

Abstract
Cognitive deficits following traumatic brain injury (TBI) remain a major cause of disability and early-onset dementia, and there is increasing evidence that chronic neuroinflammation occurring after TBI plays an important role in this process. However, little is known about the molecular mechanisms responsible for triggering and maintaining chronic inflammation after TBI. Here, we identify complement, and specifically complement-mediated microglial phagocytosis of synapses, as a pathophysiological link between acute insult and a chronic neurodegenerative response that is associated with cognitive decline. Three months after an initial insult, there is ongoing complement activation in the injured brain of male C57BL/6 mice, which drives a robust chronic neuroinflammatory response extending to both hemispheres. This chronic neuroinflammatory response promotes synaptic degeneration and predicts progressive cognitive decline. Synaptic degeneration was driven by microglial phagocytosis of complement-opsonized synapses in both the ipsilateral and contralateral brain, and complement inhibition interrupted the degenerative neuroinflammatory response and reversed cognitive decline, even when therapy was delayed until 2 months after TBI. These findings provide new insight into our understanding of TBI pathology and its management; and whereas previous therapeutic investigations have focused almost exclusively on acute treatments, we show that all phases of TBI, including at chronic time points after TBI, may be amenable to therapeutic interventions, and specifically to complement inhibition.
SIGNIFICANCE STATEMENT There is increasing evidence of a chronic neuroinflammatory response after traumatic brain injury (TBI), but little is known about the molecular mechanisms responsible for triggering and maintaining chronic inflammation. We identify complement, and specifically complement-mediated microglial phagocytosis of synapses, as a pathophysiological link between acute insult and a chronic neurodegenerative response, and further that this response is associated with cognitive decline. Complement inhibition interrupted this response and reversed cognitive decline, even when therapy was delayed until 2 months after injury. The data further support the concept that TBI should be considered a chronic rather than an acute disease condition, and have implications for the management of TBI in the chronic phase of injury, specifically with regard to the therapeutic application of complement inhibition.
Keywords: cognitive recovery, complement, neuroinflammation, therapy, traumatic brain injury
Introduction
Traumatic brain injury (TBI) remains a major cause of disability among young adults and is a major risk factor for early-onset dementia (Nordstrom et al., 2014). To date, acute management of TBI patients is limited to the treatment of cerebral edema, management of intracerebral hemorrhage and contusions, and motor rehabilitation. And although TBI patients are more likely to present with cognitive deficits later in the course of disease, little is known of the pathophysiological link between the initial acute insult and chronic neurodegeneration. Following an initial mechanical insult, TBI is characterized by a dynamic process of secondary injury that involves chronic neuroinflammation (Alawieh et al., 2018c), but the triggers of this chronic neuroinflammatory response remain unknown. Cumulative preclinical evidence suggests a role for the complement system in secondary injury acutely after TBI (Sewell et al., 2004; Stahel et al., 2009; Ruseva et al., 2015; Alawieh et al., 2018c), but a role for complement in a neuroinflammatory response that occurs in the chronic phases of injury has not been explored. Also unexplored are complement inhibitory strategies to modulate a neuroinflammatory response beyond the acute phase of TBI.
Complement can be activated via three pathways: the classical, lectin, and alternative pathway, with the latter also serving as an amplification loop. All pathways converge at C3 cleavage and activation, with the subsequent and sequential production of bioactive molecules, namely, C3 opsonins, C3a, C5a, and the cytolytic membrane attack complex (C5b-9). Using a toolbox of injury-site targeted complement inhibitors and an acute treatment paradigm (1 h after TBI), we previously demonstrated that the inhibition of all complement pathways at the C3 activation step was optimum to prevent the exacerbation of acute injury and to reduce subacute progression of neuroinflammation (Alawieh et al., 2018c).
In the current study, we used the complement inhibitor CR2Crry to investigate the role of complement in clinically relevant TBI paradigms. CR2Crry is a murine C3 inhibitor of all activation pathways that is targeted specifically to sites of complement activation (Atkinson et al., 2005). The CR2Crry construct is a fusion protein consisting of a complement receptor 2 (CR2) targeting moiety, which binds tissue-deposited C3 opsonins, linked to the complement inhibitor Crry. We investigated whether complement serves as a link between acute and chronic degenerative responses following TBI, and explored the complement-microglia-synaptic axis in the chronic phase of the disease. For this study, we used a controlled cortical impact (CCI) model of TBI to produce a moderate to severe injury, and which is a common, well-characterized, and reproducible model.
In this study, we also demonstrate that complement inhibition administered in the chronic phase after TBI interrupts a chronic neuroinflammatory response and reverses cognitive decline. These findings have significant implications in the management of the large worldwide cohort of TBI survivors who are in the early phases of post-traumatic cognitive decline. With regard to the potential clinical translation of a complement inhibitory strategy, it is worthwhile to note that multiple inhibitors that function at the C3 activation step are in various stages of clinical development, including inhibitors specifically targeted to sites of complement activation and injury.
Materials and Methods
Animals and study design
We used the CCI model to induce TBI in mice. This model has been previously described (Smith et al., 1995; Fox et al., 1998) and was implemented in our laboratory with a consistent and reliable pattern of postoperative motor and cognitive deficits (Alawieh et al., 2018c). Using a pneumatic impactor device (Infinite Horizon, Precision Scientific), contusions were delivered to the brain on intact dura. The impactor parameters included a 3 mm tip, a depth of 2.5 mm, velocity of 5.5 m/s, and dwell time of 100 ms. Impacts were delivered at a 10° angle relative to vertical axis. For sham animals, the same procedure was performed with the exception of impact deliver. Animals were excluded if the impact involves the bone rather than dura, which occurred in 2 animals; or if animals die before receiving treatment (vehicle or CR2-Crry), which occurred in 1 animal. Following impact, scalp incision was closed using sutures or clips and animals were allowed to recover in the surgical suite with access to soft food and water before being returned to their home cages. For complement inhibitor treatment, a dose of 10 mg/kg of CR2Crry diluted into 100 μl of PBS was used, as this dose was found to be optimal to suppress complement activity in acute studies (Alawieh et al., 2018c). In this work, CR2Crry (10 mg/kg) or vehicle (PBS) was administered as a single dose at 7 or 28 d after TBI, or as three total doses delivered as one dose every 48 h starting at 28 or 56 d after TBI. Treatment was administered intravenously via a tail vein injection. The recombinant protein CR2Crry was constructed, expressed, and purified as previously described (Atkinson et al., 2005). Before administration to animals, CR2Crry was tested for presence of endotoxin and for complement inhibitory activity using a zymosan assay (Atkinson et al., 2005).
Animal care
All vertebrate animal experiments were performed after approval by the Institutional Animal Care and Use Committee at the Medical University of South Carolina. Adult male C57BL/6 mice were used in all studies, and were housed at the Medical University of South Carolina Department of Laboratory Animal Research in Helicobater-negative rooms. Standard care of the animals included ad libitum access to regular chow food and water, 12 h:12 h light:dark cycles, and routine cage changes every 10-14 d. All mice were obtained from The Jackson Laboratory at 10-11 weeks of age, and were allowed to acclimate to new housing facility for 1 week. Following acclimation to housing facility, animals were randomly assigned to sham, vehicle, or CR2Crry groups. All animal behavioral testing and image quantification was performed using automated scripts applied to batch data that includes all groups, or by research personnel blinded to group assignments.
Blood–brain barrier (BBB) integrity
To assess for continued leakage of the BBB at chronic time points, a subset of animals received an intravenous injection of 2% Evans blue solution (Alawieh et al., 2018a) before death, and Evans blue fluorescence was then analyzed on histologic sections using confocal microscopy as described below.
Biodistribution studies
To study the tissue-specific distribution of CR2Crry after intravenous injection, CR2Crry was radiolabeled using 125I (PerkinElmer) using the IODO-GEN method (Pierce Biotechnology) following the manufacturer's instructions. A total of 5 mCi was used per 100 μg of CR2Crry protein as previously described (Atkinson et al., 2005). A therapeutic dose of CR2Crry (10 mg/kg) was spiked with 2% (w/w) of radiolabeled protein and administered via tail vein injection to sham or TBI animals at either 30 or 60 d after TBI. Blood and other organs were then harvested at 6 h after treatment. Organs were rinsed with PBS, weighed, and radioactivity counted using a Hewlett-Packard 5780 γ counter at the 125I window while correcting for count decays. Specific activity obtained for each tissue/organ was then corrected by the weight and reported as activity per gram of tissue.
Behavioral testing
A panel of motor and cognitive testing was used to evaluate recovery of deficits during the study period. The time points of testing varied based on the experimental design, as depicted in schematics associated with figures. Behavioral testing was staggered so only one cognitive task was performed on a given day, and the most stressful tasks were performed later. Animals were allowed to recover for at least 2 h between different tasks. The Corner task was performed as described by Zhang et al. (2002), and was used to measure forearm laterality. Laterality was assessed at baseline and then at designated time points after TBI. Normalized laterality index was then calculated as follows: [(laterality at time t) + 2]/[(baseline laterality) + 2)]. The ladder rung task was performed as described by Metz and Whishaw (2009). Animals were acclimated to run the ladder in the week before TBI induction. At the designated time points after TBI, animals were then placed on the ladder and encouraged to walk through the ladder rungs for three trials by placing the home cage at the other end of the ladder. Animals were video-recorded during the task, and videos were scored at low playback speed based on a reported scale of 1-6 per paw placement (Metz and Whishaw, 2009). Scores were obtained per forepaw and hindpaw, and then aggregate scores were reported. The Barnes maze task was performed as described by Alawieh et al. (2018a). Animals were trained on the task after their TBI to assess for the impact of TBI on the learning abilities, and then animals were tested for retention after learning to assess their retention of memory. Between animal testing, the maze was cleaned thoroughly to eliminate aromatic cues in finding the target box. The learning phase was carried over 5 d, and then animals were tested for retention at 48 h after training. The timing of the task was defined by the design of each experiment depending on the treatment group. The Passive avoidance task was performed as described by Alawieh et al. (2018a) using automated sensing and shock systems (GraphicState 4, Coulbourn Instruments). Training occurred over 2 d (one session per day) during which a shock was delivered when an animal entered the dark chamber. Animals were tested for retention of avoidance memory after 24 h of rest. The latency to enter the dark chamber was recorded as a measure of memory retention. The Novel object recognition task was performed as described by Hammond et al. (2004). Animals were assessed by video-recording and automated analysis on Noldus EthovisionXT system for the time spent exploring a new object compared with a familiar object.
Enriched environment exposure
To simulate motor and cognitive rehabilitation, a subset of animals were allocated to a rehabilitation group, which involve exposure to an enriched environment. Enriched environment cages were double-sized compared with standard housing, and included multiple ladders, running wheels, tubes, and platforms in addition to wooden toys. Toys and objects were changed twice weekly to maintain continuous exposure to novel stimuli. Animals assigned to standard housing were maintained in regular cages without access to additional enrichment.
MRI
MRI scans were acquired on day 60 after TBI for a subset of animals using a 7T/30 Bruker Biospec animal scanner as previously described (Alawieh et al., 2018b). The size of lesion was evaluated using T2-weighted images from 2-mm-thick sections. Imaging parameters included TR: 3750 ms, TE: 3750/32.5 ms, FOV: 30 mm × 30 mm, matrix: 128 × 128, and number of excitations: 2. For each animal, we acquired 15 axial slices without gaps at 2 mm thickness. Images were then exported and analyzed using National Institutes of Health ImageJ.
Histologic analyses
At designated time points of death, brains were isolated after intracardiac perfusion with ice-cold PBS followed by 4% PFA. Brains were then fixed overnight in 4% PFA, and then cryoprotected for 24 h in 30% sucrose. Brains were then cut into serial 40-µm-thick slices and stored in PBS at 4°C until analyzed. For assessment of lesion volume, serial sections of 40-µm-thick slices that were 200 µm apart were selected and stained using cresyl violet stain as previously described (Alawieh et al., 2018a). Slices were imaged at 10× using Olympus BX61 light microscope with Visiopharm image acquisition and analysis software (Visiopharm). To provide an unbiased approach to quantify lesion volume, we used our previously described approach of mapping areas of tissue loss and gliosis to the Paxinos' brain atlas, followed by 3D reconstruction of the brain with lesion volume (for additional details, see Alawieh et al., 2018a). Areas of tissue loss, as well as areas of dense gliosis on Nissl stain were outlined, mapped to the brain atlas, and 3D-reconstructed using Amira 2019 (Thermo Fisher Scientific) to visualize lesion and to compute volume.
Immunofluorescence (IF) staining
IF staining was performed on 40-µm-thick slices prepared as detailed above. Sections were chosen based on stereological coordinates (between –2.5 mm and 1.5 mm relative to bregma). From each animal, 3 or 4 full brain slices were selected from matching coordinates. We used a free-floating approach for staining. Sections were first cleaned with several rounds of PBS, and permeabilized using 3% H2O2 solution followed by 0.1% Triton-X in PBS. Sections were then blocked in donkey serum (5% in PBS), washed and incubated with primary antibody overnight followed by washing, and incubation with fluorescently tagged secondary antibody. The primary antibodies used in this study include the following: anti-C3d (R&D Systems, AF2655), anti-Iba1 (Abcam, ab5076), anti-NeuN (Abcam, ab104225), anti-Galectin-3 (R&D Systems, AF1197), anti-GFAP (Abcam, ab7260), anti-SV2 (Developmental Studies Hybridoma Bank), anti-MAP2 (Abcam, Ab32454), and anti-CD68 (Abcam, ab125212).
Epifluorescence imaging and quantification
For epifluorescence imaging (used in Fig. 1), whole-brain slices were stained as described above. Imaging was performed as tile scans at 10× magnification using Olympus BX61 light microscope with Visiopharm image acquisition and analysis software (Visiopharm). Intensity heat maps were then generated using in-house MATLAB (The MathWorks) script that normalizes the intensity at each pixel to the average in sham animals at the same location. Quantification of signal was performed using National Institutes of Health ImageJ. Full slices were imaged and quantified to avoid bias in field selection.
Figure 1.

Post-traumatic neuroinflammation persists and evolves during the chronic phase of TBI and predicts cognitive decline despite sustained motor recovery. A, Illustration of the location of brain impact in our TBI model. Red highlighted area represents area of primary impact. B, Extent of microgliosis (Galectin-3 IF) and astrocytosis (GFAP IF) in full coronal brain slices at 30 and 60 d after TBI showing evolution of the inflammatory response. C, Graphical illustration of the progression of astrocytosis in the murine brain after TBI over 60 d. Heat map reflects density of astrocytes normalized to density in sham brain. D,E,F, Quantification of (B) showing GFAP+ and Galectin-3+ cell density at different intervals after TBI. N = 4/group (6 sections each). **p < 0.01; ***p < 0.001; ANOVA. G,H, Performance on corner task (forearm laterality, N = 9-16/group) and ladder rung task (overall motor score, N = 8-10/group) at different time points after TBI in vehicle controls and sham controls. *p < 0.05 (ANOVA with Bonferroni). I, Avoidance learning was assessed using passive avoidance task by measuring the latency to enter a dark chamber associated with a shock in independent cohorts of animals at different time points after TBI or sham surgery. J, K, Barnes maze testing for spatial learning and memory performed by training the animals for 5 d to find escape hole on elevated maze, followed by assessing the retention of memory after 48 h of last session (day 7 of the task). We computed the path length (distance needed to find the escape hole) on each of the 5 training days and on the testing day (retention day). Two measures were obtained from the task: the path length on the retention day that reflects the retention of learned spatial memory, and the slope of improvement in path length during the 5 training days that reflects the difference in temporal dynamics of learning. The slope is a negative measure as it reflects the expected reduction of path length with additional training days. I, Path length to find escape hole on retention day, and (J) slope of path length during the 5 d of training, as assessed in an independent cohort of animals trained starting 27, 47, or 81 d after TBI. N = 9-12/group. *p < 0.05; **p < 0.01; ANOVA with Bonferroni. N = 6/group. *p < 0.05 (ANOVA with Bonferroni). L, To evaluate whether the performance of behavioral tasks correlated with the histologic extent of neuroinflammation, we analyzed the correlation matrix between the different behavioral tasks and the density of Galectin-3 deposition in the brain on day 60 after TBI. R2 for each correlation is shown on heat map. All correlations with Galectin-3 area were significant with p < 0.001 and |R2|>0.5.
Confocal and super-resolution microscopy
Following IF staining as detailed above, confocal microscopy was performed using the Carl Zeiss LSM 880 microscope with Airy-scan super-resolution feature (resolution of 140 nm in xy axis and 400 nm in z axis). Scanning was performed at 40× magnification for high-power fields, and at 20× magnification for tile scans. Tile scans were first acquired for perilesional and contralateral brain to determine the relative location in the slices that matched predetermined stereological coordinates for high field selection. Unbiased stereology was then used in high-power field selection by identifying zones within the tiles that match the same stereological coordinates across the different animals. Image processing and quantification were then performed using the ZEN Blue and ZEN black software (Carl Zeiss). For quantification of signal intensity, mean intensity per field was computed. An average of three fields per animal was used as individual count for statistical analysis. 3D rendering of high-power fields was performed using Amira 2019 (Thermo Fisher Scientific) using transparency to allow for overlap detection. For quantification of synaptic density, several measures were used. We defined the synaptic volume as the percentage by volume of the synaptic volume relative to the overall tissue volume in the slice. The tissue volume was computed by subtracting the empty space volume in each field from the total slice volume. Synaptic volume was computed using Amira 2019 from the reconstructed 3D volumes obtained from SV-2+ staining. The volume of Iba1+ cells was similarly computed. The phagocytic index for microglia was defined as the percentage by volume of SV-2+ material overlapping within Iba1+ cells to that of the volume of Iba+ cells. This was achieved by creating a mask using the Iba1+ volume and subtracting SV-2 signal outside this volume to detect SV-2 signal that was limited to within the Iba1+ volume. To evaluate the impact of microgliosis on perilesional synaptic density, we calculated the density of synapses at sequential distance from the edge of gliotic scar. A series of spatially adherent and nonoverlapping high-power fields were acquired starting from the edge of the gliotic scar and for an approximate 0.6 mm distance. A histogram of SV-2 signal was then generated for the distance midline of the imaged volume, and 2D-signal histograms were used to quantify the synaptic density measured via the peaks in histogram and overall intensity level. Sham animals were used to determine synaptic density in noninjured brains. We defined the recovery of synaptic density for each animal as the distance at which the density in CCI animals achieved >67% that of sham animals. This was computed from the signal histograms. Additionally, we performed continuous wavelet analysis using MATLAB (The MathWorks) to dissect the change in overall intensity and frequency of synaptic signal. The output of the algorithm is a magnitude scalogram that shows the overall difference in signal intensity (overall synaptic signal) and the frequency of synaptic density, where a shift in power (higher signal intensity on higher frequency) indicates an overall increase in synaptic density independent of signal from individual synapses.
In addition to quantification of synaptic volume, we used spots analysis in Imaris 9.3 (Bitplane) to quantify individual synaptic counts. Using Imaris script with uniform settings across all slices, we computed the overall density of spots (synaptic counts) per slice and the number of spots localizing to the Iba1+ volume. To quantify the morphology of microglia (Iba1+ cells), we performed filament analysis using Amira 2019 (Thermo Fisher Scientific) to capture the backbone architecture of the microglia by downsampling individual cells into line segments. Then the cumulative length of each segment was normalized to the volume of microglia. Based on this metric, change in spectrum from branching to amoeboid morphology can be detected via a reduction in filament length to glial volume ratio.
IF imaging of C3 deposition on synapses was performed using super-resolution microscopy via the Vutara 352 system (Bruker). Following staining, brain slices were fixed to the imaging platform with gelatin, followed by imaging of 40 µm 3D volume using Alexa-Fluor-647 and Alexa-Fluor-568 (Thermo Fisher Scientific) as secondary antibodies. Both confocal and super-resolution imaging was acquired for each section. Colocalization of signal was then identified via signal histograms at the same depth.
Statistics
Statistical analysis was performed using Prism 8 (GraphPad), SPSS version 25 (IBM), or MATLAB 2019 (The MathWorks). Power analysis for sample size estimation was performed using G*Power 3.1 (Faul et al., 2007), and effect size was estimated based on prior work from our team (Alawieh et al., 2018c) and using a power of 80% and a significance level <5% corrected for multiple comparisons whenever appropriate. Pearson's correlation coefficient was used to assess correlation of histologic and behavioral data. p value and R2 were used to evaluate each correlation. Detailed statistical tests used for each graph are presented in the figure legends.
Results
TBI induces an evolving chronic global cerebral neuroinflammatory response that is associated with worsening cognitive performance
Using moderately severe CCI targeted to the parietal lobe (refer to Fig. 1A), we assessed the progression of astrocytosis and microgliosis in the injured brain as a surrogate of the neuroinflammatory response over 60 d of recovery. Analysis of coronal sections at the level of the motor cortex and hippocampus revealed that both microgliosis and astrogliosis continue to expand over 60 d after the initial insult, as illustrated by a significant increase in the area occupied by GFAP+ astrocytes and Galectin-3+ microglia (Fig. 1B–E). Galectin-3 and Iba-1, also used as a marker in this study, are expressed by both microglia and macrophages; and while we use the term microglia, our study does not differentiate between the two cell types. Galectin-3 preferentially marks activated and proliferating microglia, compared with Iba1 that marks both activated and resting microglia (Lalancette-Hebert et al., 2012; Alawieh et al., 2018a). This neuroinflammatory response was limited to the perilesional brain acutely (7 d), but expanded over 30-60 d to involve both the cortex and hippocampus bilaterally (Fig. 1B–E). Microgliosis appears to stabilize by 30 d, whereas astrocytosis continues to expand over 60 d after TBI (Fig. 1D,E).
We performed a battery of behavioral tasks over 90 d of recovery to determine whether there was a correlation between motor or cognitive performance and the neuroinflammatory response. Motor performance, as assessed by corner task and ladder rung task, showed significant improvement over time between 14 and 90 d after TBI (Fig. 1G,H). Forearm laterality on corner task showed a rapid improvement (50%) within the first 30 d after TBI, followed by a slower recovery of residual deficits by 90 d (Fig. 1G). Motor performance on the ladder rung task showed continued steady improvement over 90 d of recovery (Fig. 1H). In contrast to the improvements seen in motor tasks over time, cognitive performance as assessed by passive avoidance and Barnes maze showed worsening cognitive deficits over time (Fig. 1I–K). Animals tested on passive avoidance task at 90 d showed a worsening of fear conditioning memory compared with 30 d after TBI (Fig. 1I). For Barnes maze, compared with 30 d after TBI, animals tested at 90 d showed significantly longer path length to find the escape hole during retention task, and had a significantly lower learning slope during the training phase (Fig. 1J,K). Similarly, performance on cognitive tasks inversely correlated with the area of galectin-3 immunostaining in the brain at 60 d after CCI (Fig. 1L; R2 = 0.52-0.62, p < 0.001). Thus, despite an improvement in motor deficits over 90 d after TBI, cognitive performance continued to worsen in recovering mice, which correlated with the extent of ongoing neuroinflammation in the brain.
Complement activation persists in the chronic phase of TBI
We next assessed whether the continued neuroinflammatory response is associated with continued complement activation in the brain by assessing deposition of the cleavage product, C3d. Minimal C3d deposition was observed in brains from sham controls, whereas there was prominent C3d deposition in the perilesional brain at 60 d after TBI (Fig. 2A). C3d colocalized with neurons and dendrites, as well as non-neuronal cell types in the perilesional hippocampus (Fig. 2A). Areas of microgliosis were also associated with a persistent breach in BBB integrity at 60 d, as demonstrated by Evans blue extravasation (Fig. 2B).
Figure 2.
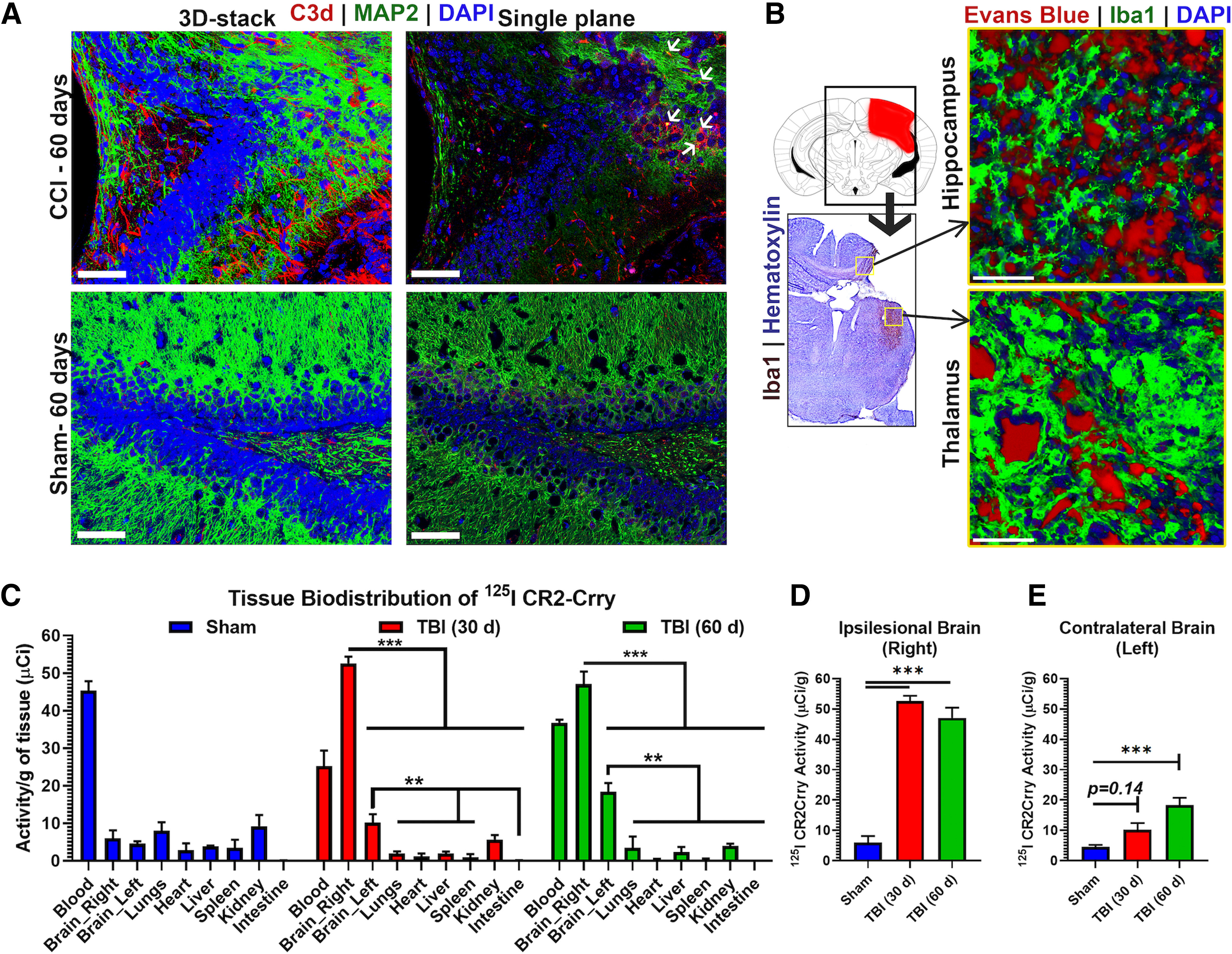
Ongoing complement deposition and BBB leakage 60 d after murine TBI. A, IF staining in the perilesional brain 60 d after TBI showing robust complement deposition (red) in injured but not sham brains. Scale bars, 50 µm. Left, Rendered 3D-stack covering 40 μm volume. Right, Single-slice view showing colocalization of C3d with MAP2+ neurons (green). B, Coimmunostaining for Iba1 (microglial marker, green) and Evans blue (red) administered intravenously before brain harvest showing BBB leakage and Evans blue extravasation in areas of microgliosis in the perilesional hippocampus and thalamus in TBI mice but not in sham controls. Scale bars, 50 µm. C, Biodistribution study for localization of CR2Crry in sham animals and animals subjected to TBI. 125I CR2Crry injected in sham animals or TBI animals at 30 or 60 d after injury, followed by tissue analysis for 125I activity at 6 h after administration. N = 3/group. **p < 0.01; ***p < 0.001; ANOVA with Bonferroni. D, E, Comparison of CR2Crry localization to the ipsilateral and contralateral hemisphere in TBI animals at 30 and 60 d after TBI compared with sham. ANOVA with Bonferroni. N = 3/group. ***p < 0.001.
We additionally performed a biodistribution study to investigate specific localization of C3d to the injured brain. CR2Crry, a targeted complement inhibitor that binds the C3d activation product, was radiolabeled to assess its localization within the brain, as well as to other organs and tissues, at chronic time points after TBI. 125I-labeled CR2Crry was injected intravenously to sham animals or to animals at either 30 or 60 d after TBI, and 125I activity in different tissues determined 6 h after administration (Fig. 2C). In sham controls, high levels of CR2Crry was present in the blood compared with all other tissues analyzed, including the brain (Fig. 2C). However, at both 30 and 60 d after TBI, CR2Crry bound at significantly higher levels in the ipsilateral (right) brain hemisphere compared with the left hemisphere and other organs (Fig. 2C). CR2Crry was also significantly higher in the left (contralateral) hemisphere compared with other organs. There was no significant difference between the amount of CR2Crry that bound in the ipsilateral hemisphere when injected at either 30 or 60 d after TBI (Fig. 2D,E). The contralateral hemisphere had significantly higher CR2Crry localization compared with sham at 60 but not 30 d after TBI, reflecting an expanding zone of complement activation and neuroinflammation over time. Collectively, these findings show that a neuroinflammatory response that includes complement activation, microgliosis, and astrogliosis continues to expand 60 d after unilateral TBI to involve both cerebral hemispheres. Furthermore, as a therapeutic consideration, CR2Crry specifically targets regions of complement deposition in the brain as long as 60 d after TBI (refer Fig. 2C).
A role for complement in the chronic neuroinflammatory response and cognitive decline
Complement inhibition has been shown to be protective in TBI models when applied acutely (up to 1 h) after injury. Based on the above data, we investigated administration of CR2Crry at 1 and 2 months after TBI. A single dose of CR2Crry administered 30 d after TBI did not significantly reduce overall perilesional C3d deposition compared with vehicle-treated animals as measured 1 week after treatment (Fig. 3A). However, when CR2Crry was administered every 48 h over a 1 week period (three doses) starting 30 d after TBI, there was a significant reduction in C3d deposition at 1 week after initiation of treatment, with C3d levels not significantly different from that seen in sham animals (Fig. 3A,C). A similar outcome was seen in mice treated with three doses of CR2Crry starting 2 months after TBI (Fig. 3B,D).
Figure 3.
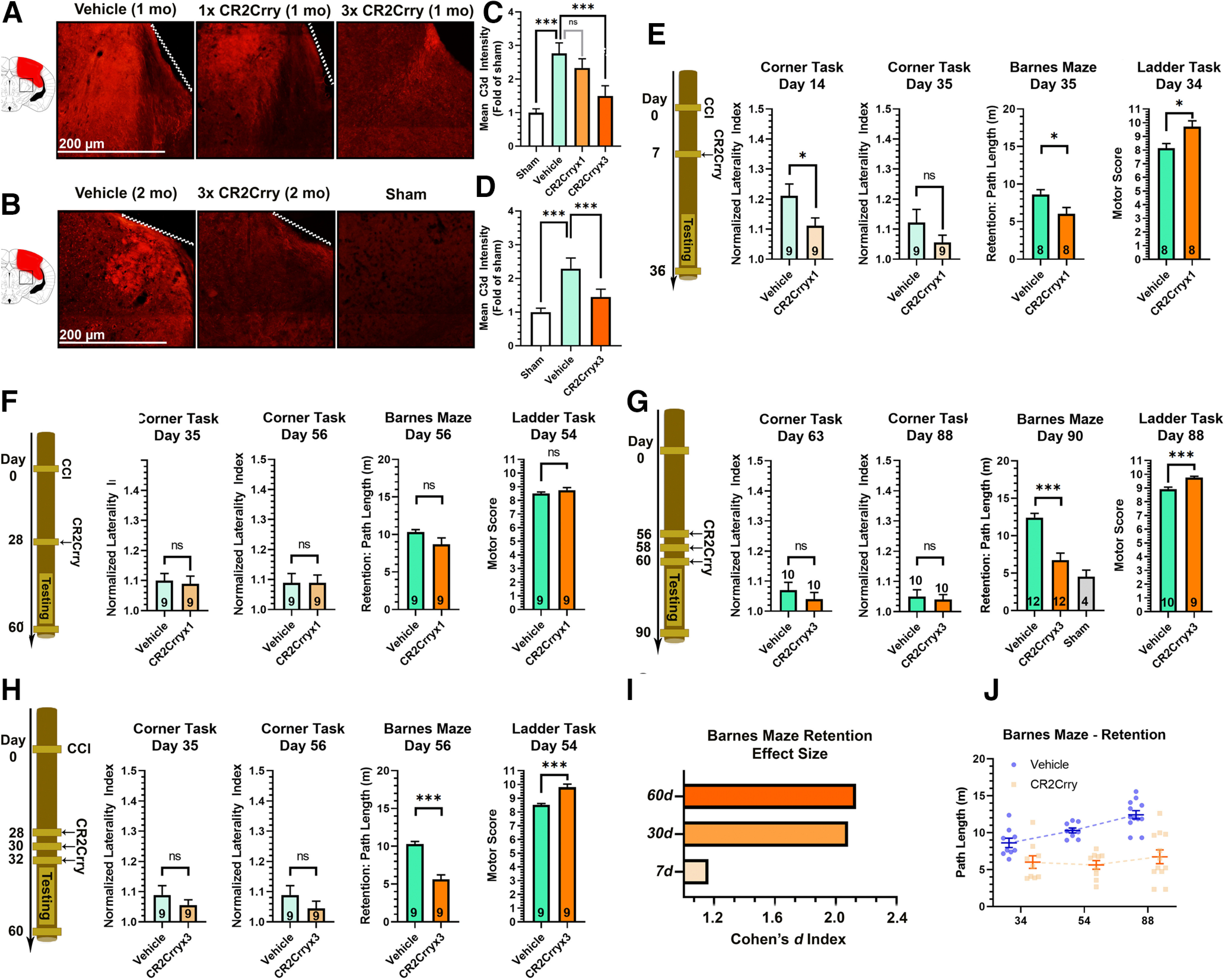
Chronically administered CR2Crry suppresses C3d deposition in the brain after TBI. A, B, IF staining for C3d deposition in the perilesional brain following TBI and administration of CR2Crry or vehicle. A, Mice were treated at 30 d after TBI with a single dose of PBS vehicle of CR2Crry, or with 3 doses of CR2Crry every 48 h. C3d deposition was measured 1 week after initiation of treatment. B, Mice were treated at 2 months after TBI with 3 doses of PBS or CR2Crry every 48 h. C3d deposition was measured 1 week after initiation of treatment. Fields are 250 µm × 250 µm, representative images. C, D, Quantification of C3d deposition in A, B, using mean IF intensity compared with sham using unbiased stereology. N = 5 animals/group (2 or 3 slices per mouse). ***p < 0.001 (ANOVA with Bonferroni). E–H, Motor and cognitive outcomes measures, as indicated, after vehicle or CR2Crry treatment. E, Single-dose CR2Crry treatment at 7 d after TBI. F, Single-dose CR2Crry treatment at 28 d after TBI. G, Three doses of CR2Crry treatment every 48 h starting 28 d after TBI. H, Three doses of CR2Crry treatment every 48 h starting 56 d after TBI. Student's t test. N values shown on bars. *p < 0.05. ***p < 0.001. I, Effect size of CR2Crry treatment on Barnes maze performance compared with vehicle at different time points of administration measured via the Cohen's d index. J, Change in path length on retention day at different time points after TBI between vehicle and CR2Crry-treated animals. N = 8-12/group. ns = p > 0.05.
We next investigated whether there was a link between complement-dependent neuroinflammation and motor and/or cognitive deficits that occur in the chronic phase after TBI. CR2Crry was administered as a single dose on day 7 (Fig. 3E) or day 28 (Fig. 3F) after TBI, or as three doses 48 h apart starting on either day 28 or day 56 after TBI (Fig. 3G,H). Outcomes were assessed over a period of 4 weeks after treatment. In terms of the therapeutic window with a single-dose treatment with CR2Crry, we showed that a single dose administered at 7 d after TBI improved corner test performance compared with vehicle at day 14, but not at day 35 (Fig. 3E). The same 7 d single-treatment paradigm significantly improved spatial learning and memory on Barnes maze task and improved overall motor score on ladder rung task (Fig. 3E). However, with the single 28 d dose of CR2Crry, there was no improvement compared with controls in any of the tests (Fig. 3F). Given the expected amplification of complement activity in the brain in the more chronic phases of injury, together with the finding that a single dose of CR2Crry was not sufficient to reduce complement activity at 30 d, we assessed whether additional dosing of CR2Crry would achieve behavioral benefit at 1 or 2 months after TBI. In this context, animals treated with three doses of CR2Crry starting on day 28 showed significant improvement on spatial memory retention and ladder task performance (Fig. 3G). The improved outcome measures with 28 d CR2Crry treatment correlated with C3d deposition, which was reduced by treatment with three doses of CR2Crry, but not with a single dose (Fig. 3A–D). There was a similar improvement in Barnes maze and ladder task performance when three doses of CR2Crry were administered starting at 56 d after TBI (Fig. 3H). Furthermore, in contrast to the decline in cognitive performance in vehicle controls over time, the effect size of CR2Crry on Barnes maze retention memory increased with more delayed administration, which was predominantly secondary to worsening deficits in the vehicle group (Fig. 3I,J). Performance on Barnes maze in animals tested at 90 d following 3 × CR2Crry treatments starting at 2 months after TBI was comparable with animals tested on day 35 following treatment at 1 week. There were no differences in survival between vehicle and CR2Crry in all treatment cohorts (p > 0.1). Finally, when comparing performance on Barnes maze at 90 d in animals treated with CR2Crry starting on day 56 with those of vehicle controls, CR2Crry-treated mice had improved performance (shorter path lengths) at 90 d compared with the vehicle controls (compare Fig. 3E and Fig. 3F; p < 0.05). These findings indicate the suppressing complement activity in the brain reversed cognitive decline following TBI in recovering mice.
Rehabilitation treatment augments but does not substitute for the efficacy of complement inhibition in the chronic phase after TBI
Rehabilitation therapy is currently the only available intervention for motor and cognitive deficits after TBI in the postacute phase (Salazar et al., 2000; Zhu et al., 2007; Cicerone et al., 2019). We therefore evaluated cognitive recovery after rehabilitation therapy, and determined whether complement inhibition could augment the response to rehabilitation therapy. To model rehabilitation, we used a combination of enriched environment exposure and passive physical activity. Animals were assigned to rehabilitation cages versus regular housing starting at 2 months after TBI to coincide with timing of complement inhibition. When performance on Barnes maze was evaluated, there was no significant difference in the slope of learning or retention of learned spatial memory between rehabilitation and standard housing groups; however, CR2Crry treatment alone or in combination with rehabilitation significantly improved outcomes (Fig. 4A–D). There were no differences in total ambulation time between the different groups on open field testing (Fig. 4E). We also used the novel-object recognition (NOR) to assess for impairment in recognition memory in the chronic phase after TBI. Compared with sham animals, there was a significant reduction in time spent near a novel object in vehicle controls or animals exposed to rehabilitation alone. In contrast, there was no significant difference between sham controls and animals treated with CR2Crry or CR2Crry with rehabilitation (Fig. 4F). Notably, complete recovery of recognition memory was only observed in animals treated with combination therapy, suggesting an additive effect of rehabilitation on recovery of recognition memory at 90 d after TBI. Thus, rehabilitation alone did not improve cognitive performance on spatial learning, spatial memory, and novel object recognition in the chronic phase after TBI, but augmented the effects of complement inhibition.
Figure 4.

Rehabilitation therapy does not reverse cognitive decline after TBI. A, Schematic of treatment and testing paradigm. Following TBI, mice were treated with 3 doses of CR2Crry or vehicle and exposed to either 30 d of enriched environment (motor and cognitive enrichment, rehabilitation) or standard housing, with assessment for performance on motor and cognitive tasks at 90 d. Dashed line indicates border of injury site. B, Spatial learning and retention of spatial memory on Barnes maze among mice treated with different combinations of CR2Crry, vehicle, and rehabilitation. N = 12/group. *p < 0.05; **p < 0.01; repeated-measures ANOVA with Bonferroni. C, D, Comparison of path length and number of error pokes (not in target hole) on retention day between the different groups. N = 12/group. **p < 0.01; ***p < 0.001; ANOVA with Bonferroni. E, Comparison of total ambulation distance on open field testing of animals from the different groups. ANOVA with Bonferroni. N = 12/group. F, Performance on NOR task between the different treatment groups compared with sham. Percentage time spent near novel versus familiar object. N = 7/group. *p < 0.05; **p < 0.01; ANOVA with Bonferroni. G, Motor score on ladder rung task at different time points following TBI. Repeated-measures ANOVA with Bonferroni. N = 9/group.
We also assessed motor function over 90 d of recovery after TBI using the ladder rung task. Both rehabilitation and CR2Crry alone similarly improved motor scores compared with vehicle, but combined treatment improved recovery compared with rehabilitation alone (Fig. 4G). CR2Crry did not have a cognitive enhancing effect in sham animals as measured by Barnes maze task (not shown).
Complement inhibition at chronic time points after TBI does not alter lesion volume but does suppress an ongoing neuroinflammatory response
Prior work suggests that acute suppression of the inflammatory response limits the expansion of tissue loss and lesion volume after TBI (Rancan et al., 2003; Ruseva et al., 2015; Rich et al., 2016; Alawieh et al., 2018b). Therefore, we investigated whether improvement in cognitive performance with complement inhibition administered at chronic time points after TBI can be explained by the extent of tissue loss. Volume reconstruction of Nissl-stained brains from mice at 90 d after TBI showed that chronically administered complement inhibition (three doses starting 56 after TBI) did not impact lesion volume (Fig. 5A–C). However, the extent of gliosis manifesting as increased Nissl density in the perilesional brain was inhibited with chronic CR2Crry treatment (Fig. 5C). MRI of brains at 60 d after TBI similarly showed no difference in the extent of tissue loss between vehicle- and CR2Crry-treated mice (Fig. 5D), but high-resolution IF staining for microglia activity demonstrated that chronic administration of CR2Crry significantly reduced the density and volume of microgliosis in the ipsilateral perilesional brain at 90 d after (Fig. 5E–G TBI. CR2Crry also reduced the extent of microgliosis in the contralateral hemisphere, thus halting the global progression of a neuroinflammatory response in the chronic phase after TBI (Fig. 5F,G). These findings further indicate that the underlying mechanism of complement-dependent cognitive decline is dependent on an evolving neuroinflammatory response.
Figure 5.

CR2Crry inhibits gliosis but not lesion volume after TBI. A, Representative Nissl immunostaining of animals treated with vehicle or CR2Crry starting day 56 (3 doses every 48 h). Brains were extracted on day 90 after TBI for analysis. Red contour represents area of tissue loss. Orange contour represents area of gliosis. B, 3D reconstruction of lesion volume from Nissl-stained serial sections (40 µm thick, 200 µm apart) using Amira to show volumes of tissue loss and gliosis. C, Quantification of volume of tissue loss and gliosis shown in B. N = 8/group. *p < 0.05 (Student's t test). D, Representative T2-weighted MRI images of animals treated as in A showing similar area of tissue loss (T2 Bright) between vehicle and CR2-Crry treated mice at 1 week after treatment. E, F, High-resolution IF staining for perilesional ipsilateral (E) and contralateral (F) cortex and hippocampus showing density of Iba1+ cells (red). G, Quantification of IF staining in E and F. N = 5 animals/group (2 or 3 sections each). *p < 0.05; **p < 0.01; Student's t test.
Complement activation promotes the loss of synaptic density in the perilesional and contralateral brain following TBI
Cognitive and motor dysfunction after TBI reflects an ongoing neurodegenerative process, and behavioral manifestations of cognitive deficits are likely secondary to loss of neurons or synaptic connections. Therefore, we investigated whether complement-driven neuroinflammation and cognitive decline was associated with the loss of synaptic density as determined by SV2 immunoreactivity, which will similarly mark excitatory and inhibitory synapses. At 90 d after TBI, there was an expected reduction in synaptic density in the perilesional area of microgliosis in both CR2Crry and vehicle control treated animals compared with sham (Fig. 6A). However, the overall synaptic density in the ipsilateral hemisphere, measured by the cumulative volume of all SV-2+ clusters, was significantly higher in CR2Crry-treated compared with vehicle-treated mice (Fig. 6A,B). Whereas areas within the perilesional scar showed loss of synaptic density in TBI animals, synaptic density increased as a function of distance from the edge of gliotic scar (Fig. 6C). To evaluate whether complement inhibition prevents the perilesional loss of synaptic density and expansion of neurodegenerative response beyond the immediate area of injury, we measured synaptic density as function of SV-2 signal intensity over distance from the edge of gliotic scar (Fig. 6C,D). Complement inhibition reduced the extent of synaptic loss as demonstrated by earlier spatial recovery of synaptic density CR2Crry-treated animals compared with sham (Fig. 6C,D). To assess the change in synaptic density over distance, we performed continuous wavelet transformation using MATLAB (see Materials and Methods) to distinguish the change in intensity of SV-2 expression versus the number of synaptic puncta as a function of distance from the perilesional scar (Fig. 6E). Compared with vehicle controls, CR2Crry-treated animals showed a significantly higher intensity of synaptic staining compared with vehicle at distances of between 100 and 500 μm from the edge of the scar (Fig. 6E). However, an increase in the number of synaptic puncta manifests on the magnitude scalogram as a shift in the intensity (yellow range) toward higher frequencies. Higher intensity in the higher-frequency range corresponded to higher density of synaptic puncta, in contrast to higher intensity of SV-2 expression. In CR2-Crry-treated brains, the SV-2 signal was distributed among a higher range of frequencies involving higher frequencies of synaptic spikes (0.5-1 kHz range) compared with vehicle, indicating an overall increase in synaptic density in CR2Crry-treated animals (Fig. 6E). When evaluating the change in synaptic density over distance from injury site defined by the edge of the scar, we also observed a faster (shorter distance) recovery of the intensity of synaptic signal in CR2Crry compared with vehicle (Fig. 6E, black arrow). Compared with vehicle-treated animals, CR2Crry-treated animals recovered 67% of normal (sham) synaptic density at a distance that was ∼250 μm closer to the perilesional scar (p < 0.01; Fig. 6F). This was associated with higher overall synaptic density in the perilesional brain of CR2Crry versus vehicle-treated mice (Fig. 6F).
Figure 6.

Complement inhibition at 2 months after TBI prevents ongoing synaptic loss. Animals were subjected to TBI at 12 weeks of age, and treated over 1 week with 3 doses of CR2Crry or vehicle every other day starting 2 months after TBI. Analyses were performed at 3 months after TBI. A, Confocal imaging of 1.2 mm × 1 mm × 0.04 mm IF fields from the perilesional brain of sham, or vehicle and CR2-Crry treated mice stained for synaptic bodies (SV-2, green), microglia/macrophages (Iba1, red), and DAPI (blue). Shown are 3D fields reconstructed using Amira from the same stereotactic lesion. Loss of hippocampal architecture is evident in post-TBI brains. B, The total synaptic volume was quantified, using Amira 3D volume reconstruction, as a percentage of synaptic volume per tissue volume. N = 5 animals/group. *p < 0.05; **p < 0.01; Welch's ANOVA test with Dunnett's multiple comparisons. C, Synapse (SV-2, green) and microglia (Iba1, red) staining of sections selected from the perilesional brain starting from the edge of the scar over a 0.5 mm length. Representative images showing 3D stack of 40-μm-thick sections, imaged using confocal microscopy. D, Top, Histogram showing changes in synaptic signal intensity as a function of distance from gliotic scar edge (red marker), from vehicle and CR2-Crry-treated animals. Shown are mean ± SEM per distance data point. N = 5 animals/group. Bottom, Smoothed curve of the change in synaptic density over distance between groups. Dashed line indicates sham density. Curve indicates earlier normalization of synaptic intensity in CR2Crry-treated mice compared with vehicle. E, Magnitude scalogram for data from D, showing the intensity (power) of synaptic signal over distance from gliotic scar in terms of different frequencies of peaks (spikes in histogram denoting individual synapses). Scalograms represent a clearly higher intensity in CR2Crry-treated animals across higher frequencies compared with vehicle. Black arrow indicates distance at which synaptic density recovers 67% intensity compared with that in sham animals. F, Quantification of synaptic recovery measured as the distance to recover 67% of synaptic density, and the overall synaptic density across the two groups. N = 5 animals/group. **p < 0.01 (Student's t test). G, High-resolution 3D reconstruction of the contralateral hippocampus from confocal imaging of SV-2 and Iba1-stained slices at 90 d after TBI. H, Quantification of Iba1 cell density and synaptic volume from vehicle and CR2-Crry-treated mice. N = 5 animals/group. **p < 0.01; ***p < 0.001; Student's t test. I–K, Correlation of ipsilateral synaptic volume with performance on Barnes maze on day 90 (I) and performance on NOR task on day 90 (J). K, Correlation of synaptic volume in contralateral hippocampus with Barnes maze performance. Shown on graph are R2, p value, and N for Pearson's coefficient for linear correlation. Error bars indicate mean ± SEM. Blue circles represent vehicle. Green circles represent CR2Crry.
Since we demonstrated that an ongoing neuroinflammatory response expands to the contralateral hemisphere following TBI (Fig. 1), we assessed how the expansion of this inflammatory response affects synaptic density in the contralateral hemisphere. The contralateral hippocampus of vehicle controls showed a significantly higher density of Iba1+ cells compared with the contralateral hippocampus in CR2Crry-treated mice, and higher density of Iba1+ cells correlated with lower overall synaptic volume (Fig. 6G,H). These findings indicate that the progression of neuroinflammation in the brain following TBI is associated with a degenerative response that is inhibited with CR2Crry treatment. We also assessed whether synaptic loss correlated with worse performance on cognitive tasks at 90 d after TBI. Ipsilateral synaptic density positively correlated with performance on NOR task, but not with Barnes maze performance (Fig. 6I,J). Performance on Barnes maze did, however, correlate with synaptic density in the contralateral hippocampus (Fig. 6K).
Complement inhibition prevents synaptic uptake by microglia in ipsilateral and contralateral brain
Given the inverse association between synaptic density and microglial activation in the perilesional brain, we investigated whether microglial activity is associated with phagocytosis of synaptic connections. Using high-resolution microscopy, we compared with occurrence of synaptic bodies (SV2+ puncta) within Iba1+ cell bodies (Fig. 7). Super-resolution imaging and reconstruction of high-resolution stacks were used to compute the phagocytic index as the volume of synaptic material per volume of microglia (Fig. 7A,B). There was a significant reduction in the phagocytic index in the perilesional hippocampus of CR2Crry-treated mice compared with vehicle-treated mice; the difference did not reach significance in the perilesional cortex (Fig. 7B). A similar effect was also observed in the contralateral hippocampus, where CR2Crry treatment reduced microglial uptake of synaptic material. Using Imaris, we performed spot analysis by automated capture of synaptic puncta in a 3D tissue volume, with measurement of overall density of synaptic puncta (Fig. 7C). There was a significant reduction in the density of SV-2 spots within microglia with CR2Crry compared with vehicle treatment (Fig. 7C,D). To correct for the total density of synapses in each field, we also calculated the percentage of total microglial spots that were localized within Iba1+ cells, and found a similar reduction in colocalized spots with CR2Crry compared with vehicle treatment (Fig. 7D). Analysis by phagocytic index (volume of synaptic material per volume of microglia) revealed similar CR2Crry versus vehicle differences (Fig. 7E,F). There was also a significant inverse correlation between phagocytic index and synaptic volume per slice (R2 = 0.64, p < 0.01; Fig. 7F), further supporting the hypothesis that synaptic uptake by microglia underlies the degenerative loss of synapses in the perilesional and contralateral brain. Since prior work suggested that amoeboid morphology is indicative of upregulated phagocytic machinery (Li and Barres, 2018), we used Amira to decode the architecture of individual microglial cells, and computed the length of branches relative to cell volume. Amoeboid cells show shorter branches and thus have a lower filament length to volume ratio. Microglia in the contralateral hippocampus of CR2Crry-treated mice showed a higher filament length to volume ratio, indicative of a more branched and less amoeboid morphology (Fig. 7G). There was also a significant inverse correlation between the phagocytic index of microglia and the filament to volume ratio (R2 = 0.44, p < 0.01; Fig. 7G–I). For further confirmation of microglial phagocytosis of synapses, we costained for CD68 (phagolysosomal marker) and SV-2, and demonstrated that, within microglial cell bodies, both signals colocalize (Fig. 7J). The above findings collectively indicate that, following TBI, synaptic phagocytosis by activated microglia within the perilesional and contralateral brain predicts cognitive impairment chronically. Although this process persisted for several months after TBI, complement inhibition, even when administered at chronic time points after TBI, suppressed microglial activity and prevented the expansion of a neuroinflammatory degenerative response, with resultant preservation of synaptic connectivity and reversal of cognitive decline.
Figure 7.

Complement guides microglial phagocytosis of synapses chronically after TBI. Animals were subjected to TBI at 12 weeks of age, and treated over 1 week with 3 doses of CR2Crry or vehicle every other day starting 2 months after TBI. Analyses were performed at 3 months after TBI. A, Super-resolution IF staining of SV-2 and Iba1 in the perilesional brain showing colocalization of synaptic material to microglial cell bodies. Shown are the 2D snapshots of the Iba1 volume (red) and SV-2 (green) signal (left) and the 3D volume of microglia showing synaptic material within the Iba1 volume (right) from both cortex and hippocampus. B, Quantification of the phagocytic index (volume of SV-2 signal within Iba1 cells to volume of Iba1 cells) from the cortex and hippocampus of both groups. N = 5 animals/group. *p < 0.05 (Student's t test). C, Confocal IF imaging of the contralateral hippocampus stained for SV-2 and Iba1 (left column) followed by automated 3D reconstruction and spot analysis by Imaris representing synapses as spots in 3D space (right column; see Materials and Methods). D, Quantification of synaptic puncta or spots within microglia cells from C. N = 7 animals/group. *p < 0.05; **p < 0.01; Student's t test. E, Representative 3D reconstruction of high-power fields from contralateral hippocampus followed by selection and reconstruction of individual microglia with internalized synaptic material: green represents SV-2; red represents Iba1; blue represents DAPI. Fields are 212 × 212 × 40 μm. F, Quantification of phagocytic index in contralateral hippocampus. N = 7 animals/group. **p < 0.01 (Student's t test). G, Representative high-power fields from the contralateral hippocampus stained for Iba1 followed by filament analysis in Imaris to assess for extent and length of branching per microglia. H, Quantification of the length of microglial branching in G, normalized to the microglial volume. N = 7 animals/group. *p < 0.05 (Student's t test). I, Correlation of length of microglial branching and phagocytic index. Pearson's correlation, R2 = 0.44, p < 0.01. J, IF staining for Iba1 (red), CD68 (yellow), and SV-2 (green) followed by reconstruction of individual microglia in Amira showing colocalization of CD68 and SV-2 signal. Clusters of CD68-SV-2 colocalization were observed in vehicle controls and not in CR2Crry-treated mice. K, L, Confocal and super-resolution imaging of SV-2 and C3d colocalization using Vutara 352 super-resolution microscopy. M, Quantification of intensity histogram for both SV-2 and C3d signals. Shaded region represents background signal. N, Quantification of colocalization of peaks in M between the two experimental groups. N = 5 animals/group. **p < 0.05 (Student's t test). Error bars indicate mean ± SEM.
Finally, to demonstrate a direct interaction between complement and synaptic bodies, we investigated C3d deposition on SV-2+ material in the perilesional brain using super-resolution imaging with Vutara 352. At 90 d after TBI, C3d deposits within the perilesional brain colocalized with SV-2 material. CR2Crry administered 2 months after TBI significantly reduced synaptic C3d deposition (Fig. 7K–N).
Discussion
TBI is a major risk factor for chronic cognitive decline (Barnes et al., 2014, 2018), and there are no available pharmacological interventions that can stop or reverse cognitive decline in TBI patients (Johnson et al., 2010; Breunig et al., 2013; Barnes et al., 2014). Previous work has established a role for complement in triggering an acute inflammatory response after TBI (Kossmann et al., 1997; Bellander et al., 2001, 2011; Stahel et al., 2001), but little is known about whether/how this response continues to expand chronically. Here we demonstrate that, 2 months after an initial insult, there is ongoing complement activation, and that complement inhibition interrupts a degenerative neuroinflammatory response and reverses cognitive decline, even when therapy is delayed until 2 months after the initial injury. These data indicate that ongoing chronic complement activation in the injured brain continues to drive a robust neuroinflammatory response. We further show that this chronic complement-dependent neuroinflammatory response correlates with synaptic degeneration and progressive cognitive decline.
Three months after murine TBI, there was an ongoing spatial expansion of a neuroinflammatory response as measured by the extent of astrogliosis in both the perilesional and contralateral brain. This astrogliotic scar also overlapped with areas of complement deposition. To investigate the role of complement in this chronic neuroinflammatory response, we used a clinically relevant approach of complement inhibition. CR2Crry specifically localizes to areas of complement activation and C3d deposition, which increases bioavailability and safety compared with systemic complement inhibition (Atkinson et al., 2005). The use of complement inhibition is clinically relevant but has additional advantages over the use of genetic deficiency models for the study of TBI in the chronic phase. First, genetic complement protein deletion has CNS developmental effects, and prior work has demonstrated that synaptic organization and the roles of CNS-resident microglia and astrocytes are altered in complement-deficient mice (see Stephan et al., 2012; Neniskyte and Gross, 2017; Magdalon et al., 2020). Second, the role of complement in propagating acute pathology after TBI is well documented. Therefore, the use of acute complement inhibition or the use of complement-deficient mice will prevent progression to chronic neuroinflammation, as has been shown previously (Alawieh et al., 2015, 2018c; Krukowski et al., 2018). Thus, it would not be possible to dissect the chronic role of complement given the acute protection provided by complement deficiency. Notably, CR2Crry gains access to the brain via a BBB breach that occurs in the proximity of the growing scar, and this is consistent with previous clinical and preclinical data showing continued BBB permeability beyond the acute phase of TBI (Tomkins et al., 2008). As expected, CR2Crry localized predominantly to the ipsilateral hemisphere, but CR2Crry binding also occurred in the contralateral hemisphere, especially at more delayed time points, which was consistent with histologic evidence showing expansion of the astrogliotic response over 60 d. Our demonstration that the BBB remains disrupted at 60 d after TBI, and that CR2Crry has access to the cerebral parenchyma, indicates that, in addition to the production of complement proteins in the brain in the chronic phase of TBI, the vasculature is also a likely source of complement. However, regardless of the source, complement is activated locally and can contribute to pathology via similar mechanisms.
Motor deficits following TBI are likely to improve spontaneously after injury because of development of adaptive compensatory strategies. Indeed, even in the absence of treatment, we observed near normalization of deficits observed on the corner task that measures gross motor deficits by 60 d after TBI. The ladder rung task also showed improvement, though with persistent deficits at 90 d after TBI. On the other hand, cognitive deficits continued to worsen in vehicle-treated controls over 90 d after TBI. Furthermore, this worsening of cognitive measures correlated with the extent of microglial activation in the brain. Consistent with these findings, recent work using an inhibitor of CSF1R to deplete microglia at 4 weeks after TBI demonstrated improvement in cognitive deficits (Henry et al., 2020). These findings are further supported by the reported clinical course of TBI, which involves delayed manifestation of neurocognitive and neuropsychiatric complaints (Moretti et al., 2012).
We demonstrate that three doses of CR2Crry administered over a week are necessary to suppress complement activity in the injured brain at 1-2 months after insult. This is in contrast to acute administration of complement inhibition where we have shown that a single dose administered immediately after injury is sufficient to suppress acute and subacute complement activation and neuroinflammation (Alawieh et al., 2018c). Further supporting the role of complement in cognitive decline, three doses of CR2Crry resulted in an improvement on cognitive tasks. When the effect size of CR2Crry treatment on cognitive performance was compared at different time points of administration, we observed a higher effect size at more delayed treatment times, which can be explained by the worsening performance in the vehicle group compared with absence of a similar decline in CR2Crry-treated groups. An interesting finding is that synaptic loss in the ipsilateral hippocampus correlated with performance on NOR, but not on Barnes maze task, whereas synaptic loss in the contralateral hippocampus better predicted performance on Barnes maze task. These findings are consistent, however, with prior data from hippocampal lesion studies that demonstrated that unilateral hippocampal ablation was sufficient to impair performance on NOR task, whereas spatial memory was preserved even with unilateral hippocampal injury (van Praag et al., 1998).
From a clinical standpoint, rehabilitative interventions are the only available management strategy for TBI to improve cognitive and motor functions. However, cumulative evidence shows only a modest effect of rehabilitation on chronic outcomes, and that rehabilitation is likely to speed up recovery rather than change long-term outcomes (Salazar et al., 2000; Zhu et al., 2007; Hallock et al., 2016; De Luca et al., 2018). This clinical outcome is supported by our findings here in mice. With motor and cognitive rehabilitation, we observed improvement in motor, but not cognitive, recovery. Interestingly, the combination of rehabilitation and CR2Crry treatment resulted in a quantitatively higher effect on ladder performance and NOR tasks, supporting the hypothesis that rehabilitation and complement inhibition involve different pathophysiological processes as previously shown in stroke (Alawieh et al., 2018b).
There was no difference in the area of tissue loss between animals treated with CR2Crry and vehicle, but the area of gliosis was significantly reduced in animals treated with CR2Crry, within both the ipsilateral brain and contralateral hippocampus. Prior clinical and preclinical studies on neurodegenerative disorders have shown that synaptic loss is an early degenerative finding that can predict cognitive impairment (Scheff et al., 2006). Our data demonstrate a similar finding, where animals exhibited a significant loss in synaptic density in both ipsilateral and contralateral hippocampus in conjunction with an expanding inflammatory response. Inhibition of complement resulted in preservation of synaptic density, an effect that was more pronounced in the contralateral than ipsilateral hippocampus. An explanation for the lower protective effect of CR2Crry on ipsilateral synaptic density is that treatment was administered at 2 months after injury, a time when significant neuronal and synaptic loss has already occurred in the ipsilateral brain. On the other hand, involvement of the contralateral brain is a delayed response resulting from chronic expansion of an inflammatory response. We also show that synaptic material was often located within microglial cell bodies, suggesting a role for microglial phagocytosis in elimination of perilesional and contralateral synapses, a finding previously reported in models of neurodegenerative disorders (Hong et al., 2016). A direct role for complement in synapse phagocytosis is supported by our finding that C3d opsonins are deposited on synaptic bodies in vehicle-treated animals, and that synaptic material colocalized with microglial phagolysosomal markers. Treatment with CR2Crry resulted in inhibition of C3d opsonization and inhibition of phagocytosis of hippocampal synapses. Collectively, these findings demonstrate that complement activity in the perilesional brain results in sustained activation of perilesional microglia. The resulting opsonization and phagocytosis of hippocampal synapses in the chronic phase of TBI are associated with cognitive decline. Inhibition of complement activation, even in the chronic phase after injury, can stabilize neurocognitive decline by suppressing microglial activation and phagocytosis of synaptic connections.
There are limitations to this study. Any specific effects of the anaphylatoxins (C3a and C5a) on the inflammatory response were not investigated. Both opsonins and anaphylatoxins can mediate microglial activation and are produced downstream of C3 cleavage, and CR2Crry inhibits the generation of both C3 opsonins and the anaphylatoxins. Also, we did not use microglia-specific markers that can distinguish microglia from macrophages. Although the majority of cells within the expanding scar are likely to be microglia, a contribution of blood-derived monocytes/macrophages cannot be excluded. Definitive distinction between these cell types is an ongoing area of investigation and beyond the scope of the current work. Finally, in this work, the interaction of complement with microglia has been the focus; however, our data also suggest a possible role for astrocytes in the ongoing neuroinflammatory response after TBI, and this represents an area for future study.
Footnotes
S.T. is an inventor on a licensed patent for CR2-targeted complement inhibition. The remaining authors declare no competing financial interests.
This work was supported by National Institutes of Health Grants 1P20GM109040, U54GM10494, and U54GM10491 to D.A. and S.T., Department of Veterans Affairs Merit Awards 1BX004256, 1RX001141, 21RX002363, and IK6BX005235 to S.T., and American Heart Association Pre-doctoral Fellowship 15PRE25250009 to A.A. Authors acknowledge the use of the MUSC Cell and Molecular Imaging Core (supported by awards from the National Institutes of Health P30 CA138313 and S10 OD018113), the Center for Oral Health Research (National Institute of General Medical Sciences Award P30GM103331), and the Josh Spruill Imaging Facility (National Institutes of Health-National Institute of General Medical Sciences P30 GM103342).
References
- Alawieh A, Elvington A, Tomlinson S (2015) Complement in the homeostatic and ischemic brain. Front Immunol 6:417. 10.3389/fimmu.2015.00417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A, Langley EF, Tomlinson S (2018a) Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci Transl Med 10:eaao6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A, Andersen M, Adkins DL, Tomlinson S (2018b) Acute complement inhibition potentiates neurorehabilitation and enhances tPA-mediated neuroprotection. J Neurosci 38:6527–6545. 10.1523/JNEUROSCI.0111-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S (2018c) Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci 38:2519–2532. 10.1523/JNEUROSCI.2197-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson C, Song H, Lu B, Qiao F, Burns TA, Holers VM, Tsokos GC, Tomlinson S (2005) Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J Clin Invest 115:2444–2453. 10.1172/JCI25208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K (2014) Traumatic brain injury and risk of dementia in older veterans. Neurology 83:312–319. 10.1212/WNL.0000000000000616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Byers AL, Gardner RC, Seal KH, Boscardin WJ, Yaffe K (2018) Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA Neurol 75:1055–1061. 10.1001/jamaneurol.2018.0815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellander BM, Singhrao SK, Ohlsson M, Mattsson P, Svensson M (2001) Complement activation in the human brain after traumatic head injury. J Neurotrauma 18:1295–1311. 10.1089/08977150152725605 [DOI] [PubMed] [Google Scholar]
- Bellander BM, Olafsson IH, Ghatan PH, Bro Skejo HP, Hansson LO, Wanecek M, Svensson MA (2011) Secondary insults following traumatic brain injury enhance complement activation in the human brain and release of the tissue damage marker S100B. Acta Neurochir (Wien) 153:90–100. 10.1007/s00701-010-0737-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig JJ, Guillot-Sestier MV, Town T (2013) Brain injury, neuroinflammation and Alzheimer's disease. Front Aging Neurosci 5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicerone KD, Goldin Y, Ganci K, Rosenbaum A, Wethe JV, Langenbahn DM, Malec JF, Bergquist TF, Kingsley K, Nagele D, Trexler L, Fraas M, Bogdanova Y, Harley JP (2019) Evidence-based cognitive rehabilitation: systematic review of the literature from 2009 through 2014. Arch Phys Med Rehabil 100:1515–1533. 10.1016/j.apmr.2019.02.011 [DOI] [PubMed] [Google Scholar]
- De Luca R, Calabro RS, Bramanti P (2018) Cognitive rehabilitation after severe acquired brain injury: current evidence and future directions. Neuropsychol Rehabil 28:879–898. 10.1080/09602011.2016.1211937 [DOI] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A (2007) G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39:175–191. 10.3758/bf03193146 [DOI] [PubMed] [Google Scholar]
- Fox GB, Fan L, Levasseur RA, Faden AI (1998) Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma 15:599–614. 10.1089/neu.1998.15.599 [DOI] [PubMed] [Google Scholar]
- Hallock H, Collins D, Lampit A, Deol K, Fleming J, Valenzuela M (2016) Cognitive training for post-acute traumatic brain injury: a systematic review and meta-analysis. Front Hum Neurosci 10:537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Tull LE, Stackman RW (2004) On the delay-dependent involvement of the hippocampus in object recognition memory. Neurobiol Learn Mem 82:26–34. 10.1016/j.nlm.2004.03.005 [DOI] [PubMed] [Google Scholar]
- Henry RJ, Ritzel RM, Barrett JP, Doran SJ, Jiao Y, Leach JB, Szeto GL, Wu J, Stoica BA, Faden AI, Loane DJ (2020) Microglial depletion with CSF1R inhibitor during chronic phase of experimental traumatic brain injury reduces neurodegeneration and neurological deficits. J Neurosci 40:2960–2974. 10.1523/JNEUROSCI.2402-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci 11:361–370. 10.1038/nrn2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossmann T, Stahel PF, Morganti-Kossmann MC, Jones JL, Barnum SR (1997) Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J Neuroimmunol 73:63–69. 10.1016/s0165-5728(96)00164-6 [DOI] [PubMed] [Google Scholar]
- Krukowski K, Chou A, Feng X, Tiret B, Paladini MS, Riparip LK, Chaumeil MM, Lemere C, Rosi S (2018) Traumatic brain injury in aged mice induces chronic microglia activation, synapse loss, and complement-dependent memory deficits. Int J Mol Sci 19:3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J (2012) Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci 32:10383–10395. 10.1523/JNEUROSCI.1498-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18:225–242. 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- Magdalon J, Mansur F, Teles E Silva AL, de Goes VA, Reiner O, Sertié AL (2020) Complement system in brain architecture and neurodevelopmental disorders. Front Neurosci 14:23. 10.3389/fnins.2020.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz GA, Whishaw IQ (2009) The ladder rung walking task: a scoring system and its practical application. J Vis Exp 28:1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti L, Cristofori I, Weaver SM, Chau A, Portelli JN, Grafman J (2012) Cognitive decline in older adults with a history of traumatic brain injury. Lancet Neurol 11:1103–1112. 10.1016/S1474-4422(12)70226-0 [DOI] [PubMed] [Google Scholar]
- Neniskyte U, Gross CT (2017) Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci 18:658–670. 10.1038/nrn.2017.110 [DOI] [PubMed] [Google Scholar]
- Nordstrom P, Michaelsson K, Gustafson Y, Nordstrom A (2014) Traumatic brain injury and young onset dementia: a nationwide cohort study. Ann Neurol 75:374–381. 10.1002/ana.24101 [DOI] [PubMed] [Google Scholar]
- Rancan M, Morganti-Kossmann MC, Barnum SR, Saft S, Schmidt OI, Ertel W, Stahel PF (2003) Central nervous system-targeted complement inhibition mediates neuroprotection after closed head injury in transgenic mice. J Cereb Blood Flow Metab 23:1070–1074. 10.1097/01.WCB.0000084250.20114.2C [DOI] [PubMed] [Google Scholar]
- Rich MC, Keene CN, Neher MD, Johnson K, Yu ZX, Ganivet A, Holers VM, Stahel PF (2016) Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci Lett 617:188–194. 10.1016/j.neulet.2016.02.025 [DOI] [PubMed] [Google Scholar]
- Ruseva MM, Ramaglia V, Morgan BP, Harris CL (2015) An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc Natl Acad Sci USA 112:14319–14324. 10.1073/pnas.1513698112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar AM, Warden DL, Schwab K, Spector J, Braverman S, Walter J, Cole R, Rosner MM, Martin EM, Ecklund J, Ellenbogen RG (2000) Cognitive rehabilitation for traumatic brain injury: a randomized trial. Defense and Veterans Head Injury Program (DVHIP) Study Group. JAMA 283:3075–3081. 10.1001/jama.283.23.3075 [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384. 10.1016/j.neurobiolaging.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z (2004) Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol 155:55–63. 10.1016/j.jneuroim.2004.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK (1995) A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 12:169–178. 10.1089/neu.1995.12.169 [DOI] [PubMed] [Google Scholar]
- Stahel PF, Morganti-Kossmann MC, Perez D, Redaelli C, Gloor B, Trentz O, Kossmann T (2001) Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma 18:773–781. 10.1089/089771501316919139 [DOI] [PubMed] [Google Scholar]
- Stahel PF, Flierl MA, Morgan BP, Persigehl I, Stoll C, Conrad C, Touban BM, Smith WR, Beauchamp K, Schmidt OI, Ertel W, Leinhase I (2009) Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J Neuroinflammation 6:2. 10.1186/1742-2094-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B (2012) The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35:369–389. 10.1146/annurev-neuro-061010-113810 [DOI] [PubMed] [Google Scholar]
- Tomkins O, Shelef I, Kaizerman I, Eliushin A, Afawi Z, Misk A, Gidon M, Cohen A, Zumsteg D, Friedman A (2008) Blood-brain barrier disruption in post-traumatic epilepsy. J Neurol Neurosurg Psychiatry 79:774–777. 10.1136/jnnp.2007.126425 [DOI] [PubMed] [Google Scholar]
- van Praag H, Qu PM, Elliott RC, Wu H, Dreyfus CF, Black IB (1998) Unilateral hippocampal lesions in newborn and adult rats: effects on spatial memory and BDNF gene expression. Behav Brain Res 92:21–30. 10.1016/S0166-4328(97)00117-4 [DOI] [PubMed] [Google Scholar]
- Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, Lu M, Chopp M (2002) A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J Neurosci Methods 117:207–214. 10.1016/S0165-0270(02)00114-0 [DOI] [PubMed] [Google Scholar]
- Zhu X, Poon W, Chan CC, Chan SS (2007) Does intensive rehabilitation improve the functional outcome of patients with traumatic brain injury (TBI)? A randomized controlled trial. Brain Inj 21:681–690. 10.1080/02699050701468941 [DOI] [PubMed] [Google Scholar]