

Abstract
Nucleic acid amplification tests (NAATs) are a crucial diagnostic and monitoring tool for infectious diseases. A key procedural step for NAATs is sample preparation: separating and purifying target nucleic acids from crude biological samples prior to nucleic acid amplification and detection. Traditionally, sample preparation has been performed with liquid- or solid-phase extraction, both of which require multiple trained user steps and significant laboratory equipment. The challenges associated with sample preparation have limited the dissemination of NAAT point-of-care diagnostics in low resource environments, including low- and middle-income countries. We report on a paper-based device for purification of nucleic acids from whole blood using isotachophoresis (ITP) for point-of-care NAATs. We show successful extraction and purification of target nucleic acids from large volumes (33 μL) of whole human blood samples with no moving parts and few user steps. Our device utilizes paper-based buffer reservoirs to fully contain the liquid ITP buffers and does not require complex filling procedures, instead relying on the natural wicking of integrated paper membranes. We perform on-device blood fractionation via filtration to remove leukocytes and erythrocytes from our sample, followed by integrated on-paper proteolytic digestion of endogenous plasma proteins to allow for successful isotachophoretic extraction. Paper-based isotachophoresis purifies and concentrates target nucleic acids that are added directly to recombinase polymerase amplification (RPA) reactions. We show consistent amplification of input copy concentrations of as low as 3×103 copies nucleic acid per mL input blood with extraction and purification taking only 30 minutes. By employing a paper architecture, we are able to incorporate these processes in a single, robust, low-cost design, enabling the direct processing of large volumes of blood, with the only intermediate user steps being the removal and addition of tape. Our device represents a step towards a simple, fully integrated sample preparation system for nucleic acid amplification tests at the point-of-care.
Keywords: Isotachophoresis, sample preparation, nucleic acids, paper-microfluidics, protein digestion
1. INTRODUCTION
There is a need to develop point-of care (POC) infectious disease diagnostic technologies to enable faster, more economic care closer to the patient [1–3]. This includes nucleic acid amplification tests (NAATs), a crucial diagnostic and monitoring tool for bacterial and viral infections [4–7]. NAATs have been developed for a variety of diseases, including HIV, hepatitis B, hepatitis C, and tuberculosis [8–10]. NAATs may be used to determine the presence of a pathogen or as a way to surveil disease progression and inform patient treatment plans. In many cases, NAATS have replaced more traditional techniques (e.g. cell culture or immunoassays) as the gold standard [11–14]. The majority of currently available NAATs require central laboratory infrastructure, including highly trained personnel, sensitive reagents, and significant logistics related to sample collection and transport [4,15]. In low- and middle-income countries (LMIC), where disease prevalence remains high, these requirements, as well as high capital costs and other required resources (e.g. stable electricity and climate-controlled facilities) may significantly complicate NAAT implementation [16–18].
Nucleic acid amplification tests require three main steps: sample preparation, amplification, and detection [4]. Sample preparation isolates and purifies target nucleic acids from potent amplification inhibitors often present in complex biological samples to allow for successful subsequent amplification and detection, and is often considered to be a primary bottleneck in nucleic acid testing [19,20]. Traditional sample preparation is performed via liquid phase extraction (LPE), such as phenol-chloroform extraction, or solid phase extraction (SPE) techniques, which preferentially bind and release nucleic acids to silica columns, beads, or membranes. These protocols utilize high-molarity toxic chaotropic salts such as guanidine thiocyanate or guanidine hydrochloride, often included in lysis buffers, which help facilitate nucleic acid adsorption onto silica materials [21,22]. Both LPE and SPE result in purified nucleic acids that are readily added to amplification reactions. These protocols require numerous manual pipetting and centrifugation steps, can take more than an hour to perform, and can be difficult to implement in a point-of-care environment primarily due to their requirements for training, refrigerated reagent storage, and necessary associated equipment (e.g. centrifuges and pipettes) [15].
Several fully integrated sample-to-answer point-of-care NAAT platforms are commercially available, which automate the sample preparation, such as the as the m-PIMA system (Abbott), the GeneXpert (Cepheid), and the Cobas Liat platform (Roche) [23]. These solutions typically rely on solid-phase extraction techniques, using complex hydraulics and electromechanical pathways and robotics to replace manual steps, significantly increasing platform costs [24–26]. Various microfluidic approaches have also been investigated, though these often require external pressure sources for sample introduction and valving [27,28]. This additional complexity complicates overall device design and implementation.
Isotachophoresis is an electrokinetic separation technique that has been applied to purification of nucleic acids from complex biological samples [29–33]. First recognized for its ability to separate and concentrate metal ions from organic solutions [34], isotachophoresis (ITP) is appealing for nucleic acid sample preparation because it uses simple buffers and requires no moving parts or valving. ITP relies on a discontinuous buffer system containing a high electrophoretic mobility leading electrolyte (LE) and a low mobility trailing electrolyte (TE). As an electric field is applied to the system, a charged sample species with an intermediate mobility, such as nucleic acids, focuses at the interface between the two electrolytes. Species with higher or lower mobilities than the respective LE and TE, for example proteins, are excluded from the interface plug region, resulting in the purification of the species of interest. Kondratova et al. were the first to show successful isotachophoretic extraction of nucleic acids from complex biological samples, including human plasma/serum and urine in agarose gel rods, though this required significant sample preparation, including overnight incubation with proteinase K/SDS and dialysis [32,33]. Since then, isotachophoresis has been demonstrated in microchannel and paper based formats, and has been used to separate and purify nucleic acids from a range of biological samples, including urine, milk, and blood [30,35–38].
Blood is a particularly compelling sample for POC NAATs as many of the world’s most widespread diseases, such as HIV, malaria, dengue, and HBV are bloodborne. Persat et al. were the first to extract and purify nucleic acids from diluted whole blood lysate using ITP [30]. They targeted genomic DNA with an estimated extraction efficiency of 30–70% and processed 2.5 nL of whole blood that had been diluted 10-fold with lysis buffer, for a total processed sample volume of 25 nL. Eid et al. extracted nucleic acids from Listeria monocytogenes cells in diluted whole blood via ITP and amplified using off-chip recombinase polymerase amplification, achieving a limit-of-detection (LoD) of 5×103 cell-equivalents per mL when using purified genomic DNA and 2×104 cells per mL when using L. monocytogenes cells [39]. In both experiments, 2.5 μL of whole blood was spiked with target and diluted 10-fold with lysis buffer and modified LE. Marshall et al. purified and detected genomic DNA from P. falciparum parasites using ITP, with a limit of detection reported as 500 parasites per μL blood [40]. In this work, 1 μL of whole blood was added to a 14 μL reservoir of TE and proteinase K, utilizing ITP with a semi-infinite sample injection scheme to separate the nucleic acids after pathogen lysis.
Much of the previous work has focused on isotachophoretic nucleic acid extraction and purification has been performed in microchannel devices manufactured via traditional photolithography or injection molding [41,42]. These devices have a number of advantages, such as precisely defined geometries, minimal channel adhesion sample loss, and readily available mass manufacturing methods. Challenges of microchannel based ITP devices include complex LE/TE buffer loading procedures (often requiring vacuum assistance) and minimally contained free-surface liquid reservoirs [42], complicating potential application at the point-of-care. Additionally, in microchannel based ITP, the sample volumes have been very limited and recovering ITP-focused target analyte can be challenging. Paper-based ITP systems are robust, extremely inexpensive to manufacture, and allow for larger sample volumes. They also fill with electrolyte solutions and samples automatically owing to the natural wicking properties of the hydrophilic porous membranes. Large scale paper-based ITP was originally used for laboratory separations of ions [43,44]. More recently, microfluidic based paper devices have been developed for point-of-care applications [37,38,45,46]. Our group first demonstrated preconcentration in a paper based microfluidic device and how this can be used to improve the limit of detection in rapid immunoassays [37,47]. Rosenfeld and Bercovici created a paper-based ITP system by patterning wax onto a nitrocellulose membrane to constrain the LE/TE buffers, and showed 20,000 fold sample focusing and amplification-free detection of target DNA in buffer [46]. Previously, our group extracted target DNA from human serum utilizing paper-based ITP, and amplified on-device via recombinase polymerase amplification with a limit of detection of 104 copies DNA per mL from an initial sample volume of 20 μL [31]. We also showed proof-of-concept data describing extraction and amplification of DNA from whole blood while using a significant concentration of target DNA.
While ITP represents an attractive solution for nucleic acid extraction and purification from complex samples such as whole blood, additional sample preparation steps such as off-chip sample dilution are often still required. Dilution is frequently used because the high salt and protein content of blood and plasma can make isotachophoretic extraction difficult due to effects on viscosity, ionic strength, pH, and conductivity of the system [48]. For applications that are primarily concerned with pathogen presence and binary diagnoses, where the concentration of the target is high, dilution may not be problematic. Dilution can be detrimental, however, for applications where low limits of detection are necessary, such as HIV viral load monitoring [49]. In applications where low detection limits are required there is often only a single target nucleic acid per microliter, or less, so large biological sample volumes are critical to ensure sufficient target is present in the device. In much of the previous work where whole blood was used as a sample in ITP, the total processed sample volume was typically 10–25 microliters, while the volume of blood processed was 1–2.5 microliters. For this reason, there have been several works that have focused on increasing ITP sample volume or biological sample concentration. For instance, van Kooten et al. focused DNA and E. coli bacteria diluted in 50 μL trailing electrolyte into a volume of 500 pL by carefully designing their microchannel-based system [41]. While this volume is impressive in an isotachophoretic system, complex biological samples were not tested. Bender et al. extracted nucleic acids from 20 μL of human serum, representing one of the largest volumes of undiluted complex sample used in isotachophoretic systems to date [31]. For bloodborne pathogen detection in the point-of-care setting, it is often necessary to process large volumes of blood to reach low limits of detection in downstream nucleic acid amplification assays. This requires an integrated approach that addresses many unique aspects of using whole blood as a sample, including leukocyte/erythrocyte depletion and endogenous protein digestion to successfully extract and purify target nucleic acids in an inexpensive, semi-automated manner with minimal user steps.
In this paper, we present a paper-based, isotachophoretic nucleic acid sample preparation platform that extracts target DNA from human whole blood. Our device directly processes relatively large volumes (33 μL) of whole blood, so no preliminary dilution or pretreatment user steps are required. We verify nucleic acid isotachophoretic extraction and purification by real-time imaging of fluorescently labeled DNA and off-device recombinase polymerase amplification (RPA). We show an optimized integrated plasma separation membrane for cell fractionation, as well as on-paper proteolytic digestion of endogenous plasma proteins via proteinase K to enable isotachophoretic extraction, verified using SDS-PAGE. We discuss design decisions used to develop a platform that decreases the number of user steps and potential sources of error, including the first integration of paper-based ITP buffer reservoirs. The nucleic acid sample preparation platform presented here represents a step towards a simple, robust, low-cost sample preparation technique for point-of-care NAATs for bloodborne diseases.
2. MATERIALS AND METHODS
2.1. DEVICE CONSTRUCTION
We used an integrated paper-based nucleic acid sample preparation device to extract target nucleic acids from whole human blood. Figure 1 shows the device that is constructed from different layers of acrylic, membranes, PCR tape, and titanium foil. The acrylic body was fabricated from 3 mm acrylic using a CO2 laser cutter (M360, Universal Laser Systems, USA), resulting in a 65 mm × 20 mm rectangle. Two 12.5 mm square ITP buffer reservoir wells were cut out of the acrylic near the ends. A 65 mm × 20 mm rectangle of PCR tape (TempPlate RT Select Optical Film, USA Scientific, USA) was cut with the laser cutter, forming the bottom tape. Two rectangles of 20 mm × 6 mm × 0.12 mm titanium foil (LiteOutdoors, CAN) were cut by hand and are placed in between the bottom tape and the acrylic body such that roughly 7.5 mm of the foil rectangles extend over the short edges of the device. Twelve 10 mm squares of glass fiber membrane (G041 Glass Fiber Conjugate Pad, Millipore Sigma, USA) were cut using a craft cutter (Silhouette Cameo 3, Silhouette America, USA), with six squares placed in each ITP buffer reservoir well, forming the paper-based ITP reservoirs. We used paper-based ITP reservoirs instead of traditional liquid reservoirs to minimize spills and leaks from the device during buffer loading and manipulation, as the entire liquid system is contained within a membrane matrix. A section of Fusion 5 membrane (GE Healthcare Life Sciences, USA) was cut with the craft cutter and placed such that the ends of the Fusion 5 were in contact with the glass fiber reservoirs. On one end of the Fusion 5 membrane is the sample pad which features a spade-like widening that is 9 mm at its widest extent. The remainder of the Fusion 5 membrane is 3 mm wide. We placed the PCR cover tape over the Fusion 5 membrane and pressed down against the acrylic body to bond to it. The cover tape has two small circular apertures that are located over the glass fiber reservoirs for buffer addition. The cover tape also has a rectangular opening that exposes part of the narrow Fusion 5 membrane, called the eluate port, and another spade shaped cut-out, called the sample port, that reveals the sample pad. Initially, PCR tape (25 mm × 15 mm) covers the eluate port.
Figure 1:
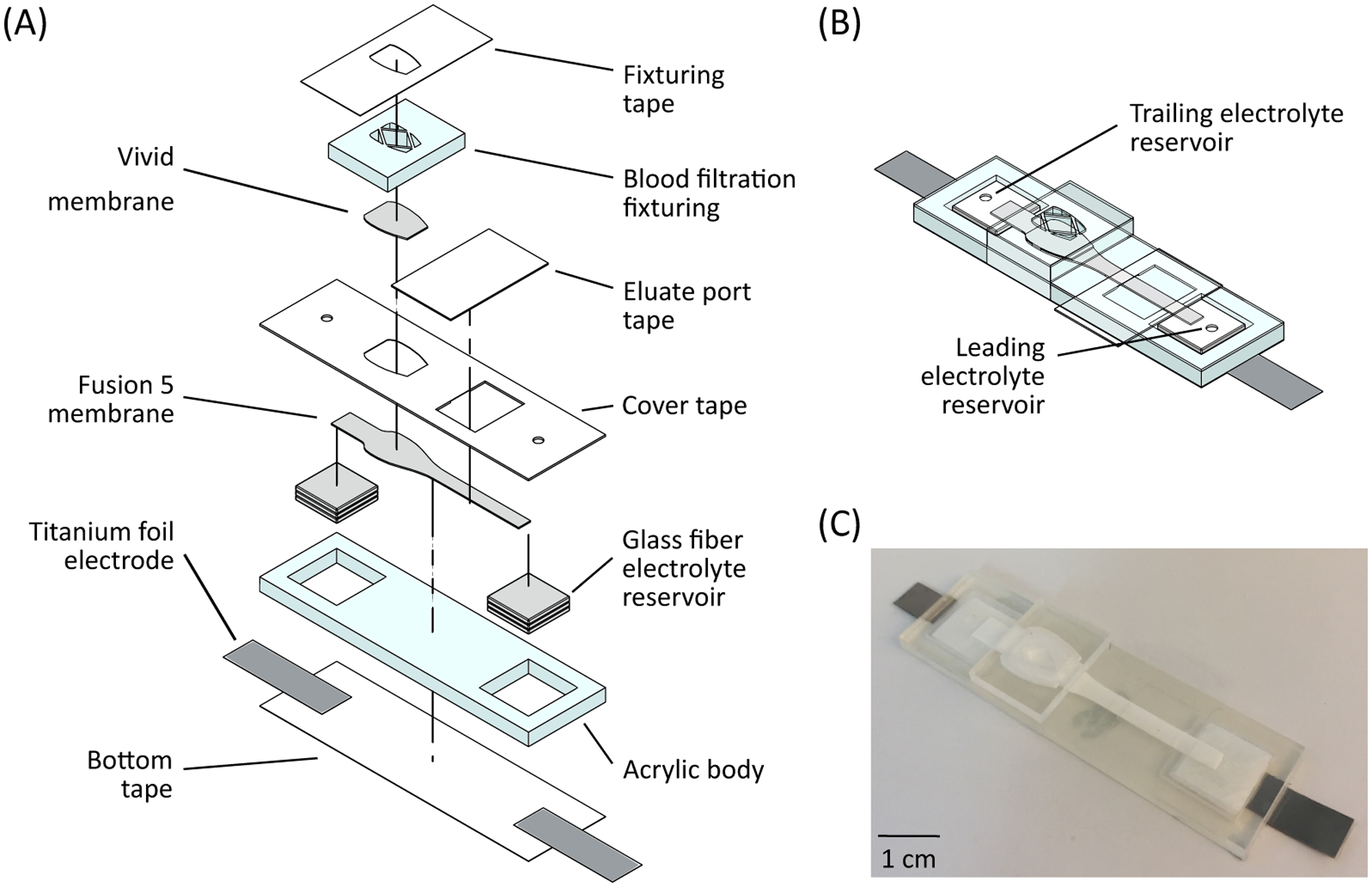
Device for extracting DNA from whole blood. (A) Device materials and construction. The device is constructed from various layers of acrylic, PCR tape, and several different membranes. The device is designed for an input sample volume of 33 μL of whole blood. (B) Schematic of assembled device, showing the glass fiber membrane reservoirs for ITP buffers. (C) Image of an assembled device.
We pipetted 20 μL of proteinase K (P8107S, New England Biolabs, USA) onto the exposed Fusion 5 sample pad and desiccated the device for 20 minutes at room temperature to dry the membrane. We then placed a spade-shaped 0.765 cm2 Vivid GR plasma separation membrane (Pall Corp., USA) in contact with the Fusion 5 membrane. A separate piece of laser-cut acrylic (referred to as the blood filtration fixturing) was placed over the Vivid membrane. The blood filtration fixturing has several holes cut through for sample addition and a laser cut relief section, which provides a compressive contact force with the Vivid membrane. Lastly, a piece of PCR tape was placed over the blood filtration fixturing and is wrapped around and attached to the sides of the acrylic body.
2.2. REAGENTS AND CHEMISTRY
The leading electrolyte consisted of 250 mM HCl (H1758, Sigma, USA) and 375 mM Tris (93362, Sigma, USA) at a calculated pH of 7.8. The trailing electrolyte consisted of 25mM serine (84959, Sigma, USA) and 25mM Tris at a calculated pH of 8.7. Using these buffers, proteinase K is positively charged in our system due to its high isoelectric point (pH 8.9) [50], allowing the ITP system to separate it from the negatively charged nucleic acids [30]. Isotachophoresis simulations were performed using SPRESSO to estimate local pH along the Fusion 5 membrane [51]. SPRESSO conditions and settings used are shown in Table A.1 in the Supporting Information.
We diluted the target DNA to various concentrations in low-EDTA TE buffer (VWR, USA) and stored the dilutions at −20 °C. The target DNA used was a synthetic strand (200 base pairs), detailed in Bender et al. [31], and derived from the proviral HIV-1 DNA pol gene, containing complementary sequences for the primers and probes adopted from Boyle et al [52]. We used a 70 base pair oligonucleotide labeled with Alexa Fluor 488 to fluorescently monitor ITP plug location and progression. Full nucleotide sequences are listed in the Supporting Information (Table A.2). All oligonucleotides were purchased from Integrated DNA Technologies (USA).
We used RPA to validate the presence of target DNA in our extracted sample after ITP. We used the TwistAmp exo kit from TwistDx (GBR) and followed standard operating procedures. The mastermix included a lyophilized pellet consisting of various proteins and enzymes, rehydration buffer, probe, and primers. Magnesium acetate was added to begin the reaction. We used 18 mM magnesium acetate to compensate for any residual EDTA that might be present in the blood sample that is stored EDTA blood tubes. The total reaction volume was 50 μL. A T16-ISO instrument (TwistDx, GBR), set at 39 °C, amplified and detected the DNA target. After 4 minutes of incubation, we removed and agitated the tubes by hand to further mix reagents and then placed them back into the T16-ISO for an additional 16 minutes.
2.3. DEVICE OPERATION
Figure 2 shows the operation of the device. We used a freshly constructed device for each experiment. We first pipetted the whole blood sample onto the Vivid plasma separation membrane. The sample consisted of 33 μL human whole blood (K2EDTA, BioIVT, USA), 1 μL target DNA at various concentrations, and 1 μL of 1 μM fluorescently labeled tracking DNA. We chose the sample volume to comply with the Vivid membrane manufacturer’s processing recommendation of 40–50 μL blood per square centimeter of membrane. Both the target DNA and fluorescently labeled tracking DNA are added to the whole blood sample immediately before sample loading onto the Vivid plasma separation membrane. After sample addition, we allowed blood filtration to occur for four minutes. During this process, plasma and the target/tracking DNA passed through the Vivid plasma separation membrane and into the underlying Fusion 5 membrane where it rehydrated the desiccated proteinase K. After four minutes, we removed the blood filtration fixturing and the Vivid plasma separation membrane and placed a rectangular piece of PCR tape over the sample port, ensuring a good seal near the sample pad to prevent evaporation and contamination.
Figure 2.
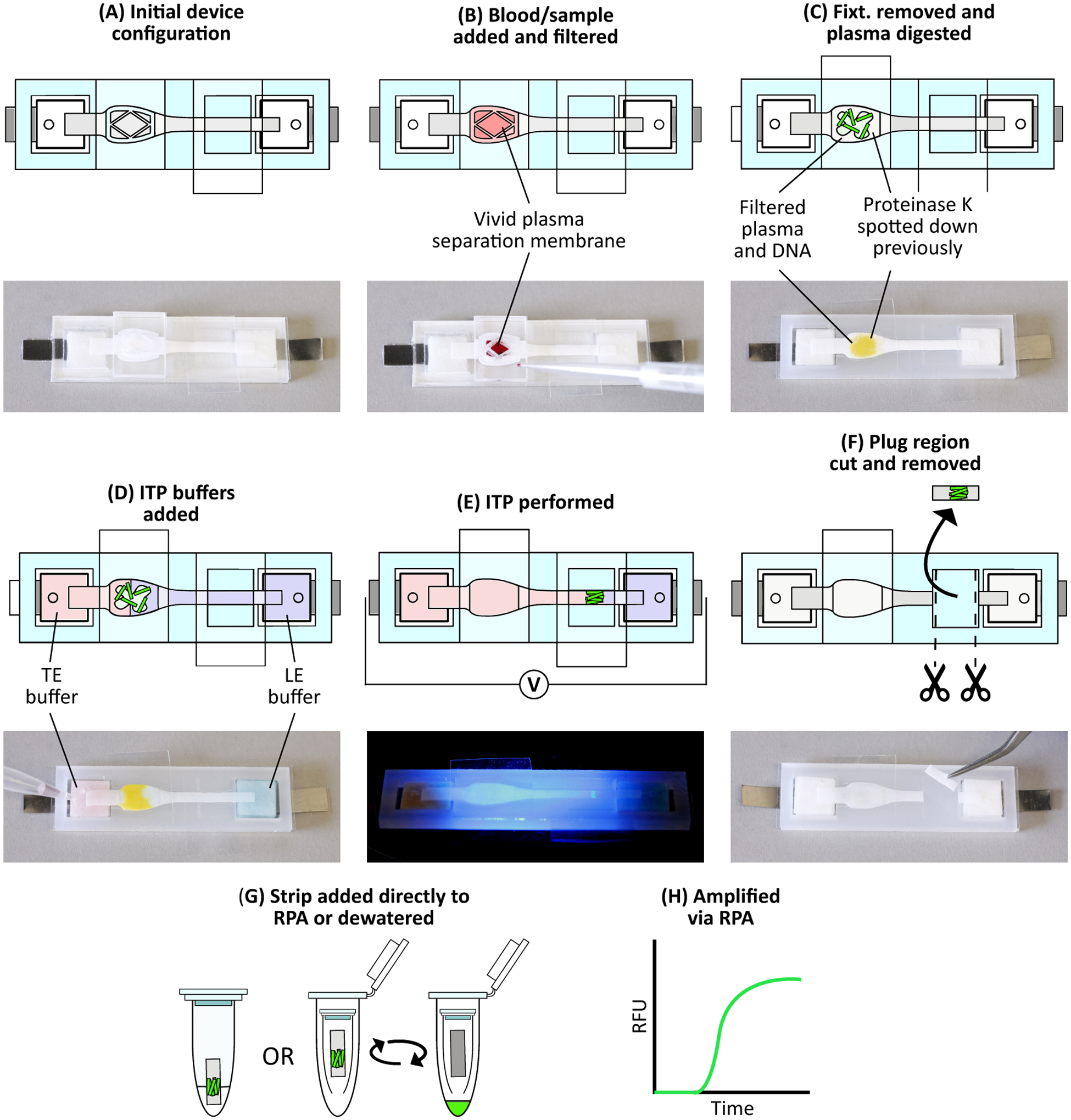
Device protocol to extract DNA from whole blood. (A) Initial device configuration. (B) We add 33 μL whole blood spiked with target DNA and fluorescently labeled tracking DNA onto the Vivid membrane. Blood filtration occurs for 4 minutes, during which plasma and nucleic acids filter through to the Fusion 5 membrane. (C) After filtration, we remove the blood filtration fixturing and Vivid and place a piece of PCR tape over the sample port. Proteolysis then occurs for 15 minutes. (D) We add ITP buffers to their respective reservoirs. Color is added to the image for clarity: red represents trailing electrolyte buffer while blue represents leading electrolyte buffer. (E) Constant current is applied across the device, beginning ITP and focusing the DNA into a narrow plug which then migrates towards the leading electrolyte reservoir. We monitor this process via fluorescent microscopy. (F) Once the ITP plug reaches the eluate port, we stop ITP, remove the eluate port tape, and cut out the exposed portion of Fusion 5 membrane containing the ITP plug. Buffer colors are removed for clarity. (G) We add the strip directly to an RPA reaction, or first dewater the strip using a centrifuge and then add the resulting liquid sample to an RPA reaction. (H) RPA is then performed with the real-time fluorescence recorded.
The plasma rehydrated the proteinase K that proteolytically digested the plasma proteins. We allowed on-paper plasma protein digestion to occur for 15 minutes at various temperatures (22 °C, 37 °C, and 55 °C). We then added 390 μL of leading electrolyte (LE) and 320 μL trailing electrolyte (TE) buffers to their respective glass fiber reservoirs through small holes in the cover tape. The liquid wicks into and saturates the glass fiber and the Fusion 5 membranes. The capillary driven flow and diffusion of the electrolytes result some spatial varying dilution of the filtered plasma across the Fusion 5 membrane. A source meter (Model 2410, Keithley, USA) then applied 1.5 mA constant current via the titanium foil electrodes, powering isotachophoresis.
We monitored ITP progression with epifluorescence microscopy of the fluorescently labeled tracking DNA. Imaging and data collection methods are discussed below. As ITP progressed, both the target and tracking DNA were focused at the ITP plug interface of the LE and TE and migrate away from the digested plasma proteins. Once the ITP plug interface reached the middle of the eluate port, we removed the eluate port tape and cut out the 5 mm exposed section of the Fusion 5 membrane containing the ITP plug with an X-Acto knife. We then added this strip directly to an RPA reaction tube, after master mix has been added to rehydrate the lyophilized reagents. The tube was then inverted several times to mix reagents and then tapped on a surface to ensure the strip sat near the bottom of the reaction tube. It was then placed into the T-16 ISO for amplification and readout.
Alternatively, we dewatered the strip using a centrifuge and added the resulting liquid sample to the RPA reactions. In this procedure, the exposed portion of Fusion 5 strip that is cut out was increased to 10 mm in length. After cutting the strip, we placed this strip into a 0.5 mL micro-centrifuge tube that had a small opening at the bottom. This micro-centrifuge tube was then placed in a larger 1.5 mL micro-centrifuge tube and both tubes were placed in a centrifuge (5415D, Eppendorf, DEU) for 3 minutes at 6,000 rcf. The resulting eluate was collected at the bottom of the 1.5 mL micro-centrifuge tube. After dewatering, we collected a total of 10 μL of eluate containing the target DNA and split this between two RPA reactions, with 5 μL of the eluate added to each reaction. We found that splitting the eluate into two separate reactions resulted in more consistent amplification, likely due to trace inhibitor dilution. After amplification, we discarded the reaction with the poorer amplification from further consideration. A list of complete user steps and their associated times are listed in Table A.3.
2.4. WHOLE BLOOD FRACTIONATION OPTIMIZATION
We measured the plasma extraction efficiency of the target DNA through the Vivid membrane and into the underlying Fusion 5 during blood fractionation. To calculate the plasma extraction efficiency, we measured the mass of the underlying Fusion 5 membrane before and after blood fractionation. We define plasma extraction efficiency as plasma in the Fusion 5 membrane divided by the total plasma in the sample as, ηP.Extract = (mP.F5/ρP)/Vblood * (1 − HCT), where mP.F5 is the mass of plasma residing in the underlying Fusion 5 membrane after fractionation, ρP is the density of human plasma, Vblood is the volume of blood applied to the Vivid plasma separation membrane, and HCT is the blood hematocrit value. We measured the blood hematocrit value using a Hb 201+ analyzer (HemoCue, USA). We also qualitatively investigated cell fractionation as well as quantified hemolysis. We visualized cell fractionation using a hemocytometer (Bright Line Hemacytometer 1492, Hausser Scientific, USA) with a TMS Inverted Microscope (Nikon, JPN). Hemolysis was measured using the Harboe method [53,54] with plasma filtered by the device and a spectrophotometer (NanoDrop 2000, ThermoFisher Scientific, USA) and compared against plasma that had been separated using a centrifuge.
2.5. SDS-PAGE
We performed sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to analyze plasma protein digestion. We used 12% Mini-PROTEAN TGX precast protein gels (#4561045, Bio-Rad, USA) and Tris/Glycine/SDS running buffer (#1610732, Bio-Rad, USA). We used the same device as described previously, though without the blood fractionation fixturing and associated steps to simplify the procedure. Pooled, sterile-filtered human serum (H4522, Sigma-Aldrich, USA) was first pipetted onto the Fusion 5 membrane sample pad region, on which we had spotted proteinase K previously. We used 20 μL of proteinase K and 20 μL of serum. We then covered the sample pad region with PCR tape, as described previously. Three different digestion temperatures were tested: 22 °C, 37 °C, and 55 °C. For experiments not at room temperature (22 °C), we placed the device on a hotplate. We determined the proper hotplate setpoint such that the sample pad reaches the desired digestion temperature by inserting a Type-K thermocouple (9251T93, McMaster-Carr, USA) under the sample port tape, ensuring good contact with the sample pad. To reach a steady-state temperature of roughly 55 °C or 37 °C, we set the hotplate to 65 °C or 45 °C, respectively. After 15 minutes, the PCR tape was removed, and the sample pad region of the Fusion 5 membrane was cut out and dewatered in a centrifuge. We also digested serum directly in tubes. In those experiments, we added proteinase K directly into human serum in a 1.5 mL tube and placed it into a water bath set at 55°C for 15 minutes. For both experiment types, we then diluted the resulting serum digest to 2.5 mg/mL of protein with a total volume of 20 μL, verified using a spectrophotometer (NanoDrop 2000, ThermoFisher Scientific, USA). The diluted proteins were added to 2x Laemmli sample buffer (S3401, Sigma-Aldrich, USA) on ice in a 1:1 ratio and immediately placed in a boiling water bath for 5 minutes. Then, 10 μL of the final solutions were added to the gel lanes. A protein ladder (Precision Plus Protein Dual Color Standards #1610374, Bio-Rad, USA) was also added. Electrophoresis was performed in a Mini-Protean Tetra cell (Bio-Rad, USA) at 200V for 45 minutes. We performed Coomassie Blue staining (Bio-Safe Coomassie G-250 #1610786, Bio-Rad, USA) after fixing the proteins in 40% ethanol and 10% acetic acid. A Perfection V370 scanner (Epson, JPN) was used for gel imaging.
2.6. DATA COLLECTION
Real-time epifluorescence imaging of the ITP system was performed to determine the plug location and ITP progression, visualized by Alexa Fluor 488 labeled tracking DNA. We used an AZ100 microscope (Nikon, JPN) with a 0.5X objective, a 488 nm excitation and 518 nm emission filter cube set (Omega Optics, USA), and a 16-bit cooled CCD camera (Cascade 512B, Photometrics, USA). Grayscale images of the evolution of the ITP system were captured every 1 second for 20 minutes. A video of a representative run is available in the Supporting Information.
We quantify the extraction efficiency of our ITP system by comparing the bulk fluorescence of the fluorescently labeled tracking DNA to the plug region after extraction. We first establish the baseline by pipetting 1 μL of 1 μM fluorescently labeled tracking DNA onto a region of Fusion 5 membrane. The fluorescence in the focused ITP plug is determined by integrating the fluorescence in a region of interest around the plug. The images are corrected for background by selecting a region of the Fusion 5 strip far from the tracking DNA to reduce any effects of autofluorescence of the membrane and/or PCR tape. We repeat this process for a total of 6 individual trials and the average experimental integrated fluorescence is divided by the average baseline integrated fluorescence to estimate the extraction efficiency of the system.
We used a T16-ISO instrument for RPA incubation and real-time fluorescent measurement. Real-time fluorescence values of the RPA reactions were recorded every 8–10 seconds. For comparison of the amplification reactions, we normalized intensity values by subtracting the absolute intensity measured at 45 seconds. We found that fluorescence values initially decrease at the beginning of an experiment – at 45 seconds, the fluorescence values have sufficiently stabilized.
3. RESULTS AND DISCUSSION
We designed this device to simplify isotachophoretic sample preparation of nucleic acids from whole blood. For instance, paper-based reservoirs for ITP buffers are used to minimize the risk of spillage while loading the liquids and manipulating the device. By using an all-paper system, the sample and buffer liquids are fully contained within the membrane matrix, and simple loading procedures can be used. The only steps required by the user are the removal and application of various tapes and the initial dispensing of ITP buffers. The applied ITP buffer volumes are chosen such that the initial LE/TE interface is naturally located at the sample pad region, resembling a traditional finite injection ITP scheme without the need for careful loading protocols. The paper-based architecture also allows for the integration of a plasma separation membrane (which functions via plasma wicking to the underlying membrane) and localized dispensing and desiccation of proteinase K, both of which would be difficult to implement in microchannel ITP formats. The device’s processes are separated into three main areas: blood filtration/fractionation, on-paper plasma protein digestion, and nucleic acid extraction. Although integrated, on-device amplification has been shown by our group previously [31], we choose to focus on integration and upstream sample preparation in this work, using off-device amplification for device validation.
Blood fractionation is important in many blood based NAAT applications to remove potent amplification inhibitors such as hemoglobin [55] and to remove sources of proviral DNA (e.g. white blood cells in the case of HIV viral load monitoring). Many current POC NAAT platforms use cell-free plasma sample and require an initial blood centrifugation step [49]. In our device, erythrocytes and leukocytes are trapped by and remain in the integrated Vivid plasma separation membrane as blood passes through, resulting in cell-free plasma. Previous publications have shown Vivid membrane integration into various devices, with plasma extraction efficiencies ranging from 25–80% [56–61]. The manufacturer, Pall, reports >80% plasma extraction efficiency in their marketing materials. We achieve a plasma extraction efficiency of 72 ± 6.8 % (n=9) when we place the Vivid plasma separation membrane on top of the Fusion 5 membrane with no additional fixturing. We refer to this as the “standard configuration” and illustrate this in the Supporting Information (Figure A.1). By adding the blood filtration fixturing which applies a compressive force to the separation membrane and the underlying Fusion 5, we achieve 88 ± 4.4 % (n=5) plasma extraction efficiency. Some optimization of the rastered relief section is required in the blood fixturing design, as we find that too little of a relief (i.e. more compressive force) results in increased hemolysis, while too much of a relief does not provide enough compressive force and overall extraction efficiency decreases. Free hemoglobin in the filtered plasma is 3.89 ± 0.86 mg/dL (n=4), well within normal reported plasma free hemoglobin levels [62]. Free hemoglobin in plasma obtained via centrifugation is 1.86 ± 0.10 mg/dL (n=4). Hemocytometer images showing whole blood before and cell-free plasma after filtration are available in the Supporting Information (Figure A.2).
We find that protein digestion is necessary for successful isotachophoretic extraction of nucleic acids from filtered plasma/serum samples. This is in agreement with other studies, which also performed some form of protein digestion/degradation prior to extraction [30,31,39,63,64]. A common explanation for this requirement is that target nucleic acids and large proteins form complexes which impede migration and extraction. The majority of isotachophoretic extractions to date have used tube-based protein digestion, prior to sample addition into the ITP system, adding another user step. Marshall et al. incorporated proteinase K into their trailing electrolyte to simplify their process [63]. Bender et al., using a paper based ITP system, spotted a small amount of proteinase K onto a glass fiber membrane before sample addition [31]. In this work, we employ a similar procedure in which proteinase K is dispensed onto a membrane prior to sample addition. To better understand this process, we perform sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to visualize protein digestion. Figure 3 shows several SDS-PAGE gels showing the results of this digestion process, at varying proteinase K concentrations and conditions.
Figure 3.
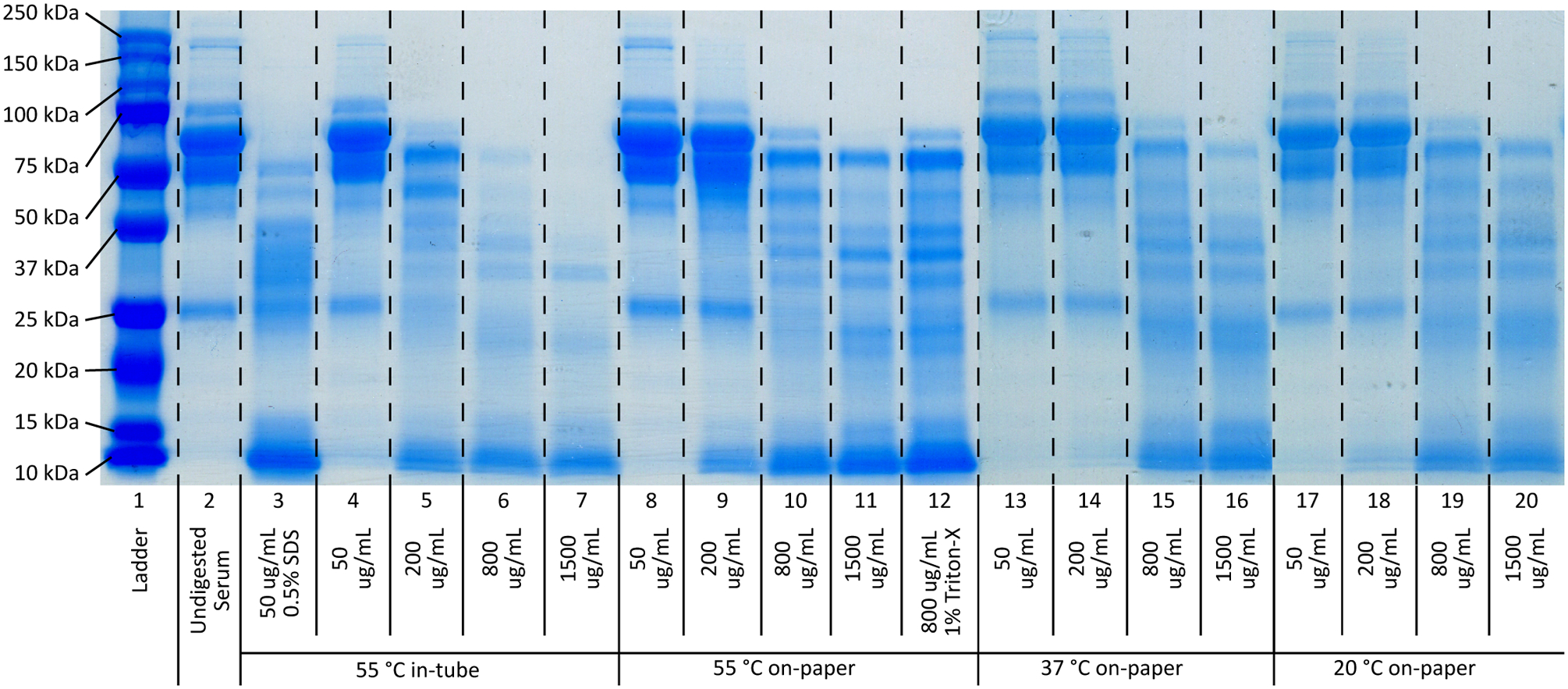
SDS-PAGE results of human serum protein digests via proteinase K. All digests were performed for 15 minutes to limit the overall protocol timespan. Lane 1 is a protein ladder (ranging from 250 kDa to 10 kDa) and lane 2 is undigested human serum. Lane 3 represents a commonly used digestion buffer for DNA extraction protocols, with 50 μg/mL proteinase K with 0.5% (w/v) SDS, digested in a microcentrifuge tube in a water bath at 55 °C. Lanes 4–7 show the effect of various proteinase K concentrations (50–1500 μg/mL) without SDS present, in a water bath at 55°C. Without SDS, the digestion is significantly diminished, though there is a clear relationship between digestion completion and proteinase K concentration. Lanes 8–11 show the same proteinase K concentration range using the on-paper digestion protocol at 55 °C. We see a lower digestion completion when compared to the in-tube protocol, though the same concentration dependent relationship is present. Lanes 12–15 and lanes 16–19 show the same proteinase K concentration range, using the on-paper digestion protocol at 37 °C and 22 °C, respectively. No significant difference is observed between the various temperature conditions.
While most traditional protein digests are performed over several hours, we chose to limit our digestion time to 15 minutes in order to reduce the overall protocol time consistent with point-of-care applications. In Figure 3, Lane 2 shows undigested human serum. The large band for albumin is visible between the 50 kDa and 75 kDa. Lane 3 represents a typical digestion buffer for DNA extraction (50 μg/mL proteinase K and 0.5% SDS) with the digestion performed in a tube in a water bath at 55 °C. The dark albumin band looks to be completely digested, with many digestion products visible between 37 kDa and 25 kDa. If this same buffer is used with the on-paper protocol, the digestion looks similar to (or even slightly worse than) on-paper digests performed without SDS present. This is shown in Figure A.3. Lanes 4–7 show digestions with various concentrations of proteinase K only, performed in a tube in a water bath at 55 °C. The data show that the level of digestion increases with proteinase K concentration. Lanes 8–11 are digests with the same proteinase K concentrations performed using the on-paper digestion protocol at 55 °C. We see the similar concentration-digestion relationship, though we generally see less overall digestion compared to the tube-based digestions. We hypothesize that the reduced effectiveness of the on-paper digestion is due to the loss of activity of the proteinase K due to the desiccation process. More sophisticated immobilization techniques, such as lyophilization, may reduce this effect. No significant difference in digestion performance is observed when compared against Lane 10. Lanes 12–15 show on-paper digestion performed at 37 °C. Lanes 16–19 are on-paper digests performed at room temperature (22 °C). We do not observe significant differences between the three temperature conditions when performing on-paper digestion. This is consistent with studies reporting proteinase K retains more than 80% of its activity between 20 °C and 60 °C [65]. We see a slight temperature dependency when in-tube digestion is performed, though only at high proteinase K concentrations (shown in Figure A.4). It is worth noting that Figure 3 represents a compilation of two different gels: Lanes 1–12 represent one gel, while Lanes 13–19 were performed on a separate gel due to gel size limitations. As such, there is some inter-gel variability due to staining/de-staining as well as inherent digestion variation, though we focus primarily on major differences between lane band structures.
We evaluate the acceptability of the digestion in our system by the ability of ITP to successfully migrate and purify the nucleic acids downstream. Spatiotemporal maps of ITP extraction progression while using various concentrations of proteinase K and on-paper digestion protocol are shown in the Figure 4. When undigested serum is used (i.e. no proteinase K, Figure 4A), the labeled DNA forms a diffuse ITP plug and does not migrate downstream (to the right). When proteinase K digests the serum (i.e. 50 μg/mL and 200 μg/mL), the labeled DNA forms a marginally concentrated ITP plug, but still fails to migrate downstream (Figure 4B and 4C). Poor spatial migration is problematic because the target nucleic acids are not sufficiently concentrated or spatially separated from inhibitors present in the sample. Figure 4D shows the ITP migration when 800 μg/mL proteinase K is used. A highly concentrated ITP plug is formed and successfully migrates down the entirety of the Fusion 5 membrane. Similar behavior is seen when 1500 μg/mL proteinase K is used (shown in Figure A.5). The SDS-PAGE results show that large proteins are still present with proteinase K concentrations less than or equal to 200 μg/mL. We hypothesize that these large proteins play a large role in ITP dynamics by modifying the mobility of nucleic acids or binding to the porous membrane and increasing the system’s electroosmotic flow. There likely lies a proteinase K concentration in our system and protocol between 200 μg/mL and 800 μg/mL that sufficiently digests the larger plasma proteins and allows for successful ITP migration and purification. We choose to use 800 μg/mL proteinase K with on-paper digestion protocols in our subsequent experiments to conserve reagents. Additional spatiotemporal maps are shown in the Supporting Information in Figure A.5, and a time series of successful ITP progression visualized by the fluorescently labeled tracking DNA in digested plasma is shown in Figure A.6.
Figure 4.
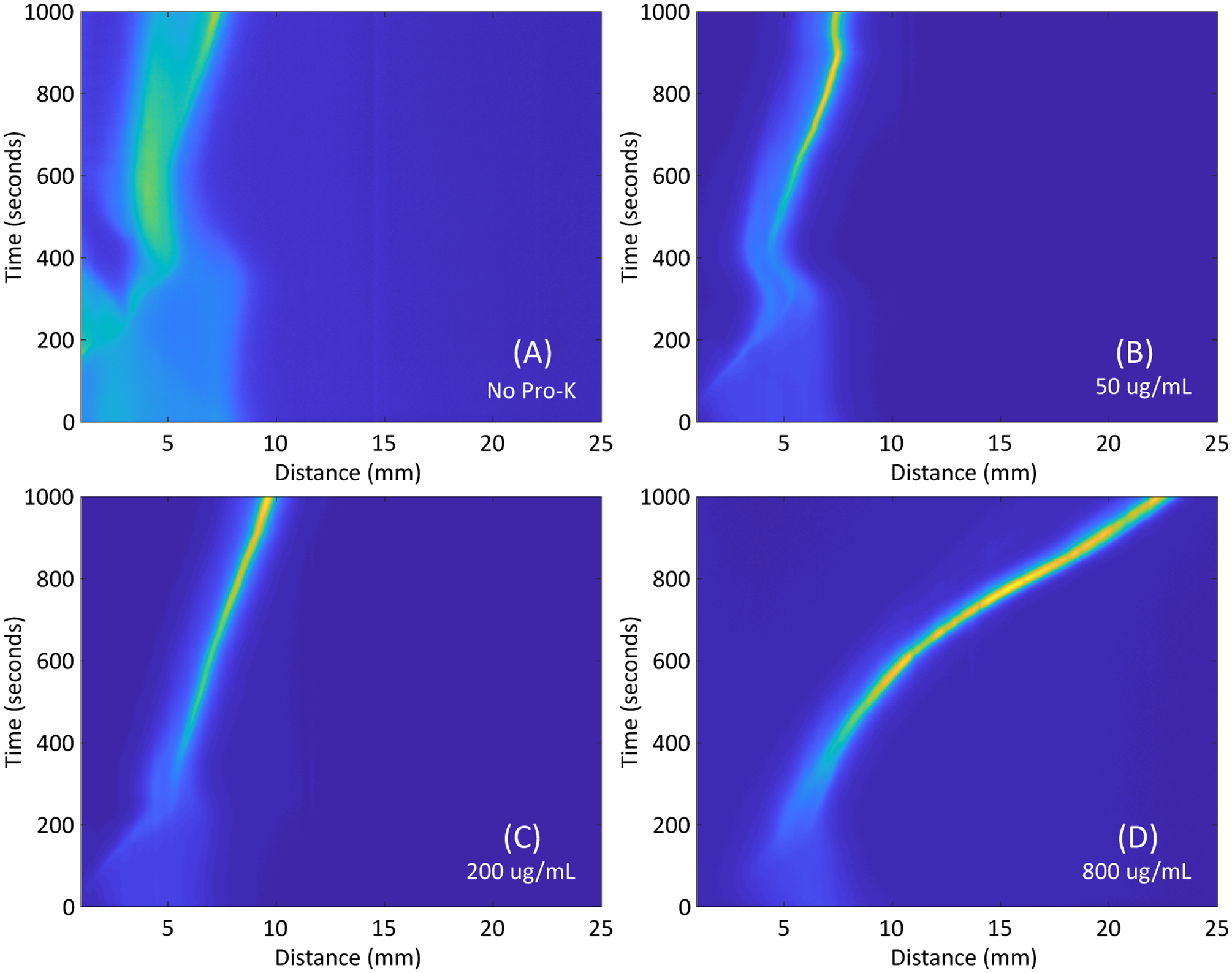
Spatiotemporal maps of representative ITP progression with varying concentrations of proteinase K, visualized via fluorescently-labeled tracking DNA with the on-paper paper digestion protocol. (A) When no proteinase K is used, the tracking DNA focuses into a diffuse region and does not migrate down the length of the Fusion 5 membrane. (B) and (C) When moderate concentrations of proteinase K are used, the fluorescently labeled DNA focuses into a concentrated plug, although the plug still does not migrate well downstream. (D) When 800 μg/mL proteinase K is used, the tracking DNA is focused into a concentrated plug and migrates down the length of the strip successfully.
By comparing the integrated fluorescence density of the isotachophoretic plug region to the bulk fluorescence of the tracking DNA, we estimate that the extraction efficiency of our system is 21%. This is comparable to other isotachophoretic systems that perform nucleic acid extraction from complex samples [66].
We present RPA amplification curves showing amplification of target DNA that has been extracted from whole blood, as shown in Figure 5. We show amplification for input target DNA copy concentrations of 3×105 copies/mL, 3×104 copies/mL, and 3×103 copies/mL in whole blood. These copy concentrations correlate to input copy numbers of 10,000 copies/trial, 1,000 copies/trial, and 100 copies/trial, respectively. Figure 5A shows results using the direct addition protocol, where the portion of Fusion 5 strip containing the ITP plug and target DNA is added directly to the RPA reaction tubes. Figure 5B shows amplification results using spun-out eluate. In both protocols, all trials successfully amplified down to 100 cps DNA/trial (3×103 cps/mL whole blood). Trials below 100 cps DNA/trial did not consistently amplify. No template controls (NTC) did not amplify. We also performed RPA amplifications with target DNA diluted in whole blood directly added to RPA reactions without processing via the described device as a control, shown in Figure A.8 and discussed in the Supplementary Information. In these experiments, the amplification is significantly inhibited, consistent with previous studies that investigated the inhibitory effect of whole blood on RPA [39,67].
Figure 5.
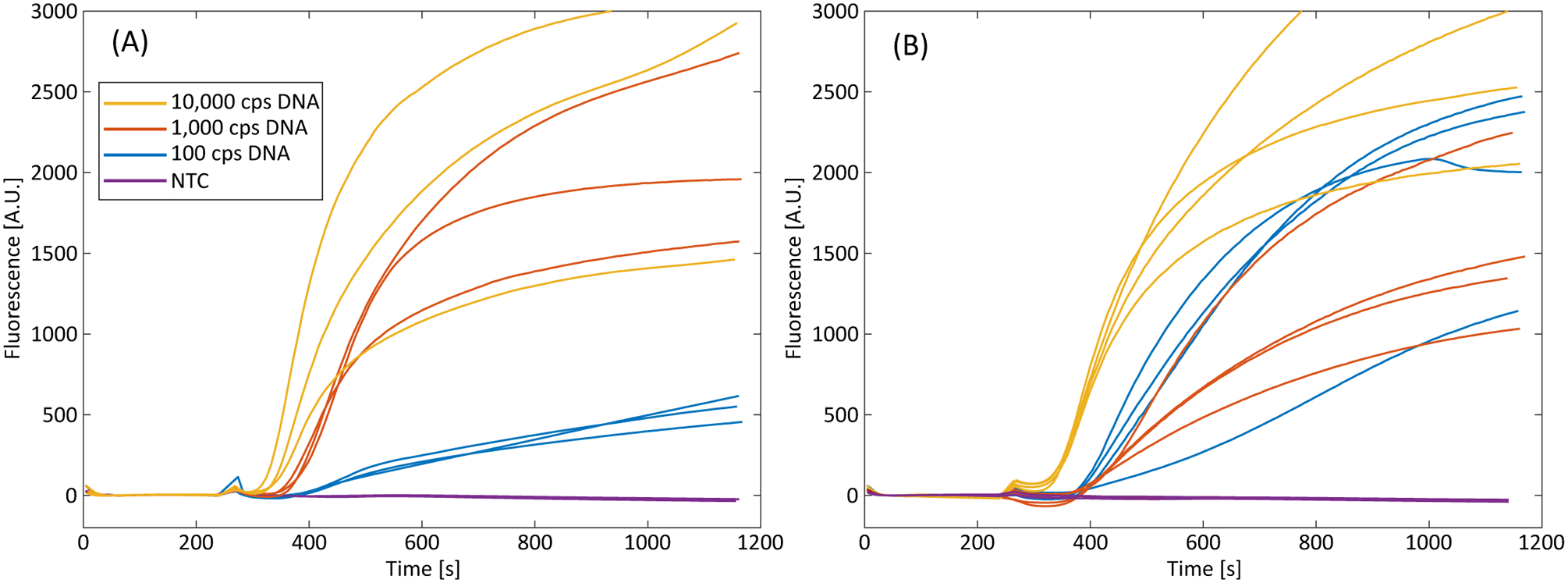
Recombinase polymerase amplification curves using DNA extracted from whole human blood. Input copy numbers of 10,000 cps/trial, 1,000 cps/trial, and 100 cps/trial correspond to input copy concentrations of 3×105 cps/mL, 3×104 cps/mL, and 3×103 copies DNA per mL of input blood, respectively. NTC is a no template control. (A) Amplification curves using spun-out eluate (n=3 for all trials). (B) Amplification curves using the direct strip addition (n=4 for all trials). All trials amplify down to 100 cps/trial (3×103 cps input DNA per mL of whole blood).
The direct addition trials’ amplification slopes, endpoint fluorescence values, and times-to-threshold exhibit more variation than those of the spun-out eluates. We believe this is partially due to the effects the cut-out portion of Fusion 5 may have in the reaction tube, such as decreased mixing, nucleic acid or amplification enzyme loss due to non-specific binding with the membrane, or sensor obstruction of the T16-ISO. Strip effects are particularly apparent when the reaction tubes are removed, agitated, and placed back into the T16-ISO at the four-minute mark. We see large deviations in normalized fluorescence immediately after replacement of the tubes, presumably due to the strip being repositioned into the excitation beam path during agitation and altering the measured fluorescence. This can be seen in several trials (e.g., 1,000 cps DNA in Figure 5B) where the normalized fluorescence is negative for a portion of time immediately after the tubes are returned to the readout machine. In preliminary experiments, we found that larger sections of Fusion 5 strip introduced into the RPA reactions had an even greater effect on fluorescence measurements, resulting in drastically different initial fluorescence values. We currently use a 5 mm × 3 mm section of Fusion 5 to introduce the ITP plug and target DNA into the RPA reaction, but a smaller strip area may mitigate strip-related interference. Other solutions may include removing the strip prior to amplification (i.e. dipping the strip into the reaction buffer) or a small insert that holds the strip away from the excitation beam path of the T16-ISO. Other sources of variability (such inconsistent mixing, reagent degradation, inhibitor presence, etc.) may be controlled for by the inclusion of a competitive internal positive control, similar to Rohrman et al. [68] or Gregory et al. [69]
4. CONCLUSIONS
We present a paper-based nucleic acid amplification test sample preparation device and protocol for the isotachophoretic extraction of target nucleic acids from whole human blood in under 30 minutes. A 33 μL volume of whole blood mixed with target nucleic acids is first fractionated and filtered via an integrated plasma separation membrane, achieving 88% plasma extraction efficiency. Our device directly processes whole blood, thereby eliminating preliminary blood fractionation or dilution steps that are typically required for POC NAATs and ITP systems [49]. We process 33 μL of whole blood which is an order of magnitude larger volume than previous microfluidic ITP devices have reported [30,42]. Using large volumes of undiluted initial samples is important in applications in which low detection limits are required, as many devices are volume-limited and dilution significantly decreases the number of total target nucleic acids present in the device. On-paper plasma protein digestion then occurs via proteinase K, reducing the average size of the plasma proteins and allowing for successful isotachophoretic extraction of the target nucleic acids. We use paper-based isotachophoresis to concentrate the target nucleic acids and separate them from amplification inhibitors present. The device presented here minimizes the number of user steps and sources of error by utilizing paper-based ITP buffer reservoirs and the self-filling characteristics of porous membranes, eliminating the need for more complex filling protocols. We then perform off-device RPA detection of the purified target nucleic acids by cutting out the portion of Fusion 5 membrane that contains the ITP plug and adding the membrane section directly to RPA reactions. Alternatively, we dewater the Fusion 5 membrane and add the resulting liquid eluate to RPA reactions. We show successfully amplification of input copy concentrations as low as 3×103 copies of DNA per mL of input blood, corresponding to 100 cps/trial. These concentrations are well within the clinical ranges of various bloodborne infections such as HIV, HCV, and HBV [11,70,71]. While this system and its current limit-of-detection are not relevant to all diseases or conditions (such as HIV+ individuals on successful antiretroviral treatment, who have viral loads of <1,000 cps/mL), there remains larger clinical relevancy for use in POC infectious disease testing. With respect to HIV, there still remains a significant population who are either not on antiretroviral treatment or who have not achieved viral suppressed. Individuals with unsuppressed HIV infections may have viral loads as high as 107 cps/mL [72]. HCV viral loads can range near 105 cps/mL [73,74], while HBV viral loads range from 300 to 106 cps/mL [75]. We use target DNA as a step towards working with hardened targets, such as viral or bacterial infections. In this work, we demonstrate on-paper plasma protein digestion using proteinase K, and we anticipate that this digestion step can also be leveraged for viral lysis, as proteinase K is a potent and well-characterized lytic agent [76]. Future work will focus on incorporating on-paper lysis in our device to enable detection of bloodborne infectious diseases. The device presented here represents a step towards sample preparation methods for NAATs well-suited for POC bloodborne disease testing. While our protocol necessitated the use of a fluorescence microscope and benchtop power supply, alternatives such as visible dyes that focus at the ITP interface and compact power sources are currently being investigated and integrated.
Supplementary Material
HIGHLIGHTS.
A paper-based device is presented for the purification of nucleic acids using isotachophoresis.
Large sample volumes of whole blood are directly used, with integrated on-device cell fractionation.
Paper-based, on-device protein digestion is incorporated and paper-based electrolyte reservoirs simplify buffer loading.
Recombinase polymerase amplification is used to verify extraction and purification.
Successful isotachophoretic extraction and amplification of as few as 3×103 cps DNA per mL is shown.
ACKNOWLEDGEMENTS
We gratefully acknowledge Jared Shadish from the Cole DeForest laboratory at the University of Washington for their assistance in the SDS-PAGE experiments. The experimental work reported in this publication was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Number R01EB022630. Mr. Bender was funded through a National Science Foundation Graduate Research Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Science Foundation.
REFERENCES
- [1].Gubala V, Harris LF, Ricco AJ, Tan MX, Williams DE. Point of Care Diagnostics: Status and Future. Anal Chem 2012;84:487–515. 10.1021/ac2030199. [DOI] [PubMed] [Google Scholar]
- [2].St John A, Price CP. Existing and Emerging Technologies for Point-of-Care Testing. Clin Biochem Rev 2014;35:155–67. [PMC free article] [PubMed] [Google Scholar]
- [3].Drain PK, Hyle EP, Noubary F, Freedberg KA, Wilson D, Bishai W, et al. Evaluating Diagnostic Point-of-Care Tests in Resource-Limited Settings. Lancet Infect Dis 2014;14:239–49. 10.1016/S1473-3099(13)70250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Niemz A, Ferguson TM, Boyle DS. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol 2011;29:240–50. 10.1016/j.tibtech.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Craw P, Balachandran W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab Chip 2012;12:2469–86. 10.1039/C2LC40100B. [DOI] [PubMed] [Google Scholar]
- [6].Calmy A, Ford N, Hirschel B, Reynolds SJ, Lynen L, Goemaere E, et al. HIV Viral Load Monitoring in Resource-Limited Regions: Optional or Necessary? Clin Infect Dis 2007;44:128–34. 10.1086/510073. [DOI] [PubMed] [Google Scholar]
- [7].El Ekiaby M, Lelie N, Allain J-P. Nucleic acid testing (NAT) in high prevalence–low resource settings. Biologicals 2010;38:59–64. 10.1016/j.biologicals.2009.10.015. [DOI] [PubMed] [Google Scholar]
- [8].Stramer SL, Glynn SA, Kleinman SH, Strong DM, Caglioti S, Wright DJ, et al. Detection of HIV-1 and HCV Infections among Antibody-Negative Blood Donors by Nucleic Acid–Amplification Testing. N Engl J Med 2004;351:760–8. 10.1056/NEJMoa040085. [DOI] [PubMed] [Google Scholar]
- [9].Saldanha J, Gerlich W, Lelie N, Dawson P, Heermann K, Heath A. An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang 2001;80:63–71. 10.1046/j.1423-0410.2001.00003.x. [DOI] [PubMed] [Google Scholar]
- [10].Pai M, Flores LL, Pai N, Hubbard A, Riley LW, Colford JM. Diagnostic accuracy of nucleic acid amplification tests for tuberculous meningitis: a systematic review and meta-analysis. Lancet Infect Dis 2003;3:633–43. 10.1016/S1473-3099(03)00772-2. [DOI] [PubMed] [Google Scholar]
- [11].Saag MS, Holodniy M, Kuritzkes DR, O’Brien WA, Coombs R, Poscher ME, et al. HIV viral load markers in clinical practice. Nat Med 1996;2:625–9. 10.1038/nm0696-625. [DOI] [PubMed] [Google Scholar]
- [12].Abe A, Inoue K, Tanaka T, Kato J, Kajiyama N, Kawaguchi R, et al. Quantitation of hepatitis B virus genomic DNA by real-time detection PCR. J Clin Microbiol 1999;37:2899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Caliendo AM, Gilbert DN, Ginocchio CC, Hanson KE, May L, Quinn TC, et al. Better Tests, Better Care: Improved Diagnostics for Infectious Diseases. Clin Infect Dis 2013;57:S139–70. 10.1093/cid/cit578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hadgu A, Dendukuri N, Hilden J. Evaluation of Nucleic Acid Amplification Tests in the Absence of a Perfect Gold-Standard Test: A Review of the Statistical and Epidemiologic Issues. Epidemiology 2005;16:604–12. [DOI] [PubMed] [Google Scholar]
- [15].Anastassova Dineva M, Mahilum-Tapay L, Lee H. Sample preparation: a challenge in the development of point-of-care nucleic acid -based assays for resource-limited settings. Analyst 2007;132:1193–9. 10.1039/B705672A. [DOI] [PubMed] [Google Scholar]
- [16].Niemz A, Boyle DS. Nucleic acid testing for tuberculosis at the point-of-care in high-burden countries. Expert Rev Mol Diagn 2012;12:687–701. 10.1586/erm.12.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee HH, Dineva MA, Chua YL, Ritchie A, Ushiro-Lumb I, Wisniewski CA. Simple amplification-based assay: A nucleic acid-based point-of-care platform for HIV-1 testing. J Infect Dis 2010;201:S65–71. 10.1086/650385. [DOI] [PubMed] [Google Scholar]
- [18].LaBarre P, Hawkins KR, Gerlach J, Wilmoth J, Beddoe A, Singleton J, et al. A Simple, Inexpensive Device for Nucleic Acid Amplification without Electricity—Toward Instrument-Free Molecular Diagnostics in Low-Resource Settings. PLOS ONE 2011;6:e19738. 10.1371/journal.pone.0019738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chin CD, Linder V, Sia SK. Lab-on-a-chip devices for global health: Past studies and future opportunities. Lab Chip 2007;7:41–57. 10.1039/B611455E. [DOI] [PubMed] [Google Scholar]
- [20].Mariella R. Sample preparation: the weak link in microfluidics-based biodetection. Biomed Microdevices 2008;10:777–84. 10.1007/s10544-008-9190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol 1990;28:495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Katevatis C, Fan A, Klapperich CM. Low concentration DNA extraction and recovery using a silica solid phase. PLOS ONE 2017;12:e0176848. 10.1371/journal.pone.0176848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Abel G Current status and future prospects of point-of-care testing around the globe. Expert Rev Mol Diagn 2015;15:853–5. 10.1586/14737159.2015.1060126. [DOI] [PubMed] [Google Scholar]
- [24].McElgunn CJ, Pereira CR, Parham NJ, Smythe JE, Wigglesworth MJ, Smielewska A, et al. A Low Complexity Rapid Molecular Method for Detection of Clostridium difficile in Stool. PLOS ONE 2014;9:e83808. 10.1371/journal.pone.0083808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chan K, Weaver SC, Wong P-Y, Lie S, Wang E, Guerbois M, et al. Rapid, Affordable and Portable Medium-Throughput Molecular Device for Zika Virus. Sci Rep 2016;6:38223. 10.1038/srep38223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Trébucq A, Enarson DA, Chiang CY, Van Deun A, Harries AD, Boillot F, et al. Xpert® MTB/RIF for national tuberculosis programmes in low-income countries: when, where and how? 2011. [DOI] [PubMed]
- [27].Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, Feldman SH, et al. A fully integrated microfluidic genetic analysis system with sample-in–answer-out capability. Proc Natl Acad Sci U S A 2006;103:19272–7. 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen D, Mauk M, Qiu X, Liu C, Kim J, Ramprasad S, et al. An integrated, self-contained microfluidic cassette for isolation, amplification, and detection of nucleic acids. Biomed Microdevices 2010;12:705–19. 10.1007/s10544-010-9423-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rogacs A, Qu Y, Santiago JG. Bacterial RNA Extraction and Purification from Whole Human Blood Using Isotachophoresis. Anal Chem 2012;84:5858–63. 10.1021/ac301021d. [DOI] [PubMed] [Google Scholar]
- [30].Persat A, Marshall LA, Santiago JG. Purification of Nucleic Acids from Whole Blood Using Isotachophoresis. Anal Chem 2009;81:9507–11. 10.1021/ac901965v. [DOI] [PubMed] [Google Scholar]
- [31].Bender AT, Borysiak MD, Levenson AM, Lillis L, Boyle DS, Posner JD. Semiquantitative Nucleic Acid Test with Simultaneous Isotachophoretic Extraction and Amplification. Anal Chem 2018. 10.1021/acs.analchem.8b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kondratova V, Serd’uk O, Shelepov V, Lichtenstein A. Concentration and isolation of DNA from biological fluids by agarose gel isotachophoresis. BioTechniques 2005;39:695–9. 10.2144/000112020. [DOI] [PubMed] [Google Scholar]
- [33].Kondratova VN, Botezatu IV, Shelepov VP, Lichtenstein AV. Isotachophoresis of nucleic acids in agarose gel rods. Biochem Mosc 2009;74:1285–8. 10.1134/S0006297909110169. [DOI] [PubMed] [Google Scholar]
- [34].Kendall J, Crittenden ED. The Separation of Isotopes. Proc Natl Acad Sci U S A 1923;9:75–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bercovici M, Kaigala GV, Mach KE, Han CM, Liao JC, Santiago JG. Rapid detection of urinary tract infections using isotachophoresis and molecular beacons. Anal Chem 2011;83:4110–7. 10.1021/ac200253x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Borysiak MD, Kimura KW, Posner JD. NAIL: Nucleic Acid detection using Isotachophoresis and Loop-mediated isothermal amplification. Lab Chip 2015;15:1697–707. 10.1039/c4lc01479k. [DOI] [PubMed] [Google Scholar]
- [37].Moghadam BY, Connelly KT, Posner JD. Isotachophoretic preconcenetration on paper-based microfluidic devices. Anal Chem 2014;86:5829–37. 10.1021/ac500780w. [DOI] [PubMed] [Google Scholar]
- [38].Li X, Luo L, Crooks RM. Low-voltage paper isotachophoresis device for DNA focusing. Lab Chip 2015;15:4090–8. 10.1039/C5LC00875A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Eid CG Santiago J. Assay for Listeria monocytogenes cells in whole blood using isotachophoresis and recombinase polymerase amplification. Analyst 2017;142:48–54. 10.1039/C6AN02119K. [DOI] [PubMed] [Google Scholar]
- [40].Marshall LA, Wu LL, Babikian S, Bachman M, Santiago JG. Integrated Printed Circuit Board Device for Cell Lysis and Nucleic Acid Extraction. Anal Chem 2012;84:9640–5. 10.1021/ac302622v. [DOI] [PubMed] [Google Scholar]
- [41].van Kooten XF, Truman-Rosentsvit M, Kaigala GV, Bercovici M. Focusing analytes from 50 μL into 500 pL: On-chip focusing from large sample volumes using isotachophoresis. Sci Rep 2017;7. 10.1038/s41598-017-10579-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Marshall LA, Rogacs A, Meinhart CD, Santiago JG. An injection molded microchip for nucleic acid purification from 25 microliter samples using isotachophoresis. J Chromatogr A 2014;1331:139–142. [DOI] [PubMed] [Google Scholar]
- [43].Taglia V, Lederer M. Isotachophoresis on paper Part I. Investigation of general conditions and separation of some inorganic anions. J Chromatogr A 1973;77:467–71. 10.1016/S0021-9673(00)92224-6. [DOI] [Google Scholar]
- [44].Abelev G, Karamova E. Counterflow immunoisotachophoresis on cellulose acetate membranes. Anal Biochem 1984;142:437–444. [DOI] [PubMed] [Google Scholar]
- [45].Schaumburg F, Kler PA, Carrell CS, Berli CLA, Henry CS. USB powered microfluidic paper-based analytical devices. Electrophoresis 2019. 10.1002/elps.201900273. [DOI] [PubMed] [Google Scholar]
- [46].Rosenfeld T, Bercovici M. Amplification-free detection of DNA in a paper-based microfluidic device using electroosmotically balanced isotachophoresis. Lab Chip 2018;18:861–8. 10.1039/C7LC01250K. [DOI] [PubMed] [Google Scholar]
- [47].Moghadam BY, Connelly KT, Posner JD. Two Orders of Magnitude Improvement in Detection Limit of Lateral Flow Assays Using Isotachophoresis. Anal Chem 2015;87:1009–17. 10.1021/ac504552r. [DOI] [PubMed] [Google Scholar]
- [48].Rogacs A, Marshall LA, Santiago JG. Purification of nucleic acids using isotachophoresis. J Chromatogr A 2014;1335:105–20. 10.1016/j.chroma.2013.12.027. [DOI] [PubMed] [Google Scholar]
- [49].Drain PK, Dorward J, Bender A, Lillis L, Marinucci F, Sacks J, et al. Point-of-Care HIV Viral Load Testing: an Essential Tool for a Sustainable Global HIV/AIDS Response. Clin Microbiol Rev 2019;32:e00097–18. 10.1128/CMR.00097-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ebeling W, Hennrich N, Klockow M, Metz H, Orth HD, Lang H. Proteinase K from Tritirachium album Limber. Eur J Biochem 1974;47:91–7. 10.1111/j.1432-1033.1974.tb03671.x. [DOI] [PubMed] [Google Scholar]
- [51].Bercovici M, Lele SK, Santiago JG. Open source simulation tool for electrophoretic stacking, focusing, and separation. J Chromatogr A 2009;1216:1008–18. 10.1016/j.chroma.2008.12.022. [DOI] [PubMed] [Google Scholar]
- [52].Boyle DS, Lehman DA, Lillis L, Peterson D, Singhal M, Armes N, et al. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. MBio 2013;4. 10.1128/mBio.00135-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Harboe M A Method for Determination of Hemoglobin in Plasma by Near-Ultraviolet Spectrophotometry. Scand J Clin Lab Invest 1959;11:66–70. 10.3109/00365515909060410. [DOI] [PubMed] [Google Scholar]
- [54].Han V, Serrano K, Devine DV. A comparative study of common techniques used to measure haemolysis in stored red cell concentrates. Vox Sang 2010;98:116–23. 10.1111/j.1423-0410.2009.01249.x. [DOI] [PubMed] [Google Scholar]
- [55].Al-Soud WA, Rådström P. Purification and Characterization of PCR-Inhibitory Components in Blood Cells. J Clin Microbiol 2001;39:485–93. 10.1128/JCM.39.2.485-493.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liu C, Liao S-C, Song J, Mauk MG, Li X, Wu G, et al. A high-efficiency superhydrophobic plasma separator. Lab Chip 2016;16:553–60. 10.1039/C5LC01235J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Im SB, Kim SC, Shim JS. A smart pipette for equipment-free separation and delivery of plasma for on-site whole blood analysis. Anal Bioanal Chem 2016;408:1391–7. 10.1007/s00216-015-9259-0. [DOI] [PubMed] [Google Scholar]
- [58].Liu C, Mauk M, Gross R, Bushman FD, Edelstein PH, Collman RG, et al. Membrane-based, sedimentation-assisted plasma separator for point-of-care applications. Anal Chem 2013;85:10463–70. 10.1021/ac402459h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hin S, Loskyll M, Klein V, Keller M, Strohmeier O, von Stetten F, et al. Membrane-based sample inlet for centrifugal microfluidic cartridges. Microelectron Eng 2018;187–188:78–83. 10.1016/j.mee.2017.12.006. [DOI] [Google Scholar]
- [60].Homsy A, van der Wal PD, Doll W, Schaller R, Korsatko S, Ratzer M, et al. Development and validation of a low cost blood filtration element separating plasma from undiluted whole blood. Biomicrofluidics 2012;6:012804–012804–9. 10.1063/1.3672188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lu Z, Rey E, Vemulapati S, Srinivasan B, Mehta S, Erickson D. High-yield paper-based quantitative blood separation system. Lab Chip 2018;18:3865–71. 10.1039/C8LC00717A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fairbanks VF, Ziesmer SC, O’brien PC. Methods for measuring plasma hemoglobin in micromolar concentration compared. Clin Chem 1992;38:132–40. [PubMed] [Google Scholar]
- [63].Marshall LA, Han CM, Santiago JG. Extraction of DNA from Malaria-Infected Erythrocytes Using Isotachophoresis. Anal Chem 2011;83:9715–8. 10.1021/ac202567j. [DOI] [PubMed] [Google Scholar]
- [64].Qu Y, Marshall LA, Santiago JG. Simultaneous Purification and Fractionation of Nucleic Acids and Proteins from Complex Samples Using Bidirectional Isotachophoresis. Anal Chem 2014;86:7264–8. 10.1021/ac501299a. [DOI] [PubMed] [Google Scholar]
- [65].Bajorath J, Saenger W, Pal GP. Autolysis and inhibition of proteinase K, a subtilisin-related serine proteinase isolated from the fungus Tritirachium album Limber. Biochim Biophys Acta 1988;954:176–82. 10.1016/0167-4838(88)90069-6. [DOI] [PubMed] [Google Scholar]
- [66].Rogacs A, Marshall LA, Santiago JG. Purification of nucleic acids using isotachophoresis. J Chromatogr A 2014;1335:105–20. 10.1016/j.chroma.2013.12.027. [DOI] [PubMed] [Google Scholar]
- [67].Kersting S, Rausch V, Bier FF, von Nickisch-Rosenegk M. Rapid detection of Plasmodium falciparum with isothermal recombinase polymerase amplification and lateral flow analysis. Malar J 2014;13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Crannell ZA, Rohrman B, Richards-Kortum R. Quantification of HIV-1 DNA Using Real-Time Recombinase Polymerase Amplification. Anal Chem 2014;86:5615–9. 10.1021/ac5011298. [DOI] [PubMed] [Google Scholar]
- [69].Gregory JB, Litaker RW, Noble RT. Rapid One-Step Quantitative Reverse Transcriptase PCR Assay with Competitive Internal Positive Control for Detection of Enteroviruses in Environmental Samples. Appl Environ Microbiol 2006;72:3960–7. 10.1128/AEM.02291-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hsu C-S, Liu C-J, Liu C-H, Wang C-C, Chen C-L, Lai M-Y, et al. High hepatitis C viral load is associated with insulin resistance in patients with chronic hepatitis C. Liver Int 2008;28:271–7. 10.1111/j.1478-3231.2007.01626.x. [DOI] [PubMed] [Google Scholar]
- [71].Iloeje UH, Yang H-I, Su J, Jen C-L, You S-L, Chen C-J, et al. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006;130:678–86. 10.1053/j.gastro.2005.11.016. [DOI] [PubMed] [Google Scholar]
- [72].Pilcher CD, Joaki G, Hoffman IF, Martinson FE, Mapanje C, Stewart PW, et al. Amplified transmission of HIV-1: comparison of HIV-1 concentrations in semen and blood during acute and chronic infection. AIDS 2007;21:1723–1730. 10.1097/QAD.0b013e3281532c82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Martinot-Peignoux M, Le Breton V, Fritsch S, Le guludec G, Labouret N, Keller F, et al. Assessment of Viral Loads in Patients with Chronic Hepatitis C with AMPLICOR HCV MONITOR Version 1.0, COBAS HCV MONITOR Version 2.0, and QUANTIPLEX HCV RNA Version 2.0 Assays. J Clin Microbiol 2000;38:2722–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hadinedoushan H, Salmanroghani H, Amirbaigy MK, Akhondi-Meybodi M. Hepatitis C Virus Genotypes and Association With Viral Load in Yazd, Central Province of Iran. Hepat Mon 2014;14. 10.5812/hepatmon.11705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Iloeje UH, Yang H-I, Su J, Jen C-L, You S-L, Chen C-J, et al. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006;130:678–86. 10.1053/j.gastro.2005.11.016. [DOI] [PubMed] [Google Scholar]
- [76].Burrell MM, editor. Enzymes of molecular biology. Totowa, N.J: Humana Press; 1993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.