

The superfamily of aldo-keto reductases (AKRs) is composed of over 190 members and can be classified into 16 different families (visit www.med.upenn.edu/akr). AKR1C3 (C3 subtype of aldosterone reductase family 1) refers to the first AKR in family 1, subfamily C, and is encoded by the AKR1C3 gene. AKR1C3 was first cloned and expressed from a human prostate cDNA library. This protein is a soluble monomeric NADP(H) (nicotinamide-adenine dinucleotide phosphate or reduced form of nicotinamide adenine dinucleotide phosphate) dependent oxidoreductase. The AKR proteins exhibit at least 40% sequence identity at the amino acid level and have an (α/β)8-barrel structure within their structure. Especially, AKR1C3 has 86% sequence identity with AKR1C1, AKR1C2, and AKR1C4 [Supplementary Figure 1]. AKR1C3 is known to be involved in the metabolism and biosynthesis of estrogen, androgen, progesterone, and prostaglandin, etc.[1]
In a previous study, Wang et al[2] demonstrated that AKR1C2 and AKR1C3 mediated the transformation of prostaglandin D2 (PGD2) to prostaglandin F2 (PGDF2), and then enhanced the proliferation of prostate cells via the activation of G-protein-coupled receptors for prostaglandin F2α (PGF2α) and phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt) signaling pathway. The overexpression of AKR1C3 is known to clear reactive oxygen species (ROS) and facilitate the accumulation of PGF2α. This not only resulted in the proliferation of prostate cancer (PCa) cells but also facilitated the resistance of PCa cells to radiation by activating the mitogen-activated protein kinase (MAPK) signaling pathway [resulting in the up-regulation of p-MEK (phosphorylated mitogen-activated protein/extracellular signal-regulated kinase) and p-ERK (extracellular signal-regulated kinase) 1/2] and by reducing the expression of peroxisome proliferator-activated receptor γ (PPARγ).[3] Moreover, the ETS-related gene (ERG) transcription factor is known to regulate the expression of AKR1C3 in PCa cells by directly combining with the AKR1C3 gene. ERG can promote cell migration and invasion, de-differentiation, epithelial-to-mesenchymal transition (EMT), and androgen receptor signal transduction. A recent study also found that the nuclear receptor, estrogen-related receptor alpha (ERRα) can regulate the expression of AKR1C3 and that both ERRα and ERG can synergistically regulate each other at the transcriptional level to promote the advanced growth of PCa.[4]
As a critical androgen synthase, AKR1C3 promotes the biosynthesis of androgens and the activation of androgen receptors in PCa. Wang et al[5] discovered that the AKR1C3 gene is overexpressed in most aggressive PCa cell lines. AKR1C3 is known to induce an EMT phenotype in PCa cells both, in vitro and in vivo, by activating extracellular-regulated protein kinases (ERK) which then up-regulates various transcription factors zinc finger box-binding homeobox 1 (ZEB1), Twist family BHLHT transcription factor 1 (Twist1), and Slug. Some studies reported that a single-nucleotide polymorphism (SNP) in the AKR1C3 is responsible for the deterioration of PCa. The polymorphism of AKR1C3 has also been associated with serum testosterone levels during androgen deprivation therapy (ADT) and may represent a prognostic factor for the progression to castration-resistant PCa in Japanese men with metastatic PCa.[6]
An increase in the levels of AKR1C3 can contribute to the transformation of PGD2 to 11b-PGF2α; this facilitates the activation of proliferative transcription factors such as nuclear factor kappa-B (NF-κB) complex.[3] Studies have also illustrated that the overexpression of AKR1C3 can boost the survival and angiogenesis of PC-3 (a form of human PCa cell line). These results also indicated that AKR1C3-mediated tumor angiogenesis is regulated by androgen and estrogen metabolism. Subsequently, the potent combination of androgen and estrogen activates the insulin-like growth factor-1 (IGF-1) and AKT signal pathway followed by high vascular endothelial growth factor (VEGF) expression in PCa cells.[7]
Breast cancer (BRC) patients who exhibit the overexpression of AKR1C3 have a worse prognosis than those with lower expression levels. AKR1C3 can increase the ratio of 17 β-estradiol to progesterone in breast tissue. Furthermore, the formation of PGF2α epimers has been shown to activate PGF receptors and deprive PPARγ of its putative anti-proliferative prostaglandin J2 (PGJ2) ligands.[8] In another study, Yoda et al.[1] reported that 11β-PGF2a, produced by the catalysis of AKR1C3, phosphorylates ERK and cAMP response element-binding protein (CREB) and then induces the overexpression of Slug in BRC cells via the PGF2α receptor. Therefore, AKR1C3 reduces the sensitivity of BRC cells to chemotherapeutic drugs. These findings confirmed that the stimulation of 11 β-PGF2 α has a powerful effect on Slug related to EMT. Zhong et al[9] demonstrated that when AKR1C3 was overexpressed, the tumor suppressor phosphatase and tensin homolog deleted on chromosome ten (PTEN) was lost, thus leading to a remarkable increase in activated AKT.
In endometrial cancer (EC), AKR1C3 is considered one of the vital key enzymes of estrogen concentration. The actions of estrogen and progestin are regulated at the receptor level via the expression of estrogen and progesterone receptors, as well as at the pre-receptor level, by the interconversion of active hormones with their inactive counterparts. The expression of AKR1C1 and AKR1C3 in cases of EC determines the ratio of pregnendione (P) to estradiol (E2), thus influencing the progression of endometrial carcinoma.[10] Furthermore, Li and Narahara[11] demonstrated that a range of EC cell lines was all sensitive to the growth-inhibitory effect of 15-deoxy-Δ 12, 14-PGJ2, known to be the ligand for PPARγ. Besides, 15d-PGJ2 significantly up-regulated the expression of AKR1C3 protein in three EC cell lines and that the cell cycle of EC was arrested in the G2 phase.
With regards to the relationship between AKR1C3 and urinary bladder carcinoma (UBC), Figueroa et al[12] reported a strong association between the risk of UBC and variations in genes that were involved in the metabolism of polycyclic aromatic hydrocarbons (PAHs) and aromatic amines (AAs). These authors analyzed 65 SNPs in 15 candidate genes that are known to be activated by tobacco carcinogens and regulate the transcription of metabolic genes or code for products that can activate or detoxify PAH or AA. Results showed that genetic variation involved in genes that participate in the metabolism of carcinogens, especially AKR1C3, could be responsible for the high risk of UBC. In a previous study, Tiryakioglu et al[13] reported a strong association between AKR1C3 variants and the risk of UBC; the homozygous variant genotype of rs12529 was negatively correlated with UBC, while rs1937920 was positively correlated with an increased risk of UBC.
AKR1C3 plays an important role in regulating the proliferation, differentiation, and apoptosis of myeloid cells. Studies of acute myeloid leukemia (AML) have illustrated that the enforced overexpression of AKR1C3 suppressed the ability of AML cells to differentiate when induced by all-trans retinoic acid (ATRA). In contrast, the down-regulation of AKR1C3 in AML cells is known to mediate the differentiation. Studies by Verma et al[14] showed that a combination therapy, featuring an AKR1C3 inhibitor, along with either etoposide or daunorubicin, elicited an effective adjuvant effect, in an AML cell line, thus potentiating the cytotoxicity of etoposide and daunorubicin by up to 6.25-fold and over 10-fold, respectively. More recently, Verma et al developed AKR1C3 inhibitors by modifying a range of natural products. These inhibitors caused more than a 100-fold reduction in dose index, thus causing the complete re-sensitization of a daunorubicin-resistant AML cell line to a chemotherapeutic agent, and over a 100-fold dose reduction of the dose of cytarabine is not only AML cell lines but also primary T-acute lymphoblastic leukemia (T-ALL) cells.[15] However, at least in leukemia, the inhibition of AKR1C3 alone is not enough to exert anti-leukemia effects. Because the leukemic properties of AML cells are consolidated by the combined activity of AKR1C1–AKR1C4 [Supplementary Table 1 and Figure 1].
Figure 1.
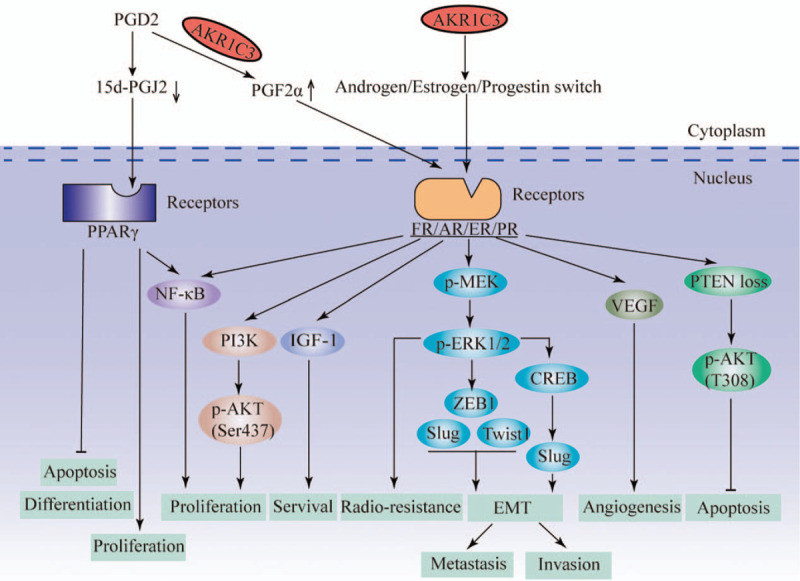
Overview of the major pathways for AKR1C3's action on tumor cells. AKR1C3 promotes the proliferation, survival, radio-resistance, EMT, metastasis, invasion, and angiogenesis and inhibits the apoptosis, differentiation of tumor cells through the above signal pathway. 5d-PGJ2: 15-d prostaglandin J2; AKR1C3: C3 subtype of aldosterone reductase family 1; AKT: Protein kinase B; AR: Androgen receptor; CREB: cAMP-response element-binding protein; EMT: Epithelial-to-mesenchymal transition; ER: Estrogen receptor; FR: Prostaglandin receptor; IGF-1: Insulin-like growth factor-1; NF-κB: Nuclear factor kappa-B; p-AKT: Phosphorylated protein kinase B; PCD2: Prostaglandin D2; p-ERK1/2: Phosphorylated extracellular-regulated protein kinases 1/2; PGF2α: Prostaglandin F2α; PI3K: Phosphatidylinositol 3 kinase; p-MEK: Phosphorylated mitogen-activated protein kinase kinase; PPARγ: Peroxisome proliferator-activated receptor γ; PR: Progesterone receptor; PTEN: Phosphatase and tensin homolog deleted on chromosome ten; Twist1: Twist family BHLHT transcription factor 1; VEGF: Vascular endothelial growth factor; ZEB1: Zinc finger box-binding homeobox 1.
In summary, an increasing body of evidence supports the fact that AKR1C3 plays a key role in malignancies. The up- or down-regulation of AKR1C3 expression occurs in both hormone-dependent and hormone-independent tumors. The former type of tumor includes PCa, bladder cancer, BRC, and EC, while the latter form includes AML, gastric cancer, esophageal cancer, lung cancer, and brain tumors. The mechanism underlying how AKR1C3 acts on malignant tumors is related to the diversity of this enzyme's characteristics; AKR1C3 is known to play roles in a range of signal pathways, including the PI3K/Akt, MAPK, ERK, NF-κB, IGF-1/AKT, PTEN/AKT, and ERK/CREB signaling pathways. However, there are still many unanswered questions, especially in hormone-independent tumors. In future research, AKR1C3 can be knockdown or overexpressed in cancer cells. And then investigate the potential influence of AKR1C3 on the biological behavior of tumors, including migration, invasion, proliferation, differentiation, cell morphology, angiogenesis, and lymphatics. Such studies should involve immunohistochemistry, signaling pathways, bioinformatics analysis, omics, clinical research, and so on.
Funding
This research was supported by the Medical Innovation Project of Fujian Province (No. 2019-CX-14), the Joint Funds for the Innovation of Science and Technology, Fujian province (No. 2019Y9083), and the Fujian Medical University Cancer Center and the Key Clinical Specialty Discipline Construction Program of Fujian, China (No. Min Wei Ke Jiao 2012 No. 149).
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: Xiao XZ, Lin LY, Zhuang MK, Zhong CM, Chen FL. Roles of AKR1C3 in malignancy. Chin Med J 2021;134:1052–1054. doi: 10.1097/CM9.0000000000001379
Supplemental digital content is available for this article.
References
- 1.Penning TM. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol Cell Endocrinol 2019; 489:82–91. doi: 10.1016/j.mce.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang S, Yang Q, Fung KM, Lin HK. AKR1C2 and AKR1C3 mediated prostaglandin D2 metabolism augments the PI3K/Akt proliferative signaling pathway in human prostate cancer cells. Mol Cell Endocrinol 2008; 289:60–66. doi: 10.1016/j.mce.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, He S, Chen Y, Liu Y, Feng F, Liu W, et al. Overview of akr1c3: inhibitor achievements and disease insights. J Med Chem 2020; 2020:1–91. doi: 10.1021/acs.jmedchem.9b02138. [DOI] [PubMed] [Google Scholar]
- 4.Xu Z, Ma T, Zhou J, Gao W, Li Y, Yu S, et al. Nuclear receptor ERRα contributes to castration-resistant growth of prostate cancer via its regulation of intratumoral androgen biosynthesis. Theranostics 2020; 10:4201–4216. doi: 10.7150/thno.35589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang B, Gu Y, Hui K, Huang J, Xu S, Wu S, et al. AKR1C3, a crucial androgenic enzyme in prostate cancer, promotes epithelial-mesenchymal transition and metastasis through activating ERK signaling. Urol Oncol 2018; 36:472.e411-472 e420. doi: 10.1016/j.urolonc.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Shiota M, Endo S, Fujimoto N, Tsukaharaa S, Ushijima M, Kashiwagi E, et al. Polymorphisms in androgen metabolism genes with serum testosterone levels and prognosis in androgen-deprivation therapy. Urol Oncol: Semin Orig Invest 2020; 2020:1–8. doi: 10.1016/j.urolonc.2020.06.033. [DOI] [PubMed] [Google Scholar]
- 7.Dozmorov MG, Azzarello JT, Wren JD, Fung KM, Yang Q, Davis JS, et al. Elevated AKR1C3 expression promotes prostate cancer cell survival and prostate cell-mediated endothelial cell tube formation: implications for prostate cancer progression. BMC Cancer 2010; 10:672.doi: 10.1186/1471-2407-10-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oduwole OO, Li Y, Isomaa VV, Mantyniemi A, Pulkka AE, Soini Y, et al. 17beta-hydroxysteroid dehydrogenase type 1 is an independent prognostic marker in breast cancer. Cancer Res 2004; 64:7604–7609. doi: 10.1158/0008-5472.CAN-04-0446. [DOI] [PubMed] [Google Scholar]
- 9.Zhong T, Xu F, Xu J, Liu L, Chen Y. Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN loss. Biomed Pharmacother 2015; 69:317–325. doi: 10.1016/j.biopha.2014.12.022. [DOI] [PubMed] [Google Scholar]
- 10.Rizner TL, Smuc T, Rupreht R, Sinkovec J, Penning TM. AKR1C1 and AKR1C3 may determine progesterone and estrogen ratios in endometrial cancer. Mol Cell Endocrinol 2006; 248:126–135. doi: 10.1016/j.mce.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Narahara H. 15-deoxy-delta(12,14)-prostaglandin J(2) induces growth inhibition, cell cycle arrest and apoptosis in human endometrial cancer cell lines. Int J Mol Med 2013; 31:778–788. doi: 10.3892/ijmm.2013.1268. [DOI] [PubMed] [Google Scholar]
- 12.Figueroa JD, Malats N, Garcia-Closas M, Real FX, Silverman D, Kogevinas M, et al. Bladder cancer risk and genetic variation in AKR1C3 and other metabolizing genes. Carcinogenesis 2008; 29:1955–1962. doi: 10.1093/carcin/bgn163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiryakioglu NO, Tunali NE. Association of AKR1C3 polymorphisms with bladder cancer. Urol J 2016; 13:2615–2621. doi: 10.22037/uj.v13i2.3384. [PubMed] [Google Scholar]
- 14.Verma K, Zang T, Gupta N, Penning TM, Trippier PC. Selective AKR1C3 inhibitors potentiate chemotherapeutic activity in multiple acute myeloid leukemia (AML) cell lines. ACS Med Chem Lett 2016; 7:774–779. doi: 10.1021/acsmedchemlett.6b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma K, Zang T, Penning TM, Trippier PC. Potent and highly selective aldo-keto reductase 1C3 (AKR1C3) inhibitors act as chemotherapeutic potentiators in acute myeloid leukemia and T-cell acute lymphoblastic leukemia. J Med Chem 2019; 62:3590–3616. doi: 10.1021/acs.jmedchem.9b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.