

Abstract
The selectivity of the glomerular filter is established by physical, chemical, and signaling interplay among its three core constituents: glomerular endothelial cells, the glomerular basement membrane and podocytes. Functional impairment or injury of any of these three components can lead to proteinuria. Podocytes are injured in many forms of human and experimental glomerular disease, including minimal change disease, focal segmental glomerulosclerosis and diabetes mellitus. One of the earliest signs of podocyte injury is loss of their distinct structure, which is driven by dis-regulated dynamics of the actin cytoskeleton. Status of the actin cytoskeleton in podocytes depends on a set of actin binding proteins, nucleators and inhibitors of actin polymerization, and regulatory GTPases. Mutations that alter protein function in each category have been implicated in glomerular diseases in humans and animal models. In addition, a growing body of studies suggest that pharmacological modifications of the actin cytoskeleton have the potential to become novel therapeutics for podocyte-dependent chronic kidney diseases. This review presents an overview of the essential proteins that establish actin cytoskeleton in podocytes and studies demonstrating the feasibility of drugging actin cytoskeleton in kidney diseases.
Keywords: glomerular diseases, podocytes, actin cytoskeleton, regulatory GTPases, actin binding proteins (ABPs), nucleators of actin polymerization
Introduction
Podocytes are terminally differentiated epithelial cells of the glomerulus which develop a characteristic architecture specialized for glomerular ultrafiltration. Their structure is traditionally divided into three distinct subcellular compartments: the cell body, primary processes which are microtubule-driven membrane extensions, and foot processes (FPs) which are actin-driven membrane extensions [1]. Adjacent podocytes interdigitate with each other at their FPs which are bridged with a specialized intercellular junction called a slit diaphragm (SD). FPs serve as an adhesive apparatus to the glomerular basement membrane (GBM), which together with endothelial cells and their glycocalyx form a filtration barrier.
Regardless of the underlying glomerular disease, the earliest signs of podocyte injury are characterized by the reorganization of FPs structure resulting in a fusion of filtration slits termed “FP effacement” [2–7]. Indeed, for over 50 years the effacement of FPs has been a main feature of proteinuria, though its significance with regard to proteinuria is still a mystery [8]. Recently, a study by Thomas Benzing and colleagues suggested that permeability of the renal filter is modulated through compression of the capillary wall [9]. By using CRISPR-Cas9-based genome editing, the authors inserted base-pair changes into the mouse genome that resulted in mutations in the NPHS2 gene that encodes the protein podocin. Podocin and nephrin are main protein components of the SD. Mice expressing mutant podocin mimic a human hereditary nephrotic syndrome with progression to focal segmental glomerulosclerosis (FSGS) [9]. By using a combination of scanning electron micrographs and a super-resolution stimulated emission depletion microscopy (STED) imaging, the authors were able to visualize nephrin and podocin at high spatial resolution, and thus investigate ultrastructural changes of FPs morphology during progressive albuminuria. By combining quantitative analyses with mathematical modelling, they showed that morphological alterations of the glomerular filtration barrier, namely FP effacement, lead to reduced compressive forces that counteract filtration pressure, thereby resulting in capillary dilatation, and ultimately albuminuria [9, 10]. This study further established the essential role of FP’s morphology with regard to selectivity of the glomerular filter.
Since changes in FP morphology are driven by changes in the underlying actin cytoskeleton, numerous studies have established that FP effacement is a direct consequence of disregulated actin cytoskeleton in podocytes. The key findings that unequivocally established the role of the actin cytoskeleton in the maintenance of podocyte function were the identification of distinct genetic mutations that underlie hereditary forms of FSGS in actin binding or regulatory proteins such as nephrin, CD2-associated protein (CD2AP), α-actinin 4, and inverted formin-2 (INF2)[11–15]. While some of these proteins and their mechanisms are podocyte-specific, such as nephrin and podocin, the majority of essential players and pathways are commonly present in other mammalian cell types (reviewed in [16, 17]). In general, the overall organization of the actin cytoskeleton is defined by the interplay between proteins that regulate actin polymerization and depolymerization, proteins that crosslink actin filaments into networks or bundles (actin binding proteins, ABPs), and proteins that coordinate those two processes together with the signaling pathways that originate at the plasma membrane such as regulatory GTPases (Figure 1).
Figure 1. Schematics of the glomerular filter.
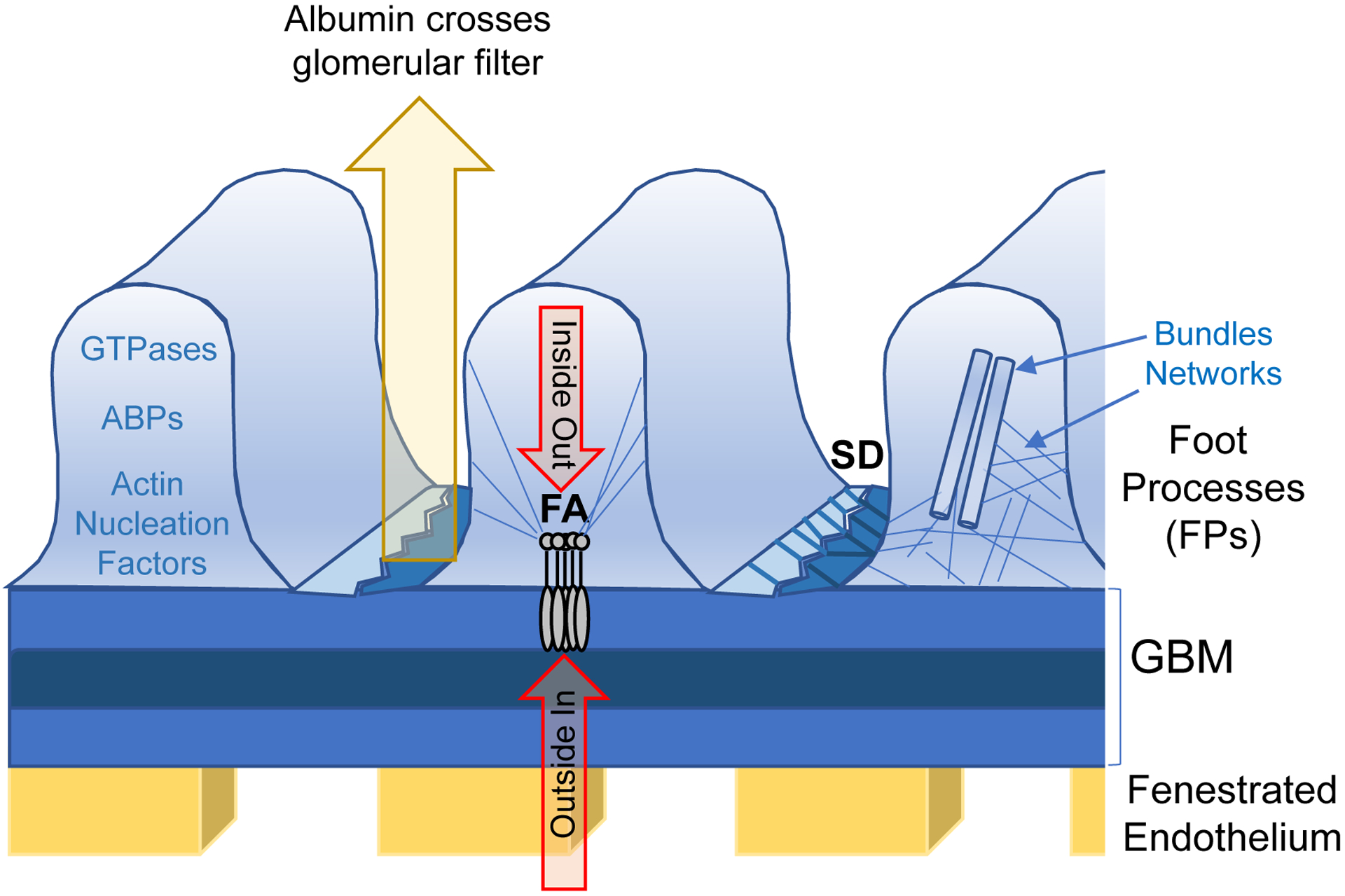
Specificity of the kidney filter is maintained by fenestrated endothelium, the glomerular basement membrane (GBM) and glomerular podocytes. The structure of podocyte foot processes (FPs) is regulated by two signaling platforms: focal adhesions (FAs) and the slit-diaphragm (SD). Those two hubs integrate ‘inside out’ and ‘outside in’ signaling, which defines the global organization of the actin cytoskeleton. The status of the actin cytoskeleton is defined by regulatory GTPases, actin binding proteins (ABPs) and actin nucleation factors. Two major actin structures are tight actin bundles and loose networks, both of which are established by actin crosslinking proteins.
Organization of the actin cytoskeleton in FPs is regulated by two separate signaling platforms, each formed by a specific set of proteins: focal adhesions (FA) which encore FPs to the GBM, and the slit diaphragm (SD) which is a modified cell-cell junction (Figure 1). The role of signaling at the SD in general and specifically with regard to nephrin has been recently reviewed [18]. Both platforms or ‘signaling hubs’ are capable of sensing signaling molecules that are circulating in the blood stream, thus initiating so called “outside in” signaling [19]. In addition, both platforms directly sense the status of the actin cytoskeleton within the FPs as a part of so called “inside out” signaling [17, 20]. Interplay between both signaling platforms ultimately defines the status of the actin cytoskeleton in podocytes [7]. This review aims to summarize current insights regarding essential proteins that establish the actin cytoskeleton in podocytes.
The canonical GTPases in podocytes
The global organization of the actin cytoskeleton is ultimately defined by the Rho family of GTPases [21]. The prototypic members of this family are RhoA, Rac1 and Cdc42. These regulatory enzymes adopt two distinct conformations dependent upon GTP binding and hydrolysis. When in a GTP-bound state, these enzymes interact with and activate a wide variety of downstream effectors, which ultimately define the global status of the actin cytoskeleton (Figure 2). GTP hydrolysis induces a conformational change within the enzyme resulting in a GDP-bound state, which in turn leads to disengagement of the GTPase and its downstream effectors. Since these GTPases are not highly active enzymes, GTP hydrolysis is typically potentiated by GTPase activating proteins (GAPs), whereas replacement of GDP for GTP and thus re-activation of the GTPase is often facilitated by GTP exchange factors (GEF). A third regulator of small GTPases is called a GDP dissociation inhibitor (GDI), whose function is to prevent the activation of the GTPase by blocking GDP-GTP exchange. While canonical GTPases are present in most cell types, the specificity of their action is determined by cell-type specific regulators and downstream effectors of their GTPase cycle.
Figure 2. The GTPase cycle of regulatory GTPases.
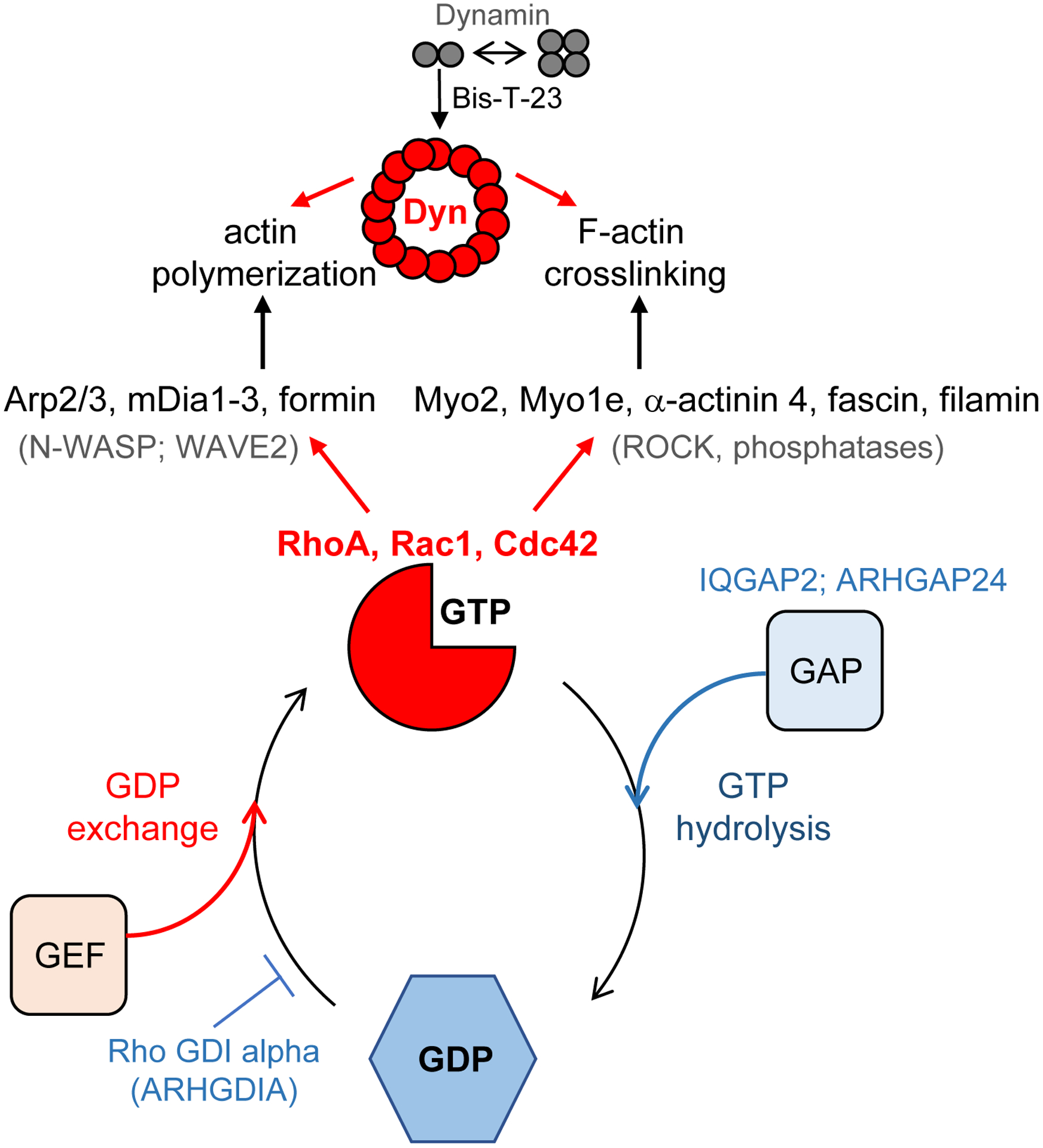
When in the GTP-bound state, these enzymes interact with cell-type specific downstream effectors, thus activating actin nucleators and actin crosslinking proteins. GTP hydrolysis requires the binding of GTPase activating proteins (GAPs), and a subsequent switch into the GDP-bound conformation disengages interactions between GTPase and upstream effectors. Interactions with GDP exchange factors (GEF) promotes re-activation of the GTPase. Examples of the regulatory proteins are indicated in the figure. GTPase cycle of large GTPase Dynamin (Dyn) is regulated by its oligomerization status, with dimers and tetramers being inactive form of the enzyme and rings being the active form of the enzyme.
Pawson and colleagues [22] were the first to examine which one of these three GTPases is essential for the podocyte’s structure and function. They showed that mice lacking Cdc42 specifically in podocytes developed congenital nephropathy and died as a result of kidney failure within 2 weeks after birth [22]. In contrast, mice lacking Rac1 or RhoA in podocytes were overtly normal and lived to adulthood [22]. Similar phenotypes were subsequently observed by Kretzler’s group which also generated podocyte-specific deletions of Cdc42 and Rac1 [23]. While the absence of Cdc42 resulted in congenital nephropathy, the absence of Rac1 had no effect on the development or the morphology of podocytes even when mice were monitored well into adulthood [23]. That said, the deletion of Rac1 prevented protamine sulfate-induced FPs effacement, thus implicating Rac1 activity in injured podocytes. However, in a chronic hypertensive damage model, the absence of Rac1 worsened proteinuria and sclerosis. Together, these data suggest that while blocking Rac1 might prevent acute FP effacement, Rac1 activity might be essential for repair and a return to homeostasis during chronic glomerular injury. In complementary studies, Shaw and colleagues proposed that dysregulation of Rac1 activity might underlie podocyte injury [24]. They showed that mutations in Arhgap24, which impair its GAP activity (Arhgap24 is Rac1-GAP) and results in increased levels of Rac1:GTP (Figure 2), were associated with FSGS [24].
Furthermore, identification of mutations in the gene ARHGDIA that cause nephrotic syndrome show that dysregulation of one of the regulatory proteins can influence multiple GTPases [25]. ARHGDIA encodes the Rho GDP dissociation factor alpha (Rho-GDI alpha). While these mutations abrogated interactions between GDI protein and RhoA, thus decreasing RhoA activity, they also indirectly increased levels of active Rac1 and Cdc42 in podocytes. Finally, Ras GTPase-activating-like protein (also known as IQGAP2 or p195) has been shown to be required for establishment of functional podocytes [26]. While the list of regulatory proteins that influence the status of the podocyte cytoskeleton is expected to grow, together these studies show that while not all canonical GTPases are essential for the structure and function of healthy podocytes, dis-regulation of their activities due to mutations in their regulatory proteins might play a role in a disease setting.
Dynamin, a large GTPase that regulates actin in podocytes
In addition to the Rho family of GTPases mentioned above, the actin cytoskeleton in podocytes is also regulated by a large GTPase dynamin [27, 28]. In contrast to its smaller family members, dynamin exists in multiple oligomerization states which include dimers, tetramers, octamers, partial and full rings and spirals [29]. Furthermore, dynamin does not require additional regulatory proteins such as GAPs, GEFs or GDIs due to its distinct biochemical characteristics. The structure of the dynamin tetramer that consists of two dimers has been elucidated [30]. Dynamin tetramers can oligomerize into partial and full rings or helices, and this oligomerization increases its GTPase activity due to the presence of the domain within dynamin itself that acts as an intramolecular GAP [31]. Dynamin has a relatively low affinity for both GTP and GDP [32], therefore release of GDP does not require the presence of designated GEFs, nor is dynamin:GDP conformation stabilized by the presence of GDIs.
Another characteristic that distinguishes dynamin from canonical small GTPases is that dynamin directly binds actin filaments [33, 34]. Dynamin-actin interactions promote GTP-dependent dynamin oligomerization, which releases capping protein gelsolin from the barbed ends resulting in potent actin polymerization from the fast, growing barbed ends which in turn leads to focal adhesion maturation [35]. The molecular mechanism by which dynamin releases gelsolin from the barbed ends is at present unknown, but fluorescence lifetime imaging microscopy suggests that it requires GTP binding and a major conformation change within dynamin tetramers [34].
The physiological relevance of dynamin’s role in regulating actin cytoskeleton has been demonstrated when mice expressing a dynamin mutant that promotes its oligomerization, specifically in podocytes, exhibited unusually long foot processes [36]. These data provide compelling proof that the length of FPs is dependent on dynamin-mediated actin polymerization. Indeed, increase in dynamin oligomerization by use of a small molecule Bis-T-23 [34], was sufficient to improve kidney health in diverse models of both transient and chronic kidney injury [36]. Specifically, Bis-T-23 restored the normal ultrastructure of foot processes, lowered proteinuria, lowered collagen IV deposits in the mesangial matrix, diminished mesangial matrix expansion, and extended lifespan [36]. Together, these findings suggest feasibility of targeting the actin cytoskeleton as therapeutics for glomerular disease via GTPase as a proxy.
Proteins that directly influence actin polymerization in podocytes
Organization of the actin cytoskeleton that underlies FPs is determined by polymerization and depolymerization rates of the actin filaments and actin binding proteins that crosslink actin filaments into bundles and networks (Table 1 and reviewed in [37]).
Table 1.
Actin binding proteins in podocytes
| Protein | Function in podocytes | Binding partners | Reference |
|---|---|---|---|
| Actin monomer (G-actin binding) | |||
| Profilin | Actin nucleotide exchange factor | Inf2 | [57] |
| Nucleators | |||
| Arp2/3-complex | Branched actin networks | WASP, WAVE | [38] |
| Formins: | |||
| Inf2 (inverted formin 2) | Unbranched actin structures | mDia | [41] |
| mDia | Unbranched actin structures | Inf2 | [41] |
| Elongation-promoting factors | |||
| Ena/VASP | Accelerate elongation and prevent capping | WAVE, Profilin | [58] |
| Barbed end capping | |||
| CapZ | Caps barbed ends of filaments | Inf2 | [57] |
| Gelsolin | Severs actin filaments, and caps barbed ends of filaments | [35] | |
| Crosslinking/Bundling | |||
| Non-muscle myosin II | Actin crosslinking and contractile properties | [59, 60] | |
| Non-muscle myosin I (Myo1e) | Actin crosslinking and contractile properties | [45] | |
| α-actinin 4 | Crosslinks actin filaments | [13] | |
Nucleators of actin polymerization
One of the nucleators of actin polymerization that controls the integrity of FPs is the Arp2/3 complex [38]. The Arp2/3 complex is unique in its capability to organize filaments into branched networks. The nucleation and branching activities of Arp2/3 are coupled and regulated by ATP, nucleation promoting factors (NPFs) and actin [39]. A recent study showed that interplay between two NPFs, N-WASP and WAVE, which in turn are downstream effectors of Cdc42 and Rac1 signaling axes, are involved in Arp2/3 mediated formation of actin networks in FPs [38]. Indeed, loss of both N-WASP and Arp2/3 in podocytes resulted in FP effacement and proteinuria, thus establishing an essential role for these proteins in establishing actin cytoskeleton in podocytes [38].
Actin polymerization in FPs is also regulated by atypical formin, Inf2 (Inverted Formin-2). Formins, which are present in almost all eukaryotes, promote actin filament assembly by accelerating filament nucleation and elongation, and by blocking filament capping (reviewed in [40]). Mutations in the INF2 gene are a common cause of familial FSGS [14]. In general, Inf2 promotes actin polymerization by staying associated with the growing barbed end of an actin filament and by directly binding mammalian diaphanous-related formin (mDia). Studies by Martin Pollak and colleagues showed that INF2 disease-causing mutations affect interactions between Inf2 and mDia [41, 42]. mDia is yet another member of the formin family and is one of the downstream effectors of RhoA-activated actin polymerization (reviewed in [43]). Since the binding of Inf2 to mDia antagonizes mDia-mediated actin polymerization in response to active Rho signaling, mutations that disrupt Inf2-mDia interactions are expected to result in disregulated actin polymerization in FPs. Unexpectedly, knock in mice bearing disease-causing INF2 mutations do not show any obvious phenotype [44]. However, stressing this system by protamine-induced kidney injury resulted in the collapse of podocyte FP architecture [44], implicating Inf2 as an important modulator of Rho/mDia signaling in FPs in injured podocytes.
Crosslinking / bundling proteins
In addition to proteins that regulate actin nucleation and polymerization, the status of the actin cytoskeleton is also defined by actin crosslinking proteins. One of the essential crosslinkers is Myo1e, non-muscle class I myosin, whose gene mutations underlie FSGS [45]. Mice lacking Myo1E in podocytes exhibit proteinuria, podocyte injury and CKD. The primary role of non-muscle myosin is to generate tension by binding F-actin, which in turn generates the contractile forces that help the glomerular capillaries to resist the high intraluminal hydrostatic pressure [9].
It is worth noting that actin dynamics in podocytes are not only regulated by GTPases or ABPs, but also by microRNAs (miRNAs) [46]. A study by Maria Pia Rastaldi and colleagues showed that Brain Derived Neurotrophic Factor (BDNF), a member of the neurotrophin family of polypeptide growth factors, binds the tropomyosin-related kinase B (TrkB) receptor on podocytes [47]. BDNF binding resulted in a reduction of microRNA-134 and an increase in microRNA-132, which subsequently increased Limk1 translation and phosphorylation. Increase in the level of Limk1 increased cofilin phosphorylation, thus ultimately increasing the overall actin polymerization in podocytes. Of note, cofilin is an actin-binding protein that disassembles actin filaments. BDNF treatment profoundly reduced albuminuria in mouse adriamycin nephropathy accompanied by a reformation of podocyte FPs. Together, these studies provide a compelling evidence of the complexity of the pathways and mechanisms that regulate podocyte structure.
In addition, point mutations in the α-actinin 4 gene (ACTN4) cause an autosomal dominant form of human FSGS [13]. α-actinin 4 protein is one of the actin crosslinking proteins in podocytes, and gain of function mutations have been associated with formation of aggregated and rapidly degraded cytoskeletal proteins.
Open questions and future directions
Given the close connection between podocyte structure and function, it has been argued that targeting actin cytoskeleton dynamics might be a valuable strategy for treating CKD [6, 48]. As of now, therapeutics that directly target dynamics of the actin cytoskeleton have not been developed. Indeed, nephrotic syndrome, which is accompanied by massive FP effacement, is treated with immune system suppressants such as corticosteroids, as well as blood pressure medications such as ACE (angiotensin-converting enzyme) inhibitors or ARBs (angiotensin receptor blockers).
While it might be impossible to pharmacologically alter actin cytoskeleton directly, a number of studies are suggesting that it might be feasible and beneficial to alter actin cytoskeleton dynamics in podocytes by targeting actin binding or regulatory proteins. In addition to the positive examples of using dynamin activators presented above [36], several studies suggested that it might be beneficial to alter RhoA-signaling in podocytes by targeting ROCK (Rho-associated protein kinase) [21]. A potent Rho-kinase inhibitor, fasudil, is used for the treatment of cerebral vasospasm, which is often due to subarachnoid hemorrhage. It is also used to treat pulmonary hypertension by attenuating vascular smooth muscle contraction [49]. Several additional ROCK inhibitors, such as Y27632 (inhibits p160ROCK) or SAR407899 have been shown to attenuate proteinuria and interstitial fibrosis in a number of rodent models including diabetic nephropathy [50], 5/6 nephrectomy [51], UUO obstruction [52], and ischemia reperfusion injury [53]. Given such diverse models of kidney injury which include both chronic as well as acute kidney injury, as of now, the mechanism of action for these ROCK inhibitors in kidney is still not elucidated.
The idea that ABPs might be novel pharmacological targets in humans is being actively explored in neurodegenerative diseases and cancer. While originally promising (NCT03784300), a Phase 2b clinical trial examining efficacy of the PTI-125 inhibitor of filamin, a classic actin bundling protein, showed that this investigational therapy failed to significantly lower the levels of Alzheimer’s relevant biomarkers present in fluid surrounding the brain and the spinal cord of patients (NCT04079803). A small molecule that inhibits fascin is currently being tested in patients with advanced or metastatic solid tumor malignancies (NCT03199586). Fascin is an actin bundling protein that is expressed at low levels in normal adult epithelial cells and its upregulated expression correlates with highly metastatic tumors. An orally available inhibitor NP-G2–044 binds fascin and thereby prevents its interactions with actin filaments [54]. Inhibition of fascin crosslinking activity in turn inhibits reorganization of the actin cytoskeleton necessary for tumor cell migration and invasion. It is important to note that filamin and fascin are expressed in podocytes. Filamin has been implicated in regulating actin dynamics upon HIV-induced nephropathy [55], whereas fascin-1 was shown to colocalize with nephrin in mouse kidney sections as well as along actin fibers in cultured podocytes [56]. In addition, levels of phosphorylated fascin-1 were found to be strongly reduced in glomeruli of patients with diabetic nephropathy, implicating its bundling activity in the organization of the actin cytoskeleton in podocytes.
In summary, a growing number of human trials focusing on actin binding and regulatory proteins, together with our growing understanding of the role of distinct players in podocytes, highlight the challenges associated with the development of novel therapeutics, and the highly active and evolving field of research in the biology of glomerular diseases.
Multiple-choice questions (5 questions) (answers are provided following the reference list)
- What is the common pathological feature of podocyte injury?
- Cell division.
- Loss of microtubule-driven major membrane extensions.
- Cell death.
- Cell cycle arrest.
- Foot processes effacement.
e
- What proteins regulate actin cytoskeleton in podocytes?
- Lysosomes.
- DNA repair enzymes.
- Mitochondrial enzymes.
- Regulatory GTPases, actin binding proteins and actin nucleators.
- DNA polymerase.
d
- What signaling platform(s) in podocytes regulate actin cytoskeleton?
- Podocytes contain two signaling platforms: focal adhesions and a modified cell-cell junction termed the slit diaphragm.
- A micronucleus. Micronucleus is usually a sign of genotoxic events and chromosomal instability.
- Multivesicular bodies (MVBs). MVBs are a specialized subset of endosomes that contain membrane-bound intraluminal vesicles.
- Lysosomes, a membrane-bound cell organelle that contains digestive enzymes.
- Microtubules, tubulin polymers that provide structure and shape to major podocyte extensions.
a
- What proteins influence the GTPase cycle of canonical small GTPases?
- The concentration of GTP in the cell.
- GTPase activating proteins (GAPs), GTPase exchange factors (GEFs) and Guanosine nucleotide dissociation inhibitors (GDIs).
- Kinases and phosphatases.
- Direct interactions with actin filaments.
- Direct interactions between different GTPases.
b
- What are standard treatments for glomerular diseases?
- Drugs that directly alter actin cytoskeleton dynamics in podocytes.
- Either diabetes medications such as metformin and sulfonylureas, or insulin therapy.
- Immune system suppressants such as corticosteroids, as well as blood pressure medications such as ACE (angiotensin-converting enzyme) inhibitors or ARBs (angiotensin receptor blockers).
- Antidepressants such as Prozac and Lexapro.
- Antibiotics.
c
Key summary points.
Podocytes contain two signaling platforms that regulate global organization of the actin cytoskeleton: focal adhesions and slit diaphragm
Dysregulation of the actin cytoskeleton by mutations in proteins that link actin to either focal adhesions or slit diaphragm underlie the genetics of hereditary nephrotic syndrome
Actin cytoskeleton in podocytes is regulated by (1) regulatory GTPases including RhoA, Rac1, Cdc42 and Dynamin; (2) regulators of actin polymerization including inverted formin, mDia and the Arp2/3 complex; (3) actin binding proteins including non-muscular myosin (Myo), a-actinin 4, fascin and filamin.
Current treatments for glomerular diseases do not directly target either podocytes or actin cytoskeleton.
Feasibility of the pharmacological alterations of the actin cytoskeleton in podocytes in human diseases has not been established.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest:
S. S. is co-founders and shareholder of Trisaq, a biotechnology company that develops novel kidney-protective therapies.
References
- 1.Pavenstädt H, Kriz W, Kretzler M (2003) Cell biology of the glomerular podocyte. Physiol Rev 83:253–307 [DOI] [PubMed] [Google Scholar]
- 2.Kerjaschki D (2001) Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J Clin Invest 108:1583–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reiser J, von Gersdorff G, Simons M, Schwarz K, Faul C, Giardino L, Heider T, Loos M, Mundel P (2002) Novel concepts in understanding and management of glomerular proteinuria. Nephrol Dial Transplant 17:951–955 [DOI] [PubMed] [Google Scholar]
- 4.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P (2002) A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol 13:630–638 [DOI] [PubMed] [Google Scholar]
- 5.Kistler AD, Singh G, Pippin J, Altintas MM, Yu H, Fernandez IC, Gu C, Wilson C, Srivastava SK, Dietrich A, Walz K, Kerjaschki D, Ruiz P, Dryer S, Sever S, Dinda AK, Faul C, Reiser J (2013) TRPC6 protects podocytes during complement-mediated glomerular disease. J Biol Chem 288:36598–36609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reiser J, Sever S (2013) Podocyte biology and pathogenesis of kidney disease. Ann Rev Med 64:357–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reiser J, Sever S, Faul C (2014) Signal transduction in podocytes--spotlight on receptor tyrosine kinases. Nat Rev Nephrol 10:104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brinkkoetter PT, Ising C, Benzing T (2013) The role of the podocyte in albumin filtration. Nat Rev Nephrol 9:328–336 [DOI] [PubMed] [Google Scholar]
- 9.Butt L, Unnersjö-Jess D, Höhne M, Edwards A, Binz-Lotter J, Reilly D, Hahnfeldt R, Ziegler V, Fremter K, Rinschen MM, Helmstädter M, Ebert LK, Castrop H, Hackl MJ, Walz G, Brinkkoetter PT, Liebau MC, Tory K, Hoyer PF, Beck BB, Brismar H, Blom H, Schermer B, Benzing T (2020) A molecular mechanism explaining albuminuria in kidney disease. Nat Metab 2:461–474 [DOI] [PubMed] [Google Scholar]
- 10.Benzing T (2020) Molecular Design of the Kidney Filtration Barrier. Trans Am Clin Climatol Assoc 131:125–139 [PMC free article] [PubMed] [Google Scholar]
- 11.Santin S, García-Maset R, Ruíz P, Giménez I, Zamora I, Peña A, Madrid A, Camacho JA, Fraga G, Sánchez-Moreno A, Cobo MA, Bernis C, Ortiz A, de Pablos AL, Pintos G, Justa ML, Hidalgo-Barquero E, Fernández-Llama P, Ballarín J, Ars E, Torra R; FSGS Spanish Study Group (2009) Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int 76:1268–1276 [DOI] [PubMed] [Google Scholar]
- 12.Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS (1999) Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 286:312–315 [DOI] [PubMed] [Google Scholar]
- 13.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodríguez-Pérez JC, Allen PG, Beggs AH, Pollak MR (2000) Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24:251–256 [DOI] [PubMed] [Google Scholar]
- 14.Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR (2010) Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 42:72–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyer O, Benoit G, Gribouval O, Nevo F, Tete MJ, Dantal J, Gilbert-Dussardier B, Touchard G, Karras A, Presne C, Grunfeld JP, Legendre C, Joly D, Rieu P, Mohsin N, Hannedouche T, Moal V, Gubler MC, Broutin I, Mollet G, Antignac C (2011) Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol 22:239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P (2007) Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 17:428–437 [DOI] [PubMed] [Google Scholar]
- 17.Sever S, Schiffer M (2018) Actin dynamics at focal adhesions: a common endpoint and putative therapeutic target for proteinuric kidney diseases. Kidney Int 93:1298–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin CE, Jones N (2018) Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front Endocrinol (Lausanne) 9:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perico L, Conti S, Benigni A, Remuzzi G (2016) Podocyte-actin dynamics in health and disease. Nat Rev Nephrol 12:692–710 [DOI] [PubMed] [Google Scholar]
- 20.Case LB, Waterman CM (2015) Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat Cell Biol 17:955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spiering D, Hodgson L (2011) Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr 5:170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basile DP, Dwinell MR, Wang SJ, Shames BD, Donohoe DL, Chen S, Sreedharan R, Van Why SK (2013) Chromosome substitution modulates resistance to ischemia reperfusion injury in Brown Norway rats. Kidney Int 83:242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, Vining C, Randolph A, Herbach N, Wanke R, Atkins KB, Gyung Kang H, Henger A, Brakebusch C, Holzman LB, Kretzler M (2013) Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int 84:920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, Antignac C, Pollak M, Kopp JB, Winn MP, Shaw AS (2011) Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest 121:4127–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, Beck BB, Gribouval O, Zhou W, Diaz KA, Natarajan S, Wiggins RC, Lovric S, Chernin G, Schoeb DS, Ovunc B, Frishberg Y, Soliman NA, Fathy HM, Goebel H, Hoefele J, Weber LT, Innis JW, Faul C, Han Z, Washburn J, Antignac C, Levy S, Otto EA, Hildebrandt F (2013) ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 123:3243–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugano Y, Lindenmeyer MT, Auberger I, Ziegler U, Segerer S, Cohen CD, Neuhauss SCF, Loffing J (2015) The Rho-GTPase binding protein IQGAP2 is required for the glomerular filtration barrier. Kidney Int 88:1047–1056 [DOI] [PubMed] [Google Scholar]
- 27.Sever S, Altintas MM, Nankoe SR, Moller CC, Ko D, Wei C, Henderson J, del Re EC, Hsing L, Erickson A, Cohen CD, Kretzler M, Kerjaschki D, Rudensky A, Nikolic B, Reiser J. (2007) Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest 117:2095–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S, Volpicelli-Daley L, Tian X, Wu Y, Ma H, Son SH, Zheng R, Moeckel G, Cremona O, Holzman LB, De Camilli P, Ishibe S (2012) Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest 122:4401–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sever S, Chang J, Gu C (2013) Dynamin rings: not just for fission. Traffic 14:1194–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reubold TF, Faelber K, Plattner N, Posor Y, Ketel K, Curth U, Schlegel J, Anand R, Manstein DJ, Noé F, Haucke V, Daumke O, Eschenburg S (2015) Crystal structure of the dynamin tetramer. Nature 525:404–408 [DOI] [PubMed] [Google Scholar]
- 31.Sever S, Muhlberg AB, Schmid SL (1999) Impairment of dynamin’s GAP domain stimulates receptor-mediated endocytosis. Nature 398:481–486 [DOI] [PubMed] [Google Scholar]
- 32.Binns DD, Helms MK, Barylko B, Davis CT, Jameson DM, Albanesi JP, Eccleston JF (2000) The mechanism of GTP hydrolysis by dynamin II: a transient kinetic study. Biochemistry 39:7188–7196 [DOI] [PubMed] [Google Scholar]
- 33.Gu C, Yaddanapudi S, Weins A, Osborn T, Reiser J, Pollak M, Hartwig J, Sever S (2010) Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J 29:3593–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu C, Chang J, Shchedrina VA, Pham VA, Hartwig JH, Suphamungmee W, Lehman W, Hyman BT, Bacskai BJ, Sever S (2014) Regulation of Dynamin Oligomerization in Cells: The Role of Dynamin-Actin Interactions and Its GTPase Activity. Traffic 15:819–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu C, Lee HW, Garborcauskas G, Reiser J, Gupta V, Sever S (2017) Dynamin Autonomously Regulates Podocyte Focal Adhesion Maturation. J Am Soc Nephrol 28:446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiffer M, Teng B, Gu C, Shchedrina VA, Kasaikina M, Pham VA, Hanke N, Rong S, Gueler F, Schroder P, Tossidou I, Park JK, Staggs L, Haller H, Erschow S, Hilfiker-Kleiner D, Wei C, Chen C, Tardi N, Hakroush S, Selig MK, Vasilyev A, Merscher S, Reiser J, Sever S (2015) Pharmacological targeting of actin-dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat Med 21:601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campellone KG, Welch MD (2010) A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 11:237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schell C, Sabass B, Helmstaedter M, Geist F, Abed A, Yasuda-Yamahara M, Sigle A, Maier JI, Grahammer F, Siegerist F, Artelt N, Endlich N, Kerjaschki D, Arnold HH, Dengjel J, Rogg M, Huber TB (2018) ARP3 Controls the Podocyte Architecture at the Kidney Filtration Barrier. Dev Cell 47:741–757 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goley ED, Welch MD (2006) The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol 7:713–726 [DOI] [PubMed] [Google Scholar]
- 40.Hegsted A, Yingling CV, Pruyne D (2017) Inverted formins: A subfamily of atypical formins. Cytoskeleton (Hoboken) 74:405–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun H, Al-Romaih KI, MacRae CA, Pollak MR (2014) Human Kidney Disease-causing INF2 Mutations Perturb Rho/Dia Signaling in the Glomerulus. EBioMedicine 1:107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun H, Schlondorff J, Higgs HN, Pollak MR (2013) Inverted formin 2 regulates actin dynamics by antagonizing Rho/diaphanous-related formin signaling. J Am Soc Nephrol 24:917–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mizuno H, Watanabe N (2012) mDia1 and formins: screw cap of the actin filament. Biophysics (Nagoya-shi) 8:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Subramanian B, Sun H, Yan P, Charoonratana VT, Higgs HN, Wang F, Lai KV, Valenzuela DM, Brown EJ, Schlöndorff JS, Pollak MR (2016) Mice with mutant Inf2 show impaired podocyte and slit diaphragm integrity in response to protamine-induced kidney injury. Kidney Int 90:363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, Buelli S, Tomasoni S, Piras R, Krendel M, Bettoni S, Morigi M, Delledonne M, Pecoraro C, Abbate I, Capobianchi MR, Hildebrandt F, Otto E, Schaefer F, Macciardi F, Ozaltin F, Emre S, Ibsirlioglu T, Benigni A, Remuzzi G, Noris M; PodoNet Consortium (2011) MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 365:295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee HW, Khan SQ, Faridi MH, Wei C, Tardi NJ, Altintas MM, Elshabrawy HA, Mangos S, Quick KL, Sever S, Reiser J, Gupta V (2015) A Podocyte-Based Automated Screening Assay Identifies Protective Small Molecules. J Am Soc Nephrol 26:2741–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li M, Armelloni S, Zennaro C, Wei C, Corbelli A, Ikehata M, Berra S, Giardino L, Mattinzoli D, Watanabe S, Agostoni C, Edefonti A, Reiser J, Messa P, Rastaldi MP (2014) BDNF repairs podocyte damage by microRNA-mediated increase of actin polymerization. J Pathol 235:731–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian X, Ishibe S (2016) Targeting the podocyte cytoskeleton: from pathogenesis to therapy in proteinuric kidney disease. Nephrol Dial Transplant 31:1577–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shi J, Wei L (2013) Rho kinases in cardiovascular physiology and pathophysiology: the effect of fasudil. J Cardiovasc Pharmacol 62:341–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komers R (2013) Rho kinase inhibition in diabetic kidney disease. Br J Clin Pharmacol 76:551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Babelova A, Jansen F, Sander K, Lohn M, Schafer L, Fork C, Ruetten H, Plettenburg O, Stark H, Daniel C, Amann K, Pavenstädt H, Jung O, Brandes RP (2013) Activation of Rac-1 and RhoA contributes to podocyte injury in chronic kidney disease. PLoS One 8:e80328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nagatoya K, Moriyama T, Kawada N, Takeji M, Oseto S, Murozono T, Ando A, Imai E, Hori M (2002) Y-27632 prevents tubulointerstitial fibrosis in mouse kidneys with unilateral ureteral obstruction. Kidney Int 61:1684–1695 [DOI] [PubMed] [Google Scholar]
- 53.Kentrup D, Reuter S, Schnöckel U, Grabner A, Edemir B, Pavenstädt H, Schober O, Schäfers M, Schlatter E, Büssemaker E (2011) Hydroxyfasudil-mediated inhibition of ROCK1 and ROCK2 improves kidney function in rat renal acute ischemia-reperfusion injury. PLoS One 6:e26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J, Dey R, Wang Y, Jakoncic J, Kurinov I, Huang XY (2018) Structural Insights into the Induced-fit Inhibition of Fascin by a Small-Molecule Inhibitor. J Mol Biol 430:1324–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tandon R, Levental I, Huang C, Byfield FJ, Ziembicki J, Schelling JR, Bruggeman LA, Sedor JR, Janmey PA, Miller RT (2007) HIV infection changes glomerular podocyte cytoskeletal composition and results in distinct cellular mechanical properties. Am J Physiol Renal Physiol 292:F701–710 [DOI] [PubMed] [Google Scholar]
- 56.Kliewe F, Scharf C, Rogge H, Darm K, Lindenmeyer MT, Amann K, Cohen CD, Endlich K, Endlich N (2017) Studying the role of fascin-1 in mechanically stressed podocytes. Sci Rep 7:9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rollason R, Wherlock M, Heath JA, Heesom KJ, Saleem MA, Welsh GI (2016) Disease causing mutations in inverted formin 2 regulate its binding to G-actin, F-actin capping protein (CapZ alpha-1) and profilin 2. Biosci Rep 36:e00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hohenstein B, Kasperek L, Kobelt DJ, Daniel C, Gambaryan S, Renné T, Walter U, Amann KU, Hugo CP (2005) Vasodilator-stimulated phosphoprotein-deficient mice demonstrate increased platelet activation but improved renal endothelial preservation and regeneration in passive nephrotoxic nephritis. J Am Soc Nephrol 16:986–996 [DOI] [PubMed] [Google Scholar]
- 59.Arrondel C, Vodovar N, Knebelmann B, Grünfeld JP, Gubler MC, Antignac C, Heidet L (2002) Expression of the nonmuscle myosin heavy chain IIA in the human kidney and screening for MYH9 mutations in Epstein and Fechtner syndromes. J Am Soc Nephrol 13:65–74 [DOI] [PubMed] [Google Scholar]
- 60.Johnstone DB, Zhang J, George B, Leon C, Gachet C, Wong H, Parekh R, Holzman LB (2011) Podocyte-specific deletion of Myh9 encoding nonmuscle myosin heavy chain 2A predisposes mice to glomerulopathy. Mol Cell Biol 31:2162–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]