

Cells shape and remodel themselves using specific transport mechanisms to locally expand their breakdown.
Abstract
The F-actin cytoskeleton drives cellular form and function. However, how F-actin-based changes occur with spatiotemporal precision and specific directional orientation is poorly understood. Here, we identify that the unconventional class XV myosin [Myosin 15 (Myo15)] physically and functionally interacts with the F-actin disassembly enzyme Mical to spatiotemporally position cellular breakdown and reconstruction. Specifically, while unconventional myosins have been associated with transporting cargo along F-actin to spatially target cytoskeletal assembly, we now find they also target disassembly. Myo15 specifically positions this F-actin disassembly by associating with Mical and using its motor and MyTH4-FERM cargo-transporting functions to broaden Mical’s distribution. Myo15’s broadening of Mical’s distribution also expands and directionally orients Mical-mediated F-actin disassembly and subsequent cellular remodeling, including in response to Semaphorin/Plexin cell surface activation signals. Thus, we identify a mechanism that spatiotemporally propagates F-actin disassembly while also proposing that other F-actin-trafficked-cargo is derailed by this disassembly to directionally orient rebuilding.
INTRODUCTION
Cells and tissue systems are built and remodeled through the ability of actin and tubulin proteins to associate and form long polymers, actin filaments (F-actin), and microtubules, respectively. These F-actin and microtubules structurally support cells and provide the tracks on which cargo is transported within them (1). Assembling and disassembling F-actin and microtubule cytoskeletal elements in precise spatiotemporal patterns and in specific directional orientations are therefore what underlie the diversity of cellular and tissue system behaviors (1). A large set of extracellular cues has now been identified that work via cell surface receptors to stimulate cytoskeletal dynamics at specific places within cells [e.g., (2)]. So too, a vast array of players have been identified that work intracellularly to organize the cytoskeleton in multiple ways [e.g., (1, 2)]. However, how these proteins come together to locally drive cellular changes in space and time and with specific directional orientations is still poorly understood.
To aid in the understanding of how extracellular signals affect the cytoskeletal elements underlying cellular behaviors, we have been using simple high-resolution single-cell in vivo genetic and cellular models and one of the largest families of extracellular cues, the Semaphorins (Semas). Semas are best known to negatively regulate movement and growth, and emerging data have shown links to numerous pathologies including brain disorders, cancer, cardiovascular abnormalities, and immunocompromising diseases (3). However, how Semas affect cellular form and function remains far from clear. We have uncovered that Semas induce F-actin disassembly and cellular remodeling through an oxidoreductase (Redox)-driven mechanism mediated by Mical (4), a conserved flavoprotein monooxygenase enzyme that binds to both the Sema receptor Plexin and F-actin (fig. S1A) (4, 5). Mical uses its enzymatic Redox activity to posttranslationally oxidize two specific methionine residues at the pointed end of actin (fig. S1A) (6). This oxidation occurs along the interface of actin filament subunits, disrupting the interactions between individual filament subunits and resulting in F-actin disassembly-induced cellular changes (fig. S1A) (6–10). The MICALs [a protein family coded for by three mammalian MICAL genes (MICAL-1, MICAL-2, and MICAL-3) and invertebrate Mical] (5) use this F-actin disassembling Redox enzymatic activity in numerous different types of cells, tissues, and events that depend on F-actin reorganization, and they are also increasingly becoming linked to various diseases and human abnormalities [reviewed in (11–14)]. However, it is still poorly understood how MICALs’ effects are spatially targeted within cells to enable such directionally critical phenomena as growth/extension, navigation, and connectivity.
Using a genetic screening approach, we now identify that Mical interacts with the class XV myosin (Myo15) and requires this unconventional myosin to direct Sema/Plexin/Mical-mediated F-actin disassembly and cellular remodeling. Genetic interaction, biochemical analyses, and cellular and live imaging studies go on to support that Myo15 binds and controls Mical’s subcellular positioning, thereby targeting the location and extent of F-actin disassembly. These effects rely on Myo15’s motor and cargo-transporting activity to position Mical within the dynamic regions of remodeling cells and subcellularly orient the dismantling and rebuilding of cellular structures. This work, therefore, defines important new mechanisms underlying how the cytoskeletal framework of cells is spatially regulated to directionally control and position the breakdown and remodeling of cells.
RESULTS
Myo15 drives Mical-mediated cellular remodeling
Mical regulates F-actin organization and cellular behaviors in numerous tissues, including Drosophila mechanosensory bristles (Fig. 1A and fig. S1B) (4), which have long provided a high-resolution single-cell model for studying the cellular, molecular, and biochemical mechanisms of cytoskeletal-driven remodeling in vivo [reviewed in (15, 16)]. In particular, Mical is required for specifying the proper curved shaping of bristles, such that unlike the slightly curved, unbranched bristles of wild-type (+/+) flies (Fig. 1A), loss of Mical (Mical−/−) results in straight and/or bent (i.e., stiffer and less curved) bristles that are the result of too much F-actin within them [fig. S1, C and D, (1)] (4, 17). So too, elevating the levels of Mical using the bristle-specific B11-GAL4 line (subsequently referred to as Bristle Mical+++) also results in robust but distinctively different cellular/cytoskeletal alterations, such that Bristle Mical+++ produces an easily observable membranous extension (cellular branch) (Fig. 1B, arrowhead) (4). This branch is similar in location and length from cell to cell/animal to animal (Fig. 1B) (4, 6–8, 18) and results from localized F-actin disassembly and subsequent remodeling/new F-actin assembly [fig. S1D, (1)] (4, 6–8, 18). These Mical-triggered cytoskeletal/cellular breakdown and remodeling events are also dependent on Sema/Plexin signaling, and they occur at sites where Mical and its activating receptor Plexin overlap in localization [fig. S1D, (2)] (4, 7, 8, 18).
Fig. 1. The unconventional class XV myosin Myo15 controls Sema/Plexin/Mical-mediated cellular remodeling.
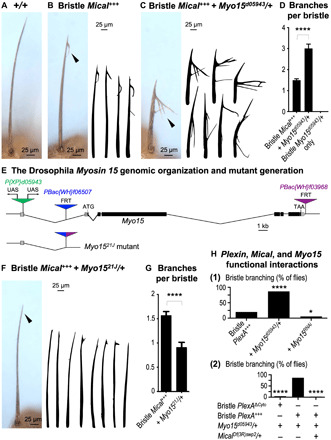
(A) Bristle cells project a long, unbranched, F-actin–driven cellular extension. (B) Elevating bristle Mical levels (Bristle Mical+++ = UAS:Mical/+, B11-GAL4/+) generates a shorter bristle and branching at the tip (arrowhead and drawings). (C and D) Mical-driven bristle effects are strongly enhanced by the P{XP}d05943 mutant, such that the bristles are shorter and more branched (arrowhead, drawings, and graph). Means ± SEM. n ≥ 40 animals (one bristle per animal) per genotype. ****P < 0.0001, unpaired t test (two-tailed). Bristle Mical+++ + Myo15d05943/+ = UAS:Mical/+, B11-GAL4/+, P{XP}d05943/+. Bristle Myo15d05943/+ only = B11-GAL4/+, P{XP}d05943/+. (E) Myo15 genomic map and mutant generation. Exons, black boxes; 5′/3′ untranslated region, gray boxes; transposons, triangles. Only a short noncoding region remains in the Myo1521J mutant we generated by deleting the region between the transposons containing FRT sites (blue and purple). (F and G) Mical-driven bristle branching is suppressed by decreasing Myo15 (arrowhead, drawings, and graph). Means ± SEM. n ≥ 47 animals (one bristle per animal) per genotype. ****P < 0.0001, unpaired t test (two-tailed). (H) Myo15 functions with Sema/Plexin/Mical-mediated cellular remodeling. (1) Elevating Myo15 increases Plexin (PlexA)–induced bristle branching, and reducing Myo15 decreases it. n ≥ 30 animals per genotype. (2) Removing PlexA’s intracellular region (PlexAΔCyto) prevents Myo15 from enhancing PlexA. Decreasing Mical (Df(3R)swp2/+) prevents Myo15 from enhancing PlexA. n ≥ 24 animals per genotype. ****P < 0.0001 and *P < 0.05, chi-square test.
We have therefore used this readily observable, reproducible, and stereotypical branching pattern that occurs in Bristle Mical+++ animals to search (screen) for proteins that increase or decrease this branching effect and are thereby likely to be involved in Sema/Plexin/Mical-mediated cellular remodeling (fig. S2A) (7). In particular, the screen itself was a simple one in which publicly available mutant and transgenic Drosophila fly lines were crossed to flies expressing Mical specifically in bristles (Bristle Mical+++) (fig. S2A). Progeny were then examined for modification (i.e., enhancement or suppression) of this Mical-dependent branching effect (fig. S2A). During the screening process, we observed a marked qualitative and quantitative enhancement of Mical-dependent bristle branching by the P element transposon mutation P{XP}d05943 (compare Fig. 1, B and C; Fig. 1D and fig. S2B). These bristles had a significant increase in the number of branches (Fig. 1, C and D), as well as noticeably longer branches (Fig. 1C). We also noted that P{XP}d05943 enhanced Mical’s effects on the length of the main bristle shaft, further shortening it (compare Fig. 1C to Fig. 1, A and B). Thus, the P{XP}d05943 mutation enhances Mical’s effects on cellular remodeling.
We next examined the genomic insertion position of the P{XP}d05943 transposon and found that it is situated in the Drosophila class XV myosin (Myo15) gene (Fig. 1E) (19–21). Drosophila Myo15 [also called Myo10A, due to its genomic position within the Drosophila genome, and Sisyphus (Syph)] codes for an unconventional member of the myosin family of actin-based motor proteins (19–21). Furthermore, the P{XP}d05943 transposon is positioned upstream of the Myo15 coding region and contains upstream activator sites (UASs) for GAL4 transcription factor drivers (Fig. 1E), indicating that P{XP}d05943, in combination with the B11-GAL4 driver, is likely to be increasing the expression of Myo15 to enhance Mical-dependent cellular remodeling. To directly test this hypothesis, we generated transgenic flies expressing Myo15 and indeed found that Myo15 significantly enhanced Mical-mediated cellular remodeling, resembling the effects on Mical we observed with P{XP}d05943 (compare fig. S2C to Fig. 1, B and C; fig. S2, B and D). Notably, we also found that this bristle expression of Myo15 on its own altered cellular remodeling (fig. S2, D to F) and that decreasing the levels of Mical suppressed these Myo15-dependent changes (fig. S2F). Together, therefore, these results indicate that the unconventional myosin Myo15 and the F-actin disassembler Mical genetically interact to regulate cellular remodeling.
Sema/Plexin/Mical-mediated cellular remodeling specifically requires Myo15
Myosin family members are actin-based molecular motors that are grouped into more than 30 classes based on the similarity of their motor domain, which binds actin and hydrolyzes adenosine triphosphate (ATP) to (i) move/hold actin filaments (i.e., myosins functioning as force generators/mechanical tethers) or (ii) move myosins processively along actin filaments (i.e., myosins functioning as intracellular transporters) (22). The 14 known Drosophila myosin genes and their protein products fall into 10 different classes: I (three different genes), II (two different genes), III, V, VI, VII (two different genes), XV, XVIII, XX, and XXII (fig. S3A) (19, 20, 23). Given that myosins work with actins, which MICALs also affect (as described above), we wondered whether Myo15’s effects were specific or whether all myosins affected Mical’s cellular actions. Our results revealed that raising the levels of other myosins such as members of classes I, II, V, VII, and XVIII did not notably modify Mical-mediated cellular effects (fig. S3B). Thus, the class XV myosin Myo15 is a myosin family member that specifically regulates Mical-mediated cellular remodeling.
Myo15 is the only known Drosophila class XV myosin, but no loss-of-function/“knockout” genetic mutant lines have been generated. Thus, to further examine the role of Myo15 in Mical-mediated cellular remodeling, we used a FLP/FRT (site-specific) recombination technique (24) to delete the entire coding region of Myo15 and generate a Myo15 mutant, Myo1521J (Fig. 1E). Characterizing these Myo1521J mutants revealed that they reach the third instar larval stages but typically die before or shortly after pupation and rarely survive to adulthood (fig. S4A). We did note, however, a few adult escapers that had variable visible defects including effects on wings, eyes, and bristles, such as alterations to bristle length and morphology (e.g., fig. S4A). Notably, we also found that loss of one copy of Myo15 (Myo1521J/+) or bristle-specific expression of a Myo15 RNAi (RNA interference) transgene strongly suppressed Mical-dependent cellular remodeling (Fig. 1, F and G, and fig. S4, B to D), thereby supporting that Myo15 is required for Mical-mediated cellular remodeling. Furthermore, Mical uses a specific Redox enzymatic activity to affect F-actin and remodel cells (fig. S1A) (11), and our results revealed that Myo15 specifically regulates enzymatically active Mical’s effects (fig. S4, E and F) and that these Myo15 effects are counteracted (fig. S4G) by the SelR/MsrB reductase enzyme that works in opposition to Mical’s enzyme activity (7, 25). Moreover, since Mical functions with Sema/Plexin repulsive guidance cues/receptors (fig. S1A) (4, 5, 11), we wondered whether Myo15 also plays a role in Sema/Plexin repulsive signaling. Our results revealed that raising the bristle levels of Myo15 increased Sema/Plexin-mediated cellular remodeling, while reducing the levels of Myo15 decreased this remodeling [Fig. 1H, (1)]. Likewise, the Myo15-induced increase in Sema/Plexin-mediated cellular remodeling was dependent not only on the Plexin cytoplasmic (intracellular) domain that interacts with Mical [Fig. 1H, (2)] but also on Mical itself [Fig. 1H, (2)]. Thus, Myo15 is required for Sema/Plexin/Mical Redox–driven cellular remodeling.
Myo15 colocalizes and coimmunoprecipitates with Mical in vivo
As a class XV myosin, Myo15, along with class VII (Myo7a and Myo7b) and class XXII myosins are the four Drosophila members of the MyTH4-FERM superclass of myosins (which also includes four members in mammals: Myo7a, Myo7b, Myo10, and Myo15). Members of the MyTH4-FERM superclass are defined by the presence of MyTH4 (myosin tail homology 4) and FERM (protein four-point-one, Ezrin, Radixin, Moesin) domains in their tail regions (Fig. 2A and fig. S5). MyTH4-FERM myosins function as intracellular transporters within actin-rich filopodia/cellular protrusions, with each MyTH4-FERM class family member differing in their localization and the particular cargo they carry [reviewed in (26)]. Specifically, Drosophila Myo15 and the mammalian Myo15 have been shown to traffic specific protein cargo along F-actin to the tips of F-actin–rich structures such as filopodia and stereocilia (21, 27–29), and we also observed Myo15 colocalizing along with F-actin (but not microtubules) in the bristle model (fig. S6, A and B). Notably, we also found that Mical and Myo15 localized together in vivo (Fig. 2B and figs. S6, C and D, and S7A). In particular, using the single-cell bristle model, we found that Myo15 had a broader cellular distribution than Mical, but Mical and Myo15 colocalized in regions where Mical-triggered cellular remodeling was occurring, including within and around extending bristle tips and branches, and within tracks emanating from robust Mical localization (Fig. 2B and figs. S6, C and D, and S7A). Furthermore, turning to coimmunoprecipitation assays, we found that Mical and Myo15 could be coimmunoprecipitated from in vivo tissues (Fig. 2C). Thus, our results demonstrate a colocalization and association between Myo15 and Mical in vivo.
Fig. 2. Myo15 associates with and distributes Mical to spatially and directionally remodel cells.
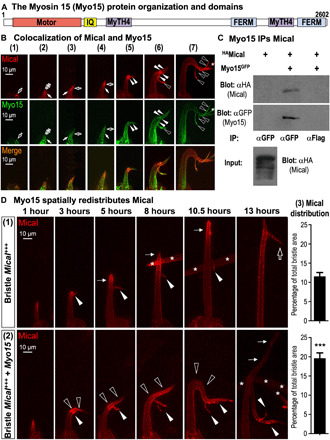
(A) Drosophila Myo15’s domain organization matches mammalian Myo15. (B) Mical (red) and Myo15 (green) colocalize in vivo (UAS:mCherryMical/+, UAS:Myo15GFP/+, B11-GAL4/+). (1) to (7) show different bristles at increasing developmental stages. Mical-Myo15 overlap/colocalize within early extending bristle tips (open arrows), tracks around these tips (closed arrows), narrow tracks emanating from Mical-enriched regions (closed arrowheads), and within Mical-triggered branches (open arrowheads). Asterisks indicate adjacent bristle. (C) Myo15 immunoprecipitates (IPs) Mical in vivo. α, antibody. (D) Myo15 spatially redistributes Mical (red) to remodel cells. Bristles begin extending at 0 hour (~31 hours after puparium formation). One to13 hours show different bristles at increasing developmental stages and typical of those stages/genotypes. (1) Bristle Mical+++ (UAS:mCherryMical/+, B11-GAL4/+): Elevating Mical levels induces a branch (3 hours, arrowhead). Bristles then (5 to 10.5 hours) extend beyond (thin arrows) this branch point. A second Mical-triggered branch (open arrow) then forms (13 hours). Mical (red) localizes at/near bristle tips at all stages (1 to 13 hours) and enters both branches (2 to 13 hours). Asterisks indicate adjacent bristle. (2) Bristle Mical+++ + Myo15 (UAS:mCherryMical/+, UAS:Myo15GFP/+, B11-GAL4/+): Myo15 increases and reorients (open arrowheads) Mical-triggered bristle remodeling. This enhanced remodeling (3 to 10.5 hours) also correlates with an increase in Mical’s (red) distribution, including within circuitous/reoriented regions (open arrowheads) and branches (closed arrowheads). Vertical growth eventually resumes (13 hours, thin arrows) but with little Mical (red) within it. (3) Elevating bristle Myo15 increases Mical’s bristle distribution. Areas of high Mical measured at 7 to 9 hours. Means ± SEM. n ≥ 11 bristles (5 to 10 animals) per genotype. ***P = 0.0001, unpaired t test (two-tailed).
Myo15 positions Mical to spatially and directionally control cellular remodeling
Mical-driven F-actin/cellular remodeling is not detectible along the entirety of cells such as the bristle process but rather at distinct loci within the cell’s dynamic regions, where it is activated to generate readily observable effects such as filopodia/branches [Fig. 1B, arrowhead; reviewed in (11–14)]. Therefore, to better understand how this branch formation is occurring, we imaged developing bristles over time to temporally follow both the remodeling bristle and the spatial localization of Mical within it. During the early stages of bristle extension, we noted that Mical-mediated F-actin reorganization generated an easily observable small branch at the tip of the growing bristle [Fig. 2D, (1), 3 hours, arrowhead; (4, 6, 7)]. Then, as development continued, the main bristle shaft elongated past this branch point [Fig. 2D, (1), 5 to 10.5 hours, thin arrows], advancing until Mical triggered the generation of a second larger branch that was spatiotemporally distinct from the first [Fig. 2D, (1), 13 hours, open arrow] (4, 6, 7). Further analysis revealed that Mical localized to bristle tips during the course of this bristle extension [Fig. 2D, (1), red], making forays into the first branch [Fig. 2D, (1), arrowheads, red] but maintaining the bulk of its localization to the main bristle shaft throughout its elongation and after formation of the second branch [Fig. 2D, (1), red].
Notably, we found that Myo15 was redistributing Mical (fig. S7, B and C), including dynamically and spatiotemporally controlling aspects of this stereotypical pattern of Mical localization and bristle remodeling [Fig. 2D, (2)]. In particular, during the earliest stages of bristle development, we found that Myo15 did not induce any observable changes to Mical or bristle development, such that bristle extension proceeded normally, and Mical localized in a normal pattern within extending bristle tips [Fig. 2D, (2), 1 hour, red]. However, as the first Mical-triggered branch began to form [Fig. 2D, (2), 3 hours, closed arrowhead], we noticed that Myo15 significantly broadened Mical’s distribution [Fig. 2D, (2), red; quantified in Fig. 2D, (3)] and induced a notable enhancement of Mical’s effects that resulted in less stability to the bristle tip [i.e., a curved/floppy appearance; Fig. 2D, (2), open arrowheads]. Furthermore, Myo15 expanded and redistributed Mical into these floppy tips/extensions and new branches [Fig. 2D, (2), 3 to 13 hours, red], which, in turn, resulted in larger and more complex and misoriented extensions/branches [Fig. 2D, (2), 10.5 hours, arrowheads, and fig. S7, B and C]. Moreover, we noticed that the typical vertical extension of the bristle was delayed in forming and then, upon forming, was devoid of appreciable Mical localization [Fig. 2D, (2), 13 hours, thin arrows]. Thus, Myo15 significantly modifies Mical’s subcellular localization (Fig. 2D and fig. S7, B and C), and this Myo15-dependent change in Mical’s localization corresponds with marked changes to cell shape, including affecting the location and extent of Mical-mediated cellular remodeling (Figs. 1, B to D, and 2D and figs. S2, B to D, and S7, B and C).
Myo15 locally redistributes Mical in vivo
Our results revealed that Myo15 was modifying Mical’s distribution, so we wondered whether Myo15 was involved in a long-range transport of Mical from the cell body to the growing bristle tip or whether Myo15 was locally altering Mical’s distribution. To address this question, we examined the localization of Mical in Myo15 “knockout/knockdown” mutants (Myo1521J/+ + Bristle Myo15 RNAi), which resulted in strong suppression of Mical-triggered cellular remodeling/branching (fig. S4, B and D). These Myo15 knockout/knockdown mutants, however, did not significantly change Mical’s localization to bristle tips [Fig. 3A, (1) and (2)]. Thus, Myo15 does not alter Mical-mediated cellular remodeling by preventing Mical’s long-range transport to the growing bristle tip. Furthermore, since we still see extensive amounts of Mical at the bristle tip when we decrease Myo15 levels [Fig. 3A, (1) and (2)], but Mical’s effects on cellular remodeling are substantially suppressed (Fig. 1, F and G, and fig. S4, B to E), these results further support that Myo15 is not simply involved in getting high levels of Mical to a given subcellular location but is required to locally broaden Mical’s effects on cellular remodeling.
Fig. 3. Myo15 and its motor and cargo-transporting regions locally expand Mical-mediated remodeling.
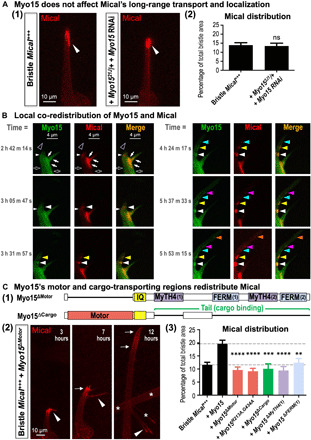
(A) Myo15 does not alter Mical’s long-range transport. (1) Mical (red) still localizes to bristle tips (arrowheads) in Myo15 knockout/knockdown. (2) Mical’s distribution measured as in Fig. 2D (3). Means ± SEM. n ≥ 12 bristles (6 to 12 animals) per genotype. ns (not significant), P = 0.8083, unpaired t test (two-tailed). (B) Mical locally redistributes with Myo15 in vivo. Same bristle imaged over time (times from start of movie S1). Myo15 (green) and Mical (red) exhibit regions of colocalization (closed arrows) and no colocalization (open arrows). During remodeling/extension (open arrowheads and similar regions at subsequent times), Mical’s (red) movement/redistribution tracks with Myo15 (different color arrowheads), resulting in coenriched areas/nodes that enlarge (get brighter) over time. Note that a slight postimaging brightness adjustment was made to the last red channel image (5 hours, 53 min, 15 s) to better highlight the most distal labeling spot. (C) Myo15’s motor and cargo-transporting regions drive Mical’s redistribution and cellular remodeling. (1) Myo15ΔMotor, lacks the motor domain; Myo15ΔCargo, lacks both cargo-binding MyTH4-FERM domains. (2) Bristles imaged over time as in Fig. 2D. Myo15ΔMotor does not notably enhance Mical-triggered remodeling nor redistribution (red) into branches [arrowheads; compare to Fig. 2D, (2)]. Instead, Mical remains prominently in the vertically extending bristle tip (arrows). Asterisks indicate adjacent bristle. (3) Mical’s bristle distribution measured as in Fig. 2D (3) and in (A) (2). Means ± SEM. n ≥ 11 bristles (6 to 12 animals) per genotype. ****P < 0.0001, ***P = 0.0002, and **P = 0.0087, one-way analysis of variance (ANOVA) (****P < 0.0001) with Dunnett’s multiple comparisons (each transgene compared to +Myo15).
These results are consistent with work in other systems where proteins such as dyneins and kinesins drive long-range movements along microtubules, whereas myosins mediate short-range movements along actin filaments (22). Previous work using the purified motor domain of class XV myosins reveals that Myo15 is a high–duty ratio molecular motor [i.e., the myosin head (motor) spends its time strongly bound to its F-actin track during each ATPase cycle, a property required for processive movement of myosins on F-actin] and that the Myo15 motor moves rapidly (400 nm/s) with a relatively short 8-nm power stroke toward the membrane-proximal barbed end of F-actin (30, 31). Therefore, these previous observations, coupled with our results in Figs. 1, 2, and 3A, raise the hypothesis that Myo15 associates with Mical and locally moves and redistributes Mical to broaden Mical’s effects on cellular remodeling. To further test this hypothesis, we initially explored using assays with purified proteins to visualize Myo15’s ability to move and change Mical’s distribution. However, Myo15s are the largest myosins in the mammalian (and fly) proteome (fig. S3A) (22), and because of their extensive size (310 to 395 kDa; Fig. 2A) (19–22), full-length Myo15s have not been purified and so are unable to be examined for their ability to move (with their cargo) along purified F-actin. However, these types of Myo15 (including Drosophila Myo15) motor-driven movements along F-actin can be visualized in vivo [e.g., (21, 27–30, 32–39)]. We therefore used the in vivo bristle model system to visualize and track Myo15 and Mical in real time. Previous work has shown that Myo15 colocalizes with and moves along F-actin in cells/in vivo [e.g., (21, 27–30, 32–39)]. Likewise, our results observing Myo15 in vivo reveal that Myo15 colocalizes with F-actin (fig. S6, A and D), relocalizes within the elongating/branching bristle (Fig. 2B), and mobilizes in real time during this remodeling (Fig. 3B and movie S1). Moreover, our results in which we directly followed fluorescently tagged forms of both Myo15 (Myo15GFP) and Mical (mCherryMical) in live imaging experiments revealed co-movement of Myo15 and Mical in vivo, including the local redistribution of Mical with Myo15 into newly projecting cellular extensions (Fig. 3B and movie S1). These live imaging results (Fig. 3B and movie S1), in combination with our still imaging at different time points (Figs. 2, B and D, and 3A and fig. S7C), paint a picture that, in conjunction with cellular remodeling, Mical moves locally from regions of robust Mical enrichment into new areas and that Mical co-mobilizes with Myo15 during this change in localization. So too, these live imaging observations, coupled with our colocalization and immunoprecipitation results, and that Myo15 is required for Mical-mediated cellular remodeling and sufficient to enhance it, support the hypothesis that Myo15 interacts with and is locally transporting Mical to expand Mical-mediated cellular remodeling.
Myo15 uses its myosin motor activity and cargo-transporting MyTH4-FERM domains to spatially target Mical-mediated cellular remodeling
The Myo15 class of myosins locally transports different cargos, which they do through the use of their motor domain and various tail domains that directly associate with specific cargo (Fig. 2A) (21, 27, 28). Therefore, to further test the hypothesis that Myo15 locally transports Mical to broaden Mical’s effects on cellular remodeling, we used site-directed mutagenesis to generate a series of deletion mutants in which we disrupted either the myosin motor activity or its cargo-binding domains. In particular, to alter the motor activity of Myo15, we (i) deleted the entire motor domain of Myo15 [Myo15ΔMotor; Fig. 3C, (1)] and (ii) made two point mutations (R213A and G434A) in the highly conserved switch I and switch II regions of Myo15’s motor (Myo15R213A,G434A; fig. S8, A and B). These point mutations are known to disrupt myosin motor activity (e.g., in mouse Myo15 and Dictyostelium discoideum Myosin II) by preventing ATP hydrolysis, thereby keeping the myosin motor in a weak actin-binding state and unable to move along actin filaments (27, 40). Further, since Myo15−/− mutants rarely survive to adulthood (fig. S4A), we designed our experiments not to remove wild-type Myo15 and replace it with Myo15 motor mutant but to compare the Myo15 motor mutants effects on Mical to wild-type Myo15 [Fig. 2D, (2) and (3)]. Notably, we found that even without a motor domain, Myo15 localized throughout the extending bristle process in a pattern similar to wild-type Myo15 (fig. S8C). However, the redistribution of Mical that was a hallmark of elevating wild-type Myo15 [Fig. 2D, (2) and (3), and fig. S7, B and C] did not occur with motor mutant forms of Myo15 [Fig. 3C, (2) and (3)]. Instead, Mical’s distribution with motor mutant forms of Myo15 resembles Mical+++ bristles without elevated Myo15 [Fig. 3C, (3), bottom dashed line]. Likewise, while some branching still occurred in response to coexpression of Mical and the motor mutant forms of Myo15 [Fig. 3C, (2), arrowheads; this was not unexpected since wild-type Myo15 is still present], the extensive Mical-mediated cellular remodeling that was induced by wild-type Myo15 was suppressed by disrupting Myo15’s myosin motor activity [compare Fig. 3C, (2), with Fig. 2D, (2); fig. S7, B and C]. We also saw results consistent with these motor mutants functioning in a dominant negative manner {e.g., notice the decrease in Mical’s distribution with these motor mutations when compared to Bristle Mical+++ only [Fig. 3C, (3), bottom dashed line]}. Since the cargo-binding tail is still present in these motor mutants, they may interact with and keep Mical from associating with endogenous Myo15. Likewise, while the oligomeric state of Myo15 in cells has not been definitively defined, myosins from different classes exist as single-headed or double-headed forms (40). Therefore, if Myo15 is double-headed, then heterodimerization of the endogenous Myo15 with a motor mutant form could produce a dominant negative Myo15. Similar discussions have arisen from identification/analysis of Myo15 mutations in mammals including humans [e.g., (27, 28, 31, 41)]. Together, these results support that Myo15 uses its motor activity-driven transport function to locally distribute Mical and thereby spatially expand Mical-mediated cellular remodeling.
We next sought to determine whether Myo15 might be using its tail domains to locally transport Mical. Myo15 class myosins use their tail domains to locally transport cargo (21, 27, 28), and structure-based amino acid sequence analysis reveals that the key residues within the tail domains and MyTH4-FERM interfaces are completely conserved in Drosophila Myo15 and all MyTH4-FERM tandem-containing proteins [e.g., fig. S5; reviewed in (42)]. We therefore disrupted the cargo-binding tail domain of Myo15 by deleting both of its MyTH4 and FERM domains [Myo15ΔCargo; Fig. 3C, (1)] and made a transgenic animal lacking all of these cargo-transporting tail regions. Our results revealed that while the MyTH4 and FERM domains, similar to the Myo15 motor domain, were dispensable for the normal localization of Myo15 to the extending bristle and its tip (fig. S8C), removing these cargo-transporting MyTH4-FERM domains prevented Myo15 from both (i) redistributing Mical and (ii) driving Mical-mediated cellular remodeling [Fig. 3C, (3)]. We also saw results consistent with this cargo-binding mutant functioning in a dominant negative manner [e.g., Fig. 3C, (3)]. Furthermore, generating and examining a series of seven additional Myo15 deletion mutant transgenic fly lines (fig. S9A) revealed that it was the first MyTH4 [MyTH4(1)] and FERM [FERM(1)] domains that were required for Myo15’s full effects of both redistributing Mical and driving Mical-mediated cellular remodeling [Fig. 3C, (3), and figs. S8C and S9B]. Thus, these results support the hypothesis that the cargo-transporting tail region of Myo15 and specifically its first MyTH4 and FERM domains work in combination with the Myo15 motor domain to locally traffic Mical along F-actin to induce cellular remodeling.
Myo15 positions Mical to spatially target F-actin disassembly
MICALs are Redox enzymes that target F-actin for disassembly and reorganization (4, 6, 11–14). Since Myo15 is necessary and sufficient for Mical’s Redox-dependent cellular remodeling, we wondered whether Myo15 might also be affecting Mical’s F-actin disassembly activity. As introduced above, the Drosophila bristle system provides a simple, high-resolution single-cell model for studying actin dynamics in vivo [reviewed in (15, 16)]. In particular, as the bristle begins to extend, actin (termed actin snarls in bristles, which have analogy to similar types of actin in other cells such as actin patches in yeast) is polymerized at the tip, and then if stabilized by cross-linking proteins and interactions with the plasma membrane, these snarls/patches function to shape, position, and be incorporated into bundles of F-actin (fig. S1B) [see (15, 43, 44) and references therein for more detail]. These F-actin bundles are well organized, have their barbed end facing distally toward the tip of the bristle, and help to elongate and stabilize the developing bristle [Fig. 4A, (1), and fig. S1B] (4, 15–17, 43, 44). In contrast, bristle expression of Myo15 on its own altered F-actin organization such that we noticed regions with more prominent F-actin/bundles of F-actin, as well as regions with less prominent F-actin [Fig. 4A, (2), and fig. S6A]. As previously defined (fig. S1D) (4, 6–8, 10, 18), bristle-specific expression of Mical also altered the organization of this F-actin/bundled F-actin, inducing small regions of F-actin disassembly and remodeling/branching [Fig. 4, A, (3), and B, (1)]. Notably, we found that loss of Myo15 (Myo1521J/+) suppressed this Mical-mediated F-actin disassembly [Fig. 4A, (4)], while increasing the levels of Myo15 markedly enhanced it [Fig. 4A, (5)]. Furthermore, tracking bristle development over time [as in Figs. 2D, (1) and (2), and 3C, (2), and fig. S9B] revealed that when the levels of Myo15 were increased with Mical, the cellular regions containing disrupted F-actin were significantly expanded, and these F-actin disruptions increased over time (Fig. 4B, (2) and compare to (1) and (3)]. Moreover, we found that those expanded regions exhibiting disrupted F-actin markedly overlapped with the broadened distribution of Mical that is triggered by Myo15 (Fig. 4C; see also Fig. 2D and fig. S7, B and C), and Mical and Myo15 also overlapped in these regions of disrupted F-actin (fig. S6, C and D). Thus, these results reveal that Myo15 functions to promote Mical-triggered F-actin disassembly, and it does so by directing Mical into new cellular regions.
Fig. 4. Myo15 positions Mical to spatially target and expand F-actin disassembly.
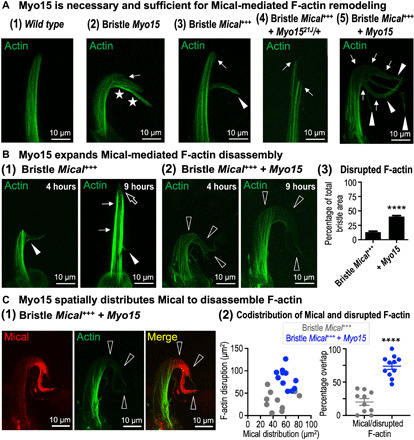
Genotypes are as previously described plus the addition of GFPactin transgene to visualize actin. (A) Myo15 is necessary and sufficient for Mical-mediated F-actin disassembly. (1) Normal bristle F-actin organization (B11-GAL4/+, UAS:GFPActin/+). (2) Elevating Myo15 induces regions of more (stars) and less (arrow) F-actin. Abnormal bristle bends, and other alterations accompanied these F-actin changes (e.g., stars; see also figs. S2, E and F, and S6A). (3) Elevating Mical induces regions of less F-actin (arrow) and branching (arrowhead) (4, 7). (4) Reducing Myo15 decreases Mical-triggered F-actin disassembly and branching (arrows). (5) Elevating Myo15 increases Mical-triggered F-actin disassembly (arrows) and branching (arrowheads). (B) Myo15 expands Mical-mediated F-actin disassembly. Bristle development tracked as in Figs. 2D [(1) and (2)] and 3C (2) and fig. S9B. (1) Elevating Mical induces F-actin disruptions and branching (4 hours, closed arrowhead). Bristles then extend past this branch point (9 hours, thin arrows), where F-actin disruptions (open arrow) and branching [13 hours; Fig. 2D, (1)] occur again. (2) Elevating Myo15 increases Mical-triggered F-actin disruptions (4 hours, open arrowheads), and these further increase with age (9 hours, open arrowheads). (3) Percentage of the bristle with disrupted F-actin. Means ± SEM. n ≥ 12 bristles (6 to 12 animals) per genotype. ****P < 0.0001, unpaired t test (two-tailed). (C) Myo15’s increased broadening of Mical’s distribution (red) overlaps with Myo15’s increased broadening of Mical-triggered F-actin (green) disruptions [open arrowheads in (1) and quantified in (2)]. Means ±SEM. n > 10 bristles (5 to 10 animals) per genotype. ****P < 0.0001, two-tailed t test.
Myo15, similar to Mical, regulates muscle actin filament organization and neural connectivity
MICALs regulate F-actin organization/cellular remodeling in numerous tissues to direct axon guidance, synaptic structure and function, dendritic pruning, and muscle morphology/organization [reviewed in (11–14)]. We therefore wondered whether Myo15, similar to Mical, might also function in some of these other tissues. To test this, we used the developing Drosophila larval neuromuscular junction (NMJ) to examine Myo15 in both the developing nervous and musculoskeletal systems (Fig. 5A). Mical is broadly expressed in the nervous and musculoskeletal systems (5, 45), and Mical mutants have an abnormal synapse structure at NMJs, including synaptic boutons that are abnormally clustered together and synapses that do not spread out normally along muscles (Fig. 5B) (7, 45, 46). Myo15, too, is expressed in the nervous and musculoskeletal systems (e.g., fig. S10, A to C) (47, 48), including localizing to growth cones (34) and presynaptic terminals at the NMJ (fig. S10D). Likewise, Myo15 mutants had an abnormal synaptic structure with defects that resembled Mical mutants (7, 45), including that NMJ synapse length was reduced in Myo15 mutants along muscle 1/9 (Fig. 5B) and muscle 6/7 (Fig. 5C). In addition, Mical mutants have disrupted actin and myofilament organization within skeletal muscles on the postsynaptic side of the NMJ (7, 45), and increasing Mical expression in muscles using a muscle-specific GAL4 driver causes F-actin organizational defects (7). Likewise, we found that muscle-specific expression of Myo15 caused significant alterations to F-actin organization (Fig. 5D), which included aggregations and abnormal clumping of F-actin that resembled the defects seen with muscle-specific expression of Mical (7). Thus, similar to our observations using the high-resolution in vivo bristle cell model, these results reveal that Myo15, similar to Mical, regulates synaptic structure and muscle actin filament organization.
Fig. 5. Mical and Myo15 regulate synaptic structure and F-actin muscle organization.
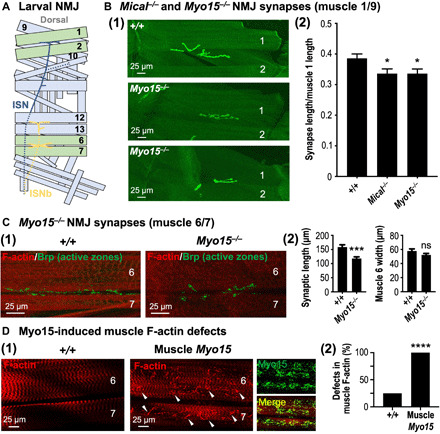
(A) Illustrated hemisegment showing the stereotypical synaptic innervation of intersegmental nerve (ISN) (blue) and ISNb (yellow) motor axons. Muscles 1, 2, 6, and 7, green; other muscles, light blue; dashed lines, nerve lying underneath/lateral to muscles. (B) Myo15−/− mutants, similar to Mical−/− mutants, have reduced synaptic innervation. Synapses visualized with CD8-GFP-Shaker (shGFP7A) (7, 45). (1) Compared to wild-type (shGFP7A/shGFP7A) synapses, which spread out along muscles 1 and 9 (top), Myo15−/− mutant synapses (Myo1521J/Y; shGFP7A/shGFP7A) are shorter (middle and bottom). Muscle 2 indicated for reference. (2) Both Mical−/− and Myo15−/− mutants have reduced synaptic innervation. Mical−/− (MicalI666, shGFP7A/MicalG56, shGFP7A). Means ± SEM. n ≥ 20 synapses/muscles (10 animals) per genotype. *P = 0.0243 (Myo15−/−) and *P = 0.0219 (Mical−/−), unpaired t test (two-tailed). (C) Synaptic innervation of muscles 6 and 7 is also shorter in Myo15−/− (Myo1521J/Y) mutants. Synapses/presynaptic active zones visualized with nc82 [Bruchpilot (Brp)] antibody. Muscles visualized with fluorophore-conjugated phalloidin. Means ± SEM. n ≥ 17 synapses/muscles (10 animals) per genotype. ***P = 0.0002 and ns, P = 0.1395, unpaired t test (two-tailed). (D) Elevating Myo15 in muscles (Muscle Myo15 = UAS:Myo15GFP/+, 24B-GAL4/+) significantly alters F-actin (red) organization [(1) and (2)], including disrupting its striated pattern (1) and inducing clumping (arrowheads). Myo15 (green) localizes with these F-actin defects (merge). n ≥ 19 synapses/muscles (10 animals) per genotype. ****P < 0.0001, chi-square test.
DISCUSSION
Numerous cellular and tissue-level behaviors involve the poorly understood spatiotemporal breakdown and reorganization of the actin cytoskeleton. Our results now support a new understanding of these events by identifying a synergistic interaction that spatiotemporally and directionally mobilizes the machinery needed for F-actin disruption and remodeling. In particular, previous results have revealed that different extracellular and intracellular signals such as Sema/Plexin repulsive guidance cues and Rab guanosine triphosphatases (GTPases), respectively, bind and activate the Redox enzyme Mical at specific places and times to posttranslationally modify and disassemble F-actin [Fig. 6, A, (1), and B, (1)] (11–14). Class XV unconventional myosins are also known to be spatiotemporally active, using their processive motor function to locally move cargo into specific cellular regions [Fig. 6B, (1)] (26, 41, 49). Here, we find a convergence of these effects such that a class XV myosin physically and functionally associates with Mical (Fig. 6, A, (2), and B, (2)]. Furthermore, our results support that Myo15 uses its motor and cargo-binding regions to locally transport Mical, thereby broadening Mical’s cellular distribution and its effects on F-actin [Fig. 6, A, (2), and B, (2) and (3)]. Moreover, our results support that these Myo15-Mical interactions serve to directionally orient and enhance F-actin disassembly and remodeling, including increasing Sema/Plexin’s repulsive effects at particular places and times [Fig. 6, A, (2), and B, (3)]. Our observations therefore support a model that Myo15 and Mical combine in vivo to spatiotemporally position and expand the breakdown and remodeling of cells and their membranous extensions.
Fig. 6. Summary/model: Myo15 propagates Mical Redox-triggered F-actin disassembly and cellular remodeling.
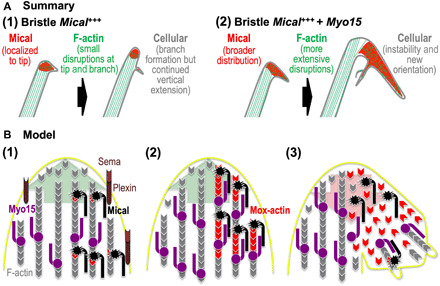
(A) Summary: (1) Left: Mical (red) localizes to the growing bristle tip and is spatiotemporally activated to disassemble F-actin and trigger cellular remodeling/branching. Right: As development continues, the bristle again assembles F-actin/bundled filaments, which elongate the bristle past this region of F-actin disassembly/branching. (2) Left and right: Elevating Myo15 levels distributes Mical more broadly around the branch point, spatially increasing F-actin disassembly. Right: Myo15’s expansion of Mical’s distribution and F-actin disassembly hinders new F-actin/bundled filament assembly, destabilizing and misdirecting the elongating bristle. Myo15 thereby transforms regions where Sema/Plexin/Mical activation typically induces limited disassembly/reorganization (1) into more expansive effects (2). (B) Model: (1) During outgrowth/extension (large green arrow), Mical (black) specifically localizes and is activated by Sema repellents (light brown) and their Plexin receptors (dark brown) (4, 5). Activated Mical oxidizes F-actin (gray) subunits on their pointed ends, generating Mical-oxidized actin (Mox-actin) subunits (red) (6, 7, 9). Myo15 (purple) also works within these growing regions, carrying cargo toward F-actin barbed ends/the membrane (26, 41, 49). (2) Mical–Myo15 associations transport Mical and its Redox-enzymatic F-actin-modifying effects toward F-actin barbed ends/the membrane. (3) This Myo15 transport expands Mical-mediated F-actin disassembly, which breaks down specific cellular regions (large red arrow). This F-actin disassembly also derails/deposits F-actin-transported cargo specifically within these disrupted regions, which positions it to assist in the local reconstruction/branching that follows Mical-triggered F-actin disassembly.
Unconventional myosins, including MyTH4-FERM class myosins, have previously been associated with using their transport functions to locally carry cargo to promote/broaden the localized assembly of cytoskeletal structures (22, 26). Our results now uncover roles for them in using their transport functions to promote/broaden the localized disassembly of the cytoskeleton. Furthermore, we believe that our results also harmonize these contrasting roles by advancing a logic for how cellular assembly/rebuilding spatiotemporally follows disassembly. In particular, our results herein support that Myo15-Mical interactions expand F-actin disassembly, but we also find that this Mical-mediated F-actin disassembly is accompanied by new filopodia/branch formation (e.g., see Figs. 1, B, C, and F, and 4A) (4, 7, 8, 18). So too, raising the levels of Myo15 on its own induces F-actin/cellular effects that are suppressed by a loss of Mical (i.e., Mical is required for at least some of these Myo15-induced changes; fig. S2, E and F). However, we also find that raising the levels of Myo15 on its own induces different effects than raising the levels of Mical on its own [e.g., compare Fig. 1B with fig. S2E and compare Fig. 4A, (2) with (3)], including generating regions of increased F-actin and regions of decreased F-actin [Fig. 4A, (2)]. Therefore, together, these results support that, in our in vivo model, Myo15 is likely to be transporting other proteins in addition to Mical to exert its effects. Both Drosophila and mammalian Myo15s are known to associate with and/or transport different cargo including cell adhesion molecules such as DE-cadherin (21), actin regulatory proteins such as Eps8 and gelsolin (28, 50), tubulin and microtubule regulatory proteins such as EB1 and Katanin (21), scaffolding proteins such as whirlin (27, 33, 51), and other regulatory proteins such as protein kinases, G proteins, and membrane-associated guanylate kinases (MAGUKs) (21, 29, 39, 52). These cargos allow Myo15s to play important roles in the building of cells including aiding the extension of actin-filled projections such as filopodia and stereocilia and promoting cellular adhesion [e.g., (21, 26, 28)]. Our results therefore support a model in which Myo15 transports additional cargo along with Mical, and Mical’s breakdown of F-actin alters this transport and specifically deposits/positions this cargo to locally drive new actin assembly, filopodial extension, and cellular rebuilding [Fig. 6B, (3)]. Thus, we hypothesize that Myo15-Mical interactions serve two purposes: (i) Myo15 carries Mical to expand local F-actin disassembly, and (ii) Myo15’s carrying of other cargo allows for cytoskeletal/cellular rebuilding in these disassembled areas.
The role of Myo15 in Mical-mediated F-actin disassembly is also interesting in light of the mechanisms of how Mical modifies actin. In particular, actin filaments are typically found in cells with their barbed ends positioned toward the plasma membrane (1). Mical is activated at specific loci to modify the pointed end of F-actin subunits [e.g., (4, 6)]. If and how Mical’s effects might be propagated from this activation region are unknown. The Myo15-Mical interactions we have uncovered herein, coupled with Myo15’s ability to function as an F-actin barbed end-directed transporter (26, 30), thus provide a means to allow Mical’s effects to be both broadened and directionally propagated toward the cell membrane. Furthermore, F-actin is often bundled together within cells (by F-actin cross-linkers), including within filopodia, microvilli, stereocilia, and bristles, and some of these bundles can be quite large (>250 nm in diameter) [e.g., (1, 15)]. While Mical is known to disassemble bundled F-actin (4), how Mical might gain access to different parts of bundled filaments is not known, but that is also likely important as F-actin is highly abundant in the growing regions of cells (1), and modifying one filament within a bundle is unlikely to cause disassembly of the filament bundle. In this regard, it is interesting to consider that some myosins including MyTH4-FERM class myosins are known to move more efficiently and even laterally on specific types of actin filaments, such as those bound to tropomyosins or bundled with specific cross-linkers [e.g., (53)]. While nothing is known of the preference of Myo15s for different types of F-actin, they are known to be associated with F-actin that is highly bundled, such as that in filopodia, microvilli, and stereocilia [e.g., (21, 27–29, 32, 34–36, 38, 54)] and now bristles. Thus, actin’s architecture may dictate where and to what extent Mical is mobilized by Myo15 and the subcellular addresses to which it is transported. These properties would allow Mical to spread its disassembly effects to multiple portions/regions of bundled filaments while also possibly protecting different architectural types of F-actin and/or cellular regions from Mical-mediated disassembly.
In light of our results with Myo15 and Mical, it is interesting to speculate that additional myosins may also play a role in MICAL-mediated F-actin disassembly. Notably, mutations in another MyTH4-FERM class myosin, Myo7a, encoded by the Drosophila crinkled gene, were initially found in the 1930s in part based on their effects on bristle morphology [e.g., see (19, 20, 55) and references therein]. While Myo7a did not enhance Mical’s effects similar to Myo15 (fig. S3B), the bristle defects in crinkled (Myo7a) mutants are complex, including having irregularly stubby, wavy, and branching bristles [see (55) and references therein], and so future work should aim to further investigate whether there is a relationship between Mical and Myo7a. Furthermore, as we were submitting this manuscript, another MICAL family member (MICAL-1) was found to interact with another myosin (MyoVa and MyoVb) (56). However, in contrast to our results with Myo15, which expands Mical’s distribution/F-actin disassembly, MICAL-1 was found not to be transported by MyoVa but to be tethered to a specific spot and derail MyoVa and its cargo as MyoVa passed through that specific locale (56). Thus, these two observations [our results and (56)] are complementary and represent different aspects of an emerging hypothesis: that interactions between myosins and negative effectors of the F-actin cytoskeleton are a critical and widely used means by which cells carry out their different behaviors. So too, these two observations [our results and (56)]—coupled with the large size and multiple protein-protein interacting regions characterizing both MICALs and unconventional myosins (11, 22), that MyoVa only interacts with MICAL-1 but not MICAL-2 and MICAL-3 (56), and our results herein that increasing Drosophila MyoV does not function similar to Myo15 to enhance/broaden Mical’s effects on cellular remodeling in the bristle model (fig. S3B)—raise the interesting possibility that multiple different types of myosins may interact with the different MICALs with the effects being used to either expand F-actin disassembly and cellular remodeling [i.e., if the myosin (similar to Myo15) carries Mical] or restrict disassembly and cargo unloading [i.e., if Mical is the one tethered to a specific locale and interacts with a myosin (similar to MyoVa) as the myosin moves through that specific region].
In conclusion, our observations provide new molecular, biochemical, and cellular mechanisms for spatiotemporally breaking down and rebuilding the form and function of cells and tissue systems. Likewise, given our results herein and that Myo15s and Mical exhibit overlapping expression patterns including within the brain, liver, kidney, lung, pancreas, and skeletal muscle [e.g., (11, 21, 29, 40, 41, 47, 48) and references therein], these proteins are positioned to interact and play important roles in different tissues, including perhaps under disease conditions. For example, Myo15s are well known for transporting cargo that is needed for structuring, elongating, and maintaining F-actin within the stereocilia of sound-transducing hair cells of the inner ear [reviewed in (26, 41, 49)], and mutations in Myo15, including within the motor and first MyTH4 and FERM domains, cause deafness (41, 57). While a role for Mical in vertebrate inner ear hair cells has not been explored, the Drosophila bristle cell is structurally and functionally analogous to these hair cells [e.g., reviewed in (58)]. Furthermore, as in bristle cells, F-actin turnover and remodeling drive the elongation and maintenance of hair cell stereocilia, and cofilin, an actin regulatory protein that synergizes with Mical in bristle remodeling (8), is also required in stereocilia (59, 60). Moreover, enzymes that directly reverse Mical’s effects on actin, methionine sulfoxide reductases (MsrBs/SelR) (7, 25), are also involved in hair cell form and function and are required for hearing (61, 62). Likewise, it is also notable that altering the levels of this unconventional myosin has such marked effects on the cytoskeletal/cellular breakdown and remodeling that occurs in response to directional (chemotaxic) signals such as Sema/Plexin repulsive guidance. In particular, we find that during the spatiotemporal course of cellular development, Myo15 markedly increases the cytoskeletal/cellular transformation of regions that typically only undergo small changes in response to Sema/Plexin/Mical repulsive guidance (e.g., Fig. 6A) (4). These observations indicate important new mechanisms controlling Sema/Plexin signaling and the chemotaxic responses of cells, including providing a means to expand and widen the cytoskeletal and cellular effects that occur following the activation of cell surface receptors by external stimuli. Given that targeting chemotaxis is an important therapeutic strategy, including for limiting the metastasis of cancer cells or for stimulating the directional extension of injured axons, these mechanisms are also of biomedical relevance.
MATERIALS AND METHODS
Experimental design
The study was initiated to search for genes that alter Mical’s (and, by extension, Mical’s upstream activator, Semaphorins/Plexins) effects on cellular behaviors. After identifying Myo15 in our search/genetic screen, we hypothesized that Myo15 is a modulator of Mical’s effects. Experiments were then designed to test this hypothesis and whether Mical could also modify Myo15’s effects. Since those experiments supported our hypothesis, experiments were then designed to ascertain Myo15’s role in regulating Mical’s effects. Since Myo15 is a myosin that is known to carry cargo along F-actin, multiple different types of experiments (coimmunostaining, coimmunoprecipitation, analyzing the distribution of Mical in response to increasing or decreasing Myo15, co-live imaging and tracking of Mical and Myo15 movements, site-directed mutagenesis of regions of Myo15 important for its transport functions, etc.) were designed to test whether Mical associates with Myo15 and whether Myo15 could alter Mical’s cellular positioning. Since these experiments supported the hypothesis that Mical is redistributed by Myo15, experiments were then designed to determine how this Mical redistribution affected cells (including looking at F-actin organization). These experiments raised and supported the hypothesis that Myo15 redistributes Mical to broaden Mical-mediated F-actin disassembly. To pursue our study and test our hypotheses, the overall design was a controlled laboratory experiment, in which research subjects for our study were animals (Drosophila melanogaster), including analysis of different cells in vivo and including in vivo analyses at the single-cell level (experimental units of investigation), and using genetic methods to raise and lower the levels of Mical, Myo15, and other tested proteins. All animal work was performed in accordance with university guidelines. For each representative image, movie, graph, or immunoblot, the experiments were based on what is published in the field and were repeated at least two separate times (for both biological and technical replicates) with similar results. For in vivo experiments, n is stated and described in the figure legend for each experiment. Animal experiments were not randomized: Animals of the correct genotype were determined, and those collected of that genotype were included as data. Blinding was not used because the genotype was determined by the experimentor based on the identification of different Drosophila genetic/chromosome markers. As defined with regard to the specifics of each experiment (genotype, condition, etc.), all data points were collected, all data were included, and none were excluded. Any outliers, such as a rare case of poor expression in a particular transgenic animal, were not used for subsequent experiments (as described in Materials and Methods). No statistical methods were used to predetermine sample size or stop data collection, which was based on what is published in the field. Details of all our experiments, including the treatments applied, the types of observations made, and the measurement techniques used, are described below, as well as in Results and/or the figure legends.
Mical activity-dependent bristle branching genetic screen
The enhancer-suppressor single-cell genetic screen based on Mical activity-dependent bristle branching was performed as described in (7), with a minor change in the scoring system. Here, bristles were scored on a 5-point scale with “0” being no enhancement or suppression of Mical activity-dependent bristle branching [i.e., bristles had a similar number and length of branches as Bristle Mical (UAS:Mical/+, B11-GAL4/+)]. Bristles with slight suppression of the Mical activity-dependent phenotype (i.e., a branch was present but was shorter than in Bristle Mical flies) were scored as “−1,” and bristles with no branch were scored as “−2.” Bristles with increased length and/or number of branches were considered as enhanced Mical activity-dependent branching and were scored as “+1” or “+2,” depending on the degree of increase in branch length and/or number (+2 equaling more increase in branch length and/or number). For simplicity and consistency, only the left posterior scutellar bristle was scored in each fly, as this is a large, easily identifiable macrochaetae (single-bristle cell) on the thorax of the fly. Flies with ethyl methanesulfonate (EMS)-induced mutations, transposable element insertions, or UAS-tagged transgenes were screened in this assay. The analysis of other myosins for effects on Mical activity-dependent bristle branching was conducted as described above and as detailed in fig. S3B.
Transgenic and transposable element fly lines and molecular biology
All Mical mutant lines were as previously described (5, 45). All Mical, PlexA, and SelR transgenic fly lines, except HAMical in pUAST (see below), were as previously described (4, 7). The Myo15 pUASP line was obtained from the Bloomington Drosophila Stock Center [w*; P{UASp-Myo10A.RFP}22; called UAS-Syph-RFP in (21)]. The Myo15 fly lines w1118 PBac{WH}f03968 and y1 sc* v1; P{TRiP.HMS02255}attP2 (Myo15 RNAi) were obtained from the Bloomington Drosophila Stock Center. P{XP}d05943 and PBac{WH}f06507 were obtained from the Exelixis Collection at Harvard Medical School.
To make transgenic animals expressing HAMical, hemagglutinin (HA) [three copies of HA (3× HA)] was polymerase chain reaction (PCR)–amplified with the forward primer 5′-GCTCTAGACAAAACATGGGATCCTACCCATACGATGTTCCTGACTATGCGG-3′ and the reverse primer 5′-CTAGCTAGCAGATCTAGCGTAATCTGGAACGTCATATGGATATC-3′. Following sequencing and then digestion with the restriction enzymes Xba I and Nhe I, the resulting PCR-amplified DNA containing HA was then ligated to the 5′ end of the complementary DNA of Mical in the vector pOT2a partially digested with Xba I and Nhe I. HAMical was then moved to pUAST following digestion with Xba I. Drosophila embryo injections to generate transgenic flies were then performed by BestGene Inc.
To make transgenic animals for Myo15, we benefited from the previous work (21) that had generated DNA constructs for Myo15 [both green fluorescent protein (GFP)–tagged and red fluorescent protein (RFP)–tagged] in the pUASP vector [both obtained as a gift from S. Parkhurst (21)]. Myo15-GFP (Syph-GFP) (21) is composed of DNA coding for GFP fused to the C terminus of the Myo15 open reading frame (ORF) (yielding a 2602–amino acid Myo15 protein) in pUASP (21). Myo15-RFP (Syph-RFP) (21) is composed of DNA coding for RFP fused to the C terminus of the Myo15 ORF in pUASP (21). Sequencing of these obtained DNA constructs revealed a nucleotide difference in the motor domain from the National Center for Biotechnology Information (NCBI) reference sequence (NM_132441.3), resulting in a histamine (H) substitution for tyrosine (Y) at amino acid 280. In addition, the Myo15-RFP (Syph-RFP) (21) construct in pUASP contained a nucleotide difference in the tail region from the reference sequence, resulting in an asparagine (N) substitution for serine (S) at amino acid 2097 [amino acid position is based on the reference sequence (NM_132441.3)]. To compare the effects of these differences with the reference sequence, site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene) was used to change the DNA coding for H280 to Y280 using forward (5′-CGGACAAGTACTTCTATCTGAACC-3′) and reverse (5′-GGTTCAGATAGAAGTACTTGTCCG-3′) primers. Note also that Myo15 appears to be heavily alternatively spliced such that the NCBI reference sequence (NM_132441.3) is a splice variant that is 452 amino acids longer than the previously described 2602–amino acid Myo15 protein construct (21). In particular, amino acids 1171 to 1622 that are listed in the NCBI reference sequence (NM_132441.3) are not included within that splice form (21). That Myo15 splice form (21) includes amino acids 1 to 1170 and 1623 to 3054 of that particular NCBI reference sequence (NM_132441.3). In addition, note that the longer splice form contains its insertion between the F1 regions of the FERM(1) domain and that there are no conserved domains predicted in this region of difference between the splice forms. Note also that these and other Myo15 splice forms have been annotated in FlyBase (including splice form D, shown in Fig. 1E).
For optimal expression in Drosophila somatic cells using the GAL4-UAS system, Myo15-RFP (construct with H280 and N2097) and Myo15-GFP [both the complete construct before (H280) and after site-directed mutagenesis (Y280), as described above] were subcloned from the pUASP vector (optimized for expression in germline cells) into the pUAST vector (optimized for expression in somatic cells) using Kpn I and Xba I restriction enzyme sites. Drosophila embryo injections to generate transgenic flies were then performed by BestGene Inc. One independent Myo15-RFP H280, N2097 transgenic line in pUAST [called Myo15-RFP (pUAST), which was similar to that described in (21) but is in the pUAST vector], five independent Myo15-GFP H280 transgenic lines in pUAST [called Myo15-GFP (pUAST), which was similar to that described in (21) but is in the pUAST vector], and eight independent Myo15-GFP Y280 transgenic lines in pUAST (called Myo15GFP) were generated, and all showed similar phenotypes when expressed in bristles. In addition, the effects of the two variants (H280 and Y280; N2097 and S2097) on Mical-mediated bristle remodeling were indistinguishable. Unless otherwise noted, all data shown use the Y280 construct (Myo15GFP) or the untagged version of this construct (Myo15; see below). To make an untagged version of Myo15 in pUAST, a fragment was PCR-amplified from Myo15GFP DNA in pUAST using a forward primer (5′-CGTGTCATCCAGGCTAGCAT-3′) that recognized a unique Nhe I site within Myo15 (amino acids 1606 to 1607) and a reverse primer (5′-GGTCTAGATTAGTTGAGCTCCCG-3′) to engineer a Stop Xba I site at the 3′ end of Myo15 and then ligated into Myo15GFP pUAST digested with Nhe I and Xba I, thereby removing the region coding for GFP. Three independent transgenic lines were made (UAS:Myo15), and they all gave similar bristle phenotypes. Bristle expression of either the GFP- or RFP-tagged [UAS:Myo15GFP, UAS:Myo15-GFP (pUAST), and UAS:Myo15-RFP (pUAST)] or untagged forms of Myo15 all produced similar effects on Mical localization and bristle morphology, indicating that the C-terminal tag did not substantially alter the function of Myo15 [see also (21, 34)] or its effects on Mical. Note, however, that we did see slight variations in enhancement of Mical [e.g., as seen in the images of the manuscript when experimental design necessitated using a tagged form of Myo15 (e.g., Figs. 2 and 3 and fig. S7B) versus an untagged form of Myo15 (e.g., Fig. 4 and fig. S7C)]. Note also that all comparisons between wild-type Myo15 and the different deletion constructs of Myo15 (see below) were performed using similarly tagged (GFP-containing) constructs.
All Myo15 domain deletion DNA and point mutations were placed in pUAST and contain a C-terminal GFP (similar to wild-type Myo15GFP). Myo15ΔMotor (amino acids 1 to 63 and amino acids 749 to 2602 of Myo15 fused to GFP) was generated by a two-step process. First, a Myo15ΔNterm-motor construct (a region coding for amino acids 749 to 2602 of Myo15 fused to GFP) was generated by PCR-amplifying a (5′)Kpn I/(3′)Sfi I–DNA fragment coding for amino acids 749 to 1847 of Myo15 (forward primer, 5′-TTTGGTACCATGACCGAGCGTCTGCG-3′; reverse primer, 5′-ATTACGGCCTGCTTGGCCT-3′) and ligating the fragment into Myo15GFP digested with Kpn I/Sfi I. Then, a (5′ and 3′)Kpn I–DNA fragment coding for amino acids 1 to 63 of Myo15 was PCR-amplified (forward primer, 5′-GAGGGTACCATGGACTG-3′; reverse primer, 5′-ATAGGTACCGCTGCCCAGATCCT-3′) and ligated into Myo15ΔNterm-motor digested with Kpn I. Myo15R213A,G434A was made using the QuikChange Lightning Site-Directed Mutagenesis Kit (Strategene) to sequentially introduce point mutations (coding for amino acid R213 to amino acid A213: forward primer, 5′-GACAATAGTTCGCGGTTTGGAAAGTATCTGG-3′; reverse primer, 5′-CCAGATACTTTCCAAACCGCGAACTATTGTC-3′; amino acid G434 to amino acid A434: forward primer, 5′-CTGGACATCTTTGCGTTCGAGGATTTGGC-3′; reverse primer, 5′-GCCAAATCCTCGAACGCAAAGATGTCCAG-3′) into Myo15GFP pUAST in the region corresponding to the conserved switch I (consensus sequence: NXNSSRFG) and switch II (consensus sequence: DIXGFE) regions of the Myo15 motor domain. Two independent Myo15R213A,G434A transgenic lines were generated, and both showed similar bristle phenotypes.
The generation of all other domain deletions was based on identification of conserved domains using the NCBI Conserved Domain Database (CD-Search tool criteria). As mentioned above, all Myo15 domain deletion DNA was in pUAST and contains a C-terminal GFP (similar to wild-type Myo15GFP). Between 2 and 10 independent lines were generated for each construct, and the bristle phenotypes resulting from expression of each line were recorded. With a few exceptions (e.g., poor expression of the construct), all independent lines of a given construct showed similar phenotypes when expressed in bristles. Outliers were not used for subsequent experiments. Myo15ΔFERM(1),MyTH4(2),FERM(2) (also called Myo15ΔPMF) codes for amino acids 1 to 1788 of Myo15 fused to GFP and lacks the DNA regions coding for the pleckstrin homology (PH)-like portion of the FERM(1), MyTH4(2), and FERM(2); it was generated in a two-step cloning process. First, a Myo15Shortened pUAST construct (coding for amino acids 1 to 1607 of Myo15 fused to GFP) was created using PCR (forward primer, 5′-CAGGCTAGCTCACAATCGAAGGC-3′; reverse primeq 5′-CCTCTAGATTATTTGTATAGTTCATCCATGC-3′) to amplify a Nhe I–GFP–Xba I fragment, which was then ligated into Myo15 pUAST digested with Nhe I/Xba I, thereby producing a Myo15 construct lacking the DNA coding for the C-terminal tail region, which contains the PH-like portion of the FERM(1), MyTH4(2), and FERM(2) domains. To complete the construct, an additional (5′)Spe I/(3′)Nhe I–DNA fragment coding for amino acids 1606 to 1788 of Myo15 was PCR-amplified (forward primer, 5′-GCACTAGTATGGACACCAC-3′; reverse primer, 5′-AATGCTAGCCAGGGCACGCA-3′) and ligated into Myo15shortened pUAST digested with Nhe I. Myo15ΔFERM(1) (also called Myo15ΔPH-like) codes for amino acids 1 to 1788 and amino acids 1895 to 2602 of Myo15 fused to GFP, and it was generated by PCR-amplifying (forward primer, 5′-CAGGCTAGCTCACAATCGAAGG-3′; reverse primer, 5′-AAAGCTAGCGTTGAGCTCCCGC-3′) a (5′ and 3′)Nhe I–DNA fragment coding for amino acids 1895 to 2602 of Myo15 and ligating the fragment into Myo15ΔFERM(1),MyTH4(2),FERM(2) digested with Nhe I. Myo15ΔMyTH4(2),FERM(2) (also called Myo15ΔMF) codes for amino acids 1 to 2019 of Myo15 fused to GFP and lacks the DNA regions coding for the MyTH4(2) and FERM(2) domains; it was generated by PCR-amplifying (forward primer, 5′-GCACTAGTATGGACACCAC-3′; reverse primer, 5′-AATAAGCTAGCGGCGTGGCCCT-3′) a (5′)Spe I/(3′)Nhe I–DNA fragment coding for amino acids 1606 to 2019 of Myo15 and ligating the fragment into the Myo15shortened pUAST construct at the Nhe I site. Myo15ΔMyTH4(2) codes for amino acids 1 to 2119 and amino acids 2269 to 2602 of Myo15 fused to GFP and was generated by PCR-amplifying (forward primer, 5′-TTAGCTAGCTCCGCTGGACGTTCA-3′; reverse primer, 5′-AAAGCTAGCGTTGAGCTCCCGC-3′) a (5′ and 3′)Nhe I–DNA fragment coding for amino acids 2269 to 2602 of Myo15 and ligating the fragment into Myo15ΔMyTH4(2),FERM(2) digested with Nhe I. Myo15ΔFERM(2) codes for amino acids 1 to 2279 of Myo15 fused to GFP and was generated by PCR-amplifying (forward primer, 5′-CAACTAGTATACGCCTGCCG-3′; reverse primer, 5′-CATGCTAGCGTAGATCTGTCG-3′) a (5′)Spe I/(3′)Nhe I–DNA fragment coding for amino acids 2120 to 2279 of Myo15 and ligating the fragment into Myo15ΔMyTH4(2),FERM(2) digested with Nhe I. Myo15ΔMyTH4(1) codes for amino acids 1 to 922 and amino acids 1081 to 2602 of Myo15 fused to GFP and was generated in a two-step process. First, a (5′)Avr II/(3′)Nhe I–DNA fragment coding for amino acids 260 to 922 of Myo15 was PCR-amplified (forward primer, 5′-TTTACGAACTCCTAGGCGG-3′; reverse primer: 5′-ATAGCTAGCCTGCAATCTCAAG-3′) and ligated into the Myo15 pUAST construct at unique Avr II and Nhe I sites within Myo15. Second, a (5′)Spe I/(3′)Nhe I–DNA fragment coding for amino acids 1081 to 1607 of Myo15 was PCR-amplified (forward primer, 5′-TTAACTAGTTGGACCAGAG-3′; reverse primer, 5′-CATGCTAGCCTGGATGACA-3′) and ligated into the vector generated in the first step digested with Nhe I. Myo15ΔMyTH4(1),MyTH4(2) (also called Myo15ΔMyTH4(1&2)) codes for amino acids 1 to 922 and amino acids 1081 to 2119 and amino acids 2269 to 2602 of Myo15 fused to GFP, and it lacks the DNA coding for both MyTH4 domains; it was generated by PCR-amplifying (forward primer, 5′-GCACTAGTATGGACACCAC-3′; reverse primer, 5′-CCTCTAGATTATTTGTATAGTTCATCCATGC-3′) a (5′)Spe I/(3′)Xba I–DNA fragment coding for amino acid 1606 to the end of GFP from the Myo15ΔMyTH4(2) construct and ligating the fragment into Myo15ΔMyTH4(1) digested with Nhe I/Xba I. Myo15ΔCargo (also called Myo15MyTH4(1),FERM(1),MyTH4(2),FERM(2) or Myo15ΔMPMF) codes for amino acids 1 to 922 and amino acids 1081 to 1788 of Myo15 fused to GFP (i.e., lacks all conserved tail domains) and was generated by PCR-amplifying (forward primer, 5′-GCACTAGTATGGACACCAC-3′; reverse primer, 5′-CCTCTAGATTATTTGTATAGTTCATCCATGC-3′) a (5′)Spe I/(3′)Xba I–DNA fragment coding for amino acid 1606 to the end of GFP from the Myo15ΔFERM(1),MyTH4(2),FERM(2) construct and ligating the fragment into Myo15ΔMyTH4(1) digested with Nhe I/Xba I. All restriction enzymes used were from New England Biolabs.
Generation of Myo15 mutant flies
FRT-containing transposable elements (PBac{WH}f03968 and PBac{WH}f06507) inserted within Myo15 were used to delete the entire coding region of Myo15 through FLP-mediated recombination [(24) and as we have previously used (7)]. Successful deletion of the targeted region was screened by PCR using WH transposon primers [WH5′ plus (reverse) and WH3′ plus (forward)] (24) together with genomic primers (forward, 5′-AGCCACGCATACACAAGCAC-3′; reverse, 5′-ATGAGGACGAGGACGACGAG-3′). Two deletion mutants were generated (Myo1514B and Myo1521J). To further confirm these mutants, additional PCR was performed using genomic primers (forward, 5′-ATCAATCAACAATACACGCACC-3′; reverse, 5′-AGGATGACAGCGATCAACTCA-3′) to amplify a fragment containing the transposable element and the genomic DNA flanking it. The DNA fragment was excised from an agarose gel using the QIAquick gel extraction kit (QIAGEN) and cloned into pCR2.1-TOPO (Thermo Fisher Scientific). Clones were screened with an Eco RI digest, and positive clones were sequenced using WH5′ plus (reverse) and WH3′ plus (forward) primers to verify the deletion. Both deletion lines gave similar results in the Mical activity-dependent bristle branching assay (e.g., Fig. 1F), but for simplicity, only Myo1521J is shown here and was used for all subsequent analysis.
Coimmunoprecipitation
Coimmunoprecipitation assays were performed using standard approaches [e.g., (5, 18)]. Briefly, lysates were prepared in detergent-free radioimmunoprecipitation assay (RIPA) buffer [50 mM tris-HCl (pH 8.0) and 150 mM NaCl] supplemented with cOmplete EDTA-free protease inhibitor cocktail tablets (Roche) and phosphatase inhibitor cocktail 3 tablets (Sigma-Aldrich) from embryos with neuronal expression of HAMical and Myo15GFP (UAS:Myo15GFP, ELAV-GAL4/Cyo; UAS:HAMical/TM3Sb Actin LacZ) or HAMical only (ELAV-GAL4/Cyo; UAS:HAMical/TM3Sb Actin LacZ). Lysates were precleared with GammaBind G Sepharose beads (GE Healthcare), and then similar amounts were incubated overnight at 4°C with a GFP antibody (5 μg/ml; A11120, Thermo Fisher Scientific) or a Flag antibody (5 μg/ml; F3165, Sigma-Aldrich), with anti-Flag incubation serving as a control for nonspecific binding of target proteins to beads or antibody. Following antibody incubation, lysates were incubated with beads for 90 min at 4°C. Beads were washed with detergent-free RIPA buffer six times and then resuspended in 2× Laemmli buffer. Coimmunoprecipitation lysates were analyzed by Western blot using standard techniques. Western blots were probed with an HA antibody conjugated to horseradish peroxidase (HRP) (1:3000; 3F10, Roche) or a GFP antibody (1:5000; A-6455, Molecular Probes/Thermo Fisher Scientific), followed by donkey anti-rabbit ECL HRP-conjugated secondary antibody (1:500; NA934VS, Amersham/GE Healthcare). SuperSignal West Pico or SuperSignal West Femto Chemiluminescent Substrates (Thermo Fisher Scientific) were used to detect HRP-conjugated antibodies. Similar results that a GFP antibody [but not a Flag (nonspecific control) antibody] immunoprecipitates HAMical along with Myo15GFP, but only in embryos expressing Myo15GFP, were seen in independent experiments.
Characterization, quantification, imaging, and drawings (adult)
The bristles of recently emerged adults were examined using a dissecting stereomicroscope (Leica S8 APO, 1.6× objective). During this examination, bristle morphology/defects were noted as described in (4). In addition, the number of branches on the left posterior scutellar bristle of each fly was counted and recorded for quantification of branches per bristle or percentage of flies with bristle branching. For quantification of bristle branching in Plexin overexpression flies, all four scutellar bristles were examined for branches. If one or more of the four scutellar bristles had branching, the fly was considered to have scutellar bristle branching. After examination, flies were stored in 70% ethanol for preservation. For imaging, the wings, legs, and abdomen were removed using Dumont #5 forceps (Ted Pella Inc.), leaving the head and thorax intact. The head of each fly was stuck to double-sided tape (3M) on a slide. Bristle images were taken and compiled with a Zeiss Discovery M2 Bio stereomicroscope with a motorized zoom and focus and Zeiss AxioVision software and Extended Focus Software (a gift from B. Lee). From these images, bristles were carefully traced in Microsoft PowerPoint using the “curve” tool to generate bristle drawings that accurately reflected bristle morphology. All images shown are of posterior scutellar bristles. All details that correspond to specific genotypes for adults are listed in the figure legends except for the following: Bristle Mical+++ + Myo1521J/+ = UAS:Mical/+, B11-GAL4/+, Myo1521J/+; Bristle PlexA+++ = UAS:HAPlexA/+, B11-GAL4/+; Bristle PlexA+++ + Myo15d05943/+ = UAS:HAPlexA/+, B11-GAL4/+, P{XP}d05943/+; Bristle PlexA+++ + Bristle Myo15 RNAi = UAS:HAPlexA/+, B11-GAL4/+, P{TRiP.HMS02255}attP2/+; Bristle PlexAΔCyto + Myo15d05943/+ = UAS:HAPlexAΔCyto/+, B11-GAL4/+, P{XP}d05943/+; Bristle PlexA+++ + Myo15d05943/+ = UAS:HAPlexA/+, B11-GAL4/+, P{XP}d05943/+; Bristle PlexA+++ + Myo15d05943/+ + MicalDf(3R)swp2/+ = UAS:HAPlexA/+, B11-GAL4/+, P{XP}d05943/+, Df(3R)swp2/+.
Characterization and imaging (pupae)
Pupae were collected and placed on double-sided tape (3M) in petri dishes. Genotyping was performed with the aid of balancer chromosomes with the Tb1 marker or using a Zeiss Discovery M2 Bio stereomicroscope and visualization of GFP balancer chromosomes or GFP or mCherry fusion proteins. Pupae were kept at 25°C until they reached the desired stage for imaging. Then, pupae were removed from their outer pupal case, and whole pupae were mounted, dorsal side up, in depression-well slides with VECTASHIELD mounting medium (Vector Laboratories) and typically imaged within 4 hours with a Zeiss LSM510 confocal microscope using the 40× or 63× oil objective. Images were compiled using ZEN lite software (Zeiss). Image background, brightness, contrast, and levels were adjusted using Adobe Photoshop.
Live imaging
Genotyping and staging were conducted as described above, and white prepupae were marked. After 33 hours of prepupa formation, pupae were collected on double-sided tape in a petri dish. Pupae were then removed from the pupal case and placed dorsal side down in a glass-bottom petri dish (part no. P35GC-1.5-14-C, MatTek Corporation) with wet paper inside for humidity. Imaging was performed on a Zeiss LSM710 confocal microscope using the 20× objective. Images were taken for 6 hours by combining both the modules of Z-stack and time series without any time interval and compiled using ZEN lite software (Zeiss). In particular, Z-projections of confocal stack acquisition was set up (10 slices each with a distance of 1.651 μm between slices, acquired every 2 min and 37 s for both the red and green channels), which was then compiled as an image, and this set up was cycled for 140 times for 6 hours using the time series module without any time interval. Titles, text, arrowheads, and timestamp were added using Adobe Premiere Pro.
F-actin and microtubule organization and cellular distribution of Myo15 and Mical in bristles
F-actin was observed as generally described previously (4) using either a transgene coding for GFPActin (either GFPActin42A or GFPActin5C), a transgene coding for LifeAct (LifeActRuby), or fluorophore-conjugated phalloidin. When using a GFPActin transgene in combination with increased levels of Myo15, untagged Myo15 or Myo15-RFP (pUAST) was used. Microtubule organization was observed using a transgene for tubulin (GFPAlpha-tubulin84B). Myo15 distribution was observed using a transgene coding for either Myo15GFP or Myo15-RFP (pUAST). Mical distribution (observed using a transgene coding for mCherryMical) and F-actin organization (observed using a transgene coding for GFPActin42A) in bristles were calculated as a percentage of the total bristle process using images acquired from the Zeiss LSM510 confocal microscope. ImageJ software was used to trace (using the “polygon selection” tool) and measure the area of the bristle process (including branches but excluding the cell body). Similarly, the area of the bristle with high Mical levels (Mical distribution) or low levels of F-actin (F-actin disruption) was traced and measured [Fig. 4C, (2)]. In particular, within the bristle tip and its branches, areas with high Mical levels (Mical distribution) were measured from single-bristle cells and plotted on the x axis, and areas with low levels of F-actin (F-actin disruption) were measured from single-bristle cells and plotted on the y axis. The percentage of overlap between this Mical distribution and disrupted F-actin (Mical/disrupted F-actin) is also presented. As a means to delineate the regions of the bristle with high Mical expression, the “Fire” Lookup-table in ImageJ was used to highlight where Mical was strongly expressed. All quantifications were performed on posterior scutellar bristles and bristles of similar age [7 to 9 hours (~38 to 40 hours after puparium formation)], unless otherwise specified. Triple labeling of Mical, Myo15, and F-actin was performed by genotyping, staging, marking of white prepupae, removing pupae from their pupal case, and placing pupae dorsal side up on double-sided tape in a petri dish as described above. The epithelia on the dorsal side of the thorax were then dissected in phosphate-buffered saline (PBS) using fine scissors. The interior organs and fat bodies were then removed carefully using fine forceps and using care to avoid damaging the epidermis containing the bristles. Dissected fillets were then fixed in 4% paraformaldehyde in PBS for 10 min, followed by another 10 min of fixation in 4% paraformaldehyde containing 0.3% PBST (Triton X-100 in PBS). Fixed fillets were then washed three times in 0.3% PBST for 10 min each, incubated overnight with iFluor 405–conjugated phalloidin (1:500 in 0.3% PBST; ab176752, Abcam), and then washed again three times in 0.3% PBST. Slides were then prepared with VECTASHIELD mounting medium (Vector Laboratories) and imaged with the Zeiss LSM710 confocal microscope using a 20× objective. Images were compiled from all slices using ZEN lite software (Zeiss). All details that correspond to specific genotypes for pupae are listed above or in the figure legends except for the following: Bristle Mical+++ + Myo15R213A,G434A = UAS:mCherryMical/+, B11-GAL4/+, UAS:Myo15R213A,G434A/+; Bristle Mical+++ + Myo15Δ transgenes = UAS:mCherryMical/+, B11-GAL4/+, UAS:Myo15Δ transgenes/+.
Larval NMJ innervation and muscle analysis
Third instar larvae were collected from vial walls and dissected using standard approaches to allowing for visualization of the nervous system and muscles (7, 45). Myo1521J mutants from a Myo1521J/FM7c, P{2xTb1 RFP}FM7c, sn+ stock were identified on the basis of the lack of the Tb1 marker (balancer from Bloomington Drosophila Stock Center). Myo1521J mutants were all males due to the lack of viable mutant males in the stock to produce homozygous females. After dissection, all larvae were fixed in methanol for 20 min and then washed 3× in PBS. For synaptic studies using CD8-GFP-Shaker to label synapses (7, 45), larvae were then mounted on glass slides using VECTASHIELD mounting medium (Vector Laboratories). For other synaptic studies in which F-actin and the synaptic marker Brp (Bruchpilot) were used, after fixation and washing, larvae were blocked with 5% goat serum (Sigma-Aldrich) in PBS with 0.1% Triton X-100 (Thermo Fisher Scientific) for 1 hour at room temperature. Larvae were then incubated in a Brp (nc82) antibody (1:500; Developmental Studies Hybridoma Bank) in PBS for 1 to 2 hours at room temperature or overnight at 4°C and then washed 5× in PBS with 0.1% Triton X-100. Next, larvae were incubated in Alexa Fluor 635–conjugated phalloidin (1:100; Thermo Fisher Scientific) and goat anti-mouse Alexa Fluor 488–conjugated immunoglobulin G (IgG) (1:500; Thermo Fisher Scientific) in PBS for 1 to 2 hours at room temperature or overnight at 4°C and then washed in PBS with 0.1% Triton X-100 before mounting on glass slides using VECTASHIELD mounting medium (Vector Laboratories). For analysis of F-actin organization in muscles, larvae were incubated in Alexa Fluor 635–conjugated phalloidin (1:100; Thermo Fisher Scientific). Larvae were imaged with the Zeiss LSM510 confocal microscope using the 40× oil [nc82 (Brp)–labeled synapses] and 20× (muscles and CD8-GFP-Shaker–labeled synapses) objectives. Images were compiled using ZEN lite software (Zeiss). The length of synaptic innervation at muscles 1/9 and 6/7 was measured in ImageJ as previously described (7). Further, NMJ synapse length is correlated with the size of the muscle being innervated [e.g., see (7, 45) and references therein]. Therefore, to account for any size differences in larvae [e.g., Myo15−/− mutant larvae (Myo1521J/Y) are smaller than normal in overall size], the length of muscle 1 was measured in ImageJ using the “straight” tool to make a vertical line from the anterior end of the muscle to the posterior end, and the length of the line was measured. The ratio of synapse length/muscle 1 length is therefore shown in Fig. 5B (2). In addition, for images of synapses at muscles 6 and 7 taken at higher magnification in which the entire length of the muscle was not measurable, larvae of similar size were chosen for analysis, muscle width was measured to make sure the muscle size of all samples and genotypes was similar, and synaptic innervation was comparable. Muscle width was measured in ImageJ using the straight tool to make a horizontal line from one side of muscle 6 to the other side, and the length of the line was measured and is presented in Fig. 5C [(2), right graph] to show that the width of muscle 6 is not significantly different in wild type and Myo15−/− mutants. Quantification of F-actin organization defects was performed by identifying the presence of abnormalities in the internal layers of muscles 6 and 7 [as previously described in (7, 45)]. Muscles were considered to have F-actin organization defects if any deviation in F-actin such as F-actin accumulation or disruptions in the normal striation pattern were present. Myo15 localization to presynaptic terminals was observed by crossing UAS:Myo15GFP to the neuronal ELAV-GAL4 driver then processing and imaging the larvae as described above except using antibodies to HRP conjugated to Alex Fluor 594 (1:500; Jackson ImmunoResearch) to label neurons and their synaptic terminals and antibodies to GFP (rabbit anti-GFP; 1:1000; Invitrogen) and goat anti-rabbit Alexa Fluor 488–conjugated IgG (1:500; Thermo Fisher Scientific) to label Myo15GFP.
Statistical analysis
For each graph, the statistical test used, the value of n, what n represents, and the P value for each comparison are stated in the figure legend or in Materials and Methods. Graphs show means ± SEM, or in some cases, as indicated in the figure legend (e.g., Figs. 1H and 5D and fig. S2F), we scored for either the presence or absence of a defect (i.e., either the animals had that defect or they did not have that defect and so there are no error bars) and so that data are presented as a percentage of animals with that defect [and appropriate statistical tests for those types of data (e.g., chi-square test) were used]. These and all other statistical methods used were based on what is standard in the field, and no statistical tests were used to determine whether the data met assumptions of the statistical approach. All statistical analyses were performed using GraphPad Prism 7 or 8. A P value of P > 0.05 is not considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Acknowledgments
We thank H. Kramer, J. Johnson, N. Alto, L. Alto, and Terman laboratory members for comments, discussions, and assistance and S. Parkhurst, J. Merriam, A. Ephrussi, B. Lee, H. Aberle, C. Spaeth, Bloomington Drosophila Stock Center, Drosophila Genetic Resource Center, and FlyBase for reagents, materials, support, and/or resources. Funding: This work was supported by grants from the NIH (MH085923) and Welch Foundation (I-1749) to J.R.T. Author contributions: Conceptualization: S.K.R. and J.R.T. Formal analysis: S.K.R. and R.B. Funding acquisition: J.R.T. Methodology: S.K.R. and R.B. Investigation: S.K.R. and R.B. Resources: J.R.T. Supervision: J.R.T. Visualization: S.K.R., R.B., and J.R.T. Writing—original draft: S.K.R. Writing—review and editing: S.K.R., R.B., and J.R.T. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabg0147/DC1
REFERENCES AND NOTES
- 1.T. D. Pollard, R. D. Goldman, The Cytoskeleton (Cold Spring Harbor Laboratory Press, 2017). [Google Scholar]
- 2.M. Tessier-Lavigne, A. L. Kolodkin, Neuronal Guidance: The Biology of Brain Wiring (Cold Spring Harbor Laboratory Press, 2011). [Google Scholar]
- 3.A. Kumanogoh, Semaphorins: A Diversity of Emerging Physiological and Pathological Activities (Springer Japan, 2015). [Google Scholar]
- 4.Hung R.-J., Yazdani U., Yoon J., Wu H., Yang T., Gupta N., Huang Z., van Berkel W. J., Terman J. R., Mical links semaphorins to F-actin disassembly. Nature 463, 823–827 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terman J. R., Mao T., Pasterkamp R. J., Yu H. H., Kolodkin A. L., MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109, 887–900 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Hung R. J., Pak C. W., Terman J. R., Direct redox regulation of F-actin assembly and disassembly by Mical. Science 334, 1710–1713 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung R. J., Spaeth C. S., Yesilyurt H. G., Terman J. R., SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 15, 1445–1454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grintsevich E. E., Yesilyurt H. G., Rich S. K., Hung R. J., Terman J. R., Reisler E., F-actin dismantling through a redox-driven synergy between Mical and cofilin. Nat. Cell Biol. 18, 876–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grintsevich E. E., Ge P., Sawaya M. R., Yesilyurt H. G., Terman J. R., Zhou Z. H., Reisler E., Catastrophic disassembly of actin filaments via Mical-mediated oxidation. Nat. Commun. 8, 2183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu H., Yesilyurt H. G., Yoon J., Terman J. R., The MICALs are a family of F-actin dismantling oxidoreductases conserved from drosophila to humans. Sci. Rep. 8, 937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alto L. T., Terman J. R., MICALs. Curr. Biol. 28, R538–R541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanoni M. A., Structure-function studies of MICAL, the unusual multidomain flavoenzyme involved in actin cytoskeleton dynamics. Arch. Biochem. Biophys. 632, 118–141 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Fremont S., Romet-Lemonne G., Houdusse A., Echard A., Emerging roles of MICAL family proteins - from actin oxidation to membrane trafficking during cytokinesis. J. Cell Sci. 130, 1509–1517 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Manta B., Gladyshev V. N., Regulated methionine oxidation by monooxygenases. Free Radic. Biol. Med. 109, 141–155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tilney L. G., DeRosier D. J., How to make a curved Drosophila bristle using straight actin bundles. Proc. Natl. Acad. Sci. U.S.A. 102, 18785–18792 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rich S. K., Terman J. R., Axon formation, extension, and navigation: Only a neuroscience phenomenon? Curr. Opin. Neurobiol. 53, 174–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hung R.-J., Terman J. R., Extracellular inhibitors, repellents, and Semaphorin/Plexin/MICAL-mediated actin filament disassembly. Cytoskeleton 68, 415–433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon J., Kim S. B., Ahmed G., Shay J. W., Terman J. R., Amplification of F-actin disassembly and cellular repulsion by growth factor signaling. Dev. Cell 42, 117–129.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashita R. A., Sellers J. R., Anderson J. B., Identification and analysis of the myosin superfamily in Drosophila: A database approach. J. Muscle Res. Cell Motil. 21, 491–505 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Tzolovsky G., Millo H., Pathirana S., Wood T., Bownes M., Identification and phylogenetic analysis of Drosophila melanogaster myosins. Mol. Biol. Evol. 19, 1041–1052 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Liu R., Woolner S., Johndrow J. E., Metzger D., Flores A., Parkhurst S. M., Sisyphus, the Drosophila myosin XV homolog, traffics within filopodia transporting key sensory and adhesion cargos. Development 135, 53–63 (2008). [DOI] [PubMed] [Google Scholar]
- 22.Masters T. A., Kendrick-Jones J., Buss F., Myosins: Domain organisation, motor properties, physiological roles and cellular functions. Handb. Exp. Pharmacol. 235, 77–122 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Sebé-Pedrós A., Grau-Bové X., Richards T. A., Ruiz-Trillo I., Evolution and classification of myosins, a paneukaryotic whole-genome approach. Genome Biol. Evol. 6, 290–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parks A. L., Cook K. R., Belvin M., Dompe N. A., Fawcett R., Huppert K., Tan L. R., Winter C. G., Bogart K. P., Deal J. E., Deal-Herr M. E., Grant D., Marcinko M., Miyazaki W. Y., Robertson S., Shaw K. J., Tabios M., Vysotskaia V., Zhao L., Andrade R. S., Edgar K. A., Howie E., Killpack K., Milash B., Norton A., Thao D., Whittaker K., Winner M. A., Friedman L., Margolis J., Singer M. A., Kopczynski C., Curtis D., Kaufman T. C., Plowman G. D., Duyk G., Francis-Lang H. L., Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36, 288–292 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Lee B. C., Peterfi Z., Hoffmann F. W., Moore R. E., Kaya A., Avanesov A., Tarrago L., Zhou Y., Weerapana E., Fomenko D. E., Hoffmann P. R., Gladyshev V. N., MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol. Cell 51, 397–404 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weck M. L., Grega-Larson N. E., Tyska M. J., MyTH4-FERM myosins in the assembly and maintenance of actin-based protrusions. Curr. Opin. Cell Biol. 44, 68–78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belyantseva I. A., Boger E. T., Naz S., Frolenkov G. I., Sellers J. R., Ahmed Z. M., Griffith A. J., Friedman T. B., Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 7, 148–156 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Manor U., Disanza A., Grati M. H., Andrade L., Lin H., Di Fiore P. P., Scita G., Kachar B., Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr. Biol. 21, 167–172 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mauriac S. A., Hien Y. E., Bird J. E., Carvalho S. D., Peyroutou R., Lee S. C., Moreau M. M., Blanc J. M., Geyser A., Medina C., Thoumine O., Beer-Hammer S., Friedman T. B., Ruttiger L., Forge A., Nurnberg B., Sans N., Montcouquiol M., Defective Gpsm2/Gαi3 signalling disrupts stereocilia development and growth cone actin dynamics in Chudley-McCullough syndrome. Nat. Commun. 8, 14907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bird J. E., Takagi Y., Billington N., Strub M.-P., Sellers J. R., Friedman T. B., Chaperone-enhanced purification of unconventional myosin 15, a molecular motor specialized for stereocilia protein trafficking. Proc. Natl. Acad. Sci. U.S.A. 111, 12390–12395 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang F., Takagi Y., Shams A., Heissler S. M., Friedman T. B., Sellers J. R., Bird J. E., The ATPase mechanism of myosin 15, the molecular motor mutated in DFNB3 human deafness. J. Biol. Chem. 296, 100243 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belyantseva I. A., Boger E. T., Friedman T. B., Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. U.S.A. 100, 13958–13963 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delprat B., Michel V., Goodyear R., Yamasaki Y., Michalski N., El-Amraoui A., Perfettini I., Legrain P., Richardson G., Hardelin J.-P., Petit C., Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum. Mol. Genet. 14, 401–410 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Sanchez-Soriano N., Goncalves-Pimentel C., Beaven R., Haessler U., Ofner-Ziegenfuss L., Ballestrem C., Prokop A., Drosophila growth cones: A genetically tractable platform for the analysis of axonal growth dynamics. Dev. Neurobiol. 70, 58–71 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Zheng L., Zheng J., Whitlon D. S., García-Añoveros J., Bartles J. R., Targeting of the hair cell proteins cadherin 23, harmonin, myosin XVa, espin and prestin in an epithelial cell model. J. Neurosci. 30, 7187–7201 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yochelis A., Ebrahim S., Millis B., Cui R., Kachar B., Naoz M., Gov N. S., Self-organization of waves and pulse trains by molecular motors in cellular protrusions. Sci. Rep. 5, 13521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang Q., Indzhykulian A. A., Mustapha M., Riordan G. P., Dolan D. F., Friedman T. B., Belyantseva I. A., Frolenkov G. I., Camper S. A., Bird J. E., The 133-kDa N-terminal domain enables myosin 15 to maintain mechanotransducing stereocilia and is essential for hearing. eLife 4, e08627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacquemet G., Stubb A., Saup R., Miihkinen M., Kremneva E., Hamidi H., Ivaska J., Filopodome mapping identifies p130Cas as a mechanosensitive regulator of filopodia stability. Curr. Biol. 29, 202–216.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tadenev A. L. D., Akturk A., Devanney N., Mathur P. D., Clark A. M., Yang J., Tarchini B., GPSM2-GNAI specifies the tallest stereocilia and defines hair bundle row identity. Curr. Biol. 29, 921–934.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.M. Preller, D. J. Manstein, Myosin motors: Structural aspects and functionality, in Comprehensive Biophysics, Vol 4, Molecular Motors and Motility, Y. E. Goldman, E. M. Ostap, Eds. (Elsevier, 2012), pp. 118–150. [Google Scholar]
- 41.Rehman A. U., Bird J. E., Faridi R., Shahzad M., Shah S., Lee K., Khan S. N., Imtiaz A., Ahmed Z. M., Riazuddin S., Santos-Cortez R. L. P., Ahmad W., Leal S. M., Riazuddin S., Friedman T. B., Mutational spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum. Mutat. 37, 991–1003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu Q., Li J., Zhang M., Cargo recognition and cargo-mediated regulation of unconventional myosins. Acc. Chem. Res. 47, 3061–3070 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Frank D. J., Hopmann R., Lenartowska M., Miller K. G., Capping protein and the Arp2/3 complex regulate nonbundle actin filament assembly to indirectly control actin bundle positioning during Drosophila melanogaster bristle development. Mol. Biol. Cell 17, 3930–3939 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu J., Wang H., Guo X., Chen J., Cofilin-mediated actin dynamics promotes actin bundle formation during Drosophila bristle development. Mol. Biol. Cell 27, 2554–2564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beuchle D., Schwarz H., Langegger M., Koch I., Aberle H., Drosophila MICAL regulates myofilament organization and synaptic structure. Mech. Dev. 124, 390–406 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Orr B. O., Fetter R. D., Davis G. W., Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 550, 109–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knowles-Barley S., Longair M., Armstrong J. D., BrainTrap: A database of 3D protein expression patterns in the Drosophila brain. Database 2010, baq005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aradska J., Bulat T., Sialana F. J., Birner-Gruenberger R., Erich B., Lubec G., Gel-free mass spectrometry analysis of Drosophila melanogaster heads. Proteomics 15, 3356–3360 (2015). [DOI] [PubMed] [Google Scholar]
- 49.McGrath J., Roy P., Perrin B. J., Stereocilia morphogenesis and maintenance through regulation of actin stability. Semin. Cell Dev. Biol. 65, 88–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mburu P., Romero M. R., Hilton H., Parker A., Townsend S., Kikkawa Y., Brown S. D., Gelsolin plays a role in the actin polymerization complex of hair cell stereocilia. PLOS ONE 5, e11627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kikkawa Y., Mburu P., Morse S., Kominami R., Townsend S., Brown S. D. M., Mutant analysis reveals whirlin as a dynamic organizer in the growing hair cell stereocilium. Hum. Mol. Genet. 14, 391–400 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Mburu P., Kikkawa Y., Townsend S., Romero R., Yonekawa H., Brown S. D. M., Whirlin complexes with p55 at the stereocilia tip during hair cell development. Proc. Natl. Acad. Sci. U.S.A. 103, 10973–10978 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Santos A., Shauchuk Y., Cichon U., Vavra K. C., Rock R. S., How actin tracks affect myosin motors. Adv. Exp. Med. Biol. 1239, 183–197 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Rzadzinska A. K., Schneider M. E., Davies C., Riordan G. P., Kachar B., An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J. Cell Biol. 164, 887–897 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiehart D. P., Franke J. D., Chee M. K., Montague R. A., Chen T.-l., Roote J., Ashburner M., Drosophila crinkled, mutations of which disrupt morphogenesis and cause lethality, encodes fly myosin VIIA. Genetics 168, 1337–1352 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niu F., Sun K., Wei W., Yu C., Wei Z., F-actin disassembly factor MICAL1 binding to Myosin Va mediates cargo unloading during cytokinesis. Sci. Adv. 6, eabb1307 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J., Guan J., Wang H., Yin L., Wang D., Zhao L., Zhou H., Wang Q., Genotype-phenotype correlation analysis of MYO15A variants in autosomal recessive non-syndromic hearing loss. BMC Med. Genet. 20, 60 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muller U., Littlewood-Evans A., Mechanisms that regulate mechanosensory hair cell differentiation. Trends Cell Biol. 11, 334–342 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Narayanan P., Chatterton P., Ikeda A., Ikeda S., Corey D. P., Ervasti J. M., Perrin B. J., Length regulation of mechanosensitive stereocilia depends on very slow actin dynamics and filament severing proteins. Nat. Commun. 6, 6855 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGrath J., Tung C.-Y., Liao X., Belyantseva I. A., Roy P., Chakraborty O., Li J., Berbari N. F., Faaborg-Andersen C. C., Barzik M., Bird J. E., Zhao B., Balakrishnan L., Friedman T. B., Perrin B. J., Actin at stereocilia tips is regulated by mechanotransduction and ADF/cofilin. Curr. Biol. 31, 1141–1153.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmed Z. M., Yousaf R., Lee B. C., Khan S. N., Lee S., Lee K., Husnain T., Rehman A. U., Bonneux S., Ansar M., Ahmad W., Leal S. M., Gladyshev V. N., Belyantseva I. A., Van Camp G., Riazuddin S., Friedman T. B., Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am. J. Hum. Genet. 88, 19–29 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kwon T.-J., Cho H.-J., Kim U.-K., Lee E., Oh S.-K., Bok J., Bae Y. C., Yi J.-K., Lee J. W., Ryoo Z.-Y., Lee S. H., Lee K.-Y., Kim H.-Y., Methionine sulfoxide reductase B3 deficiency causes hearing loss due to stereocilia degeneration and apoptotic cell death in cochlear hair cells. Hum. Mol. Genet. 23, 1591–1601 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Yang Y., Kovacs M., Xu Q., Anderson J. B., Sellers J. R., Myosin VIIB from Drosophila is a high duty ratio motor. J. Biol. Chem. 280, 32061–32068 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Hammer J. A. III, Sellers J. R., Walking to work: Roles for class V myosins as cargo transporters. Nat. Rev. Mol. Cell Biol. 13, 13–26 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Kwon M., Godinho S. A., Chandhok N. S., Ganem N. J., Azioune A., Thery M., Pellman D., Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 22, 2189–2203 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bretscher A., Edwards K., Fehon R. G., ERM proteins and merlin: Integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3, 586–599 (2002). [DOI] [PubMed] [Google Scholar]
- 67.Planelles-Herrero V. J., Blanc F., Sirigu S., Sirkia H., Clause J., Sourigues Y., Johnsrud D. O., Amigues B., Cecchini M., Gilbert S. P., Houdusse A., Titus M. A., Myosin MyTH4-FERM structures highlight important principles of convergent evolution. Proc. Natl. Acad. Sci. U.S.A. 113, E2906–E2915 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabg0147/DC1