

Exome sequencing and Drosophila modeling revealed variants in SMARCA5 as a novel cause of a neurodevelopmental disorder.
Abstract
Intellectual disability encompasses a wide spectrum of neurodevelopmental disorders, with many linked genetic loci. However, the underlying molecular mechanism for more than 50% of the patients remains elusive. We describe pathogenic variants in SMARCA5, encoding the ATPase motor of the ISWI chromatin remodeler, as a cause of a previously unidentified neurodevelopmental disorder, identifying 12 individuals with de novo or dominantly segregating rare heterozygous variants. Accompanying phenotypes include mild developmental delay, frequent postnatal short stature and microcephaly, and recurrent dysmorphic features. Loss of function of the SMARCA5 Drosophila ortholog Iswi led to smaller body size, reduced sensory dendrite complexity, and tiling defects in larvae. In adult flies, Iswi neural knockdown caused decreased brain size, aberrant mushroom body morphology, and abnormal locomotor function. Iswi loss of function was rescued by wild-type but not mutant SMARCA5. Our results demonstrate that SMARCA5 pathogenic variants cause a neurodevelopmental syndrome with mild facial dysmorphia.
INTRODUCTION
Regulation of gene expression by transcription requires a highly coordinated and precise process that involves chromatin regulators and modifiers, transcription factors, cohesins, and mediators. The basic and dynamic unit of chromatin is the nucleosome, onto which DNA is wrapped around an octameric histone complex. Subsequent high-order DNA structure requires chromatin remodelers to make DNA accessible to alter nucleosome position and, in turn, regulate gene transcription using the energy released by adenosine triphosphate (ATP) hydrolysis. ATP-dependent nucleosome remodeling not only provides an open and accessible chromatin state but also is involved in chromatin assembly, nucleosome sliding and spacing, and gene repression, which are crucial for a wide range of biological processes in normal development including early embryonic development stages (1). ATP-dependent nucleosome remodelers, which share similar adenosine triphosphatase (ATPase) domains and have an affinity to the nucleosome, are categorized into four subfamilies based on their unique domains and associated subunits: SWI/SNF, ISWI (imitation switch), CHD (chromatin helicase DNA binding), and INO80/SWR1.
A number of neurodevelopmental disorders have been linked to pathogenic variants in genes encoding chromatin remodelers and associated subunits, including the SWI/SNF subunits ARID1A [Mendelian Inheritance in Man (MIM): 603024], ARID1B (MIM: 614556), ARID2 (MIM: 609539), SMARCA4 (MIM: 603254), SMARCB1 (MIM: 601607), SMARCE1 (MIM: 603111), SMARCD1 (MIM: 601735), SMARCC2 (MIM: 601734), and DPF2 (MIM: 601671) in Coffin-Siris syndrome (2–8); SMARCA2 (MIM: 600014) in Nicolaides-Baraitser syndrome (MIM: 601358) (9, 10); BCL11A (MIM: 606557) in Dias-Logan syndrome (MIM: 617101) (11); ACTL6B in intellectual developmental disorder with severe speech and ambulation defects (MIM: 618487) (12); ACTB (MIM: 102630) and ACTG1 (MIM: 102560) in Baraitser-Winter syndrome (13); CHD subunits CHD4 (MIM: 603277) in Sifrim-Hitz-Weiss syndrome (MIM: 617159) (14, 15); CHD7 (MIM: 608892) in CHARGE syndrome (MIM: 214800) (16); CHD3 (MIM: 602120) in Snijders Blok-Campeau syndrome (MIM: 618205) (17); ISWI subunits BPTF (MIM: 601819) in neurodevelopmental disorder with dysmorphic facies and distal limb anomalies (18); and, last, SMARCA1 in Coffin-Siris syndrome–like and Rett syndrome–like phenotypes (19, 20).
ISWI is one of the well-studied chromatin remodeling complexes in animal models. Iswi was first identified in Drosophila melanogaster, which has a single Iswi ATPase motor protein compared to two in humans (21–23). In vertebrates, the ISWI complex is typically composed of two to four subunits, including an ATPase, a catalytic subunit, and a scaffolding component involved in target recognition or stabilization. Eight different ISWI-containing chromatin remodeling complexes have been identified in humans, including ACF, CHRAC, NoRC, RSF, WICH, SNF2H/NURD/Cohesin, CERF, and NURF (24–31). All of these complexes contain either the SNF2L or SNF2H ATPase subunit encoded by SMARCA1 or SMARCA5, respectively. The two proteins share 83% amino acid similarity, and they are interchangeable in these complexes (32). Morpholino knockdown of the zebrafish SMARCA5 ortholog results in decreased body size and reduced cardiac ventricle size (33). A critical role for SMARCA5 in mammalian development has also been demonstrated in mice, where knockout embryos die during preimplantation stages and conditional knockouts lead to substantial reductions in body weight, body size, and brain size with cerebellar hypoplasia (34). However, a specific syndrome associated with SMARCA5 rare germline variants has not yet been described. In addition, whereas SMARCA5 has been implicated in neural stem cell proliferation and differentiation (26, 34, 35), the spectrum of its functions in the nervous system remains undetermined, with the underlying mechanisms largely underexplored.
Here, we report 12 individuals (6 females and 6 males) from 10 unrelated families assembled through international collaborations with germline variants in SMARCA5. These individuals share the core presentations of mild developmental delay, postnatal microcephaly, short stature, and mild facial dysmorphia. Using a Drosophila model, we found that Iswi loss-of-function (LoF) results in a spectrum of neurodevelopmental abnormalities including reduced body/brain size, dendrite and axon mispatterning, and behavioral deficits. These findings mirror the key growth and cognitive phenotypes present in affected individuals. More importantly, human wild-type (WT) SMARCA5 but not affected individuals’ variants rescue these phenotypes, confirming their pathogenic role in these models. Together, these data describe a novel syndromic neurodevelopmental disorder caused by pathogenic variants in the SMARCA5 gene.
RESULTS
Clinical presentations of individuals with SMARCA5 variants
After identifying de novo or dominantly inherited variants in SMARCA5 in each family, we assembled the cohort of 12 individuals through the assistance of GeneMatcher (36). Individual 3 was recruited from the Deciphering Developmental Disorders (DDD) study. Clinical phenotypes overlapped and are summarized in Table 1, and detailed data are compiled in table S1. The majority of the probands (8 of 10; 80%) had neurodevelopmental disorders with developmental delays in some area, but most delays were mild. Because detailed developmental history information was inadequate for the two adult individuals (mother and maternal grandmother of individual 4), their degree of neurodevelopmental delay is unclear. While all probands have normal hearing, mild (N = 4) or severe (N = 2) speech delay was present in six probands (60%; 6 of 10), with the age of first words ranging from 12 to 27 months. Motor delay was reported in six probands (60%), with the age of walking ranging from 15 to 48 months. Hypotonia was found in five probands (50%), and peripheral hypertonia was noted in individual 4 during infancy. Regarding cognition, four probands had mild intellectual disability and one had severe intellectual disability (50%). A formal diagnosis of autism spectrum disorder was made in only individual 3, and attention deficit disorder/attention-deficit/hyperactivity disorder was detected in three probands (30%). A brain magnetic resonance imaging scan was performed for seven probands and brain computed tomography scan or head ultrasound for two additional probands, showing generally normal morphology, although individual 11 showed mildly delayed myelination and periventricular occipital gliosis and individual 12 showed Chiari malformation type 1. Seizures occurred in two probands (20%). Postnatal microcephaly [head circumference (HC) ≤ −2 SD], short stature (height ≤ −2 SD), and failure to thrive, which were concomitant with feeding difficulties, were present in eight probands (80%), among whom two individuals had birth weight below −2 SD, one had birth length below −2 SD, and three had birth HC less than −2.5 SD. The mother (individual 5) of proband 4 also had short stature and microcephaly when evaluated at age of 39 years old, and her mother (individual 6) also had short stature. Two individuals in the cohort, individuals 10 and 11, had normal postnatal height and weight, with individual 11 having a normal head circumference and individual 10 having macrocephaly. Eight of nine probands in this cohort (89%) had some type of abnormalities of extremities, including brachydactyly (two of five), clinodactyly (three of seven), 2-3 toe syndactyly (two of seven), and hallux valgus/sandal gap (six of seven), whereas cardiac, gastrointestinal, and genitourinary defects were uncommon (table S1). Individual 2 had multiple cardiac septal defects and a noncompaction dilated cardiomyopathy, with no causative variants in known cardiac genes identified by her clinical exome sequencing. A dermatologic finding, café au lait macule, was identified in two individuals (individuals 3 and 9), who had the recurrent variant, c.1301_1306del, p.(Ile434_Leu435del). Photographs from all participants show a number of shared craniofacial features, including blepharophimosis and/or short palpebral fissures with or without almond-shaped eyes (9 of 11), periorbital fullness (8 of 9), epicanthal folds (4 of 11), a wide/prominent nasal bridge (8 of 9), a short/smooth philtrum (8 of 10), a thin or tented upper lip (8 of 10), broad, arched, sparse, or horizontal eyebrows (11 of 11), and prominent ears or attached ear lobes (9 of 10) (Fig. 1 and table S1). Vision abnormalities are reported in 5 of 12 cases, including myopia, hyperopia, strabismus, and astigmatism. A search of the ClinVar database noted one additional individual (ID 599594) with short stature and a de novo SMARCA5 variant, c.940A>C, p.(Lys314Gln), although no detailed clinical information was available for this subject. Together, these data indicated that variants in SMARCA5 are associated with a variable neurodevelopmental phenotype with predominantly mild developmental delay and intellectual disability, short stature, and microcephaly.
Table 1. Summary of clinical characteristics associated with heterozygous SMARCA5 variant.
| Clinical findings |
Number of individuals/assessed |
Percentage |
| Gender | 6 female and 6 male | |
| Neurodevelopment | ||
| Prenatal microcephaly |
3/7 | 43% |
| Postnatal microcephaly |
9/12 | 75% |
| Postnatal short stature |
10/12 | 83% |
| Failure to thrive | 8/10 | 80% |
| Hypotonia | 5/10 | 50% |
| Developmental delays |
8/10 | 80% |
| Motor delay | 6/10 | 60% |
| Speech/language delay |
6/10 | 60% |
| Behavioral problems | 3/10 | 30% |
| Facial feature | ||
| Blepharophimosis/ short palpebral fissures |
9/11 | 82% |
| Epicanthal folds | 4/11 | 36% |
| Periorbital fullness | 8/9 | 89% |
| Wide/prominent nasal bridge |
8/9 | 89% |
| Short/smooth philtrum |
8/10 | 80% |
| Thin or tented upper lip | 8/10 | 80% |
| Broad, arched, sparse, or horizontal eyebrows |
11/11 | 100% |
| Prominent ears or attached ear lobes |
9/10 | 90% |
| Other abnormalities | ||
| Limb abnormalities | 10/11 | 91% |
| Eye/vision | 5/12 | 42% |
| Seizures | 2/12 | 17% |
Fig. 1. Facial photographs of affected individuals with SMARCA5 variants showing dysmorphisms.

Individual identifiers correlate with those in table S1. Note the similarities in facial appearance with blepharophimosis, short palpebral fissures, periorbital fullness, epicanthal folds, wide and/or high nasal bridge, short and/or flat philtrum, thin upper lip, and arched eyebrows. Individual 1: 3.7-year-old male; individual 4: 18-year-old male; individual 5: 39-year-old female; individual 7: 9.5-year-old female; individual 10: 3-year-old male; individual 11: 3.6-year-old male. Photo credits: M. A. Deardorff, Children’s Hospital Los Angeles; A. Kurolap and D. Tiosano, Ruth Rappaport Children’s Hospital; M. J. M. Nowaczyk, McMaster University; Y. Huang, University of California, Los Angeles; N. Boy, University Hospital Heidelberg.
Identification of SMARCA5 variants
Exome sequencing of the 10 families revealed nine different heterozygous novel variants in SMARCA5 (NM_003601.3), including eight variants confirmed to be de novo in nine individuals. Notably, two unrelated individuals were found to carry the same de novo variant, c.1301_1306del, p.(Ile434_Leu435del). One missense variant, c.1711G>C, p.(Ala571Pro), was identified in a three-generation pedigree with an affected proband, mother, and maternal grandmother. These nine variants include one splice-altering variant, c.802-2A>G; one in-frame deletion, c.1301_1306del, p.(Ile434_Leu435del); and seven missense variants, c.1130A>T, p.(Asp377Val); c.1682C>A, p.(Ala561Asp); c.1711G>C, p.(Ala571Pro); c.1775G>A, p.(Arg592Gln); c.1855G>C, p.(Glu619Gln); c.2207G>A, p.(Arg736Gln); and c.2677G>A, p.(Glu893Lys) (Fig. 2A). All variants were absent in population genomics resources (i.e., 1000 Genomes Project, ESP6500SI, and gnomAD) and our in-house dataset of >10,000 exomes. All seven missense variants were predicted to be deleterious by multiple bioinformatic prediction algorithms (SIFT, LRT, MutationTaster, CADD, etc.) (table S2). Reverse transcription polymerase chain reaction (RT-PCR) followed by Sanger sequencing of complementary DNA (cDNA) from fibroblasts and a lymphoblastoid cell line (LCL) obtained from individual 1 with the splice-altering variant demonstrated that it causes exon 7 skipping, leading to an in-frame deletion of 52 amino acids (p.Ala268_Lys319del) in the highly conserved ATPase domain (Fig. 2B). These missense variants (including the ClinVar variant) and the small deletions were mapped to a protein schematic, and eight variants appear to cluster within or around the helicase domains, whereas p.(Arg736Gln) and p.(Glu893Lys) are located in the HAND-SANT-SLIDE (HSS) domains (Fig. 2A). Of note, all the variants reside in protein locations intolerant to change as calculated by the MetaDome server (Fig. 2A and table S2).
Fig. 2. Molecular genetic findings.

(A) An intolerance landscape plot generated by MetaDome for SMARCA5 variant analysis (top panel) and a schematic outline of SMARCA5 protein showing conserved variants identified in individuals 1 to 12 and ClinVar at the bottom. (B) RT-PCR analysis of individual 1’s fibroblasts and lymphoblastoid cell line showing the SMARCA5 exon 7 skipping in both cell types because of a splice-altering variant.
To explore the structural impact of the six missense variants within or around the helicase domains, the recently published cryogenic electron microscopy three-dimensional structure (37) of WT SMARCA5 incorporated with the nucleosome complex was obtained [Protein Data Bank (PDB): 6ne3]. SMARCA5 binds to DNA in the nucleosome through its ATPase helicase domains, which form a positively charged cleft on the binding surface (fig. S1A). The p.(Lys314Gln) and p.(Asp377Val) changes result in loss of the positive charge, and the p.(Ala561Asp) change introduces a negatively charged side chain, which may diminish its anchor of DNA binding (table S2 and fig. S1, B and C). Similarly, the p.(Ala571Pro), p.(Arg592Gln), and p.(Glu619Gln) changes may disturb hydrogen bonds and structural stability, therefore possibly affecting interaction with DNA, histone, or adenosine diphosphate (ADP) ligand binding (table S2 and fig. S1, B and C). Recent studies have demonstrated that the acidic patch, a negatively charged structure on the surface of the nucleosome, is critically important for recruiting chromatin remodelers, and an acidic patch binding (APB) motif in SMARCA5 has been discovered (38). Notably, the substitution of Arg736, one of the five residues in the APB motif, with alanine in SMARCA5 disrupted its nucleosome sliding activity (39). It seems reasonable to speculate that the replacement of Arg736 with glutamine, similar loss of positive charge, and the side chain changing from a large residue to a small one could similarly alter its remodeler activity. The three-dimensional structure of the HSS domains of WT human SMARCA5 was obtained by using homology modeling with SWISS-MODEL. The x-ray structure of Drosophila Iswi at a 1.9-Å resolution was used as a template, as it demonstrates 81.25% identity with human SMARCA5 (PDB: 1ofc) (40). As shown in fig. S1D, residue Glu893 was mapped onto the SANT domain helix 3 (SANT3), a recognition helix that makes contacts with unmodified histone tails and has an overall negatively charged surface. A change from a negatively charged side chain to a positively charged one [p.(Glu893Lys)] in the SANT motif, which is predicted to disrupt a local salt bridge, may affect SANT motif substrate recognition ability to contact histone, and, in turn, to hinder ATPase binding.
SMARCA5’s ortholog Iswi plays a key role in Drosophila larval development
To gain independent support for pathogenicity of the variants identified in humans and investigate whether SMARCA5 is required for normal development, we studied the Drosophila gene Iswi (Imitation SWI), which is orthologous to human SMARCA1 or SMARCA5. We first assessed the functional effects of an Iswi LoF mutant, Iswi2, on larvae development (41) and found that Iswi2 homozygous larvae died before the end of larval stage. For those that survived to third instar, body size was substantially smaller than WT larvae (Fig. 3, A and B). It was previously reported that Iswi is important for chromosome condensation during mitosis, and Iswi LoF leads to mitotic recombination defects (41, 42). To investigate whether Iswi affects cell mitosis, we performed immunostaining with phosphorylated histone H3 (pH3), a proliferation marker for mitotic cells. We observed that in the brain lobes, the number of pH3+ positive cells was significantly decreased in Iswi2 larvae (Fig. 3, C and D). We next asked whether Iswi may play a role in neuronal development. To accomplish this, we used Drosophila dendritic arborization (da) sensory neurons, which provide a suitable system to study neuronal development and dendritic morphology. There are four classes of da neurons, with class IV da (C4da) neurons extending the most complex and highly branched dendrites (43). We found that in Iswi2, the dendrite complexity of C4da neurons was decreased. Both the total dendrite length and dendritic branch number exhibited a significant reduction (fig. S2, B to D), indicating that Iswi is important for dendrite morphogenesis. Furthermore, Iswi LoF also caused a tiling defect between C4da neurons ddaC and v’ada. Tiling refers to a phenomenon in which the dendrites of adjacent neurons establish complete but nonredundant interaction to occupy the space between the cell bodies as observed in WT larvae (44). In Iswi2 larvae, however, the space is not fully covered because there is a significantly enlarged gap in the dendritic field between ddaC and v’ada (Fig. 3, E and F). These results suggest that Iswi plays a pleiotropic role during fly larval development, highlighting its involvement in neural proliferation and morphogenesis.
Fig. 3. Iswi LoF leads to developmental defects in Drosophila larvae.
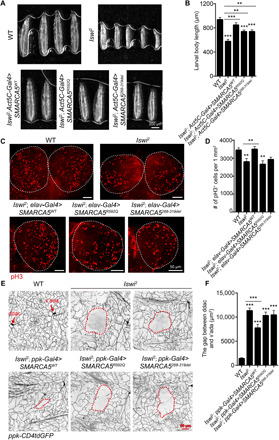
(A and B) Compared with WT, larval body size is significantly smaller in Iswi2 third-instar larvae, which can be rescued by the expression of SMARCA5WT by Act5C-Gal4. (A) Images showing larvae of different genotypes. Scale bar, 200 μm. (B) Quantification of larval body length. N = 30 to 41 larvae. (C and D) In the brain lobes of Iswi2 larvae, the number of pH3+ positive mitotic cells is significantly decreased. N = 12 to 17 brain lobes. Expressing SMARCA5WT by elav>Gal4 is sufficient to rescue the decreased proliferation in Iswi LoF larvae. (C) The brains were dissected from third-instar larvae and stained with pH3. Scale bar, 50 μm. (D) Quantification of pH3+ positive mitotic cells; P = 0.0090. (E and F) In Iswi2, C4da ddaC and v’ada neurons exhibit dendritic tiling defects. SMARCA5WT, but not patient variants, partially rescues the tiling defects. Scale bar, 50 μm. (E) The red dashed circles outline the gap area between C4da ddaC and v’ada neurons. (F) Quantification of the gap in C4da neuron dendritic field; N = 17 to 34 dendritic fields from 4 to 5 larvae. All data are means ± SEM. The data were analyzed by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test; **P < 0.01, ***P < 0.001.
We next expressed Iswi RNAi under the control of elav-Gal4, a pan-neuronal driver (45), to determine whether Iswi is cell-autonomously required in neurons. Neural-specific knockdown of Iswi leads to significantly smaller brain size in adult flies (Fig. 4, A and B). Moreover, in the Iswi knockdown brains, we observed severe structural deficits of the mushroom body, the learning and memory center of the fly (46). Compared to control, the volume of both the vertical lobe and the horizontal lobe is markedly reduced after Iswi knockdown, with bilaterally missing vertical lobe and reduction of the horizontal lobe (Fig. 4, C to E). Consistent with these findings, Gong et al. (47) reported that Iswi knockdown in flies results in mushroom body morphologic abnormalities, as well as disrupted sleep, circadian rhythmicity, memory, and social behaviors, suggesting that Iswi is required during development for numerous adult behaviors.
Fig. 4. Iswi neural-specific knockdown causes abnormal brain structure and motor deficits in adult files.

(A and B) Iswi neural-specific knockdown leads to smaller brain size compared with control flies. (A) Representative images of fly brains of control and Iswi knockdown flies. Scale bar, 100 μm. (B) Quantification of adult fly brain volume. N = 6, P = 0.0002. (C to E) Iswi neural-specific knockdown affects mushroom body structure in adult flies. While expressing SMARCA5WT partially rescues the structure, the patient variants are not able to rescue mushroom body morphology. Cyan and magenta dashed lines outline the vertical lobe and horizontal lobe of the mushroom body in the right hemisphere, respectively. N = 11 to 35 brains. (C) Representative images of fly brains from different groups. Mushroom bodies are marked by FasII staining. Scale bar, 100 μm. (D) Quantification of vertical lobe volume. (E) Quantification of horizontal lobe volume. SMARCA5WT rescue significantly increases the horizontal lobe volume in Iswi knockdown flies. (F) The negative geotaxis test shows that Iswi knockdown leads to locomotion deficit in flies. Expression of SMARCA5WT in Iswi knockdown flies is able to rescue the climbing deficit to control levels, while SMARCA5R592Q and SMARCA5268-319del mutants fail to rescue. N = 3 to 12 groups, with each group containing 10 flies. All data are means ± SEM. The data were analyzed by unpaired t test or one-way ANOVA followed by Tukey’s multiple comparison test; ***P < 0.001.
As motor delay was reported in several patients, we next examined the locomotor function in Iswi knockdown flies by the negative geotaxis assay (a climbing test). We found that while ~80% of control flies climbed to or past the target distance, only a small portion of Iswi knockdown flies (less than 20%) showed normal climbing ability (Fig. 4F), indicating that neural knockdown of Iswi severely impaired the motor skills in flies. Together, these data indicate that Iswi is necessary for brain development and function in flies.
SMARCA5 WT but not mutants rescue the defects observed after Iswi LoF
To determine the functional consequences of the SMARCA5 variants identified in individuals 1 and 2, we generated transgenic flies with inducible expression of WT SMARCA5 (SMARCA5WT), SMARCA5R592Q and SMARCA5268-319del, respectively. We found that these variants did not change SMARCA5’s localization in the nucleus (fig. S2A). We investigated whether human SMARCA5WT and variants could rescue the defects observed in Iswi2 larvae and neural-specific Iswi knockdown flies. First, we expressed SMARCA5WT and the two human variants in Iswi2 larvae under the control of Act5C-Gal4, which drives the expression ubiquitously (48), and found that SMARCA5WT expression was sufficient to rescue the larval body size similar to WT. In contrast, SMARCA5R592Q and SMARCA5268-319del only partially rescued the body length in Iswi2 larvae (Fig. 3, A and B). Second, we examined whether the expression of WT or mutant SMARCA5 rescued the decreased cellular proliferation observed in Iswi2 larval brain lobes. When using the elav-Gal4 driver to express SMARCA5WT in Iswi LoF larvae, we found that SMARCA5WT robustly rescued the proliferation deficits, while no rescue was observed when expressing the two patient variants.
Third, we expressed SMARCA5WT in C4da neurons of Iswi2 larvae using the C4da neuron–specific ppk-Gal4 driver (43) and found that SMARCA5WT substantially rescued the tiling deficit between ddaC and v’ada, resulting in significantly decreased gap area in the dendritic field. Perhaps because Drosophila Iswi is the ortholog of the human SMARCA1 and SMARCA5, expressing SMARCA5 only partially restored the neurodevelopmental defects in Iswi2 (Fig. 3, E and F). Notably, both human SMARCA5 variants completely failed to rescue the tiling phenotype in Iswi2 (Fig. 3, E and F). Overexpressing SMARCA5WT in WT larvae reduced dendritic complexity (fig. S2, B to D), similar to the phenotype observed in Iswi2, suggesting that the precise control of Iswi expression is crucial for normal dendrite morphogenesis. While the expression of SMARCA5R592Q also caused impaired dendrite outgrowth and branching, the dendrite length and branch number in SMARCA5268-319del–overexpressing neurons were comparable to WT flies without transgenic expression of human SMARCA5 (fig. S2, B to D). We did not observe dendritic tiling defects in any of the three overexpression transgenic lines. These results suggest that although both variants jeopardized normal function of SMARCA5, they appear to differ in their mutagenic strength and may not be complete LoF.
Last, human WT SMARCA5 partially reversed the mushroom body morphologic abnormalities and fully rescued locomotion defects induced by neural-specific Iswi knockdown. We found that the expression of SMARCA5WT partially restored horizontal lobe volume of the mushroom body, although it had little effect on the vertical lobes (Fig. 4, C to E). Comparatively, expressing either SMARCA5R592Q or SMARCA5268-319del was unable to rescue the morphological defects. In the negative geotaxis test, SMARCA5WT fully rescued the climbing deficit in Iswi knockdown flies, resulting in similar motor performance as control flies, while the patient variants failed to improve climbing capacity (Fig. 4F).
DISCUSSION
We describe a novel syndromic neurodevelopmental disorder caused by de novo or dominantly inherited variants in SMARCA5, a gene that encodes an ATPase motor protein in the ISWI chromatin remodeler complex and that is extremely intolerant to both LoF (pLI = 1) and missense (Z = 5.42) variants based on the gnomAD population database. We identified seven missense variants, one splice-altering variant that led to exon skipping and in-frame deletion, and one recurrent in-frame deletion in 12 individuals from 10 unrelated families from around the world. The individuals in our study demonstrated a broad range of clinical features, as is not uncommon in neurodevelopmental disorders associated with pathogenic variants in aforementioned chromatin remodeling complexes (i.e., SWI/SNF, CHD, and ISWI) that vary from isolated autism to syndromic intellectual disability (5, 17, 49, 50). The most consistent clinical findings in our cohort are postnatal microcephaly and short stature observed in 10 of 12 individuals as well as developmental delay in 8 of 10 individuals, although, again, the severity of these delays varies widely from mild to severe (individual 7). Individual 10 has macrocephaly with a variant outside the helicase C-terminal domain, and individual 11 is normocephalic with a variant in HSS domains; both of these individuals have delays in motor and speech development but have normal stature, illustrating phenotypic variability. Conversely, individuals 1 and 8 have postnatal microcephaly and short stature but normal development with variants in the helicase ATP-binding domain. Varying degrees of developmental delay have been consistently reported in neurodevelopmental disorders associated with pathogenic variants involved in chromatin remodeling complexes. Similarly, varying head size ranging from microcephalic to macrocephalic is also frequently observed, for example, in SMARCB1 and CHD3 (50, 51). It is widely acknowledged that different variants within different functional domains of a gene can cause different phenotypes. Although the numbers are relatively small, when comparing phenotypes to identified variants, no clear genotype-phenotype correlation was established in this cohort. It is noteworthy that variants in SMARCA1, which is highly homologous to SMARCA5, have been associated with distinct phenotypes such as Rett syndrome, Coffin-Siris–like syndrome, and sporadic schizophrenia (19, 20, 52). This can be explained by different temporal and spatial expression patterns of both genes, although SMARCA1 and SMARCA5 could be interchangeably present in the different ISWI complexes that often have overlapping and complex-specific functions.
SMARCA5 is located at 4q31.21, a region overlapping with the deletions observed in the 4q deletion syndrome, which is characterized by a broad clinical spectrum and phenotypic variability (53). Review of the literature revealed that two subjects have interstitial deletions encompassing the SMARCA5 gene. One patient with 14-Mb deletion (4q28.3-31.23) has development delay, growth failure, ventricular septum defect, and craniofacial dysmorphisms (54), whereas the other subject with a 6-Mb deletion has only mild speech delay (55). Searching the Decipher database returned 14 participants that have deletions containing SMARCA5 with a range of heterogeneity in clinical findings. Because all of these deletions are larger than 6-Mb and include many other genes, it is unclear how haploinsufficiency of SMARCA5 contributes to their specific phenotypes, but it suggests that heterozygous SMARCA5 deletion is compatible with life. On the basis of our findings, we propose that SMARCA5 should be considered in the context of 4q deletion syndrome.
In general, this novel SMARCA5-associated syndrome reported here appears to result in a less severe phenotype when compared to other neurodevelopmental disorders due to pathogenic variants in ARID, SMARC, and CHD gene families, where almost all of the variants occur de novo and parent-child transmission is rarely documented (50, 56). We present here a three-generation pedigree (for individuals 4 to 6) segregating a deleterious missense variant. It is interesting to note that individual 4’s maternal grandfather also has short stature (indicated in the pedigree; fig. S3); however, he has a pathogenic variant in TLK2 [NM_006852.3:c.364C>T, p.(Arg122*)] that is associated with “Mental retardation, autosomal dominant 57” (OMIM no. 618050), which also includes short stature. This variant is not inherited by individuals 5 and 6, as confirmed by exome sequencing. Although SMARCA5-associated neurological complications are generally mild, postnatal proportionate short stature and microcephaly are rather predominant, with SDs ranging from −2 to −4.78 for height, and −2 to −6.21 for head circumference, in 8 of 10 probands. It has been widely appreciated that many rare diseases of the epigenetic machinery demonstrate significant phenotype overlap. Individuals presented here have craniofacial dysmorphisms, and some facial features overlap with SMARCA2-associated blepharophimosis intellectual disability syndrome (57), including blepharophimosis, short palpebral fissures, epicanthal folds, high arched and sparse eyebrows, and short metacarpals. Recently, pathogenic variants of BPTF, encoding a noncatalytic subunit of ISWI complex, have been implicated in a neurodevelopmental disorder (18). Phenotype comparison of BPTF- and SMARCA5-associated disorders reveals additional similarities, including developmental delay, intellectual disability, motor delay, speech delay, failure to thrive, dysmorphisms, and limb abnormalities. Overall, individuals with the SMARCA5-associated phenotypes appear to share similar dysmorphic features and clinical histories, including blepharophimosis, short palpebral fissures, periorbital fullness, wide/high nasal bridge, a thin or tented upper lip, short/smooth philtrum, high arched, sparse, or broad eyebrows, and hallux valgus, but each of the shared clinical findings is relatively nonspecific. Additional individuals and further assessment will be required to further clarify the clinical spectrum and constellation of features associated with this novel disorder to subsequently assist in the clinical diagnosis; however, SMARCA5 gene sequencing should be considered in patients with failure to thrive, short stature, microcephaly, and dysmorphic features.
Our phenotypic analyses in the fly Iswi LoF models and human patients emphasize the pleiotropic and critical role of SMARCA5 and the SNF2H-containing chromatin remodeling complexes in nervous system development. We observed that LoF mutants of its Drosophila ortholog Iswi led to decreased larval body size and sensory dendrite complexity, together with a tiling defect. Such dendritic defects have also been described in cerebellar Purkinje neurons in conditional knockout of Snf2h in mice (34). In addition, analyses of pan-neuronal knockout and telencephalon-specific knockout of Snf2h in mice have both revealed a smaller brain phenotype (34, 58). Apart from its previously reported function in cell proliferation/stem cell self-renewal (59) and hence smaller head/microcephaly, we identified its requirement in postmitotic neurons. Specifically, we found that Iswi LoF leads to defects in sensory neuron dendrite tiling and mushroom body development, highlighting its underappreciated role in neural circuit assembly (47). This likely contributes to the behavioral deficits such as impaired locomotion in the Iswi knockdown flies and intellectual disability, hypotonia, and speech delay in patients. Nevertheless, the patient variants, SMARCA5R592Q and SMARCA5268-319del, largely failed to replace Iswi in flies, highlighting the functional effect of the variants that we identified. Given that both variants could partially rescue the reduced body size in Iswi2, and that the overexpression of SMARCA5R592Q in neurons affects their dendrite morphology, these two variants may not be complete LoF alleles and may retain partial function, allowing it to interact with the dendrite morphogenesis machinery. Modeling variants in the three-dimensional structure suggests that they may disrupt interactions of SMARCA5 with the nucleosome and, in turn, affect ATPase binding. An emerging question is how SMARCA5 and, more generally, the chromatin remodelers exert their diverse functions under various developmental scenarios. Therefore, an imminent task is to identify the DNA substrates processed by the remodelers at a particular time point and in a specialized tissue niche, which could be potential therapeutic targets. Together, our functional studies of the SMARCA5 missense variants suggest that they are hypomorphic alleles, although a dominant effect mechanism through competitive inhibition of the WT functional allele in the ISWI complex is also possible.
In summary, we identify and phenotypically characterize a novel neurodevelopmental syndrome that overlaps with conditions caused by variants in different subunits of chromatin remodeling complexes. Frequent features include postnatal short stature and microcephaly, in addition to shared dysmorphia. Our findings in a fly in vivo model highlight the important and previously underappreciated role of the ISWI family proteins in dendrite morphogenesis, neural circuit formation, and diverse behaviors. In conclusion, this study expands the spectrum of ISWI-related disorders and extends the tally of causative genes in neurodevelopmental disorders.
METHODS AND MATERIALS
Research participants
Informed consent was obtained from all the families according to protocols approved by local institutional review boards and human research ethics committees. Permission for clinical photographs was given separately. Individual 2 was recruited to the Murdoch Children’s Research Institute’s Undiagnosed Diseases Project Victoria for gene discovery after a nondiagnostic clinical exome. Trio exome sequencing was undertaken at the Broad Center for Mendelian Genomics.
Genetic analysis
Genomic DNA was extracted from whole blood from the affected children and their parents. Exome sequencing was performed with a variety of standard capture kits, and data analysis was performed independently. In patients 9 and 11, exome sequencing was performed in the framework of the German project “TRANSLATE NAMSE,” an initiative from the National Action League for People with Rare Diseases [Nationales Aktionsbündnis für Menschen mit Seltenen Erkrankungen (NAMSE)] facilitating innovative genetic diagnostics for individuals with suggested rare diseases. RNA was extracted from fibroblasts, and an LCL was obtained from individual 1 and control LCLs using the RNeasy Mini Kit with on-column DNaseI digestion (Qiagen, catalog nos. 74104 and 79254), according to the manufacturer’s instructions. Total RNA (2 μg) was reverse-transcribed to cDNA with the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, catalog no. 4368814) using random primers. PCR was performed with the cDNA as template using primers 5′-TTGCACTCGATTTGAAGACTCT-3′ and 5′-AGTTAAGAAGTGACCACAGCTC-3′. cDNA from a commercial pooled tissue cDNA library (qPCR Human Reference cDNA, random primed; Clontech, catalog no. 639654) was included as a control. Subsequent Sanger sequencing was performed using the same primers.
Fly stocks
Ppk-CD4tdGFP, ppk-Gal4, UAS-Iswi RNAi, elav-Gal4, UAS-mCD8::GFP, and P{CaryP}attP2 have been previously described (47, 60). To generate the UAS- SMARCA5WT, UAS-SMARCA5R592Q, and UAS-SMARCA5268-319del stocks, the coding sequences were cloned into a pACU2 vector, and the constructs were then injected (Rainbow Transgenic Flies Inc). The experimental procedures have been approved by the Institutional Biosafety Committee at the Children’s Hospital of Philadelphia.
Live imaging in flies
Live imaging was performed as described (61). In brief, embryos were collected for 2 to 24 hours on yeasted grape juice agar plates and were aged at 25°C. At 96 hours after egg laying, larvae were mounted in 90% glycerol under coverslips sealed with grease and imaged using a Zeiss LSM 880 microscope. Neurons were reconstructed with Neurostudio for dendrite morphology analyses.
Immunohistochemistry
Third-instar larvae were dissected, and tissues were subjected to immunostaining as described according to standard protocols. The following antibodies were used: mouse anti-FLAG antibody (F3156, 1:1000, Sigma-Aldrich), rat anti–histone H3 (phospho-S28) antibody (ab10543, Abcam, 1:500), and fluorescence-conjugated secondary antibodies (1:1000, Jackson ImmunoResearch).
Negative geotaxis test
Negative geotaxis was performed as described previously (62). Three days after eclosion, 10 flies of the same genotype were placed in a 20-cm-high vial. After 2-min adaptation, the vial was gently tapped against the table so the flies would be knocked to the bottom of the vial. The pass rate is defined as the percentage of flies climbing beyond the 8-cm mark within 10 s. Each vial was assayed 10 times, and the average pass rate is defined as the average across 10 trials. Male and female flies were picked randomly.
Acknowledgments
We thank all of the families involved in this study for their participation. Funding: Research reported in this publication was supported, in part, by the Roberts Collaborative Functional Genomics Rapid Grant (to D.L. and M.A.D.) from CHOP and Institutional Development Funds (to H.H.) from CHOP. This work was supported by NIH grants DP2 NS111996 (to M.S.K.) and T32 HL07953 (to N.N.G.) and funding from the Burroughs Wellcome Fund (to M.S.K.). We are grateful for funding support for the Undiagnosed Diseases Program Victoria from the Harbig Family Foundation and the Murdoch Children’s Research Institute. The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program. Sequencing and analysis of individual 2 were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and were funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung, and Blood Institute (grant UM1 HG008900) and, in part, by National Human Genome Research Institute grant R01 HG009141. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant HICF-1009-003), a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute (grant WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of Wellcome or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12, granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research through the Comprehensive Clinical Research Network. Author contributions: D.L., A.K., H.B.F., M.B., K.M., M.J.G.S., E.W., E.J.B., M.J.M.N., C.G.-J., M.M., A.D.-W., H.L., J.A.M.-A., L.S.P., S.M.W., J.C., and H.H. contributed to the molecular evaluation of the affected individuals. N.B., K.G., E.W., J.H., M.J.M.N., A.S., D. Bartholomew, Y.H., E.M.C.S., T.B., D.C., D. Brown, K.L., T.G., D.T., T.Y.T., and M.A.D. contributed to the clinical evaluation of the affected individuals. D.L., Q.W., N.N.G., M.E.M., M.S.K., and Y.S. contributed to the functional investigations. All authors read, edited, and approved the manuscript. Competing interests: K.M. and M.J.G.S. are employees of GeneDx Inc. C.G.-J. is an employee of Regeneron. The authors declare no other competing interests. Data and materials availability: Patient variants are available in the Leiden Open Variation Database (LOVD; www.LOVD.nl/SMARCA5). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf2066/DC1
REFERENCES AND NOTES
- 1.Clapier C. R., Cairns B. R., The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Santen G. W. E., Aten E., Sun Y., Almomani R., Gilissen C., Nielsen M., Kant S. G., Snoeck I. N., Peeters E. A. J., Hilhorst-Hofstee Y., Wessels M. W., den Hollander N. S., Ruivenkamp C. A. L., van Ommen G.-J. B., Breuning M. H., den Dunnen J. T., van Haeringen A., Kriek M., Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 44, 379–380 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Nixon K. C. J., Rousseau J., Stone M. H., Sarikahya M., Ehresmann S., Mizuno S., Matsumoto N., Miyake N., Study D. D. D., Baralle D., McKee S., Izumi K., Ritter A. L., Heide S., Heron D., Depienne C., Titheradge H., Kramer J. M., Campeau P. M., A syndromic neurodevelopmental disorder caused by mutations in SMARCD1, a core SWI/SNF subunit needed for context-dependent neuronal gene regulation in flies. Am. J. Hum. Genet. 104, 596–610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsurusaki Y., Okamoto N., Ohashi H., Kosho T., Imai Y., Hibi-Ko Y., Kaname T., Naritomi K., Kawame H., Wakui K., Fukushima Y., Homma T., Kato M., Hiraki Y., Yamagata T., Yano S., Mizuno S., Sakazume S., Ishii T., Nagai T., Shiina M., Ogata K., Ohta T., Niikawa N., Miyatake S., Okada I., Mizuguchi T., Doi H., Saitsu H., Miyake N., Matsumoto N., Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–378 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Machol K., Rousseau J., Ehresmann S., Garcia T., Nguyen T. T. M., Spillmann R. C., Sullivan J. A., Shashi V., Jiang Y.-h., Stong N., Fiala E., Willing M., Pfundt R., Kleefstra T., Cho M. T., Laughlin H. M., Piera M. R., Orellana C., Martínez F., Caro-Llopis A., Monfort S., Roscioli T., Nixon C. Y., Buckley M. F., Turner A., Jones W. D., van Hasselt P. M., Hofstede F. C., van Gassen K. L. I., Brooks A. S., van Slegtenhorst M. A., Lachlan K., Sebastian J., Madan-Khetarpal S., Sonal D., Sakkubai N., Thevenon J., Faivre L., Maurel A., Petrovski S., Krantz I. D., Tarpinian J. M., Rosenfeld J. A., Lee B. H.; Undiagnosed Diseases Network, Campeau P. M., Expanding the spectrum of BAF-related disorders: De novo variants in SMARCC2 cause a syndrome with intellectual disability and developmental delay. Am. J. Hum. Genet. 104, 164–178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wieczorek D., Bögershausen N., Beleggia F., Steiner-Haldenstätt S., Pohl E., Li Y., Milz E., Martin M., Thiele H., Altmüller J., Alanay Y., Kayserili H., Klein-Hitpass L., Böhringer S., Wollstein A., Albrecht B., Boduroglu K., Caliebe A., Chrzanowska K., Cogulu O., Cristofoli F., Czeschik J. C., Devriendt K., Dotti M. T., Elcioglu N., Gener B., Goecke T. O., Krajewska-Walasek M., Guillén-Navarro E., Hayek J., Houge G., Kilic E., Simsek-Kiper P. Ö., López-González V., Kuechler A., Lyonnet S., Mari F., Marozza A., Dramard M. M., Mikat B., Morin G., Morice-Picard F., Özkinay F., Rauch A., Renieri A., Tinschert S., Utine G. E., Vilain C., Vivarelli R., Zweier C., Nürnberg P., Rahmann S., Vermeesch J., Lüdecke H.-J., Zeschnigk M., Wollnik B., A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 22, 5121–5135 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Shang L., Cho M. T., Retterer K., Folk L., Humberson J., Rohena L., Sidhu A., Saliganan S., Iglesias A., Vitazka P., Juusola J., O’Donnell-Luria A. H., Shen Y., Chung W. K., Mutations in ARID2 are associated with intellectual disabilities. Neurogenetics 16, 307–314 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Vasileiou G., Vergarajauregui S., Endele S., Popp B., Büttner C., Ekici A. B., Gerard M., Bramswig N. C., Albrecht B., Clayton-Smith J., Morton J., Tomkins S., Low K., Weber A., Wenzel M., Altmüller J., Li Y., Wollnik B., Hoganson G., Plona M.-R., Cho M. T.; Deciphering Developmental Disorders Study, Thiel C. T., Lüdecke H.-J., Strom T. M., Calpena E., Wilkie A. O. M., Wieczorek D., Engel F. B., Reis A., Mutations in the BAF-complex subunit DPF2 are associated with Coffin-Siris syndrome. Am. J. Hum. Genet. 102, 468–479 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Houdt J. K. J., Nowakowska B. A., Sousa S. B., van Schaik B. D. C., Seuntjens E., Avonce N., Sifrim A., Abdul-Rahman O. A., van den Boogaard M.-J. H., Bottani A., Castori M., Cormier-Daire V., Deardorff M. A., Filges I., Fryer A., Fryns J.-P., Gana S., Garavelli L., Gillessen-Kaesbach G., Hall B. D., Horn D., Huylebroeck D., Klapecki J., Krajewska-Walasek M., Kuechler A., Lines M. A., Maas S., MacDermot K. D., Kee S. M., Magee A., de Man S. A., Moreau Y., Morice-Picard F., Obersztyn E., Pilch J., Rosser E., Shannon N., Stolte-Dijkstra I., Van Dijck P., Vilain C., Vogels A., Wakeling E., Wieczorek D., Wilson L., Zuffardi O., van Kampen A. H. C., Devriendt K., Hennekam R., Vermeesch J. R., Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat. Genet. 44, 445–449 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Wolff D., Endele S., Azzarello-Burri S., Hoyer J., Zweier M., Schanze I., Schmitt B., Rauch A., Reis A., Zweier C., In-frame deletion and missense mutations of the C-terminal helicase domain of SMARCA2 in three patients with Nicolaides-Baraitser syndrome. Mol. Syndromol. 2, 237–244 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dias C., Estruch S. B., Graham S. A., Rae J. M., Sawiak S. J., Hurst J. A., Joss S. K., Holder S. E., Morton J. E. V., Turner C., Thevenon J., Mellul K., Sánchez-Andrade G., Ibarra-Soria X., Deriziotis P., Santos R. F., Lee S.-C., Faivre L., Kleefstra T., Liu P., Hurles M. E.; D. D. D. Study, Fisher S. E., Logan D. W., BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am. J. Hum. Genet. 99, 253–274 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bell S., Rousseau J., Peng H., Aouabed Z., Priam P., Theroux J.-F., Jefri M., Tanti A., Wu H., Kolobova I., Silviera H., Manzano-Vargas K., Ehresmann S., Hamdan F. F., Hettige N., Zhang X., Antonyan L., Nassif C., Ghaloul-Gonzalez L., Sebastian J., Vockley J., Begtrup A. G., Wentzensen I. M., Crunk A., Nicholls R. D., Herman K. C., Deignan J. L., Al-Hertani W., Efthymiou S., Salpietro V., Miyake N., Makita Y., Matsumoto N., Østern R., Houge G., Hafström M., Fassi E., Houlden H., Klein Wassink-Ruiter J. S., Nelson D., Goldstein A., Dabir T., van Gils J., Bourgeron T., Delorme R., Cooper G. M., Martinez J. E., Finnila C. R., Carmant L., Lortie A., Oegema R., van Gassen K., Mehta S. G., Huhle D., Jamra R. A., Martin S., Brunner H. G., Lindhout D., Au M., Graham J. M. Jr., Coubes C., Turecki G., Gravel S., Mechawar N., Rossignol E., Michaud J. L., Lessard J., Ernst C., Campeau P. M., Mutations in ACTL6B cause neurodevelopmental deficits and epilepsy and lead to loss of dendrites in human neurons. Am. J. Hum. Genet. 104, 815–834 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivière J.-B., van Bon B. W. M., Hoischen A., Kholmanskikh S. S., O’Roak B. J., Gilissen C., Gijsen S., Sullivan C. T., Christian S. L., Abdul-Rahman O. A., Atkin J. F., Chassaing N., Drouin-Garraud V., Fry A. E., Fryns J.-P., Gripp K. W., Kempers M., Kleefstra T., Mancini G. M. S., Nowaczyk M. J. M., van Ravenswaaij-Arts C. M. A., Roscioli T., Marble M., Rosenfeld J. A., Siu V. M., de Vries B. B. A., Shendure J., Verloes A., Veltman J. A., Brunner H. G., Ross M. E., Pilz D. T., Dobyns W. B., De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 44, 440–444 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sifrim A., Hitz M.-P., Wilsdon A., Breckpot J., Al Turki S. H., Thienpont B., Rae J. M., Fitzgerald T. W., Singh T., Swaminathan G. J., Prigmore E., Rajan D., Abdul-Khaliq H., Banka S., Bauer U. M. M., Bentham J., Berger F., Bhattacharya S., Bu’Lock F., Canham N., Colgiu I.-G., Cosgrove C., Cox H., Daehnert I., Daly A., Danesh J., Fryer A., Gewillig M., Hobson E., Hoff K., Homfray T.; INTERVAL Study, Kahlert A.-K., Ketley A., Kramer H.-H., Lachlan K., Lampe A. K., Louw J. J., Manickara A. K., Manase D., McCarthy K. P., Metcalfe K., Moore C., Newbury-Ecob R., Omer S. O., Ouwehand W. H., Park S.-M., Parker M. J., Pickardt T., Pollard M. O., Robert L., Roberts D. J., Sambrook J., Setchfield K., Stiller B., Thornborough C., Toka O., Watkins H., Williams D., Wright M., Mital S., Daubeney P. E. F., Keavney B., Goodship J.; UK10K Consortium, Abu-Sulaiman R. M., Klaassen S., Wright C. F., Firth H. V., Barrett J. C., Devriendt K., Patrick D. R. F., Brook J. D.; Deciphering Developmental Disorders Study, Hurles M. E., Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 48, 1060–1065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiss K., Terhal P. A., Cohen L., Bruccoleri M., Irving M., Martinez A. F., Rosenfeld J. A., Machol K., Yang Y., Liu P., Walkiewicz M., Beuten J., Gomez-Ospina N., Haude K., Fong C.-T., Enns G. M., Bernstein J. A., Fan J., Gotway G., Ghorbani M.; D. D. D. Study, van Gassen K., Monroe G. R., van Haaften G., Basel-Vanagaite L., Yang X.-J., Campeau P. M., Muenke M., De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am. J. Hum. Genet. 99, 934–941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vissers L. E. L. M., van Ravenswaaij C. M. A., Admiraal R., Hurst J. A., de Vries B. B. A., Janssen I. M., van der Vliet W. A., Huys E. H. L. P. G., de Jong P. J., Hamel B. C. J., Schoenmakers E. F. P. M., Brunner H. G., Veltman J. A., van Kessel A. G., Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Blok L. S., Rousseau J., Twist J., Ehresmann S., Takaku M., Venselaar H., Rodan L. H., Nowak C. B., Douglas J., Swoboda K. J., Steeves M. A., Sahai I., Stumpel C. T. R. M., Stegmann A. P. A., Wheeler P., Willing M., Fiala E., Kochhar A., Gibson W. T., Cohen A. S. A., Agbahovbe R., Innes A. M., Au P. Y. B., Rankin J., Anderson I. J., Skinner S. A., Louie R. J., Warren H. E., Afenjar A., Keren B., Nava C., Buratti J., Isapof A., Rodriguez D., Lewandowski R., Propst J., van Essen T., Choi M., Lee S., Chae J. H., Price S., Schnur R. E., Douglas G., Wentzensen I. M., Zweier C., Reis A., Bialer M. G., Moore C., Koopmans M., Brilstra E. H., Monroe G. R., van Gassen K. L. I., van Binsbergen E., Newbury-Ecob R., Bownass L., Bader I., Mayr J. A., Wortmann S. B., Jakielski K. J., Strand E. A., Kloth K., Bierhals T.; The DDD study, Roberts J. D., Petrovich R. M., Machida S., Kurumizaka H., Lelieveld S., Pfundt R., Jansen S., Deriziotis P., Faivre L., Thevenon J., Assoum M., Shriberg L., Kleefstra T., Brunner H. G., Wade P. A., Fisher S. E., Campeau P. M., CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat. Commun. 9, 4619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stankiewicz P., Khan T. N., Szafranski P., Slattery L., Streff H., Vetrini F., Bernstein J. A., Brown C. W., Rosenfeld J. A., Rednam S., Scollon S., Bergstrom K. L., Parsons D. W., Plon S. E., Vieira M. W., Quaio C. R. D. C., Baratela W. A. R., Guio J. C. A., Armstrong R., Mehta S. G., Rump P., Pfundt R., Lewandowski R., Fernandes E. M., Shinde D. N., Tang S., Hoyer J., Zweier C., Reis A., Bacino C. A., Xiao R., Breman A. M., Smith J. L.; Deciphering Developmental Disorders Study, Katsanis N., Bostwick B., Popp B., Davis E. E., Yang Y., Haploinsufficiency of the chromatin remodeler BPTF causes syndromic developmental and speech delay, postnatal microcephaly, and dysmorphic features. Am. J. Hum. Genet. 101, 503–515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karaca E., Harel T., Pehlivan D., Jhangiani S. N., Gambin T., Akdemir Z. C., Gonzaga-Jauregui C., Erdin S., Bayram Y., Campbell I. M., Hunter J. V., Atik M. M., Van Esch H., Yuan B., Wiszniewski W., Isikay S., Yesil G., Yuregir O. O., Bozdogan S. T., Aslan H., Aydin H., Tos T., Aksoy A., De Vivo D. C., Jain P., Geckinli B. B., Sezer O., Gul D., Durmaz B., Cogulu O., Ozkinay F., Topcu V., Candan S., Cebi A. H., Ikbal M., Gulec E. Y., Gezdirici A., Koparir E., Ekici F., Coskun S., Cicek S., Karaer K., Koparir A., Duz M. B., Kirat E., Fenercioglu E., Ulucan H., Seven M., Guran T., Elcioglu N., Yildirim M. S., Aktas D., Alikaşifoğlu M., Ture M., Yakut T., Overton J. D., Yuksel A., Ozen M., Muzny D. M., Adams D. R., Boerwinkle E., Chung W. K., Gibbs R. A., Lupski J. R., Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron 88, 499–513 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopes F., Barbosa M., Ameur A., Soares G., de Sá J., Dias A. I., Oliveira G., Cabral P., Temudo T., Calado E., Cruz I. F., Vieira J. P., Oliveira R., Esteves S., Sauer S., Jonasson I., Syvänen A.-C., Gyllensten U., Pinto D., Maciel P., Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 53, 190–199 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Tsukiyama T., Daniel C., Tamkun J., Wu C., ISWI, a member of the SWI2/SNF2 ATPase family, encodes the 140 kDa subunit of the nucleosome remodeling factor. Cell 83, 1021–1026 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Ito T., Bulger M., Pazin M. J., Kobayashi R., Kadonaga J. T., ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 90, 145–155 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Varga-Weisz P. D., Wilm M., Bonte E., Dumas K., Mann M., Becker P. B., Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature 388, 598–602 (1997). [DOI] [PubMed] [Google Scholar]
- 24.LeRoy G., Orphanides G., Lane W. S., Reinberg D., Requirement of RSF and FACT for transcription of chromatin templates in vitro. Science 282, 1900–1904 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Bochar D. A., Savard J., Wang W., Lafleur D. W., Moore P., Côté J., Shiekhattar R., A family of chromatin remodeling factors related to Williams syndrome transcription factor. Proc. Natl. Acad. Sci. U.S.A. 97, 1038–1043 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barak O., Lazzaro M. A., Lane W. S., Speicher D. W., Picketts D. J., Shiekhattar R., Isolation of human NURF: A regulator of Engrailed gene expression. EMBO J. 22, 6089–6100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poot R. A., Dellaire G., Hulsmann B. B., Grimaldi M. A., Corona D. F., Becker P. B., Bickmore W. A., Varga-Weisz P. D., HuCHRAC, a human ISWI chromatin remodelling complex contains hACF1 and two novel histone-fold proteins. EMBO J. 19, 3377–3387 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banting G. S., Barak O., Ames T. M., Burnham A. C., Kardel M. D., Cooch N. S., Davidson C. E., Godbout R., McDermid H. E., Shiekhattar R., CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum. Mol. Genet. 14, 513–524 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Strohner R., Nemeth A., Jansa P., Hofmann-Rohrer U., Santoro R., Längst G., Grummt I., NoRC—A novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. 20, 4892–4900 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakimi M.-A., Bochar D. A., Schmiesing J. A., Dong Y., Barak O. G., Speicher D. W., Yokomori K., Shiekhattar R., A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 418, 994–998 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Bozhenok L., Wade P. A., Varga-Weisz P., WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J. 21, 2231–2241 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oppikofer M., Bai T., Gan Y., Haley B., Liu P., Sandoval W., Ciferri C., Cochran A. G., Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep. 18, 1697–1706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguirre A., Montserrat N., Zacchigna S., Nivet E., Hishida T., Krause M. N., Kurian L., Ocampo A., Vazquez-Ferrer E., Rodriguez-Esteban C., Kumar S., Moresco J. J., Yates J. R. III, Campistol J. M., Sancho-Martinez I., Giacca M., Izpisua Belmonte J. C., In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 15, 589–604 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvarez-Saavedra M., De Repentigny Y., Lagali P. S., Raghu Ram E. V., Yan K., Hashem E., Ivanochko D., Huh M. S., Yang D., Mears A. J., Todd M. A., Corcoran C. P., Bassett E. A., Tokarew N. J., Kokavec J., Majumder R., Ioshikhes I., Wallace V. A., Kothary R., Meshorer E., Stopka T., Skoultchi A. I., Picketts D. J., Snf2h-mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat. Commun. 5, 4181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazzaro M. A., Picketts D. J., Cloning and characterization of the murine Imitation Switch (ISWI) genes: Differential expression patterns suggest distinct developmental roles for Snf2h and Snf2l. J. Neurochem. 77, 1145–1156 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Sobreira N., Schiettecatte F., Valle D., Hamosh A., GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armache J. P., Gamarra N., Johnson S. L., Leonard J. D., Wu S., Narlikar G. J., Cheng Y., Cryo-EM structures of remodeler-nucleosome intermediates suggest allosteric control through the nucleosome. eLife 8, e46057 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldman J. A., Garlick J. D., Kingston R. E., Chromatin remodeling by imitation switch (ISWI) class ATP-dependent remodelers is stimulated by histone variant H2A.Z. J. Biol. Chem. 285, 4645–4651 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dao H. T., Dul B. E., Dann G. P., Liszczak G. P., Muir T. W., A basic motif anchoring ISWI to nucleosome acidic patch regulates nucleosome spacing. Nat. Chem. Biol. 16, 134–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grüne T., Brzeski J., Eberharter A., Clapier C. R., Corona D. F. V., Becker P. B., Müller C. W., Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol. Cell 12, 449–460 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Deuring R., Fanti L., Armstrong J. A., Sarte M., Papoulas O., Prestel M., Daubresse G., Verardo M., Moseley S. L., Berloco M., Tsukiyama T., Wu C., Pimpinelli S., Tamkun J. W., The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol. Cell 5, 355–365 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Corona D. F. V., Siriaco G., Armstrong J. A., Snarskaya N., McClymont S. A., Scott M. P., Tamkun J. W., ISWI regulates higher-order chromatin structure and histone H1 assembly in vivo. PLOS Biol. 5, e232 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grueber W. B., Ye B., Yang C.-H., Younger S., Borden K., Jan L. Y., Jan Y.-N., Projections of Drosophila multidendritic neurons in the central nervous system: Links with peripheral dendrite morphology. Development 134, 55–64 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Grueber W. B., Jan L. Y., Jan Y. N., Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development 129, 2867–2878 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Luo L., Liao Y. J., Jan L. Y., Jan Y. N., Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 8, 1787–1802 (1994). [DOI] [PubMed] [Google Scholar]
- 46.Aso Y., Hattori D., Yu Y., Johnston R. M., Iyer N. A., Ngo T.-T., Dionne H., Abbott L. F., Axel R., Tanimoto H., Rubin G. M., The neuronal architecture of the mushroom body provides a logic for associative learning. eLife 3, e04577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gong N. N., Dilley L. C., Williams C. E., Moscato E. H., Szuperak M., Wang Q., Jensen M., Girirajan S., Tan T. Y., Deardorff M. A., Li D., Song Y., Kayser M. S., The chromatin remodeler ISWI acts during Drosophila development to regulate adult sleep. Sci. Adv. 7, eabe2597 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito K., Awano W., Suzuki K., Hiromi Y., Yamamoto D., The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development 124, 761–771 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Bögershausen N., Wollnik B., Mutational landscapes and phenotypic spectrum of SWI/SNF-related intellectual disability disorders. Front. Mol. Neurosci. 11, 252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drivas T. G., Li D., Nair D., Alaimo J. T., Alders M., Altmüller J., Barakat T. S., Bebin E. M., Bertsch N. L., Blackburn P. R., Blesson A., Bouman A. M., Brockmann K., Brunelle P., Burmeister M., Cooper G. M., Denecke J., Dieux-Coëslier A., Dubbs H., Ferrer A., Gal D., Bartik L. E., Gunderson L. B., Hasadsri L., Jain M., Karimov C., Keena B., Klee E. W., Kloth K., Lace B., Macchiaiolo M., Marcadier J. L., Milunsky J. M., Napier M. P., Ortiz-Gonzalez X. R., Pichurin P. N., Pinner J., Powis Z., Prasad C., Radio F. C., Rasmussen K. J., Renaud D. L., Rush E. T., Saunders C., Selcen D., Seman A. R., Shinde D. N., Smith E. D., Smol T., Blok L. S., Stoler J. M., Tang S., Tartaglia M., Thompson M. L., van de Kamp J. M., Wang J., Weise D., Weiss K., Woitschach R., Wollnik B., Yan H., Zackai E. H., Zampino G., Campeau P., Bhoj E., A second cohort of CHD3 patients expands the molecular mechanisms known to cause Snijders Blok-Campeau syndrome. Eur. J. Hum. Genet. 28, 1422–1431 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mannino E. A., Miyawaki H., Santen G., Schrier Vergano S. A., First data from a parent-reported registry of 81 individuals with Coffin-Siris syndrome: Natural history and management recommendations. Am. J. Med. Genet. A 176, 2250–2258 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Homann O. R., Misura K., Lamas E., Sandrock R. W., Nelson P., McDonough S. I., DeLisi L. E., Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol. Psychiatry 21, 1690–1695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strehle E.-M., Yu L., Rosenfeld J. A., Donkervoort S., Zhou Y., Chen T.-J., Martinez J. E., Fan Y.-S., Barbouth D., Zhu H., Vaglio A., Smith R., Stevens C. A., Curry C. J., Ladda R. L., Fan Z. J., Fox J. E., Martin J. A., Abdel-Hamid H. Z., McCracken E. A., McGillivray B. C., Masser-Frye D., Huang T., Genotype-phenotype analysis of 4q deletion syndrome: Proposal of a critical region. Am. J. Med. Genet A. 158A, 2139–2151 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Duga B., Czako M., Komlosi K., Hadzsiev K., Torok K., Sumegi K., Kisfali P., Kosztolanyi G., Melegh B., Deletion of 4q28.3-31.23 in the background of multiple malformations with pulmonary hypertension. Mol. Cytogenet. 7, 36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rim J. H., Kim S. W., Han S.-H., Yoo J., Clinical and molecular delineation of a novel de novo 4q28.3-31.21 interstitial deletion in a patient with developmental delay. Yonsei Med. J. 56, 1742–1744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santen G. W. E., Aten E., Vulto-van Silfhout A. T., Pottinger C., van Bon B. W. M., van Minderhout I. J. H. M., Snowdowne R., van der Lans C. A. C., Boogaard M., Linssen M. M. L., Vijfhuizen L., van der Wielen M. J. R., Vollebregt M. J. E.; Coffin-Siris Consortium, Breuning M. H., Kriek M., van Haeringen A., den Dunnen J. T., Hoischen A., Clayton-Smith J., de Vries B. B. A., Hennekam R. C. M., van Belzen M. J., Coffin-Siris syndrome and the BAF complex: Genotype-phenotype study in 63 patients. Hum. Mutat. 34, 1519–1528 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Cappuccio G., Sayou C., Tanno P. L., Tisserant E., Bruel A.-L., Kennani S. E., Sá J., Low K. J., Dias C., Havlovicová M., Hančárová M., Eichler E. E., Devillard F., Moutton S., Van-Gils J., Dubourg C., Odent S., Gerard B., Piton A., Yamamoto T., Okamoto N., Firth H., Metcalfe K., Moh A., Chapman K. A., Aref-Eshghi E., Kerkhof J., Torella A., Nigro V., Perrin L., Piard J., Guyader G. L., Jouan T., Thauvin-Robinet C., Duffourd Y., George-Abraham J. K., Buchanan C. A., Williams D., Kini U., Wilson K.; Telethon Undiagnosed Diseases Program, Sousa S. B., Hennekam R. C. M., Sadikovic B., Thevenon J., Govin J., Vitobello A., Brunetti-Pierri N., De novo SMARCA2 variants clustered outside the helicase domain cause a new recognizable syndrome with intellectual disability and blepharophimosis distinct from Nicolaides-Baraitser syndrome. Genet. Med. 22, 1838–1850 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Alvarez-Saavedra M., Yan K., De Repentigny Y., Hashem L. E., Chaudary N., Sarwar S., Yang D., Ioshikhes I., Kothary R., Hirayama T., Yagi T., Picketts D. J., Snf2h drives chromatin remodeling to prime upper layer cortical neuron development. Front. Mol. Neurosci. 12, 243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xi R., Xie T., Stem cell self-renewal controlled by chromatin remodeling factors. Science 310, 1487–1489 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Han C., Jan L. Y., Jan Y.-N., Enhancer-driven membrane markers for analysis of nonautonomous mechanisms reveal neuron-glia interactions in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 108, 9673–9678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song Y., Li D., Farrelly O., Miles L., Li F., Kim S. E., Lo T. Y., Wang F., Li T., Thompson-Peer K. L., Gong J., Murthy S. E., Coste B., Yakubovich N., Patapoutian A., Xiang Y., Rompolas P., Jan L. Y., Jan Y. N., The mechanosensitive ion channel piezo inhibits axon regeneration. Neuron 102, 373–389.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ali Y. O., Escala W., Ruan K., Zhai R. G., Assaying locomotor, learning, and memory deficits in Drosophila models of neurodegeneration. J. Vis. Exp. 11, 2504 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf2066/DC1