activities. Previous researches have reported that slow inward currents drove action potentials and even burst firing in adjacent neurons causing synchronous neuronal activities in nucleus accumbens and hippocampal CA1 region [28, 41, 58–60]. Although the frequency of slow inward currents is relatively lower in comparison to sEPSC, the peak amplitude of slow inward currents is larger than sEPSC which indicates a larger quantity of glutamate release. And it has been shown that the astrocytic vesicles that generate slow inward currents are much larger than synaptic vesicles [61]. All of the evidence above hinted that increasing slow inward currents frequency increased extracellular glutamate concentration.
Neuronal mGluR5 contributes to the modulation of synaptic glutamate release and drug-seeking behavior. In the NAcore, mGluR5 antagonist inhibits cocaine seeking behavior through increasing extracellular glutamate concentration [51, 62, 63]. Genetic deletion of mGluR5 in the dopamine D1 medium spiny neurons of NAc inhibited cocaine seeking behavior and abolished 2-AG-mediated long-term depression [64]. Therefore, the inhibitory effect of methAEA on reinstatement of cocaine seeking might be an overall consequence of stimulating both neuronal and astrocytic mGluR5 via astrocytic glutamate release.
Disrupted endocannabinoid AEA modulation of synaptic glutamate release in rats after cocaine self-administration and extinction training
As retrograde signaling, eCBs are released with both tonic and activity-dependent phasic modes [65–69]. 2-AG is a high-affinity agonist for CB1Rs and CB2Rs, whereas AEA is a low-affinity agonist for CB1Rs [70]. While AEA is responsible for maintaining basal endocannabinoid signaling, 2-AG mediates strong and rapid signaling for inhibitory feedback via presynaptic CB1R receptors [19]. These different actions are likely related to the fact that AEA is a partial agonist at CB1Rs, while 2-AG acts as full agonist at both CB1Rs and CB2Rs [71]. Direct application of 2-AG or enhancement of 2-AG tone via inhibiting its hydrolysis inhibits synaptic glutamate release [72–74]. In contrast, the effects of AEA on presynaptic release are complicated as it acts not only as a CB1R agonist but also as an agonist for TRPV1 [75, 76]. AEA has been shown to decrease presynaptic glutamate release in the presence of the FAAH inhibitor URB597 in the midbrain periaqueductal gray [13]. However, AEA increased synaptic glutamate release when CB1Rs are blocked by AM251 in wild type mice, but not in TRPV1 knock-out mice [13].
Notably, a recent research demonstrated that TRPV1 specifically potentiated synaptic connectivity of the glutamatergic projections from basolateral amygdala to NAcore D1-positive MSNs in adult mice [77]. However, in the NAc, the relative contributions of presynaptic CB1Rs and TRPV1 to modulating synaptic function remained unclear. Direct activation of CB1Rs with CP55,940 remarkably decreased sEPSC frequencies in MSNs within the NAcore, indicating a CB1R-mediated downregulation of the release probability of glutamate (Fig. 2A). Intriguingly, JZL184 that accumulates extracellular 2-AG via inhibiting MAGL [78], reduced sEPSC frequencies in MSNs within the NAcore of saline rats (Fig. 2C). In contrast, URB597 or methAEA significantly increased the sEPSC frequencies in MSNs within the NAcore from saline rats (Fig. 2B, D). Given the dual targets of AEA on presynaptic terminals, we attempted to discriminate the differential contributions of CB1Rs and TRPV1 to AEA-induced presynaptic glutamatergic modulation. Despite pre-incubating NAc slices with the CB1R antagonist AM251, methAEA still elevated presynaptic glutamate release in the saline group (Fig. 2E). However, when TRPV1 was blocked with SB366791, methAEA significantly reduced presynaptic glutamate release (Fig. 2F). These results suggest that the net effect of methAEA through CB1Rs and TRPV1 activation is to increase presynaptic glutamate release in the NAcore, which is consistent with previous findings in other brain regions [75, 76, 79].
In addition to impaired mGluR2/3 function, endocannabinoid-mediated presynaptic glutamatergic modulation was also disrupted in cocaine group (Fig. 3). Few studies have reported that CB1Rs or TRPV1 are involved in animal models of cocaine self-administration. CB1Rs expression was shown to be downregulated in the dorsal striatum after cocaine self-administration [80]. In addition, increasing TRPV1 signaling was shown to facilitate priming-induced cocaine reinstatement [81]. Our findings further provide direct evidence of CB1Rs and TRPV1 dysfunction in regulating presynaptic glutamate release in animal models for drug addiction.
Capacity for endocannabinoid AEA-induced modulation of astrocytic glutamate release in rats after cocaine self-administration and extinction training
In the central nervous system, CB1Rs are distributed in both neurons and astrocytes. Unlike that of mGluR5, the role of CB1Rs on astrocytic glutamate release remained intact in cocaine group (Fig. 4D), as both CP55,940 and methAEA were able to promote astrocytic glutamate release. It has previously been shown that increases in slow inward currents frequency concomitantly increase extracellular glutamate concentrations [82]. Also, slow inward currents mediate astrocyte-neuron crosstalk through modulating synaptic plasticity, which has been observed in the NAc, cortex, thalamus, and hippocampus [28, 83–85]. In line with these previous findings, our present results suggested that CB1Rs stimulation may represent an alternative pathway for enhancing astrocytic glutamate release to restore impaired glutamate homeostasis in cocaine group.
Sustained methAEA infusion in the NAcore restores impaired glutamate homeostasis in rats after cocaine self-administration and extinction training
To take advantage of intact CB1Rs-mediated astrocytic glutamate release in cocaine group, we next sought to restore impaired glutamate homeostasis via sustained methAEA infusions into the NAcore. The restored mGluR2/3 function (Fig. 5A) might be related to co-localized TRPV1 [86] activated by methAEA stimulation. Calcium influx through TRPV1 causes a calcium/calmodulin-dependent translocation of G-protein-coupled receptor kinase 5 away from the plasma membrane [87]. Several studies have demonstrated that G-protein-coupled receptor kinases (GRKs) are key proteins that phosphorylate GPCRs (e.g., mGluR2/3) to initiate receptor internalization [88–90]. Alternatively, methAEA may also elevate Ca2+ levels in astrocytes via CB1Rs-mediated Gq/11 signaling, which might induce Ca2+-dependent translocation of GRKs and inhibit mGluR5 desensitization/internalization from the astrocytic membrane [91]. Therefore, we hypothesize that methAEA restores impaired glutamate homeostasis in cocaine-experienced rats via two parallel ways: (1) normalizing presynaptic modulation via TRPV1; and (2) increasing the supplement of extrasynaptic glutamate via activation of astrocytic CB1Rs. However, our present study did not provide any direct evidence of whether or not methAEA regulated mGluR desensitization and/or internalization, which requires further investigation. A working model that summarized the findings of the current study and hypothesis is shown in Fig. 6.
Fig. 6. A working model shows the involvement of endocannabinoid signaling in glutamate homeostasis in the NAcore based on the finding of the current study.
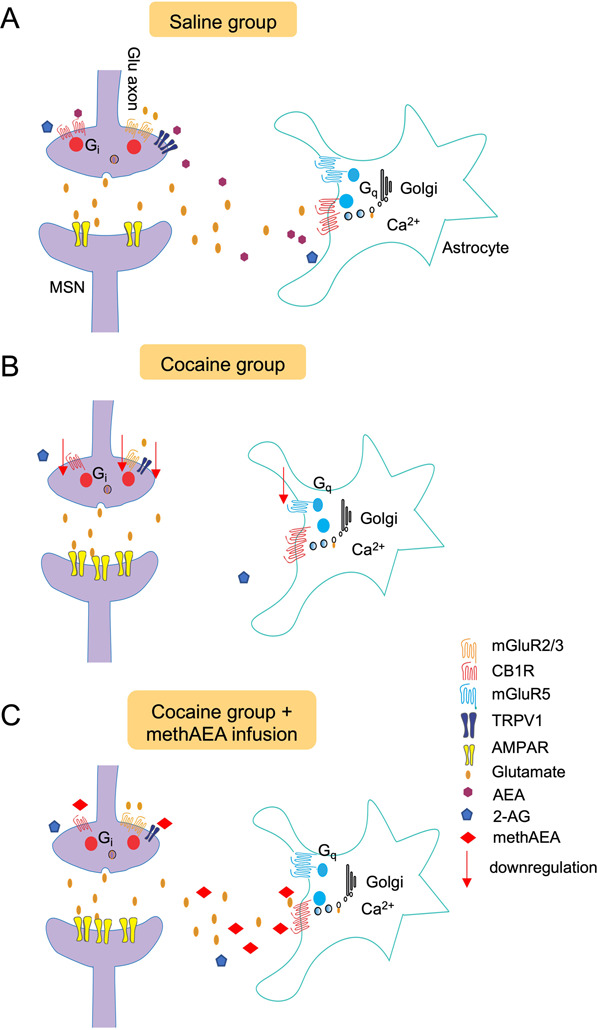
A Presynaptic CB1R and mGluR2/3 activation inhibit synaptic glutamate release, while TRPV1 promotes synaptic glutamate release. Endocannabinoid AEA is agonist for both CB1R and TRPV1, while 2-AG targets CB1R. Endogenous AEA, but not 2-AG, presynaptically increased glutamatergic drive to MSNs within the NAcore. Astrocytic glutamate release is mediated by mGluR5 and CB1R located on astrocyte, which reach mGluR2/3 to modulate presynaptic release. B After extinction of cocaine self-administration, glutamate homeostasis was interrupted. Extrasynaptic glutamate is depleted, and the functions of CB1R, mGluR2/3 and TRPV1 for regulating synaptic glutamate release were impaired. Although mGluR5-mediated astrocytic glutamate release was decreased, the astrocytic CB1R remained functional and could be a potential target for methAEA. C methAEA restores impaired glutamate homeostasis in cocaine-experienced rats via two parallel ways: (1) normalizing presynaptic modulation via TRPV1; and (2) increasing the supplement of extrasynaptic glutamate via activation of astrocytic CB1Rs.
Application of endocannabinoid-mimetic drugs in preventing relapse to cocaine seeking
Compensating for reduced extrasynaptic glutamate via supplying the substrate (i.e., N-acetylcysteine) of the Xc- glutamate-cystine exchanger has been shown to be effective in preventing reinstatement to drug seeking [4, 31, 92]. Our present results demonstrated that chronic methAEA infusion in the NAcore inhibited either priming- or cue-induced reinstatement of cocaine seeking. Our collective findings suggest that astrocytic CB1Rs, rather than presynaptic CB1Rs, may have mediated the restoration of presynaptic mGluR2/3 and astrocytic mGluR5 signaling and corresponding inhibition of relapse to cocaine seeking. Our conclusion is based on the following findings: (1) no response of sEPSCs to the CB1R agonist, CP55,940 suggests that presynaptic CB1Rs remained dysfunctional after chronic methAEA treatment; and (2) AEA induced a more robust effect on astrocytic CB1Rs than on presynaptic CB1Rs.
The restorations of mGluR2/3 function and mGluR5-mediated astrocytic glutamate release via astrocytic CB1Rs activation potentially contributed to the inhibited reinstatement of cocaine seeking. Previous studies have reported that activating NAc mGluR2/3 signaling was sufficient to reduce cocaine seeking behavior [5, 93–95]. It is noteworthy that CB1Rs have been recognized as pharmacotherapeutic targets for neuropsychiatric disorders [96–100]. Our present findings provided novel evidence that endocannabinoid-mimetic drugs may ameliorate addictive behavior via previously unrecognized mechanisms in terms of promoting astrocytic and presynaptic glutamate release via CB1Rs and TRPV1, respectively.
Supplementary information
Acknowledgements
The project was funded by the National Key Research and Development Program of China (2017YFC0803605) and the Natural Science Foundation of China (grant numbers 81271472 and 31571094) to H-WS. The authors have no financial interests relating to the work described and declare no conflict of interest.
Author contributions
ZLY and SHW were responsible for the study concept and design and drafted the manuscript. ZLY and ZYQ contributed to the acquisition of animal data. ZLY contributed to the acquisition of electrophysiology data. ZLY, YZP and ZXQ assisted with the data analysis and interpretation of the data. SJ helped with the experimental design. Specially, we acknowledge Dr. Joseph F. Cheer for his insightful suggestions on the manuscript. All of the authors critically reviewed the content and approved the final version for publication.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Lan-Yuan Zhang, Yue-Qing Zhou
Contributor Information
Jie Shi, Email: shijie@bjmu.edu.cn.
Hao-Wei Shen, Email: shenhaowei@nbu.edu.cn.
Supplementary information
Supplementary Information The online version contains supplementary material available at (10.1038/s41386-021-00955-1).
References
- 1.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–72.. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 2.Scofield MD, Kalivas PW. Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist. 2014;20:610–22.. doi: 10.1177/1073858413520347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghasemzadeh MB, Nelson LC, Lu XY, Kalivas PW. Neuroadaptations in ionotropic and metabotropic glutamate receptor mRNA produced by cocaine treatment. J Neurochem. 1999;72:157–65.. doi: 10.1046/j.1471-4159.1999.0720157.x. [DOI] [PubMed] [Google Scholar]
- 4.Shen HW, Scofield MD, Boger H, Hensley M, Kalivas PW. Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J Neurosci. 2014;34:5649–57.. doi: 10.1523/JNEUROSCI.4564-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–7. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, Kalivas PW. Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron. 2013;77:867–72.. doi: 10.1016/j.neuron.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB, et al. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci USA. 2011;108:385–90.. doi: 10.1073/pnas.1011265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pendyam S, Mohan A, Kalivas PW, Nair SS. Computational model of extracellular glutamate in the nucleus accumbens incorporates neuroadaptations by chronic cocaine. Neuroscience. 2009;158:1266–76.. doi: 10.1016/j.neuroscience.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry. 2010;67:81–4. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–25.. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Marzo V, De Petrocellis L. Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R Soc Lond B Biol Sci. 2012;367:3216–28.. doi: 10.1098/rstb.2011.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Jia D, Wang Y, Qu L, Wang X, Song J, et al. Enhanced ability of TRPV1 channels in regulating glutamatergic transmission after repeated morphine exposure in the nucleus accumbens of rat. Brain Res. 2017;1660:47–57. doi: 10.1016/j.brainres.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Kawahara H, Drew GM, Christie MJ, Vaughan CW. Inhibition of fatty acid amide hydrolase unmasks CB1 receptor and TRPV1 channel-mediated modulation of glutamatergic synaptic transmission in midbrain periaqueductal grey. Br J Pharm. 2011;163:1214–22.. doi: 10.1111/j.1476-5381.2010.01157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metna-Laurent M, Marsicano G. Rising stars: modulation of brain functions by astroglial type-1 cannabinoid receptors. Glia. 2015;63:353–64.. doi: 10.1002/glia.22773. [DOI] [PubMed] [Google Scholar]
- 15.Di Marzo V, Stella N, Zimmer A. Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci. 2015;16:30–42. doi: 10.1038/nrn3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parsons LH, Hurd YL. Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci. 2015;16:579–94.. doi: 10.1038/nrn4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sloan ME, Gowin JL, Ramchandani VA, Hurd YL, Le, Foll B. The endocannabinoid system as a target for addiction treatment: Trials and tribulations. Neuropharmacology. 2017;124:73–83. doi: 10.1016/j.neuropharm.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 18.Solinas M, Justinova Z, Goldberg SR, Tanda G. Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J neurochemistry. 2006;98:408–19.. doi: 10.1111/j.1471-4159.2006.03880.x. [DOI] [PubMed] [Google Scholar]
- 19.Mateo Y, Johnson KA, Covey DP, Atwood BK, Wang HL, Zhang S, et al. Endocannabinoid actions on cortical terminals orchestrate local modulation of dopamine release in the nucleus accumbens. Neuron. 2017;96:1112–26 e5. doi: 10.1016/j.neuron.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xi ZX, Spiller K, Pak AC, Gilbert J, Dillon C, Li X, et al. Cannabinoid CB1 receptor antagonists attenuate cocaine’s rewarding effects: experiments with self-administration and brain-stimulation reward in rats. Neuropsychopharmacology. 2008;33:1735–45. doi: 10.1038/sj.npp.1301552. [DOI] [PubMed] [Google Scholar]
- 21.Xi ZX, Gilbert JG, Peng XQ, Pak AC, Li X, Gardner EL. Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine-primed relapse in rats: role of glutamate in the nucleus accumbens. J Neurosci. 2006;26:8531–6. doi: 10.1523/JNEUROSCI.0726-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fattore L, Martellotta MC, Cossu G, Mascia MS, Fratta W. CB1 cannabinoid receptor agonist WIN 55, 212-2 decreases intravenous cocaine self-administration in rats. Behav Brain Res. 1999;104:141–46.. doi: 10.1016/S0166-4328(99)00059-5. [DOI] [PubMed] [Google Scholar]
- 23.Navarrete M, Araque A. Endocannabinoids mediate neuron-astrocyte communication. Neuron. 2008;57:883–93.. doi: 10.1016/j.neuron.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 24.Navarrete M, Perea G, Maglio L, Pastor J, Garcia de Sola R, Araque A. Astrocyte calcium signal and gliotransmission in human brain tissue. Cereb Cortex. 2013;23:1240–6. doi: 10.1093/cercor/bhs122. [DOI] [PubMed] [Google Scholar]
- 25.Ma YY, Henley SM, Toll J, Jentsch JD, Evans CJ, Levine MS, et al. Drug-primed reinstatement of cocaine seeking in mice: increased excitability of medium-sized spiny neurons in the nucleus accumbens. ASN Neuro. 2013;5:257–71.. doi: 10.1042/AN20130015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corbit LH, Chieng BC, Balleine BW. Effects of repeated cocaine exposure on habit learning and reversal by N-acetylcysteine. Neuropsychopharmacology. 2014;39:1893–901. doi: 10.1038/npp.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–63.. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, et al. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci USA. 2007;104:1995–2000. doi: 10.1073/pnas.0609408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi ZX, Ramamoorthy S, Baker DA, Shen H, Samuvel DJ, Kalivas PW. Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharm Exp Ther. 2002;303:608–15.. doi: 10.1124/jpet.102.039735. [DOI] [PubMed] [Google Scholar]
- 30.Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 2010;33:391–8. doi: 10.1016/j.tins.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, et al. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–9. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghasemzadeh MB, Mueller C, Vasudevan P. Behavioral sensitization to cocaine is associated with increased glutamate receptor trafficking to the postsynaptic density after extended withdrawal period. Neuroscience. 2009;159:414–26.. doi: 10.1016/j.neuroscience.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 33.Yang HJ, Zhang HY, Bi GH, He Y, Gao JT, Xi ZX. Deletion of type 2 metabotropic glutamate receptor decreases sensitivity to cocaine reward in rats. Cell Rep. 2017;20:319–32.. doi: 10.1016/j.celrep.2017.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, et al. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–9. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- 35.McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–60.. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miguens M, Del Olmo N, Higuera-Matas A, Torres I, Garcia-Lecumberri C, Ambrosio E. Glutamate and aspartate levels in the nucleus accumbens during cocaine self-administration and extinction: a time course microdialysis study. Psychopharmacology. 2008;196:303–13.. doi: 10.1007/s00213-007-0958-x. [DOI] [PubMed] [Google Scholar]
- 37.Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–93.. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirischuk S, Kirchhoff F, Matyash V, Kettenmann H, Verkhratsky A. Glutamate-triggered calcium signalling in mouse bergmann glial cells in situ: role of inositol-1,4,5-trisphosphate-mediated intracellular calcium release. Neuroscience. 1999;92:1051–9. doi: 10.1016/S0306-4522(99)00067-6. [DOI] [PubMed] [Google Scholar]
- 39.Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. 2000;12:2333–44.. doi: 10.1046/j.1460-9568.2000.00131.x. [DOI] [PubMed] [Google Scholar]
- 40.Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–43.. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Martin R, Bajo-Graneras R, Moratalla R, Perea G, Araque A. GLIAL CELL SIGNALING. Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science. 2015;349:730–4. doi: 10.1126/science.aaa7945. [DOI] [PubMed] [Google Scholar]
- 42.Hoffmann HM, Crouzin N, Moreno E, Raivio N, Fuentes S, McCormick PJ, et al. Long-lasting impairment of mGluR5-activated intracellular pathways in the striatum after withdrawal of cocaine self-administration. Int J Neuropsychopharmacol. 2017;20:72–82. doi: 10.1093/ijnp/pyw086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knackstedt LA, Trantham-Davidson HL, Schwendt M. The role of ventral and dorsal striatum mGluR5 in relapse to cocaine-seeking and extinction learning. Addict Biol. 2014;19:87–101. doi: 10.1111/adb.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCutcheon JE, Loweth JA, Ford KA, Marinelli M, Wolf ME, Tseng KY. Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J Neurosci. 2011;31:14536–41.. doi: 10.1523/JNEUROSCI.3625-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pomierny-Chamiolo L, Miszkiel J, Frankowska M, Pomierny B, Niedzielska E, Smaga I, et al. Withdrawal from cocaine self-administration and yoked cocaine delivery dysregulates glutamatergic mGlu5 and NMDA receptors in the rat brain. Neurotox Res. 2015;27:246–58.. doi: 10.1007/s12640-014-9502-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scheyer AF, Christian DT, Wolf ME, Tseng KY. Emergence of endocytosis-dependent mGlu1 LTD at nucleus accumbens synapses after withdrawal from cocaine self-administration. Front Synaptic Neurosci. 2018;10:36. doi: 10.3389/fnsyn.2018.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xi ZX, Baker DA, Shen H, Carson DS, Kalivas PW. Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens. J Pharm Exp Ther. 2002;300:162–71.. doi: 10.1124/jpet.300.1.162. [DOI] [PubMed] [Google Scholar]
- 48.Cai Z, Schools GP, Kimelberg HK. Metabotropic glutamate receptors in acutely isolated hippocampal astrocytes: developmental changes of mGluR5 mRNA and functional expression. Glia. 2000;29:70–80. doi: 10.1002/(SICI)1098-1136(20000101)29:1<70::AID-GLIA7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 49.Devaraju P, Sun MY, Myers TL, Lauderdale K, Fiacco TA. Astrocytic group I mGluR-dependent potentiation of astrocytic glutamate and potassium uptake. J Neurophysiol. 2013;109:2404–14.. doi: 10.1152/jn.00517.2012. [DOI] [PubMed] [Google Scholar]
- 50.Morel L, Higashimori H, Tolman M, Yang Y. VGluT1+ neuronal glutamatergic signaling regulates postnatal developmental maturation of cortical protoplasmic astroglia. J Neurosci. 2014;34:10950–62.. doi: 10.1523/JNEUROSCI.1167-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Peng X-Q, Jordan CJ, Li J, Bi G-H, He Y, et al. mGluR5 antagonism inhibits cocaine reinforcement and relapse by elevation of extracellular glutamate in the nucleus accumbens via a CB1 receptor mechanism. Sci Rep. 2018;8:1–14.. doi: 10.1038/s41598-018-22087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bushong EA, Martone ME, Ellisman MH. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci. 2004;22:73–86. doi: 10.1016/j.ijdevneu.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, et al. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci. 2011;14:1276–84.. doi: 10.1038/nn.2929. [DOI] [PubMed] [Google Scholar]
- 54.Lavialle M, Aumann G, Anlauf E, Prols F, Arpin M, Derouiche A. Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc Natl Acad Sci USA. 2011;108:12915–9. doi: 10.1073/pnas.1100957108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Panatier A, Vallee J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–98.. doi: 10.1016/j.cell.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 56.Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006;29:547–53.. doi: 10.1016/j.tins.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 57.Gomez-Gonzalo M, Martin-Fernandez M, Martinez-Murillo R, Mederos S, Hernandez-Vivanco A, Jamison S, et al. Neuron-astrocyte signaling is preserved in the aging brain. Glia. 2017;65:569–80.. doi: 10.1002/glia.23112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirase H, Qian L, Bartho P, Buzsaki G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2004;2:E96. doi: 10.1371/journal.pbio.0020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pal B. Astrocytic actions on extrasynaptic neuronal currents. Front Cell Neurosci. 2015;9:474. doi: 10.3389/fncel.2015.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998;18:6822–9. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu J, Peng H, Kang N, Zhao Z, Lin JH, Stanton PK, et al. Glutamate-induced exocytosis of glutamate from astrocytes. J Biol Chem. 2007;282:24185–97.. doi: 10.1074/jbc.M700452200. [DOI] [PubMed] [Google Scholar]
- 62.Novak M, Halbout B, O’Connor EC, Rodriguez Parkitna J, Su T, Chai M, et al. Incentive learning underlying cocaine-seeking requires mGluR5 receptors located on dopamine D1 receptor-expressing neurons. J Neurosci. 2010;30:11973–82.. doi: 10.1523/JNEUROSCI.2550-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X, Moussawi K, Knackstedt L, Shen H, Kalivas PW. Role of mGluR5 neurotransmission in reinstated cocaine-seeking. Addict Biol. 2013;18:40–9. doi: 10.1111/j.1369-1600.2011.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bilbao A, Neuhofer D, Sepers M, Wei SP, Eisenhardt M, Hertle S, et al. Endocannabinoid LTD in Accumbal D1 neurons mediates reward-seeking behavior. iScience. 2020;23:100951. doi: 10.1016/j.isci.2020.100951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee SH, Ledri M, Toth B, Marchionni I, Henstridge CM, Dudok B, et al. Multiple forms of endocannabinoid and endovanilloid signaling regulate the tonic control of GABA Release. J Neurosci. 2015;35:10039–57.. doi: 10.1523/JNEUROSCI.4112-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baldi R, Ghosh D, Grueter BA, Patel S. Electrophysiological measurement of cannabinoid-mediated synaptic modulation in acute mouse brain slices. Curr Protoc Neurosci. 2016;75:62921–19.. doi: 10.1002/cpns.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramikie TS, Nyilas R, Bluett RJ, Gamble-George JC, Hartley ND, Mackie K, et al. Multiple mechanistically distinct modes of endocannabinoid mobilization at central amygdala glutamatergic synapses. Neuron. 2014;81:1111–25.. doi: 10.1016/j.neuron.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kellogg R, Mackie K, Straiker A. Cannabinoid CB1 receptor-dependent long-term depression in autaptic excitatory neurons. J Neurophysiol. 2009;102:1160–71.. doi: 10.1152/jn.00266.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–63.. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- 70.Pertwee RG. Endocannabinoids and their pharmacological actions. Handb Exp Pharm. 2015;231:1–37. doi: 10.1007/978-3-319-20825-1_1. [DOI] [PubMed] [Google Scholar]
- 71.Sugiura T, Kishimoto S, Oka S, Gokoh M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res. 2006;45:405–46.. doi: 10.1016/j.plipres.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 72.Xu C, Hermes DJ, Nwanguma B, Jacobs IR, Mackie K, Mukhopadhyay S, et al. Endocannabinoids exert CB1 receptor-mediated neuroprotective effects in models of neuronal damage induced by HIV-1 Tat protein. Mol Cell Neurosci. 2017;83:92–102. doi: 10.1016/j.mcn.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Centanni SW, Morris BD, Luchsinger JR, Bedse G, Fetterly TL, Patel S, et al. Endocannabinoid control of the insular-bed nucleus of the stria terminalis circuit regulates negative affective behavior associated with alcohol abstinence. Neuropsychopharmacology. 2019;44:526–37. doi: 10.1038/s41386-018-0257-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rossi S, De Chiara V, Musella A, Sacchetti L, Cantarella C, Castelli M, et al. Preservation of striatal cannabinoid CB1 receptor function correlates with the antianxiety effects of fatty acid amide hydrolase inhibition. Mol Pharm. 2010;78:260–8. doi: 10.1124/mol.110.064196. [DOI] [PubMed] [Google Scholar]
- 75.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–7. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 76.Chavez AE, Chiu CQ, Castillo PE. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat Neurosci. 2010;13:1511–8. doi: 10.1038/nn.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deroche MA, Lassalle O, Castell L, Valjent E, Manzoni OJ. Cell-Type- and endocannabinoid-specific synapse connectivity in the adult nucleus accumbens core. J Neurosci. 2020;40:1028–41.. doi: 10.1523/JNEUROSCI.1100-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buczynski MW, Parsons LH. Quantification of brain endocannabinoid levels: methods, interpretations and pitfalls. Br J Pharmacol. 2010;160:423–42.. doi: 10.1111/j.1476-5381.2010.00787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Musella A, De Chiara V, Rossi S, Prosperetti C, Bernardi G, Maccarrone M, et al. TRPV1 channels facilitate glutamate transmission in the striatum. Mol Cell Neurosci. 2009;40:89–97. doi: 10.1016/j.mcn.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 80.Bystrowska B, Frankowska M, Smaga I, Pomierny-Chamiolo L, Filip M. Effects of cocaine self-administration and its extinction on the rat brain cannabinoid CB1 and CB2 receptors. Neurotox Res. 2018;34:547–58.. doi: 10.1007/s12640-018-9910-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.You IJ, Hong SI, Ma SX, Nguyen TL, Kwon SH, Lee SY, et al. Transient receptor potential vanilloid 1 mediates cocaine reinstatement via the D1 dopamine receptor in the nucleus accumbens. J Psychopharmacol. 2019;33:1491–500. [DOI] [PubMed]
- 82.Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA. 2000;97:8629–34.. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, et al. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–84.. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR- mediated neuronal excitation. Nat Neurosci. 2001;4:803–12.. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- 85.Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–7. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carlton SM, Du J, Zhou S. Group II metabotropic glutamate receptor activation on peripheral nociceptors modulates TRPV1 function. Brain Res. 2009;1248:86–95. doi: 10.1016/j.brainres.2008.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scherer PC, Zaccor NW, Neumann NM, Vasavda C, Barrow R, Ewald AJ, et al. TRPV1 is a physiological regulator of mu-opioid receptors. Proc Natl Acad Sci USA. 2017;114:13561–66.. doi: 10.1073/pnas.1717005114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mao LM, Liu XY, Zhang GC, Chu XP, Fibuch EE, Wang LS, et al. Phosphorylation of group I metabotropic glutamate receptors (mGluR1/5) in vitro and in vivo. Neuropharmacology. 2008;55:403–8. doi: 10.1016/j.neuropharm.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dale LB, Bhattacharya M, Anborgh PH, Murdoch B, Bhatia M, Nakanishi S, et al. G protein-coupled receptor kinase-mediated desensitization of metabotropic glutamate receptor 1A protects against cell death. J Biol Chem. 2000;275:38213–20.. doi: 10.1074/jbc.M006075200. [DOI] [PubMed] [Google Scholar]
- 90.Iacovelli L, Molinaro G, Battaglia G, Motolese M, Di Menna L, Alfiero M, et al. Regulation of group II metabotropic glutamate receptors by G protein-coupled receptor kinases: mGlu2 receptors are resistant to homologous desensitization. Mol Pharm. 2009;75:991–1003. doi: 10.1124/mol.108.052316. [DOI] [PubMed] [Google Scholar]
- 91.Ribeiro FM, Ferreira LT, Paquet M, Cregan T, Ding Q, Gros R, et al. Phosphorylation-independent regulation of metabotropic glutamate receptor 5 desensitization and internalization by G protein-coupled receptor kinase 2 in neurons. J Biol Chem. 2009;284:23444–53.. doi: 10.1074/jbc.M109.000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramirez-Nino AM, D’Souza MS, Markou A. N-acetylcysteine decreased nicotine self-administration and cue-induced reinstatement of nicotine seeking in rats: comparison with the effects of N-acetylcysteine on food responding and food seeking. Psychopharmacology. 2013;225:473–82.. doi: 10.1007/s00213-012-2837-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–60. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, et al. Activator of G protein signaling 3: a gatekeeper of cocaine sensitization and drug seeking. Neuron. 2004;42:269–81.. doi: 10.1016/S0896-6273(04)00159-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reissner KJ, Kalivas PW. Using glutamate homeostasis as a target for treating addictive disorders. Behav Pharm. 2010;21:514–22.. doi: 10.1097/FBP.0b013e32833d41b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McGuire P, Robson P, Cubala WJ, Vasile D, Morrison PD, Barron R, et al. Cannabidiol (CBD) as an Adjunctive Therapy in Schizophrenia: A Multicenter Randomized Controlled Trial. Am J Psychiatry. 2018;175:225–31. [DOI] [PubMed]
- 97.Bergamaschi MM, Queiroz RH, Chagas MH, de Oliveira DC, De Martinis BS, Kapczinski F, et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naive social phobia patients. Neuropsychopharmacology. 2011;36:1219–26. doi: 10.1038/npp.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Costa B, Trovato AE, Comelli F, Giagnoni G, Colleoni M. The non-psychoactive cannabis constituent cannabidiol is an orally effective therapeutic agent in rat chronic inflammatory and neuropathic pain. Eur J Pharmacol. 2007;556:75–83. doi: 10.1016/j.ejphar.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 99.Adamczyk P, Miszkiel J, McCreary AC, Filip M, Papp M, Przegalinski E. The effects of cannabinoid CB1, CB2 and vanilloid TRPV1 receptor antagonists on cocaine addictive behavior in rats. Brain Res. 2012;1444:45–54. doi: 10.1016/j.brainres.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 100.Mayo LM, Asratian A, Lindé J, Morena M, Haataja R, Hammar V, et al. Elevated Anandamide, Enhanced Recall of Fear Extinction, and Attenuated Stress Responses Following Inhibition of Fatty Acid Amide Hydrolase: A Randomized, Controlled Experimental Medicine Trial. Biol Psychiatry. 2020;87:538–47. doi: 10.1016/j.biopsych.2019.07.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.