

Abstract
Dyslipidemia is a chronic metabolic disease characterized by elevated levels of lipids in plasma. Recently, various studies demonstrate that the increased activity of adenosine 5′-monophosphate-activated protein kinase (AMPK) causes health benefits in energy regulation. Thus, great efforts have been made to develop AMPK activators as a metabolic syndrome treatment. In the present study, we investigated the effects of the AMPK activator C24 on dyslipidemia and the potential mechanisms. We showed that C24 (5–40 μM) dose-dependently increased the phosphorylation of AMPKα and acetyl-CoA carboxylase (ACC), and inhibited lipogenesis in HepG2 cells. Using compound C, an AMPK inhibitor, or hepatocytes isolated from liver tissue-specific AMPK knockout AMPKα1α2fl/fl;Alb-cre mice (AMPK LKO), we demonstrated that the lipogenesis inhibition of C24 was dependent on hepatic AMPK activation. In rabbits with high-fat and high-cholesterol diet-induced dyslipidemia, administration of C24 (20, 40, and 60 mg · kg−1· d−1, ig, for 4 weeks) dose-dependently decreased the content of TG, total cholesterol (TC), and low-density lipoprotein cholesterol (LDL-C) in plasma and played a role in protecting against hepatic dysfunction by decreasing lipid accumulation. A lipid-lowering effect was also observed in high-fat and high-cholesterol diet-fed hamsters. In conclusion, our results demonstrate that the small molecular AMPK activator C24 alleviates hyperlipidemia and represents a promising compound for the development of a lipid-lowering drug.
Keywords: adenosine 5′-monophosphate-activated protein kinase, AMPK activator, C24, liver, triglycerides, cholesterol, VLDL, hypolipidemic drug, metabolic syndrome
Introduction
Elevated living standards accompanied by increasing human overnutrition have increased the incidence of dyslipidemia and cardiovascular diseases (CVDs), which cause significant social burdens worldwide. Dyslipidemia is a hallmark of metabolic syndrome, and it is characterized by elevated triglyceride (TG), low-density lipoprotein cholesterol (LDL-C), and apolipoprotein B (apoB) values and reduced high-density lipoprotein cholesterol (HDL-C) values in plasma [1]. Dyslipidemia is one of the leading causes of CVDs [2]. Although lifestyle improvements and existing pharmaceutical therapies (such as statins, ezetimibe, and bile acid sequestrants) partially effectively control disease progression [3], many individuals do not achieve their lipid-lowering targets and remain at a high risk of developing CVDs [4]. These individuals may need novel drugs as alternative therapies to prevent cardiovascular events and related comorbidities.
Adenosine 5′-monophosphate-activated protein kinase (AMPK) is a Ser/Thr protein kinase that consists of a catalytic subunit (α) and two regulatory subunits (β and γ) [5]. AMPK plays a pivotal role in energy metabolism, and it senses multiple physiological and pathogenetic processes, such as exercise, heat shock, ischemia, and hypoxia [6]. AMPK is activated by energy shortages (e.g., upregulation of the ratio of AMP/ADP:ATP), increases the rate of catabolic pathways and decreases the rate of anabolic pathways to control energy homeostasis via α-subunit T172 phosphorylation [7–10]. AMPK has been proposed as a potential therapeutic target for metabolic disorders based on these roles [11–13].
The liver is one of the most important organs for lipid synthesis and output. AMPK activation in the liver inhibits the process of lipogenesis and induces fatty acid oxidation by mediating the phosphorylation of downstream substrates, which are the rate-limiting proteins for lipid metabolism [14, 15]. Acetyl-CoA carboxylase (ACC) is an enzyme that converts acetyl-CoA to malonyl-CoA to provide a precursor for fatty acid synthesis. AMPK phosphorylates ACC (S79) and inhibits the production of precursors to suppress fatty acid synthesis. A decrease in cellular malonyl-CoA relieves the inhibitory effect on carnitine palmitoyl transferase-1 (CPT-1) and accelerates fatty acid oxidation [16, 17]. 3-Hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) is a key enzyme in cholesterol biosynthesis. AMPK activation inhibits the activity of HMGCR via the phosphorylation site at Ser872, which reduces cholesterol biosynthesis [18–20]. Sterol regulatory-element binding proteins (SREBPs) are the key transcription factors in regulating the activities of gene promoters involved in maintaining sterol and fatty acid homeostasis [21]. AMPK phosphorylates and inhibits SREBP transcriptional activity to reduce lipogenesis and lipid accumulation [22]. Therefore, the role of AMPK activation in lipid metabolism processes in the liver reveals the potential of AMPK activators for dyslipidemia treatment [23].
C24 is a small-molecule AMPK activator based on PT1 structure modification. Compared to PT1, it has better oral bioavailability, dissolution and potential application in drug discovery [24, 25]. We previously identified that C24 activated AMPK by relieving α subunit autoinhibition, which decreased gluconeogenesis and exerted beneficial effects in diabetic db/db mice [26]. However, little is known about the role of C24 in lipid regulation. The present study aims to evaluate C24 function in lipid-lowering and clarify the potential mechanism to provide more evidence for the activation of AMPK as a dyslipidemia treatment.
Materials and methods
Animals
Male C57BL/6J mice approximately 8 weeks old were purchased from Shanghai Model Organisms (Shanghai, China). Male Golden Syrian hamsters approximately 8 weeks old were purchased from Charles River (Beijing, China). New Zealand rabbits were purchased from Shanghai Jambo Biological Technology (Shanghai, China). Animal welfare and animal experimental procedures were performed in accordance with the current guide of the Animal Ethics Committee of the Shanghai Institute of Materia Medica. Animals were maintained at 22 ± 2 °C under a 12-h light–dark cycle. Food and water were available ad libitum. Age-matched animals were used for all experiments. To obtain liver tissue-specific AMPKα1/α2 double knockout mice, AMPKα1/α2-floxed mice were crossed with Alb-cre mice to generate the liver tissue-specific AMPK knockout AMPKα1α2fl/fl;Alb-cre mice (AMPK LKO) [27].
C24 formulation for in vivo study
The compound C24 (C29H20ClNO3) is an AMPK activator that we identified previously [25, 26]. C24 was prepared as an orange solid according to the synthetic method disclosed in CN 201710208352. For the in vivo study, C24 and N-methylglucamine were dissolved at a mass ratio of 2:1 in deionized water. This solution was subjected to spray drying, which led to a water-soluble powder as the formulation of C24 with N-methylglucamine (content of C24 in formulated powder, 66.7%). This formation was readily dissolved in 0.5% carboxymethyl cellulose (0.5% CMC-Na) for oral administration to animals.
Cell culture
HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific, catalog number: 12800017, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, catalog number: 10091-148, Waltham, MA, USA) and incubated under 5% CO2 at 37 °C. Cells were passaged at 80%–90% confluence and plated for experiments.
Hepatocyte isolation
Hepatocyte isolation was performed as reported previously [28]. Briefly, C57BL/6J mice were anesthetized and then perfused with calcium-free buffer containing calcium chelators from the inferior vena cava to the liver and exiting from the portal vein. This step removed the blood and caused destruction of some of the intercellular junctions due to calcium loss. A collagenase enzyme (Worthington-Biochem., catalog number: LS004196, Lakewood, NJ, USA) was added to the buffer to breakdown the extracellular matrix and the tight junctions. The liver was stripped and shredded mechanically, and then the hepatocytes were obtained by Percoll (Sigma-Aldrich, catalog number: P1644-1L, St. Louis, MO, USA) density-gradient centrifugation. Cells were plated for follow-up experiments.
Measurement of lipogenesis using [1,2-14C] acetic acid (sodium salt) in vitro
HepG2 cells or hepatocytes were cultured in six-well plates and treated with C24 at the indicated concentrations for 20 h. [1,2-14C] Acetic acid (sodium salt) (Perkin Elmer, catalog number: NEC553001MC, Waltham, MA, USA) (0.1 μCi/well) was added for another 4 h. The medium was removed, and the cells were washed gently three times with cold PBS. Cells were lysed with 0.5 M KOH, and [14C]-labeled de novo lipogenesis in cells was examined using petroleum ether and measured in scintillation liquids.
Western blotting analyses of cells and animal tissue samples
Cell sample preparation: at the end of treatment, the culture medium was removed, 100 μL of loading buffer was added to each well to lyse cells. The samples were processed with heating to denature the protein at 100 °C for 10 min.
Tissue sample preparation
liver tissues were lysed in RIPA buffer (Beyotime, catalog number: P0013B, Haimen, China) with the addition of protease inhibitors and phosphatase inhibitors. The protein concentrations of the samples were measured using the Bradford assay. Approximately, 20 μg of protein from the samples was used for loading.
Samples were loaded into 10% SDS-PAGE lanes and transferred to NC membranes (GE Healthcare, catalog number: 10600002, Little Chalfont, Bukinghamshire, UK). The membranes were blocked in 5% fat-free milk and incubated with primary antibodies on a shaker at 4 °C overnight. The membranes were washed in TBST three times for 10 min and incubated with secondary anti-mouse or anti-rabbit antibodies for 1 h. The membranes were washed three times, and the protein contents were measured using ECLTM prime Western blotting detection reagents (GE Healthcare, catalog number: RPN2232, Little Chalfont, Bukinghamshire, UK) on a ChemiDocTM Touch image system (Bio-Rad, Hercules, CA, USA). Total AMPKα (catalog number: 2532L), phosphorylated AMPKα (catalog number: 2535L), total ACC (catalog number: 3662S), and phosphorylated ACC (catalog number: 3661S) antibodies were purchased from Cell Signaling Technology (Trask Lane Danvers, MA, USA). The LDLR antibody (catalog number: 66414-1-lg) was obtained from Proteintech (Rosemont, IL, USA). β-Actin (catalog number: AM1021B) was purchased from ABGENT (San Diego, CA, USA). Peroxidase-affinipure goat anti-rabbit IgG (H + L) (catalog number: 111-035-003) and peroxidase-affinipure goat anti-mouse IgG (H + L) (catalog number: 115-035-003) were purchased from Jackson ImmunoResearch Laboratories (Pike West Grove, Pennsylvania, PA, USA).
Relative quantitative PCR
RNA was extracted from treated cells or mouse liver tissues by using TRIzol (TaKaRa, catalog number: 9019, Japan). Then, RNA (1000 ng) was transcribed to cDNA. cDNA mixed with SYBR Green (Bimake, catalog number: B21702, Houston, TX, USA) and gene primers were used to detect gene expression under the Mx30005PTM Q-PCR Systems (Agilent Technologies, Santa Clara, CA, USA). GAPDH was used as the reference gene. The relative quantity of mRNA was normalized to the control group using the ΔΔCT method, and the primer sequences are shown in the Supplementary material.
Relative AMPK activities measured in vivo
Male C57BL/6J mice weighing 20–25 g at 8–10 weeks were randomly assigned to two groups (n = 6). Mice were treated with 0.5% CMC-Na (Vehicle) or C24 (100 mg· kg−1) via gavage. After 2 h, the mice were sacrificed after anesthetization, and the livers were collected as soon as possible and frozen in liquid nitrogen. The phosphorylation of hepatic AMPKα (T172) and ACC (S79) levels was detected using Western blotting analysis to reflect AMPK activity.
Measurement of lipogenesis using [1,2-14C] acetic acid (sodium salt) in vivo
The lipogenesis effect of C24 in vivo was measured using previously described experimental methods [29]. Briefly, eight male C57BL/6J mice weighing 20–25 g at 8–10 weeks of age were divided into two groups (n = 4). Mice were fasted for 48 h followed by refeeding a high-fat and high-carbohydrate diet (Research diets, catalog number: D17010102, NJ, USA) for another 48 h. Animals received one dose of vehicle or C24 (100 mg · kg−1). 1 h later, all mice were intraperitoneally injected with 1 μCi [1,2-14C] acetate. 1 h after [1,2-14C] acetate injection, the mice were sacrificed after anesthetization. Approximately 100 mg of liver was homogenized in cold PBS. The [14C]-labeled lipids were extracted and detected in the same manner as the cell lysates.
VLDL-TG secretion experiment
Male C57BL/6J mice weighing 20–25 g at 8–10 weeks were fasted for 48 h. The mice were divided into two groups randomly (n = 9–10), refed a high-carbohydrate diet (Research Diets, catalog number: D10012G, NJ, USA) and treated with vehicle (0.5% CMC-Na, n = 10) or C24 (100 mg · kg−1 · d−1, n = 9) for 3 d. The lipogenesis ability of livers is elevated during refeeding [30]. After the last administration, the mice were injected with tyloxapol (600 mg · kg−1) via the tail vein to inhibit endogenous lipoprotein lipase [31]. The content of very-LDL-TG (VLDL-TG) secreted into the circulation in 4 h was measured. The collected blood from the tail vein was subjected to centrifugation at 12,000×g for 2 min, and plasma was obtained. Plasma TG was measured using a TG determination kit (Shanghai Changzheng, catalog number: 1.02.1801, Shanghai, China).
New Zealand rabbit experiment
Diet-enriched fat- and cholesterol-fed rabbits are often used to study metabolic disease [32]. New Zealand rabbits weighing 2.0–2.5 kg were fed a high-fat high-cholesterol diet or NC after a 5-d adaptation. A NC diet was obtained from the Shanghai Xinhui Animal Feed Co., Ltd. The high-fat and high-cholesterol diet containing 5% lard oil and 0.5% cholesterol (referred to as the HC diet) was purchased from Jiangsu Xietong Organism Co., Ltd. Dyslipidemia in rabbits was imposed by feeding New Zealand rabbits an HC diet for 75 d. Plasma TC ≥ 10 mmol · L−1 was used as the filter criterion. Hyperlipidemic rabbits were randomly assigned to different groups (n = 9). The vehicle group was treated with CMC-Na. C24 was administered at 20, 40, and 60 mg· kg−1· d−1 for 4 weeks via gavage. Atorvastatin (0.5 mg · kg−1· d−1) and fenofibrate (10 mg · kg−1 · d−1) were the positive control drugs. Body weights and food intake were recorded regularly. The concentrations of plasma TG, TC, LDL-C, HDL-C, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured every two weeks. The TG assay kit (catalog number: A110-1-1), TC assay kit (catalog number: A111-1-1), LDL-C assay kit (catalog number: A113-1-1), HDL-C assay kit (catalog number: A112-1-1), AST assay kit (catalog number: A010-2-1), and ALT assay kit (catalog number: A009-2-1) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Golden Syrian hamster experiment
All hamsters at 6–8 weeks were maintained on a standard rodent chow diet during a 7-d adaptive period and then fed a high-fat and high-cholesterol diet containing 11.5% coconut oil, 11.5% corn oil, 5% fructose, and 0.5% cholesterol (referred to as the HFHC diet) (Research Diets, catalog number: C11953, New Brunswick, NJ, USA) for 14 d. The HFHC diet rapidly induced hyperlipidemia in response to the added cholesterol and fat [33, 34]. The hamsters were randomly assigned to different groups (n = 8) and treated with CMC-Na (vehicle group) or C24 (20, 40, and 60 mg· kg−1 · d−1) for 2 weeks via gavage. Fenofibrate (100 mg· kg−1· d−1) was the positive-control drug. Food intake and body weights were monitored during the experiment. Blood was collected from the retro-orbital sinus and subjected to centrifugation at 12,000×g for 2 min at 4 °C to obtain plasma. The concentration of lipid profiles (TC, TG, LDL-C, and HDL-C), AST, and ALT were measured using commercial kits, as described in the kit’s instructions. At the end of the experiment, hamsters were anesthetized and sacrificed. Liver samples were collected and stored at −80 °C for further analyses. The TG assay kit (catalog number: 1.02.1801) and TC assay kit (catalog number: 1.02.0401) were purchased from Shanghai Changzheng Co., Ltd. (Shanghai, China). The LDL-C assay kit (catalog number: E0410) and HDL assay kit (catalog number: E0310) were purchased from Xinjiankangcheng Co., Ltd. (Sichuang, China). The AST assay kit (catalog number: 290705 and 290706) and ALT assay kit (catalog number: 290703 and 290704) were purchased from Sysmex (Shanghai, China).
Statistical analysis
The results are represented as the means ± S.D. Differences between two groups were examined using a two-tailed unpaired Student’s t test. Differences in multiple groups were compared using one-way ANOVA or two-way ANOVA. P < 0.05 was considered statistically significant.
Results
C24 activates AMPK in HepG2 cells
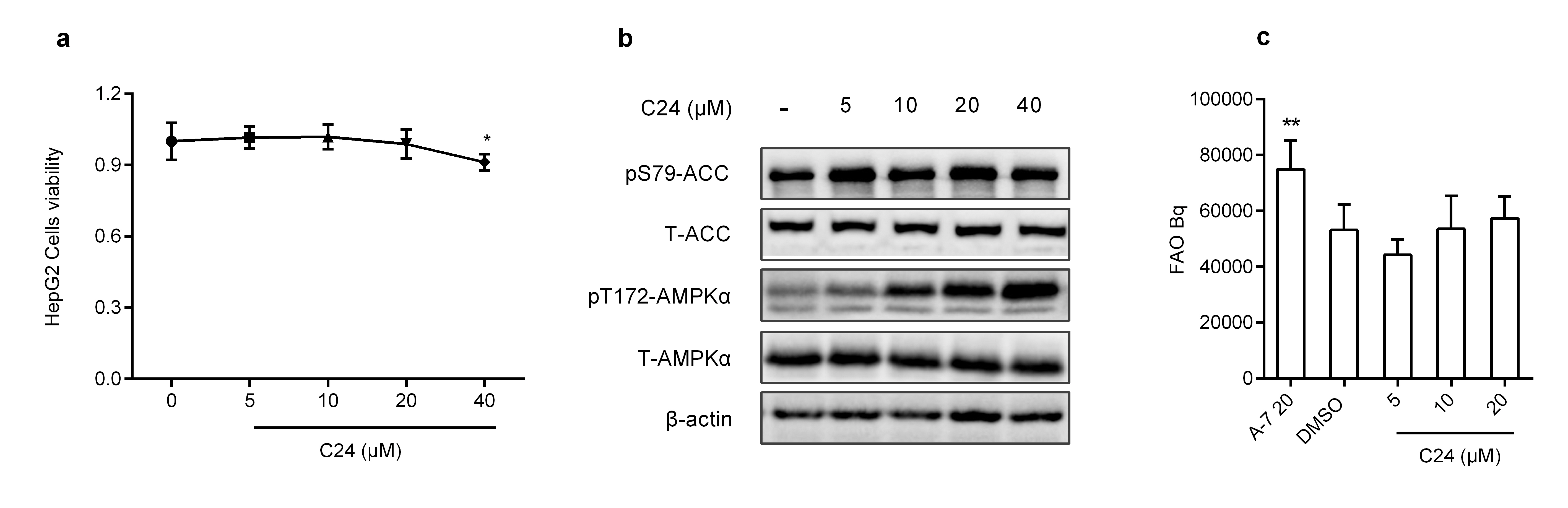
Human liver carcinoma HepG2 cells are widely used as an in vitro model to evaluate lipid metabolism. We detected the viability of HepG2 cells cultured in C24-containing medium. The CCK-8 assay demonstrated that C24 had almost no effect on cell viability after 24 h of treatment at doses less than 20 μM (Supplementary Fig. S1a). We evaluated the effects of C24 on AMPK activation in HepG2 cells. C24 treatment increased the phosphorylation of AMPKα (T172) and its substrate ACC (S79) in a time- and dose-dependent manner (Fig. 1a, b). Intracellular AMPK remained activated after 24 h of C24 treatment (Supplementary Fig. S1b). These data revealed that C24 activated AMPK in HepG2 cells, which was accompanied by an increase in ACC phosphorylation.
Fig. 1. C24 activates AMPK in HepG2 cells.

a, b Western blotting analysis and relative integrated density of indicated proteins at different times (a) or doses (b) of C24 treatment in HepG2 cells. Data are presented as the means ± S.D. Multiple groups were analyzed using one-way ANOVA, *P < 0.05 vs. DMSO; **P < 0.01 vs. DMSO; ***P < 0.001 vs. DMSO.
The effects of C24 on lipogenesis in vitro and in vivo
AMPK activation in the liver inhibits lipogenesis [35, 36]. Our previous data indicated that C24 decreased the content of TG and cholesterol in HepG2 cells [26]. To characterize the effect of C24 on de novo lipogenesis, [1,2-14C] acetate was used to label the de novo lipogenesis process. In HepG2 cells, we found that C24 moderately but significantly inhibited the biosynthesis of lipids (Fig. 2a). In vivo, we treated fasted-refed mice with a single dose of C24 (100 mg · kg−1) and sacrificed the mice after 2 h. We found that C24 had a tendency to reduce the incorporation of [1,2-14C] acetate into lipids in the liver (Fig. 2d). Consistent with C24-induced AMPK activation in HepG2 cells, we also identified that the phosphorylation levels of hepatic AMPKα (T172) and ACC (S79) were much higher in C24-treated mice than in vehicle-treated mice (Fig. 2b, c).
Fig. 2. The effects of C24 on lipogenesis in vitro and in vivo.

a The effect of 14C-labeled de novo lipogenesis in HepG2 cells after different doses of C24 treatment. b, c Western blotting analysis (b) and relative integrated density (c) of the indicated proteins in livers of C57BL/6J mice after 2 h of treatment with or without C24 (100 mg · kg−1) (n = 6). d The effect of 14C-labeled de novo lipogenesis in liver of fasted-refed mice after 2 h of treatment (n = 4). e, f After tyloxapol (600 mg · kg−1) tail vein injection, 0–4 h of the plasma TG concentration (e) and the area under curve (AUC) (f) were measured in high-carbohydrate diet-fed C57BL/6J mice after 3 d of treatment (n = 9/10). Data are presented as the means ± S.D. Multiple groups were analyzed using one-way ANOVA. Two groups were analyzed using two-tailed unpaired Student’s t test or two-way ANOVA, *P < 0.05 vs. control; **P < 0.01 vs. control; ***P < 0.001 vs. control.
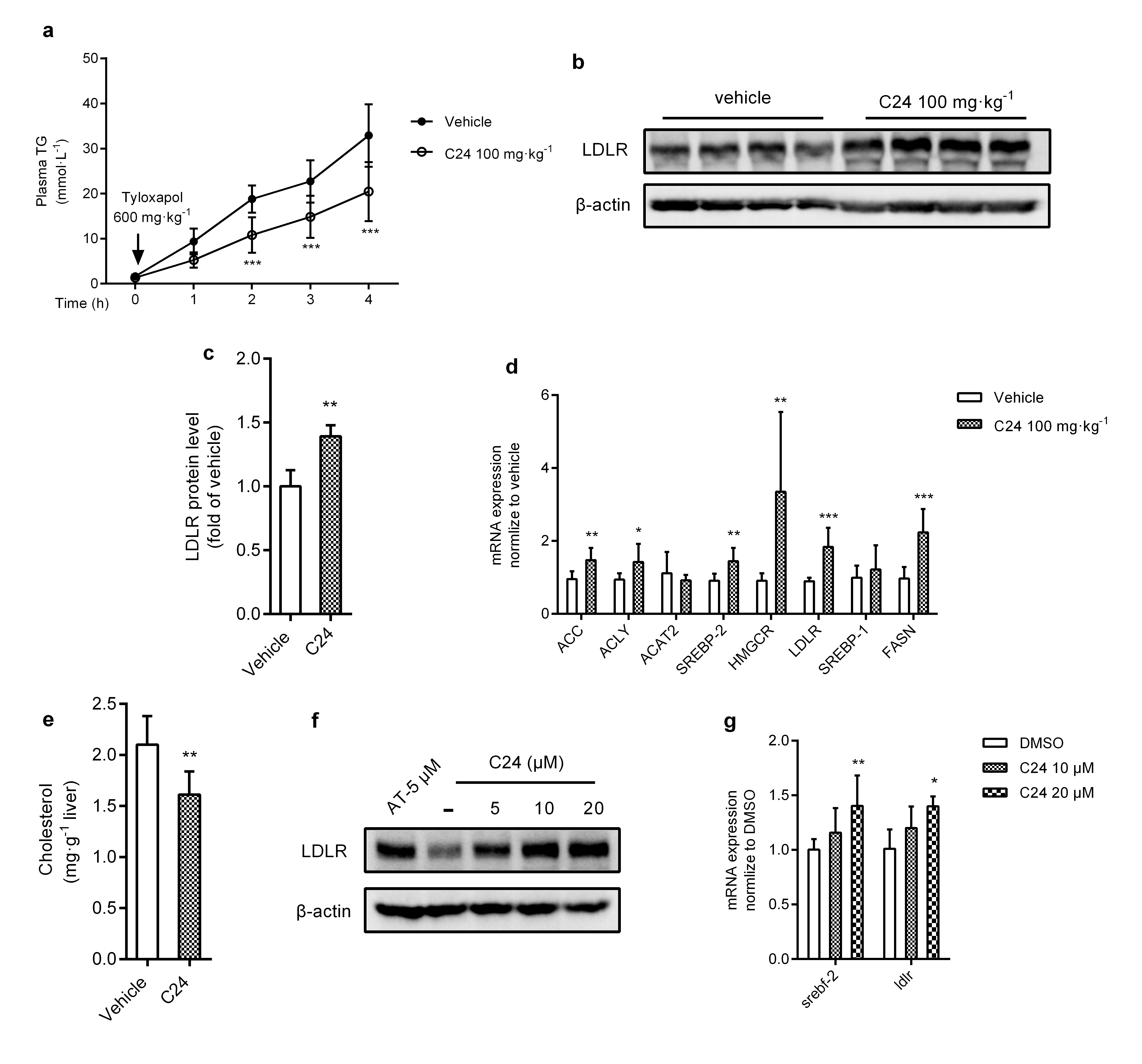
VLDL-TG secretion is an important mechanism for liver lipid output. Dyslipidemia is partially due to overproduction or increased secretion of VLDL by the liver. To test whether C24 treatment reduced VLDL-TG secretion in vivo, C57BL/6J mice were fasted for 48 h, fed a high-carbohydrate diet and treated with or without C24 (100 mg · kg−1) for 3 d via gavage. After the final administration, mice were injected with tyloxapol (600 mg · kg−1) to reduce TG degradation and clearance. VLDL-TG secretion was determined as the increase in TG content in plasma. Our findings showed that TG secretion in C24-treated mice was significantly decreased compared to that in the vehicle group (Fig. 2e, f). Taken together, these results indicated that lipogenesis inhibition by C24 treatment contributed to hepatic lipid regulation.
C24 inhibits lipogenesis via the AMPK pathway in hepatocytes
Compound C is a pan-kinase inhibitor. We used compound C to inhibit cellular AMPK activity. Compound C treatment blocked the effect of C24 on the phosphorylation level of AMPKα (T172) and its downstream ACC (S79) in HepG2 cells (Fig. 3a). Consistently, the inhibition of lipogenesis induced by C24 was weakened by cotreatment with Compound C (Fig. 3b). Our results suggested that AMPK activation is required for the lipogenesis inhibition of C24.
Fig. 3. C24 inhibits lipogenesis via AMPK pathway.

a, b Western blotting analysis of the indicated proteins (a) and 14C-labeled de novo lipogenesis (b) in HepG2 cells after compound C and C24 treatment. CC represents compound C, ns means no significant difference. c, d Western blotting analysis of the efficiency of AMPKα knockout and effects of C24 on AMPK activation in hepatocytes isolated from AMPK FLOX and AMPK LKO mice. e, f Effects of C24 on lipogenesis in hepatocytes isolated from AMPK FLOX (e) and AMPK LKO (f) mice. Data are presented as the means ± S.D. Multiple groups were analyzed using one-way ANOVA, two groups were analyzed using two-tailed unpaired Student’s t test, *P < 0.05 vs. control; **P < 0.01 vs. control.
We used hepatocytes isolated from AMPKα1α2flox/flox mice (AMPK FLOX) and AMPKα1α2fl/fl;Alb-cre mice (AMPK LKO) to confirm the dependence of AMPK activation on the effects of C24 on lipogenesis. The protein and phosphorylation levels of AMPKα (T172) in AMPK FLOX- and AMPK LKO-isolated hepatocytes were confirmed using immunoblotting analysis. C24 treatment activated the phosphorylation of AMPKα (T172) and ACC (S79) in a dose- and time-dependent manner, and AMPKα deficiency blocked C24-induced phosphorylation of ACC (S79) in hepatocytes (Fig. 3c, d). C24 (10 μM) treatment inhibited de novo lipogenesis in hepatocytes isolated from AMPK FLOX mice but not in hepatocytes isolated from AMPK LKO mice. These results suggested that the inhibition of lipogenesis by C24 required intracellular AMPK activation.
Chronic C24 treatment ameliorates lipid profiles in HC diet-induced New Zealand rabbits
The rabbit is a classical species in studies of dyslipidemia [37]. We fed rabbits an HC diet for 75 d to induce preexisting hyperlipidemia to evaluate the lipid-lowering effects of C24. The rabbits were randomly divided into different groups for different treatments for 4 weeks. Our findings showed that C24 did not cause weight loss (Supplementary Fig. S2a) but exhibited different degrees of improvement in the HC diet-induced accumulation of TC, TG, and LDL-C concentrations in plasma (Fig. 4b–d); HDL-C did not show significant changes (Fig. 4e). The positive control drug groups atorvastatin and fenofibrate showed significant lipid-lowering effects.
Fig. 4. Chronic C24 treatment improves lipid profiles in HC-fed New Zealand rabbits.

a Schematic diagram of the C24 pharmacodynamic experiment in HC diet-fed New Zealand rabbits. b–g The plasma concentrations of TG (b), TC (c), LDL-C (d), HDL-C (e), ALT (f), and AST (g) in HC diet-fed New Zealand rabbits after treatment for 4 weeks. NC represents rabbits fed normal chow and treated with CMC-Na. h The contents of triglycerides in livers of HC diet-fed New Zealand rabbits after treatment for 4 weeks. The data are presented as the means ± S.D., n = 9. Multiple groups were analyzed using one-way ANOVA, **P < 0.01 vs. Vehicle; ***P < 0.001 vs. Vehicle.
Long-term high-fat diet feeding induces lipid metabolism disorder in New Zealand rabbits, and the livers of these animals tend to develop hepatic steatosis and increase liver injury susceptibility [38]. The administration of C24 reduced the accumulation of lipids in the liver (Fig. 4h) and the liver index (liver/body weight) (Supplementary Fig. S2b). HC diet-fed rabbits had accelerated liver injury and increased serum AST and ALT activities compared to the normal chow (NC) group. In contrast, the serum active units of ALT and AST in C24-treated rabbits were obviously lower than those in the vehicle group (Fig. 4f, g).
These results demonstrated that chronic treatment with C24 ameliorated dyslipidemia and provided a certain degree of protection against liver injury in the fatty liver disease process.
Chronic C24 treatment improves dyslipidemia in HFHC hamsters
The hamster is another classical animal model in the evaluation of hypolipidemic effects [39]. We also verified the effect of C24 on lipid regulation in golden Syrian hamsters. Before treatment, animals were fed an HFHC diet for 14 d to induce hyperlipemia. After treatment with C24 (20, 40, and 60 mg · kg−1 · d−1) for 2 weeks, there were no differences in body weight (Fig. 5b) or food intake (Fig. 5c) among the groups, but the plasma TC, TG, and LDL-C contents in the treatment groups decreased strikingly compared to the vehicle group (Fig. 5d–f). HDL-C was also decreased in parallel with the reduction in plasma cholesterol (Fig. 5g). These results showed that C24 improved lipid metabolism in dyslipidemia hamsters.
Fig. 5. C24 treatment improves dyslipidemia in HFHC-fed hamsters.

a Schematic diagram of the pharmacodynamic experiment in HFHC diet-fed hamsters. b, c The body weight and food intake during treatment in HFHC diet-fed hamsters. NC represents hamsters fed normal chow and treated with CMC-Na. d–g The concentrations of plasma TC (d), TG (e), LDL-C (f), and HDL-C (g) after treatment with or without C24 for 2 weeks. Data are presented as the means ± S.D., n = 8. Body weights and food intake were analyzed using two-way ANOVA, multiple groups were analyzed using one-way ANOVA, *P < 0.05 vs. Vehicle; **P < 0.01 vs. Vehicle; ***P < 0.001 vs. Vehicle.
Discussion
AMPK activation exerts beneficial effects on diabetes, dyslipidemia, and metabolic syndrome. Many published studies have provided a solid basis for the development of an AMPK activator to treat a series of related diseases [23, 40]. C24 is an AMPK activator based on alkene oxindole scaffold modification, and it exhibits favorable pharmacokinetic characteristics after oral administration in Wistar rats [25]. We previously identified that C24 decreased hepatic gluconeogenesis and exerted antidiabetic effects in db/db mice [26]. In the present study, we found that C24 downregulated hepatic lipogenesis, decreased VLDL-TG secretion into the circulation and ameliorated dyslipidemia in preclinical animal models. The AMPK activator C24 showed beneficial effects and potency as an orally administered compound for dyslipidemia treatment.
Overnutrition induces dysfunction in glucose and lipid metabolism, which is a driving factor of metabolic syndrome, including dyslipidemia. As mentioned above, hepatic AMPK activation improves lipid metabolism via multiple pathways [22, 23]. Our data showed that C24 inhibited lipogenesis, and the effect of lipogenesis inhibition depended on hepatic AMPK activation. Notably, treatment with C24 did not affect the fatty acid oxidation rate in HepG2 cells (Supplementary Fig. S1c). This result may be due to the slight inhibition of complex I in the mitochondrial respiratory chain [26]. We found that the efficiency of TG-rich VLDL secretion was much lower after 3 d of C24 treatment than after vehicle treatment. We speculated that C24 reduced hepatic lipid biosynthesis and accumulation, and this effect may be involved in decreasing the secretion of VLDL-TG. Seven days of high-fat feeding followed by 7 d of oral administration of C24 (100 mg· kg−1) elicited hyposecretion of VLDL-TG compared to CMC-Na (0.5%) administration (Supplementary Fig. S3a). However, multiple regulatory factors contribute to VLDL assembly and secretion, and the detailed mechanisms of C24 modulation of VLDL-TG secretion require further investigation.
Statins are the first-line clinical lipid-lowering drugs that inhibit HMGCR activity to restrict cholesterol synthesis, which reduces intracellular cholesterol and feeds back to regulate the protein and gene levels of LDLR [41, 42]. We demonstrated that C24 treatment upregulated LDLR mRNA and protein levels (Supplementary Fig. S3b–d) and the lipid metabolism-related factors, such as transcriptional target genes of SREBPs, exhibited different levels of increase (Supplementary Fig. S3d). These results may be related to the hepatic cholesterol-lowering effect of C24 treatment (Supplementary Fig. S3e). To confirm this effect, we investigated the effect of C24 on LDLR expression in HepG2 cells and observed that LDLR mRNA and protein levels were elevated (Supplementary Fig. S3f, g), which was consistent with the results obtained from the in vivo study (Supplementary Fig. S3b–d).
Evidence from genetic, epidemiological and clinical studies indicates that the overload of LDLs in plasma causes atherosclerotic CVDs [43]. AMPK activation inhibits cholesterol synthesis in the liver and lowers LDL-C in plasma to reduce lipid load; these effects have attracted great attention to the targeting of AMPK to interfere with the development of atherosclerosis [22, 44]. C24 showed significant lipid-lowering effects in diet-enriched high-fat and high-cholesterol-induced hypertriglyceridemia and hypercholesterolemia in New Zealand rabbits and hamsters. Dietary lipid components regulate lipogenesis [45], and a high-fat and high-cholesterol diet suppressed hepatic de novo lipogenesis to a certain extent. There may be some limitations to evaluate the role of C24 in lipogenesis to alleviate dyslipidemia. Atherosclerosis is a progressive vascular disease with abnormal lipid metabolism and impaired endothelial function [46–48]. Administration of AMPK activators (such as salicylate, A769662, AICAR, and IMM-H007) modulates macrophage cholesterol homeostasis, inflammation and vascular dysfunction to delay the progression of atherosclerosis [49–52]. Our unpublished data also showed that C24 treatment activated AMPK in macrophages, and oral C24 treatment ameliorated atherosclerosis development in Western diet-fed ApoE-/- mice. The role that C24 plays in regulating atherosclerosis is worthy of further study.
In summary, we examined the AMPK activator C24, which decreased hepatic lipogenesis and VLDL-TG secretion and improved dyslipidemia. This work provides a promising lead compound for new lipid-lowering drug development and supports the concept that the activation of the hepatic AMPK pathway may be a useful strategy for the treatment of dyslipidemia.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81673493, 81273566, and 81803596) and the National Key New Drug Creation and Manufacturing Program, Ministry of Science and Technology (2018ZX09711002).
Author contributions
JYL, FJN, ZFX, and YMZ participated in the research design. SMS, ZFX, YMZ, XWZ, CDZ, YYY, and TTL performed the experiments and data analysis; JPY, YMZ, FJN, SCC, HWJ, and JL contributed new reagents and analytic tools; SMS, ZFX, JYL, and FJN contributed to the preparation of the paper.
Competing interest
The authors declare no competing interests
Footnotes
These authors contributed equally: Shui-mei Sun, Zhi-fu Xie, Yang-ming Zhang
Contributor Information
Fa-jun Nan, Email: fjnan@simm.ac.cn.
Jing-ya Li, Email: jyli@simm.ac.cn.
Supplementary information
The online version of this article (10.1038/s41401-020-0472-9) contains supplementary material, which is available to authorized users.
References
- 1.Kolovou GD, Anagnostopoulou KK, Cokkinos DV. Pathophysiology of dyslipidaemia in the metabolic syndrome. Postgrad Med J. 2005;81:358–66. doi: 10.1136/pgmj.2004.025601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genest JG., Jr Dyslipidemia and coronary artery disease. Can J Cardiol. 2000;16(Suppl A):3A–4A. [PubMed] [Google Scholar]
- 3.Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. J Am Coll Cardiol. 2019;73:e285–350. doi: 10.1016/j.jacc.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Zhang M, Deng Q, Wang LH, Huang ZJ, Zhou MG, Li YC, et al. Prevalence of dyslipidemia and achievement of low-density lipoprotein cholesterol targets in Chinese adults: a nationally representative survey of 163,641 adults. Int J Cardiol. 2018;260:196–203. doi: 10.1016/j.ijcard.2017.12.069. [DOI] [PubMed] [Google Scholar]
- 5.Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. 2016;48:e224. doi: 10.1038/emm.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carling D. The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–78. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 8.Hardie DG. AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu Rev Nutr. 2014;34:31–55. doi: 10.1146/annurev-nutr-071812-161148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31–7. doi: 10.1016/j.ceb.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019;18:527–51. doi: 10.1038/s41573-019-0019-2. [DOI] [PubMed] [Google Scholar]
- 12.Hardie DG, Ross FA, Hawley SA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol. 2012;19:1222–36. doi: 10.1016/j.chembiol.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardie DG. AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol. 2015;33:1–7. doi: 10.1016/j.ceb.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Henin N, Vincent MF, Gruber HE, Van, den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9:541–6. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- 15.Muoio DM, Seefeld K, Witters LA, Coleman RA. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem J. 1999;338(Pt 3):783–91. [PMC free article] [PubMed] [Google Scholar]
- 16.Wakil SJ, Stoops JK, Joshi VC. Fatty acid synthesis and its regulation. Annu Rev Biochem. 1983;52:537–79. doi: 10.1146/annurev.bi.52.070183.002541. [DOI] [PubMed] [Google Scholar]
- 17.Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19:1649–54. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–22. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 19.Ingebritsen TS, Geelen MJ, Parker RA, Evenson KJ, Gibson DM. Modulation of hydroxymethylglutaryl-CoA reductase activity, reductase kinase activity, and cholesterol synthesis in rat hepatocytes in response to insulin and glucagon. J Biol Chem. 1979;254:9986–9. [PubMed] [Google Scholar]
- 20.Loh K, Tam S, Murray-Segal L, Huynh K, Meikle PJ, Scott JW, et al. Inhibition of adenosine monophosphate-activated protein kinase-3-hydroxy-3-methylglutaryl coenzyme a reductase signaling leads to hypercholesterolemia and promotes hepatic steatosis and insulin resistance. Hepatol Commun. 2018;3:84–98. doi: 10.1002/hep4.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology—divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710–30. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Xu SQ, Mihaylova MM, Zheng B, Hou XY, Jiang BB, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–88. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esquejo RM, Salatto CT, Delmore J, Albuquerque B, Reyes A, Shi YJ, et al. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine. 2018;31:122–32. doi: 10.1016/j.ebiom.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008;283:16051–60. doi: 10.1074/jbc.M710114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu LF, Li YY, Su MB, Zhang M, Zhang W, Zhang LN, et al. Development of novel alkene oxindole derivatives as orally efficacious AMP-activated protein kinase activators. ACS Med Chem Lett. 2013;4:475–80. doi: 10.1021/ml400028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li YY, Yu LF, Zhang LN, Qiu BY, Su MB, Wu F, et al. Novel small-molecule AMPK activator orally exerts beneficial effects on diabetic db/db mice. Toxicol Appl Pharmacol. 2013;273:325–34. doi: 10.1016/j.taap.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Wu LY, Zhang LN, Li BH, Jiang HW, Duan YN, Xie ZF, et al. AMP-activated protein kinase (AMPK) regulates energy metabolism through modulating thermogenesis in adipose tissue. Front Physiol. 2018;9:122. doi: 10.3389/fphys.2018.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li WC, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol Biol. 2010;633:185–96. doi: 10.1007/978-1-59745-019-5_13. [DOI] [PubMed] [Google Scholar]
- 29.Cramer CT, Goetz B, Hopson KL, Fici GL, Ackermann RM, Brown SC, et al. Effects of a novel dual lipid synthesis inhibitor and its potential utility in treating dyslipidemia and metabolic syndrome. J Lipid Res. 2004;45:1289–301. doi: 10.1194/jlr.M400018-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Tepperman HM, Tepperman J. The hexosemonophosphate shunt and adaptive hyperlipogenesis. Diabetes. 1958;7:478–85. doi: 10.2337/diab.7.6.478. [DOI] [PubMed] [Google Scholar]
- 31.Lam TK, Gutierrez-Juarez R, Pocai A, Bhanot S, Tso P, Schwartz GJ, et al. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med. 2007;13:171–80. doi: 10.1038/nm1540. [DOI] [PubMed] [Google Scholar]
- 32.Lozano WM, Arias-Mutis OJ, Calvo CJ, Chorro FJ, Zarzoso M. Diet-induced rabbit models for the study of metabolicsyndrome. Animal. 2019;9:463. doi: 10.3390/ani9070463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sullivan MP, Cerda JJ, Robbins FL, Burgin CW, Beatty RJ. The gerbil, hamster, and guinea pig as rodent models for hyperlipidemia. Lab Anim Sci. 1993;43:575–8. [PubMed] [Google Scholar]
- 34.Bhatia G, Rizvi F, Saxena R, Puri A, Khanna AK, Chander R, et al. In vivo model for dyslipidemia with diabetes mellitus in hamster. Indian J Exp Biol. 2003;41:1456–9. [PubMed] [Google Scholar]
- 35.Ford RJ, Fullerton MD, Pinkosky SL, Day EA, Scott JW, Oakhill JS, et al. Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem J. 2015;468:125–32. doi: 10.1042/BJ20150125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boudaba N, Marion A, Huet C, Pierre R, Viollet B, Foretz M. AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine. 2018;28:194–209. doi: 10.1016/j.ebiom.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinberg D. In celebration of the 100th anniversary of the lipid hypothesis of atherosclerosis. J Lipid Res. 2013;54:2946–9. doi: 10.1194/jlr.R043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, et al. Recommendation on design, execution, and reporting of animal atherosclerosis studies: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. doi: 10.1161/ATV.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 39.Dillard A, Matthan NR, Lichtenstein AH. Use of hamster as a model to study diet-induced atherosclerosis. Nutr Metab. 2010;7:89. doi: 10.1186/1743-7075-7-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. 2017;357:507–11. doi: 10.1126/science.aah5582. [DOI] [PubMed] [Google Scholar]
- 41.Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, et al. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010;51:1486–95. doi: 10.1194/jlr.M003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brautbar A, Ballantyne CM. Pharmacological strategies for lowering LDL cholesterol: statins and beyond. Nat Rev Cardiol. 2011;8:253–65. doi: 10.1038/nrcardio.2011.2. [DOI] [PubMed] [Google Scholar]
- 43.Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–72. doi: 10.1093/eurheartj/ehx144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pinkosky SL, Filippov S, Srivastava RAK, Hanselman JC, Bradshaw CD, Hurley TR, et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J Lipid Res. 2013;54:134–51. doi: 10.1194/jlr.M030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishikata N, Shikata N, Kimura Y, Noguchi Y. Dietary lipid-dependent regulation of de novo lipogenesis and lipid partitioning by ketogenic essential amino acids in mice. Nutr Diabetes. 2011;1:e5. doi: 10.1038/nutd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rader DJ, Puré E. Lipoproteins, macrophage function, and atherosclerosis: beyond the foam cell? Cell Metab. 2005;1:223–30. doi: 10.1016/j.cmet.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med. 2014;5:927–46. [PMC free article] [PubMed] [Google Scholar]
- 48.Tabas I, Garcíacardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, Zhong LP, Wang FZ, Zhu HB. Dissecting the role of AMP-activated protein kinase in human diseases. Acta Pharm Sin B. 2017;7:249–59. doi: 10.1016/j.apsb.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li D, Wang D, Wang Y, Ling WH, Feng X, Xia M. Adenosine monophosphate-activated protein kinase induces cholesterol efflux from macrophage-derived foam cells and alleviates atherosclerosis in apolipoprotein E-deficient mice. J Biol Chem. 2010;285:33499–509. doi: 10.1074/jbc.M110.159772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fullerton MD, Ford RJ, McGregor CP, LeBlond ND, Snider SA, Stypa SA, et al. Salicylate improves macrophage cholesterol homeostasis via activation of AMPK. J Lipid Res. 2015;56:1025–33. doi: 10.1194/jlr.M058875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu J, Ming H, Li HY, Yu B, Chu M, Zhu H, et al. IMM-H007, a novel small molecule inhibitor for atherosclerosis, represses endothelium inflammation by regulating the activity of NF-κB and JNK/AP1 signaling. Toxicol Appl Pharmacol. 2019;381:114732. doi: 10.1016/j.taap.2019.114732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.