

Abstract
DNA methylation has become increasingly recognized in the etiology of complex diseases, including thrombotic disorders. Blood is often collected in epidemiological studies for genotyping and has recently also been used to examine DNA methylation in epigenome-wide association studies. DNA methylation patterns are often tissue-specific, thus, peripheral blood may not accurately reflect the methylation pattern in the tissue of relevance. Here, we collected paired liver and blood samples concurrently from 27 individuals undergoing liver surgery. We performed targeted bisulfite sequencing for a set of 35 hemostatic genes primarily expressed in liver to analyze DNA methylation levels of >10,000 cytosine-phosphate-guanine (CpG) dinucleotides. We evaluated whether DNA methylation in blood could serve as a proxy for DNA methylation in liver at individual CpGs. Approximately 30% of CpGs were nonvariable and were predominantly hypo- (<25%) or hypermethylated (>70%) in both tissues. While blood can serve as a proxy for liver at these CpGs, the low variability renders these unlikely to explain phenotypic differences. We therefore focused on CpG sites with variable methylation levels in liver. The level of blood–liver tissue correlation varied widely across these variable CpGs; moderate correlations (0.5 ≤ r < 0.75) were detected for 6% and strong correlations ( r ≥ 0.75) for a further 4%. Our findings indicate that it is essential to study the concordance of DNA methylation between blood and liver at individual CpGs. This paired blood–liver dataset is intended as a resource to aid interpretation of blood-based DNA methylation results.
Keywords: DNA methylation, hemostasis, Factor VII, bisulfite sequencing, epigenetic
Introduction
Procoagulant mechanisms can play a pathophysiological role in arterial and venous thrombosis, as can anticoagulant mechanisms in hemorrhagic disorders. Genome-wide association studies (GWASs) have identified several variants within hemostatic genes that associate with circulating levels of the respective proteins, 1 2 3 4 thrombotic diseases such as coronary artery disease (CAD), 5 ischemic stroke, 6 and venous thrombosis, 7 as well as bleeding disorders. 8 9 In addition to genetic variants, there is increasing evidence for the involvement of epigenetic mechanisms, such as DNA methylation, in many human diseases, 10 and those caused by arterial or venous thrombosis are no exception. 11 12 13 14
Over the last few years, technological advances have made it possible to perform so-called epigenome-wide association studies (EWASs) by the use of bead arrays that allow for genome-wide mapping of DNA methylation at a large number of cytosine-phosphate-guanine (CpG) dinucleotide sites. With regards to arterial thrombotic diseases, EWASs have identified several CpGs that are differentially methylated in cases with myocardial infarction (MI) or ischemic stroke compared with controls. 11 12 13 14 A large EWAS also identified CpGs showing differential methylation in relation to age-independent cardiovascular risk factors. 15 A methylation risk score, created based on these differentially methylated CpGs, was significantly associated with incident cardiovascular events in the Framingham offspring study, and this association was independent of age, sex, and classical vascular risk factors. 15 Another recent EWAS, including nine population-based cohorts from the United States and Europe, found that methylation levels at 52 CpG sites were significantly associated with incident MI or coronary heart disease (CHD). 16 Mendelian randomization analyses supported a causal effect of DNA methylation on incident CHD. 16 The identified differentially methylated CpGs mainly map to genes related to inflammation, calcium regulation, and kidney function. 12 14 16 Thus, in contrast to GWASs of thrombotic and bleeding disorders that have identified genetic variants associated with several hemostatic genes, epigenetic studies have so far not implicated a role for altered DNA methylation levels in hemostatic genes for these traits.
Most EWASs analyze methylation in DNA isolated from peripheral blood, as it is readily available in large epidemiological studies. However, for many diseases, peripheral blood may not accurately reflect the methylation pattern in the tissue of pathophysiological relevance that is less accessible. Several studies have analyzed genome-wide methylation data in different cells and tissues across individuals, and generally found a high overall correlation in methylation levels between different cell lines and tissues. 17 18 19 However, there are also studies showing that the differences in methylation between tissues within one person are much larger than the interindividual differences within a specific tissue, 20 21 indicating that epigenetic signatures are often tissue-specific.
In line with this, results from more recent studies on methylation correlations have shown that it is important to compare methylation levels in paired samples from the same subject as such analyses are less sensitive to confounding due to environmental, clinical, and technical factors. 22 23 So far there have been few studies using within-subject analyses and, as far as we are aware, they have compared DNA methylation signatures in blood versus the brain 22 23 24 or the eye. 25 These studies focused on variable CpG sites, and investigated whether interindividual variation in methylation in peripheral blood could be used as a proxy for interindividual variation in brain or eye methylation. The main results from these studies are that for the majority of variable DNA methylation sites, DNA methylation in peripheral blood cells is not a strong predictor for the variation either in brain or in ocular tissue. 22 23 24 25 Thus, one of the challenges in epigenetic studies is to increase knowledge of when DNA methylation data from peripheral blood cells can be used in the prediction of the methylation state in hard-to-access tissues that are more relevant for the pathology of interest.
We hypothesized that the lack of evidence for an association between methylation patterns in hemostatic genes in epigenetic studies on thrombotic and bleeding traits performed so far may, at least in part, be explained by the fact that methylation patterns in blood do not reflect methylation signatures in the tissue of relevance. Several hemostatic factors are synthesized in the liver, and we recently investigated allele-specific expression and methylation of 35 of these genes. 26 27 Here, we use targeted bisulfite sequencing to allow unbiased detection of all (>10,000) CpGs within these 35 genes. We profiled the per CpG methylation pattern in both liver and blood samples collected from the same subjects at the same point of time. We focused on CpG sites with variable methylation levels in the liver. The aim of the present study was to evaluate whether DNA methylation levels for some of these CpG sites in blood can be used as a proxy for DNA methylation of the same CpG sites in the liver.
Methods
Sample Collection
Liver tissue and venous blood samples were obtained from 27 unrelated individuals (11 women and 16 men) of European descent undergoing liver surgery at the Sahlgrenska University Hospital, Gothenburg, Sweden. The mean age at the time of sample collection was 66 years for women (range: 50–77 years) and 70 years for men (range: 55–83 years). The reason for surgery was secondary malignant tumor of the liver or the intrahepatic bile ducts or primary liver cancer. The liver tissue samples collected for this study were as distant as possible to the tumor or metastasis, and were all noncirrhotic and nontumorous on macroscopic examination by the surgeon. For each participant, blood and liver samples were collected on the same day, no more than 4 hours apart. The liver samples were immediately placed in RNAlater at 4°C and held for 3 to 4 days, then aliquoted and stored at −80°C. The blood samples were anticoagulated with EDTA, aliquoted, and stored at −80°C within 24 hours. This study was approved by the Ethics Committee at the University of Gothenburg (ref. 665–13) and written informed consent was obtained from all subjects prior to participation.
DNA Extraction and Methylation Sequencing
We selected 35 genes with a role in the hemostatic system and with predominant expression in the liver based on data from the Genotype-Tissue Expression (GTEx) project. 28 DNA extraction and the target sequencing design for DNA methylation analysis of these genes have been described in detail elsewhere. 26 27 In brief, genomic DNA (gDNA) was isolated from the liver samples using the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen, Hilden, Germany) and from the blood samples using the QIAamp DNA Blood Midi Kit (Qiagen). DNA sequencing libraries were prepared using the SureSelect custom capture kit (Agilent, Santa Clara, California, United States) targeting the 35 genes, including upstream (5,000 bp) and downstream (500 bp) regions. Prepared libraries were bisulfite-converted with the EZ DNA methylation kit (Zymo Research, Irvine, California, United States) and all blood and liver samples were multiplexed in one lane on the Illumina HiSeq 2500 (Illumina, San Diego, California, United States) with High Output mode and 100 bp paired-end reads. Raw sequence reads were quality-controlled using FastQC (Babraham Bioinformatics, Cambridge, United Kingdom). Low-quality bases were removed and the reads were trimmed with TrimGalore! v. 0.4.1. ( http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ ). The methylation sequence reads were aligned to an in silico bisulfite-converted version of the human reference genome hg19 with Bowtie2 v. 2.2.6. 29 Deduplication and CpG methylation were then called using the Bismark v. 0.14.5 pipeline ( https://www.bioinformatics.babraham.ac.uk/projects/bismark/ ) with default parameters. All genes and regions included in this design are presented in Table 1 . SeqMonk (v. 1.46.0, https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/ ) was used for visualizations.
Table 1. The 35 hemostatic genes included in the targeted methylation sequencing design.
| Gene name | Region included in the capture design | Protein name |
|---|---|---|
| A2M | chr12:9,219,804–9,280,558 | Alpha2-macroglobulin |
| C4BPB | chr1:207,257,584–207,273,837 | Complement component 4 binding protein, β |
| CPB2 | chr13:46,626,822–46,684,211 | Carboxypeptidase B2 (TAFI) |
| F10 | chr13:113,774,613–113,804,343 | Factor X |
| F11 | chr4:187,180,618–187,211,335 | Factor XI |
| F12 | chr5:176,828,639–176,844,077 | Factor XII |
| F13B | chr1:197,007,821–197,041,397 | Factor XIII B |
| F2 | chr11:46,732,043–46,761,556 | Prothrombin (factor II) |
| F5 | chr1:169,480,692–169,560,769 | Factor V |
| F7 | chr13:113,755,105–113,775,495 | Factor VII |
| F9 | chrX:138,607,895–138,646,117 | Factor IX |
| FGA | chr4:155,503,780–155,516,897 | Fibrinogen α chain |
| FGB | chr4:155,479,132–155,494,415 | Fibrinogen β chain |
| FGG | chr4:155,525,228–155,539,402 | Fibrinogen γ chain |
| GGCX | chr2:85,771,478–85,798,657 | Vitamin K-dependent gamma-carboxylase |
| HABP2 | chr10:115,305,590–115,349,860 | Factor VII activating protease (FSAP) |
| HRG | chr3:186,378,798–186,396,523 | Histidine-rich glycoprotein |
| KLKB1 | chr4:187,143,672–187,180,125 | Plasma kallikrein |
| KNG1 | chr3:186,415,098–186,461,178 | Kininogen-1 |
| LMAN1 | chr18:56,994,556–57,031,508 | Lectin, mannose-binding, 1 |
| MCFD2 | chr2:47,128,509–47,154,215 | Multiple coagulation factor deficiency protein 2 |
| PLG | chr6:161,118,225–161,175,585 | Plasminogen |
| PROC | chr2:128,142,996–128,187,322 | Vitamin K-dependent protein C |
| PROS1 | chr3:93,591,381–93,697,934 | Vitamin K-dependent protein S |
| PROZ | chr13:113,805,468–113,827,194 | Vitamin K-dependent protein Z |
| SERPINA1 | chr14:94,842,584–94,862,029 | Alpha1-antitrypsin |
| SERPINA10 | chr14:94,749,150–94,764,608 | Protein Z-related protease inhibitor |
| SERPINA5 | chr14:95,041,731–95,059,957 | Plasma serine protease inhibitor |
| SERPINC1 | chr1:173,872,442–173,891,516 | Antithrombin III |
| SERPIND1 | chr22:21,123,383–21,142,508 | Heparin cofactor 2 |
| SERPINE1 | chr7:100,764,879–100,783,047 | Plasminogen activator inhibitor-1 |
| SERPINF2 | chr17:1,636,130–1,659,059 | Alpha2-antiplasmin |
| SERPING1 | chr11:57,360,027–57,382,826 | Plasma protease C1 inhibitor |
| TFPI | chr2:188,328,458–188,424,219 | Tissue factor pathway inhibitor |
| VKORC1 | chr16:31,101,675–31,111,276 | Vitamin K epoxide reductase complex subunit 1 |
Note: Locations are given according to the human reference genome hg19.
Annotation
Genomic and functional annotation of all CpGs was performed based on Ensembl annotation v. 67 30 for the following categories: (1) upstream regions (defined as +5 to +2 kbp from transcription start site [TSS]), (2) promoters (defined as +2 to −1 kbp from TSS), (3) first exon, (4) all other exons (i.e., excluding first exon), (5) exon/intron boundaries, (6) first intron, (7) all other introns (i.e., excluding first intron), (8) downstream (defined as up to 500 bp downstream of the transcription termination site), (9) CpG islands (CGI), and (10) CGI shores. CpGs were annotated as methylation quantitative trait loci (mQTL) or displaying allele-specific methylation (ASM) based on bisulfite-sequencing studies in the liver 27 or blood. 31
Statistical Analyses
A principle component analysis (PCA) of the methylation data was performed to look for potential confounders and/or deviating samples. Variable CpGs were defined based on DNA methylation levels in liver samples as those with a methylation range differing at least 5% across individuals after exclusion of methylation values above the 90th and below the 10th percentile for each CpG, as previously described. 32 For variable CpGs, methylation levels in liver and blood samples isolated from the same individuals were compared using one-sided Pearson's correlation test. Negligible correlations were defined as r < 0.25, weak as 0.25 ≤ r < 0.5, moderate as 0.5 ≤ r < 0.75, and strong as r ≥ 0.75. To account for multiple testing, we applied a false discovery rate (FDR) of 0.05. Enrichment tests on strongly correlated CpGs were performed using Fisher's exact test to assess whether these CpGs appeared to be overrepresented in specific genomic or functional contexts compared with all variable CpGs. Grouping of strongly correlated CpGs into different correlation patterns was performed by manual inspection of the correlation plots. Enrichment tests on continuous CpGs were performed in comparison with all variable CpGs. All statistical analyses were performed using R software (v. 3.6.1; https://www.r-project.org/ ; The R Project, Vienna, Austria).
Results
Data Quality Control
All blood samples clustered distinctly from liver samples in PCA analyses ( Supplementary Fig. S1 , available in the online version), indicating that tissue type is the main driver of DNA methylation within the dataset, as expected. No confounding covariation was observed in the PCA plots with regard to sex or age of the subjects ( Supplementary Fig. S1A, B , respectively, available in the online version).
A total of 10 to 20 M sequence reads were generated per sample, with an average read depth of approximately 50×. The targeted 35 hemostatic genes in our design contained a total of 19,059 CpGs. For the downstream analyses, we only included CpGs with available high-quality methylation calls in five or more paired liver–blood samples. A total of 10,483 CpG sites met this criterion. For the vast majority, i.e., 83% of these CpGs, methylation calls were present for all of the 27 paired tissue samples. For the remaining samples, 14% had methylation calls in 20 to 26 paired samples, 3% in 10 to 19 paired samples, and 0.1% in 5 to 10 paired samples.
Patterns of Variability in CpG Methylation
As expected, methylation values were bimodally distributed in both blood and liver, with a large number of CpG sites at the extremes ( Fig. 1A, B ). We used standard deviation (SD) to index methylation variability and observed that a relatively large number of CpGs displayed low levels of variability in both blood and liver ( Fig. 1C, D ). Approximately 70% of all CpGs ( n = 7,499) met the present definition of variable (see Methods). The remaining nonvariable CpGs ( n = 2,984) were hypo- or hypermethylated in all samples, or were predominantly hypo- or hypermethylated but with one outlier (data not shown). In total, 777 CpGs were concurrently hypomethylated (mean %methylation ± SD; liver: 1.5 ± 1.2%; blood: 2.1 ± 2.3%; mean difference in methylation between paired samples ± SD: 0.7 ± 1.4%) and 2,206 were concurrently hypermethylated (mean ± SD; liver: 95.8 ± 1.9%; blood: 94.8 ± 2.9%; mean difference in methylation between paired samples ± SD: 1.6 ± 1.8%). While blood can be used as a proxy for liver for these fixed methylated CpGs, the fact that they have low variability between individuals likely limits their utility to describe phenotypic differences in EWASs. Therefore, the remaining analyses will be restricted to variable CpGs. However, for the interested readers, details for the nonvariable CpGs can be found in Supplementary Table S1 (available in the online version).
Fig. 1.
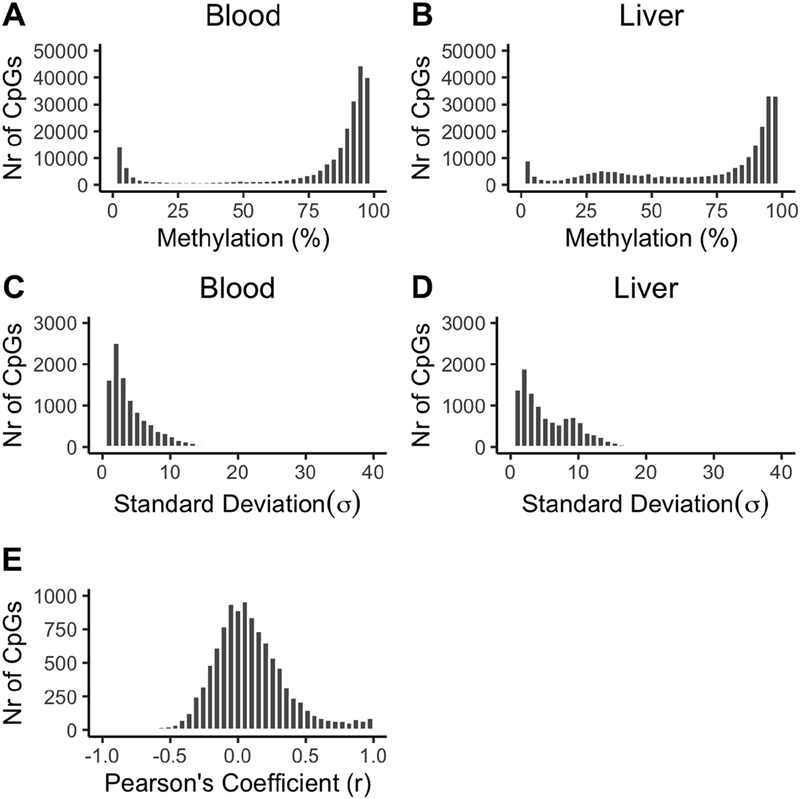
Frequency distribution of the percent methylation, standard deviation, and Pearson's correlation coefficient for the investigated CpGs. ( A, B ) Percent methylation for all 10,483 CpGs in blood and liver, respectively; ( C, D ) standard deviation as an index of methylation variability for all 10,483 CpGs in blood and liver, respectively; ( E ) Pearson's correlation coefficient ( r ) for the 7,499 variable CpGs.
Correlations in CpG Methylation Levels between Liver and Blood
We assessed within-subject correlations in methylation levels between liver and blood for each of the variable CpGs. The majority of the correlation coefficients were close to 0 (mean and median r = 0.11 and 0.07, respectively), but with a clearly visible right skewness. The distribution for these correlation coefficients is displayed in Fig. 1E . According to our definitions in the Methods section, negligible correlations were observed for 4,993 (67%), weak correlations for 1,726 (23%), moderate correlations for 451 (6%), and strong correlations for 329 (4%) of the variable CpGs. A summary of these results is presented in Table 2 , and detailed in Supplementary Table S2 (available in the online version). Included in the supplementary table is information on CpG genomic location (chromosome number and position), gene name, number of paired samples included in the analysis, Pearson's correlation coefficients, r 2 as a measure of the percent variance explained, unadjusted p -value and whether significant at the FDR threshold, mean percent methylation and standard deviation in both blood and liver, mean percent difference in methylation between paired blood–liver samples, previous evidence of genetic association (i.e., mQTL or ASM), and genomic annotation.
Table 2. Summary of the total number of nonvariable CpGs and variable CpGs divided into strength of correlation between liver and blood.
| Type | Nonvariable | Variable | |||
|---|---|---|---|---|---|
| Negligible | Weak | Moderate | Strong | ||
| r < 0.25 | (0.25 ≤ r < 0.5) | (0.5 ≤ r < 0.75) | ( r ≥ 0.75) | ||
| Number of CpGs | 2,984 | 4,993 | 1,726 | 451 | 329 |
| Difference in methylation (median) | 0.7% | 17% | 14% | 8% | 2.4% |
| r (median) | NA | 0.02 | 0.29 | 0.6 | 0.88 |
| r 2 (median) | NA | 0.01 | 0.11 | 0.37 | 0.77 |
| Nominal p < 0.05 | NA | 0 | 458 | 415 | 324 |
| FDR < 0.05 | NA | 0 | 361 | 315 | |
| Association with genetic variant (e.g., mQTL or ASM) |
975 | 1,647 | 560 | 127 | 30 |
Abbreviations: ASM, allele-specific methylation; CpG, cytosine-phosphate-guanine dinucleotide; FDR, false discovery rate; mQTL, methylation quantitative trait loci; NA, not applicable.
Note: Variable CpGs were defined based on DNA methylation levels in liver tissue samples as those with a methylation range differing at least 5% across individuals after exclusion of methylation values above the 90th and below the 10th percentile for each CpG, as previously described. 32 Difference in methylation (%DNA methylation in liver minus that in blood, per paired sample set); r, Pearson's correlation; r 2 , percent variance explained; nominal p -value < 0.05; FDR < 0.05; previous mQTL or ASM based on publically available bisulfite-sequencing studies in blood 31 and liver. 27 Detailed results per CpG are provided for nonvariable CpGs in Supplementary Table S1 and for variable CpGs in Supplementary Table S2 .
All of the 35 genes harbored at least one CpG displaying a strong correlation between liver and blood, with the exception of SERPINC1 . In total, blood was able to explain >80% of the variance in liver DNA methylation at 151 CpGs across 30 genes, and >90% of the variance in liver at 69 CpGs across 24 genes (for details, see Supplementary Table S2 , available in the online version). The largest number of strongly correlated CpGs ( n = 41) was found in the gene encoding factor VII ( F7) . The locations of these CpGs are displayed in a schematic of F7 and F10 together with moderately correlated CpGs and the average percent methylation at each CpG in blood and liver ( Fig. 2 ). Similar schematics for the remaining 33 genes can be found in Supplementary Fig. S2 (available in the online version).
Fig. 2.
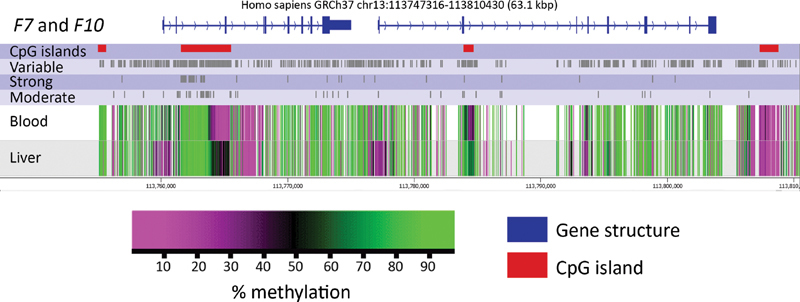
Schematic illustrations of F7 and F10 depicting DNA methylation patterns and location of correlated CpGs in blood and liver tissue. At least 5 kbp upstream and 0.5 kbp downstream of each gene are included: blue indicates the gene structure and red CpG islands. Variable CpGs and those displaying moderate and strong correlations are shown in gray . The average methylation status of the 27 samples for each CpG in blood and liver is also presented; purple represents a low degree of methylation (hypomethylation) and green a high degree of methylation (hypermethylation). All other genes can be found in Supplementary Figure S2 (available in the online version).
Enrichment of Strongly Correlated CpGs in Genomic or Functional Contexts
We next evaluated whether CpGs with a strong correlation between blood and liver were over- or underrepresented in specific genomic or functional contexts. CpG sites with a strong correlation between the two tissues were enriched in introns (excluding the first intron; p < 0.01, Fisher's exact test). Contrarily, in the first intron, CGI shores, and exons (excluding the first exon), the liver–blood correlation appeared at a lesser extent as compared with all variable CpGs ( p < 0.05, Fisher's exact test). No clear over- or underrepresentation was observed in the remaining genomic contexts (promoter, downstream, CGIs, first exon, and upstream; Fig. 3 ).
Fig. 3.
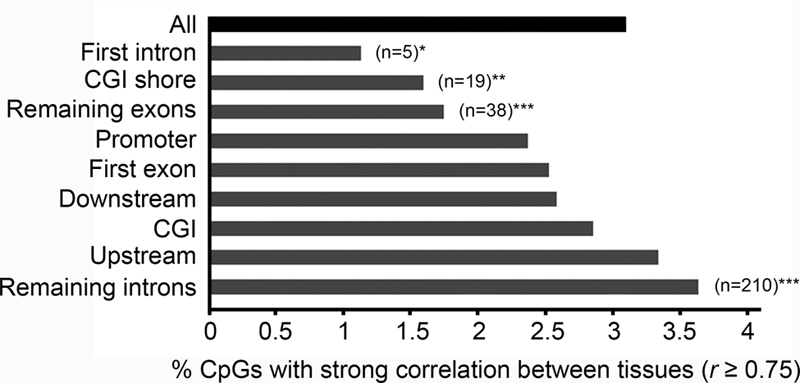
Enrichment analysis for variable CpGs with strong within-individual correlations in methylation levels between liver and blood in different genomic contexts. Data are presented as percent CpGs correlating between tissues. ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ p < 0.001 (Fisher’s exact test).
Correlation Patterns for CpG Methylation between Liver and Blood
We manually inspected a large number of scatter plots for CpGs displaying moderate and strong correlations in methylation levels between liver and blood, and identified two main correlation patterns: (1) continuous (exemplified in Fig. 4A ) and (2) trimodal/bimodal, with methylation levels close to 0, 50, and 100% (i.e., trimodal), or 0 and 50%, or 50 and 100% (i.e., bimodal; exemplified in Fig. 4B–D ). The remaining CpGs were not clearly represented by either of these two main patterns. Many of these were bimodal-like and were predominantly fully methylated or nonmethylated, but with several outliers that affected the correlation coefficient (data not shown). It is of note that some CpGs displayed negative correlations ( n = 59 moderate and 5 strong) and were due to fully hypo- or hypermethylated DNA methylation clusters with a very small mean percent difference in methylation between paired blood and liver samples ( Supplementary Table S2 [available in the online version]; data not shown).
Fig. 4.
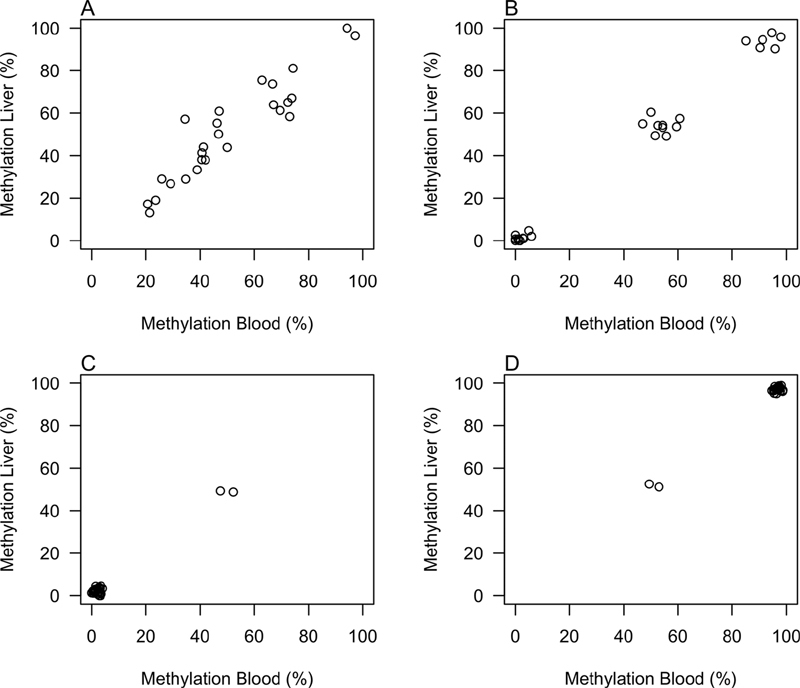
Representative examples of the two main CpG methylation correlation patterns between liver and blood samples; ( A ) continuous correlation, here exemplified by CpG position chr17:1642181 in SERPINF2 , and ( B ) trimodal correlation pattern, exemplified by CpG position chr4:155515417 in FGA ; ( C, D ) bimodal correlation patterns, exemplified by CpG position chr12:9253895 in A2M and chr1:169533986 in F5 , respectively.
CpGs with Evidence of Genetic Regulation
Genetic variation at single nucleotide polymorphisms (SNPs) is a contributor to DNA methylation variability between individuals. Previous studies have demonstrated that there is a significant overlap of mQTLs between tissue types, and for these CpGs DNA methylation in blood may be a reliable correlate of DNA methylation in other more relevant tissues. 33 34 However, there is also a large portion of mQTL-associated CpGs that show tissue-specific effects. 33 34 We therefore next used publically available mQTL and ASM data from a whole genome bisulfite-sequencing study in blood 31 and from our previous ASM study in liver 27 to determine the concordance of these genotype-associated CpGs in blood and liver. We found that 127 CpGs with moderate and 30 CpGs with strong correlations were previously reported to associate with genetic variants in either blood or liver (see Supplementary Table S2 , available in the online version). It is also of note that approximately 30% of CpGs classified as nonvariable in our study were previously associated with genotype. These may represent CpGs associated with low-frequency SNPs for which our sample size may have been too restrictive to capture any genetic diversity and/or for which our definition of variable may have been too constricting.
CpGs Displaying a Continuous Correlation Pattern
CpG sites that are concordant in liver and blood and linked to genotype could, theoretically, be assessed through genotyping methods. Therefore, arguably, the most interesting novel findings entail the CpGs classified as both variable in liver and showing a continuous correlation pattern. In this study, 75 (23%) of the CpGs displaying a strong correlation of methylation levels between liver and blood exhibited both of these properties. An apparent enrichment of these CpGs was observed in CGIs ( p < 0.001, Fisher's exact test).
Of the 35 genes, 18 did not harbor any CpG with a continuous methylation correlation pattern. However, A2M , F10 , F11 , F2 , F5 , HABP2 , KLKB1 , KNG1 , LMAN1 , PLG , PROC , PROS1 , SERPINA5 , and SERPING1 all harbored 1 to 5 CpG sites with these properties, and SERPINF2 and F9 each had 7 and 9 of these sites, respectively ( Table 3 ). The largest number ( n = 24) of CpGs with these properties was found in F7 , and these CpGs were all located in CGIs and/or in the promoter ( Table 3 ).
Table 3. Number and genomic locations of variable CpGs displaying strong and continuous correlation in methylation levels between liver and blood.
| Number of informative CpGs per region | |||||||
|---|---|---|---|---|---|---|---|
| Gene name | Nr CpGs | Upstream | Promoter | Exon | Intron | CGI | CGI shore |
| A2M | 1 | Chr12:9230732 | |||||
| F10 | 4 | Chr13:113776950 | Chr13:113784015 Chr13:113786948 Chr13:113793118 |
Chr13:113784015 | |||
| F11 | 2 | Chr4:187200776 Chr4:187205797 |
|||||
| F2 | 1 | Chr11:46748284 | |||||
| F5 | 3 | Chr1:169504830 Chr1:169514790 Chr1:169515916 |
|||||
| F7 | 24 | Chr13:113761619 Chr13:113761653 Chr13:113761670 Chr13:113761687 Chr13:113761721 Chr13:113761755 Chr13:113761789 Chr13:113761857 Chr13:113761891 Chr13:113761925 Chr13:113761959 Chr13:113762033 |
Chr13:113761619 Chr13:113761653 Chr13:113761670 Chr13:113761687 Chr13:113761721 Chr13:113761755 Chr13:113761789 Chr13:113761857 Chr13:113761891 Chr13:113761925 Chr13:113761959 Chr13:113762033 Chr13:113762509 Chr13:113762554 Chr13:113762577 Chr13:113762623 Chr13:113762657 Chr13:113762708 Chr13:113762844 Chr13:113763310 Chr13:113763372 Chr13:113763406 Chr13:113763474 |
Chr13:113765997 | |||
| F9 | 9 | ChrX:138633272 ChrX:138643737 |
ChrX:138621897 ChrX:138624419 ChrX:138634842 ChrX:138640263 ChrX:138641008 ChrX:138641086 ChrX:138642197 |
||||
| HABP2 | 4 | Chr10:115306262 | Chr10:115313586 Chr10:115318439 Chr10:115343587 |
||||
| KLKB1 | 2 | Chr4:187149540 | Chr4:187172280 | ||||
| KNG1 | 5 | Chr3:186427341 Chr3:186427950 Chr3:186429140 Chr3:186432858 |
Chr3:186440243 | ||||
| LMAN1 | 2 | Chr18:57002439 Chr18:57002464 |
|||||
| PLG | 5 | Chr6:161122707 | Chr6:161134274 | Chr6:161131516 Chr6:161137990 Chr6:161152449 |
|||
| PROC | 2 | Chr2:128146272 Chr2:128169899 |
Chr2:128146272 | ||||
| PROS1 | 1 | Chr3:93685812 | |||||
| SERPINA5 | 1 | Chr14:95045497 | |||||
| SERPINF2 | 7 | Chr17:1642181 Chr17:1643012 Chr17:1643709 Chr17:1643934 Chr17:1644992 |
Chr17:1645152 | Chr17:1648295 | |||
| SERPING1 | 2 | Chr11:57362618 | Chr11:57381989 | ||||
Abbreviations: CGI, CpG island; Nr, number.
Note: “Exon” and “intron” annotations are based only on the second to last exon/intron in the transcript (i.e., omitting the first exon/intron). For the remaining contexts, if one CpG overlaps more than one annotated region, both are presented. Genes are listed alphabetically. For further details for a specific CpG, see online Supplementary Table S2 .
Discussion
Several hemostatic factors are produced in the liver and DNA methylation may contribute to regulation of the expression of these genes. Given the difficulty in obtaining liver tissue samples, it is of great importance to know whether DNA methylation in blood could serve as a proxy for DNA methylation in liver. Therefore, we used liver tissue and blood samples obtained from the same individuals at the same point in time to explore the degree of correlation in DNA methylation levels at individual CpG sites between liver and blood for 35 hemostatic genes. We used a within-subject approach and found that concordance of DNA methylation between blood and liver varied widely at individual CpGs. Approximately 30% of CpGs were nonvariable comprising concurrently hypo- or hypermethylated CpGs. Of the approximately 7,500 variable CpGs, 6% were moderately correlated and 4% were strongly correlated. The DNA methylation level in blood could serve as a proxy for the methylation level in liver at these sites.
To the best of our knowledge, this is the first study analyzing DNA methylation correlations between human liver and blood. A few other studies with similar within-subject design have been performed on other tissues. 22 23 24 25 However, these assessed genome-wide DNA methylation patterns with BeadChip arrays rather than specific genes of interest with targeted sequencing. In one study paired blood and brain samples were obtained from 27 patients with epilepsy undergoing neurosurgery, and significant correlations in methylation levels between brain and blood were detected for less than 2% of the variable CpGs after a Bonferroni correction. 22 Two other smaller studies ( n = 12 and n = 16) used paired blood and brain samples and identified correlation for approximately 10% of the analyzed variable CpGs when applying less stringent correction for multiple testing. 23 34 Lastly, one study compared paired blood and eye tissue collected postmortem from eight donors, and only a small proportion of the CpG sites showed correlation in DNA methylation levels between blood and different ocular tissues. 25 The results from these four studies are thus in line with our present findings, that DNA methylation correlation at individual variable CpGs between liver and blood is generally low.
Among the CpGs displaying strong correlation between liver and blood in the present study, we observed two correlation patterns. The first main pattern was a trimodal/bimodal correlation pattern, with methylation levels close to 0, 50, and 100% (i.e., trimodal), or 0 and 50%, or 50 and 100% (i.e., bimodal). A similar trimodal pattern among correlated CpGs was reported in a study on DNA methylation in blood versus eye 25 and in another study on blood versus postmortem brain tissue, 32 and these authors concluded that this pattern is likely to be mediated by DNA sequence variation. In line with this, 9% of CpGs with strong correlations between blood and liver had previously been demonstrated to be mQTL or ASM-associated CpGs in two bisulfite-sequencing studies in blood 31 and liver. 27 For these CpGs, this supports the use of blood-based studies to explore DNA methylation as a mechanistic link between noncoding SNPs and variations observed in gene expression, circulating protein levels, and disease traits. However, from a biomarker perspective, genetically associated CpGs could potentially be assayed using regular DNA sequencing or genotyping, which is less labor-intensive than bisulfite-conversion sequencing.
The second pattern was a continuous correlation pattern, i.e., methylation levels spanning a wide range. This pattern was observed for 75 CpGs. We observed an enrichment of continuous CpGs in regulatory elements (CGIs, promoters, and upstream regions), whereas the enrichment analysis of all strongly correlated CpGs showed an enrichment in introns. This could indicate that the continuous CpGs are more relevant for gene regulation. However, evidence is now mounting that methylation in introns may also play a role in regulation, 35 and more studies are clearly needed to unravel the regulatory mechanisms of gene body methylation. Furthermore, it should be noted that due to the small number of both variable and continuous CpGs in relation to all CpGs analyzed, the results for the enrichment of this subset should be interpreted with caution.
The gene containing the largest number of CpGs that were both variable in liver and displayed strong and continuous correlations in methylation between the tissues was F7 . These CpGs were located in a CGI, which overlaps the F7 promoter. This gene encodes the vitamin K-dependent serine protease coagulation factor VII (FVII), which is one of the key initiators of the coagulation cascade. Plasma concentrations of FVII have been shown to associate with incident MI and ischemic stroke. 36 37 38 39 Interestingly, F7 promoter methylation levels in peripheral blood mononuclear cells have been linked to plasma concentrations of activated FVII and CAD. 40 Taken together with the findings in our study, it is possible that F7 promoter methylation in the liver contributes to the regulation of plasma concentrations of FVII, and that blood may well be used as a proxy tissue for liver in this particular case.
One important strength of this study is that we used a within-subject approach, i.e., the two tissues were derived from the same individual at the same point in time, meaning that comparisons between tissues were done at the same biological age, within the same sex, and under the same study-specific and environmental circumstances. Furthermore, all 54 samples were processed at the same time and run on the same sequencing lane to minimize technical confounding. Finally, we used targeted bisulfite sequencing as opposed to commercially available methylation arrays, which provides a much higher resolution of individual CpGs. Current arrays cover <4% of CpGs and have poor coverage over the gene bodies of hemostatic genes, meaning that potentially informative CpG sites are currently overlooked in the majority of array-based DNA methylation studies. In fact, of the 10,483 CpGs included in our study, only one variable methylation site with strong correlation between liver and blood is represented. Therefore, as DNA methylation studies, such as EWAS, move away from array-based to whole-genome bisulfite-sequencing-based, this paired dataset will be a valuable resource.
With this being said, we recognize that there are several limitations to this study. First, as both liver tissue and whole blood are complex tissues consisting of heterogeneous cellular populations, our results reflect the mean DNA methylation level at each CpG site. Therefore, we acknowledge that possible confounding due to variability in cell-type composition between individuals is an important limitation of our study. Although we would have liked to adjust for this by using cell-type deconvolution algorithms, we were constrained by the targeted design employed in this study. However, while we recognize that this is a limitation, it is of note that accounting for cell heterogeneity is of much greater importance in EWAS when studying disease states, where changes in cellular composition often strongly associate with the disease phenotype. Second, our target design was limited to 5,000 bp upstream and 500 bp downstream of each gene, which means we could have overlooked CpGs in relevant regulatory elements outside of these regions. However, as described above, it still provides a great enrichment compared with commonly used array-based analyses. Third, the study participants were patients undergoing resection of liver/hepatic bile duct metastasis or primary liver cancer, which may limit the generalizability of the findings. Fourth, our study included only 27 individuals. While large from a within-subject analysis point of view, it is likely that we did not have enough genetic diversity to capture all potential mQTL or ASM-CpGs. Thus it is possible that additional CpG sites may be strongly correlated in a larger study. Finally, we recognize that this study should preferably be replicated in an independent sample for confirmation.
In conclusion, our data show that it is essential to examine the concordance of DNA methylation between blood and liver at individual CpGs when interpreting blood-based EWAS results for the investigated 35 hemostatic genes that are predominantly expressed in liver. Approximately 30% of 10,483 CpGs analyzed in this study were nonvariable and were predominantly hypomethylated or hypermethylated in both tissues. Among the CpGs with variable methylation levels in liver, only 4% displayed a strong correlation to methylation levels in blood and 6% displayed a moderate correlation. This paired blood–liver dataset, derived from high-resolution targeted bisulfite sequencing, is intended as a resource for the thrombosis and hemostasis research community to aid interpretation of blood-based DNA methylation results. These markers may enhance the identification of meaningful epigenetic differences in these 35 hemostatic genes for thrombotic and bleeding disorders.
Acknowledgments
The authors thank the Transplant Centre at the Sahlgrenska University Hospital for technical and administrative assistance in providing the liver and blood specimens. We thank the sequencing service provided by Science for Life Laboratory (SciLifeLab), Stockholm. The computations were performed using resources provided by the Swedish National Infrastructure for Computing (SNIC) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX).
Funding Statement
Funding This study was supported by the Swedish Heart and Lung Foundation (20190203), the Swedish Research Council (2018-02543), the Swedish state under the agreement between the Swedish government and the county councils (the ALF-agreement, ALFGBG-720081), the Bioinformatics Long-term Support (WABI), SciLifeLab (Stockholm and Uppsala, Sweden), the Swedish Foundation for Strategic Research (RIF14–0081), the Rune and Ulla Amlövs Foundation for Neurologic Research, the John and Brit Wennerström Foundation for Neurologic Research, the Marcus Borgströms Foundation for Neurologic Research, and the Nilsson-Ehle Endowments.
Conflict of Interest None declared.
Authors' Contributions
M.O.L., T.M.S., and C.J. conceived the research design of the present study. C.J. provided funding and was responsible for sample contribution. M.O.L. isolated gDNA and prepared sequencing libraries. M.O.L., L.H., and M.D.L. acquisitioned and processed the data. A.A., B.A., and S.N. performed the statistical analyses. M.O.L., A.A., and B.A. drafted the figures. M.O.L., A.A., C.J., and T.M.S. interpreted the data. M.O.L., C.J., and T.M.S. drafted the manuscript. All authors intellectually reviewed the manuscript, contributed to the last revision process, and approved the version to be published.
These authors contributed equally to this work.
These authors jointly supervised this work.
What is known about this topic?
DNA methylation has been recognized in the etiology of several thrombotic diseases.
Epigenetic studies, such as epigenome-wide association studies (EWASs) have not yet implicated a role for altered DNA methylation levels in hemostatic genes for thrombotic disorders.
DNA methylation patterns are often tissue-specific, and most EWASs are performed on DNA isolated from peripheral blood which may not accurately reflect the methylation pattern in the tissue of relevance.
What does this paper add?
Here 35 hemostatic genes with predominant expression in the liver were examined by targeted bisulfite sequencing to evaluate whether DNA methylation levels in blood can be used as a proxy for liver at 10,483 individual CpGs.
Approximately 30% CpGs were nonvariable and were predominantly hypomethylated (<25%) or hypermethylated (>70%) in both tissues. For the remaining variable CpGs, moderate correlations (0.5 ≤ r < 0.75) were detected for 6% and strong correlations ( r ≥ 0.75) for a further 4% of CpGs.
Concordance of DNA methylation between blood and liver at individual CpGs varied widely. This paired blood–liver dataset is intended as a resource for the thrombosis and hemostasis research community to aid interpretation of blood-based DNA methylation results.
Supplementary Material
References
- 1.de Vries P S, Chasman D I, Sabater-Lleal M. A meta-analysis of 120 246 individuals identifies 18 new loci for fibrinogen concentration. Hum Mol Genet. 2016;25(02):358–370. doi: 10.1093/hmg/ddv454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olsson M, Stanne T M, Pedersen A. Genome-wide analysis of genetic determinants of circulating factor VII-activating protease (FSAP) activity. J Thromb Haemost. 2018;16(10):2024–2034. doi: 10.1111/jth.14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellcome Trust Case Control Consortium . Smith N L, Chen M H, Dehghan A. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: the CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121(12):1382–1392. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanne T M, Olsson M, Lorentzen E. A Genome-wide study of common and rare genetic variants associated with circulating thrombin activatable fibrinolysis inhibitor. Thromb Haemost. 2018;118(02):298–308. doi: 10.1160/TH17-04-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikpay M, Goel A, Won H H. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.AFGen Consortium ; Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium ; International Genomics of Blood Pressure (iGEN-BP) Consortium ; INVENT Consortium ; STARNET ; BioBank Japan Cooperative Hospital Group ; COMPASS Consortium ; EPIC-CVD Consortium ; EPIC-InterAct Consortium ; International Stroke Genetics Consortium (ISGC) ; METASTROKE Consortium ; Neurology Working Group of the CHARGE Consortium ; NINDS Stroke Genetics Network (SiGN) ; UK Young Lacunar DNA Study ; MEGASTROKE Consortium . Malik R, Chauhan G, Traylor M. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(04):524–537. doi: 10.1038/s41588-018-0058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.INVENT Consortium . Klarin D, Emdin C A, Natarajan P, Conrad M F, Kathiresan S. Genetic analysis of venous thromboembolism in UK Biobank identifies the ZFPM2 locus and implicates obesity as a causal risk factor. Circ Cardiovasc Genet. 2017;10(02):e001643. doi: 10.1161/CIRCGENETICS.116.001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falcone G J, Woo D. Genetics of spontaneous intracerebral hemorrhage. Stroke. 2017;48(12):3420–3424. doi: 10.1161/STROKEAHA.117.017072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.My Life Our Future programme . Konkle B A, Johnsen J M, Wheeler M, Watson C, Skinner M, Pierce G F. Genotypes, phenotypes and whole genome sequence: approaches from the My Life Our Future haemophilia project. Haemophilia. 2018;24 06:87–94. doi: 10.1111/hae.13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Do C, Shearer A, Suzuki M. Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era. Genome Biol. 2017;18(01):120. doi: 10.1186/s13059-017-1250-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis Armstrong N M, Chen W M, Brewer M S. Epigenome-wide analyses identify two novel associations with recurrent stroke in the Vitamin Intervention for Stroke Prevention clinical trial. Front Genet. 2018;9:358. doi: 10.3389/fgene.2018.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krupinski J, Carrera C, Muiño E. DNA methylation in stroke. Update of latest advances. Comput Struct Biotechnol J. 2017;16:1–5. doi: 10.1016/j.csbj.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakatochi M, Ichihara S, Yamamoto K. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. Clin Epigenetics. 2017;9:54. doi: 10.1186/s13148-017-0353-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rask-Andersen M, Martinsson D, Ahsan M. Epigenome-wide association study reveals differential DNA methylation in individuals with a history of myocardial infarction. Hum Mol Genet. 2016;25(21):4739–4748. doi: 10.1093/hmg/ddw302. [DOI] [PubMed] [Google Scholar]
- 15.Fernández-Sanlés A, Sayols-Baixeras S, Curcio S, Subirana I, Marrugat J, Elosua R. DNA methylation and age-independent cardiovascular risk, an epigenome-wide approach: the REGICOR study (REgistre GIroni del COR) Arterioscler Thromb Vasc Biol. 2018;38(03):645–652. doi: 10.1161/ATVBAHA.117.310340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agha G, Mendelson M M, Ward-Caviness C K. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation. 2019;140(08):645–657. doi: 10.1161/CIRCULATIONAHA.118.039357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byun H M, Siegmund K D, Pan F. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet. 2009;18(24):4808–4817. doi: 10.1093/hmg/ddp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lokk K, Modhukur V, Rajashekar B. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15(04):r54. doi: 10.1186/gb-2014-15-4-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varley K E, Gertz J, Bowling K M. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23(03):555–567. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang R, Jones M J, Chen E. Discordance of DNA methylation variance between two accessible human tissues. Sci Rep. 2015;5:8257. doi: 10.1038/srep08257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slieker R C, Bos S D, Goeman J J. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6(01):26. doi: 10.1186/1756-8935-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun P R, Han S, Hing B. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry. 2019;9(01):47. doi: 10.1038/s41398-019-0376-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walton E, Hass J, Liu J. Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research. Schizophr Bull. 2016;42(02):406–414. doi: 10.1093/schbul/sbv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar R D, Jones M J, Meaney M J, Turecki G, Kobor M S. BECon: a tool for interpreting DNA methylation findings from blood in the context of brain. Transl Psychiatry. 2017;7(08):e1187. doi: 10.1038/tp.2017.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hewitt A W, Januar V, Sexton-Oates A. DNA methylation landscape of ocular tissue relative to matched peripheral blood. Sci Rep. 2017;7:46330. doi: 10.1038/srep46330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olsson Lindvall M, Hansson L, Klasson S, Davila Lopez M, Jern C, Stanne T M. Hemostatic genes exhibit a high degree of allele-specific regulation in liver. Thromb Haemost. 2019;119(07):1072–1083. doi: 10.1055/s-0039-1687879. [DOI] [PubMed] [Google Scholar]
- 27.Olsson Lindvall M, Davila Lopez M, Klasson S. A comprehensive sequencing-based analysis of allelic methylation patterns in hemostatic genes in human liver. Thromb Haemost. 2020;120(02):229–242. doi: 10.1055/s-0039-3401824. [DOI] [PubMed] [Google Scholar]
- 28.GTEx Consortium . The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(06):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg S L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(04):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kersey P J, Allen J E, Armean I.Ensembl Genomes 2016: more genomes, more complexity Nucleic Acids Res 201644(D1):D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung W A, Shao X, Morin A. Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome. Genome Biol. 2017;18(01):50. doi: 10.1186/s13059-017-1173-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10(11):1024–1032. doi: 10.1080/15592294.2015.1100786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith A K, Kilaru V, Kocak M. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics. 2014;15:145. doi: 10.1186/1471-2164-15-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin D, Chen J, Perrone-Bizzozero N. Characterization of cross-tissue genetic-epigenetic effects and their patterns in schizophrenia. Genome Med. 2018;10(01):13. doi: 10.1186/s13073-018-0519-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jjingo D, Conley A B, Yi S V, Lunyak V V, Jordan I K. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3(04):462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Folsom A R, Wu K K, Rosamond W D, Sharrett A R, Chambless L E. Prospective study of hemostatic factors and incidence of coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 1997;96(04):1102–1108. doi: 10.1161/01.cir.96.4.1102. [DOI] [PubMed] [Google Scholar]
- 37.Meade T W, Mellows S, Brozovic M.Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study Lancet 19862(8506):533–537. [DOI] [PubMed] [Google Scholar]
- 38.Tracy R P, Arnold A M, Ettinger W, Fried L, Meilahn E, Savage P. The relationship of fibrinogen and factors VII and VIII to incident cardiovascular disease and death in the elderly: results from the cardiovascular health study. Arterioscler Thromb Vasc Biol. 1999;19(07):1776–1783. doi: 10.1161/01.atv.19.7.1776. [DOI] [PubMed] [Google Scholar]
- 39.Zakai N A, Lange L, Longstreth W T., Jr Association of coagulation-related and inflammation-related genes and factor VIIc levels with stroke: the Cardiovascular Health Study. J Thromb Haemost. 2011;9(02):267–274. doi: 10.1111/j.1538-7836.2010.04149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friso S, Lotto V, Choi S W. Promoter methylation in coagulation F7 gene influences plasma FVII concentrations and relates to coronary artery disease. J Med Genet. 2012;49(03):192–199. doi: 10.1136/jmedgenet-2011-100195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.