

ABSTRACT
Simlukafusp alfa (FAP-IL2v, RO6874281/RG7461) is an immunocytokine comprising an antibody against fibroblast activation protein α (FAP) and an IL-2 variant with a retained affinity for IL-2Rβγ > IL-2 Rβγ and abolished binding to IL-2 Rα. Here, we investigated the immunostimulatory properties of FAP-IL2v and its combination with programmed cell death protein 1 (PD-1) checkpoint inhibition, CD40 agonism, T cell bispecific and antibody-dependent cellular cytotoxicity (ADCC)-mediating antibodies. The binding and immunostimulatory properties of FAP-IL2v were investigated in vitro and compared with FAP-IL2wt. Tumor targeting was investigated in tumor-bearing mice and in a rhesus monkey. The ability of FAP-IL2v to potentiate the efficacy of different immunotherapies was investigated in different xenograft and syngeneic murine tumor models. FAP-IL2v bound IL-2 Rβγ and FAP with high affinity in vitro, inducing dose-dependent proliferation of natural killer (NK) cells and CD4+/CD8+ T cells while being significantly less potent than FAP-IL2wt in activating immunosuppressive regulatory T cells (Tregs). T cells activated by FAP-IL2v were less sensitive to Fas-mediated apoptosis than those activated by FAP-IL2wt. Imaging studies demonstrated improved tumor targeting of FAP-IL2v compared to FAP-IL2wt. Furthermore, FAP-IL2v significantly enhanced the in vitro and in vivo activity of therapeutic antibodies that mediate antibody-dependent or T cell-dependent cellular cytotoxicity (TDCC) and of programmed death-ligand 1 (PD-L1) checkpoint inhibition. The triple combination of FAP-IL2v with an anti-PD-L1 antibody and an agonistic CD40 antibody was most efficacious. These data indicate that FAP-IL2v is a potent immunocytokine that potentiates the efficacy of different T- and NK-cell-based cancer immunotherapies.
KEYWORDS: FAP-il2v, rg7461, immunocytokine, interleukin-2, fibroblast activation protein
Introduction
Interleukin-2 (IL-2) is a cytokine produced primarily by activated T cells that plays a critical function in the generation, differentiation, survival, and homeostasis of immune effector cells.1,2 IL-2 signaling is mediated by binding to the IL-2 receptor (IL-2 R), which consists of up to three individual subunits, α (CD25), β (CD122), and γ (CD132).1 The low-affinity dimeric IL-2 Rβγ form is expressed on natural killer (NK) cells, monocytes, macrophages, and resting CD4+ and CD8+ T cells.1,2 The high-affinity trimeric IL-2 Rαβγ is transiently induced on activated NK cells and CD4+ and CD8+ T cells. IL-2 functions to expand T cell populations in an autocrine fashion, differentiate antigen-activated CD4+/CD8+ T cells into effector T cell subsets, and activate NK cells.2 Activation of innate and adaptive immune effector cells in this manner is the basis for using IL-2 to stimulate an anti-tumor response.3–5 However, to counteract autoimmunity, IL-2 also has immunosuppressive properties and is involved in peripheral immune tolerance mediated by CD4+ FOXP3+ regulatory T cells (Tregs), which constitutively express high levels of IL-2 Rα.2,6,7 Tregs suppress T cell activity, thereby compromising anti-tumor immunity.8 IL-2 is also essential for activation-induced cell death (AICD) of activated T cells by upregulating the expression of Fas ligand and downregulating apoptosis inhibitors.9,10
High-dose IL-2 treatment (aldesleukin) is an effective therapy for some patients with metastatic renal cell carcinoma and malignant melanoma.11–14 The therapeutic use of IL-2 as a cancer therapy is limited by its short half-life and severe cardiovascular events, as well as pulmonary, hepatic, gastrointestinal, neurological, and hematological toxicity.15 The systemic and untargeted application of IL-2 may also compromise anti-tumor immunity by activating Tregs and inducing AICD. As such, there is a renewed interest in IL-2-based therapies that are better tolerated and preferentially activate anti-tumor immune responses.16,17
Several attempts have been made to selectively deliver IL-2 to the tumor environment by fusing IL-2 to antibodies directed against common tumor-associated antigens.18–26 These immunocytokines have been shown to increase the circulating half-life of IL-220,25 with modest efficacy in early-phase clinical trials.19,24,26 However, no molecule has progressed beyond Phase 2 due to a number of limitations, including preferential affinity for IL-2 Rαβγ on peripheral immune cells and pulmonary vascular endothelium that can override tumor targeting,25 similar severe toxicity as free IL-2 therapy,22,26 and continued activation of Tregs due to the use of wild-type (wt) IL-2.18,26
Cergutuzumab amunaleukin (CEA-IL2v) is a novel immunocytokine that comprises an IL-2 variant (IL2v) moiety fused to a carcinoembryonic antigen (CEA)-targeting antibody.27 The IL2v part is engineered by structure-based mutation of key residues in the IL-2 Rα interface (F42A, Y45A, L72G) to abolish binding to IL-2 Rα while maintaining affinity for IL-2 Rβγ. These properties enable targeted delivery of immunostimulatory IL-2 to the site of the tumor without preferential activation of Tregs over immune effector cells. Abolished binding of CEA-IL2v to IL-2 Rα was confirmed in vitro.27 In vivo, CEA-IL2v treatment strongly expanded NK cell and CD8+ T cell populations in murine models of human cancer and was efficacious as single-agent, and potentiated the efficacy of antibody-dependent cellular cytotoxicity (ADCC)-competent antibodies and an anti-programmed death-ligand 1 (PD-L1) checkpoint inhibitor.27
Fibroblast activation protein α (FAP) is a dimeric Type II transmembrane glycoprotein with proteolytic activity.28,29 FAP is expressed scarcely in normal, healthy adult tissues, but is highly expressed on the surface of cancer-associated fibroblasts and pericytes in >90% of human epithelial malignancies.28–33 The high and restricted expression on reactive stroma in the tumor microenvironment makes FAP a promising target for tumor treatment. The basic design of simlukafusp alfa (FAP-IL2v) is shown in Figure 1. FAP-IL2v contains the IL2v mutein and a (G4S)3 connector fused to the high-affinity FAP human IgG1 antibody 4B9 that was generated and affinity matured by phage display. FcγR binding is abolished by the introduction of P329G LALA mutations whereas binding to the neonatal Fc receptor (FcRn) is maintained to retain IgG-like antibody half-life.34
Figure 1.
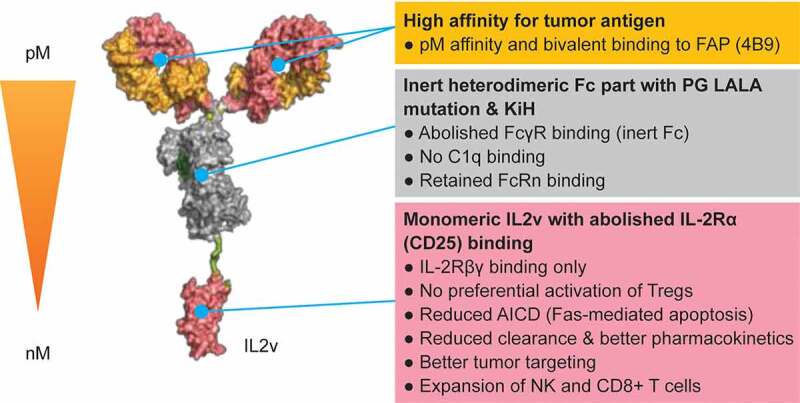
Structure of FAP-IL2v
Abbreviations: AICD, activation−induced cell death; FAP, fibroblast activation protein-α; FAP-IL2v, FAP-targeted interleukin 2 variant (IL2v) immunocytokine; FcγR, Fc gamma receptor; FcRn, neonatal Fc receptor; IL-2Rβγ, interleukin 2 receptor β and γ; KiH, “knobs-into-holes” technology; NK, natural killer (cell); Tregs, regulatory T cells.
FAP-IL2v is designed to increase the pool of activated immune effector cells and is intended to be a combination partner to enhance the efficacy of cancer immunotherapies that work through the patient’s immune effector cells. In this study, we show that FAP-IL2v targets the site of the tumor and activates NK cells and CD4+/CD8+ T cells, without preferentially activating Tregs, leading to up-regulation of IL-2 Rα (CD25) and CD69 and phosphorylation of the IL-2 receptor signaling molecule STAT5 (signal transducers and activators of transcription 5). Furthermore, we demonstrate that increasing the pool of activated NK cells potentiates the efficacy of ADCC-competent antibodies while increasing the pool of T cells potentiates the efficacy of T cell bispecific antibodies. Furthermore, preexisting T cell responses are enhanced by combining FAP-IL2v with checkpoint inhibitors (e.g., PD-L1) and CD40 agonism in a variety of xenograft and syngeneic murine tumor models.
Results
FAP-IL2v binds FAP and IL-2Rβγ in vitro with high affinity, but not IL-2Rα
The IL2v portion of FAP-IL2v was designed not to bind to IL-2R, but to retain the affinity and functional activity for the intermediate-affinity dimeric IL-2Rβγ. Surface plasmon resonance experiments demonstrated that human IL-2Rα binds to human IL2wt bound to the IL-2Rβγ complex, but not to IL2v bound to IL-2Rβγ complex (Supplementary Figure 1a). These data were confirmed in Tag-lite competition binding assays, which showed that IL2wt bound to IL-2R better than IL2v bound IL-2R on cells expressing IL-2R containing all three chains (IL-2Rα, β and γ), whereas on cells expressing IL-2R containing only IL-2Rβ- and γ-chains (no IL-2Rα-chain), the binding of IL2v and IL2wt to IL-2R was similar (right panel) (Supplementary Figure 1b).
Supplementary Table 1 shows the respective affinities of FAP-IL2v to human, mouse and cynomolgus monkey IL-2Rα, IL-2Rβγ, and FAP. A murinized version of FAP-IL2v (muFAP-muIL2v) was generated for use in fully immunocompetent mice to minimize potential anti-drug antibody formation; this variant showed comparable binding properties to FAP-IL2v. The ability of FAP-IL2v to bind simultaneously to FAP and the IL-2 receptor was assessed in a “sandwich” binding assay on FAP expressing fibroblasts. Binding of FAP-IL2v to FAP was detected with recombinant IL-2Rβγ-Fc heterodimer, confirming the ability of bispecific binding (Supplementary Figure 2).
FAP-IL2v activates CD4+/CD8+ T cells and NK cells in vitro, but not preferentially Tregs
FAP-IL2v induced a dose-dependent proliferation of resting NK cells and resting and activated CD4+ and CD8+ T cells within peripheral blood mononuclear cells (PBMCs) derived from healthy volunteers (Supplementary Figures 3 and 4 and Supplementary Table 2). In general, NK cells responded promptly to FAP-IL2v treatment, and most cells underwent proliferation after 5 days of treatment. Similar to NK cells, most CD8+ T cells underwent proliferation after 5 days of treatment, whereas CD4+ T cells responded more slowly, with only a proportion of cells proliferating after the same period. Compared with NK cells, higher concentrations of FAP-IL2v were required to stimulate resting T cells. Activation of IL-2R signaling in immune cells was demonstrated by a dose-dependent increase of STAT5 phosphorylation (Figure 2a and Supplementary Table 3). Treatment with FAP-IL2v activated CD4+ T cells, CD8+ T cells, and NK cells to a similar extent as treatment with FAP-IL2wt. Importantly, FAP-IL2v was significantly less potent than FAP-IL2wt in activating Tregs, which express high levels of IL-2Rα. Activation of CD4+ and CD8+ T cells was also demonstrated by the upregulation of the early activation marker CD69 and late activation marker IL-2Rα (CD25) (Supplementary Figure 5). FAP-IL2v and FAP-IL2wt induced a similar upregulation of CD69, whereas upregulation of IL-2Rα was much stronger following treatment with FAP-IL2v. Notably, T cells activated by FAP-IL2v were less sensitive to Fas-mediated apoptosis (AICD) than T cells activated by FAP-IL2wt or aldesleukin (Supplementary Figure 6).
Figure 2.

Activation of CD4+/CD8+ T cells and NK cells and cell killing in vitro. (a) Phosphorylation of STAT5 on CD4 T cells (CD3+ CD4+ CD25-), NK cells (CD3-CD56+), CD8 T cells (CD3+ CD8+ CD25-), and Tregs (CD4+ CD25+ FOXP3+) 20 min after treatment with different IL2/IL2v-containing constructs. (b) Tumor cell lysis and upregulation of NK cell activation markers following co-culture of LS174T colon cancer cells, GM05389 FAP-expressing fibroblasts, and human PBMCs with FAP-IL2v alone, cetuximab (1000 ng/mL fixed concentration) alone or the combination of FAP-IL2v and cetuximab, for 48 hours. (c) Tumor cell lysis, IL-2Rα (CD25) upregulation on CD8+ T cells, and cytokine release upon treatment of MKN45 cells with FAP-IL2v alone or in combination with CEA-TCB (0.2 nM fixed concentration) in the presence of PBMCs. Data are mean and standard deviation of triplicate experiments
Abbreviations: CEA-TCB; carcinoembryonic antigen T-cell bispecific antibody; FAP-IL2v, fibroblast activation protein-α -targeted interleukin 2 variant (IL2v) immunocytokine (the lead molecule); FAP-IL2wt, the lead molecule containing wild-type IL-2 instead of IL2v; MFI, mean fluorescent intensity; NK, natural killer (cell); (p)STAT5, (phosphorylated) signal transducer and activator of transcription 5A; PBMCs, peripheral blood mononuclear cells; Tregs, regulatory T cells.
FAP-IL2v enhances cetuximab-mediated ADCC and CEA-TCB-mediated T-cell-dependent cellular cytotoxicity (TDCC) in vitro
Cell-killing assays were used to determine if the activating effect of FAP-IL2v on NK cells and CD4+/CD8+ T cells could enhance cell killing by antibodies that mediate ADCC or TDCC. LS174T colon cancer cell killing with FAP-IL2v alone was limited, but enhanced considerably when combined with the ADCC-competent antibody cetuximab (Figure 2b). NK cell activation (as measured by upregulation of IL-2Rα and CD69) was also enhanced considerably with the combination compared with FAP-IL2v alone. Similarly, CEA-TCB enhanced the FAP-IL2v-mediated killing of MKN45 gastric cancer cells compared with FAP-IL2v treatment alone (Figure 2c). This was accompanied by increased activation of T cells and cytokine release when combining FAP-IL2v with CEA-TCB. Interestingly, a drop-off in IL-8 levels was observed at higher doses of FAP-IL2v in combination with cetuximab (Figure 2c). The reason for this is unknown.
FAP-IL2v pharmacokinetics and pharmacodynamics in the mouse
Exposure of human FAP-IL2v in CD-1 mice was dose proportional (Supplementary Table 4). Dose normalized AUC0–16h ranged from 479 to 557 μg · h/mL following intravenous (IV) dosing (1–4 mg/kg) FAP-IL2v clearance was 38.7–46.8 mL/d/kg, which is faster than a normal IgG, but this result can be expected because of the target-mediated drug disposition following IL2v binding to IL-2 receptors on T cells and NK cells, as well as binding to low levels of FAP in normal tissues.35 When tumor-free severe combined immunodeficiency (SCID) beige mice were administered a single IV dose of 0.3 mg/kg human FAP-IL2v or human FAP-IL2wt, it became obvious that FAP-IL2wt is much more rapidly cleared than FAP-IL2v through CD25 mediated drug disposition (Supplementary Figure 7b). Importantly, a dose-dependent exposure could be observed in C57BL/6 mice treated with 0.5, 1.0, or 2.0 mg/kg FAP-IL2v (Supplementary Figure 7b).
Blood exposure of FAP-IL2v in tumor-free C57BL/6 mice and in mice bearing Panc02-H7-Fluc subcutaneous tumors is shown in Supplementary Figure 8a. Lower levels of peripheral activation were observed in tumor-bearing mice compared with tumor-free mice, as seen by reduced increases of CD3 and CD8 T cells, but not CD4 T cells, in the plasma 3 days after administration of FAP-IL2v (Supplementary Figure 8b). FAP-targeted IL2v accumulation in the tumors induced higher levels of CD3 cells, CD8 T cells, NKp46 NK cells, and CD68-positive macrophages 3 days post-treatment compared with the vehicle group (Supplementary Figure 8 c).
FAP-IL2v targets the tumor in vivo
Biodistribution in mice
Single-photon emission computed tomography (SPECT)/computed tomography (CT) imaging and biodistribution studies were performed in immunocompetent BALB/c mice bearing syngeneic Renca renal cancer carcinoma tumors in the right kidney that showed a relatively high number of FAP-positive fibroblasts in the tumor stroma by immunohistochemistry (Supplementary Figure 9). The highest tumor uptake of 111In-labeled antibodies injected IV was observed with FAP-IgG (17.9 ± 1.1%ID/g of tissue; Figure 3a). Tumor uptake observed with FAP-IL2v (13.8 ± 1.3%ID/g) was higher than that with FAP-IL2wt (11.5 ± 0.8%ID/g; p = .04). As observed in the SPECT/CT images, some accumulation of FAP-IL2v was observed in the lymphoid tissues of the spleen, but this was significantly lower compared with FAP-IL2wt (10.8 ± 0.9%ID/g vs 38.0 ± 3.6%ID/g, respectively; p = .0002). FAP-IL2wt mainly accumulated in the spleen and in various lymph nodes. Tumor uptake of untargeted DP47-IL2v (5.3 ± 0.8%ID/g) was twofold to threefold lower than that of FAP-targeted antibodies. As DP47-IL2v has lower clearance than FAP-IL2v (data not shown), the lower uptake of DP47-IL2v is not related to faster clearance. In addition, enlargement of the spleen was observed in these mice. The diffuse signal seen in other organs of DP47-IL2v-treated mice likely represents binding to immune cells in the blood pool in these mice and the longer half-life of DP47-IL2v.
Figure 3.

Biodistribution in animal models. (a) SPECT/CT imaging of 111In-labeled FAP-IgG, FAP-IL2v, FAP-IL2wt and DP47-IL2v in immunocompetent BALB/c mice (n = 3 per treatment) bearing renal carcinoma in the right kidney. Imaging was performed 3 days after IV injection of 0.3 μg 111In-labeled antibodies co-injected with 25 μg of corresponding unlabeled antibodies. (b) FDG-PET/CT axial view imaging of 89Zr-FAP-IL2v localization in lesions of a single rhesus monkey with breast carcinoma. The monkey was treated with 0.5 mg/kg of FAP-IL2v IV mixed with tracer amounts of 89Zr-labeled FAP-IL2v. PET scans were performed on the day of administration and 3 days (67 hours) and 6 days (154 hours) after IV injection. On-treatment biopsy was performed on Day 3 following the PET scan
Abbreviations: FAP, fibroblast activation protein-α; FDG-PET/CT, fluorodeoxyglucose-positron emission tomography/computed tomography; IL2v, interleukin 2 variant; IL2wt, interleukin 2 wild-type; 111In, indium-111; PET, positron emission tomography; SPECT/CT, single-photon emission computed tomography/computed tomography; 89Zr, zirconium-89.
There was a trend for dose dependence in spleen accumulation of FAP-IL2v; 0.1 mg/kg had the highest spleen accumulation, followed by 0.3 mg/kg and 1 mg/kg, suggestive of a saturable IL-2R sink in the spleen (Supplementary Figure 10). Tumor accumulations were identical between 0.3 mg/kg and 1 mg/kg.
Biodistribution in a rhesus monkey with breast carcinoma
Positron-emission tomography (PET) imaging in a tumor-bearing rhesus monkey was conducted to demonstrate the superior tumor targeting of FAP-IL2v in a fully immunocompetent and relevant animal model. FAP expression in tumor stroma was confirmed by immunohistochemistry (Supplementary Figure 11a). Three fluorodeoxyglucose (FDG)-avid lesions were identified on PET scans. Based on postmortem histology, two-third FDG-avid lesions were confirmed as tumor lesions, while the FDG-avid lesion in the axillary lymph node was most like an inflammatory lesion. ImmunoPET scans showed that 89Zr-FAP-IL2v selectively accumulated in all three lesions identified on FDG PET scans (Figures 3b; 0.04–0.06%ID/mL). 89Zr-FAP-IL2v was primarily excreted through the hepatobiliary pathway, with minimal uptake in the lungs, spleen, and lymphoid tissues (Supplementary Figure 11b-d). Analysis of tumor biopsies revealed immune cell infiltration (Supplementary Figure 11e). Transient lymphocytosis and peripheral expansion of immune cells was observed (Supplementary Table 5) alongside an increase of CD25+ cells (indicative of lymphocyte activation in T and NK cells) and the percentage of CD3+, CD4+, CD25+, and FoxP3+ cells at Day 4 (Supplementary Table 6).
FAP-IL2v is efficacious in combination with therapeutic antibodies in murine models of human cancer
Combination with ADCC-competent antibodies
muFAP-muIL2v potentiated the efficacy of an ADCC-competent antibody against human epidermal growth factor receptor 2 (HER2) in BALB-neuT genetically engineered mice that spontaneously develop breast tumors because of the mammary-specific expression of a constitutively active variant of ratHER2 (neuT). In this model, single-agent muFAP-muIL2v (2 mg/kg) or an equivalent dose of untargeted muDP47-muIL2v (0.7 mg/kg) was given once weekly for 5 weeks, starting on week 19/21 after birth when mammary tumors were already measurable (average size 200 mm3). Single-agent muFAP-muIL2v did not exhibit therapeutic efficacy compared with the vehicle control group (Figure 4a). Only anti-ratHER2 muIgG2a significantly prolonged survival as a single-agent therapy compared with vehicle alone, with a median survival of 90 days (p = .0011). This effect appeared to be more than additive: the combination of anti-ratHER2 and muFAP-muIL2v significantly extended the median survival to 114 days compared with single-agent muFAP-muIL2v (p = .0016) and single-agent anti-ratHER2 (p = .042). Anti-ratHER2 combined with untargeted muDP47-muIL2v was not superior to single-agent anti-ratHER2 (p = .08). This efficacy was accompanied by a significant increase in tumor-infiltrating CD3 T cells, NK cells, and CD68-positive cells (Figure 4b-d and Supplementary Figure 12). The highest increase of NK cells and activated macrophages was observed in the combination group. The kinetics of baseline immune infiltration in BALB-neuT mouse model are shown in Supplementary Figure 13.
Figure 4.

Efficacy in combination with an ADCC-competent antibody. (a) Targeted muFAP-muIL2v (2 mg/kg), untargeted muDP47-muIL2v (0.7 mg/kg), and anti-ratHER2 muIgG2a (20 mg/kg) were administered as single agents and in combination in the BALB-neuT genetically engineered mouse mammary tumor model (n = 6 per group). Treatments were given IV once weekly for 5 weeks starting at Week 19 or Week 21. (b-c) Mammary fat pad tumors from these mice were harvested 5 days after the last treatment and analyzed by immunohistochemistry for CD3 T cells, NK cells, and CD68-expressing cells. Representative immunohistochemistry staining images are shown in Supplementary Figure 12 and 13. * p < .05; ** p < .01; *** p < .001 vs vehicle control (unpaired t-test)
Abbreviations: EGFR, epidermal growth-factor receptor; FAP-IL2v, fibroblast activation protein-α -targeted interleukin 2 variant (IL2v) immunocytokine (the lead molecule); HER2, human epidermal growth factor 2; hu humanized; mu, murinized; muFAP-muIL2v, murinized version of the lead molecule; muDP47-muIL2v, murinized untargeted interleukin 2 variant immunocytokine SC, subcutaneous; SCID, severe combined immunodeficient.
Similar efficacy was demonstrated in combination with cetuximab in the lung orthotopic xenograft A549 model in huCD16-transgenic SCID mice (which express the human FcγRIIIα receptor on NK cells and the homologous muFcγRIV receptor on macrophages/monocytes; Supplementary Figure 14a) and in the syngeneic B16-huEGFR lung metastatic model in fully immunocompetent C57BL/6 huEGFR huCD16-transgenic mice (Supplementary Figure 14b). Finally, combining FAP-IL2v (1 mg/kg QW for 5 weeks) with the ADCC-competent anti-CD20 antibody obinutuzumab (25 mg/kg QW for 5 weeks) in the subcutaneous WSU-DLCL2 diffuse large B-cell lymphoma xenograft model in SCID/huCD16-transgenic mice achieved greater tumor control than either agent given as monotherapy (Supplementary Figure 14 c). A reduction in tumor volume of 28% (p = .0303), 63% (p < .0001), and 98% (p < .0001) in tumor growth was observed in the FAP-IL2v, obinutuzumab, and the combination groups, respectively, compared with controls at study Day 39.
Combination with TDCC-competent antibody
muFAP-muIL2v also demonstrated synergy with a murinized T cell bispecific antibody targeting CEA on tumor cells and CD3 on T cells (muCEA-TCB). Median survival in Panc02-huCEA-transgenic C57BL/6 mice was significantly longer with single-agent muFAP-muIL2v (2 mg/kg IV QW; 38.5 days; p = .0231) and single-agent muCEA-TCB (2.5 mg/kg IV weekly; 41.5 days; p = .0003) compared with vehicle alone (28 days; Figure 5). However, combination therapy with muFAP-muIL2v plus muCEA-TCB achieved significantly longer median survival (61 days) compared with single-agent muFAP-muIL2v (p < .0001), muCEA-TCB (p = .0033), and vehicle (p < .0001).
Figure 5.

Efficacy in combination with a TDCC-competent antibody. muFAP-muIL2v (2 mg/kg IV once weekly) was administered in combination with the TDCC-competent antibody muCEA-TCB (2.5 mg/kg IV twice weekly) in the inflamed orthotopic Panc02-CEA model in CEA transgenic C57BL/6 mice (n = 8 mice per group). Treatments (four in total) started 7 days after tumor cell injection
Abbreviations: muCEA-TCB, murinized carcinoembryonic antigen-T cell bispecific; muFAP-muIL2v, murinized version of fibroblast activation protein-α-targeted interleukin 2 variant immunocytokine; TDCC, T cell-dependent cellular cytotoxicity.
Combination with PD-L1 checkpoint inhibition
The combination of FAP-IL2v with checkpoint inhibitor anti-PD-L1 was tested using muFAP-muIL2v and anti-muPD-L1 (a murine IgG2a surrogate antibody against murine PD-L1) in a Panc02 pancreatic orthotopic syngeneic model in fully immunocompetent C57BL/6 mice. This model is characterized by inflamed tumors with preexisting antigen-specific T cells. Panc02 cells express PD-L1 and are susceptible to PD-L1-targeting therapies.36,37 Median survival with muFAP-muIL2v (2 mg/kg) administered with anti-muPD-L1 (10 mg/kg) given via intraperitoneal (IP) injection weekly for 5 weeks was significantly longer compared with the respective single agents (p = .0003 vs. muFAP-muIL2v and p = .05 vs. anti-muPD-L1; Figure 6a).
Figure 6.
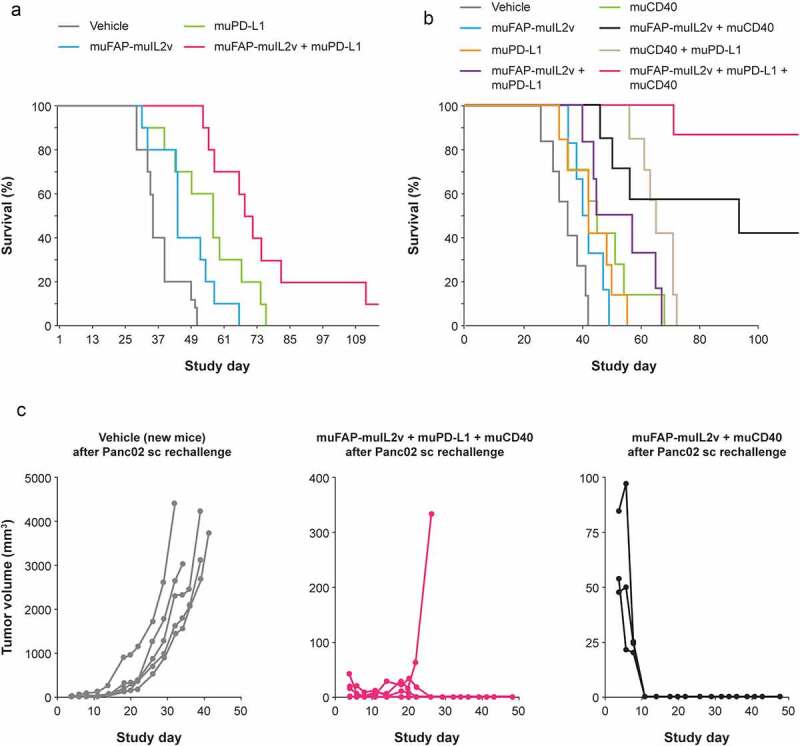
Efficacy in combination with PD-L1 checkpoint inhibition. (a) Efficacy of muFAP-muIL2v (2 mg/kg IP) and anti-muPD-L1 (10 mg/kg IP) as single agents and in combination given once weekly for 5 weeks starting on Day 7 in the Panc02 pancreatic orthotopic syngeneic model in C57BL/6 mice (n = 10 mice per group). (b) Efficacy in the same mouse model with the addition of muCD40 (10 mg/kg IP given once on Day 7) as single agent and in combination with muFAP-muIL2v (2 mg/kg IP) and anti-muPD-L1 (10 mg/kg IP) given once weekly for 3 weeks (n = 6–8 mice per group). (c) Tumor growth in surviving mice from experiments shown in part (b) that were re-challenged subcutaneously with Panc02 cells (5 × 105) on Day 121 (Day 0 in Figure 6c). No additional antibody therapy was given in these experiments. A new group of vehicle-only treated mice was used for comparison in this experiment
Abbreviations: IP, intraperitoneally; muFAP-muIL2v, murinized version of fibroblast activation protein-α-targeted interleukin 2 variant immunocytokine; muPD-L1, murinized anti-PD-L1 antibody; muCD40, murinized agonistic CD40 antibody.
Triple combination with PD-L1/CD40 antibodies
FAP-IL2v also strongly enhanced the activity of an agonistic CD40 antibody (anti-muCD40; 10 mg/kg given IP once at Day 7) in the Panc02 model (Figure 6b). Median survival was 40 days with muFAP-IL2v monotherapy, 45 days with anti-muCD40 monotherapy, and 93 days (p = .0005 vs vehicle control) with the combination. The triplet of muFAP-muIL2v, anti-muPD-L1, and anti-muCD40 achieved the greatest efficacy (median survival not reached vs 35 days with vehicle control, p < .0001). Long-term surviving animals from this study who received the triplet or doublet combinations were protected from tumor re-challenge with Panc02 cells (Figure 6c).
Discussion
To increase the benefit–risk ratio of the currently available systemic cytokine therapy, it is desirable to develop novel immunocytokines that combine antibodies with superior tumor-targeting ability and cytokines with broad-based immune-modulatory activities to effectively mobilize or sustain anti-tumor immune responses at the tumor site. Various approaches are currently in development to achieve immunostimulation through IL-2 without activating suppressive Tregs, including NKTR-214 (bempegaldesleukin, Nektar Therapeutics) and ALT-803 (Altor BioScience). The prodrug NKTR-214 is a modified form of IL-2 conjugated to releasable polyethylene glycol chains that mask the part of IL-2 that interacts with the IL-2Rα subunit to reduce activation of Tregs.38 Preclinical data show that NKTR-214 treatment results in rapid expansion of effector T cells and is well tolerated in non-human primates.38 ALT-803 is a supercomplex of modified IL-15 and IL-15Rα fused to an IgG1 Fc. IL-15 stimulates NK cells and effector T cells, but unlike IL-2, IL-15 does not stimulate Tregs. Initial clinical data show that ALT-803 is well tolerated and leads to dose-dependent increases in CD8+ T cells and NK cells.39,40
In contrast to these engineered IL-2Rβγ agonists that act in a “untargeted” manner, FAP-IL2v is a novel immunocytokine designed to bind FAP on tumor stroma cells to deliver an immunostimulatory IL-2 variant to the tumor site without preferential activation of immunosuppressive Tregs. Here, we investigated the pharmacodynamic and pharmacokinetic properties of FAP-IL2v in vitro and in vivo and show how the immunostimulatory properties of FAP-IL2v can potentiate the efficacy of a variety of cancer immunotherapies.
FAP-IL2v is a species of cross-reactive and bound human, mouse, rhesus, and cynomolgus monkey FAP and IL-2Rβγ, but not IL-2α, with high affinity in vitro. FAP-IL2v was significantly less potent than FAP-IL2wt in activating immunosuppressive Tregs, which express high levels of IL-2Rα. Because the binding to IL-2Rβγ was retained, FAP-IL2v was able to activate NK cells and CD4+/CD8+ T cells in vitro, as demonstrated by up-regulation of classical activation markers, such as CD69 and IL-2Rα (CD25), and induction of proliferation and phosphorylation of the IL-2R signaling maker STAT5, in a dose-dependent manner that did not differ from that of FAP-IL2wt. This is expected to translate into significantly lower expansion of Tregs than immune effector cells in vivo, which is desirable when attempting to restore immune cell activation and attenuate local tumor immunosuppression. These data are consistent with the proposed mode of action of FAP-IL2v, which was engineered to abolish binding to IL-2Rα without affecting binding to IL-2Rβγ on immune effector cells. The binding properties of the IL2v part of FAP-IL2v are consistent with those of the IL2v part of CEA-IL2v.27
Strongly activated T cells upregulate Fas and are sensitive to Fas-mediated apoptosis.41 This was observed with aldesleukin treatment and in cells treated with FAP-IL2wt. FAP-IL2v-treated T cells, however, were considerably less sensitive to Fas-mediated apoptosis, suggesting that IL2v-stimulated T cells are more resistant to activation-induced cell death.
Data from in vivo imaging studies on tumor-bearing immunocompetent mice and a rhesus monkey with breast carcinoma demonstrated that FAP-IL2v preferentially localized within tumor tissue over lymphoid and other tissues. While some accumulation was seen in the spleen in mice, splenic accumulation was not observed in the rhesus monkey, which likely reflects species differences in the response to FAP-IL2v. High liver uptake of FAP-IL2v was observed in the monkey, but not in mice. This different localization of FAP-IL2v between mice and monkeys may also be explained by the lower dose administered to the monkey as compared to the dose applied to mice, and potentially the older age of the monkey and generally lower pharmacodynamic response in monkeys versus mice, for example, regarding absolute increase in T cell numbers.
The fact that FAP-IL2wt showed more distribution in the spleen than FAP-IL2v indicates that the distribution in the wild-type setting is driven by binding to IL-2R; however, with lower affinity to the IL-2R complex, the anti-Fap domain takes over the targeting. Tumor localization of FAP-IL2v is consistent with FAP binding on tumor stromal cells, the limited expression of FAP in healthy tissues, and the lack of IL-2Rα binding on immune cells. The lack of IL-2Rα binding by FAP-IL2v is expected to lead to the relative absence of a peripheral IL-2Rα sink on immune cells. This will likely overcome the poor tumor targeting properties seen with conventional wild-type IL-2-based immunocytokines.25 However, it should be noted that FAP-IL2v still has functional activity on immune cells in the peripheral blood and lymphoid tissues through its monovalent and intermediate affinity to IL-2Rβγ. These observations are supported by pharmacokinetic studies that showed an approximately twofold reduction in clearance of an untargeted DP47-IL2v immunocytokine when compared with the analogous wild-type DP47-IL2wt immunocytokine, confirming that the introduction of the IL2v indeed reduces the IL-2Rα -mediated clearance and results in a longer half-life.42
Biodistribution studies in the breast cancer-bearing rhesus monkey confirmed that FAP-IL2v targets the tumor in vivo and, importantly, showed that uptake of FAP-IL2v in lung tissue was minimal (lung metastases were not present in this animal). No signs of pulmonary injury were seen in this animal. Pulmonary edema is a key and severe side effect of IL-2 therapy and is mediated by the direct binding of IL-2 to IL-2Rα on lung endothelial cells.16 The lack of binding of FAP-IL2v to IL-2Rα and its limited distribution to lung tissue should translate into reduced pulmonary toxicity compared with other IL-2-based therapies. Biodistribution studies in the rhesus monkey ran for 6 days, which is sufficient to ensure that the observed targeting is specific. The murine biodistribution studies were limited to 3 days, which may not be sufficient time for complete distribution and targeting. This limitation was due to the aggressive tumor growth in the murine models and the need for recurrent anesthesia/imaging-related procedures; later, scans were not considered practical to minimize the discomfort to the mice.
A possible clinical application of FAP-IL2v would be in combination with ADCC-competent antibodies of the IgG1 isotype to enhance their role in ADCC. Our data showed that FAP-IL2v increased the pool of activated NK cells in vitro, which significantly potentiated the ADCC activity of cetuximab in cell killing assays. This translated into strongly enhanced anti-tumor efficacy mediated by ADCC-competent or -enhanced antibodies in the lung orthotopic xenograft A549 model in huCD16 transgenic SCID mice, in the genetically engineered BALB-neuT mouse model developing spontaneous breast tumors, in the syngeneic lung metastatic B16-EGFR model in fully immunocompetent C57BL/6 huEGFR huCD16 transgenic mice, and in the aggressive non-Hodgkin’s lymphoma model WSU-DLCL2 in human CD16 transgenic SCID mice. While FAP-IL2v (or the murine equivalent muFAP-muIL2v) monotherapy significantly extended median survival compared with vehicle alone in some models, combination therapy with FAP-IL2v significantly potentiated the efficacy of all tested ADCC-competent antibodies (cetuximab, a ratHER2-specific antibody, and obinutuzumab).
FAP-IL2v also potentiated the efficacy of immunotherapies that work through T cells. FAP-IL2v is synergized with the T cell bispecific antibody CEA-TCB in vitro, which translated into enhanced efficacy in the inflamed syngeneic orthotopic pancreatic Panc02 model in human CEA transgenic C57BL/6 mice vs either treatment alone. CEA-TCB binds CEA on tumor cells and the CD3ε chain within the TCR complex on T cells and triggers an increase in tumor-infiltrating T cells, T cell activation, secretion of cytotoxic granules, and tumor cell lysis and regression.43,44
FAP-IL2v was also shown to synergize with a therapeutic antibody targeting the PD-L1 checkpoint inhibitor and an agonistic CD40 antibody. Most notably, the triple combination of FAP-IL2v with PD-L1 checkpoint inhibition and CD40 agonism resulted in long-term survival of the majority of animals, and this therapeutic effect persisted when surviving animals were re-challenged with tumor cells but without further administration of therapeutic antibodies. In this setting, we believe that FAP-IL2v in combination with the checkpoint inhibitor PD-L1 strongly enhances the activation of preexisting antigen-specific T cells in the tumor, whereas the combination with the CD40 agonistic antibody can further enhance the generation of a long-lasting anti-tumor efficacy in the majority of animals.
Taken together, these data indicate that FAP-IL2v is a versatile combination partner for a diverse range of cancer immunotherapies including ADCC-competent antibodies, T cell bispecific antibodies, and checkpoint inhibitors. The immunostimulatory properties of FAP-IL2v may be particularly useful in combination with PD-L1 checkpoint inhibition and/or CD40 agonism for the treatment of inflamed tumors, but also in poorly inflamed tumors when combined with ADCC- or TDCC-mediating antibodies. Furthermore, our data show that FAP-IL2v overcomes many of the limitations of aldesleukin and other IL-2–based immunocytokines, by superior tumor targeting, lack of preferential activation of Tregs, and lack of binding to pulmonary epithelium. FAP-IL2v is currently in Phase 1b/2 clinical development as a monotherapy and in combination with trastuzumab and cetuximab (NCT02627274), atezolizumab (anti-PD-L1; NCT03386721), and atezolizumab plus bevacizumab (NCT03063762). Study NCT02627274 in patients with metastatic solid tumors has identified a recommended dose and schedule for FAP-IL2v of 20 mg IV once weekly.45 Rapid expansion of CD8 cells and NK cells, but not Tregs, was observed in patients treated at this dose. Three long-lasting (>6 months) objective responses with FAP-IL2v monotherapy were observed across the dose escalation range (in patients with head and neck cancer, penile squamous cell carcinoma, and checkpoint inhibitor-resistant malignant melanoma). Tumor shrinkage occurred in four melanoma patients. The most adverse events were Grade 1/2 and included (>30%) pyrexia, infusion-related reactions, fatigue/asthenia, nausea, diarrhea, decreased appetite and elevated aspartate and/or alanine transaminase.45 At the dose range applied to mice in this study, we did not observe any clinical signs of toxicity due to FAP-IL2v, but, in general, mice are less sensitive to IL-2 mediated toxicities.
In conclusion, FAP-IL2v is a novel and potent immunocytokine that targets the site of the tumor and activates immunostimulatory CD4+/CD8+ T cells and NK cells without preferentially activating immunosuppressive Tregs. It is a versatile combination partner for various cancer immunotherapies and has the potential to be developed for clinical use in combination with checkpoint inhibitors, T cell bispecific antibodies, ADCC-mediating antibodies, and other novel immunomodulatory agents.
Materials and methods
The cell lines used in these studies and their origin are described in the supplementary methods, along with details of the surface plasmon resonance experiments, sandwich binding assays, and pharmacokinetic experiments.
Immunocytokines and antibodies
For in vitro studies, studies in immunodeficient mice, and short-term studies in immunocompetent mice, human IgG1-based immunocytokines (FAP-IL2v and FAP-IL2wt) were used. To reduce the risk of immunogenicity and loss of exposure in long-term pharmacodynamic studies in fully immunocompetent C57BL/6 mice, murinized surrogate immunocytokines (muFAP-muIL2v, muDP47-muIL2v) were used. These surrogates were based on the respective human(ized) variable regions and mouse IgG1 constant regions with DAPG mutations to abolish FcγR and C1q interaction and a mutated murine IL-2 variant carrying homologous mutations to abolish muIL-2Rα binding. The design details are given in Supplementary Tables 7 and 8. All immunocytokines were produced by transient production using HEK293 or stable Chinese hamster ovary clones followed by purification using Protein A affinity chromatography and size-exclusion/ion-exchange chromatography. STAT5 phosphorylation, proliferation, and AICD with FAP-IL2v are described elsewhere.27 The anti-ratHER2 muIgG2a and a murinized version of cetuximab (based on the clone C225) were generated in house. Commercial cetuximab was obtained from Merck and obinutuzumab was obtained from Roche Pharmaceuticals.
In vitro combination with ADCC-competent antibodies
A total of 30,000 LS174T human colon tumor cells and 5000 GM05389 human fibroblasts were seeded into 96-well flat-bottom plates and co-cultured with 300,000 human PBMCs at an effector-to-target cell ratio (E:T) of 10:1. Cells were incubated with increasing concentrations of FAP-IL2v with or without 1000 ng/mL of the anti- epidermal growth-factor receptor (EGFR) antibody cetuximab. Cytotoxicity was determined after 24 hours by measuring lactate dehydrogenase (LDH) release into the supernatant (Roche Applied Science). PBMCs were collected from the supernatant and stained with CD3 BUV359 (clone UCHT1, BD Bioscience), CD56 BV711 (clone HCD56, BioLegend), CD25 BUV421 (clone BC96, BioLegend), and CD69 PE (clone FN50, BioLegend) to determine NK cell (CD3-CD56+) activation by flow cytometry.
In vitro combination with CEA-TCB
The effect of combining FAP-IL2v with a T cell bispecific antibody that binds CEA on tumor cells and CD3 on T cells (CEA-TCB) was tested on MKN45 human gastric cancer cells co-cultured with PBMCs. Briefly, 30,000 MKN45 tumor cells were seeded into 96-well plates with 300,000 PBMCs (E:T 10:1) and incubated with increasing concentrations of FAP-IL2v in the presence or absence of 0.2 nM CEA-TCB. After 48 hours, cytotoxicity was determined by measuring LDH release as before. The release of Granzyme B, tumor necrosis factor, interferon γ, RANTES, IL-6, and IL-8 into the supernatant was determined by Cytokine Bead Array (BD Bioscience). Cells were stained with CD3 FITC (clone UCHT1, BioLegend), CD8 APC (clone SK1, BioLegend) and CD25 PE-Cy7 (clone BC-95, BioLegend) to determine CD8+ T cell (CD3+ CD8+) activation by flow cytometry.
Mouse xenograft and syngeneic models
Subcutaneous and orthotopic xenograft, syngeneic, and genetically engineered mice models were used to assess the in vivo efficacy of single-agent FAP-IL2v and combination therapies. This study was reviewed and approved by the Institutional Animal Care and Use Committee of the Preclinical Pharmacology Department, Roche Innovation Center Zurich, Schlieren, Switzerland, and performed in accordance with the animal research protocols approved by the local government (Kantonale Verwaltung Veterinäramt kant. Zürich, Switzerland; license ZH193/2014). All animals were handled according to guidelines of the Federation of European Laboratory Animal Science Associations, Gesellschaft für Versuchstierkunde/Society of Laboratory Animal Science (GV-Solas) and the Tierschutzgesetz (TierSchG). Mice were maintained under specific-pathogen-free conditions with daily cycles of 12 hours light/darkness. Food and water were provided ad libitum. Continuous health monitoring was carried out.
All mice were female and bred by Charles River Laboratories. Models comprised: human (hu) CD16-transgenic SCID mice IV inoculated with 3 × 106 A549-Fluc cells (non-small cell lung cancer xenograft lung model) or subcutaneously inoculated with 2 × 106 WSU-DLCL2 cells (non-Hodgkin lymphoma model); huEGFR huCD16-transgenic C57BL/6 mice inoculated IV with 0.1 × 106 B16-huEGFR cells (syngeneic lung metastatic model); C57BL/6 mice intrapancreatically inoculated with 1 × 105 Panc02-H7 cells; CEA-transgenic C57BL/6 mice intrapancreatically inoculated with 1 × 105 Panc02-CEA cells (pancreatic syngeneic models); SCID beige mice; and BALB-neuT mice (19–21 weeks old; genetically engineered mouse model). BALB-neuT mice overexpress the rat HER2/neu oncogene and develop mammary hyperplasia within the first month of life, which progresses to aggressive carcinomas in all the 10 mammary glands. Up to 10 mammary tumors can be found at week 22–24.
Mice were randomized and therapy started when evidence of tumor growth was visible in the target organ of sacrificed scout animals (days indicated in figure legends). All treatments were administered IV unless indicated otherwise. The termination criteria for euthanizing animals were sickness with locomotion impairment. Median overall survival was defined as the experimental day by which 50% of animals had been sacrificed. Kaplan-Meier survival curves and the pairwise log-rank test were used to compare survival between animals.
Biodistribution and SPECT/CT imaging in mice
Fully immunocompetent BALB/c mice bearing orthotopic Renca tumors (n = 3 per group) were injected IV with tracer amounts (0.3 µg) of 111In-labeled antibodies FAP-IgG (4B9 IgG), FAP-IL2v, FAP-IL2wt, and untargeted-IL2v (DP47-IL2v) along with 25 µg of corresponding unlabeled antibodies. SPECT/CT and terminal biodistribution studies were conducted 3 days after antibody injection. In addition, SPECT/CT images were acquired in CD-1 and C57BL/6 mice at 4, 24, and 72 hours at different FAP-IL2v dose levels (0.1, 0.3, and 1.0 mg/kg) to determine whole-body tissue pharmacokinetics and the impact of IL-2R sink.
Mice were anesthetized (2% isoflurane/O2) and placed in prone position in the SPECT/CT scanner (U-SPECT-II, MILabs). SPECT images were acquired over 60 minutes using a 1.0-mm-diameter pinhole mouse collimator tube (3–4 frames of 15 minutes, 99 bed-positions per frame) followed by a CT scan (615 µA, 65 kV). Scans were reconstructed with MILabs reconstruction software. Mice were euthanized (CO2/O2 asphyxiation) immediately after the last SPECT/CT scan and relevant tissues were dissected. The radioactivity concentration in the tissues was determined and uptake (%ID/g) was calculated. Unpaired t-tests were used to compare groups.
Imaging in rhesus monkey with breast carcinoma
The PET/CT procedure is described in detail in the supplement. All studies involving this animal were approved by the Wake Forest University Institutional Animal Care and Use Committee and conducted in accordance with the US Animal Welfare Act and the Department of Health and Human Services regulations (Office of Laboratory Animal Welfare assurance number #A-3391-01). Wake Forest University is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. This rhesus monkey was provided with appropriate veterinary care and social and environmental enrichment. Briefly, an 11-year-old rhesus monkey with confirmed spontaneously occurring invasive ductal carcinoma with vascular invasion was treated with 0.5 mg/kg of FAP-IL2v mixed with tracer amounts of FAP-IL2v radiolabeled with zirconium-89 (89Zr) as previously described.46 PET scans were conducted on the day of administration and 3 days (67 hours) and 6 days (154 hours) after injection. On-treatment tumor biopsy for assessment of immune cell infiltration was performed on Day 3 following the PET scan.
Supplementary Material
Acknowledgments
The authors wish to thank all contributors and project team members from Roche Innovation Centers Zurich, Basel, and Munich for their support of this project. Support for third-party writing assistance for this article, furnished by Jamie Ashman, was provided by Prism Ideas.
Funding Statement
This study and editorial support for the preparation of this manuscript is funded by Roche Pharma Research and Early Development. Edwin J. W. Geven was supported by a Roche Postdoctoral Fellowship. Nijmegen and Wake Forest Research groups received research funding from Roche. Studies at Wake Forest were supported in part by the Wake Forest Baptist Comprehensive Cancer Center National Cancer Institute’s Cancer Center Support Grant, award number P30CA012197.
Disclosure of interests
I Waldhauer, V Gonzalez-Nicolini, A Freimoser-Grundschober, L Fahrni, RJ Hosse, J Sam, E Bommer, V Steinhart, E Hussar, S Colombetti, E van Puijenbroek, M Neubauer, G Acuna, J Charo, V Teichgräber, S Evers, M Bacac, E Moessner, P Umana, and C Klein are employees of Roche and hold stock/stock options with Roche. TK Nayak and S Lang were employees of Roche at the time the study was conducted. Authors I Waldhauer, VG Nicolini, A Freimoser-Grundschober, RJ Hosse, S Colombetti, J Charo, M Bacac, E Moessner, P Umana, and C Klein hold patents related to FAP-IL2v and related cancer immunotherapies. D Gerrits, OC Boerman, J Mark Cline, and G Dugan received research funding from Roche for the work under consideration. EJW Geven received grants from Roche for the work under consideration and other research. PK Garg had “grant-in-aid” from Roche to conduct PET/CT imaging studies on tumor bearing non-human primates for this study. F Cavallo has nothing to disclose.
Abbreviations
ADCC, antibody-dependent cellular cytotoxicity; AICD, activation-induced cell death; CEA, Carcinoembryonic antigen; CEA-IL2v, Cergutuzumab amunaleukin; CT, Computed tomography; E:T, effector-to-target cell ratio; FAP, fibroblast activation protein α; FAP-IL2v, simlukafusp alfa; FcRn, neonatal Fc receptor; IL-2, Interleukin-2; IL-2R, IL-2 receptor; IL2v, IL-2 variant; IV, intravenously; LDH, lactate dehydrogenase; NK, natural killer; PBMC, peripheral blood mononuclear cells; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PET, positron-emission tomography; SCID, severe combined immunodeficiency; SPECT, single-photon emission computed tomography; TDCC, T cell-dependent cellular cytotoxicity; Tregs, regulatory T cells; wt, wild-type.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Arenas-Ramirez N, Woytschak J, Boyman O.. Interleukin-2: biology, design and application. Trends Immunol. 2015;36(12):763–13. doi: 10.1016/j.it.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–90. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 3.Antony GK, Dudek AZ. Interleukin 2 in cancer therapy. Curr Med Chem. 2010;17(29):3297–302. doi: 10.2174/092986710793176410. [DOI] [PubMed] [Google Scholar]
- 4.Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology. 2016;5(6):e1163462. doi: 10.1080/2162402X.2016.1163462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol. 2014;192(12):5451–58. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Cruz LM, Klein L. Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol. 2005;6(11):1152–59. doi: 10.1038/ni1264. [DOI] [PubMed] [Google Scholar]
- 7.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–51. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 8.Maloy KJ, Powrie F. Fueling regulation: IL-2 keeps CD4+ Treg cells fit. Nat Immunol. 2005;6(11):1071–72. doi: 10.1038/ni1105-1071. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen T, Russell J. The regulation of FasL expression during activation-induced cell death (AICD). Immunology. 2001;103(4):426–34. doi: 10.1046/j.1365-2567.2001.01264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8(5):615–23. doi: 10.1016/S1074-7613(00)80566-X. [DOI] [PubMed] [Google Scholar]
- 11.Chow S, Galvis V, Pillai M, Leach R, Keene E, Spencer-Shaw A, Shablak A, Shanks J, Liptrot T, Thistlethwaite F, et al. High-dose interleukin 2 – a 10-year single-site experience in the treatment of metastatic renal cell carcinoma: careful selection of patients gives an excellent outcome. J Immunother Cancer. 2016;4:67. doi: 10.1186/s40425-016-0174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klapper JA, Downey SG, Smith FO, Yang JC, Hughes MS, Kammula US, Sherry RM, Royal RE, Steinberg SM, Rosenberg S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: a retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer. 2008;113(2):293–301. doi: 10.1002/cncr.23552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott DF, Cheng S-C, Signoretti S, Margolin KA, Clark JI, Sosman JA, Dutcher JP, Logan TF, Curti BD, Ernstoff MS, et al. The high-dose aldesleukin “select” trial: a trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2015;21(3):561–68. doi: 10.1158/1078-0432.CCR-14-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith FO, Downey SG, Klapper JA, Yang JC, Sherry RM, Royal RE, Kammula US, Hughes MS, Restifo NP, Levy CL, et al. Treatment of metastatic melanoma using interleukin-2 alone or in conjunction with vaccines. Clin Cancer Res. 2008;14(17):5610–18. doi: 10.1158/1078-0432.CCR-08-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prometheus Laboratories, Inc . San Diego, CA 92121. Proleukin® (aldesleukin) Package Insert. 2012. [Google Scholar]
- 16.Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci USA. 2010;107(26):11906–11. doi: 10.1073/pnas.1002569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ledford H. Old cancer drug gets fresh look. Nature. 2014;509(7502):541–42. doi: 10.1038/509541a. [DOI] [PubMed] [Google Scholar]
- 18.Connor JP, Cristea MC, Lewis NL, Lewis LD, Komarnitsky PB, Mattiacci MR, Felder M, Stewart S, Harter J, Henslee-Downey J, et al. A phase 1b study of humanized KS-interleukin-2 (huKS-IL2) immunocytokine with cyclophosphamide in patients with EpCAM-positive advanced solid tumors. BMC Cancer. 2013;13(1):20. doi: 10.1186/1471-2407-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danielli R, Patuzzo R, Di Giacomo AM, Gallino G, Maurichi A, Di Florio A, Cutaia O, Lazzeri A, Fazio C, Miracco C, et al. Intralesional administration of L19-IL2/L19-TNF in stage III or stage IVM1a melanoma patients: results of a phase II study. Cancer Immunol Immunother. 2015;64(8):999–1009. doi: 10.1007/s00262-015-1704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillies SD, Lo K-M, Burger C, Lan Y, Dahl T, Wong W-K. Improved circulating half-life and efficacy of an antibody-interleukin 2 immunocytokine based on reduced intracellular proteolysis. Clin Cancer Res. 2002;8:210–16. [PubMed] [Google Scholar]
- 21.Holden SA, Lan Y, Pardo AM, Wesolowski JS, Gillies SD. Augmentation of antitumor activity of an antibody-interleukin 2 immunocytokine with chemotherapeutic agents. Clin Cancer Res. 2001;7:2862–69. [PubMed] [Google Scholar]
- 22.King DM, Albertini MR, Schalch H, Hank JA, Gan J, Surfus J, Mahvi D, Schiller JH, Warner T, Kim K, et al. Phase I clinical trial of the immunocytokine EMD 273063 in melanoma patients. J Clin Oncol. 2004;22(22):4463–73. doi: 10.1200/JCO.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwager K, Hemmerle T, Aebischer D, Neri D. The immunocytokine L19-IL2 eradicates cancer when used in combination with CTLA-4 blockade or with L19-TNF. J Invest Dermatol. 2013;133:751–58. doi: 10.1038/jid.2012.376. [DOI] [PubMed] [Google Scholar]
- 24.Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, Reynolds CP, Kimball J, Albertini MR, Wagner B, et al. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children’s Oncology Group (COG) phase II study. J Clin Oncol. 2010;28:4969–75. doi: 10.1200/JCO.2009.27.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci USA. 2015;112:3320–25. doi: 10.1073/pnas.1416159112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weide B, Eigentler TK, Pflugfelder A, Zelba H, Martens A, Pawelec G, Giovannoni L, Ruffini PA, Elia G, Neri D, et al. Intralesional treatment of stage III metastatic melanoma patients with L19-IL2 results in sustained clinical and systemic immunologic responses. Cancer Immunol Res. 2014;2:668–78. doi: 10.1158/2326-6066.CIR-13-0206. [DOI] [PubMed] [Google Scholar]
- 27.Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017;6(3):e1277306. doi: 10.1080/2162402X.2016.1277306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brennen WN, Isaacs JT, Denmeade SR. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol Cancer Ther. 2012;11(2):257–66. doi: 10.1158/1535-7163.MCT-11-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamson EJ, Keane FM, Tholen S, Schilling O, Gorrell MD. Understanding fibroblast activation protein (FAP): substrates, activities, expression and targeting for cancer therapy. Proteomics Clin Appl. 2014;8(5–6):454–63. doi: 10.1002/prca.201300095. [DOI] [PubMed] [Google Scholar]
- 30.Liu R, Li H, Liu L, Yu J, Ren X. Fibroblast activation protein: a potential therapeutic target in cancer. Cancer Biol Ther. 2012;13(3):123–29. doi: 10.4161/cbt.13.3.18696. [DOI] [PubMed] [Google Scholar]
- 31.Altmann A, Haberkorn U, Siveke J. The latest developments in imaging of fibroblast activation protein. J Nucl Med. 2021;62(2):160–67. doi: 10.2967/jnumed.120.244806. [DOI] [PubMed] [Google Scholar]
- 32.Kratochwil C, Flechsig P, Lindner T, Abderrahim L, Altmann A, Mier W, Adeberg S, Rathke H, Röhrich M, Winter H, et al. 68Ga-FAPI PET/CT: tracer uptake in 28 different kinds of cancer. J Nucl Med. 2019;60:801–05. doi: 10.2967/jnumed.119.227967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laverman P, Van Der Geest T, Terry SYA, Gerrits D, Walgreen B, Helsen MM, Nayak TK, Freimoser-Grundschober A, Waldhauer I, Hosse RJ, et al. Immuno-PET and immuno-SPECT of rheumatoid arthritis with radiolabeled anti-fibroblast activation protein antibody correlates with severity of arthritis. J Nucl Med. 2015;56(5):778–83. doi: 10.2967/jnumed.114.152959. [DOI] [PubMed] [Google Scholar]
- 34.Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs. 2011;3(5):422–30. doi: 10.4161/mabs.3.5.16983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng R, Iyer S, Theil F-P, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs. 2011;3(1):61–66. doi: 10.4161/mabs.3.1.13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, Nordquist E, Cruz-Monserrate Z, Yu L, Young G, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67(2):320–32. doi: 10.1136/gutjnl-2016-311585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rivera IO, Iclozan C, McGill A, Ghansah T. PD-L1/PD-1 Immunotherapy modulates effector T cells homeostasis and function in murine pancreatic cancer. J Immunol. 2016;196:72.11–72.11. [Google Scholar]
- 38.Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res. 2016;22(3):680–90. doi: 10.1158/1078-0432.CCR-15-1631. [DOI] [PubMed] [Google Scholar]
- 39.Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, Miller JS, Farhad M, Anderton K, Lindsey K, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19(5):694–704. doi: 10.1016/S1470-2045(18)30148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller JS, Cooley S, Holtan S, Arora M, Ustun C, Jeng E, Wong HC, Verneris MR, Wagner JE, Weisdorf DJ, et al. First-in-human phase I dose escalation trial of IL-15N72D/IL-15Rα-Fc superagonist complex (ALT-803) demonstrates immune activation with anti-tumor activity in patients with relapsed hematological malignancy. Blood. 2015;126:1957. doi: 10.1182/blood.V126.23.1957.1957. [DOI] [Google Scholar]
- 41.Akimzhanov AM, Wang X, Sun J, Boehning D. T-cell receptor complex is essential for Fas signal transduction. Proc Natl Acad Sci USA. 2010;107(34):15105–10. doi: 10.1073/pnas.1005419107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klein C, Waldhauer I, Nicolini V, Dunn C, Freimoser-Grundschober A, Herter S, Geven E, Boerman O, Nayak T, Puijenbroek E, et al. Tumor-targeted, engineered IL-2 variant (IL-2v)-based immunocytokines for the immunotherapy of cancer. Cancer Res. 2013;73:486–486. [Google Scholar]
- 43.Bacac M, Klein C, Umana P. CEA TCB: a novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. 2016;5(8):e1203498. doi: 10.1080/2162402X.2016.1203498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22(13):3286–97. doi: 10.1158/1078-0432.CCR-15-1696. [DOI] [PubMed] [Google Scholar]
- 45.Melero I, Castanon Alvarez E, Mau-Sorensen M, Lassen UN, Lolkema MP, Robbrecht DG, Gomez-Roca CA, Martin-Liberal J, Tabernero J, Ros W, et al. Clinical activity, safety, and PK/PD from a phase I study of RO6874281, a fibroblast activation protein (FAP) targeted interleukin-2 variant (IL-2v) (Abstract 412PD). Ann Oncol. 2018;29:viii134. doi: 10.1093/annonc/mdy279.400. [DOI] [Google Scholar]
- 46.Nayak TK, Garmestani K, Milenic DE, Brechbiel MW. PET and MRI of metastatic peritoneal and pulmonary colorectal cancer in mice with human epidermal growth factor receptor 1-targeted 89Zr-labeled panitumumab. J Nucl Med. 2012;53(1):113–20. doi: 10.2967/jnumed.111.094169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.