

New exosome-laden scaffolds shed light on the immune cell activities that benefit tissue repair.
Abstract
Designing scaffolds capable of inducing and guiding appropriate immune responses holds promise for tissue repair/regeneration. Biofunctional scaffolds were here prepared by immobilizing mesenchymal stromal exosomes onto fibrous polyester materials and allowed cell-mediated delivery of membrane-bound vesicles. Quantitative cell-level analyses revealed that immune cells dominated the uptake of exosomes from scaffolds in vivo, with materials and exosomes acting as the recruiter and trainer for immune cells, respectively, to synergistically promote beneficial macrophage and regulatory T cell responses in skin wounds in mice. Adaptive T helper cell responses were found active in remote immune organs, and exosome-laden scaffolds facilitated tissue repair in large skin injury models. This study demonstrated important mechanisms involved in local and systemic immune responses to biological implants, and understanding tissue-reparative immunomodulation may guide the design of new biofunctional scaffolds.
INTRODUCTION
The immune system not only is our body’s first line of defense against the invasion of pathogens but also plays pivotal roles in mediating tissue development, homeostasis, and reparative processes. Through a variety of orchestrated events, immune cells and mediators affect tissue repair and regeneration processes by being involved in inflammation, angiogenesis, and stem/progenitor cell activities such as proliferation and differentiation (1–3). Among the innate immune responses, the macrophage (MΦ) polarization from the proinflammatory M1-like toward the reparative M2-like phenotypes was found abundant to promote tissue repair (4, 5). The adaptive immune cells are also active in their reciprocal communications with the innate cells: CD4+ T helper 2 (TH2) cells and regulatory T cells (Tregs) were capable of secreting cytokines such as interleukin-4 (IL-4) and IL-10 and transforming growth factor–β (TGF-β) to favor the MΦ transition toward the immunomodulatory M2-like phenotype (1–3, 6). Tregs also produce growth factors that can directly induce the proliferation and differentiation of the tissue-resident progenitors/stem cells (7, 8). On the other hand, CD8+ T cells and CD4+ TH1 cells secrete tumor necrosis factor–α (TNF-α) and interferon-γ (IFN-γ), which would prolong the existence of proinflammatory MΦ and induce stem cell apoptosis (9).
Designing biomaterials to modulate the immune system toward pro-regenerative responses can shed light on the future direction of regenerative medicine. Investigations into the innate and adaptive immune responses within the context of biomaterials began to emerge in recent years, showing that biofunctional scaffolds could promote tissue regeneration by modulating local MΦs and their cross-talk with T cells (10–12). Understanding and engineering the respective roles of the scaffold materials and biologics for initiating the beneficial immune responses particularly on the systemic level, however, is still to be explored. Toward this end, a new type of cell-free biological scaffolds was constructed here by immobilizing exosomes derived from mesenchymal stromal cells (MSCs) onto the fibrous polymer meshes; the innate and adaptive immune responses in the local tissue as well as in the remote lymphatic organs in the skin wound models were investigated.
In the form of membrane-surrounded extracellular vesicles, exosomes shed from cells shuttle the messaging cargos of proteins and nucleic acids to mediate intercellular communications. In particular, MSCs harvested from various tissues are known to have immunomodulatory functions and offer promising therapeutic options in transplant surgeries and treatment of graft-versus-host diseases (13, 14). Similar to the parent MSCs, the immunomodulation function associated with MSC exosomes involves promoting the M2-like MΦ phenotype, Treg population, and TH2 immune responses (15–18). As an alternative to live cells, MSC exosomes are promising for use as biological entities to develop therapies with equivalent or superior efficacies (19–21). Here, it is of great interest to understand whether scaffolds and exosomes could be developed as a combinatorial cell-free system to initiate synergistic immune responses on local and systemic levels. This study could potentially facilitate the development of new biofunctional scaffolds on the basis of the action mechanisms of these systems within the complex biological environment in the host.
RESULTS
Preparation of exosome-functionalized scaffolds and cellular uptake of exosomes from scaffolds in vitro and in vivo
To prepare exosome-functionalized scaffolds, polycaprolactone (PCL) fibers were fabricated through an electrospinning process followed by a multistep chemical treatment to conjugate polyethylenimine (PEI) molecules onto the porous structure (fig. S1A). After chemical modification, the nitrogen element and amine groups corresponding to the conjugated PEI on the fiber surface were detected (fig. S1, B and C); the diameters of PCL fibers remained unchanged, yet the hydrophilicity of the material surfaces was increased (fig. S1, D and E). MSC exosomes were analyzed for purity to show exosome makers and the absence of cellular or serum contaminants (tables S1 and S2). Vesicles had the classic “cup-shaped” morphology (Fig. 1B), with an average diameter of 76.7 ± 25.4 nm and zeta potential of −30.8 ± 9.8 mV. The PEI-modified electrospun fibers (PEFs) became positively charged and enabled tethering of MSC exosomes to PCL fiber surfaces as a result of the negative potential of exosome membranes (Fig. 1, A to D). The amounts of exosomes immobilized onto PEF were controllable and plateaued to reach a maximum level that the PCL scaffold could carry (Fig. 1E). The binding between MSC exosomes and PCL scaffolds was found to be stable in the buffer solutions, only showing a minimal initial detachment of the exosome residuals (fig. S1F). PEF bearing MSC exosomes (Exo-PEF) was then compared to suspended exosome vesicles. Notably, being tethered to the polyester surface did not compromise the function of MSC exosomes to modulate MΦ toward the immunomodulatory M2-like phenotype in vitro. It was noted that the gene expression of the M2 marker CD206 and the anti-inflammatory cytokines including IL-4 and IL-10 were enhanced by exosome-functionalized scaffolds, possibly because of the added influence of the scaffold’s microenvironment (Fig. 1, F to H) (22, 23).
Fig. 1. MSC exosome–functionalized fibrous scaffolds showed immunomodulatory effects on MΦs cultured in vitro.
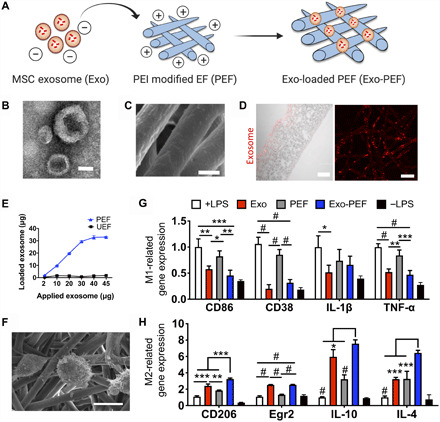
(A) Schematic of immobilizing exosomes to polycaprolactone (PCL) scaffolds through electrostatic interaction. (B) TEM imaging of MSC exosomes. (C) Scanning electron microscopy (SEM) imaging of Exo-PEF fibers. (D) Transverse (left) and vertical (right) imaging of PCL fibers modified with DI-labeled exosomes visualized via red fluorescence. (E) Quantities of exosomes immobilized onto PEF in variation with the initial exosome quantities added for reaction and comparison to the unmodified substrates (UEF). (F) Lipopolysaccharides (LPS)–stimulated bone marrow–derived MΦs (BMDMs) cultured on Exo-PEF. (G) Expression of M1 MΦ-related genes (CD86, CD38, IL-1β, and TNF-α) and (H) M2 MΦ-related genes (CD206, Egr2, IL-10, and IL-4) in the LPS-stimulated MΦs treated with suspended exosomes (Exo), PEF, or Exo-PEF; BMDMs with or without LPS stimulation cultured on tissue plates serving as +LPS and –LPS controls, respectively. Exosome dosage: 10 μg of protein mass (n = 5). Scale bars, 50 nm (B), 2 μm (C), and 100 μm (D, left)/20 μm (D, right). *P < 0.05, **P < 0.01, ***P < 0.001, and #P < 0.0001.
To understand the delivery of exosomes from scaffolds to cells, MΦ cell line RAW 264.7 or human umbilical vein endothelial cells (HUVECs) were seeded on Exo-PEF scaffolds and compared to the counterpart system where cells were cultured in the bottom chambers in separation with the scaffold materials placed in the upper transwell (Fig. 2A). When the labeled exosomes were examined, only those cells that were cultured directly on the surface of Exo-PEF scaffolds showed exosome+ signals (Fig. 2, B and C). The results indicated that exosomes immobilized onto scaffolds would not be released unless they were in direct contact with cells and then sequestered by them, suggesting a possible interactive process involved in exosomes reaching to cells. The two control groups, with exosomes applied to the upper chamber with or without plain unmodified electrospun fibers (UEFs), indicated that exosomes were able to permeate the transwell membrane and reach cells at the bottom chamber if exosomes were not immobilized to scaffolds.
Fig. 2. The uptake of exosomes from scaffolds was mediated through cell contact, and Exo-PEF prolonged exosome retention, showing dominant exosome uptake by MΦs in wounds.
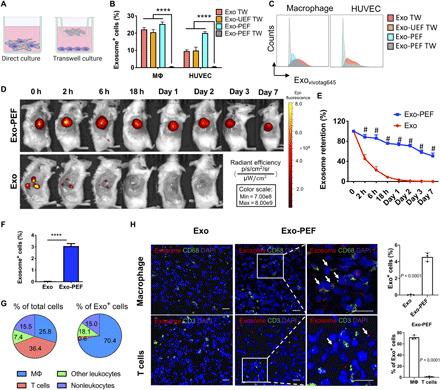
(A) Schematic of studying the exosome uptake where cells were either directly seeded on or separated from Exo-PEF through the transwell culture. (B and C) Using liquid exosomes directly supplied to the upper chamber of the transwells with or without UEF as control groups (Exo TW and Exo-UEF TW), the exosome uptake by MΦs and HUVECs cultured on Exo-PEF or without the contact of Exo-PEF (Exo-PEF TW) was compared: (B) percent exosome-positive cells and (C) representative flow cytometry histograms. (n = 5) (D) In vivo imaging of fluorophore-labeled exosomes in tissues receiving subcutaneous exosome injection (Exo) or Exo-PEF implantation and (E) comparison of exosome retention over 7 days based on quantitative analysis. Exosome dosage: 15 μg of protein mass. (F) Percentages of exosome+ cells in tissues harvested on day 7 after injury. (G) Average percent distributions of different cell types in total cells or in pregated exosome+ cells as analyzed by flow cytometry (n = 5). (H) Fluorescent imaging and statistic calculation of the exosome (red) uptake by MΦ (CD68 in green) or T cells (CD3 in green) in the wounds on day 7; arrows show the exosome colocalization with CD68 receptors on MΦ cell surfaces but no overlap with T cells. Scale bar, 20 μm. # or ****P < 0.0001. Blue, DAPI showing cell nuclei.
To investigate the cellular uptake of exosomes in vivo, the wounded skin tissues in mice were treated with Exo-PEF and harvested on day 7 after injury. The exosome retention was monitored through the labeled fluorescent exosomes. It was shown that the subcutaneously injected exosomes were undetectable after 24 hours in vivo, whereas the exosome signals persisted in the Exo-PEF group, with 50.8 ± 5.8% fluorescence remaining on day 7 versus day 0 (Fig. 2, D and E). Specific cell types in exosome+ cells in the Exo-PEF group were divided and identified in four groups: MΦ (CD45+F4/80+), T cells (CD45+CD3+), other leukocytes (CD45+CD3–F4/80–), and nonleukocytes (CD45–). In general, about 3% of the cells in the harvested tissue carried the exosome fluorescent signals (Fig. 2F). Notably, MΦs accounted for 70.4 ± 4.8% of the exosome+ cells, with the MΦ population at the level of 25.8 ± 2.5% within the wounds on day 7. With a total cell population comparable to MΦ, T cells carrying the exosome signals, however, were at a much lower level, making only 3% of the exosome+ cell population (Fig. 2G). Immunostaining and microscopic imaging validated the flow cytometry identification results: Exosomes were found to colocalize with CD68+ MΦs rather than CD3+ T cells (Fig. 2H). Other types of leukocytes and nonleukocytes had similar levels on taking up exosomes from Exo-PEF scaffolds according to quantitative analyses, and each category contributed to lower than 20% of the exosome+ cells (Fig. 2G).
Immunomodulatory responses induced in local tissue
To understand the immunomodulatory effects of Exo-PEF scaffolds in vivo, Exo-PEF materials were studied in skin wounds in mice with the sham, injected exosomes (Exo) and blank PEF controls for over 2 weeks (Fig. 3A). As shown in Fig. 3 (B and E), the implantation of the scaffold materials, PEF or Exo-PEF, increased the number of MΦs (Fig. 3B), which was most pronounced on day 7. The MΦ response was differentiated through the analysis of MΦ subtypes: PEF treatment significantly led to the accumulation of the proinflammatory, CD86+ M1-like MΦs at all three time points (Fig. 3C), while Exo-PEF resulted in a onefold greater number of immunomodulatory, CD206+ M2-like MΦs compared to wound controls on days 3 and 7 (Fig. 3D). In comparison to M2/M1-like MΦ ratios, MΦ polarization from the M1-like (day 3) to the M2-like (day 7 and beyond) subtype was evident in all groups, with the Exo-PEF group inducing an early and most significant dominance of immunomodulatory M2-like MΦs on day 7 (Fig. 3E). To validate the effects of Exo-PEF, scaffolds were also studied in a subcutaneous implantation model in the nonpathological skin tissue in mice. Analyses showed similar proinflammatory- and immunomodulatory-biased MΦ responses in vivo resulting from PEF and Exo-PEF implantation, respectively (fig. S2, A to D). In that model, Exo-PEF also mitigated foreign body reaction to the implanted polyester scaffolds (fig. S2E).
Fig. 3. Exo-PEF promoted the M2 MΦ subtype and Treg populations in the skin wound model.
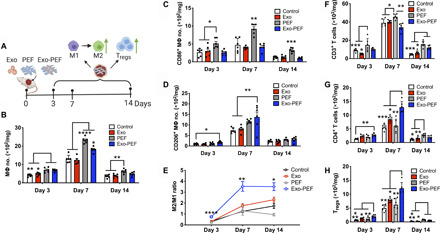
(A) Full excisional skin wounds were treated with injected exosomes, PEF, or Exo-PEF. Exosome dosage: 30 μg of protein mass. Analyses of immune cell numbers per milligram of tissue (with untreated wounds as the negative control): (B) total MΦs, (C) CD86+ M1 MΦs, (D) CD206+ M2 MΦs, (E) ratio of CD206+ M2 versus CD86+ M1 MΦ, (F) total T cells, (G) CD4+ T cells, and (H) CD4+CD25+FoxP3+ Tregs on days 3, 7, and 14 after injury. n = 6. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
T cells constitute an active group of adaptive immune cells during the skin wound-healing process. Tregs were particularly reported to be essential for the hair follicle regeneration (24, 25). By looking at the pan-T cell subsets in different groups, it was observed that CD4+ TH2 and CD4+CD25+FoxP3+ Tregs were two pronounced groups in the Exo-PEF–treated wounds. Notably, Exo-PEF promoted the presence of Tregs in higher than twofold quantity compared to untreated wounds on day 7 (Fig. 3, G and H). Moreover, at this time point, Exo-PEF also led to a higher portion of the T cells (18.6 ± 6.4%) secreting the anti-inflammatory cytokine IL-10 (fig. S3A), with most CD4+ cells and all Tregs identified to be IL-10+ (fig. S3, B and C). Without exosomes, the blank PEF slightly increased the total number of T cells (Fig. 3F) that did not contain the beneficial CD4+ T cells or Tregs (Fig. 3, G and H), suggesting the possible presence of other T cell subtypes, such as CD8 or γδ T cells in the wounds treated with PEF alone.
Remote adaptive immune responses induced in lymphatic organs
The TH1 and TH2 lymphocytes expressing the CD4 co-receptors are important adaptive cells by releasing T cell cytokines to mediate inflammation and tissue remodeling. In analyzing inguinal lymph nodes (LNs) near skin wounds, IL-4–secreting CD4+ TH2 cells seemed to emerge in the Exo-PEF group on day 7, albeit at a low level (Fig. 4A); the injected or immobilized exosomes both induced higher populations of CD4+IL4+ TH2 and CD4+CD25+FoxP3+ Treg responses on day 14 (Fig. 4, A and B). The Exo-PEF treatment also demonstrated a trend to mitigate the IFN-γ+ TH1 cell number in the wounds, with the effect most noticeable on day 7, showing a more than 50% reduction of this cell population compared to the wound control (Fig. 4C).
Fig. 4. Exo-PEF activated the TH2 immune response in lymphatic organs.
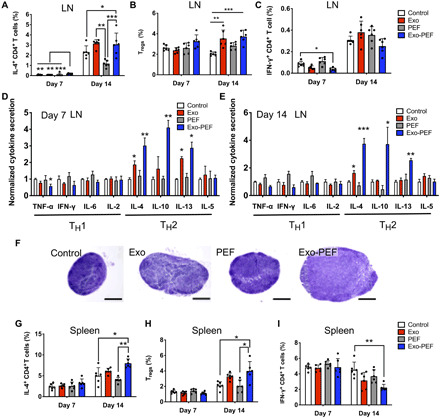
T cell subtypes were analyzed in lymph nodes (LN) and spleen following skin excisional wounds and treatments. (A to C and G to I) Percent analyses of IL-4+CD4+ T cells, CD4+CD25+FoxP3+ Tregs, and IFN-γ+CD4+ T cells in the inguinal LNs (A, B, and C, respectively) and spleen (G, H, and I, respectively) (n = 6; *P < 0.05, **P < 0.01, and ***P < 0.001). (D and E) Cytokine array studies on supernatants produced by ex vivo LN cells isolated from the LNs harvested on day 7 (D) and day 14 (E) after injury. (F) Representative hematoxylin and eosin (H&E)–stained images of the LNs harvested from mice (scale bar, 500 μm).
The cytokine array experiments further suggested the activation of the TH2 immune responses induced by the immobilized or liquid MSC exosomes, with the stronger effects observed in the former. Specifically, in comparison to untreated wounds on day 7, Exo-PEF promoted the secretion of IL-4 (3.0-fold), IL-10 (2.9-fold), and IL-13 (2.9-fold) and decreased that of TNF-α (1.8-fold) and IFN-γ (1.6-fold); the trend continued on day 14, showing the elevated secretion of IL-4 (3.8-fold), IL-10 (3.7-fold), and IL-13 (2.6-fold) (Fig. 4E). The TH2 cytokine responses were less potent in the Exo group on day 7 and almost unnoticeable on day 14 except for the secretion of the IL-4 cytokine (Fig. 4D). The hypertrophy of the LNs in the Exo and Exo-PEF groups was observed on day 14, indicating immune activation in the LNs (Fig. 4F).
In the spleen, only the Exo-PEF treatments induced the responses on day 14 similar to those in the LNs, showing IL-4–secreting CD4+ T cells and Tregs in higher numbers and IFN-γ+CD4+ T cells being decreased (Fig. 4, G to I). To understand the origin of TH2 immunoactivation and the possible immunogenic effects of any exosome components diffusing into the tissue or organs, T cells were isolated from the LNs harvested from the Exo and Exo-PEF hosts at week 2. Dosing MSC exosomes directly in vitro failed to stimulate the proliferation peak from these T cells ex vivo, suggesting that the adaptive immunomodulation in vivo induced by Exo-PEF was not due to any inherent immunogenicity of MSC exosomes (fig. S4).
Effects of exosome-laden scaffolds on healing large skin wounds
To understand the pro-regenerative therapeutic effects of Exo-PEF scaffolds, large square skin excisional wounds with an area of 225 mm2 were created on the dorsal skin of mice. By measuring the wound areas from day 0 to week 2, we found that Exo-PEF scaffolds significantly accelerated wound closure (65.1 ± 5.3%) compared to untreated wounds (18.2 ± 4.7%), treatments of injected exosomes (29.2 ± 10.5%), and blank PEF scaffolds (16.8 ± 7.2%) (Fig. 5, A and B). Histological analysis was performed to evaluate wound reepithelialization and collagen deposition through hematoxylin and eosin (H&E) and Masson’s trichrome (MT) staining, respectively. Histology images were acquired near the wound margin as shown in Fig. 5C. Reconstitution of epidermal covering was observed in the Exo-PEF group, while the Exo group exhibited a fibrin-like granulocyte-rich tissue surface. In comparison, other groups showed no epidermal covering. Granulation tissues (GTs) were formed in the wound bed in all groups, but morphology varied. GT in Exo-PEF was much thicker than that in other groups and exhibited pinkish color in H&E staining, indicating a healthier GT formation. In addition, Exo-PEF allowed collagenous extracellular matrix deposition, with elongated fibroblasts resembling tissue organization in the normal skin. In comparison, in control and PEF groups, there was minimal collagen deposition (Fig. 5C). The wound beds in the Exo-PEF group also contained a significantly higher number of blood vessels that were stained positive for α-smooth muscle actin (α-SMA) than that in the other groups (Fig. 5, D and E).
Fig. 5. Exo-PEF healed large skin wounds in 2 weeks, showing wound closure, reepithelialization, collagen deposition, and blood vessel formation.
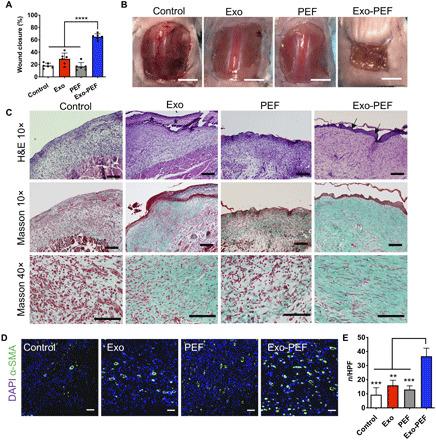
(A) Percent wound closures and (B) representative images of the wounds after 2-week treatments. (C) H&E and Masson’s trichrome (MT) histological staining of wound tissues after 2-week treatments. Asterisks, fibrin-like granulocyte-rich layer in the Exo group; arrows, neo-formed epidermis in the Exo-PEF group. Scale bars, 200 μm (10×) and 100 μm (40×). (D) Fluorescent staining of α-smooth muscle actin (α-SMA) (green) and DAPI (blue) in the wound bed. Scale bar, 50 μm. (E) Statistical calculation of α-SMA–positive blood vessel numbers per high-power field (HPF). Exosome dosage: 50 μg of protein mass. n = 6. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
DISCUSSION
Investigation of immune responses following implantation of biomaterials in the body is a long-standing topic in developing biocompatible medical implants. Incorporation of biological components, such as proteins, exosomes, and cells, into material scaffolds for application in tissue engineering and regenerative medicine has added another layer of complexity with respect to understanding the host response and how it would affect the integration of the implants. In essence, the primary function of the immune system is to defend the host and eliminate foreign invaders. Developing bioactive constructs appropriately interacting with the immune system therefore faces formidable challenges (26). In our experiments, it was found that most of the cells absorbing exosomes from scaffolds were immune cells (Fig. 2, F to H). This may also be relevant to other types of biological scaffolds, implying the importance of immune responses in tissue reactions and the necessity to understand and use this the mechanism to guide the biological communication network.
Given the diversity of their components from proteins to microRNAs, exosomes carry a multitude of signals (27). Through proteomic profiling, both immunomodulatory factors and proangiogenic factors could be detected in MSC exosomes (table S1). Binding exosomes to fibrous scaffolds were shown to store, localize, and deliver membranous vesicles (Fig. 2). One direct benefit is the prolonged retention of exosomes in effecting their function in the hostile injury environment. Tethering exosomes through electrostatic interactions seemed to be reversible, but in an interactive way involving the active uptake by cells (Fig. 2, A to C). The delivery may therefore be considered a dynamic “on-demand” process, and this is attractive and in contrast with the passive transport through diffusion, as shown in the previous exosome-laden or many drug delivery systems (28–30). Another interesting property of Exo-PEF scaffolds was the synergy between materials and exosomes, as observed in promoting the beneficial immunomodulation. In the experiments, the standalone PEF scaffolds increased the expression of anti-inflammatory cytokines and the M2 marker in vitro (Fig. 1H), presumably because of the topological cues of fibrous materials (31, 32). The effects, however, were not translated at the wound sites, as PEF scaffolds recruited MΦ and T cells locally, but leaning toward the M1-biased MΦ and T cells likely in favor of inflammation. Increasing the population of immune cells nevertheless is pivotal in the circumstance of exosomes with the presence of PEF, as immune cells recruited by materials could be turned around showing the beneficial responses (Fig. 3). Exo-PEF scaffolds therefore synergized the respective effects of scaffolds and exosomes as the “recruiter” and “trainer” for immune cells in the process (Fig. 6).
Fig. 6. Possible immunomodulation mechanisms involved in Exo-PEF materials promoting tissue repair may include (i) the recruitment and training of immune cells, (ii) the action of anti-inflammatory mediators, and (iii) the effects from remote immune organs.
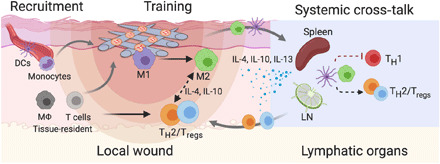
M1, M1-like MΦ; M2, M2-like MΦ; Tregs, regulatory T cells; TH1/2, T helper 1/2 cells; DCs, dendritic cells; LN, lymph nodes.
In the context of tissue repair and regeneration, the immune responses across different organisms and tissues are divergent in terms of suppression and activation. This would affect the inflammatory and angiogenic processes as well as the progenitor activities and the regeneration outcome of a tissue (33–35). In our study, it was shown that the tissue reaction to Exo-PEF scaffolds started with the modulation of the MΦ response manifested in the M2-biased polarization (Fig. 2, B to E). The increased population of the M2-like MΦ subtype has been recognized as a crucial event (35), and a delayed transition from the proinflammatory M1-like to immunomodulatory M2-like phenotypes would cause fibrosis and scarring reactions to inhibit the regeneration of bone, muscle, heart, and the skin tissue (33–35). Exo-PEF scaffolds also notably increased the adaptive immune responses both locally and remotely (Figs. 3, F to H, and 4). On the time scale, the adaptive responses were generally active on day 14 in a phase later than the peak of the macrophage response occurring on day 7. Treg differentiation and other remote adaptive responses could be induced by the cytokines secreted from the innate immune cells, e.g., IL-10 and TGF-β that communicate to T cells (36, 37). It was worth mentioning that increases in dendritic cells (DCs), MΦs, and IL-10–secreting cells were detectable within the LNs (fig. S5, A to D) and were accompanied by decreases in T cells including CD4+ T cells (fig. S5, E and F) in mice treated by Exo-PEF scaffolds. Innate and adaptive cells may thereby exist trafficking in and out of the lymphatic organs (fig. S5). Using technologies such as molecular imaging to trace immune cell trafficking may be carried out to further investigate the cell migration pattern.
Scaffolds carrying therapeutic exosomes may potentially offer a promising category of cell-free regenerative system as demonstrated by the effects of Exo-PEF on healing large skin wounds in mice (Fig. 5). Skin wounds as large as 225 mm2 in size are considered a critical skin injury that cannot self-heal properly despite the regeneration capability of skin tissue. The prohealing effects observed for Exo-PEF are therefore substantial. Accelerated MΦ transition from proinflammatory to immunomodulatory phenotypes, increased local Treg population, and activation of TH2 response all favor anti-inflammatory events to benefit the healing. In addition, the signals conveyed through the anti-inflammatory cytokines including IL-4, IL-10, and IL-13 have recently been expanded in the regenerative biology by maintaining the trophic factors such as insulin-link growth factor, vascular endothelial growth factor, TGF-β, and amphiregulin, which are essential to promoting progenitor activity (38–40).
Despite the potential of exosome-functionalized scaffolds providing an effective and potent immunomodulatory system, the cellular communication network between the immunomodulation and regeneration processes is still to be elucidated in detail (Fig. 6). In this regard, high-throughput cell-level analyses need to be adopted to identify the complex and versatile populations of immune cells, as well as other niche cells such as endothelial cells, fibroblasts, and progenitor cells. The techniques may include mass cytometry with the heavy metal–labeled antibodies or single-cell sequencing to reveal the dynamics of the cell transcriptome. Especially, high-throughput techniques can be applied to characterize the plasticity and dynamics of the MΦ population in different stages of tissue healing, as it has been recognized that the classically activated M1 phenotype and the alternatively activated M2 phenotype cannot accurately reflect the polarization and complexity of MΦ subtypes during tissue responses (41). Moving forward to clinical applications, key components in MSC exosomes contributing to the immunomodulation effects need to be further identified. Studies should also be conducted to further rule out the possibility of exosome immunogenicity, especially among individuals or even across species.
To conclude, immobilizing exosomes to polymeric fibrous scaffolds generated multifunctional scaffolds capable of storing biologics and delivering exosome signals. The synergy between scaffold materials and exosomes was found effective to proactively recruit immune cells and modulate orchestrated M2/TH2/Treg responses locally and on the systemic level to benefit tissue repair. The study lays the foundation for establishing the principles of designing biological scaffolds with immunomodulatory functions for regenerative medicine.
MATERIALS AND METHODS
Materials and reagents
The ultrapure water (≥18 megohms) was prepared from the deionized (DI) water through the Milli-Q system (Millipore, Billerica, MA). PCL (CAS: 24980-41-4) and lipopolysaccharide (LPS) were obtained from Sigma-Aldrich (Milwaukee, WI). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were purchased from Pierce Biotechnology (Rockford, IL, USA). The branched PEI with the average Mw of 70 kDa was from Macklin (Shanghai, China). The TRIzol kit, Dulbecco’s phosphate-buffered saline (DPBS), RPMI 1640 medium, Dulbecco’s modified Eagle’s medium (DMEM), and other cell culture reagents were purchased from Invitrogen (CA, USA) unless otherwise specified. 4′,6-Diamidino-2-phenylindole (DAPI) was obtained from Thermo Fisher Scientific (MA, USA). HUVECs, endothelial cell medium (ECM), and endothelial cell growth supplements (ECGS) were purchased from ScienCell (Carlsbad, CA). The mouse bone marrow MSCs (BM-MSCs) and the basal culture medium were purchased from Cyagen Biosciences Inc. (CA, USA). The quality control report provided by the vendor for this MSC product included the absence of endotoxin, >98% cell purity, and the adipogenic, osteogenic, and chondrogenic differentiation potential. The murine macrophage cell line RAW 264.7 was obtained from the Cell Culture Center of the Institute of Basic Medical Sciences (Beijing, China). Growth factors including macrophage colony-stimulating factors (M-CSFs) and cytokine IL-4 were purchased from PeproTech (Canada) unless otherwise specified. The flow cytometry antibodies and reagents were purchased from BioLegend (USA) unless otherwise specified. The suppliers of other chemicals, biological reagents, and equipment were specified below.
Cell isolation and culture
All cells were cultured at 37°C under humidified atmosphere with 5% CO2. The MSCs were cultured in basal medium supplemented with 10% fetal bovine serum (FBS) and 0.4% penicillin-streptomycin (P/S). MSCs between passages 5 and 9 were used for the following experiments. HUVECs were maintained in ECM supplemented with 5% FBS, 1% P/S solution, and 1% ECGS. RAW 264.7 MΦs were cultured in DMEM supplemented with 10% FBS and 1% P/S. To isolate the primary macrophages, the bone marrow cells from the femur and tibia of Balb/c mice were harvested and cultured in the RPMI medium supplemented with 0.25 mM β-mercaptoethanol, 1× nonessential amino acid, 10% heat-inactivated FBS (incubated at 56°C in water bath for 30 min), 1% P/S, and M-CSF (20 ng/ml). The cell culture media were changed every other day. The bone marrow–derived macrophages (BMDMs) were stained by the CD11b and F4/80 antibody and analyzed by flow cytometry at day 7, showing >90% purity (fig. S6).
Isolation and characterization of MSC exosomes
Exosomes were isolated from BM-MSCs from Balb/c mice. Cells at 80% confluency were washed with PBS and changed to serum-free medium. Supernatants were collected after 24 hours of incubation, followed by centrifugation at 1200g for 15 min and 10,000g for another 30 min to remove cell debris. Exosomes were precipitated by ultracentrifugation at 100,000g for 1 hour and then washed twice with PBS. The precipitated exosomes were resuspended in 200 μl of PBS and stored at −80°C until further use. The protein contents of the collected exosomes were measured using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific). For morphology observation, exosomes were fixed with 4% paraformaldehyde (PFA), stained with 2% (w/v) uranyl acetate (Sigma-Aldrich) for 5 min, washed three times with DI water, and air-dried overnight. The exosome morphologies were observed with a transmission electron microscope (TEM) (Tecnai G2 T20, Thermo Fisher Scientific). The purity of the collected exosomes was analyzed on the basis of the standards suggested by the International Society for Extracellular Vesicles (42) and showed the presence of positive exosomal markers without contamination because of other cellular organelles or culture medium. (tables S1 and S2).
For mass spectrometry (MS), exosomes were treated by lysing buffer solutions [6% SDS, 200 mM dithiothreitol, and 200 mM tris-HCl (pH 7.6)] at 95°C for 5 min followed by SDS–polyacrylamide gel electrophoresis. For protein identification, Coomassie-stained total aggregated proteins from each sample were cut out of the gel and destained with a solution of 100 mM ammonium bicarbonate in 50% acetonitrile. After dithiothreitol reduction and iodoacetamide alkylation, the proteins were digested with porcine trypsin (Promega, Madison, WI) overnight at 37°C. The resulting tryptic peptides were extracted from the gel pieces with 80% acetonitrile and 0.1% formic acid (FA). The samples were dried in a vacuum centrifuge concentrator and resuspended in 0.1% FA. The resulting peptides were loaded to Thermo Orbitrap Fusion Lumos, and full MS spectra were acquired in an Orbitrap mass analyzer. The MS data were aligned with the Escherichia coli Reviewed Swiss-Prot database by the Proteome Discoverer 2.2 software.
Fabrication and characterization of exosome-laden PEF scaffolds
The electrospun fibrous scaffolds were prepared from 21% (w/v) PCL solutions in a mixture of CHCl3 and dimethylformamide at a volume ratio of 9:1. After stirring for 3 hours until the PCL was completely dissolved, the solution was loaded into a syringe with a blunt-ended stainless 21-gauge steel needle. The feeding rate and the applied voltage were controlled at 2 ml/hour and 10 kV, respectively. The electrospun fibers were collected on a flat steel plate covered with a foil paper at a distance of 14 cm from the needle tip. The EF membranes with a thickness of 80 to 90 μm were collected and manually trimmed into round shapes with a diameter of 12 mm and mass at 2.7 ± 0.3 mg. Scaffolds of this size were used in all experiments except those for experiments in the large skin injury model. To immobilize exosomes onto PCL EF scaffolds, the tailored scaffold membrane was immersed in a mixture of 1 M aqueous NaOH solution and 75% ethanol solution at a ratio of 1:1 (v/v) for 30 min with gentle agitation at room temperature. The treatment was to enhance the PCL EF hydrophilicity and to generate carboxyl groups on the fiber surface via hydrolysis. Scaffolds were washed thoroughly afterward with DI water.
To covalently conjugate PEI, 60 pieces of NaOH-treated circular scaffolds were incubated in 25 ml of 0.1 M MES buffer (pH 5.1) containing 9.3 mM EDC and 5.8 mM NHS for 2 hours on a rotary shaker at room temperature. The 70-kDa branched PEI was dissolved in 25 ml of 0.2 M MES buffer (pH 6.5) to prepare a 3.5 mM PEI solution, which was added to the EFs and EDC/NHS reaction and shaken for another 2 hours. The EDC:NHS:PEI molar ratio was at 8:5:3. PEF was washed four times using excessive 1 M NaCl solutions to remove the physically adsorbed PEI and then multiple times with water for 2 days.
To confirm the success of PEI conjugation, the 2,4,6-trinitrobenzene sulfonic acid assay (Sigma-Aldrich) was performed to detect amine groups on PEF fiber surfaces by forming of a highly chromogenic (orange) product. Conjugation to EF scaffolds was also characterized by x-ray photoelectron spectroscopy (AXIS Supra, Kratos Analytical Ltd., UK). The UEF and PEF samples (d = 12 mm) were directly mounted on the test stage with a total acquisition time of 15 min for each sample. The overlapping peaks were resolved using the peak synthesis method by applying the Gaussian peak components after Shirley-type background subtraction. The morphology of the EF materials in each modification steps was characterized with a scanning electron microscope (SEM) (Quanta FEG450, FEI, USA). Water contact angle measurements of PCL EF scaffolds before and after PEI modification were conducted at room temperature under 60% relative humidity using the sessile drop method on a contact angle analyzer (USA Kino Industry Co. Ltd.).
For sterilization, the EF membranes were rinsed with 75% ethanol solutions for 30 min, washed with sterilized DI water three times, and air-dried in a biosafety cabinet (Thermo Fisher Scientific, Germany). EF scaffolds were further sterilized by exposure to ultraviolet radiation for 30 min on each side in a biosafety cabinet and were kept at 4°C until use. To immobilize MSC exosomes, the prescribed amount of exosomes was suspended in 1 ml of PBS, added to PEF scaffolds pretailored into appropriate sizes and shapes, and incubated for 30 min at 4°C on a rotator. The mass of exosomes immobilized onto scaffolds was determined by measuring the difference of protein contents in PBS solutions before and after the binding reaction. The amount of exosome added in each experiment was lower than the maximal loading capacity of PEF to ensure the full binding of exosomes in the solution to the scaffold to reduce the waste of exosomes. To visualize exosome loaded to PEF scaffolds, exosomes were incubated with 0.1 mM 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) solutions (Invitrogen) and washed with PBS three times to remove excessive dye molecules. The DiI-labeled exosomes were loaded to PEF scaffold and imaged via confocal microscopy (Zeiss) after embedding in the Tissue-Tek O.C.T. compound (VWR) and cryosection.
Exosome uptake and retention
Transwell culture
RAW 264.7 cells or HUVECs were seeded on the bottom chamber of the transwell microplates, which contained a 0.4-μm porous polyester membrane between the two chambers. Ten micrograms of fluorophore-labeled exosomes were added to the upper chamber with or without the unmodified EF scaffolds as two control groups. One purpose of including the UEF scaffold in the upper chamber was to determine whether the presence of EF scaffolds would affect the permeation of free exosomes through the transwell membrane. The same amount of exosome was loaded to PEF scaffolds, which were then placed in the upper chamber. RAW 264.7 cells or HUVECs were also directly seeded to the Exo-PEF. After culture for 24 hours, cells were collected and flow cytometry analysis was performed to determine cellular uptake of exosome.
Exosome retention and uptake in vivo
The full excisional wounds with a diameter of 8 mm were created with a biopsy puncher on the dorsal skin of Balb/c mice under isoflurane anesthesia (more details were described in the “Animal wound models and wound treatments” section). Exosomes were covalently labeled with Vivo Tag 645 (PerkinElmer), and 15 μg of fluorescence-labeled exosomes was either subcutaneously injected at three points around the wound or immobilized onto round PEF (d = 12 mm) and applied on top of the wound. The 3M Tegaderm transparent wound dressing was used to cover the wound. Mice under isoflurane anesthesia were imaged at multiple time points up to day 7 using the IVIS 200 Optical Imaging System (Xenogen, Caliper Life Sciences). The wound area was selected as the region of interest (ROI), and the radiant efficiency was calculated within the ROIs. Exosome retention (%) at a time (t) was calculated by comparing the radiant efficiency at t to that at 0 h (t0) according to the following formula: [Radiant efficiency (t)]/[Radiant efficiency (t0)] × 100%. At day 7, wound tissues were harvested, digested into single-cell suspensions, stained with F4/80, CD45, and CD3 antibodies, and analyzed by flow cytometry to determine exosome uptake by macrophages (CD45+F4/80+) and T cells (CD45+CD3+). (Details for single-cell preparation and flow cytometry staining were described in the “Flow cytometry analyses” section.) The skin wound tissues were embedded in O.C.T., cryosectioned, and stained with primary antibody rabbit anti-mouse CD68 or rabbit anti-mouse CD3 followed by fluorescein isothiocyanate (FITC) goat anti-rabbit secondary antibody staining. Nuclei were stained with DAPI. The colocalization of exosome and cell markers was examined to identify cells that sequestered exosomes.
Macrophage modulation assays in vitro
BMDMs were activated with LPS (100 ng/ml; Sigma-Aldrich, L2880), seeded on 48-well tissue culture plates at a density of 2 × 105 cells per well, and then supplemented with or without 10 μg of MSC exosomes. An equal number of BMDM were also seeded on the pretailored PEF or Exo-PEF scaffolds (d = 12 mm) loaded with 10 μg of MSC exosomes. After 12 hours, cells were treated with a TRIzol kit and the total RNA was extracted and reverse-transcribed using TransScript First-Strand cDNA Synthesis SuperMix (Transgene, Beijing, China). Gene expression levels of cytokines (IL-10, IL-4, IL-1β, and TNF-α) and phenotypic markers (CD206, Egr2, CD86, and CD38) were analyzed by SYBR Green real-time polymerase chain reaction (PCR) using the CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA). The primers for qPCR were ordered from Sangon (Shanghai, China), and the primer sequence was listed in table S3. The gene expression values in one sample were all normalized to the β-actin level. Relative expressions were calculated using the comparative Ct method. The mean minimal cycle threshold values were calculated from triplicate reactions.
Animal wound models and wound treatments
All procedures were performed according to the regulations approved by the Institutional Animal Care and Use Committee of Peking University. Balb/c female mice (Vital River, Beijing, PR China) at 6 weeks old were anesthetized by isoflurane during procedure.
Subcutaneous implantation in nonpathological tissue
The scaffold membranes (d = 12 mm) of UEF, PEF, or Exo-PEF (with 30 μg of exosomes loaded) were implanted into the subcutaneous space beneath the abdomen skin. Free exosomes (30 μg) were also injected subcutaneously. The injection sites were marked with an indelible marker pen for the tissue harvest at later time points. Scissions and pockets were created in all mice regardless of the scaffold implantation. After euthanizing the mice at day 14, implanted scaffolds, inguinal LNs, and spleen were harvested for immune cell analysis using flow cytometry. The mice with implants were also kept for up to 5 weeks and analyzed by H&E histology staining.
Excisional wound model
For mechanistic immunomodulation studies, round full-thickness excisional skin wounds with a diameter of 8 mm were created using a biopsy puncher after hair removal from the dorsal skin. The scaffold membranes (d = 12 mm) of UEF, PEF, or Exo-PEF (with 30 μg of exosomes loaded) were applied on top of the wounds. Free exosomes (30 μg) were injected subcutaneously around the wounds for the Exo group. Untreated skin wounds were included as a control. 3M Tegaderm transparent wound dressing was used to cover the wounds. Wound tissues, inguinal LNs, and spleens were harvested on days 3, 7, and 14 after euthanizing the mice. Analyses of immune cell populations were performed using flow cytometry.
For wound-healing studies, the 225-mm2 square wounds were created, with four corners sutured to the dorsal muscles to prevent skin contraction. The Exo-PEFs and exosome controls for the large wound treatments were prepared similarly as described above except that a higher dose of 50 μg of exosomes loaded to 230-mm2 square EF scaffolds was applied to each wound. The digital photographs of wounds were taken at day 14. The area of the wounds was measured and calculated by ImageJ software. The percent of wound closures was calculated on the basis of the average wound area (A) measured on days 0 and 14 and the following equation: (Aday0 − Aday14)/Aday0 × 100%. Wound tissue was harvested for histological analyses after mice were euthanized at day 14.
Flow cytometry analyses
All flow cytometry data were collected using BD FACSAria II and analyzed using the FlowJo software. Gating strategies were listed in figs. S7 and S8.
Explanted scaffolds or skin wound tissues were harvested, and the attached fat tissues were first removed before weighing the wound. The skin samples were diced and digested with the Opti-MEM (Invitrogen) solutions containing Liberase TM (100 ug/ml) (Roche), collagenase D (1 mg/ml) (Roche), and hyaluronidase (1 mg/ml) (Sigma-Aldrich) for 30 min at 37°C on a shaker. The digested cell suspensions were poured through a 70-μm cell strainer. The remaining tissue chunks were minced with a syringe plunger and washed with PBS containing 2% FBS, followed by lysis treatment of the red blood cells (BioLegend).
The inguinal LNs were harvested, minced with a syringe plunger, and filtered through a 70-μm cell strainer. The spleen was diced and digested in collagenase D (2 mg/ml) (Roche) for 30 min at 37°C on a rotator. Cell suspensions were filtered through a 70-μm cell strainer followed by red blood cell lysis (BioLegend). After washing, cells were directly processed for flow cytometry staining.
Cell surface staining for flow cytometry experiments was performed on ice in the dark. Cell viability staining was conducted using the Zombie Violet Fixable Viability Kit (BioLegend), followed by nonspecific binding blockage by anti-CD16/32 incubation. Cells were then stained with surface antibody cocktail for 45 min on ice. The surface markers of the macrophage population included CD45, F4/80, and CD86, and those of T cells included CD45, CD3, CD4, and CD25. Cell suspension were then fixed and permeabilized with the Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences) according to the manufacturer’s instructions. Intracellular markers, including CD206 for M2 macrophage or FoxP3 for Tregs, were stained for 45 min on ice. To analyze immune cell populations in the wounds, the cell numbers were normalized to wound weight, reported as cell number per milligram.
To perform intracellular cytokine staining, cells from LNs were stimulated and prevented from cytokine secretion with the Cell Activation Cocktail containing brefeldin A (BioLegend) for 5 hours at 37°C in RPMI medium supplemented with 10% FBS. Cell surfaces were stained for CD3 and CD4, followed by intracellular staining of IL-4 and IFN-γ. For cytokine array analysis, cells isolated from LNs were cultured in RPMI medium without FBS for 24 hours. Supernatants were collected and analyzed to determine the level of TH1 (TNF-α, IFN-γ, IL-6, and IL-2) and TH2 (IL-4, IL-10, IL-13, and IL-5) cytokines by LEGENDplex bead-based immunoassays according to the manufacturer’s instructions (BioLegend).
Histological analyses
Skin wound tissues were harvested and cut into two parts from the middle line, fixed with 4% PFA, embedded in paraffin, and sectioned into 5-μm-thick slices. Tissue slices were stained with H&E or MT staining, and images were captured with a light optical microscope (Olympus, Japan).
For immunohistochemical staining, the harvested skin wound tissues were embedded in O.C.T., cryosectioned at a thickness of 8 μm, and stained with primary antibody and rabbit anti-mouse α-SMA, followed by the FITC goat anti-rabbit secondary antibody. Nuclei were stained with DAPI. Fluorescent images were taken with a confocal microscope, and positive signals per high-power field were counted. For quantitative image analysis, data from three animals, eight sections per animal, and four high-magnification images per section for each group were analyzed and compared.
Statistical analyses
Quantitative results were presented as means ± SD. Statistical comparisons were performed by one-way analysis of variance (ANOVA) followed by Holm-Sidak tests to compare the selected data pairs. The values of P < 0.05 were considered statistically significant.
Acknowledgments
We thank the Core Facilities at School of Life Sciences, Peking University that offer the platform for MS experiments and data analyses. We thank D. Liu for help in sample preparation and data analysis. We also thank BioRender.com for the support of generating schematics. Funding: This research was financially supported by the National Key Research and Development Program of China (project no. 2016YFC1101301) and the National Natural Science Foundation of China (project nos. 31771107 and 31322021). Author contributions: N.S., H.C., and Y.L. conceived and designed the study. N.S., Y.H., and H.C. prepared and characterized materials and samples. N.S., Y.H., F.W., and W.H. conducted biological assays and animal studies. All authors contributed to the development of methods, analysis of data, and writing of the manuscript, with N.S. and Y.L. taking the lead. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf7207/DC1
REFERENCES AND NOTES
- 1.Chung L., Maestas D. R. Jr., Housseau F., Elisseeff J. H., Key players in the immune response to biomaterial scaffolds for regenerative medicine. Adv. Drug Deliv. Rev. 114, 184–192 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Mescher A. L., Neff A. W., King M. W., Inflammation and immunity in organ regeneration. Dev. Comp. Immunol. 66, 98–110 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Karin M., Clevers H., Reparative inflammation takes charge of tissue regeneration. Nature 529, 307–315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chazaud B., Macrophages: Supportive cells for tissue repair and regeneration. Immunobiology 219, 172–178 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Spiller K. L., Koh T. J., Macrophage-based therapeutic strategies in regenerative medicine. Adv. Drug Deliv. Rev. 122, 74–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Julier Z., Park A. J., Briquez P. S., Martino M. M., Promoting tissue regeneration by modulating the immune system. Acta Biomater. 53, 13–28 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Burzyn D., Kuswanto W., Kolodin D., Shadrach J. L., Cerletti M., Jang Y., Sefik E., Tan T. G., Wagers A. J., Benoist C., Mathis D., A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dombrowski Y., O’Hagan T., Dittmer M., Penalva R., Mayoral S. R., Bankhead P., Fleville S., Eleftheriadis G., Zhao C., Naughton M., Hassan R., Moffat J., Falconer J., Boyd A., Hamilton P., Allen I. V., Kissenpfennig A., Moynagh P. N., Evergren E., Perbal B., Williams A. C., Ingram R. J., Chan J. R., Franklin R. J. M., Fitzgerald D. C., Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 20, 674–680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y., Wang L., Kikuiri T., Akiyama K., Chen C., Xu X., Yang R., Chen W., Wang S., Shi S., Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nat. Med. 17, 1594–1601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadtler K., Estrellas K., Allen B. W., Wolf M. T., Fan H., Tam A. J., Patel C. H., Luber B. S., Wang H., Wagner K. R., Powell J. D., Housseau F., Pardoll D. M., Elisseeff J. H., Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science 352, 366–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z., Chen X., Zhang Z., Zhang X., Saunders L., Zhou Y., Ma P. X., Nanofibrous spongy microspheres to distinctly release miRNA and growth factors to enrich regulatory T cells and rescue periodontal bone loss. ACS Nano 12, 9785–9799 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raimondo T. M., Mooney D. J., Functional muscle recovery with nanoparticle-directed M2 macrophage polarization in mice. Proc. Natl. Acad. Sci. U.S.A. 115, 10648–10653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontaine M. J., Shih H., Schäfer R., Pittenger M. F., Unraveling the mesenchymal stromal cells’ paracrine immunomodulatory effects. Transfus. Med. Rev. 30, 37–43 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Bader P., Kuçi Z., Bakhtiar S., Basu O., Bug G., Dennis M., Greil J., Barta A., Kállay K. M., Lang P., Lucchini G., Pol R., Schulz A., Sykora K. W., von Luettichau I., Herter-Sprie G., Uddin M. A., Jenkin P., Alsultan A., Buechner J., Stein J., Kelemen A., Jarisch A., Soerensen J., Salzmann-Manrique E., Hutter M., Schäfer R., Seifried E., Klingebiel T., Bonig H., Kuçi S., Effective treatment of steroid and therapy-refractory acute graft-versus-host disease with a novel mesenchymal stromal cell product (MSC-FFM). Bone Marrow Transplant. 53, 852–862 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lo Sicco C., Reverberi D., Balbi C., Ulivi V., Principi E., Pascucci L., Becherini P., Bosco M. C., Varesio L., Franzin C., Pozzobon M., Cancedda R., Tasso R., Mesenchymal stem cell–derived extracellular vesicles as mediators of anti-inflammatory effects: Endorsement of macrophage polarization. Stem Cells Transl. Med. 6, 1018–1028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen W., Huang Y., Han J., Yu L., Li Y., Lu Z., Li H., Liu Z., Shi C., Duan F., Xiao Y., Immunomodulatory effects of mesenchymal stromal cells-derived exosome. Immunol. Res. 64, 831–840 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Crain S. K., Robinson S. R., Thane K. E., Davis A. M., Meola D. M., Barton B. A., Yang V. K., Hoffman A. M., Extracellular vesicles from Wharton’s jelly mesenchymal stem cells suppress CD4-expressing T cells through transforming growth factor beta and adenosine signaling in a canine model. Stem Cells Dev. 28, 212–226 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Zhang B., Yeo R. W. Y., Lai R. C., Sim E. W. K., Chin K. C., Lim S. K., Mesenchymal stromal cell exosome-enhanced regulatory T-cell production through an antigen-presenting cell-mediated pathway. Cytotherapy 20, 687–696 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Zhang S., Chuah S. J., Lai R. C., Hui J. H. P., Lim S. K., Toh W. S., MSC exosomes mediate cartilage repair by enhancing proliferation, attenuating apoptosis and modulating immune reactivity. Biomaterials 156, 16–27 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Li T., Yan Y., Wang B., Qian H., Zhang X., Shen L., Wang M., Zhou Y., Zhu W., Li W., Xu W., Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 22, 845–854 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu L., Wang J., Zhou X., Xiong Z., Zhao J., Yu R., Huang F., Zhang H., Chen L., Exosomes derived from human adipose mensenchymal stem cells accelerates cutaneous wound healing via optimizing the characteristics of fibroblasts. Sci. Rep. 6, 32993 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goonoo N., Modulating immunological responses of electrospun fibers for tissue engineering. Adv. Biosyst. 1, (2017). [DOI] [PubMed] [Google Scholar]
- 23.Schoenenberger A. D., Tempfer H., Lehner C., Egloff J., Mauracher M., Bird A., Widmer J., Maniura-Weber K., Fucentese S. F., Traweger A., Silvan U., Snedeker J. G., Macromechanics and polycaprolactone fiber organization drive macrophage polarization and regulate inflammatory activation of tendon in vitro and in vivo. Biomaterials 249, 120034 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Mathur A. N., Zirak B., Boothby I. C., Tan M., Cohen J. N., Mauro T. M., Mehta P., Lowe M. M., Abbas A. K., Ali N., Rosenblum M. D., Treg-cell control of a CXCL5-IL-17 inflammatory axis promotes hair-follicle-stem-cell differentiation during skin-barrier repair. Immunity 50, 655–667.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nosbaum A., Prevel N., Truong H. A., Mehta P., Ettinger M., Scharschmidt T. C., Ali N. H., Pauli M. L., Abbas A. K., Rosenblum M. D., Cutting edge: Regulatory T cells facilitate cutaneous wound healing. J. Immunol. 196, 2010–2014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dellacherie M. O., Seo B. R., Mooney D. J., Macroscale biomaterials strategies for local immunomodulation. Nat. Rev. Mater. 4, 379–397 (2019). [Google Scholar]
- 27.Gao F., Chiu S. M., Motan D. A. L., Zhang Z., Chen L., Ji H. L., Tse H. F., Fu Q. L., Lian Q., Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 7, e2062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang K., Zhao X., Chen X., Wei Y., du W., Wang Y., Liu L., Zhao W., Han Z., Kong D., Zhao Q., Guo Z., Han Z., Liu N., Ma F., Li Z., Enhanced therapeutic effects of mesenchymal stem cell-derived exosomes with an injectable hydrogel for hindlimb ischemia treatment. ACS Appl. Mater. Interfaces 10, 30081–30091 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Lv K., Li Q., Zhang L., Wang Y., Zhong Z., Zhao J., Lin X., Wang J., Zhu K., Xiao C., Ke C., Zhong S., Wu X., Chen J., Yu H., Zhu W., Li X., Wang B., Tang R., Wang J.’., Huang J., Hu X., Incorporation of small extracellular vesicles in sodium alginate hydrogel as a novel therapeutic strategy for myocardial infarction. Theranostics 9, 7403–7416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennan M. Á., Layrolle P., Mooney D. J., Biomaterials functionalized with msc secreted extracellular vesicles and soluble factors for tissue regeneration. Adv. Funct. Mater. 30, 1909125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Won J. E., Lee Y. S., Park J. H., Lee J. H., Shin Y. S., Kim C. H., Knowles J. C., Kim H. W., Hierarchical microchanneled scaffolds modulate multiple tissue-regenerative processes of immune-responses, angiogenesis, and stem cell homing. Biomaterials 227, 119548 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Jain N., Vogel V., Spatial confinement downsizes the inflammatory response of macrophages. Nat. Mater. 17, 1134–1144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mirza R., DiPietro L. A., Koh T. J., Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 175, 2454–2462 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahdavian Delavary B., van der Veer W. M., van Egmond M., Niessen F. B., Beelen R. H., Macrophages in skin injury and repair. Immunobiology 216, 753–762 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Shook B., Xiao E., Kumamoto Y., Iwasaki A., Horsley V., CD301b+ macrophages are essential for effective skin wound healing. J. Invest. Dermatol. 136, 1885–1891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin Q., Gui L., Niu F., Yu B., Lauda N., Liu J., Mao X., Chen Y., Macrophages in keloid are potent at promoting the differentiation and function of regulatory T cells. Exp. Cell Res. 362, 472–476 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Nascimento D. C., Melo P. H., Piñeros A. R., Ferreira R. G., Colón D. F., Donate P. B., Castanheira F. V., Gozzi A., Czaikoski P. G., Niedbala W., Borges M. C., Zamboni D. S., Liew F. Y., Cunha F. Q., Alves-Filho J. C., IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat. Commun. 8, 14919 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaiss D. M., Minutti C. M., Knipper J. A., Immune- and non-immune-mediated roles of regulatory T-cells during wound healing. Immunology 157, 190–197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe S., Alexander M., Misharin A. V., Budinger G. R. S., The role of macrophages in the resolution of inflammation. J. Clin. Invest. 129, 2619–2628 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abnave P., Ghigo E., Role of the immune system in regeneration and its dynamic interplay with adult stem cells. Semin. Cell Dev. Biol. 87, 160–168 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Locati M., Curtale G., Mantovani A., Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G. K., Ayre D. C., Bach J.-M., Bachurski D., Baharvand H., Balaj L., Baldacchino S., Bauer N. N., Baxter A. A., Bebawy M., Beckham C., Zavec A. B., Benmoussa A., Berardi A. C., Bergese P., Bielska E., Blenkiron C., Bobis-Wozowicz S., Boilard E., Boireau W., Bongiovanni A., Borràs F. E., Bosch S., Boulanger C. M., Breakefield X., Breglio A. M., Brennan M. Á., Brigstock D. R., Brisson A., Broekman M. L., Bromberg J. F., Bryl-Górecka P., Buch S., Buck A. H., Burger D., Busatto S., Buschmann D., Bussolati B., Buzás E. I., Byrd J. B., Camussi G., Carter D. R., Caruso S., Chamley L. W., Chang Y.-T., Chen C., Chen S., Cheng L., Chin A. R., Clayton A., Clerici S. P., Cocks A., Cocucci E., Coffey R. J., Cordeiro-da-Silva A., Couch Y., Coumans F. A., Coyle B., Crescitelli R., Criado M. F., D’Souza-Schorey C., Das S., Chaudhuri A. D., de Candia P., De Santana E. F., De Wever O., Del Portillo H. A., Demaret T., Deville S., Devitt A., Dhondt B., Vizio D. D., Dieterich L. C., Dolo V., Rubio A. P. D., Dominici M., Dourado M. R., Driedonks T. A., Duarte F. V., Duncan H. M., Eichenberger R. M., Ekström K., Andaloussi S. E., Elie-Caille C., Erdbrügger U., Falcón-Pérez J. M., Fatima F., Fish J. E., Flores-Bellver M., Försönits A., Frelet-Barrand A., Fricke F., Fuhrmann G., Gabrielsson S., Gámez-Valero A., Gardiner C., Gärtner K., Gaudin R., Gho Y. S., Giebel B., Gilbert C., Gimona M., Giusti I., Goberdhan D. C., Görgens A., Gorski S. M., Greening D. W., Gross J. C., Gualerzi A., Gupta G. N., Gustafson D., Handberg A., Haraszti R. A., Harrison P., Hegyesi H., Hendrix A., Hill A. F., Hochberg F. H., Hoffmann K. F., Holder B., Holthofer H., Hosseinkhani B., Hu G., Huang Y., Huber V., Hunt S., Ibrahim A. G.-E., Ikezu T., Inal J. M., Isin M., Ivanova A., Jackson H. K., Jacobsen S., Jay S. M., Jayachandran M., Jenster G., Jiang L., Johnson S. M., Jones J. C., Jong A., Jovanovic-Talisman T., Jung S., Kalluri R., Kano S.-I., Kaur S., Kawamura Y., Keller E. T., Khamari D., Khomyakova E., Khvorova A., Kierulf P., Kim K. P., Kislinger T., Klingeborn M., Klinke D. J. II, Kornek M., Kosanović M. M., Kovács Á. F., Krämer-Albers E.-M., Krasemann S., Krause M., Kurochkin I. V., Kusuma G. D., Kuypers S., Laitinen S., Langevin S. M., Languino L. R., Lannigan J., Lässer C., Laurent L. C., Lavieu G., Lázaro-Ibáñez E., Lay S. L., Lee M.-S., Lee Y. X. F., Lemos D. S., Lenassi M., Leszczynska A., Li I. T., Liao K., Libregts S. F., Ligeti E., Lim R., Lim S. K., Linē A., Linnemannstöns K., Llorente A., Lombard C. A., Lorenowicz M. J., Lörincz Á. M., Lötvall J., Lovett J., Lowry M. C., Loyer X., Lu Q., Lukomska B., Lunavat T. R., Maas S. L., Malhi H., Marcilla A., Mariani J., Mariscal J., Martens-Uzunova E. S., Martin-Jaular L., Martinez M. C., Martins V. R., Mathieu M., Mathivanan S., Maugeri M., McGinnis L. K., McVey M. J., Meckes D. G. Jr., Meehan K. L., Mertens I., Minciacchi V. R., Möller A., Jørgensen M. M., Morales-Kastresana A., Morhayim J., Mullier F., Muraca M., Musante L., Mussack V., Muth D. C., Myburgh K. H., Najrana T., Nawaz M., Nazarenko I., Nejsum P., Neri C., Neri T., Nieuwland R., Nimrichter L., Nolan J. P., Hoen E. N. N., Hooten N. N., O’Driscoll L., O’Grady T., O’Loghlen A., Ochiya T., Olivier M., Ortiz A., Ortiz L. A., Osteikoetxea X., Østergaard O., Ostrowski M., Park J., Pegtel D. M., Peinado H., Perut F., Pfaffl M. W., Phinney D. G., Pieters B. C., Pink R. C., Pisetsky D. S., von Strandmann E. P., Polakovicova I., Poon I. K., Powell B. H., Prada I., Pulliam L., Quesenberry P., Radeghieri A., Raffai R. L., Raimondo S., Rak J., Ramirez M. I., Raposo G., Rayyan M. S., Regev-Rudzki N., Ricklefs F. L., Robbins P. D., Roberts D. D., Rodrigues S. C., Rohde E., Rome S., Rouschop K. M., Rughetti A., Russell A. E., Saá P., Sahoo S., Salas-Huenuleo E., Sánchez C., Saugstad J. A., Saul M. J., Schiffelers R. M., Schneider R., Schøyen T. H., Scott A., Shahaj E., Sharma S., Shatnyeva O., Shekari F., Shelke G. V., Shetty A. K., Shiba K., Siljander P. R.-M., Silva A. M., Skowronek A., Snyder O. L. II, Soares R. P., Sódar B. W., Soekmadji C., Sotillo J., Stahl P. D., Stoorvogel W., Stott S. L., Strasser E. F., Swift S., Tahara H., Tewari M., Timms K., Tiwari S., Tixeira R., Tkach M., Toh W. S., Tomasini R., Torrecilhas A. C., Tosar J. P., Toxavidis V., Urbanelli L., Vader P., van Balkom B. W., van der Grein S. G., Van Deun J., van Herwijnen M. J., Van Keuren-Jensen K., van Niel G., van Royen M. E., van Wijnen A. J., Vasconcelos M. H., Vechetti I. J. Jr., Veit T. D., Vella L. J., Velot É., Verweij F. J., Vestad B., Viñas J. L., Visnovitz T., Vukman K. V., Wahlgren J., Watson D. C., Wauben M. H., Weaver A., Webber J. P., Weber V., Wehman A. M., Weiss D. J., Welsh J. A., Wendt S., Wheelock A. M., Wiener Z., Witte L., Wolfram J., Xagorari A., Xander P., Xu J., Yan X., Yáñez-Mó M., Yin H., Yuana Y., Zappulli V., Zarubova J., Žėkas V., Zhang J.-Y., Zhao Z., Zheng L., Zheutlin A. R., Zickler A. M., Zimmermann P., Zivkovic A. M., Zocco D., Zuba-Surma E. K., Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf7207/DC1