

Abstract
Inflammasomes are filamentous signaling platforms integral to innate immunity. Currently, little is known about how these structurally similar filaments recognize and distinguish one another. A cryo-EM structure of the AIM2PYD filament reveals that the architecture of the upstream filament is essentially identical to that of the adaptor ASCPYD filament. In silico simulations using Rosetta and molecular dynamics followed by biochemical and cellular experiments consistently demonstrate that individual filaments assemble bidirectionally. By contrast, the recognition between AIM2 and ASC requires at least one to be oligomeric and occurs in a head-to-tail manner. Using in silico mutagenesis as a guide, we also identify specific axial and lateral interfaces that dictate the recognition and distinction between AIM2 and ASC filaments. Together, the results here provide a robust framework for delineating the signaling specificity and order of inflammasomes.
Subject terms: Cryoelectron microscopy, Computational models
AIM2-ASC inflammasomes are filamentous signalling platforms that play a central role in host innate defence. Here, the authors present the filament cryo-EM structure of the inflammasome receptor AIM2, which is very similar to the adaptor ASC filament structure. By employing Rosetta and Molecular Dynamics simulations the authors provide further insights into the directionality and recognition mechanisms of the individual AIM2 and ASC filaments, which is further validated with biochemical and cellular experiments.
Introduction
Inflammasomes are filamentous signaling platforms and play key roles in the metazoan innate immune system1. These supra-structures assemble upon detecting molecular signatures arising from various intracellular catastrophes, such as genomic instability, dysfunctional organelles, and pathogen invasion1. Mammals have at least 15 different receptors that lead to the assembly of inflammasomes whose ultimate goal is to induce the polymerization of procaspase-1, activating the zymogen protease by proximity-induced autoproteolysis1. Caspase-1 then executes two key innate immune responses: the cleavage/maturation of pro-inflammatory cytokines, such as interleukin-1β and -18, and the initiation of pyroptosis1. Inflammasomes play essential roles in host defense against pathogen invasion (e.g., coronaviruses, herpesviridae, and Listeria monocytogene)1–6. In addition, malfunctioning inflammasomes promote acute and chronic autoinflammatory diseases (e.g., severe COVID-19, rheumatoid arthritis, and systemic lupus erythematosus (SLE))7–9, metabolic disorders (type 2 diabetes)10,11, and even tumorigenesis (colon cancer, lung cancer, and oral cancer)12,13.
Inflammasome receptors contain multiple functional domains for autoinhibition, signal recognition, and oligomerization1,14. Importantly, the N-terminal pyrin domain (PYD) acts as the primary signal transduction module in the vast majority of inflammasomes1,14,15. PYDs are six-helix bundles that belong to the death-domain (DD) superfamily and can assemble into helical filaments. For instance, incoming signals such as viral nucleic acids induce the assembly of a receptor PYD filament14,16–18. The upstream PYD filaments then nucleate the filamentation of the PYD of central adaptor ASC (ASCPYD)14,18–20, leading to the oligomerization of the CARD of ASC (ASCCARD) to recruit and trigger the polymerization (activation) of procaspase-1 (ASC: apoptosis-associated speck-forming protein containing caspase-recruiting domain (CARD); CARDs are also six-helix bundles that belong to the DD family)14,18,21,22.
Although the structural mechanisms by which inflammasomes assemble are increasingly better understood14,17–25, little is known about the mechanisms that direct the signaling order (sequence) and specificity. For instance, all published cryo-electron microscopy (cryo-EM) structures of PYD filaments show essentially the same helical architectures (six subunits per helical turn)18,20; all CARD filaments also show the same helical architectures (four subunits per helical turn)14,21,23. These observations then led to a well-accepted model, in which the architectural complementarity between upstream and downstream filaments underpins the recognition14,17,18,20,21,26. However, it raises a considerably more complex problem as to how these similar helical filaments built from homologous protomers distinguish and recognize one another within respective subfamilies. Here, we address this fundamental mechanistic issue in the cytosolic double-stranded (ds)DNA-sensing AIM2-ASC inflammasome27. AIM2 (absent in melanoma 2) is a bipartite protein composed of the N-terminal PYD followed by the dsDNA-binding HIN domain (hematopoietic interferon-inducible nuclear antigen). Upon binding cytosolic dsDNA via its HIN domain, AIM2PYD assembles into filaments, inducing the polymerization of ASC12,16,17,19. AIM2 is essential for the host defense against numerous pathogenic viruses and bacteria6,12,16,27–30. AIM2 also plays vital roles in neuronal development by regulating timely cell death31. However, dysregulated AIM2 leads to various maladies, such as SLE, chronic kidney diseases, and lung cancer12,27,32–34.
We present a cryo-EM structure of the AIM2PYD filament at 3.2 Å resolution, which reveals that its architecture is indeed identical to that of the ASCPYD filament. Using our structure, we then investigate how AIM2PYD and ASCPYD filaments recognize, and distinguish each other by Rosetta and molecular dynamics (MD) simulations. Our in silico analyses consistently suggest that the energy landscapes that underpin the assembly of individual filaments do not impose directionality. By contrast, the energy landscape that governs the recognition between AIM2PYD and ASCPYD is polarized in a head-to-tail manner. Multiple biochemical experiments corroborate that individual filaments assemble bidirectionally. Moreover, AIM2PYD and ASCPYD filaments do not co-assemble, and the signal transduction from AIM2 to ASC occurs unidirectionally only when at least one is oligomeric. Using Rosetta-based in silico mutagenesis as a guide, our biochemical and cellular experiments consistently show that lateral interfaces of AIM2PYD drive its bidirectional assembly. We also identify specific axial interfaces that mediate the recognition between AIM2PYD and ASCPYD. Together, we demonstrate that distinct interfaces within homologous filaments direct signaling order and specificity of inflammasomes. We also set forth a broadly applicable multidisciplinary platform for delineating the signal transduction order, specificity, and directionality of filamentous assemblies.
Results
The cryo-EM structure of AIM2PYD
Using EM of negatively stained samples (nsEM), we previously found that the helical symmetry of the AIM2PYD filament is consistent with that of the ASCPYD filament17, and thus proposed that architectural complementarity is important for their recognition. However, the published high-resolution cryo-EM structure of the AIM2PYD filament displays an altered helical architecture because the N-terminal green fluorescence protein (GFP)-tag interferes with assembly35. Thus, we first determined the cryo-EM structure of the AIM2PYD filament using an untagged recombinant protein.
Cryo-EM images showed that AIM2PYD filaments are straight helical rods (Fig. 1A). The average power spectrum of 512-pixel-long nonoverlapping filament segments showed that the AIM2PYD filament displays a six-start, C3 helical symmetry of 54.4° rotation (~6 subunits per helical turn) and an axial rise of 14 Å (Fig. 1B). These parameters are remarkably similar to those of the ASCPYD filament18, further solidifying the concept that the upstream receptors provide structural templates for downstream assemblies in inflammasomes14,17,26. We fit the crystal structure of AIM2PYD into the EM map for initial modeling36, and the refined high-resolution map allowed us to model in most bulky and aliphatic side chains (Fig. 1C). The resolution of the final model was 3.2 Å according to the gold standard method (Supplementary Fig. 1A). The diameter of the outer rim is ~94 Å and that of the inner cavity is ~25 Å (Fig. 1D). The structure of individual AIM2PYD protomers is identical to the crystal structure of AIM2PYD monomer (Supplementary Fig. 1B), thus indicating that, unlike the PYD of NLRP6 (ref. 20), an AIM2PYD monomer does not undergo any conformational changes during activation. As seen from the ASCPYD filament, each AIM2PYD subunit contributes three unique protein–protein interaction interfaces (Fig. 1E). The type 1a:1b interface is largely composed of side-chain interactions, while the type 2a:2b and type 3a:3b interfaces involved both side-chain and backbone interactions (Fig. 1E). We also noted several side chains previously implicated in filament assembly throughout different interfaces (e.g., L11, D19, F27, and I46; Fig. 1E)17. Aligning the new AIM2PYD filament to the GFP-AIM2PYD filament demonstrates that although the lateral interactions are largely conserved, the axial positions are significantly different due to the altered helical symmetry (five subunits per turn in the GFP-tagged filament vs. six subunits per turn in the untagged filament; Supplementary Fig. 1C). On the other hand, aligning the cryo-EM structures of AIM2PYD and ASCPYD filaments demonstrates their congruent architectures (Fig. 1F). The subtle difference in subunit positions between AIM2PYD and ASCPYD filaments along the helical axis could reflect the unique side-chain interactions that mediate their respective filament assembly or the inherent flexibility of biomolecular structures (Fig. 1F). Nevertheless, the near perfect architectural complementarity between AIM2PYD and ASCPYD filaments supports the idea that upstream filaments provide structural templates for the assembly of downstream filaments14,17,26,37.
Fig. 1. AIM2PYD assembles into an architecturally congruent filament as the ASCPYD filament.

A A sample cryo-electron micrograph of the AIM2PYD filament (total 976 micrographs were taken). B An average power spectrum of the AIM2PYD filament from 512 px-long nonoverlapping segments. C The AIM2PYD filament model built into the EM map. A subunit with visible side chains is shown. D The model of AIM2PYD filament. Each subunit is colored differently. E A cartoon representation of three unique filament interface types. Side chains at each filament interface are shown as a stick configuration. F Overlays between AIM2PYD and ASCPYD (PDB ID: 3J63) filaments.
Deciphering the specificity and directionality of the AIM2-ASC inflammasome using Rosetta and MD
AIM2PYD and ASCPYD monomers are homologous and structurally highly conserved (root-mean-squared-deviation, RMSD 0.5 Å), and our new cryo-EM structure shows that they indeed assemble into essentially identical filaments (Fig. 1F). These observations raise significantly more complex questions as to whether and how these supra-structures distinguish and recognize each other. Importantly, such questions are germane to all filamentous signaling platforms employing PYDs or CARDs21,22,26,37–39. Thus, to establish a broadly relevant method for tackling these questions, we employed a computational approach using Rosetta. First, we tested whether RosettaDock40 could recapitulate the cryo-EM structures by docking an AIM2PYD monomer into our AIM2PYD filament structure (also an ASCPYD monomer to the ASCPYD filament (PDB ID: 3J63)18). For instance, each PYD protomer provides three unique interfaces in AIM2PYD and ASCPYD filaments (i.e., six distinct surfaces; Figs. 1E and 2A). To facilitate docking experiments, we generated a honeycomb-like side view of AIM2PYD and ASCPYD filaments, in which the center protomer makes all six possible contacts (Fig. 2A). We then divided the honeycomb into six unique subsections consisting of one ligand docked into a pocket created by three adjacent subunits (Fig. 2B). Using the local docking method in Rosetta40, we performed 5000 independent docking simulations between a ligand–pocket pair from each subsection, then compared the interface energy and RMSD from the cryo-EM structures.
Fig. 2. In silico studies suggest that homotypic filaments assemble bidirectionally and the recognition between AIM2 and ASC occurs unidirectionally.

A Cartoon representations of honeycombs. Each interface type is labeled in the hexagons which represent PYD monomers. B Rosetta docking strategy. Top docking indicates that a ligand monomer docks onto the top surface of the pocket, while bottom docking is the opposite. C Plots of Rosetta interface energy scores vs. RMSD for top and bottom docking results for AIM2PYD assembly. The B represents simulations conducted in C. D Plots of Rosetta interface energy scores vs. RMSD for docking an ASCPYD monomer on the top or bottom of AIM2PYD pockets. E A model of the AIM2PYD filament recognition of the ASCPYD filament. F, G Rosetta interface energy scores at individual filament interfaces for homotypic and heterotypic assemblies. AIM2PYD-AIM2PYD, ASCPYD-ASCPYD, ASCPYD-AIM2PYD, and AIM2PYD-ASCPYD. Each hexagon represents AIM2PYD or ASCPYD monomer. H A plot for the difference in free energy (∆∆G) for dissociating ligand PYDs from the top or bottom pockets.
For each filament, both parameters decreased concurrently, while displaying uniform energy scores from all subsections (Fig. 2C (arrow) and Supplementary Fig. 2A), indicating that RosettaDock can recapitulate the cryo-EM structures. The more favorable energy scores from the AIM2PYD filament suggest that it is is more stable than the ASCPYD filament (typically −70 s for AIM2PYD complexes vs. −50 s for ASCPYD complexes). Importantly, the uniform energy scores throughout the top and bottom subsections (Fig. 2C and Supplementary Fig. 2A) suggest that individual filaments would assemble bidirectionally. Next, we docked an ASCPYD monomer (ligand) onto all six pockets of the AIM2PYD filament and vice versa (Fig. 2D and Supplementary Fig. 2B). We first noted that the interface energies are not as favorable as the AIM2PYD•AIM2PYD complexes (−60 or worse for ASCPYD•AIM2PYDcomplxes; Fig. 2D and Supplementary Fig. 2B). Moreover, docking ASCPYD on the top pockets of the AIM2PYD filament was significantly more favorable than docking at the bottom. (Fig. 2D orange vs. gray). Docking AIM2PYD on the ASCPYD filament also showed that AIM2PYD prefers the bottom half of the ASCPYD filament with the energy scores as favorable as the homotypic ASCPYD assembly (Supplementary Fig. 2A, B (2B orange vs. gray)). These results suggest that individual filaments assemble bidirectionally, while the recognition between AIM2PYD and ASCPYD occurs unidirectionally, where the top of the AIM2PYD filament recognizes the bottom of the ASCPYD filament (Fig. 2E).
Next, we used Rosetta InterfaceAnalyzer41 to evaluate the interaction energies between homotypic and heterotypic interactions at the individual interfaces of the honeycomb (Fig. 2F). The AIM2PYD complex also showed the most favorable overall interface energy scores (Fig. 2G). Moreover, for the respective homotypic assembly of AIM2PYD and ASCPYD, the type 1 interface contributed most significantly with the top and bottom halves displaying symmetric energy scores (Fig. 2F). On the other hand, the interface energy scores were consistently worse, when ASCPYD was placed at the center of the AIM2PYD honeycomb, except for the one between the type 2a of ASCPYD and 2b of AIM2PYD (Fig. 2F vs. 2G). The overall energy scores between the top and bottom halves were again asymmetric, preferring a head-to-tail-like direction, in which the top of AIM2PYDs favoring the bottom of ASCPYD and vice versa (Fig. 2E, G; the small difference in energy scores between Fig. 2F, G likely stemmed from the subtle architectural differences in two filaments).
To test whether the simulation results are not biased by a particular algorithm, we then used MD to calculate the free energy required to dissociate a ligand PYD from each pocket described in Fig. 2B (i.e., stability; Supplementary Fig. 2C, D). Consistent with the results from RosettaDock, MD simulations suggested the AIM2PYD filament complex to be most stable, followed by ASCPYD•AIM2PYD then ASCPYD complexes. (Supplementary Fig. 3B; see also Supplementary Fig. 3C for images showing the dissociation of each monomer before and after the simulation). We compared the sum of energies required to dissociate a ligand PYD from the top vs. bottom halves (Fig. 2H; Supplementary Fig. 2C). Individually, AIM2PYD and ASCPYD complexes showed mostly uniform energy landscapes from either filament pole, with both filaments showing more stable interactions at the bottom (Fig. 2H and Supplementary Fig. 3A, B). The moderate asymmetry suggests that the bottom interfaces might be preferred for homotypic assembly, or it could also reflect the intrinsic noise from sampling multiple conformations in an all-atom MD simulation. Nevertheless, consistent with Rosetta simulations, significantly more energy was required to dissociate ASCPYD from the top of the AIM2PYD filament than the bottom (Fig. 2H and Supplementary Fig. 3B). Together, our in silico analyses consistently suggest that individual filaments assemble bidirectionally, AIM2PYD strongly prefers to assemble homotypically, and the recognition between AIM2PYD and ASCPYD occurs via the type 2 interface.
In vitro experiments corroborate in silico predictions
To test our simulation results, we first tracked the assembly of fluor-labeled recombinant AIM2PYD and ASCPYD filaments via confocal fluorescence microscopy. When we mixed two populations of differentially labeled maltose-binding-protein-tagged (MBP)-AIM2PYD at 1:1 ratio and triggered polymerization by cleaving MBP via Tobacco Etch Virus protease (TEVp)19, the two colors colocalized in the same filaments (Fig. 3A, AIM2PYD•AIM2PYD); differentially labeled ASCPYD populations also colocalized in the same filaments (Fig. 3A, ASCPYD•ASCPYD). Importantly, when we preassembled the AIM2PYD filament labeled with one color and added AIM2PYD monomers labeled with a different color, nascent filaments extended from both axial poles of existing filaments (Fig. 3A, (AIM2PYD filament) + AIM2PYD); ASCPYD filaments also displayed random bidirectional assembly (Fig. 3A, (ASCPYD filament) + ASCPYD). These results corroborate that homotypic filaments assemble bidirectionally. Next, we mixed differentially labeled MBP-AIM2PYD and MBP-ASCPYD at 1:1 ratio, and monitored their filament assembly upon triggering polymerization via TEVp. Here, each protein appeared to be oligomerized separately without colocalizing on the same filament, and two distinct filaments interacted only at one specific axial pole (Fig. 3B, AIM2PYD•ASCPYD). Such a unidirectional interaction was even more evident when we added excess nascent proteins over preformed filaments (Fig. 3B, (AIM2PYD filament) + ASCPYD and (ASCPYD filament) + AIM2PYD). In addition, no significant Förster resonance energy transfer (FRET) signals were observed when we triggered the assembly of a donor-labeled AIM2PYD and acceptor-labeled ASCPYD (Fig. 3C. see also ref. 19), indicating that AIM2PYD and ASCPYD do not co-assemble, yet the recognition entails at least one to be oligomeric. Overall, our biochemical experiments agree with the computational predictions.
Fig. 3. Individual filaments assemble bidirectionally and the recognition between AIM2 and ASC occurs at a specific pole.

A Fluorescent confocal microscopy images of Alexa488- and Dylight550-labeled AIM2PYD, Alexa488- and Dylight550-labeled ASCPYD, preassembled Alexa488-labeled AIM2PYD filament and nascent Dylight550-labeled AIM2PYD, and preassembled Alexa488-labeled ASCPYD filament and nascent Dylight550-labeled ASCPYD. B Fluorescent confocal microscopy images Alexa488-labeled AIM2PYD and Dylight550-labeled ASCPYD, preassembled Alexa488-labeled AIM2PYD filament and nascent Dylight550-labeled ASCPYD, and preassembled Alexa488-labeled ASCPYD filament and nascent Dylight550-labeled AIM2PYD. Images shown in A and B are representatives of at least three independent experiments. C The time-dependent changes in FRET ratios between donor or acceptor-labeled MBP-AIM2PYD and/or MBP-ASCPYD were monitored upon cleaving the MBP tag with TEVp.
Simulations to identify key interfaces that govern the recognition and distinction between AIM2PYD and ASCPYD
Our observations thus far suggest that there exist distinct interactions that underpin individual assemblies and those that mediate the recognition between AIM2PYD and ASCPYD, such as the type 2 interface for the heterotypic recognition. The side chains at the filament interfaces are poorly conserved between AIM2PYD and ASCPYD (Supplementary Fig. 4A), indicating that diverse side-chain interactions can support the assembly of homologous supra-structures (i.e., redundancy in assembly code). Thus, we decided to forego an alanine-scanning-like approach for identifying and validating key interfaces. Instead, we used Rosetta to suggest mutations that would selectively disrupt homotypic or heterotypic interactions without precluding filament assembly.
Here, we in silico mutated each side chain of pocket protomers that interfaces ligand PYDs to all other a.a. using PyRosetta42 (e.g., Fig. 4A top). We then obtained interface energy scores (∆Gs) for WT_ligand•WT_pocket and WT_ligand•mt_pocket complexes (Fig. 4A top. WT: wild type, mt: mutant); subtracting the ∆G of each WT•mt pair from that of the WT•WT pair then provided the effect of a given mutation (∆∆G). We then plotted ∆∆Gs for AIM2PYD•AIM2PYD (both top and bottom docking; Fig. 4A) vs. ∆∆Gs for AIM2•ASC complexes (ASCPYD docking on the top pockets of AIM2PYD; Fig. 4A). We found that the vast majority of mutations are deleterious for both AIM2PYD •AIM2PYD and AIM2PYD •ASCPYD interactions (the upper right quadrant in Fig. 4A), suggesting that the a.a. selection has already been optimized for the self-assembly and recognition. Nonetheless, we identified 88 mutations at nine unique side chains, resulting in ∆∆G(AIM2•AIM2) > 10 and ∆∆G(AIM2•ASC) < 10 (i.e., mutations that would selectively disrupt AIM2PYD•AIM2PYD interactions without abolishing AIM2PYD•ASCPYD interactions; boxed area in Fig. 4B and listed in Supplementary Fig. 4B). Interestingly, all these side chains were found on the lateral type 1 and type 3 interfaces, but none at the axial type 2 interfaces (Fig. 4B and Supplementary Fig. 4B). Next, to identify mutations that would selectively disrupt AIM2PYD•ASCPYD interactions, we looked for those resulted in ∆∆G(AIM2•AIM2) to be <10 and ∆∆G(AIM2•ASC) to be >10. Here, we identified 49 mutations at ten unique AIM2PYD side-chain positions, all but one located on the type 2b surface (Fig. 4B and Supplementary Fig. 4B). These results are consistent with the mechanism, in which the lateral interfaces drive the assembly of the AIM2PYD filament without biasing any directions, while the recognition between AIM2PYD and ASCPYD occurs at the type 2 interface.
Fig. 4. in silico mutagenesis for identifying side chains that dictate the specificity of the AIM2-ASC inflammasome assembly.
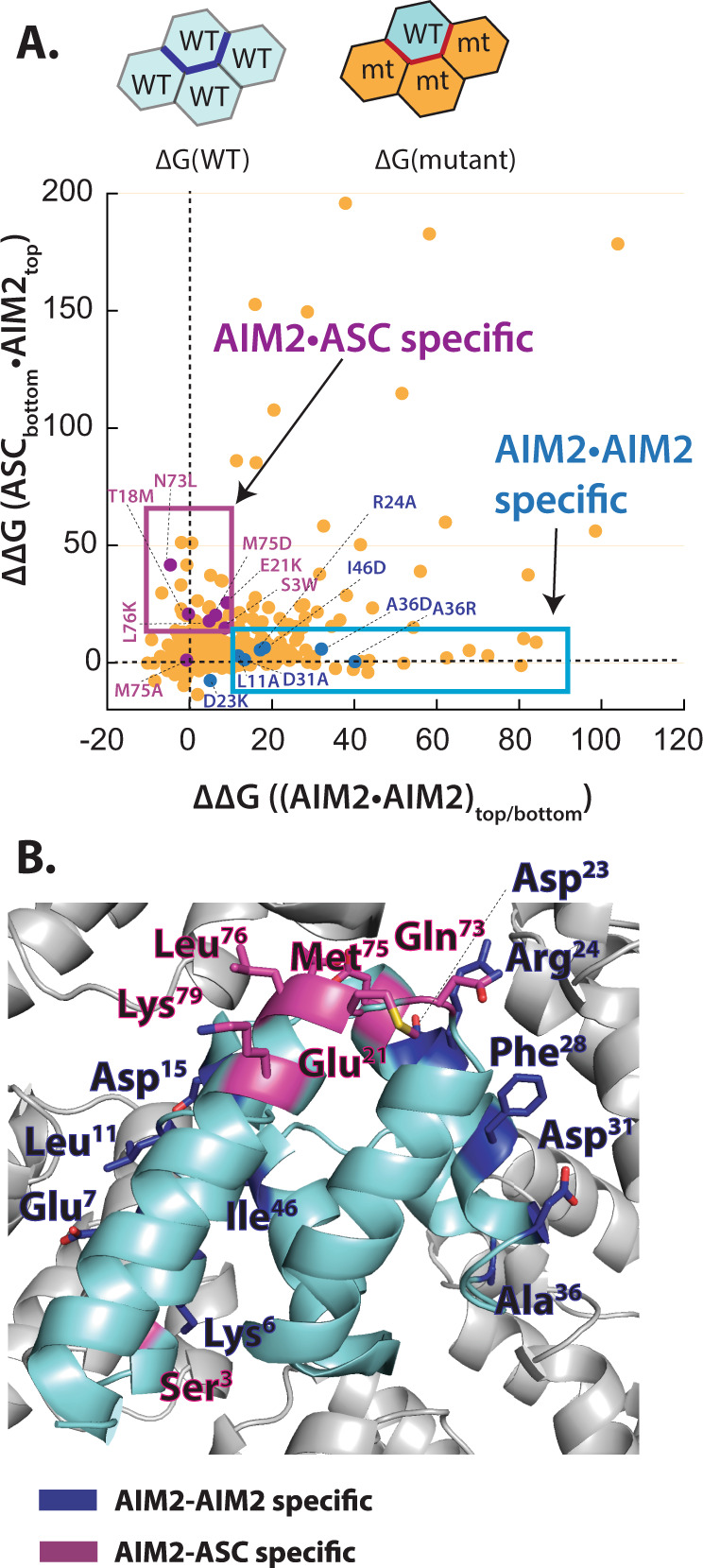
A Top: cartoons describing in silico mutagenesis strategy. The ligand monomer was kept WT and the interface residues of pocket monomers were mutated. The ∆∆G value for each mutated residue was obtained by ∆G(WT_ligand•WT_pocket)–∆G(WT_ligand•mt_pocket). One pocket is shown as an example, and we applied the same strategy to all six pockets described in Fig. 2 (mt: mutant). Bottom: a plot of AIM2PYD mutations that would interfere with AIM2PYD•AIM2PYD or AIM2PYD•ASCPYD interaction. Selected mutations for follow-up biochemical and cellular studies are indicated. B A cartoon of the AIM2PYD filament indicating the residues that Rosetta predicts to interfere with either homotypic assembly or ASCPYD recognition when mutated.
In vitro and in cellulo experiments corroborate in silico predictions
Mutations that abolish the self-assembly of AIM2PYD decreases the dsDNA-binding activity of AIM2FL, as oligomerization is couple to signal recognition17. Nevertheless, most of such AIM2FL mutants still assemble into filaments on the dsDNA scaffold17. Also of note, AIM2 filaments can self-perpetuate its assembly by accelerating the polymerization of nascent monomers19. Thus, to test our simulation results, we generated Rosetta predicted mutations on AIM2FL and first confirmed filament formation on dsDNA (Supplementary Fig. 5; all mutants formed filaments except N73L). We then determined whether dsDNA-bound AIM2FL mutants could accelerate the polymerization of FRET donor/acceptor-labeled AIM2PYD or ASCPYD (Fig. 5A–C and Supplementary Fig. 6; see also ref. 19)
Fig. 5. biochemical and cellular experiments support Rosetta predictions.

A Testing AIM2FL mutants predicted to interfere with homotypic assembly. B Testing AIM2FL mutants predicted to interfere ASCPYD recognition. C Testing ASCFL mutants predicted to interfere with AIM2PYD recognition. For A–C, sample plots show the time-dependent polymerization of FRET donor- and acceptor-labeled AIM2PYD or ASCPYD in the presence or absence of dsDNA-bound (linear plasmid ~5-kilo base-pairs (kbps)) WT or mutant AIM2FL. Dot plots summarizing the effect of Rosetta predicted AIM2 mutants that would selectively interfere with its own assembly. Polymerization half-times were normalized using WT AIM2FL-induced assembly (set to 1) and labeled PYDs alone (set to 0). *p < 0.05; **p < 0.01; ***p < 0.001. P values were calculated using Student’s t test for paired samples. D Confocal microscope images of HEK293T cells (co)-transfected with WT ASCFL-mCherry alone. WT ASCFL-mCherry plus WT or mutant AIM2FL-eGFP. E Confocal microscope images of HEK293T cells (co)-transfected with mutant ASCFL-mCherry plus WT AIM2FL-eGFP. The nucleus is stained with DAPI in both D and E. Images shown in D and E are representative of at least three independent experiments. .
As predicted from our simulation, L11A, A36D/R, and I46D were significantly more defective in accelerating the polymerization of AIM2PYD than that of ASCPYD (Fig. 5A and Supplementary Fig. 6) We also tested D23K as it appeared to enhance the interaction with ASC, while disrupting AIM2–AIM2 interactions (Fig. 4A). We found that D23K-AIM2FL did not enhance the interaction with ASCPYD, but was more defective in inducing the polymerization of AIM2PYD (Fig. 5A and Supplementary Fig. 6). These results consistently suggest that the lateral surface residues of AIM2PYD preferentially, but not exclusively, promote homotypic assembly. Next, again consistent with Rosetta predictions, E21K and M75D were significantly more defective in inducing the polymerization of ASCPYD than that of AIM2PYD (Fig. 5B and Supplementary Fig. 6), corroborating that the type 2b surface of the AIM2PYD filament recruits ASCPYD (N73L-AIM2FL failed to induce any filament formation consistent with the lack of self-assembly (Fig. 5, and Supplementary Figs. 5 and 6)). Notably, M75A-AIM2FL (null in Rosetta mutagenesis) retained the WT-like activity (Supplementary Fig. 6), supporting the idea that a simple alanine-scanning approach is inadequate due to the redundancy in assembly code.
We then used the above Rosetta-based approach to identify mutations at the type 2a surface of ASCPYD that would selectively disrupt the interaction with AIM2PYD (Supplementary Fig. 7A–C). Previously, we found that the ASCPYD filament accelerates the assembly of AIM2PYD via a positive feedback loop19. Thus, we generated Rosetta predicted mutations on full-length ASC (ASCFL) and tested their capacity for inducing the polymerization of FRET-labeled ASCPYD or AIM2PYD. We used ASCFL, as the C-terminal CARD would promote the polymerization of ASCPYD even if mutations were too deleterious. We found that L61S and G37E were significantly more defective in accelerating the assembly of AIM2PYD than that of ASCPYD, corroborating that the type 2a surface of ASCPYD recognizes AIM2PYD (Fig. 5C and Supplementary Fig. 7D).
We next probe the interactions among AIM2FL and ASCFL WT and mutants in HEK293T cells. WT AIM2FL (ASCFL) tagged with C-terminal eGFP or mCherry colocalized in the same complexes as expected (Supplementary Fig. 8). AIM2FL showed filamentous complexes that often tangled up into speck-like clusters, while ASCFL displayed large puncta, as previously reported43 (Fig. 5D left, Supplementary Fig. 8A–C). AIM2FL-eGFP mutants colocalized with WT AIM2FL-mCherry (Supplementary Fig. 8B), likely due to assembling/binding on the same (transfected) dsDNA strands as WT. Interestingly, when AIM2FL-eGFP and ASCFL-mCherry were co-transfected, ASCFL filaments further expanded as if ASCFL assembles from multiple AIM2FL foci (Fig. 5D, (+WT AIM2FL)). When we co-transfected AIM2FL-eGFP mutants and WT ASCFL-mCherry, those that preferentially decreased the ability to interact with AIM2PYD still resulted in expanded ASCFL complexes as observed from WT (Fig. 5D, L11A, D23K, and A36D). By contrast, ASCFL stayed as a single punctum when co-transfected with AIM2FL mutants that failed to accelerate the polymerization of ASCPYD (E21K and M75D; Fig. 5D, E21K and M75D); N73L-AIM2FL-eGFP, which cannot oligomerize (Supplementary Figs. 5 and 8A), also failed to interact with ASCFL-mCherry (Fig. 5D, N73L). We also imaged G37E- and L61S-ASCFL-mCherry with eGFP-tagged WT AIM2FL and ASCFL. The ASCFL mutants still showed large puncta and also colocalized with WT (Supplementary Fig. 8C). However, WT AIM2FL-eGFP failed to colocalize or induce the expansion of these mutants when co-transfected (Fig. 5E). Together, our in vitro and in cellulo experiments consistently support our in silico predictions, in which unique lateral and axial interfaces within homologous filaments dictate their recognition and distinction.
Discussion
Inflammasomes transduce signals by assembling supramolecular structures14,17–20,22,23. It is well accepted that architectural complementarity is important for the recognition between primary signaling components21–23,28,30,34. Indeed, our AIM2PYD filament structure further cements this concept. Nevertheless, these observations further highlight a long-standing question as to how such homologous filaments distinguish and recognize one another. Here, we set forth a broadly applicable research platform for answering these questions and propose design principles that define the signaling mechanisms of inflammasomes.
Strategies for homotypic filament assembly
Our experiments consistently show that the assembly of individual filaments occur bidirectionally, with lateral type 1 and type 3 interfaces (especially type 1) of AIM2PYD favoring homotypic interactions, while still supporting the recognition of ASCPYD. The lateral type 1 surfaces are the largest in any PYDs (Fig. 6A left), which would be ideal for recognizing other homotypic protomers to initiate assembly without any prescribed directionalities. The lack of directionality in homotypic assembly would then allow AIM2 to maximally benefit from one-dimensional random diffusion on pathogenic dsDNA19, resulting in a timely response by the upstream receptor (Fig. 6A right). Interestingly, the bidirectional assembly is in contrast to other cytoskeletal and signaling filaments such as actin44 and B-cell lymphoma 10 (BCL10)39. Considering that both actin and BCL10 filaments originate from cell membranes/defined borders39,44, it is tempting to speculate that the bidirectional assembly of inflammasome filaments have evolved to take full advantage of no immediate boundaries in the cytosol.
Fig. 6. Strategies for signal transduction by the AIM2-ASC inflammasome.
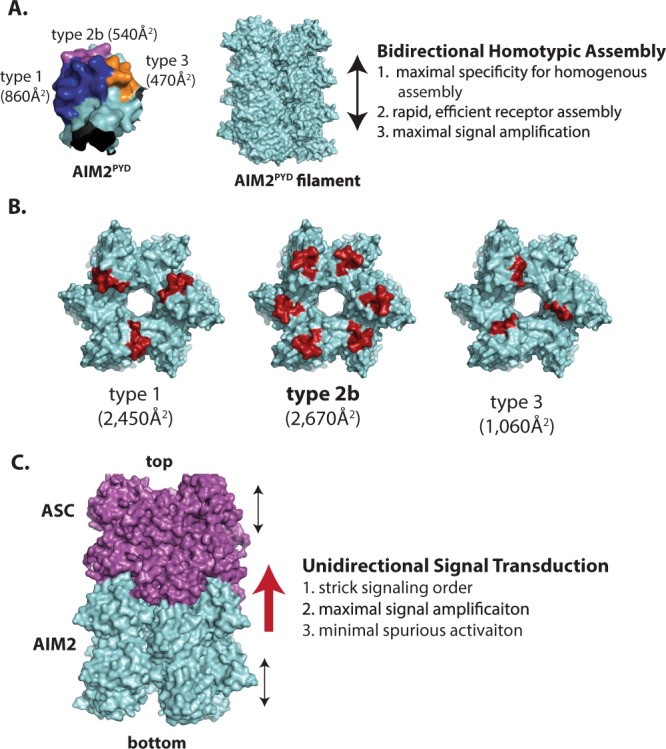
A Left: a surface representation of AIM2PYD monomer. The buried surface area in the filament for each interface type is indicated. Right: a scheme describing the advantages of bidirectional homotypic assembly. B The top surface view of the AIM2PYD filament. Each solvent accessible interface is colored in red with the calculated surface area. C A scheme describing the advantages of unidirectional signal transduction by the AIM2-ASC inflammasome.
Strategies for signaling by assembly
The AIM2PYD filament displays higher stability than either the ASCPYD filament or AIM2PYD•ASCPYD complex (Fig. 2), which would ensure homotypic assembly of the receptor filament especially within the dsDNA scaffold. On the other hand, the interaction between AIM2PYD and ASCPYD is more favorable than homotypic ASCPYD interactions at a specific axial pole (Fig. 2). In addition, AIM2PYD and ASCPYD recognize each other only when at least one is oligomeric (Fig. 3B). Of note, electrostatic surface analyses suggest that the charge complementary is reversed at the type 1 interface for AIM2 and ASC, likely indicating that the heterologous interactions between the monomers are not favorable (i.e., the type 1a surface of AIM2PYD is largely basic, whereases that of ASCPYD is acidic; Supplementary Fig. 9A). Importantly, our in silico, in vitro, and in cellulo experiments consistently demonstrate that the directional interaction at the type 2 interface is most critical (Figs. 2–5). The surface area of the type 2 interface is much smaller than that of the type 1 interface in monomeric PYDs (Fig. 6A). However, because of the axial location, the type 2b surfaces become as accessible as the type 1 surfaces once AIM2PYD assembles into a filament (Fig. 6B). Moreover, electrostatic surface analyses suggest that the bottom of the AIM2PYD filament is unfavorable for interacting with the ASCPYD filament due to highly positively charged surfaces (Supplementary Fig. 9B). We propose that such conditional scaffolding by the upstream filament not only ensures proper signal transduction orders, but also maximizes signal amplification (Fig. 6C). For instance, inflammasomes assemble in a digital fashion and entail cell death14,19. Thus, it must be imperative that ASC does not polymerize unless upstream receptors are fully activated by correct signals. Thus, the sequential/conditional assembly of the AIM2-ASC inflammasome would minimize its spurious activity in the absence of bona fide danger signals. Of note, not only the assembly, but also the signaling activity of AIM2 depends on dsDNA length, which regulates the probability of assembling the intact axial base of its filament19. Our results here further explain the dsDNA length-dependent mechanism, as the intact filament base would conditionally maximize the presence of the type 2b surfaces to recruit ASC (Fig. 6B). Subsequently, such a stringent recognition mechanism would then ensure that ASC polymerizes homogenously via its prion-like assembly mechanism45, resulting in maximal signal amplification (Fig. 6C). Alternatively, if AIM2 and ASC were to either co-assemble or interact without distinct energetic hierarchies, we envision that upstream and downstream oligomers would either alternate or even cap their respective assemblies, resulting in signal attenuation.
Future directions
Our successful implementation of Rosetta to decode the specificity of the AIM2-ASC inflammasome suggests that our approach can be broadly applied to other homologous signaling filaments. However, we noted that Rosetta was correct at ~50% in predicting energetically important mutations (Fig. 5), indicating that there is room for improvement. Nonetheless, given that precisely pinpointing the role of individual residues is intrinsically challenging, we find the Rosetta suite to be an excellent tool for decoding the specificity of the filamentous assemblies.
It was recently postulated that the directionality of the NLRP6PYD-ASCPYD interaction would be the same as what we found here for AIM2PYD-ASCPYD (ref. 20). Thus, it is tempting to speculate that recruiting ASC via its type 2a surface by the type 2b surface of upstream receptors is the universal strategy in inflammasome signaling. Other than NLRP6 and AIM2, there are at least 14 other upstream receptors that signals through ASC1,14. Future investigations will reveal to what extent the mechanisms we found for AIM2 apply in other receptors and how well Rosetta fares in answering these questions.
Methods
Protein expression and purification
Human AIM2FL (residues 1–343), AIM2PYD (residues 1–94) S94C for fluorophore labeling, ASCPYD (residues 1–92) were cloned into the pET28b vector (Novagen) with an N-terminal MBP tag and TEVp recognition site. For cryo-EM, we and found a construct including ~20 a.a. in the unstructured linker region (residues 1–117) resulted in well-separated filaments (denoted as AIM2IRND; Fig. 1A). ASCFL was cloned with the MBP tag at both N- and C-termini with the TEVp recognition site flanking MBP and ASCFL. All proteins were expressed in Escherichia coli BL21 Rosetta2DE3 and purified using affinity (MBP/amylose), cation exchange, and followed by size-exclusion chromatography. Proteins were then concentrated and stored at −80 °C, see also refs. 17,19 (all primers generated for this study are listed in Supplementary Table 2).
Cryo-EM sample preparation
A total of 5 µl sample of 2.75 µM AIM2IRND (cleaved for 30 min with 6 µM TEVp) was applied to Lacey grids, followed by automatically blotting for 1.5 s and plunge freezing, using the FEI Vitrobot Mark IV operated at 100% humidity and room temperature (Johns Hopkins University). Cryo-EM data were collected at the National Cancer Institute National cryo-EM facility (NCI NCEF, Frederick, MD) using the FEI Titan equipped with the Gatan K2 direct electron detector operating at 300 keV, using the super-resolution mode (0.66 Å/pixel). A total of 2700 micrographs were collected from one grid at a defocus range from −1.0 to −2.5 µm. Each micrograph was equally fractioned into 40 frames with a total exposure time of 12 s and a total dose of 42 electrons/Å2. Data collection statistics are listed in Supplementary Table 1.
Helical reconstruction and model building
The 2700 micrographs were binned by two (to 1.32 Å/px) and frames were aligned using MotionCor2 (ref. 46). The defocus values and astigmatism of the micrographs were determined by CTFFIND3 for the aligned full-dose micrographs47. A total of 976 micrographs were selected (images with good CTF determination and defocus <3 μm) for subsequent image processing. CTF was corrected by multiplying the micrographs (only first 20 frames were aligned with a total dose of ~20 electrons/Å2) with the theoretical CTF, which both corrects the phases and improves the signal-to-noise ratio. The e2helixboxer program in EMAN2 software package48 was used for boxing long filaments from the full-dose images. The CTF-corrected micrographs were used for the segment extraction, with a total of 116,285 384 px-long overlapping segments (with a shift of 1.5 times of axial rise) generated. The SPIDER software package49 was used for subsequent processing and reconstruction. Using a featureless cylinder as an initial reference, 99,237 segments were used in IHRSR program for the final reconstruction after the helical parameters (an azimuthal rotation of 53.3° and an axial rise of 14 Å per subunit) converged. The resolution of the final reconstruction was estimated by the FSC between two independent half maps, which shows 3.2 Å at FSC = 0.143.
We used the crystal structure of AIM2PYD (PDB ID: 4O7Q) as an initial template to dock into the AIM2PYD cryo-EM map by rigid body fitting, and then manually edited the model in Chimera50 and Coot51. We then used the modified model as the starting template to further refine against the segmented cryo-EM map using RosettaCM52. The refined monomeric model of AIM2PYD was then rebuilt by RosettaCM with helical symmetry and real-space refined, using Phenix53 to improve the stereochemistry, as well as the model-map correlation coefficient. The AIM2PYD filament model was validated with MolProbity54. Refinement statistics are listed in Supplementary Table 1.
Rosetta docking
The Rosetta Local Docking protocol40,55 was applied to Rosetta symmetry relaxed structures of the AIM2PYD cryo-EM structure and ASCPYD cryo-EM structure (PDB ID: 3J63). Each complex contained a pocket consisting of three PYDs, and one ligand PYD. The initial position of the ligand was already in the pocket to minimize the search space, as suggested by the local docking protocol. The docking simulation was done 5000 times for each fragment, and the results were analyzed by looking at the interface energy and RMSD from the initial position.
Interface energy analysis and in silico mutagenesis
We used the InterfaceAnalyzer script in Rosetta at individual interfaces of the honeycomb to obtain interaction energies.
Using PyRosetta, we first in silico mutated each interface residue of all AIM2PYD/ASCPYD protomers in the honeycomb into all other possible a.a.42. We then removed those that resulted in energy scores >2 standard deviations from the mean (e.g., prolines that would distort backbone geometry and cause potential folding issues). We then applied remaining mutants (942 possibilities) to the pocket protomers of all six subsections, leaving the ligand protomers as WT. We then used the dG_separate subroutine in Rosetta InterfaceAnalyzer to obtain ∆Gs for (WT_ligand•WT_pocket) and (WT_ligand•mutant_pocket) interfaces; subtracting the ∆G of each mutant from that of WT then provides the changes in interface energy caused by the mutation (∆∆G). Each mutant was tested at least three times and the average values were used for analyses.
Molecular dynamics
The coordinates for each protein–ligand complex were obtained from Rosetta docking experiments such that the interface energy score and interface RMSD were minimized. All MD simulations were performed using GROMACS 5.1.3 (gromacs.org) with an all-atom CHARMM36 (ref. 56) force field. Simulation scripts were created using CHARMM-GUI57,58. Initial coordinates were energy minimized using the steepest descent algorithm and subsequently equilibrated for 4.75 ns, starting in the NVT ensemble and transitioning to the NPT ensemble. Neutralizing ions were added with ~200 Cl and ~200 K to a box of 13 nm × 13 nm × 13 nm.
Following initial energy minimization and equilibration, a second step of equilibration was performed in the NPT ensemble with a 2 fs timestep for 50 ns. A Nose-Hoover59 thermostat was used to maintain a reference temperature of 300 K with a 1 ps coupling time constant. The protein and solvent were coupled to separate temperature baths. A Parrinello-Rahman6 isotropic barostat with a 5 ps coupling time constant was used to maintain a pressure of 1 bar. Particle Mesh Ewald (PME) with a 1.2 nm cutoff radius, a 0.12 nm Fourier spacing, and cubic interpolation of 4 were used for electrostatics60. Van der Waals interactions had a 1.2 nm cutoff radius. A LINCS algorithm was used for bond constraints and XYZ periodic boundary conditions were enforced61.
Following the second step of equilibration, well-tempered Metadynamics (MetaD) simulations were performed using GROMACS 5.1.3 patched with PLUMED2 (ref. 62), using a CHARMM36 force field. The collective variable (CV) was the distance between the center-of-mass of the pocket and the center-of-mass of the ligand (residue 60). The backbone RMSD stayed mostly constant during the course of simulation (Supplementary Fig. 3A). Gaussians of energy were deposited along the trajectory in this CV space. Gaussians had an initial hill height of 1 kJ/mol and a width of 0.05 nm. Gaussians were deposited every 400 fs. A bias factor of 4 was used to adjust the hill heights according to the well-tempered MetaD scheme. Gaussians were saved to a grid with a bin spacing 0.01 nm. Simulations were considered complete when the ligand completely dissociated from the pocket, i.e., the CV distance exceeded 5 nm. Positional restraints were placed on every alpha-carbon in the pocket to prevent dissociation of the pocket protomers during the entirety of the MetaD simulations. The sum of all deposited Gaussians was computed to represent the dissociation free energy.
Polymerization assays
A total of 100 nM of AIM2FL was cleaved by 6 µM TEVp for 20 min in a 384-well plate. After cleavage, 150 nM of linearized plasmid dsDNA (~5-kbps, binding site normalized) was added and allowed to bind for 30 min. To start the assay, 2 µM FRET mix of MBP-tagged AIM2PYD or ASCPYD was added to the same well containing TEVp. Each experiment consisted of a control well with no AIM2FL, one with AIM2FL WT, and multiple AIM2FL mutants for both AIM2PYD and ASCPYD wells. AIM2 and ASC samples were run at the same time to ensure proper statistical analyses. Half-times for polymerization were calculated and converted to apparent kinetic rates19,63. The no AIM2FL control and AIM2FL WT control were used to normalize the kinetics rates for each mutant into an activity ratio scaling from 0 (no AIM2FL present) to 1 (AIM2FL WT activity). P values were calculated using Student’s t test for paired samples. The same strategy was used for ASCFL WT and mutants (0.5 µM, precleaved by TEVp for 30 min) inducing the polymerization of FRET-labeled AIM2PYD or ASCPYD.
nsEM
AIM2FL bound to dsDNA was prepared in the same manner as for the polymerization assays (100 nM protein, 150 nM dsDNA, linear plasmid ~5 kilo-bps). The samples were applied to carbon coated grids and imaged19,64.
Imaging recombinant AIM2PYD and ASCPYD filaments
Filament assembly of Alexa488- or Dylight550-labeled MBP-AIM2PYD and MBP-ASCPYD (1 µM each or 3 µM of nascent proteins for Fig. 3A) was induced by removing MBP by TEVp as indicated in figure legends. For preassembly, the AIM2PYD or ASCPYD filament was cleaved and incubated for 30 min prior to adding nascent proteins. Images were then taken using a Zeiss Axioskop 50 with a Zeiss Axiocam HRC camera and an Axio Observer inverted microscope with LSM700 confocal module.
Imaging AIM2FL-eGFP/mCherry and ASCFL-eGFP/mCherry in HEK293T cells
AIM2FL and ASCFL variants were cloned into the pCMV6 vector harboring eGFP or mCherry. To preserve native PYD–PYD interactions, the fluorescent proteins were positioned at the C-terminus of AIM2FL or ASCFL. Plasmids were then transiently transfected into HEK293T cells using lipofectamine (0.5 µg each plasmid; Invitrogen). After 12 h, cells were fixed with 4% paraformaldehyde and mounted on glass slides using ProLong Gold Antifade Mountant with DAPI (Thermo Fisher). Cells were then imaged using the same confocal microscope as the recombinant proteins.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Drs. Jeffry Gray and L. Mario Amzel for discussion, and Drs. Brendan Antiochos and Shuai Wu for cell culture experiments. This work was supported by American Cancer Society Research Scholars Grant (RSG-15-224-01DMC), NSF CAREER award (MCB1845003), and NIH R01GM129342 to J.S., NIH R35GM122510 to E.H.E. Computational resources were provided by Maryland Advanced Research Computing Center at Johns Hopkins University.
Source data
Author contributions
M.M. and J.S. conceived the project and designed experiments. M.M., W.Z., Z.M., J.L., and N.M. performed experiments. M.M., W.Z., Z.M., J.L., N.M., A.Y.L., E.H.E., and J.S. interpreted data. M.M., W.Z., N.M., and J.S. wrote the paper which other authors commented on.
Data availability
The cryo-EM structure has been deposited to the Protein Data Bank, PDB ID: 7K3R. The corresponding cryo-EM map was deposited in the EMDB with access code EMD-22656. The datasets generated during and/or analyzed during this study are available from the corresponding author on request. Source data are provided with this paper.
Code availability
All MD scripts can be found at https://github.com/nav610/a2dissocation65.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Qian Yin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-23045-8.
References
- 1.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 2.Bandera A, et al. The NLRP3 inflammasome is upregulated in HIV-infected antiretroviral therapy-treated individuals with defective immune recovery. Front. Immunol. 2018;9:214. doi: 10.3389/fimmu.2018.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maruzuru Y, et al. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe. 2018;23:254–265 e7. doi: 10.1016/j.chom.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 4.Kim S, et al. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 2010;40:1545–1551. doi: 10.1002/eji.201040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reyes Ruiz VM, et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl Acad. Sci. USA. 2017;114:13242–13247. doi: 10.1073/pnas.1710433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandes-Alnemri T, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang CA, Chiang BL. Inflammasomes and human autoimmunity: a comprehensive review. J. Autoimmun. 2015;61:1–8. doi: 10.1016/j.jaut.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Yi YS. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J. Physiol. Pharm. 2018;22:1–15. doi: 10.4196/kjpp.2018.22.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J. Immunol. 2020;205:307–312. doi: 10.4049/jimmunol.2000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA. Inflammasomes and metabolic disease. Annu. Rev. Physiol. 2014;76:57–78. doi: 10.1146/annurev-physiol-021113-170324. [DOI] [PubMed] [Google Scholar]
- 11.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–379. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016;46:269–280. doi: 10.1002/eji.201545839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin C, Zhang J. Inflammasomes in inflammation-induced cancer. Front. Immunol. 2017;8:271. doi: 10.3389/fimmu.2017.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu A, Wu H. Structural mechanisms of inflammasome assembly. FEBS J. 2015;282:435–444. doi: 10.1111/febs.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bami, S. et al. The use of anakinra in the treatment of secondary hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer67, e28581 (2020). [DOI] [PubMed]
- 16.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrone SR, et al. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 2015;6:7827. doi: 10.1038/ncomms8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu A, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matyszewski M, Morrone SR, Sohn J. Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl Acad. Sci. USA. 2018;115:E1963–E1972. doi: 10.1073/pnas.1712860115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen C, et al. Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc. Natl Acad. Sci. USA. 2019;116:2052–2057. doi: 10.1073/pnas.1817221116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li, Y. et al. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl Acad. Sci. USA115, 10845–10852 (2018). [DOI] [PMC free article] [PubMed]
- 22.Lu A, et al. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 2016;23:416–425. doi: 10.1038/nsmb.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matyszewski M, et al. Cryo-EM structure of the NLRC4(CARD) filament provides insights into how symmetric and asymmetric supramolecular structures drive inflammasome assembly. J. Biol. Chem. 2018;293:20240–20248. doi: 10.1074/jbc.RA118.006050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science. 2015;350:404–409. doi: 10.1126/science.aac5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tenthorey JL, et al. The structural basis of flagellin detection by NAIP5: a strategy to limit pathogen immune evasion. Science. 2017;358:888–893. doi: 10.1126/science.aao1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 2014;14:821–826. doi: 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumari, P., Russo, A.J., Shivcharan, S. & Rathinam, V.A. AIM2 in health and disease: inflammasome and beyond. Immunol. Rev.297, 83–95 (2020). [DOI] [PMC free article] [PubMed]
- 28.Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 2010;30:693–702. doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denes A, et al. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl Acad. Sci. USA. 2015;112:4050–4055. doi: 10.1073/pnas.1419090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lammert CR, et al. AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature. 2020;580:647–652. doi: 10.1038/s41586-020-2174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu B, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science. 2016;354:765–768. doi: 10.1126/science.aaf7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komada T, et al. Macrophage uptake of necrotic cell DNA activates the AIM2 inflammasome to regulate a proinflammatory phenotype in CKD. J. Am. Soc. Nephrol. 2018;29:1165–1181. doi: 10.1681/ASN.2017080863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi, M. et al. AIM2 promotes the development of non-small cell lung cancer by modulating mitochondrial dynamics. Oncogene39, 2707–2723 (2020). [DOI] [PubMed]
- 35.Lu A, et al. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 2015;1:15013. doi: 10.1038/celldisc.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu A, Kabaleeswaran V, Fu T, Magupalli VG, Wu H. Crystal structure of the F27G AIM2 PYD mutant and similarities of its self-association to DED/DED interactions. J. Mol. Biol. 2014;426:1420–1427. doi: 10.1016/j.jmb.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sohn J, Hur S. Filament assemblies in foreign nucleic acid sensors. Curr. Opin. Struct. Biol. 2016;37:134–144. doi: 10.1016/j.sbi.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu B, et al. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell. 2014;55:511–523. doi: 10.1016/j.molcel.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David L, et al. Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc. Natl Acad. Sci. USA. 2018;115:1499–1504. doi: 10.1073/pnas.1721967115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray JJ, et al. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003;331:281–299. doi: 10.1016/S0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 41.Stranges PB, Kuhlman B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013;22:74–82. doi: 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaudhury S, Lyskov S, Gray JJ. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt FI, et al. A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J. Exp. Med. 2016;213:771–790. doi: 10.1084/jem.20151790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katsuno H, et al. Actin migration driven by directional assembly and disassembly of membrane-anchored actin filaments. Cell Rep. 2015;12:648–660. doi: 10.1016/j.celrep.2015.06.048. [DOI] [PubMed] [Google Scholar]
- 45.Cai X, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156:1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003;142:334–347. doi: 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 48.Tang G, et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 49.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 50.Pettersen EF, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comp.ut Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang, R. Y. et al. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife5, e17219 (2016). [DOI] [PMC free article] [PubMed]
- 53.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyskov S, Gray JJ. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008;36:W233–W238. doi: 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang J, MacKerell AD., Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 58.Qi Y, et al. CHARMM-GUI martini maker for coarse-grained simulations with the martini force field. J. Chem. Theory Comput. 2015;11:4486–4494. doi: 10.1021/acs.jctc.5b00513. [DOI] [PubMed] [Google Scholar]
- 59.Morriss GP, Dettmann CP. Thermostats: analysis and application. Chaos. 1998;8:321–336. doi: 10.1063/1.166314. [DOI] [PubMed] [Google Scholar]
- 60.Sagui C, Darden TA. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu, Rev. Biophys. Biomol. Struct. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 61.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 62.Bussi G, Tribello GA. Analyzing and biasing simulations with PLUMED. Methods Mol. Biol. 2019;2022:529–578. doi: 10.1007/978-1-4939-9608-7_21. [DOI] [PubMed] [Google Scholar]
- 63.Mazanek Z, Sohn J. Tracking the polymerization of DNA sensors and inflammasomes using FRET. Methods Enzymol. 2019;625:87–94. doi: 10.1016/bs.mie.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 64.Matyszewski M, Sohn J. Preparation of filamentous proteins for electron microscopy visualization and reconstruction. Methods Enzymol. 2019;625:167–176. doi: 10.1016/bs.mie.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 65.Matyszewski, M. et al. Distinct axial and lateral interactions within homologous filaments dictate the signaling specificity and order of the AIM2-ASC inflammasome. Nature Commun. Github 10.5281/zenodo.4667936 (2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM structure has been deposited to the Protein Data Bank, PDB ID: 7K3R. The corresponding cryo-EM map was deposited in the EMDB with access code EMD-22656. The datasets generated during and/or analyzed during this study are available from the corresponding author on request. Source data are provided with this paper.
All MD scripts can be found at https://github.com/nav610/a2dissocation65.