

Abstract
Three-dimensional (3D) multicomponent metal oxides with complex architectures could enable previously impossible energy storage devices, particularly lithium-ion battery (LIB) electrodes with fully controllable form factors. Existing additive manufacturing approaches for fabricating 3D multicomponent metal oxides rely on particle-based or organic-inorganic binders, which are limited in their resolution and chemical composition, respectively. In this work, aqueous metal salt solutions are used as metal precursors to circumvent these limitations, and provide a platform for 3D printing multicomponent metal oxides. As a proof-of-concept, architected lithium cobalt oxide (LCO) structures are fabricated by first synthesizing a homogenous lithium and cobalt nitrate aqueous photoresin, and then using it with digital light processing printing to obtain lithium and cobalt ion containing hydrogels. The 3D hydrogels are calcined to obtain micro-porous self-similar LCO architectures with a resolution of ~100μm. These free-standing, binder- and conductive additive-free LCO structures are integrated as cathodes into LIBs, and exhibit electrochemical capacity retention of 76% over 100 cycles at C/10. This facile approach to fabricating 3D LCO structures can be extended to other materials by tailoring the identity and stoichiometry of the metal salt solutions used, providing a versatile method for the fabrication of multicomponent metal oxides with complex 3D architectures.
Keywords: additive manufacturing, hydrogels, multicomponent metal oxides, lithium ion batteries, lithium cobalt oxide
Graphical Abstract
3D architected multicomponent metal oxides are fabricated using metal-ion containing aqueous photoresins. These photoresins are used with digital light processing printing to fabricate 3D metal-ion hydrogels, which are then calcined to give self-similar metal oxide structures. As proof-of-concept, 3D lithium cobalt oxide lattices are designed and used as lithium-ion battery cathodes.
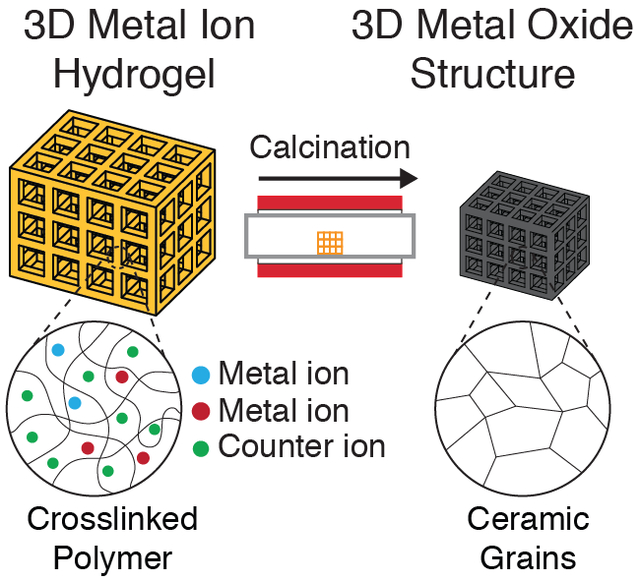
1. Introduction
Metal oxide ceramics are of significant technological importance in modern society. These materials exhibit a variety of functional properties, such as superconductivity,[1] ferroelectricity,[2] ferromagnetism,[3] and electrochemical activity.[4] As a result, they are used in almost every scientific and engineering field, ranging from catalysis,[5] implantable biomedical devices[6,7] to large-scale energy generation[8] and storage.[9] A notable application of these materials have been as lithium-ion battery (LIB) cathodes, with materials such as lithium cobalt oxide (LCO, LiCoO2), lithium nickel cobalt aluminum oxide (NCA, LiNi0.8Co0.15Al0.05O2), and lithium nickel manganese cobalt oxide (NMC, LiNi0.33Mn0.33Co0.33O2) being extensively used in both research and commercial applications.[10,11]
An ongoing challenge in the use of these cathode materials is that they are difficult to process and shape due to their high melting points and brittle nature.[12] Consequently, these materials often have simple form factors, and have largely been utilized as thin films that are cast from a slurry.[13] In recent years, there has been a surge of interest in the use of 3D architected metal oxides in LIBs as they could simultaneously enable high energy and power densities.[14] Multiple advanced techniques have been developed to fabricate these 3D materials, including solution synthesis,[15] templated growth,[16] and porosity control.[17] For example, Lu et al. used a wood template approach with sol-gel chemistry to fabricate 3D LCO cathodes.[18] Jeong et al. laminated multiple sheets of LCO/graphite together to fabricate 3D cathodes with aligned microchannels.[19] The main limitations with these approaches are that they often require multiple processing steps or are limited in the geometries or dimensions that can be achieved.
Additive manufacturing (AM) has recently emerged as the most enabling technology for the fabrication of 3D architected metal oxides — its layer-by-layer approach to fabrication circumvents many of the geometric limitations associated with the other fabrication techniques, allowing for the construction of structures with almost arbitrary geometries.[23] Material-extrusion based processes such as direct ink writing (DIW) have seen particular use due to their low cost and compatibility with metal oxide ceramics.[20] Since Sun et al. pioneered the use of DIW with lithium iron phosphate (LFP) containing aqueous-based inks for the fabrication of 3D interdigitated microbattery architectures,[21] there have been multiple demonstrations of extrusion-based methods in the fabrication of these 3D LIB electrodes.[22]
The use of vat photopolymerization (VP) processes in the fabrication of LIB cathodes has not been as extensively explored, although it is capable of achieving sub-micron resolutions[23] and volumetric throughputs of up to 100 liters per hour,[24] superior to that of material-extrusion processes. This has largely been due to the fact that the current methods for fabricating multicomponent oxides have some shortcomings: with the slurry approach, a photosensitive slurry that contains the desired multicomponent metal oxide micro/nanoparticles is used to fabricate architected nanocomposite structures, which are then calcined to obtain the final metal oxide structure.[25] For example, Hostaša et al. used a photosensitive slurry containing Al2O3, Y2O3, and Yb2O3 nanoparticles with SLA to obtain Yb0.3Y2.7Al5O12 parts after calcination.[26] The slurry method is capable of fabricating a wide range of metal oxides, but have limited resolutions due to the scattering of light by the particles.[20,27] Furthermore, agglomeration of the particles within the slurry often leads to structural inhomogeneity within the fabricated structures.[28] Reacting different metal oxides particles together in a solid-state reaction requires high temperatures, and can often result in compositional inhomogeneity.[29]
An alternative VP approach that circumvents these issues is to use nanoparticle-less organic-inorganic photoresins that contain metal heteroatoms within the polymer instead. These homogeneous photoresins are capable of producing 3D metal oxides with sub-micron features after thermal treatment. For example, Gailevičius et al. were able to use a zirconium and silicon organic-inorganic photoresin with two-photon lithography (TPL) to fabricate mixed silica/zirconia 3D structures with sub-100nm features.[30] The drawback to this method is that the materials compatible with it are often limited in composition to silicon-based oxides,[20] binary transition metal oxides,[23] or oxide glasses.[31]
The use of aqueous metal salt solutions as metal oxide precursors combines the advantages of both existing approaches — the compositional versatility afforded by using particles and the high resolutions achievable with the organic-inorganic photoresins. We recently demonstrated this concept by using an aqueous zinc nitrate–poly(ethylene glycol) diacrylate (PEGda) photoresin with TPL to fabricate 3D architected zinc-nitrate containing tetrakaidecahedron hydrogels. These hydrogels were then calcined to give self-similar piezoelectric zinc oxide (ZnO) tetrakaidecahedron structures with features on the order of 250 nm.[32] The use of metal salt solutions provides a facile and unique opportunity for the fabrication of more complex multicomponent metal oxides — the multicomponent metal salt precursor solution can be simply prepared by dissolving the constituent metal salt components in the appropriate stoichiometric ratios. The use of solutions ensures that the precursors are mixed at a molecular level, improving the compositional homogeneity of the synthesized metal oxides.[33]
To demonstrate this, we synthesized an aqueous photoresin using lithium nitrate and cobalt nitrate solutions and used it to 3D print lithium and cobalt nitrate containing hydrogels architectures using digital light processing (DLP) printing (Figure 1). Calcination of these hydrogels resulted in self-similar LCO architectures. LCO was chosen because it is one of the most commonly used lithium-ion battery (LIB) cathode materials, and has well-understood electrochemical properties.[34] Several studies have demonstrated the fabrication of 3D LCO using techniques such as direct ink writing[35] and electroplating,[36] but none has been able to fabricate complex LCO structures of arbitrary design using VP processes. Electrochemical cycling of binder- and conductive additive-free LCO lattices fabricated using this aqueous photoresin approach exhibited a first cycle specific discharge capacity of 121mAh/g at C/40, with capacity retention of 76% over 100 cycles at C/10. This work demonstrates an accessible pathway for the fabrication of multicomponent metal oxides for LIB applications and beyond.
Figure 1. DLP printing of aqueous metal salt containing photoresins.

(a) Resin formulation and schematic of the solvated metal ions with the PEGda in the aqueous photoresin. (b) Schematic of DLP printing of a Li+/Co2+ hydrogel 3D structure. (c) The 3D Li+/Co2+ hydrogel is calcined to form a self-similar LCO structure. (d) Optical images of Li+/Co2+ hydrogel (left) and calcined LCO (right) cubic lattices. Scale bars, 5mm.
2. Results and Discussion
2.1. AM of LCO Architectures
We first prepared 2.67M aqueous solutions of lithium nitrate and cobalt nitrate hexahydrate and combined them together in a 1:1 ratio. The metal precursors were then mixed with PEGda (Mn = 575g/mol) at a prescribed ratio to form a maroon solution. A UV-absorbing dye (tartrazine) and a photoinitiator (lithium phenyl-2,4,6-trimethylbenzoylphosphinate, LAP), were added to the metal nitrate-PEGda solution to yield a homogenous orange solution (Figure 1a). The amount of photoinitiator and UV-absorbing dye were decided using an iterative approach where the goal was to maximise the print speed and z-layer resolution of the photoresin. The prepared resins had viscosities similar to that of water, and were observed to be stable for upwards of six months. We used a commercial DLP printer (Ember, Autodesk) to directly fabricate cubic lattices 16 mm in diameter and 3 mm tall, with unit cells of length 850 μm and a beam thickness of approximately 150 μm, from the LCO photoresin (Figure 1b). In terms of resolution, the 150 μm achieved in this study is well below the 250 – 400 μm resolutions that are typically obtained using the slurry method with DLP printing,[37–39] and is comparable to the 85 – 200 μm resolutions that have been reported for DLP printing of organic-inorganic photoresins.[40–42] Cubic lattices were chosen as they exemplified a design that could be easily fabricated with DLP printing, but challenging with other manufacturing techniques. The simultaneous formation of an entire layer allows for the fabrication of even 90° overhangs, allowing for the fabrication of complex geometries.
The fabricated Li+/Co2+ hydrogel architectures were calcined at 700°C to obtain the LCO cubic lattices (Figure 1c). Calcination was performed in a tube furnace using a slow multi-stage thermal process at a reduced pressure of 20 Torr to impede the combustion reaction and to minimize the rate of outgassing volatiles. Figure 1d shows optical images of the lattices before and after calcination. Calcination resulted in black lattices, which was consistent with the color of LCO, and had self-similar architectures as its hydrogel precursors. A substantial amount of linear shrinkage (~40 – 50%) and mass loss (~90 – 95%) was also observed, which was dependent on the composition of the photoresin, as will be discussed in the proceeding section.
2.2. Microstructure of LCO Architectures
We found the amount of linear shrinkage, mass loss, and porosity of the final material to depend on the ratio of metal salt to polymer binder. The general equation for the combustion synthesis of LCO with PEGda (C26H46O13) in this system can be represented by Equation 1:
| (1) |
where nPEGda is the mol ratio of PEGda to Co(NO3)2.6H2O. In this model, we assume that the only decomposition products are H2O, N2, and CO2. Under these assumptions, the model shows that for every mol of LCO formed, 49nPEGda + 7.5 mols of gas are evolved.
To determine the impact of nPEGda on the microstructure of the lattices, we used resins with different volume ratios of metal salt solutions to PEGda, corresponding to nPEGda ~ 2.2, 1.0, 0.5, and 0.4 (Figure 2). We found that for nPEGDA ~ 2.2 and 1.0, the surface of the lattices were significantly cracked (Figure S1), as would be expected from the relatively large amount of gases evolved (Figure 2a). With lower nPEGDa values, ~ 0.5 and 0.4, little to no surface cracking was observed (Figure 2b). Periodic “diamond” arrays spaced ~ 21μm apart were observed on all the lattices, likely due to the pixel pattern of the DLP printer (Figure S2). Further analysis of the surface features indicated that they were comprised of partially sintered faceted particles approximately 153 ± 88nm (mean ± standard deviation) in size. To determine the level of porosity within the structure, we used a gallium focused ion beam (FIB) to mill away and expose representative cross-sections of the nodes of the lattice. Large pores at least 80μm in size were found in the nPEGda ~ 2.2 lattices (Figure 2c), which were replaced with large ~ 10μm wide cracks as nPEGda decreased to 1.0. With further nPEGda reduction, the number of large cracks decreased qualitatively, with the largest cracks having crack widths of ~ 5μm. These results indicated that resin formulations with lower nPEGda resulted in better quality LCO structures. To compensate for the volatilization of Li during the combustion synthesis,[43,44] lattices were also fabricated from nPEGda ~ 0.4, but with 5 mol% excess Li precursor resins. Cross-sections of the nodes revealed a similar microstructure (Figure 2d) to its stoichiometric counterparts, with the presence of sub-micron and micropores. Based on FIB cross-section analysis results, we carried out further characterization tests with both the stoichiometric (LCO-s) and Li excess (LCO-e) nPEGda ~ 0.4 resins. The linear shrinkage of lattices made using these resins after calcination was ~44%, with a material porosity of ~56%. The mass loss was determined to be ~92% (See Supporting Information for additional discussion).
Figure 2. Microstructure of 3D LCO structures.

(a) The gas evolved per mol of LCO formed as a function of nPEGda as determined from the general combustion synthesis equation. Points I, II, III, IV denote resins with different nPEGda values. (b) Surface microstructure of lattices made from low nPEGda resins (~0.5 and 0.4), showing a periodic “diamond” array comprised of partially sintered faceted nanoparticles. Scale bars, 200μm (left), 10μm (center), 500nm (right). (c) Cross-sections of lattice nodes made from resins with different nPEGda values. The extent of cracking and porosity decreases qualitatively with nPEGda value. Scale bars, 20μm. (d) Cross-section of lattice node made from a resin with nPEGda ~ 0.4, but with 5mol% excess Li precursor. The microstructure is qualitatively similar to its nPEGda ~0.4 stoichiometric counterpart, with the presence of submicron and micropores. Scale bars, 20μm (left) and 5μm (right).
The presence of porosity is a consequence of the size of the structures made using DLP printing. This is evidenced from our previous work with ZnO, where we used TPL with a zinc nitrate aqueous photoresin to fabricate monolithic ZnO microstructures with sub-micron feature sizes.[32] The high surface area to volume ratio of these microstructures allowed the gases formed during the combustion synthesis process to diffuse out of the material without getting trapped in them and forming pores. In contrast, the centimeter-sized lattices fabricated in this study using DLP have features ~100 μm in size. The comparatively smaller surface area to volume ratio of these lattices prevents the gases produced from completely diffusing out of the material, resulting in the formation of pores.
2.3. Material Characterization of LCO Architectures
Powder X-ray diffraction (XRD) spectra of pulverized lattices made from LCO-s and LCO-e resins indicated that the materials synthesized had peaks consistent with the hexagonal α-NaFeO2 layered structure expected of LCO (Figure 3a). The c/a ratio of 4.98 for both LCO-s and LCO-e samples also corroborated this (Table S2), close to the 4.99 value expected for compositionally stoichiometric LCO.[45] A common metric used to estimate the degree of cation mixing within the layers is the peak intensity ratio, R, of I(003)/I(104), with higher R values corresponding to less mixing.[46] The R values for the LCO-s and LCO-e samples were determined to be 1.42 and 1.52, respectively, signaling ordering of the cations. We chose to conduct further experiments on the LCO-e lattices because of their higher R value. Energy-dispersive X-ray spectroscopy (EDS) elemental mapping also showed the homogenous distribution of cobalt and oxygen throughout the structure (Figure 3b). Lithium was not mapped as it could not be detected in our EDS system. Impurities from various elements were also detected, at a combined 7.5at% (Figure S3), likely due to the incomplete removal of the polymer binder and photoactive molecules, as well as from the quartz tube used during the calcination process.
Figure 3. Characterization of LCO structures.

(a) XRD from pulverized lattices made from LCO-s and LCO-resins. The diffraction peaks are consistent with that of the ICSD reference for LCO. R denotes the peak intensity ratio of I(003)/I(104), and is a metric used to estimate the degree of cation mixing. Higher R values indicate less cation mixing. (b) EDS element maps indicate the homogenous distribution of cobalt and oxygen throughout the LCO-e lattice. Carbon, the major impurity at 6.5at%, was also detected. The combined impurities totaled 7.5at%.
2.4. Electrochemical Performance of LCO Architectures
To demonstrate the potential of these architected LCO-e lattices as battery cathodes, we performed cyclic voltammetry (CV) on slurry electrodes fabricated from pulverized LCO lattices (Figure 4a). The CV of the first cycle showed three pairs of redox peaks that closely matched that found in the literature for LCO. The first major pair of peaks at 3.948 and 3.886V was observed for the anodic (Li extraction) and cathodic (Li insertion) scans respectively, corresponding to the first order phase transition between two hexagonal phases. The two smaller pairs observed at 4.077 / 4.064V and 4.181 / 4.160V correspond to an order / disorder transition of Li ions with a distortion to a monoclinic phase.[47,48] The results from the CV scans further confirmed the identity of the synthesized material as LCO, and also indicated that it was electrochemically active.
Figure 4. Electrochemical performance of architected LCO lattices.

(a) Representative first cycle scan of a cyclic voltammogram performed on a slurry electrode from pulverized LCO-e lattices at a scan rate of 5μV/s. (b) Electrochemical cycling of three nominally identical LCO lattices, LCO-e1 (red circles), LCO-e2 (blue circles), and LCO-e3 (black circles), between 3.0 and 4.2 V at different C-rates. The lattices are assembled in a coin cell using a conductive adhesive (See Methods, Figure S5).
Coin cells with LCO-e lattices as the cathode were subjected to electrochemical cycling. Since these porous lattices were relatively brittle, a polypropylene washer was used in the coin cell to support the stress from the coin cell spring (Figure S5). The specific discharge capacities and Coulombic efficiencies (CE) for three separate coin cells (LCO-e1, LCOe2, LCO-e3), each with a mass loading of 50mg/cm2, at different C-rates (C/10 implies discharge or charge in 10 hours) are shown in Figure 4b. The average first cycle specific discharge capacity at C/40 was 121mAh/g, which is close to the lower bound of the 120 – 154 mAh/g range of discharge capacities reported in the literature for LCO materials prepared via solution combustion synthesis.[45,49–51] The decreased capacity was possibly due to parts of the lattice breaking and becoming electrically isolated during the coin cell fabrication process. The Ohmic impedance across the electrode is also relatively large because of the height of the electrode and the lack of a conductive additive.
The capacity also decreased as C rate increased: at C/20, C/10, C/5, 2C/5, and 1C, the average first cycle specific discharge capacities were 115, 107, 94, 23, and 2.6mAh/g, respectively. The lattices did not show any capacity at 2C. The CE at 2C/5, 1C, and 2C deviated significantly from 100% due to the near zero capacities at those C rates. Of the three samples, only LCO-e3 (black circles) exhibited a significant specific discharge capacity at 2C/5, possibly due to its ~10x lower resistance of the first arc in its Nyquist plot compared to the others (Figure S4). This could be due to the sample-to-sample variation in the adherence of the lattice to the current collector, with LCO-e3 having better adhesion. Extended cycling at C/10 showed a linear capacity decay of ~0.24% per cycle, resulting in a capacity retention of 76% after 100 cycles. The average CE for the final 100 cycles was determined to be 98.5 ± 0.5%. Taken together, these results clearly indicate that these 3D architected LCO lattices are electrochemically active and are able to operate as LIB cathodes.
There are a few approaches that can be taken to improve the electrochemical performance of these architected LCO cathodes. For optimal electrochemical performance, the design of the cathode should allow for high mass loadings while still maintaining thin features to reduce the ion diffusion path within the cathode.[52] To obtain these, the unit cell geometry of the lattices could be optimized to have a larger relative density so as to increase the mass loading of the cathode. The beam sizes could also be decreased to reduce the ion diffusion paths and improve the discharge capacities at higher currents.
3. Conclusion
We demonstrate that aqueous metal salt photoresins are a unique and facile platform for the digital fabrication of 3D architected multicomponent metal oxides. The homogeneity and compositional versatility of these aqueous metal salt photoresins addresses some of the limitations faced by the current state-of-the-art slurry and organic-inorganic resin approaches, as highlighted in our fabrication of LCO cubic lattices. The demonstration of these architected materials as 3D LIB cathodes further highlights the potential of this platform in enabling the production of previously impossible or impractical devices. Given that most modern LIB cathode materials are multicomponent metal oxides, there is significant potential in utilizing this technique to fabricate more advanced cathode materials like NCA and NMC, which have higher specific capacities and are more environmentally friendly. Additive manufacturing of complex multicomponent metal oxides by the simple preparation of a metal salt solution with the right stoichiometry has immense technological implications beyond energy storage — superconducting, multiferroric, and even more exotic oxide materials can potentially now be 3D printed using this facile and accessible method, paving the way for the next generation of smart devices.
4. Experimental Section/Methods
Materials:
Lithium nitrate (ReagentPlus, Sigma-Aldrich), cobalt(II) nitrate hexahydrate (≥98%, Sigma-Aldrich), poly(ethylene glycol) diacrylate Mn = 575 (PEGda) (Sigma-Aldrich), tartrazine (≥85%, Sigma-Aldrich), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) (≥95%, Sigma-Aldrich), isopropanol (Sigma-Aldrich), Timical Super C65 carbon black (MTI Corporation), polyvinylidene fluoride Mw ~ 534kDa (PVDF) (Sigma-Aldrich), 1-methyl-2-pyrrolidinone (NMP) (anhydrous, 99.5%, Sigma-Aldrich), lithium (ribbon, thickness x W 0.38mm x 23mm, 99.9%, Sigma-Aldrich), and lithium hexafluorophosphate solution (1M LiPF6 in ethylene carbonate (EC) and diethyl carbonate (DEC) 1:1 volume ratio, Selectilyte LP 40, BASF) were used as received without further purification.
LCO Resin Preparation:
2.67M solutions of lithium nitrate and cobalt nitrate were first prepared separately with deionized water. The lithium solution and cobalt solution were then mixed together in the appropriate volume ratio to obtain the aqueous lithium and cobalt metal salt precursor solution. PEGda and the metal salt precursor solution were then mixed together in a prescribed amount to form a maroon solution. LAP and tartrazine were dissolved in deionized water (10vol% of the metal salt-PEGda solution) and then added to the metal salt–PEGda solution to give an orange solution. The amount of LAP was fixed at 2.5mg of LAP per mL of metal salt-PEGda solution prepared. Similarly, the tartrazine was fixed at 0.5mg of tartrazine per mL of metal salt-PEGda solution prepared (See Supporting Information Table S4 for the compositions of all the resins made). The prepared resins had viscosities similar to that of water, and were used directly after mixing. The resins were observed to be stable for upwards of six months.
3D Printing of LCO Resin:
A commercial DLP 3D printer (Autodesk Ember) was used for the fabrication of the Li+/Co2+ hydrogel structures. Cubic lattices with unit cell length of 1mm and beam thicknesses of approximately 150μm were designed. The unit cells were arranged in a circular array to best fit the area of the coin cell after calcination. The size of the lattice was approximately 16mm in diameter and 3mm thick. The exposure parameters for these lattices varied based on the value of nPEGda in the resin, with the exposure needed decreasing as nPEGda increased. The exposure parameters for the LCO-e resin can be found in the Supporting Information Table S5.
Calcination of Hydrogel Lattices:
The Li+/Co2+ hydrogel lattices were calcined in a vacuum furnace (MTI OTF-1500X) in a quartz tube (1m in length, 92mm inner diameter) at a reduced air pressure of 20Torr to give black LCO lattices. The temperature profile was as follows: a ramp to 500°C at 0.25°C/min followed by a ramp to 700°C at 2C°/min. The samples were held at 700°C for 3 hours, before cooling to room temperature at 2C°/min. The shrinkage and mass losses are discussed in the Supporting Information.
Scanning Electron Microscope (SEM) Imaging and Focused Ion Beam (FIB) Milling:
The LCO lattices were imaged in an SEM (FEI Versa 3D DualBeam) at an accelerating voltage of 10kV. Cross-sections of the lattices were made using a gallium FIB within the same SEM at an accelerating voltage of 30kV and a current of 65nA. FIB cleaning of the cross-section was conducted at a lower accelerating voltage of 16kV and a current of 42nA.
Powder X-ray Diffraction (XRD):
XRD (PANalytical X’Pert Pro) data was collected at 45kV and 20mA using a Cu source. LCO-s and LCO-e powders were obtained by pulverizing their respective lattices between two glass slides.
Energy-dispersive X-ray Spectroscopy (EDS):
EDS maps were generated in a Zeiss 1550 VP FESEM equipped with an Oxford X-Max SDD EDS system. The applied voltage was 10kV.
Cyclic Voltammetry (CV):
LCO-e lattices were pulverized into a fine powder between glass slides, and then further crushed in a vortex mixer in a polyethylene vial with isopropanol and zirconia ball-mill balls for 2 hours. The resulting mixture was dried in a glass dish at 60 °C for 2 hours then at 100 °C in vacuum for 18 hours. The electrode slurry was prepared with the following composition: 80wt% LCO powder, 10wt% Timical Super C65 carbon black, and 10wt% binder polyvinylidene fluoride (PVDF) in N-methyl-2-pyrrolidone (NMP). The slurry was vortexed in a polyethylene vial with zirconia ball-mill balls for 5 hours, before being cast onto an Al foil using a film applicator blade and dried for 12 hours at 50 °C. 11.1 mm diameter electrode discs were punched out of the foil and the electrodes were dried for 18 hours at 100°C under vacuum. Samples were tested in CR2032 coin cells that were assembled in an argon-filled glovebox. 30μL of Selectilyte LP40 was used as the electrolyte. Li foil, was used as the counter electrode with a 25μm thick polypropylene separator. CV was performed using a BioLogic BCS-805 battery cycler scanning between 3 V and 4.2 V at a scan rate of 5μV/s.
Electrochemical Cycling of 3D LCO Structures:
A conductive adhesive was first prepared by mixing 75wt% conductive Timical Super C65 carbon black and 25wt% PVDF binder in NMP. 1.28mL of NMP was used for every 100mg of the carbon black/PVDF mixture. The solution was mixed in a vortex mixer in a polyethylene vial with zirconia ball-mill balls for 24 hours. The mixture was drop-casted onto a 0.2mm thick stainless steel spacer and thinned to 0.5mm thickness using a film applicator blade. The LCO lattice was then carefully placed into the middle of the adhesive and the whole stack was dried at 35°C for 4 hours. A polypropylene ring 0.18mm thick with inner and outer diameters of 11mm and 15.9mm, respectively, was placed around the LCO structure to support the stress of the coin cell spring. Cells were then assembled in an argon-filled glovebox using a lithium foil as the counter electrode, and Selectilyte LP 40 as the electrolyte. 400μL of electrolyte was added into the cell cavity prior to the addition of the lithium foil, although some electrolyte spilled out of the cell during crimping. A schematic of the cell stack is shown in Figure S5 of the Supporting Information.
Coin cells were analyzed by a Neware BTS4000 cycler and were cycled galvanostatically between 3.0 and 4.2 V at C/40 for two cycles, then at 5 cycles each at C/20, C/10, C/5, 2C/5, 1C, and 2C. Following these, the cells were cycled for 100 cycles at C/10. A rate of 1C corresponds to a discharge or charge in 1 hour based on a 155mAh/g capacity of LCO. 2C corresponds to a 30 minute half-cycle, etc.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the following people: K. Narita and A. Vyatskikh for assistance with the vacuum furnace setup. A. Kwong for assistance with the FIB experiments. C. Ma for assistance with the EDS experiments. J.R.G acknowledges the financial support from the National Institutes of Health (Grant No. 1R01CA194533), the Caltech SURF office, and the Vannevar-Bush Faculty Fellowship from the US Department of Defence. D.W.Y. and M.A.C. contributed equally to the work. D.W.Y., M.A.C., and J.R.G. conceived and designed the experiments. D.W.Y., Z.W.T., and V.T. developed the polymer chemistry and fabricated the structures. D.W.Y. and M.A.C. developed the heating profile and performed SEM/FIB analyses. D.W.Y. performed the XRD and EDS data collection. M.A.C. and M.A.S. performed the electrochemical experiments. D.W.Y., M.A.C., and J.R.G. wrote the manuscript.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library.
Contributor Information
Daryl W. Yee, Division of Engineering and Applied Science, California Institute of Technology, CA 91125, USA
Michael A. Citrin, Division of Engineering and Applied Science, California Institute of Technology, CA 91125, USA
Zane W. Taylor, Division of Engineering and Applied Science, California Institute of Technology, CA 91125, USA
Max A. Saccone, Division of Chemistry and Chemical Engineering, California Institute of Technology, CA 91125, USA
Victoria L. Tovmasyan, Department of Materials Science and Engineering, University of California, Berkeley, CA 94720, USA
Julia R. Greer, Division of Engineering and Applied Science, California Institute of Technology, CA 91125, USA
References
- [1].Yan MF, Rhodes WW, Gallagher PK, J. Appl. Phys. 1988, 63, 821. [Google Scholar]
- [2].Bowen CR, Kim HA, Weaver PM, Dunn S, Energy Environ. Sci. 2014, 7, 25. [Google Scholar]
- [3].Rao CNR, Annu. Rev. Phys. Chem. 1989, 40, 291. [Google Scholar]
- [4].Maduraiveeran G, Sasidharan M, Jin W, Prog. Mater. Sci. 2019, 106, 100574. [Google Scholar]
- [5].Gawande MB, Pandey RK, Jayaram RV, Catal. Sci. Technol. 2012, 2, 1113. [Google Scholar]
- [6].Chen YW, Moussi J, Drury JL, Wataha JC, Expert Rev. Med. Devices 2016, 13, 945. [DOI] [PubMed] [Google Scholar]
- [7].Andreescu S, Ornatska M, Erlichman JS, Estevez A, Leiter JC, in Fine Particles in Medicine and Pharmacy, (Eds: Matijević E), Springer, New York, USA: 2012, Ch. 3. [Google Scholar]
- [8].Li Z, Klein TR, Kim DH, Yang M, Berry JJ, van Hest MF, Zhu K, Nat. Rev. Mater. 2018, 3, 1. [Google Scholar]
- [9].Arteaga J, Zareipour H, Thangadurai V, Curr. Sustainable/Renewable Energy Rep. 2017, 4, 197. [Google Scholar]
- [10].Rozier P, Tarascon JM, J. Electrochem. Soc. 2015, 162, A2490. [Google Scholar]
- [11].He P, Yu H, Li D, Zhou H, J. Mater. Chem. 2012, 22, 3680 [Google Scholar]
- [12].Rahaman MN, Ceramic Processing and Sintering, Marcel Dekker, New York, USA: 2003. [Google Scholar]
- [13].Kraytsberg A, Ein-Eli Y, Adv. Energy Mater. 2016, 6, 1600655. [Google Scholar]
- [14].Sun H, Zhu J, Baumann D, Peng L, Xu Y, Shakir I, Huang Y, Duan X, Nat. Rev. Mater. 2019, 4, 45. [Google Scholar]
- [15].Xia Q, Ni M, Chen M, Xia H, J. Mater. Chem. A 2019, 7, 6187. [Google Scholar]
- [16].Huang C, Grant PS, J. Mater. Chem. A 2018, 6, 14689. [Google Scholar]
- [17].Li L, Erb RM, Wang J, Wang J, Chiang YM, Adv. Energy Mater. 2019, 9, 1802472. [Google Scholar]
- [18].Lu LL, Lu YY, Xiao ZJ, Zhang TW, Zhou F, Ma T, Ni Y, Yao HB, Yu SH, Cui Y, Adv. Mater. 2018, 30, 1706745. [DOI] [PubMed] [Google Scholar]
- [19].Jeong H, Lim SJ, Chakravarthy S, Kim KH, Lee J, Heo JS, Park H, J. Power Sources 2020, 451, 227764. [Google Scholar]
- [20].Chen Z, Li Z, Li J, Liu C, Lao C, Fu Y, Liu C, Li Y, Wang P, He Y, J. Eur. Ceram. Soc. 2019, 39, 661. [Google Scholar]
- [21].Sun K, Wei TS, Ahn BY, Seo JY, Dillion SJ, Lewis JA, Adv Mater. 2013, 25, 4539. [DOI] [PubMed] [Google Scholar]
- [22].Cheng M, Deivanayagam R, Shahbazian-Yassar R, Batteries Supercaps 2020, 3, 130. [Google Scholar]
- [23].Vyatskikh A, Ng RC, Edwards B, Briggs RM, Greer JR, Nano Lett. 2020, 20, 3513. [DOI] [PubMed] [Google Scholar]
- [24].Walker DA, Hedrick JL, Mirkin CA, Science 2019, 366, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang W, Sun J, Guo B, Chen X, Ananth KP, Bai J, J. Eur. Ceram. Soc. 2020, 40, 682. [Google Scholar]
- [26].Hostaša J, Schwentenwein M, Toci G, Esposito L, Brouczek D, Piancastelli A, Pirri A, Patrizi B, Vannini M, Biasini V, Scr. Mater. 2020, 187, 194. [Google Scholar]
- [27].Halloran JW, Annu. Rev. Mater. Res. 2016, 46, 19. [Google Scholar]
- [28].Ding G, He R, Zhang K, Xia M, Feng C, Fang D, Ceram. Int. 2020, 46, 4720. [Google Scholar]
- [29].Stein A, Keller SW, Mallouk TE, Science 1993, 259, 1558. [DOI] [PubMed] [Google Scholar]
- [30].Gailevičius D, Padolskytė V, Mikoliūnaitė L, Šakirzanovas S, Juodkazis S, Malinauskas M, Nanoscale Horiz. 2019, 4, 647. [Google Scholar]
- [31].Moore DG, Barbera L, Masania K, Studart AR, Nat. Mater. 2020, 19, 212. [DOI] [PubMed] [Google Scholar]
- [32].Yee DW, Lifson ML, Edwards BW, Greer JR, Adv. Mater. 2019, 31, 1901345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Varma A, Mukasyan AS, Rogachev AS, Manukyan KV, Chem. Rev. 2016, 116, 14493. [DOI] [PubMed] [Google Scholar]
- [34].Nitta N, Wu F, Lee JT, Yushin G, Mater. Today 2015, 18, 252. [Google Scholar]
- [35].Kohlmeyer RR, Blake AJ, Hardin JO, Carmona EA, Carpena-Núñez J, Maruyama B, Berrigan JD, Huang H, Durstock MF, J. Mater. Chem. A 2016, 4, 16856. [Google Scholar]
- [36].Zhang H, Ning H, Busbee J, Shen Z, Kiggins C, Hua Y, Eaves J, Davis J, Shi T, Shao YT, Zuo JM, Sci. Adv. 2017, 3, e1602427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guo J, Zeng Y, Li P, Chen J, Ceram. Int. 2019, 45, 23007. [Google Scholar]
- [38].Shuai X, Zeng Y, Li P, Chen J, J. Mater. Chem. 2020, 55, 6771. [Google Scholar]
- [39].Komissarenko DA, Sokolov PS, Evstigneeva AD, Shmeleva IA, Dosovitsky AE, Materials 2018, 11, 2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schmidt J, Colombo P, J. Eur. Ceram. Soc. 2018, 38, 57. [Google Scholar]
- [41].Vyatskikh A, Kudo A, Delalande S, Greer JR, Mater. Today Commun. 2018, 15, 288. [Google Scholar]
- [42].Zanchetta E, Cattaldo M, Franchin G, Schwentenwein M, Homa J, Brusatin G, P. Colombo. Adv. Mater. 2016, 28, 370. [DOI] [PubMed] [Google Scholar]
- [43].Sathiya M, Prakash AS, Ramesha K, Shukla AK, Materials, 2009, 2, 857. [Google Scholar]
- [44].Li H, Zhang S, Wei X, Yang P, Jian Z, Meng J, RSC Adv. 2016, 6, 79050. [Google Scholar]
- [45].Zhuravlev V, Shikhovtseva AV, Ermakova LV, Evshchik E, Sherstobitova EA, Novikov DV, Bushkova OV, Dobrovolsky YA, Int. J. Electrochem. Sci, 2019, 14, 2965. [Google Scholar]
- [46].Aziz NAA, Abdullah TK, Mohamad AA, Ionics, 2018, 24, 403. [Google Scholar]
- [47].Reimers JN, Dahn J, J. Electrochem. Soc. 1992, 139, 2091. [Google Scholar]
- [48].Shin HC, Pyun SI, Electrochim. Acta 2001, 46, 2477. [Google Scholar]
- [49].Kalyani P, Kalaiselvi N, Muniyandi N, J. Power Sources, 2002, 111, 232. [Google Scholar]
- [50].Rodrigues S, Munichandraiah N, Shukla AK, J. Power Sources, 2001, 102, 322. [Google Scholar]
- [51].Santiago EI, Andrade AVC, Paiva-Santos CO, Bulhoes LOS, Solid State Ionics, 2003, 158, 91. [Google Scholar]
- [52].Roberts M, Johns P, Owen J, Brandell D, Edstrom K, El Enany G, Guery C, Golodnitsky D, Lacey M, Lecoeur C, Mazor H, Peled E, Perre E, Shaijumon MM, Simon P, Taberna P-L, J. Mater. Chem. 2011, 21, 9876. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.