

Abstract
Big data abound in microbiology, but the workflows designed to enable researchers to interpret data can constrain the biological questions that can be asked. Five years after anvi’o was first published, this community-led multi-omics platform is maturing into an open software ecosystem that reduces constraints in ‘omics data analyses.
Generating hundreds of millions of sequences from a microbial habitat is now commonplace for many microbiologists1. While the massive data streams offer detailed snapshots of the lifestyles of microorganisms, this data revolution in microbiology means that a new generation of computational tools is needed to empower life scientists in the era of multi-omics.
To meet the growing computational needs of the life sciences, computer scientists and bioinformaticians have created thousands of software tools2. These software fall into two general categories: ‘essential tools’ that implement functions fundamental to most bioinformatics tasks, and ‘workflows’ that make specific analytic strategies accessible.
If a comprehensive microbial ‘omics investigation is a sophisticated dish, then essential tools are the kitchenware needed to cook. A chef can combine them in unique ways to answer any question, yet such freedom in data analysis not only requires the mastery of each essential tool but also demands experience in data wrangling and fluency in the command line environment to match the output format of one tool to the input requirements of the next. This barrier is overcome by workflows, which implement popular analysis strategies and make them accessible to those who have limited training in computation. If a comprehensive microbial ‘omics investigation is a sophisticated dish, then each ‘omics workflow is a recipe that turns raw material into a specific meal. For instance, a workflow for ‘pangenomics’ would typically take in a set of genomes and (1) identify open reading frames in all input genomes, (2) reciprocally align all translated amino acid sequences, (3) identify gene clusters by resolving pairwise sequence homology across all genes, and (4) report the distribution of gene clusters across genomes. By doing so, a software that implements pangenomics, such as Roary3, would seamlessly run multiple essential tools consecutively, resolve input and output requirements of each and address various ad hoc computational challenges to concoct a pangenome. Popular efforts to make accessible workflows that form the backbone of ‘omics-based microbiological studies include the Galaxy platform4, bioBakery software collection5, M-Tools (which includes GroopM6 and CheckM7) and KBase8. While ‘omics workflows conveniently summarize raw data into tables and figures, the ability to analyse data beyond predefined strategies they implement continues to be largely limited to ‘master chefs’, presenting the developers of ‘omics workflows with a substantial responsibility: predetermining the investigative routes their software enables users to traverse, which can influence how researchers interact with their data, conceivably affecting biological interpretations.
We introduced anvi’o (an analysis and visualisation platform for ‘omics data) as an alternative solution for microbiologists who wanted more freedom in research questions they could ask of their data9. We started with what we regarded as the most pressing need at the time: a platform that enabled the reconstruction and interactive refinement of microbial genomes from environmental metagenomes. The fundamentals of this strategy were already established by those who pioneered genome-resolved metagenomics10, but interactive visualisation and editing software that would enable microbiologists to intimately work with metagenome-assembled genomes was lacking. During the past five years, anvi’o has become a community-driven software platform that currently stands upon more than 90,000 lines of open-source code and supports interactive and fully integrated access to state-of-the-art ‘omics strategies including genomics, genome-resolved metagenomics and metatranscriptomics, pangenomics, metapangenomics, phylogenomics and microbial population genetics (Fig. 1).
Fig. 1 |. Integrated ‘omics with anvi’o.
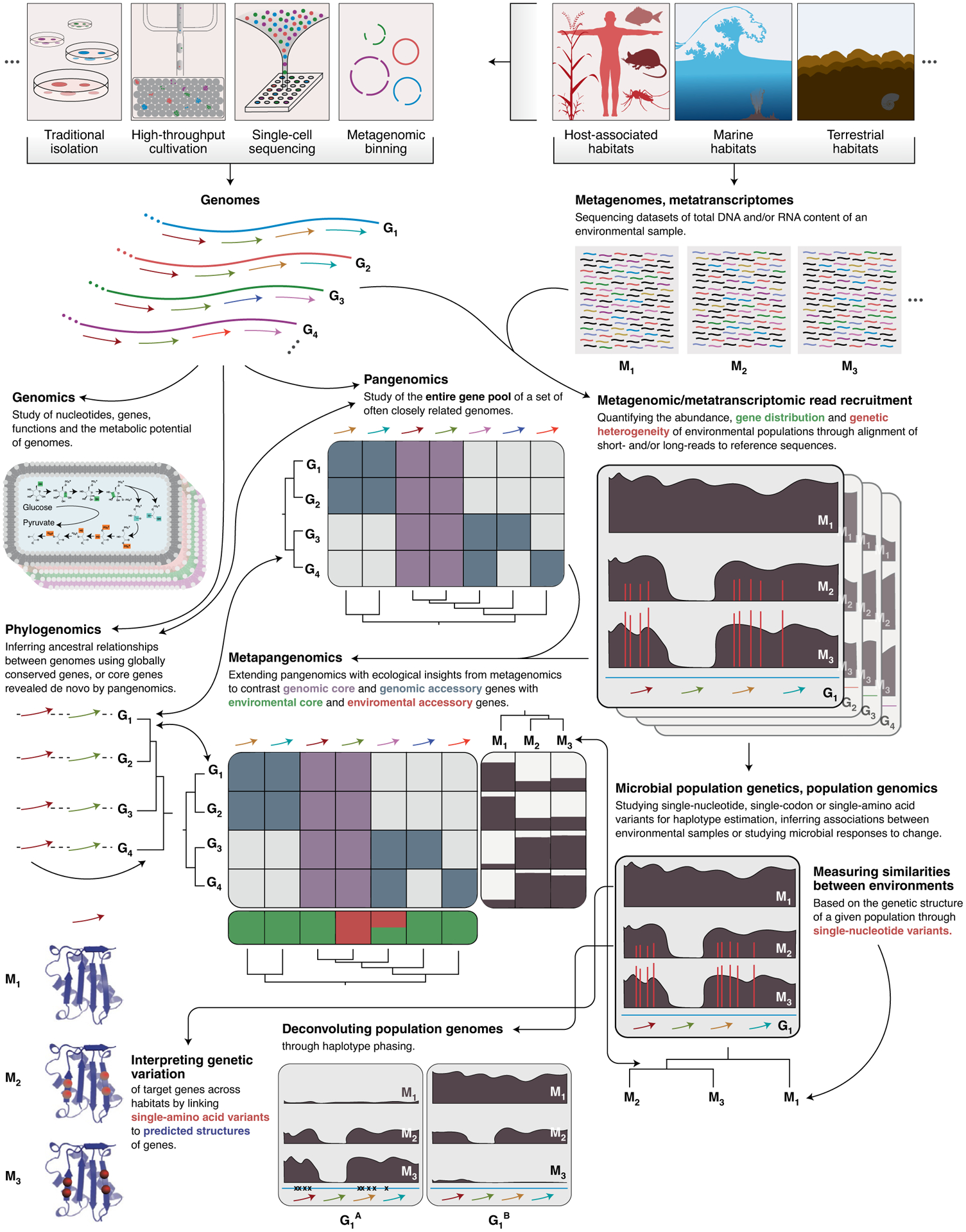
A glimpse of the interconnected nature of ‘omics analysis strategies anvi’o makes accessible, and their potential applications.
Anvi’o differs from existing bioinformatics software due to its modular architecture, which enables flexibility, interactivity, reproducibility and extensibility. To achieve this, the platform contains more than 100 interoperable programmes, each of which performs individual tasks that can be combined to build new and unique analytical workflows. Anvi’o programmes generate, modify, query, split and merge anvi’o projects, which are in essence a set of extensible, self-contained SQLite databases. The interconnected nature of anvi’o programmes that are glued together by these common data structures yields a network (http://merenlab.org/nt) rather than predetermined, linear paths for analysis. Through this modularity, anvi’o empowers its users to navigate through ‘omics data without imposing rigid workflows.
Integrated interactive visualisation is at the centre of anvi’o and helps researchers to engage with their data in all stages of analysis. Within the same interface, an anvi’o user can visualise amino acid sequence alignments between homologous genes across multiple genomes, investigate nucleotide-level coverage patterns and variants on the same DNA segment across metagenomes, interrogate associations between the genomic abundance and transcriptomic activity of environmental microbes, display phylogenetic trees and clustering dendrograms, and more. Furthermore, users can extend anvi’o displays with project-specific external data, increasing the utility of the interactive interfaces for holistic descriptions of complex systems. The anvi’o interactive interface also provides its users with the artistic freedom to change colours, sizes and drawing styles of display objects, add annotations or reorder data layers for detailed communication of intricate observations. Because each anvi’o project is self-contained, researchers can easily make their analyses available online either as a complete or partial package, thereby enabling the integration, reusability and reproducibility of their findings beyond static figures or tables. This strategy promotes transparency by permitting community validation and scrutiny through full access to data that underlie final conclusions.
Several key studies that used anvi’o during the past few years have demonstrated the integrative capabilities of the platform by implementing a combination of ‘omics strategies to facilitate in-depth analysis of naturally occurring microbial habitats. For instance, Reveillaud and Bordenstein et al. reconstructed new genomes of Wolbachia, a fastidious endosymbiont11, from individual insect ovary metagenomes, and computed a pangenome to compare these novel genomes to an existing reference12. They were then able to characterize the ecology of gene clusters in the environment by effectively combining metagenomics and pangenomics, discovering new members of the Wolbachia mobilome12. Yeoman et al. combined phylogenomics and pangenomics to infer ancestral relationships between a set of cultivar and metagenome-assembled genomes through a newly identified set of single-copy core genes13. They demonstrated the correspondence among these genomes based on gene cluster membership patterns, phylogenomic inference and average nucleotide identity in a single display13. Delmont and Kiefl et al. characterized the population structure of a subclade of SAR11, one of the most abundant microbial populations on Earth, by describing the environmental core genes of a single genome across surface ocean metagenomes14. By linking single amino acid variants in the environment to the predicted tertiary structures of these genes, they combined microbial population genetics with protein biochemistry to shed light on distinct evolutionary processes shaping the population structures of these bacteria14. Each of these studies employs unique approaches beyond well-established ‘omics workflows to create rich, reproducible and shareable data products (see http://merenlab.org/data).
Anvi’o does not implement strategies that take in raw data and produce summary tables or figures via a single command. As a result, anvi’o has a relatively steep learning curve. To address this, we have written extensive online tutorials that currently exceed 120,000 words, organized free workshops for hands-on anvi’o training, and created open educational resources to teach microbial ‘omics. To interact with anvi’o users we set up an online forum and messaging service. During the past two years, over 750 registered members of these services have engaged in technical and scientific discussions via more than 9,000 messages. But even when resources for learning are available, the journey from raw ‘omics data to biological insights often takes a significant number of atomic steps of computation. To ameliorate the burden of scale and reproducibility in big data analyses we have also introduced anvi’o workflows, which automate routine computational steps of commonly used analytical strategies in microbial ‘omics (http://merenlab.org/anvio-workflows). The anvi’o workflows are powered by Snakemake15, which ensures relatively easy deployment to any computer system and automatic parallelization of independent analysis steps. By turning raw input into data products to be analysed in the anvi’o software ecosystem, anvi’o workflows reduce the barriers for advanced use of computational resources and processing of large data streams for microbial ‘omics.
As the developers of anvi’o who strive to create an open community resource, our next big challenge is to attract bioinformaticians to consider anvi’o as a software development ecosystem they can use for their own science. Any programme that reads from or writes to anvi’o projects either directly (in any modern programming language) or through anvi’o application programmer interfaces (in Python) will immediately become accessible to anvi’o users, and such applications will benefit from the data integration, interactive data visualisation and error-checking assurances anvi’o offers.
As an open-source platform that empowers microbiologists by offering them integrated yet uncharted means to steer through complex ‘omics data, anvi’o welcomes its new users and contributors.
Acknowledgements
The URL http://anvio.org/authors serves as a dynamic list of anvi’o developers. We thank the creators of other open-source software tools for their generosity, anvi’o users for their patience with us, and K. Lolans for her critical reading of the manuscript and suggestions. We gratefully acknowledge support for anvi’o from the Simons Foundation and Alfred P. Sloan Foundation.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.White RA, Callister SJ, Moore RJ, Baker ES & Jansson JK Nat. Protoc 11, 2049–2053 (2016). [Google Scholar]
- 2.Callahan A, Winnenburg R & Shah NH Sci. Data 5, 180043 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Page AJ et al. Bioinformatics 31, 3691–3693 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jalili V et al. Nucleic Acids Res. 48, W395–W402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McIver LJ et al. Bioinformatics 34, 1235–1237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imelfort M et al. PeerJ 2, e603 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW Genome Res. 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arkin AP et al. Nat. Biotechnol 36, 566–569 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eren AM et al. PeerJ 3, e1319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tyson GW et al. Nature 428, 37–43 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Werren JH, Baldo L & Clark ME Nat. Rev. Microbiol 6, 741–751 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Reveillaud J et al. Nat. Commun 10, 1051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeoman CJ et al. PeerJ 7, e7548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delmont TO et al. eLife 8, 46497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köster J & Rahmann S Bioinformatics 28, 2520–2522 (2012). [DOI] [PubMed] [Google Scholar]