

SUMMARY
Metabolic fuels regulate insulin secretion by generating second messengers that drive insulin granule exocytosis, but the biochemical pathways involved are incompletely understood. Here we demonstrate that stimulation of rat insulinoma cells or primary rat islets with glucose or glutamine + 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (Gln + BCH) induces reductive, “counter-clockwise” tricarboxylic acid (TCA) cycle flux of glutamine to citrate. Molecular or pharmacologic suppression of isocitrate dehydrogenase-2 (IDH2), which catalyzes reductive carboxylation of 2-ketoglutarate to isocitrate, results in impairment of glucose- and Gln + BCH-stimulated reductive TCA cycle flux, lowering of NADPH levels, and inhibition of insulin secretion. Pharmacologic suppression of IDH2 also inhibits insulin secretion in living mice. Reductive TCA cycle flux has been proposed as a mechanism for generation of biomass in cancer cells. Here we demonstrate that reductive TCA cycle flux also produces stimulus-secretion coupling factors that regulate insulin secretion, including in non-dividing cells.
Graphical Abstract
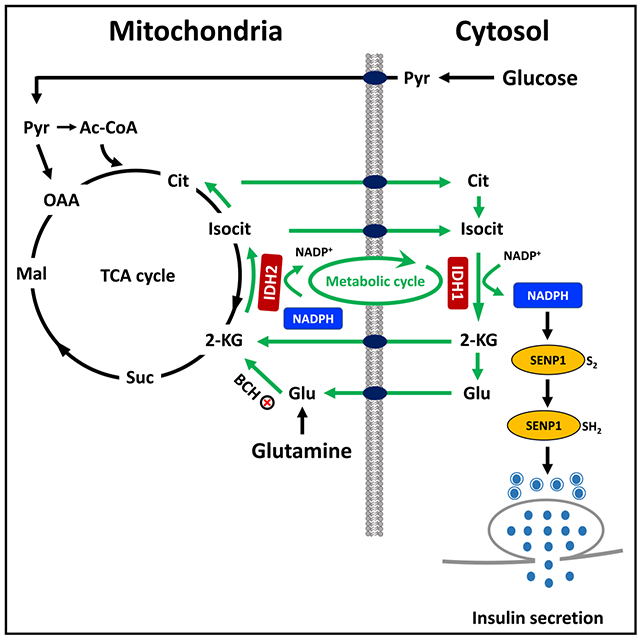
In Brief
Zhang et al. report that reductive “counter-clockwise” TCA cycle flux via isocitrate dehydrogenase-2 (IDH2) is critical for glucose- and glutamine-stimulated insulin secretion. This pathway serves as a source of citrate and isocitrate to drive cytosolic NADPH production by IDH1, resulting in activation of insulin granule exocytosis.
INTRODUCTION
Regulation of insulin secretion in response to circulating metabolites, particularly glucose, is central to control of mammalian fuel homeostasis. A core biochemical mechanism controlling glucose-stimulated insulin secretion (GSIS) involves metabolism of glucose through glycolysis and the tricarboxylic acid (TCA) cycle to yield ATP, which suppresses ATP-sensitive potassium (KATP) channels, leading to activation of voltage-gated Ca2+ channels and Ca2+ influx to trigger release of insulin-containing secretory granules (Ashcroft et al., 1984; Cook and Hales, 1984; Newgard and Matschinsky, 2001). This now decades-old mechanism is thought to be critical for the relatively brief “triggering” phase of insulin secretion, whereas other metabolic coupling factors appear to play important roles in activating the second or “amplifying” phase of the response, comprising approximately 60%–70% of insulin secreted in response to a carbohydrate-containing meal (Henquin, 2000). Indeed, significant GSIS responses are observed even when KATP channel expression or activity is suppressed by pharmacologic or molecular tools (Gembal et al., 1993; Komatsu et al., 2001; Sato et al., 1992).
Our laboratory and others have implicated anaplerotic metabolism of glucose and other fuels as a source of coupling factors for GSIS (Alves et al., 2015; Farfari et al., 2000; Lu et al., 2002). In particular, we have outlined a pathway in which anaplerotic entry of pyruvate into the TCA cycle via pyruvate carboxylase provides substrate for mitochondrial citrate and isocitrate synthesis, allowing export of these metabolites from the mitochondria to the cytosol through the mitochondrial citrate/isocitrate carrier (Joseph et al., 2006). Cytosolic isocitrate then engages with the NADP-dependent, cytosolic isoform of isocitrate dehydrogenase (ICDc or IDH1, hereafter IDH1) to generate 2-ketoglutarate (2-KG) and NADPH. The importance of this reaction is illustrated by the finding that small interfering RNA (siRNA)-mediated suppression of IDH1 expression impairs GSIS (Ronnebaum et al., 2006). A more recent study links cytosolic NADPH produced by the IDH1 reaction to glutaredoxin-mediated activation of sentrin/SUMO-specific protease-1 (SENP1), a de-SUMOylase enzyme that activates insulin granule exocytosis (Ferdaoussi et al., 2015). Consistent with these findings, direct infusion of isocitrate or NADPH into the interior of patch-clamped human β cells, obtained either from non-diabetic or type 2 diabetic donors, activates exocytosis, whereas knockout of SENP1 in mouse β cells abrogates the exocytotic response to glucose or NADPH (Ferdaoussi et al., 2015).
Glutamine (Gln) is another potent insulin secretagogue in rodent islets and cell lines, but its ability to stimulate secretion is dependent upon activation of glutamate dehydrogenase (GDH) activity with leucine or its analog 2-aminobicyclo[2,2,1]-heptane-2-carboxylic acid (BCH). We demonstrated previously that siRNA-mediated suppression of IDH1 expression impairs stimulation of insulin secretion by Gln + BCH (Odegaard et al., 2010). We interpreted these findings to mean that glutamate generated from Gln through the glutaminase reaction is converted to 2-KG by GDH, which is metabolized in the TCA cycle in the oxidative or “clockwise” direction to generate citrate and isocitrate. These metabolites would then leave the mitochondria to engage with IDH1 to generate cytosolic NADPH as a secretion coupling factor, seemingly explaining the effect of IDH1 knockdown to impair Gln + BCH-induced secretion. However, recent work in the cancer metabolism field has brought attention to the potential importance of an alternate reductive or “counter-clockwise” TCA cycle pathway for Gln and glutamate metabolism, involving NADPH-dependent reductive carboxylation of 2-KG by mitochondrial isocitrate dehydrogenase-2 (IDH2) to yield isocitrate, citrate, and acetyl-CoA, which serve as substrates to generate lipids and other biomass in rapidly dividing cancer cells (Holleran et al., 1995; Jiang et al., 2016; Metallo et al., 2011; Mullen et al., 2011; Wise et al., 2011).
To further investigate the metabolic pathway(s) that link Gln to insulin secretion, we herein have traced the metabolic fate of 13C-labeled Gln in islet β cells during stimulation with glucose or Gln + BCH using gas chromatography-mass spectrometry (GC-MS)-based analysis of mass isotopomers of citrate and other metabolic products. Surprisingly, we find that stimulatory glucose or Gln + BCH activate reductive metabolism of three different forms of 13C-labeled Gln to citrate in rat insulinoma (INS-1-derived 832/13) cells and primary rat islets. Moreover, labeling of citrate by 13C-labeled Gln, as well as glucose- and Gln + BCH-stimulated insulin secretion, NADPH production, and protein de-SUMOylation, are impaired in response to suppression of mitochondrial IDH2 activity. Finally, acute inhibition of IDH2 in living mice suppresses the normal glucose-induced increment in insulin secretion. These studies demonstrate that in addition to its role in cancer cell growth, reductive metabolism of Gln and 2-KG in the TCA cycle has an important cell signaling function in β cells, including in non-dividing primary islet cells.
RESULTS
Significant Metabolism of [13C5]Gln by the Reductive (Counter-Clockwise) TCA Cycle Pathway in 832/13 Cells and Primary Rat Islets
Gln-stimulated insulin secretion requires co-incubation with the GDH activator BCH, a leucine analog (Gylfe, 1976; Odegaard et al., 2010). Prior work from our laboratory demonstrated that siRNA-mediated knockdown of the cytosolic, NADP-dependent isoform of IDH1 caused a strong impairment of insulin secretion elicited by Gln + BCH (Odegaard et al., 2010). We interpreted these findings to mean that glutamate is metabolized in the TCA cycle in the oxidative or clockwise direction to generate citrate and isocitrate. Citrate and isocitrate can then be transported out of the mitochondria to engage with IDH1 to generate cytosolic NADPH as a secretion coupling factor (Figure 1A). However, an alternate model is also possible, in which 2-KG produced from glutamate in the GDH reaction is used for reductive/counter-clockwise metabolism in the TCA cycle to generate citrate (Figure 1B). Here we sought to distinguish between these possibilities by tracing the metabolic fate of 13C-labeled Gln in islet β cells.
Figure 1. Metabolism of [13C5]Gln by the reductive (counter-clockwise) TCA cycle pathway in 832/13 cells and primary rat islets.

832/13 cells or primary rat islets were incubated for 2 h in secretion buffer in the presence of 5 mM [13C5]Gln ± BCH prior to insulin secretion and GC-MS analyses.
(A and B) Conceptual scheme of the metabolic fate of [13C5]Gln metabolized via oxidative TCA cycle (A; clockwise) or reductive TCA cycle (B; counter-clockwise) pathways. Only the first cycle is shown for clarity.
(C and D) Insulin secretion from 832/13 cells (C) and rat islets (D) in response to Gln + BCH or stimulatory glucose (12 mM for 832/13 cells, 16.7 mM for primary islets).
(E and G) Simulated citrate isotopologue labeling from [13C5]Gln in 832/13 cells (E) or rat islets (G), based on measured labeling of pyruvate and malate (Figure S1), and assuming oxidative (clockwise) metabolism only.
(F and H) Measured citrate isotopologue labeling in 832/13 cells (F) or rat islets (H), treated with 5 mM [13C5]Gln ± BCH.
Data represent three independent aliquots of 832/13 cells and two independent islet aliquots, assayed in triplicate (cells) or duplicate (islets). ** denotes that the M5 isotopologue of citrate (blue bar) is the only one that fulfills the dual criteria of being significantly increased in the observed versus simulated data comparison, as well as being significantly increased in the presence relative to the absence of BCH, in both 832/13 cells and rat islets, with p < 0.01 for both comparisons in each cellular setting.
We performed these studies in the robustly glucose-responsive rat insulinoma INS 832/13 cell line (Hohmeier et al., 2000) and in primary rat islets. Treatment of 832/13 cells (Figure 1C) or rat islets (Figure 1D) with 5 mM Gln + BCH caused an increase in insulin secretion similar to that induced by a maximally stimulatory concentration of glucose. We investigated Gln metabolism by incubating 832/13 cells or rat islets with 5 mM [13C5]Gln ± BCH for 2 h, followed by measurement of 13C enrichment of the several possible mass isotopomers of citrate by GC-MS. We first simulated label enrichment in citrate based on direct measurement of labeling of malate and pyruvate (Figures S1A and S1B), with the assumption that TCA cycle flux is entirely oxidative (clockwise), resulting in the predicted citrate mass isotopomer distributions shown in Figure 1E (832/13 cells) and Figure 1G (primary islets). Details of the calculations used for this simulation are shown in Figure S1C. If one assumes that metabolism of Gln in the TCA cycle is exclusively oxidative, the simulation predicts that the major labeled isotopomers of citrate will be the M4 and M6 species in both the cell line and in primary cells. In contrast, if glutamate engages in significant reductive (counter-clockwise) TCA cycle metabolism, involving conversion of 2-KG to isocitrate via the mitochondrial NADPH-dependent isoform of isocitrate dehydrogenase (IDH2), then the M5 isotopomer of citrate should be a major labeled product. In fact, when label enrichment was directly measured by GC-MS in 832/13 cells incubated with [13C5]Gln, BCH treatment increased labeling of several citrate isotopologues. Surprisingly, the BCH-mediated increase of M5 citrate was greater than for any other isotopologue, relative either to the level predicted from the simulation (Figure 1E) or to its abundance in the absence of BCH (Figure 1F). No other citrate isotopologue fulfilled these dual criteria. This suggests that reductive TCA cycle metabolism of Gln makes a major contribution to the citrate pool during stimulation of insulin secretion with Gln + BCH in 832/13 cells. In cultured rat islets, [13C5]Gln metabolism to M4, M5, and M6 citrate was active even in the absence of BCH, suggesting a higher basal rate of flux through both the reductive and oxidative pathways under unstimulated conditions in islets compared to the β cell line (Figure 1H). Incubation of primary islet cells with [13C5]Gln + BCH clearly and significantly increased the level of M5 citrate, relative either to the level predicted from the simulation (Figure 1G) or to that measured in the absence of BCH (Figure 1H). M5 citrate was again the most abundant isotopologue, and the only one fulfilling these dual criteria. These data demonstrate operation of both the oxidative and reductive TCA cycle pathways in primary islet cells during stimulation of insulin secretion with Gln + BCH, but also a clear and specific induction of the reductive pathway.
[1-13C]Gln flux Analyses Confirm Active Reductive TCA Cycle Activity in 832/13 Cells and Primary Rat Islets
In order to further investigate the reductive pathway in β cells, we employed [1-13C]Gln in our flux analyses. In the clockwise/oxidative pathway, the [1-13C] label of Gln will be lost as 13CO2 in the reaction catalyzed by 2-KG dehydrogenase. Therefore, metabolites downstream of 2-KG in the oxidative TCA cycle pathway, including citrate, will not be labeled with 13C (Figure 2A). In contrast, if [1-13C]Gln engages in reductive TCA cycle metabolism, citrate will be labeled in carbon 1 to form the M1 isotopomer (Figure 2B). In 832/13 cells, incubation with [1-13C] Gln + BCH caused a strong stimulation of insulin secretion relative to treatment with [1-13C]Gln alone (Figure 2E). This was accompanied by a striking increase in M1 citrate in cells treated with [1-13C] Gln + BCH (Figure 2F). Labeling of M1 glutamate was decreased slightly, but significantly, in the presence of BCH (Figure 2G), indicating that conversion of [1-13C]Gln to M1 glutamate by glutaminase was largely unaffected by BCH. Similarly, Gln + BCH caused a robust stimulation of insulin secretion in primary rat islets (Figure 2Q). In primary islets, significant labeling of M1 citrate from [1-13C]Gln occurred in the absence of BCH, but this labeling was doubled when BCH was added (Figure 2R), again under conditions where labeling of M1 glutamate was slightly reduced by BCH (Figure 2S). These findings confirm active reductive TCA cycle flux of Gln to citrate, which is correlated with insulin secretion in both 832/13 cells and primary rat islets.
Figure 2. Confirmation of reductive pathway activity in 832/13 cells and islets using [1-13C] and [5-13C]Gln.

832/13 cells or primary rat islets were incubated for 2 h in secretion buffer containing [1-13C]Gln or [5-13C]Gln prior to insulin secretion and GC-MS analyses.
(A and B) Conceptual scheme of the metabolic fate of [1-13C]Gln metabolized via oxidative TCA cycle (A; clockwise) or reductive TCA cycle (B; counter-clockwise) pathways.
(C and D) Conceptual scheme of the metabolic fate of [5-13C]Gln metabolized via oxidative TCA cycle (C; clockwise) or reductive TCA cycle (D; counter-clockwise) pathways.
(E–V) 832/13 cells or primary rat islets were stimulated with 5 mM [1-13C]Gln ± BCH (E–G, 832/13; Q–S, islets) or 5 mM [5-13C]Gln ± BCH (H–J, 832/13 cells) for 2 h prior to tracer analyses. Insulin secretion (E, H, and Q), labeling of M1 citrate (F, I, and R), and labeling of M1 glutamate (G, J, and S). 832/13 cells or primary rat islets were stimulated with basal and stimulatory glucose for 2 h in the presence of 2 mM [1-13C]Gln (K–M, 832/13 cells; T–V, islets) or 2 mM [5-13C]Gln (N–P, 832/13 cells), and subjected to GC-MS-mediated mass isotopomer analyses. Insulin secretion (K, N, and T), labeling of M1 citrate (L, O, and U), and labeling of M1 glutamate (M, P, and V).
Data represent three independent cell or islet aliquots each assayed in duplicate (islets) ortriplicate (832/13). “p < 0.01, significant differences between indicated groups. Related supplemental data shown in Figure S2.
Glucose Stimulates Reductive Metabolism of Gln in 832/13 Cells and Primary Rat Islets
Glucose is the predominant physiological regulator of insulin secretion. We therefore investigated if glucose influences the reductive metabolism of Gln to citrate. In 832/13 cells, treatment with 12 mM glucose in the presence of 2 mM [1-13C]Gln caused a coordinate increase in insulin secretion and flux of [1-13C]Gln to M1 citrate relative to cells treated with 2.5 mM glucose + 2 mM [1-13C]Gln (Figures 2K and 2L), while labeling of glutamate was decreased by approximately 20%, probably due to dilution by unlabeled glucose-derived metabolites (Figure 2M). Similarly, treatment of primary rat islets with 16.7 mM glucose caused a robust stimulation of insulin secretion, and a significant increase in labeling of M1 citrate from [1-13C]Gln compared to islets incubated at 2.5 mM glucose (Figures 2T and 2U), again associated with a trend for decrease in M1 glutamate labeling (Figure 2V). As expected, metabolites downstream of 2-KG in the oxidative pathway, including succinate and malate, had no detectable 13C labeling. These data clearly demonstrate that glucose stimulation activates conversion of Gln to citrate via reductive TCA cycle metabolism in 832/13 cells and primary islet cells.
We also performed studies with [5-13C]Gln, which can form M+1 citrate by both the reductive and oxidative pathways (Figures 2C and 2D). M+1 citrate labeling from [5-13C]Gln is shown side-by-side with labeling from [1-13C]Gln in studies performed in 832/13 cells (Figures 2F versus 2I; Figures 2L versus 2O). We find that M+1 citrate labeling is much more pronounced under basal conditions (Gln in the absence of BCH or 2.5 mM glucose) when [5-13C]Gln is used as the substrate than when [1-13C]Gln is used, presumably reflecting the unique ability of [5-13C]Gln to label citrate by oxidative metabolism. In contrast, the increment of increase in M+1 citrate labeling in response to stimulatory conditions is much larger when [1-13C]Gln is used for labeling compared to [5-13C]Gln (22.4-fold versus 2.5-fold with [1-13C]Gln compared to [5-13C]Gln in response to stimulation with Gln + BCH, and 2.3-fold versus 40% with [1-13C]Gln compared to [5-13C]Gln in response to stimulation with 12 mM glucose). These data are consistent with simultaneous flux of Gln through oxidative and reductive pathways, but also demonstrate that reductive flux is preferentially activated under conditions where insulin secretion is stimulated, findings well aligned with the [13C5]Gln studies summarized in Figure 1.
Calculation of Relative Oxidative and Reductive TCA Cycle Fluxes during Glucose- or Gln-Stimulated Insulin Secretion
It is well understood from studies by our group (Joseph et al., 2006; Ronnebaum et al., 2006) and others (Farfari et al., 2000; Alves et al., 2015) that exposure of pancreatic islet β cells to increases in extracellular glucose levels causes an increase in oxidative TCA cycle metabolism. This does not preclude simultaneous metabolism of fuels in the oxidative and reductive directions, nor does it exclude the possibility that the relative fluxes through the two pathways can change according to the nutritional milieu. To investigate this directly, we calculated relative fluxes through the two pathways in 832/13 cells or primary rat islets exposed to [1-13C]Gln at low and high glucose, or Gln ± BCH. As shown in Figures 3A-3D, oxidative flux of [1-13C]Gln constitutes ≥ 85% of its metabolism in the TCA cycle under basal conditions (either low glucose or Gln absent BCH), whereas relative flux through the reductive pathway is ≤ 10% under these conditions. While oxidative flux of [1-13C]Gln clearly increases in response to stimulatory glucose or Gln + BCH, its relative contribution to total TCA cycle flux actually declines to an average of 73% during fuel-stimulated insulin secretion, whereas reductive flux increases to constitute an average of 27% of total flux. Schematic figure panels are provided in Figures 3E and 3F to help illustrate this point. We note that similar changes in relative fluxes are observed in response to fuel stimulation in the 832/13 cell line (Figures 3A and 3B) and in primary rat islets (Figures 3C and 3D). We also note that our calculations of relative fluxes of [1-13C]Gln support and are concordant with the findings and conclusions advanced in studies with [U-13C]Gln (Figure 1), or when comparing fluxes of [1-13C]Gln and [5-13C]Gln (Figure 2).
Figure 3. Fuel stimulation of β cells increases reductive relative to oxidative TCA cycle flux.

(A–D) Relative oxidative (black bars) and reductive (red bars) TCA cycle flux in 832/13 cells (A and B) or primary rat islets (C and D), exposed to [1-13C]Gln at low or stimulatory glucose levels (A and C) or [1-13C]Gln ± BCH (B and D).
(E and F) Conceptual rendering of changes in oxidative and reductive fluxes under basal and stimulatory conditions based on all [1-13C ]Gln experiments conducted with 832/13 cells and rat islets. Reductive flux was calculated as fractional 13C labeling of M1 citrate divided by fractional 13C labeling of M1 glutamate × 100, and oxidative flux was calculated as 100-reductive flux, assuming that all unlabeled glutamate is used in the oxidative pathway.
Data represent mean ± SEM; **p < 0.01.
Reductive TCA Cycle Flux in Islets Is Not an Artifact of Culture in Nutrient-Rich Media
Recent work demonstrates that cellular metabolism can be strongly influenced by the nutrient composition of tissue culture media, particularly those containing high concentrations of Gln and cystine (Muir et al., 2017). To test the possibility that operation of the reductive pathway could be a consequence of islets being cultured in a rich artificial media, we repeated tracer experiments with freshly isolated islets that were not exposed to media. Freshly isolated islets showed similar insulin secretion responses to Gln + BCH (Figure S2A) or high glucose (Figure S2D) as observed in cultured islets. Exposure of these freshly isolated islets to [1-13C]Gln + BCH or stimulatory glucose + 2 mM [1-13C]Gln resulted in clear enhancement of [1-13C]Gln incorporation into M1 citrate (Figures S2B and S2E), associated with a decrease in M1 glutamate labeling (Figures S2C and S2F), very similar to what we observed in cultured islets (Figure 2). These data demonstrate that active reductive TCA cycle flux in primary rat islets is not an artifact of exposure to nutrient-rich tissue culture media.
siRNA-mediated Suppression of IDH2 in 832/13 Cells Suppresses Reductive Metabolism of Gln to Citrate and Attenuates Glucose- and Gln + BCH-Stimulated Insulin Secretion
Reductive metabolism of Gln to citrate requires carboxylation of 2-KG to isocitrate. In addition to the cytosolic, NADP-dependent form of isocitrate dehydrogenase, IDH1, mammalian cells express two mitochondrial IDH isoforms. IDH3 is thought to be the classical NAD-linked “clockwise” TCA cycle enzyme that catalyzes the oxidative decarboxylation of isocitrate to 2-KG. IDH2 is the isoform thought to be responsible for “counter-clockwise” NADPH-dependent reductive carboxylation of 2-KG to isocitrate in mitochondria (Figure 4A) (Mullen et al., 2011). To investigate the possible role of IDH2 in reductive flux of Gln to citrate in β cells, we tested two siRNA duplexes specific for IDH2 by transfection into 832/13 cells, and achieved an approximate 90% suppression of IDH2 mRNA levels with both duplexes relative to levels in mock-transfected cells (No) or cells treated with a control siRNA duplex with no known gene target (siCont) (Figure 4B). A corresponding ~50% reduction of IDH2 protein levels was observed in cells treated with the IDH2 siRNAs (Figure 4C). Treatment of 832/13 cells with either of the IDH2-specific duplexes caused a significant decrease in insulin secretion in response to Gln + BCH (Figure 4D) or stimulatory glucose (12 mM) (Figure 4E), relative to mock-transfected cells or cells transfected with the siCont duplex. We then treated 832/13 cells with the IDFI2-specific duplexes or the siCont duplex, or performed mock transfection, and exposed the cells to either [1-13C]Gln or [13C5]Gln ± BCH. Consistent with findings reported in Figure 2, Gln + BCH stimulation caused a large increase in labeling of M1 citrate in mock-transfected cells and cells treated with the siCont duplex, whereas labeling of M1 citrate was decreased by approximately 50% in cells treated with the IDFI2-specific siRNAs (Figure 4F). Labeling of glutamate from [1-13C]Gln during Gln + BCH stimulation was unchanged among the cell groups (Figure 4G). Glucose stimulation of reductive metabolism of [1-13C]Gln to citrate was also attenuated by an average of 42% by IDFI2 knockdown (Figure 4H), while glutamate labeling was unaffected (Figure 4I). Confirming these findings, cells exposed to [13C5]Gln + BCH exhibited a specific and significant decrease in labeling of M5 citrate, the isotopomer generated from reductive pathway flux, in response to IDFI2 knockdown, with no effect on labeling of the M4 or M6 citrate species, the products of oxidative TCA cycle metabolism (Figure S3A). Again, labeling of glutamate and other metabolites from [13C5]Gln was unaffected by transfection of the siCont or siIDH2 duplexes (Figures S3B-S3H), and metabolites expected to be generated in the oxidative pathway remained unlabeled when [1-13C]Gln was administered (data not shown). Importantly, IDH2 knockdown had no effect on labeling of TCA cycle intermediates from [13C6]glucose (Figure S4), showing that suppression of IDH2 has a specific effect on reductive metabolism of Gln rather than a general effect on glycolysis (pyruvate labeling is not affected) or mitochondrial glucose metabolism. Taken together, the experiments summarized in Figures 4, S3, and S4 demonstrate that labeling of citrate by 13C-labeled Gln occurs at least in part via an IDH2-catalyzed reductive TCA cycle pathway, and that operation of this pathway is necessary for the full insulin secretion response to Gln + BCH or stimulatory glucose.
Figure 4. siRNA-mediated suppression of IDH2 attenuates reductive metabolism of Gln and impairs glucose- and Gln + BCH-stimulated insulin secretion.

832/13 cells were mock transfected (No) or transfected with either a control siRNA (siCont) or one of two siRNA duplexes targeting IDH2 (siIDH2A and siIDH2B) 72 h prior to analyses.
(A) Proposed role of IDH2 in reductive metabolism of Gln to isocitrate and citrate.
(B and C) qPCR (B) and immunoblot (C) analyses of IDH2 mRNA and protein levels, respectively.
(D and E) siIDH2-mediated effects on Gln + BCH- (D) and glucose- (E) stimulated insulin secretion. Secretion data are expressed as fold change relative to basal secretion from mock-transfected cells (No) set to 1.0.
(F and G) M1 citrate (F) and M1 glutamate (G) labeling in response to 5 mM [1-13C]Gln ± BCH treatment (2 h) in siIDH2-treated compared to siCont-treated or mock-transfected cells.
(H and I) M1 citrate (H) and M1 glutamate (I) labeling after 2 h treatment with low and high glucose in the presence of 2 mM [1-13C]Gln.
Data represent at least three independent cell or islet aliquots, each assayed in triplicate. Significant differences between indicated groups, *p < 0.05 and **p < 0.01.
The IDH2 inhibitor AGI6780 suppresses insulin secretion and reductive flux in 832/13 cells and rat islets
The siRNA duplexes used for knockdown of IDH2 in 832/13 cells achieved an approximate 50% suppression of IDH2 protein expression (Figure 4C). Anticipating a lesser efficiency of siRNA-mediated knockdown of IDH2 in primary rat islets, we sought an alternative tool for suppressing IDH2 activity in primary cells. We chose a small molecule, AGI6780, that inhibits both mutated and wild-type IDH2, with little effect on IDH1 activity (Urban et al., 2017; Wang et al., 2013) (Figure 5A). Treatment of either 832/13 cells (Figures 5B-5G) or primary rat islets (Figures 5H-5M) with 10 μM AGI6780 caused strong suppression of Gln + BCH- and glucose-stimulated insulin secretion in both cellular settings (Figures 5B, 5E, 5H, and 5K). In parallel with the suppression of insulin secretion, reductive flux of [1-13C]Gln to M1 citrate was sharply attenuated by AGI6780 treatment, both in 832/13 cells (Figures 5C and 5F) and primary rat islets (Figures 5I and 5L), with little effect on glutamate labeling (Figures 5D, 5G, 5J, and 5M). AGI6780 caused a small increase (7%) in labeling of all carbons of citrate (Figures S5A-S5D), and either had no impact or caused a similar small increase in labeling of other glycolytic or TCA cycle intermediates from [13C6]glucose in 832/13 cells (Figures S5E-S5L), demonstrating that the effects of AGI6780 on insulin secretion were not due to a general effect to perturb glucose metabolism. Combined, data in Figures 4 and 5 and related supplemental figures demonstrate a central and specific role for IDH2-mediated reductive flux in regulation of insulin secretion, both in the 832/13 cell line and in primary rat islets.
Figure 5. The IDH2 inhibitor AGI6780 suppresses insulin secretion and reductive flux in 832/13 cells and primary rat islets.

(A) Site of action of the IDH2 inhibitor AGI6780 to inhibit reductive metabolism of Gln. 832/13 cells were treated with AGI6780 for a total of 3.5 h.
(B–D) Effect of AGI6780 on Gln + BCH-stimulated insulin secretion (B), labeling of M1 citrate by [1-13C]Gln (C), and labeling of M1 glutamate by [1-13C]Gln (D) in 832/13 cells.
(E–G) Effect of AGI6780 on glucose-stimulated insulin secretion (E), labeling of M1 citrate by [1-13C]Gln during glucose stimulation (F), and labeling of M1 glutamate by [1-13C]Gln during glucose stimulation (G) in 832/13 cells.
(H–J) Islets were treated with the IDH2 inhibitor AGI6780 for a total of 3 h prior to measurement of reductive pathway flux and insulin secretion. Effect of AGI6780 on Gln + BCH-stimulated insulin secretion (H), labeling of M1 citrate by [1-13C]Gln (I), and labeling of M1 glutamate by [1-13C]Gln (J) in rat islets.
(K–M) Effect of AGI6780 on glucose-stimulated insulin secretion (K), labeling of M1 citrate by [1-13C]Gln during glucose stimulation (L), and labeling of M1 glutamate by [1-13C]Gln during glucose stimulation (M) in rat islets.
Significant differences between indicated groups, **p < 0.01.
Cytosolic IDH (IDH1) plays a minor role in reductive flux in β cells
In theory, NADPH-dependent reductive carboxylation of 2-KG to isocitrate could also be catalyzed by cytosolic IDH1, contributing to labeling of citrate from [1-13C] and [13C5]Gln, as illustrated schematically in Figures S6A and S6H. To test this possibility, we measured labeling of M1 citrate from [1-13C]Gln in the presence and absence of siRNA-mediated suppression of cytosolic IDH1. Treatment of 832/13 cells with an siRNA duplex specific for IDH1 caused an ~80% suppression in IDH1 mRNA levels. Consistent with our prior reports (Odegaard et al., 2010; Ronnebaum et al., 2006), suppression of IDH1 expression caused ~60% and ~55% inhibition of Gln + BCH- and glucose-stimulated insulin secretion, respectively (Figures S6B and S6E). Treatment with the IDH1-specific siRNA (siIDH1) caused a small (17%) but significant decrease in labeling of M1 citrate from [1-13C]Gln in response to stimulation with Gln + BCH (Figure S6C), and a nonsignificant trend to decrease M1 citrate labeling during stimulation with 12 mM glucose (Figure S6F) relative to siCont-treated or mock-transfected cells. IDH1 knockdown also caused a small but significant decrease in labeling of M1 glutamate during stimulation with either Gln + BCH (Figure S6D) or 12 mM glucose (Figure S6G). IDH1 knockdown had a very small effect to decrease labeling of M5 citrate (7%) and M5 glutamate (2%) relative to siCont-treated cells from [13C5]Gln in the presence of BCH (Figures S6I and S6J). All of these effects of IDH1 knockdown were minor compared to the effects observed with knockdown or pharmacologic inhibition of IDH2. These experiments therefore demonstrate that reductive metabolism of Gln to citrate is primarily mediated by IDH2 rather than IDH1.
Inhibition of IDH2 lowers cellular NADPH levels and attenuates de novo lipogenesis from [13C6]glucose
Results presented so far are consistent with the operation of a mitochondria to cytosol “shuttle” for generation of cytosolic NADPH via the combined actions of IDH2 and IDH1. Consistent with this, we have previously shown that knockdown of IDH1 lowers cellular NADPH levels and the NADPH:NADP ratio in glucose-stimulated 832/13 cells (Ronnebaum et al., 2006). To determine if IDH2 inhibition has a similar effect, we treated 832/13 cells with the IDH2 inhibitor AGI6780 or vehicle during treatment with basal (2.5 mM) or stimulatory (12 mM) glucose. Vehicle-treated 832/13 cells exhibited a large increase in NADPH, a decrease in NADP, and a strong increase in NADPH: NADP ratio as levels of glucose are raised from 2.5 to 12 mM (Figures 6A-6C). Treatment with AGI6780 caused a sharp decrease in NADPH levels and NADPH:NADP ratio compared to vehicle treatment (DMSO), both at 2.5 and 12 mM glucose, and eliminated the glucose-mediated decline in NADP levels (Figures 6A-6C). These data demonstrate a clear link between IDH2-mediated reductive TCA cycle flux and NADPH production in a robustly glucose-responsive β cell line.
Figure 6. Effect of IDH2 inhibition on NADPH production, de novo lipogenesis, and protein SUMOylation.

(A–C) Effect of the IDH2 inhibitor AGI6780 on NADPH (A), NADP (B), and NADPH:NADP ratio (C) at basal and stimulatory glucose in 832/13 cells.
(D) Simplified labeling patterns obtained during de novo synthesis of palmitate from [13C6]glucose.
(E) Palmitate isotopologues measured after 24 h of incubation of 832/13 cells with 12 mM [13C6]glucose ± AGI6780.
(F) Secretory granule-enriched subcellular fractions were prepared from 832/13 cells following 90 min of preincubation with 10 μM AGI6780 or vehicle, followed by 2 h of stimulation with Gln + BCH in the presence (+) or absence (−) of AGI6780. Samples were resolved on an SDS gel and probed with an antibody for SUMO1.
Significant differences between indicated groups, *p < 0.05 and **p < 0.01.
Another pathway that might be influenced by the proposed mitochondria/cytosol NADPH shuttle is de novo lipogenesis from glucose. To test this idea, we treated 832/13 cells with 12 mM [13C6]glucose for 24 h in the presence and absence of AGI6780. Generation of labeled acetyl-CoA for new lipid synthesis from glucose requires mitochondrial metabolism, and is expected to generate mass isotopologues of palmitate ranging from M2 to M16 (Figure 6D) that can be detected by GC-MS. Treatment of cells with AGI6780 caused a significant decrease in incorporation of 13C into all medium palmitate isotopologues (M2, 6, 8, 10) relative to vehicle-treated cells (Figure 6E). Thus, the decline in NADPH levels caused by AGI6780 treatment is accompanied by a decrease in de novo lipogenesis, as might be expected due to the obligate role of NADPH as a cofactor in the cytosolic fatty acid synthase reaction. We note that AGI6780 did not decrease labeling of citrate from [13C6]glucose, and in fact caused a small increase in labeling (Figure S5C). Thus, the AGI6780-induced decrease in total palmitate labeling by [13C6]glucose is likely due to suppressed activity of the metabolic cycle catalyzed by cytosolic IDH1 and mitochondrial IDH2.
The IDH2 inhibitor AGI6780 affects protein SUMOylation in 832/13 cells
Prior studies provide evidence for a model in which stimulatory glucose increases cytosolic NADPH to trigger glutaredoxin-mediated activation of SENP1, resulting in removal of SUMO peptides from β cell proteins and activation of insulin exocytosis (Dai et al., 2011; Ferdaoussi et al., 2015). These studies used patch-clamped β cells from SENP1 knockout mice or normal human β cells to show that glucose, glutathione, or NADPH enhances insulin granule exocytosis in a SENP1-dependent manner. Here we sought to determine if inhibition of reductive TCA cycle metabolism also affects protein SUMOylation. We pretreated 832/13 cells with AGI6780 or vehicle for 90 min, and then treated with Gln + BCH in the presence and absence of AGI6780 for an additional 120 min, followed by preparation of a secretory granule-enriched subcellular fraction that was probed with an antibody specific for the SUMO1 peptide. In two separate experiments in which Gln + BCH-treated cells were studied in the presence and absence of AGI6780, the inhibitor increased SUMOylation of two proteins in the 25–30 kD range (Figure 6F). Taken together, the data in Figure 6 demonstrate that IDH2-mediated reductive TCA cycle flux regulates β cell NADPH levels and protein SUMOylation during nutrient stimulation.
The IDH2 inhibitor AGI6780 suppresses GSIS in living mice
To determine if IDH2-catalyzed reductive TCA cycle flux also plays a role in the in vivo setting, we tested the impact of the selective IDH2 inhibitor AGI6780 on insulin secretion during an intraperitoneal (i.p.) glucose tolerance test in healthy young C57 BL6 mice. We first tested the pharmacokinetic properties of the drug by i.p. injection of 60 mg/kg body weight, followed by measurement of its levels in plasma by liquid chromatography-tandem mass spectrometry (LC-MS/MS) at multiple time points over the ensuing 90 min (Figure 7A). Drug levels climbed to a maximum level of about 15 μM over the first 30 min after injection, and then remained at that level from 30 to 90 min post-injection (Figure 7B). Thus, the circulating levels of AGI6780 achieved in vivo (15 μM) were very similar to the levels shown to suppress reductive TCA cycle flux and glucose- and Gln + BCH-stimulated insulin secretion in our in vitro studies with 832/13 cells and primary rat islets (10 μM). With this understanding of the pharmacodynamics of AGI6780 in hand, we designed our glucose tolerance test such that the drug was administered 30 min before the injection of 1.5 g glucose/kg body weight. Blood samples were taken immediately before the injection of AGI6780 at time −30 min, just before i.p. glucose injection at time 0, and then at various time points after the glucose bolus for measurement of plasma glucose, insulin, and glucagon levels (Figure 7A). Limitations in the amount of blood that could be safely and ethically collected from these mice restricted measurements of glucagon and insulin to a subset of time points.
Figure 7. Acute inhibition of IDH2 with AGI6780 in living mice impairs insulin and glucagon secretion.

(A) Schematic summary of in vivo experiment. Mice were injected with AGI6780 or DMSO (vehicle) as a control at time 30 min before (−30) injection of an i.p. glucose bolus.
(B–E) Blood samples were taken at the times indicated for measurement of circulating AGI6780 levels (B), blood glucose (C), circulating insulin (D; expressed as percent change relative to levels at −30), and glucagon (E; expressed as percent change relative to time 0). Data represent the mean ± SEM for ≥ 14 mice per group. *p < 0.02, significant increases in insulin secretion in the DMSO control group, but not the AGI6780-treated group, in response to the glucose bolus, and a significant decrease in glucagon secretion in the AGI6780-treated group, but not the DMSO control group.
(F and G) Batches of 25 rat islets were pretreated with 10 μM AGI6780 for 1 h followed by glucose stimulation in the presence of AGI6780 for 1 h. Media samples were collected and used for glucagon (F) and insulin (G) assays. **p < 0.01, significant differences between indicated groups.
Mice injected with AGI6780 had no significant insulin secretion response to the glucose bolus, whereas insulin rose significantly at 10 and 30 min after glucose bolus in control mice injected with vehicle (DMSO) (p < 0.02) (Figure 7D). Despite the absence of significant GSIS in the AGI6780-treated group, blood glucose levels were identical to those in the vehicle-treated control group (Figure 7C). This may be explained by the interesting finding that AGI6780 caused a significant decrease in circulating glucagon at 10 min post-injection (p < 0.02), whereas injection of DMSO had no effect on glucagon levels (Figure 7E). The AGI6780-induced lowering of glucagon may have nullified its hyperglycemic effect, thereby offsetting the presumed decrease in insulin-mediated glucose clearance engendered by AGI6780-induced suppression of GSIS. The interesting finding of an effect of AGI6780 on glucagon secretion in vivo led us to question if the drug would also affect glucagon secretion in isolated islets in vitro. Accordingly, we treated 3 independent batches of rat islets with AGI6780 or vehicle, at basal (2.5 mM) and stimulatory (16.7 mM) glucose. AGI6780 had no effect on glucagon secretion at low glucose, but decreased glucagon secretion by about 40% (p = 0.01) at 16.7 mM glucose (Figure 7F). AGI6780 also caused a dramatic decrease in insulin secretion at stimulatory glucose in this set of experiments (Figure 7G), consistent with data presented in Figure 5. Overall, these findings are consistent with an important role of IDH2 and reductive TCA cycle flux in regulation of both insulin and glucagon secretion.
DISCUSSION
Pancreatic islet β cells respond to changes in glucose concentration by increasing their rate of glucose metabolism, leading to increases in ATP:ADP ratio that drive the classical KATP channel-dependent pathway of insulin secretion. However, glucose metabolism also produces KATP channel-independent signals for insulin secretion, thought to be particularly important for the sustained “amplifying” or second phase of the insulin secretion response. Growing evidence suggests that these ancillary signals are produced by integration of mitochondrial and cytosolic pathways of glucose metabolism. Our work and that of others have consistently pointed to an important role of anaplerotic TCA cycle substrates for generation of metabolic intermediates that deliver signals to the cytosolic compartment for regulation of insulin granule exocytosis (Alves et al., 2015; Farfari et al., 2000; Jensen et al., 2008; Lu et al., 2002),
In the current study, we have investigated metabolic fates of Gln in islet β cells to gain a better mechanistic understanding for our prior observation that suppression of IDH1 expression impairs Gln + BCH-stimulated insulin secretion (Odegaard et al., 2010). We discovered that following conversion to glutamate and 2-KG, Gln is actively metabolized to citrate by reductive, “counter-clockwise” TCA cycle metabolism. Moreover, this pathway plays an essential role in regulation of insulin secretion, as siRNA-mediated or pharmacological suppression of mitochondrial, NADPH-linked IDH2 impairs insulin release in response to Gln + BCH or stimulatory glucose, in both 832/13 rat insulinoma cells and primary rat islets. We propose that the reductive pathway is part of a metabolic cycle that salvages products of the IDH1 reaction to help maintain high citrate and isocitrate levels, which are then available for IDH1-mediated NADPH production to support sustained insulin secretion. Supporting this model, our prior work shows that siRNA-mediated knockdown of the citrate/isocitrate carrier (CIC; SLC25A1), which transports citrate and isocitrate across the mitochondrial membrane, decreases the cytosolic pool of citrate and impairs GSIS (Joseph et al., 2006). This demonstrates that mitochondrial citrate and isocitrate production is key to the replenishment of these metabolites in the cytosol for IDH1-mediated signaling. The current study expands upon these findings by revealing that reductive TCA cycle flux contributes to mitochondrial citrate and isocitrate synthesis in islet endocrine cells, thereby supporting fuel-stimulated insulin secretion. Operation of this pathway may compensate for limitations in β cell glucose oxidation capacity in the postprandial state, possibly analogous to activation of reductive TCA cycle metabolism in tumor cells in response to hypoxia. Notably, glucose elicits a rapid rise in cytosolic Ca2+ in islets within a few minutes of stimulation, and Ca2+ is known to activate dehydrogenase enzymes, including IDH1 and IDH2 (Xu et al., 2004).
We demonstrate that the reductive pathway is active in the INS-1-derived rat insulinoma cell line 832/13 and in primary rat islets, and that its activity regulates insulin secretion in response to Gln + BCH and stimulatory glucose in both cellular settings. However, differences between the cell line and primary rat islet cells are noted. Most prominently, significant labeling of citrate from [13C5]Gln and [1-13C]Gln is evident in primary islets under basal conditions, defined either as treatment with Gln alone in the absence of BCH or with low levels of glucose. Several explanations for the higher rate of reductive metabolism of Gln under basal conditions in primary islets can be proposed. First, primary islets may have higher basal activity of GDH, or alternatively may more actively convert glutamate to 2-KG via transamination. Second, primary islets represent a mixture of cell types, including insulin-secreting β cells, but also glucagon-producing α cells and somatostatin-secreting δ cells. Gln and glutamate metabolism may be more active in the non-β cell types present in primary islet preparations under basal conditions. Interestingly, Gln has a specific effect to stimulate α cell replication relative to other amino acids (Dean et al., 2017), possibly reflecting a heightened proclivity of α cells for Gln metabolism and signaling.
The concept of reductive TCA cycle flux recently came to the fore based on stable isotope labeling studies in cancer cells exposed to hypoxia or with genetic deficiencies in mitochondrial function (Holleran et al., 1995; Metallo et al., 2011; Mullen et al., 2011; Wise et al., 2011). These studies demonstrated reductive flux of 13C-labeled Gln to citrate and acetyl-CoA, presumably via IDH-catalyzed reductive carboxylation of 2-KG to isocitrate and citrate, followed by citrate transport out of mitochondria to engage with cytosolic ATP-citrate lyase to form acetyl-CoA and oxaloacetate. This pathway for Gln metabolism was suggested to provide substrates for expansion of biomass during cancer cell proliferation, including acetyl-CoA for lipogenesis. A later paper challenged this idea with data suggesting that both reductive and oxidative flux through IDH-catalyzed reactions are active in cancer cells, but with net flux favoring the oxidative direction (Fan et al., 2013).
Using pancreatic islet cells, we demonstrate labeling of isotopomers of citrate (M4, M6) that would be expected as products of active oxidative catabolism of [13C5]Gln. However, if one assumes that all Gln metabolism occurs via the oxidative pathway, computer simulation of the citrate isotopomer distribution in cells treated with [13C5]Gln predicts that M4 and M6 citrate should be the dominant isotopomers, with very little M5 citrate (Figures 1E and 1G). When citrate isotopomers are actually measured by GC-MS, M4 and M6 citrate are labeled, but to a lesser extent than M5 citrate (Figures 1F and 1H). Moreover, in response to Gln + BCH treatment, labeling of M5 citrate is strongly induced in 832/13 cells and primary islets relative to the other isotopomers. A second set of studies with [1-13C]Gln and [5-13C] Gln align firmly with the [13C5]Gln data. Thus, both stimulatory glucose and Gln + BCH invoke a strong increase in labeling of M1 citrate from [1-13C]Gln, which can only label citrate via the reductive pathway, in both 832/13 cells and primary rat islets. Moreover, the increment of induction of M1 citrate labeling by stimulatory glucose or Gln + BCH is much larger when 1-[13C] Gln is used than when [5-13C]Gln is used; the latter labels M1 citrate by both the oxidative and reductive pathways. Finally, calculation of relative rates of flux of [1-13C]Gln through the oxidative and reductive pathways reveals that secretagogue stimulation (either high glucose or Gln + BCH) selectively activates flux through the reductive pathway while reducing relative oxidative flux (Figure 3), consistent with the proposed role of reductive flux in regulation of insulin secretion.
Prior studies in cancer cells have suggested that one explanation for the apparent simultaneous operation of oxidative and reductive pathways could be their partitioning in the mitochondrial and cytosolic compartments, respectively (Fan et al., 2013). Indeed, in studies involving suppression of the mitochondrial citrate/isocitrate carrier, a pathway for labeling of citrate from 13C-labeled Gln involving transport of labeled 2-KG to the cytosol and its reductive carboxylation by IDH1 to yield citrate was demonstrated, including experiments demonstrating blockade of this flux by suppression of IDH1 expression (Jiang et al., 2017). However, in the current study in β cells, molecular suppression of IDH2 expression or pharmacologic inhibition of its activity strongly attenuated labeling of citrate from 13C-labeled Gln (Figures 4 and 5), whereas knockdown of IDH1 expression had only a very modest impact (Figure S6). Taken together, these findings argue for net reductive flux of Gln to citrate via mitochondrial NADPH-dependent IDH2 in islet cells. The data also support a model in which this pathway regulates Gln + BCH- and glucose-stimulated insulin secretion by maintaining a constant supply of citrate and isocitrate for NADPH generation via cytosolic, NADP-dependent IDH1.
Also supporting this model, we find that suppression of IDH2 with AGI6780 causes a sharp decrease in NADPH and NADPH:- NADP ratio, and a seemingly related decline in de novo lipogenesis (Figure 6). NADPH can be derived from multiple sources, but our data are most consistent with a key role for the proposed mitochondrial/cytosolic IDH2/IDH1 NADPH shuttle, based on the following considerations: (1) The main IDH isoform used for classical oxidative TCA cycle flux is the NAD-linked IDH3. Thus, oxidative TCA flux will not be a major consumer of NADP to create a futile cycle. (2) Other mitochondrial enzymes could be producing NADPH, including nicotinamide nucleotide transhydrogenase (NNT) and malic enzyme 3 (ME3). We have previously demonstrated that siRNA-mediated suppression of ME3 expression in rat islets or its absence in mouse islets does not impair GSIS (Ronnebaum et al., 2008). (3) The C57/BL6 “J” and “N” mouse substrains differ in their expression of NNT, with the former strain lacking NNT due to a gene deletion. J strain mice have modestly reduced but still significant insulin secretion in response to a glucose challenge compared to N strain mice (Attané et al., 2016). (4) Finally, we show that AGI6780 significantly decreases de novo lipogenesis from [13C6]glucose, which requires the mitochondrial/cytosolic citrate shuttle and cytosolic NADPH as a cofactor for fatty acid synthase, thus supporting a key role of IDH2 in these pathways. In sum, while more than one enzyme can contribute to an increase in mitochondrial NADPH in response to stimulation of islets with glucose or Gln + BCH, IDH2-mediated reductive flux is unique in that it generates products (citrate and isocitrate) that serve as the oxidative substrates for NADPH generation in the cytosol via IDH1. The importance of this flux is emphasized by our finding that stimulation of insulin secretion by Gln + BCH is strongly impaired by knockdown of either mitochondrial IDH2 (the current study) or cytosolic IDH1 (Odegaard et al., 2010).
In our prior work, glucose-induced increases in NADPH and NADPH:NADP ratio were shown to be linked to insulin granule exocytosis via the protein deSUMOylase SENP1 (Ferdaoussi et al., 2015). This included studies demonstrating that mice with β cell-specific knockout of SENP1 have impaired GSIS, as well as experiments showing that addition of isocitrate or NADPH to patch-clamped human β cells stimulates insulin granule exocytosis to a similar extent as stimulatory glucose. The effects of glucose or isocitrate to stimulate exocytosis were blocked by siRNA-mediated knockdown of IDH1, but the stimulatory effect of NADPH was retained and subsequently shown to be linked to activation of SENP1 via reduction of glutathione and glutaredoxin (Ferdaoussi et al., 2015). Here we demonstrate that inhibition of IDH2 increases SUMO1 modification of two protein bands in a secretory granule-enriched subcellular fraction from Gln + BCH-treated 832/13 cells. Thus, the current data and our prior work are consistent with a model in which IDH2-catalyzed reductive TCA flux is required for maintenance of a high NADPH:NADP ratio during secretagogue stimulation, which is in turn linked to secretagogue-driven secretory protein de-SUMOylation and insulin secretion.
Importantly, the connection between reductive TCA cycle flux and regulation of insulin secretion elucidated in our in vitro studies of a robustly glucose responsive β cell line (INS-1-derived 832/13 cells) and primary rat islets also seems to apply to islets in the in vivo setting. Thus, administration of the small-molecule IDH2 inhibitor AGI6780 to normal mice 30 min prior to a glucose challenge abrogated the normal GSIS response. Interestingly, this suppression of insulin secretion was not associated with an increase in circulating glucose, probably because glucagon secretion was also suppressed by the drug. The latter observation may suggest that reductive TCA cycle flux not only regulates insulin secretion from β cells, but may also play a role in regulation of glucagon secretion from α cells. Supporting this idea, AGI6780 also inhibits glucagon secretion from rat islets studied in vitro (Figure 7F). As mentioned earlier, primary rat islets have more active reductive TCA flux under basal conditions than observed in the 832/13 rat insulinoma cell line. One possible explanation for this difference is that α cells within primary islet preparation may drive the increase in basal reductive flux. If so, this might suggest that active reductive flux in α cells under basal conditions helps maintain glucagon secretion at a relatively high rate appropriate for the fasted state, whereas suppression of this flux could contribute to attenuated glucagon secretion. This idea will require further investigation.
Our findings may have implications for understanding of insulin secretory defects that contribute to development of type 2 diabetes (T2D). T2D is often associated with obesity and insulin resistance, and in early stages of T2D development, the islets attempt to compensate for blunted insulin action by increasing insulin production. Eventually, the ability to produce insulin at high rates is lost, and the islets also lose their normal ability to respond to changes in blood glucose across the physiological range. Here we have implicated IDH2-catalyzed reductive metabolism of Gln as an essential metabolic pathway for Gln + BCH- and glucose-stimulated insulin secretion, suggesting that components of this flux may become defective in dysfunctional islets in T2D. Consistent with this idea, we recently demonstrated that patch-clamped β cells from human donors with T2D completely lack an exocytotic response to infusion of glucose to the cell interior via the patch pipette, but these same cells have a robust exocytotic response to isocitrate or NADPH, which bypass the IDH2-catalyzed reaction to serve as direct stimuli for insulin granule exocytosis (Ferdaoussi et al., 2015). In addition, global knockout of IDH2 in mice results in decreased insulin secretion in both the fasted and fed states (Lee et al., 2016). Finally, IDH2 mRNA and protein expression are decreased in two separate mouse models of T2D, the βV59M mouse, which exhibits impaired insulin secretion due to its expression of a constitutively open KATP channel (Haythorne et al., 2019), and the MKR mouse, which expresses a dominant-negative IGF-1 receptor in skeletal muscle that causes early-life insulin resistance leading to β cell dysfunction (Lu et al., 2010). Future studies will be needed to explore the potential role of altered IDH2-catalyzed reductive flux in the development of β cell dysfunction in diabetes.
Limitations of Study
Although all data pertaining to measurement of reductive TCA cycle flux obtained in the 832/13 insulinoma cell line were confirmed in primary rat islets, operation of the pathways described herein remain to be tested in human islets, studies that are feasible but challenging in light of the functional heterogeneity of human islet aliquots obtained from different donors. Also, although our studies with the IDH2 inhibitor AGI6780 provide clear evidence for inhibition of GSIS in living mice, a finding entirely consistent with all of our in vitro data, these results should be corroborated in the future in mice with inducible, β cell-specific IDH2 knockout. We plan studies of this nature as the next chapter of this work. Finally, deeper exploration of the potential role of dysregulated IDH2-catalyzed reductive TCA cycle in development of β cell failure in T2D remains to be explored in genetic and dietary animal models, as well as in human islets from T2D and non-diabetic subjects.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christopher B. Newgard (chris.newgard@duke.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique dataset or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary Rat Islets
Rat islets were harvested from chow fed, dual-housed male Wistar rats (Envigo), weighing ~300 g, under a protocol approved by the Duke University Institutional Animal Care and Use Committee (Ronnebaum et al., 2008). Islets were cultured for 1-3 days prior to experiments in RMPI-1640 media containing 8 mM glucose, supplemented with 10% fetal bovine serum, 20 units/mL penicillin, 20 μg/mL streptomycin, and 0.05 μg/mL amphotericin B. A subset of studies was performed in freshly isolated rat islets, which were harvested in Hanks media, washed in PBS, and used for metabolic flux and insulin secretion studies as described below, with no exposure to media.
Mouse Studies
Experiments were performed in 9- to 13-week-old male mice of the C57BL 6J background backcrossed in house for at least 10 generations. Mice were group housed under a 12 h light/12 h dark cycle and provided free access to a normal chow diet.
Cell Line
The rat insulinoma cell line, INS-832/13 was maintained as described (Hohmeier et al., 2000). Briefly, the cells were cultured in RPMI-1640 with 11.1 mM glucose supplemented with 10% fetal bovine serum, 10 mM HEPES, 2 mM L-glutamine, 1 mM sodium pyruvate, and 50 μM β-mecaptoethanol and passaged every 5 days using 0.05% trypsin-EDTA.
METHOD DETAILS
Insulin and Glucagon Secretion, Flux Studies
Rat islet insulin and glucagon secretion assays were performed on groups of ~15 or 25 rat islets, respectively. For [13C5]Gln or [13C6] glucose tracer experiments, ~250 rat islets were utilized per sample whereas ~125 islets were used for [1-13C]glutamine experiments. 832/13 cells were cultured to confluency in 12 well plates (~2-million cells per well). Prior to experiments, cells and islets were washed in PBS and incubated in secretion buffer (114 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.16 mM MgSO4, 20 mM HEPES, 2.5 mM CaCl2, 0.2% BSA, pH 7.2) containing 2.5 mM glucose. After 1 h (islets) or 1.5 h (cells) pre-incubation, buffer was replaced with secretion buffer containing [13C6]glucose, [5-13C]Gln, [1-13C]Gln (all Cambridge Isotope Laboratories) or [13C5]Gln (MiliporeSigma), as indicated, and cultured for 2 h. For experiments involving glucagon measurements, the incubation time was 1 h. The GDH activator 2-Aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH) was applied at 6 mM (832/13 cells) and 10 mM (islets) during secretion when indicated. A 30 mM stock solution of the IDH2 inhibitor AGI6780 (Calbiochem or MedChemExpress) was prepared in DMSO and added to cells or islets at a final concentration of 10 μM in 0.2% DMSO during the pre-incubation and secretion phases. Following the stated incubation periods, supernatants were collected for insulin (STELLUX Chemi Rodent Insulin ELISA; ALPCO) or glucagon (Lumit Glucagon Immunoassay Kit; Promega) measurements, and when stable isotope-labeled fuels were included in an experiment, cells and islets were subjected to mass spectrometry analyses as described below.
Sample Preparation for GC-MS Analysis
Following incubation with tracers, multi-well plates of 832/13 cells, or islets in microcentrifuge tubes, were immersed in liquid N2 and transferred to wet ice, prior to addition of 250 μL ice-cold MeOH per sample. 0.1 nmol norvaline was added to cells or islets in methanol as an internal standard, followed by sonication of the suspension for 1 min and collection of supernatants. After an additional methanol (500 μl) extraction, the two methanol fractions were combined and extracts were centrifuged at 20,000 x g for 30 min. The supernatant was transferred and dried completely with nitrogen gas. The dried residues were resuspended in 25 μL of methoxylamine hydrochloride (2% (w/v) in pyridine) and incubated at 40°C for 1.5 h in a heating block. After brief centrifugation, 35 μL of MTBSTFA + 1% TBDMS was added, and the samples were incubated at 60°C for 30 min. The derivatized samples were centrifuged for 5 min at 20,000 x g, and the supernatants were transferred to GC vials for GC-MS analysis. A modified GC-MS method was employed (Wang et al., 2018). The injection volume was 1 μL, and samples were injected in splitless mode. GC oven temperature was held at 80°C for two minutes, increased to 280°C at 7°C/min, and held at 280°C for a total run time of forty minutes. GC-MS analysis was performed on an Agilent 7890B GC system equipped with a HP-5MS capillary column (30 m, 0.25 mm i.d., 0.25 μm-phase thickness; Agilent J&W Scientific, Santa Clara, CA), connected to an Agilent 5977A Mass Spectrometer operating under ionization by electron impact (EI) at 70 eV. Helium flow was maintained at 1 mL/min. The source temperature was maintained at 230°C, the MS quad temperature at 150°C, the interface temperature at 280°C, and the inlet temperature at 250°C. Mass spectra were recorded in mass scan mode with m/z from 50 to 700.
13C-based Stable Isotope Analysis
M0, M1, …, Mn refer to the isotopologues containing n heavy atoms in a molecule. The stable isotope distribution of individual metabolites was measured by GC-MS as described above. A list of metabolites, their isotopomers, and retention times is provided in Table S1. The isotopologue enrichment or labeling in this work refers to the corrected isotope distribution (Fernandez et al., 1996; Tomcik et al., 2011). Briefly, the labeling of all measured metabolites in cells/islets treated with 13C-labeled substrates was corrected based on the natural isotope distribution assayed from cell samples treated with unlabeled carbon sources. Three replicates of 832/13 cells treated with all unlabeled substrates were processed and profiled by GC-MS. The natural isotope distribution of each measured metabolite was assayed and averaged to build a matrix for correcting isotope labeling of metabolite in the tracer-treated samples. Details of these methods have been published previously (Fernandez et al., 1996; Tomcik et al., 2011).
Calculation of Relative Oxidative and Reductive Fluxes
To calculate relative oxidative and reductive fluxes under basal and stimulatory conditions, we compiled all data from four groups of experiments included in this study: 1) 832/13 cells treated with [1-13C]Gln in the presence or absence of BCH; 2) rat islets treated with [1-13C]Gln in the presence or absence of BCH; 3) 832/13 cells treated with [1-13C]Gln + 2.5 mM or 12 mM glucose; 4) rat islets treated with [1-13C]Gln + 2.5 or 16.7 mM glucose. For each of these experimental groups, relative reductive flux was calculated as % M1 citrate labeling/% M1 glutamate labeling × 100. Relative oxidative flux was then derived by subtracting the relative reductive flux from 100, with the assumption that all unlabeled citrate is derived from oxidative flux, as expected when [1-13C]Gln is the only tracer.
RNAi-mediated Gene Silencing of IDH1 and IDH2
832/13 cells seeded at 30% confluence were transfected with siRNA duplexes specific for IDH1 or IDH2 for 24 h at a final concentration of 20 nM using Dharmafect Transfection Reagent 1 (Dharmacon). After an additional 48 h in culture, cells were used for insulin secretion, mass spectrometry, and qRT-PCR analyses. The IDH1 targeting sequence was GTA TGA TGG ACG CTT CAA AGA (Ronnebaum et al., 2006) and the IDH2 targeting sequences were CCA TTA TGA AGG CCT ATG A (siIDH2 A) and GAA CTA TGA CGG AGA TGT A (siIDH2 B) corresponding to siRNA id#s s167607 and s166708, respectively from Thermo Fisher Scientific. Mock transfected cells (No duplex) as well as cells transfected with a scrambled duplex, GAG ACC CTA TCC GTG ATT, with no known RNA target (siCont) were used as controls (Jensen et al., 2006; Ronnebaum et al., 2006, 2008). For qRT-PCR analyses of gene expression, RNA was purified from 832/13 cells using the RNAeasy Mini kit (QIAGEN) and reverse transcribed with the iScript cDNA synthesis kit (Bio-Rad). qRT-PCR analyses were performed on QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific) using iTaq or SYBR (Bio-Rad). The primers used to measure IDH1 transcript levels were AAA ATA TCC CCC GGC TAG TGA and CGT GTC GGC CAA TGA TGA. For IDH2 expression, Taqman Assay ID Rn01478119_m1 (Thermo Fisher Scientific) was applied. Changes in IDH1 and IDH2 mRNA levels were normalized to Cyclophilin B mRNA, amplified with primers CGG ACA GCC GGG ACA C and TTC GAT CTT GCC ACA GTC TAC AA.
NADPH and NADP Analyses
Confluent 832/13 cells in 6 well plates were treated with 10 μM AGI6790 for a total of 3.5 h during pre-incubation and secretion, and levels of nicotinamide adenine dinucleotide phosphates were measured using an enzymatic cycling method as earlier described (Jensen et al., 2006). In short, following GSIS, cells were scraped, centrifuged for 10 s at 10,000 g, 4°C, and cell pellets quick frozen in liquid N2. Prior to assay, samples were thawed on ice, resuspended in 40 mM NaOH/5 mM cysteine, sonicated for 10 s, and re-centrifuged for 1 min at 10,000 g at 4°C. 0.3 M HCl (NADPH is destroyed at low pH) or 40 mM NaOH/5 mM cysteine (NADP is destroyed at high pH) were added to fractions of the cleared extract for NADP and NADPH determinations, respectively, followed by incubation at 60°C for 15 min. NADPH standards (0.25– 4 μM in 40 mM NaOH/5 mM cysteine) were treated in parallel. Subsequently, cycling reagent (50 mM imidazole, 50 mM imidazole-HCl, 5 mM glucose 6-phosphate, 7.5 mM 2-KG, 0.1 mM ADP, 25 mM ammonium acetate, 0.1% BSA, 1.5 units/mL of Leuconostoc glucose-6- phosphate dehydrogenase (MiliporeSigma), 1.5 units/mL of beef liver glutamine dehydrogenase (MiliporeSigma) was added, and after 3 h of incubation at 37°C, samples and standards were boiled for 3 min and centrifuged at 10,000 g for 10 min. The amount of 6-phosphogluconolactone was determined by adding indicator reaction buffer (50 mM imidazole, 50 mM imidazole-HCl, 30 mM ammonium acetate, 2 mM MgCl2, 0.1 mM EDTA, 300 μM NADP, 0.5 units/mL of Torula yeast gluconate dehydrogenase (MiliporeSigma) to the cleared cycling reactions, and absorbance at 340 nm was read when the reaction was completed.
Measurement of de novo Lipid Synthesis
832/13 cells at 90% confluency were cultured for 24 h in complete media containing 11 mM [13C6]glucose in the presence of 10 μM AGI6780 or DMSO. The cells were subsequently washed in PBS, quick frozen in liquid N2 and transferred to wet ice prior to addition of 250 μL ice-cold MeOH per sample. The cells were scraped off the plate and transferred to an Eppendorf vial followed by 2-additional transfers with 800 μl MeOH each time. The combined MeOH extracts were briefly centrifuged and dried completely under nitrogen gas. The dried residue was dissolved in 25% KOH ethanol solution with brief sonication. The total lipid was hydrolyzed into free fatty acids at 85°C for 3 h. The hydrolyzed fatty acid solution (200 μl) was acidified with 300 μl 6 M HCl. Fatty acids were extracted by adding 600 μl chloroform. After centrifugation at 800 × g for 10 min, the chloroform phase (400 μl) was transferred to a new Eppendorf via and was completely dried under nitrogen gas. The dried residue was derivatized with 50 μl MTBSTFA + 1% TBDMS at 60°C for 30 min. Samples were transferred to GC-MS vials and were ready for GC-MS analysis after brief centrifugation at 800 × g for 5 min. A modified GC-MS method was employed (Wang et al., 2018). The injection volume was 1 μL, and samples were injected in split mode with a split ratio of 5:1. GC oven temperature was held at 80°C for one minute, increased to 280°C at 14°C/min, and held at 280°C for 9.5 min with a total run time of twenty five minutes. GC-MS analysis was performed on an Agilent 7890B GC system equipped with a HP-5MS capillary column (30 m, 0.25 mm i.d., 0.25 μm-phase thickness; Agilent J&W Scientific, Santa Clara, CA), connected to an Agilent 5977A Mass Spectrometer operating under ionization by electron impact (EI) at 70 eV. Helium flow was maintained at 1 mL/min. The source temperature was maintained at 230°C, the MS quad temperature at 150°C, the interface temperature at 280°C, and the inlet temperature at 250°C. Mass spectra were recorded in mass scan mode with m/z from 50 to 700. The retention time of palmitic acid derivative was at 14.2 min with detected m/z 313, 314, 315, 316, 317, 318, 319, 320, 321, 322, 323, 324, 325, 326, 327, 328, and 329 for M0, M1, M2, M3, M4, M5, M6, M7, M8, M9, M10, M11, M12, M13, M14, M15, and M16 palmitate.
Secretory Granule-Enriched Subcellular Fractions
832/13 cells were cultured to a confluent state in 6-well tissue culture dishes. Cells were preincubated with 10 μM AGI6780 or DMSO vehicle for 1.5 h in secretion buffer, followed by treatment for 2 h in the presence of Gln + BCH, in the continued presence of AGI6780 or DMSO. Cells from 3 wells were scraped and combined in one microcentrifuge tube and washed in 1 mL ice-cold MSH buffer (220 mM mannitol, 70 mM sucrose, 5 mM potassium-HEPES, pH 7.5, supplemented with HALT protease and phosphatase-inhibitor cocktail (Thermo Fisher Scientific)). After centrifugation and resuspension in 400 μL ice-cold MSH buffer, the cells were homogenized by passing the solution through a 25 gauge needle 15 times. The homogenate was incubated for 10 min on ice, and the cell mixture was centrifuged for 5 min at 5,000 x g at 4°C. The supernatant was transferred to a new tube and saved. The cell pellet was re-homogenized in 400 μL ice-cold MSH buffer, this time through a 30 gauge needle 15 times and centrifuged for 5 min, 5,000 x g at 4°C to remove cell debris, nuclei and most of the mitochondria. The corresponding supernatant was combined with the initial supernatant and centrifuged for 20 min at 21,000 x g, 4°C. The pellet containing insulin granules was resuspended in SUMO buffer containing 100 mM NaCl, 40 mM KCl, 1 mM EDTA, 20 mM HEPES pH 7.4, 10% glycerol, 1% Triton X-100, 25 mM N-Ethylmaleimide supplemented with HALT protease and phosphatase-inhibitor cocktail (Thermo Fisher Scientific) and used for immunoblot analysis as described below.
Immunoblot Analyses
832/13 cells transfected with siRNA duplexes or mock-transfected were cultured as described above. Extracts were prepared using lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% IGEPAL630) supplemented with HALT protease and phosphatase-inhibitor cocktail (Thermo Fisher Scientific). For experiments with secretory granule-enriched fractions, extracts were prepared in SUMO buffer as described above. 20 μg protein/sample was resolved on 15 well Mini-Protean TGX 4%–16%, Stain-Free precast gels (Bio-Rad) and transferred to LF-PVDF membranes using the Trans-blot turbo system (Bio-Rad). Membranes were blocked in 5% Milk and probed with IDH2 Polyclonal antibody (ABclonal; A7190), or an anti-SUMO-1 antibody (Santa Cruz Biotechnologies; SC-5308). Antibodies specific for γ tubulin (MiliporeSigma; T5326) or carboxypeptidase-E (Proteintech; 13710-1-AP) were used as loading controls for the IDH2 knockdown and SUMO1 experiments, respectively. Bands were visualized using Li-Cor Odyssey CLX and densitometry was performed with Image Studio Lite (LI-COR Biosciences).
In Vivo Studies with IDH2 Inhibitor AGI6780
To test the impact of the selective IDH2 inhibitor AGI6780 on insulin secretion in living animals we first injected 60 mg drug/kg body weight into healthy young C57 BL6 mice, followed by measurement of its levels in plasma by LC-MS/MS at multiple time points over the ensuing 90 min. Briefly, a 2 μl plasma sample was spiked with 2 μl of 0.01 mM [2H3]natifine as internal standard followed by precipitation of proteins by addition of 25 μl methanol. Quantification of AGI6780 was based on an external calibration curve with a constant amount of [2H3]natifine as internal standard using control plasma as the biological matrix (Figure S7). The chromatographic separation was performed on an Alltima HP C18-HL column (100 mm × 2.1 mm, 3 μm, Thermo Fisher Scientific, USA) and run with a gradient elution of 98% H2O/2% acetonitrile containing 0.1% formic acid (mobile phase A) and 98% acetonitrile / 2% H2O containing 0.1% formic acid (mobile phase B). The gradient started with 10% B for 0.1 min and increased to 90% B within 6.9 min. Mobile phase B was further increased to 95% within 0.2 min and maintained at 95% B for 4 min. Then gradient was returned to initial conditions (10% B) within 0.4 min and equilibrated for 9.4 min before the next injection. The flow rate was 0.4 mL/min, the column oven temperature was set at 40°C, and the injection volume was 5 μl. Samples were analyzed on a Sciex ExioLC AD UHPLC and QTRAP 6500+ mass spectrometer. Ion transitions for AGI6780 and [2H3]natifine were m/z 291/117 and 482/295, respectively.
With an understanding of the pharmacodynamics of AGI6780 in hand, we designed our glucose tolerance test such that the drug was administered 30 min before the injection of 1.5 g glucose/kg body weight after an overnight fast. Blood samples were taken immediately before the injection of AGI6780 at time −30 min, just before glucose injection at time 0, and then at various times after the glucose bolus for measurement of plasma glucose, insulin (Mercodia Mouse Insulin ELISA) and glucagon (Mercodia Glucagon ELISA) levels.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are typically expressed as mean ± SE and representing the number of independent biological and technical replicates described in Figure Legends (most often, three independent experiments performed in triplicate). Statistical significance was determined using a two-tailed Student’s t test, assuming equal variances, and p < 0.05 was considered as significant. Graphpad Prism and Excel software was used for these analyses.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| IDH2 | ABclonal | Cat# A7190; RRID: AB_2767740 |
| γ-tubulin | MiliporeSigma | Cat# T5326; RRID: AB_532292 |
| SUMO-1 | Santa Cruz Biotechnologies | Cat# SC5308; RRID: AB_628300 |
| Carboxypeptidase E | Proteintech | Cat# 13710-1-AP; RRID: AB_2083913 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| [1-13C]glutamine | Cambridge Isotope Laboratories | Cat# CLM-3612 |
| [13C5]glutamine | MiliporeSigma | Cat# 605166 |
| [13C6]glucose | Cambridge Isotope Laboratories | Cat# CLM-1396 |
| [5-13C]glutamine | Cambridge Isotope Laboratories | Cat# CLM-1166 |
| 2-Aminobicyclo[2.2.1]heptane-2-carboxylic acid | MiliporeSigma | Cat# A7902 |
| [2H3]natifine | TLC Pharmaceutical Standards | Cat# N-053002 |
| AGI6780 | MedChemExpress | Cat# HY-15734 |
| AGI6780 | CalBiochem | Cat#1432660-47-3 |
| Critical Commercial Assays | ||
| STELLUX Chemi Rodent Insulin ELISA | ALPCO | Cat# 80-INSMR-CH01 |
| Glucagon ELISA | Mercodia | Cat# 10-1281-01 |
| Mouse Insulin ELISA | Mercodia | Cat# 10-1247-01 |
| iScript cDNA Synthesis kit | Bio-Rad | Cat# 1708890 |
| Dharmafect Transfection Reagent 1 | Thermo Scientific | Cat# T-2001 |
| Lumit Glucagon Immunoassay Kit | Promega | Cat# CS3037A02 |
| Experimental Models: Cell Lines | ||
| INS-1 832/13 | MiliporeSigma | Cat# SCC207 |
| Experimental Models: Organisms/Strains | ||
| Wistar rats | Envigo | Cat# 110-M |
| C57BL 6J | In house colony at Duke University | N/A |
| Oligonucleotides | ||
| siRNA anti rIDH2 A | Thermo Fisher Scientific | s167607 |
| siRNA anti rIDH2 B | Thermo Fisher Scientific | s167608 |
| siRNA scramble (siCont) GAG ACC CTA TCC GTG ATT | Integrated DNA Technologies | N/A |
| siRNA anti rIDH1 target sequence GTA TGA TGG ACG CTT CAA AGA | Integrated DNA Technologies | N/A |
| Taqman Probe rIDH2 | Applied Biosystems | Rn01478119_m1 |
| SYBR green primer set rIDH1; F: AAA ATA TCC CCC GGC TAG TGA, R: CGT GTC GGC CAA TGA TGA | Integrated DNA Technologies | N/A |
| SYBR green primer set rPPIB; F: CGG ACA GCC GGG ACA C, R: TTC GAT CTT GCC ACA GTC TAC AA | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| Stable isotope enrichment calculation | Fernandez et al., 1996; Tomcik et al., 2011 | N/A |
| GraphPad Prism8 software | GraphPad | N/A |
| MassHunter | Agilent | https://www.agilent.com/en/product/software-informatics/mass-spectrometry-software |
| Analyst 1.6.3 | Sciex | https://sciex.com/products/software/analyst-software |
Highlights.
Glutamine is metabolized via reductive TCA cycle metabolism in pancreatic islet cells
Reductive TCA cycle flux via IDH2 is required for glucose-stimulated insulin secretion
Reductive flux is used for biomass in cancer, and for signaling in endocrine cells
Inhibition of IDH2 impairs GSIS in living mice
ACKNOWLEDGMENTS
These studies were supported by NIH, United States grants R01 DK42583 (to C.B.N.) and R01 DK123075 and an American Diabetes Association, United States faculty development award, 1-18-JDF-075 (to J.E.C.). K.E. and S.M.G. received NIH, United States fellowship support from grants T32 DK007012 and F32 DK121420, respectively. The authors are grateful to Helena Winfield for expert assistance with islet isolation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflicts of interest in conduct of this research. C.B.N. is a paid consultant for Eli Lilly, Axcella Health, Boehringer Ingelheim, and Sigilon. Whereas all of these companies have interests in diabetes therapy, they have no involvement or competing interests in the research described in this paper.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.11.020.
REFERENCES
- Alves TC, Pongratz RL, Zhao X, Yarborough O, Sereda S, Shirihai O, Cline GW, Mason G, and Kibbey RG (2015). Integrated, step-wise, mass-isotopomeric flux analysis of the TCA cycle. Cell Metab. 22, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Harrison DE, and Ashcroft SJ (1984). Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312, 446–448. [DOI] [PubMed] [Google Scholar]
- Attané C, Peyot ML, Lussier R, Zhang D, Joly E, Madiraju SR, and Prentki M (2016). Differential insulin secretion of high-fat diet-fed C57BL/6NN and C57BL/6NJ mice: implications of mixed genetic background in metabolic studies. PLoS One 11, e0159165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DL, and Hales CN (1984). Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 311, 271–273. [DOI] [PubMed] [Google Scholar]
- Dai XQ, Plummer G, Casimir M, Kang Y, Hajmrle C, Gaisano HY, Manning Fox JE, and MacDonald PE (2011). SUMOylation regulates insulin exocytosis downstream of secretory granule docking in rodents and humans. Diabetes 60, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean ED, Li M, Prasad N, Wisniewski SN, Von Deylen A, Spaeth J, Maddison L, Botros A, Sedgeman LR, Bozadjieva N, et al. (2017). Interrupted glucagon signaling reveals hepatic α cell axis and role for L-glutamine in α cell proliferation. Cell Metab. 25, 1362–1373.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Rabinowitz JD, and Shlomi T (2013). Fatty acid labeling from glutamine in hypoxia can be explained by isotope exchange without net reductive isocitrate dehydrogenase (IDH) flux. J. Biol. Chem 288, 31363–31369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfari S, Schulz V, Corkey B, and Prentki M (2000). Glucose-regulated anaplerosis and cataplerosis in pancreatic beta-cells: possible implication of a pyruvate/citrate shuttle in insulin secretion. Diabetes 49, 718–726. [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, Ilkayeva O, Smith N, Miller N, Hajmrle C, et al. (2015). Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Invest 125, 3847–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez CA, Des Rosiers C, Previs SF, David F, and Brunengraber H (1996). Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom 31, 255–262. [DOI] [PubMed] [Google Scholar]
- Gembal M, Detimary P, Gilon P, Gao ZY, and Henquin JC (1993). Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Invest 91, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylfe E (1976). Comparison of the effects of leucines, non-metabolizable leucine analogues and other insulin secretagogues on the activity of glutamate dehydrogenase. Acta Diabetol. Lat 13, 20–24. [DOI] [PubMed] [Google Scholar]
- Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, Sachse G, Silva Dos Santos M, Terron Exposito R, Davis S, et al. (2019). Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun 10, 2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC (2000).Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760. [DOI] [PubMed] [Google Scholar]
- Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, and Newgard CB (2000). Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430. [DOI] [PubMed] [Google Scholar]
- Holleran AL, Briscoe DA, Fiskum G, and Kelleher JK (1995). Glutamine metabolism in AS-30D hepatoma cells. Evidence for its conversion into lipids via reductive carboxylation. Mol. Cell. Biochem 152, 95–101. [DOI] [PubMed] [Google Scholar]
- Jensen MV, Joseph JW, Ilkayeva O, Burgess S, Lu D, Ronnebaum SM, Odegaard M, Becker TC, Sherry AD, and Newgard CB (2006). Compensatory responses to pyruvate carboxylase suppression in islet beta-cells. Preservation of glucose-stimulated insulin secretion. J. Biol. Chem 281, 22342–22351. [DOI] [PubMed] [Google Scholar]
- Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, and Newgard CB (2008). Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab 295, E1287–E1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. (2016). Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Boufersaoui A, Yang C, Ko B, Rakheja D, Guevara G, Hu Z, and DeBerardinis RJ (2017). Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Metab. Eng 43 (Pt B), 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JW, Jensen MV, Ilkayeva O, Palmieri F, Alárcon C, Rhodes CJ, and Newgard CB (2006). The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem 281, 35624–35632. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Sato Y, Aizawa T, and Hashizume K (2001). KATP channel-independent glucose action: an elusive pathway in stimulus-secretion coupling of pancreatic beta-cell. Endocr. J 48, 275–288. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Kim SH, Park KM, Lee JH, and Park JW (2016). Increased obesity resistance and insulin sensitivity in mice lacking the isocitrate dehydrogenase 2 gene. Free Radic. Biol. Med 99, 179–188. [DOI] [PubMed] [Google Scholar]
- Lu D, Mulder H, Zhao P, Burgess SC, Jensen MV, Kamzolova S, Newgard CB, and Sherry AD (2002). 13C NMR isotopomeranalysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc. Natl. Acad. Sci. USA 99, 2708–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Koshkin V, Allister EM, Gyulkhandanyan AV, and Wheeler MB (2010). Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes. Diabetes 59, 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA, and Vander Heiden MG (2017). Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 6, e27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, and DeBerardinis RJ (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, and Matschinsky FM (2001). Substrate control of insulin release. In Handbook of Physiology, Newgard CB and Matschinsky FM, eds. (Oxford University Press; ), pp. 125–152. [Google Scholar]
- Odegaard ML, Joseph JW, Jensen MV, Lu D, Ilkayeva O, Ronnebaum SM, Becker TC, and Newgard CB (2010). The mitochondrial 2-oxoglutarate carrier is part of a metabolic pathway that mediates glucose- and glutamine-stimulated insulin secretion. J. Biol. Chem 285, 16530–16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, Stevens RD, Becker TC, Sherry AD, Newgard CB, and Jensen MV (2006). A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J. Biol. Chem 281, 30593–30602. [DOI] [PubMed] [Google Scholar]
- Ronnebaum SM, Jensen MV, Hohmeier HE, Burgess SC, Zhou YP, Qian S, MacNeil D, Howard A, Thornberry N, Ilkayeva O, et al. (2008). Silencing of cytosolic or mitochondrial isoforms of malic enzyme has no effect on glucose-stimulated insulin secretion from rodent islets. J. Biol. Chem 283, 28909–28917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Aizawa T, Komatsu M, Okada N, and Yamada T (1992). Dual functional role of membrane depolarization/Ca2+ influx in rat pancreatic B-cell. Diabetes 41, 438–443. [DOI] [PubMed] [Google Scholar]
- Tomcik K, Ibarra RA, Sadhukhan S, Han Y, Tochtrop GP, and Zhang GF (2011). Isotopomer enrichment assay for very short chain fatty acids and its metabolic applications. Anal. Biochem 410, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban DJ, Martinez NJ, Davis MI, Brimacombe KR, Cheff DM, Lee TD, Henderson MJ, Titus SA, Pragani R, Rohde JM, et al. (2017). Assessing inhibitors of mutant isocitrate dehydrogenase using a suite of preclinical discovery assays. Sci. Rep 7, 12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. (2013). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626. [DOI] [PubMed] [Google Scholar]
- Wang Y, Christopher BA, Wilson KA, Muoio D, McGarrah RW, Brunengraber H, and Zhang GF (2018). Propionate-induced changes in cardiac metabolism, notably CoA trapping, are not altered by l-carnitine. Am. J. Physiol. Endocrinol. Metab 315, E622–E633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, and Thompson CB (2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 108, 19611–19616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, and Ding J (2004). Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem 279, 33946–33957. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique dataset or code.