

Abstract
Fragile X syndrome (FXS) is the most common inherited source of intellectual disability in humans. FXS is caused by mutations that trigger epigenetic silencing of the Fmr1 gene. Loss of Fmr1 results in increased activity of the mitogen-activated protein kinase (MAPK) pathway. An important downstream consequence is activation of the mitogen-activated protein kinase interacting protein kinase (MNK). MNK phosphorylates the mRNA cap-binding protein, eukaryotic initiation factor 4E (eIF4E). Excessive phosphorylation of eIF4E has been directly implicated in the cognitive and behavioral deficits associated with FXS. Pharmacological reduction of eIF4E phosphorylation is one potential strategy for FXS treatment. We demonstrate that systemic dosing of a highly specific, orally available MNK inhibitor, eFT508, attenuates numerous deficits associated with loss of Fmr1 in mice. eFT508 resolves a range of phenotypic abnormalities associated with FXS including macroorchidism, aberrant spinogenesis, and alterations in synaptic plasticity. Key behavioral deficits related to anxiety, social interaction, obsessive and repetitive activities, and object recognition are ameliorated by eFT508. Collectively, this work establishes eFT508 as a potential means to reverse deficits associated with FXS.
Electronic supplementary material
The online version of this article (10.1007/s13311-020-00932-4) contains supplementary material, which is available to authorized users.
Keywords: Fragile X syndrome, FMRP, eFT508, Behavior, Spinogenesis
Introduction
Fragile X syndrome (FXS) is the most common inherited source of intellectual disability in humans and is the leading cause of autism spectrum disorder (ASD) [1, 2]. Affected individuals display numerous behavioral deficits that include deficits in learning and memory, hyperactivity, increased anxiety, impaired social interactions, and repetitive behavioral patterns [1, 3]. Physiological abnormalities such as circuit hyperexcitability and abnormal spinogenesis are observed in FXS [4–7]. The most common genetic change associated with FXS is a trinucleotide (CGG) expansion in the 5′ untranslated region of the Fmr1 gene [8, 9]. This mutation promotes epigenetic silencing and loss of the corresponding polypeptide. Fmr1 encodes the fragile X mental retardation protein (FMRP). FMRP is a polysome-associated RNA-binding protein that governs translation of a large subset of mRNAs [10–13]. Multiple targets perform functions linked to synaptic plasticity [11, 14]. Exaggerated protein synthesis in the brain is a hallmark of FXS and is recapitulated in animal models of Fmr1 deficiency [6, 15–17]. Restoration of translational homeostasis has emerged as a potential means to correct neuronal dysfunction and rescue behavioral deficits associated with FXS [6, 15, 18].
All mRNAs possess a 5′ cap that enables signal-dependent control of translation initiation [19]. eIF4E reversibly binds to the cap and fosters translation initiation through recruitment of the scaffold eIF4G and the helicase eIF4A forming the tripartite eIF4F complex [19–21]. eIF4E is the least abundant component of eIF4F, and its activity is meticulously controlled [22, 23]. eIF4E is further regulated by eIF4E binding proteins (4EBPs) that prevent eIF4E from interacting with eIF4G. Phosphorylation of 4EBP by mechanistic target of rapamycin (mTOR) prevents binding to eIF4E, and thus promotes formation of the eIF4F complex [20]. An additional mechanism that regulates eIF4E is post-translational modification by MNK1 and MNK2 (collectively referred to as MNK) at a single site, S209 [24, 25]. eIF4E is also regulated through dephosphorylation. The main phosphatase that regulates eIF4E is protein phosphatase 2A [26–28]. Loss of FMRP results in elevated levels of eIF4E phosphorylation due to increased MAPK signaling upstream of MNK [6, 15]. Increased levels of eIF4E phosphorylation are correlated with core phenotypes associated with FXS, both in human patients and Fmr1-deficient mice [6, 29, 30]. Genetic reduction of MNK resolves behavioral deficits and restores synaptic activity in Fmr1-deficient animals [6]. Furthermore, many of the phenotypes observed in Fmr1−/− animals are rescued through introduction of a single amino acid substitution in eIF4E (S209A) that renders the protein incapable of being phosphorylated by MNK [6].
Multiple pharmacological approaches that modulate eIF4E activity have been applied to FXS. One strategy to decrease eIF4E levels makes use of the antidiabetic drug, metformin. Metformin is a translational inhibitor that affects multiple pathways including mTOR [31]. At high doses, metformin blocks 4EBP phosphorylation by mTOR resulting in a decrease of eIF4E availability. Metformin resolves behavioral deficits observed in Fmr1-deficient animals [15]. A second approach involves direct inhibition of the physical interaction between eIF4E and eIF4G with the small molecule 4EGI-1 [18]. Defects in memory and spinogenesis are corrected by 4EGI-1 in Fmr1-/- mice. The third and final approach involves inhibition of eIF4E phosphorylation by MNK using the antifungal phytotoxin natural product, cercosporamide [6]. This strategy rescues behavioral and neurological function in a mouse model of FXS. eFT508, also known as tomivosertib, is a highly specific MNK inhibitor that has emerged as an effective pharmacological tool for reduction of eIF4E phosphorylation in the context of cancer [32, 33]. eFT508 is currently in phase 2 clinical trials for cancer as it promotes tumor recognition by the immune system through repression of immune checkpoint receptors and immunosuppressive cytokines [33, 34]. Additionally, eFT508 has shown promise in the nervous system as it reverses chemotherapy-induced neuropathy and certain forms of inflammatory pain [35, 36].
Here, we sought to determine if eFT508 resolves the physiological and behavioral defects associated with FXS. We report that eFT508 crosses the blood–brain barrier and reduces cortical eIF4E phosphorylation in the Fmr1−/− model of FXS. We identify an effective dose of eFT508 sufficient to rescue hyperactive and anxiety-like behaviors, impaired social interactions, and deficits in learning and memory. Similarly, we demonstrate that eFT508 restores abnormal neuronal maturation and synaptic activity. Collectively, the data establish inhibition of MNK by eFT508 as a potential therapeutic strategy for FXS.
Methods
Animals
Wild-type (C57BL6) and Fmr1−/− mice were obtained from The Jackson Laboratory and bred at the University of Texas at Dallas to generate mice for experimental testing. Mice were housed at a maximum occupancy of 4 animals in each cage and weaned at postnatal day 21. Mice were housed on a 12-h light/dark cycle. Food and water were provided ad libitum. All behavioral assays and experiments were conducted on male mice between 3 and 6 weeks of age from wild-type or Fmr1−/− genetic backgrounds. The genotype of animals was confirmed by collected DNA from tail clips at the time of weaning. Experiments were conducted with age-matched litter mates in the wild-type and Fmr1−/− backgrounds. Wild-type C57BL6 mice and Fmr1−/− mice were obtained from an internally maintained colony at the University of Texas at Dallas after a minimum of 3 generations of breeding. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Texas at Dallas.
eFT508 Treatment
eFT508 was provided by eFFECTOR via a material transfer agreement with the University of Texas at Dallas. Mice were treated with eFT508 at varying doses 1 h prior to conducting behavioral assays or tissue collection. Doses were chosen based on previously published data on the in vivo tolerance of the compound as well as studies that used the compound to successfully reduce eIF4E phosphorylation levels in the brain and other tissues [35–37]. eFT508 was prepared as described for in vivo dosing and diluted in 0.9% saline [35]. Animals were subjected to injections of either 1 mg/kg, 3 mg/kg, 5 mg/kg, or 8 mg/kg doses of eFT508 intraperitoneally (i.p.) or a vehicle dose (saline). Mice were injected each day of the behavioral pipeline, for a total of 4 days. Tissues were collected for immunoblotting, immunohistochemistry, macroorchidism, and Golgi–Cox staining following completion of behavioral assays.
Behavioral Assays
From 3 to 5 weeks of age, mice were subjected to a battery of behavioral tests for a total of 4 days. All mice used for behavioral testing were subjected to all behavioral assays in the same order. Mice were first subjected to the open field test, followed by the novel object recognition test, social interaction assay, marble burying, and nest-building tests with a 24-h latency period between each assay. Prior to each day of testing, mice were given the relevant intraperitoneal dose of eFT508 or the vehicle control.
Open Field
Animals were habituated in the room where experiments were conducted for 30 min. They were then subjected to treatment with either eFT508 or vehicle and were individually placed in the open field area after 1 h. The open field arena consisted of a 5-sided white chamber (50 cm × 50 cm × 50 cm). Mice were allowed to explore the area for 30 min after a 1-min habituation period [15, 17, 38]. The arena was cleaned after each mouse to remove olfactory cues. Total distance traveled and time mobile were video recorded and scored electronically using Noldus EthoVision XT software (Leesburg, VA).
Novel Object Recognition
Mice were habituated in the experimental room for 30 min prior to eFT508 or vehicle treatment. One hour after intraperitoneal injection, animals were placed in the bottom left corner of the open field-testing arena (50 cm × 50 cm × 50 cm) for a 10-min habituation period. Following the habituation phase, mice were returned to their home cage while 2 identical inanimate objects were placed 20 cm away from each other on the right side of the arena. The mice were allowed to explore the objects and arena freely for 15 min before being returned to their home cages. After a 30-min latency phase, one familiar object was replaced by a novel object in the arena. Mice were placed back into the testing arena for 15 min and allowed to roam freely. Novel and familiar objects were placed in alternate corners for each test mouse to counterbalance for spatial preference. To remove olfactory cues, objects and the testing arena were cleaned after each mouse. Mice were video recorded during all testing phases and total interactions with each object (novel and familiar) were scored. Interactions were electronically scored using Noldus EthoVision XT software by defining the area of each object as a zone in the software and scoring total number of nose-point entries in each object zone.
Social Approach and Social Interaction
Animals were habituated to the experimental room for 30 min prior to eFT508 or vehicle treatment. Following intraperitoneal injections, mice were habituated to the open field testing arena (50 cm × 50 cm × 50 cm) for 15 min. Mice were returned to their home cage for 15 min. Two cylindrical wire cages were placed 20 cm apart on the right side of arena, and the mice were allowed to roam freely in the arena for 15 min. Mice were returned back to their home cage for 15 min and 1 male mouse, aged 5 weeks (familiar male 1), was placed inside 1 wire cage in the testing arena. The mice were placed back in the testing arena and allowed to explore the arena for 15 min. Following the 15-min social approach testing, the experimental mice were placed back in their home cage for 30 min and a novel male mouse, aged 5 weeks (novel male 1), was placed under the second, empty wire cage. Experimental mice were returned back to the testing arena and allowed to roam freely for 15 min for social interaction testing phase I. Following phase I, the experimental mice were returned back to their home cage for 30 min and the familiar male I mice were returned to their home cage as well. A female, aged matched mouse was placed (novel female I) in the now empty wire cage and experimental mice were placed back in the arena for social interaction testing phase II. The experimental group was placed back in the testing arena for 15 min and allowed to explore the area. All mice were returned to their home cages following testing. Cages and the testing arena were cleaned in between phase of testing and after each mouse to prevent confounding results from olfactory signals. Wire cages and novel and familiar mice were placed in alternate corners for each test to account for spatial preference. Mice were video recorded for all phases of testing and total interactions were automatically scored using Noldus EthoVision XT software (Leesburg, VA) by defining the location of each wire cage as a “novel” or “familiar” zone. Nose-point entries by experimental mice were scored when the mouse entered either zone. The total area of each zone remained fixed throughout all trials, and minute changes in location of the zones were accounted for through manual adjustment of zone placement between trials.
Marble Burying and Nest Building
Habituation in the experimental room took place for 30 min prior to eFT508 or vehicle treatment. In the marble burying assay, marbles were placed in a 5 × 3 array, evenly spaced over half of the clear plexiglass cage (19 cm × 38 cm × 13 cm). Each cage contained sufficient corncob bedding to cover the marbles. One hour after injection, the mouse was placed in the back of the cage and allowed to roam freely for 30 min. After the testing phase, a picture of the cage was taken for scoring by a blinded observer. Marbles were counted as buried if they were 2/3 covered by bedding [39, 40]. After the marble burying assay, mice were returned to their home cage for 30 min. Marbles were removed from the testing area and replaced with half of an undisturbed paper nestlet. Mice were placed back into their respective testing arenas and given 30 min to roam freely. After 30 min, mice were returned back to their home cages and pictures were taken of the nestlet. The nest shredding assay was scored on a scale of 0–5 by a blinded scorer. The nest building scale is as follows: 0—untouched nestlet, 1—20% dissociated nestlet, 2—40% dissociated nestlet, 3—60% dissociated nestlet, 4—80% dissociated nestlet, and 5—100% dissociated nestlet [41]. After both behavioral assays were run for each mouse, the cages were cleaned and corncob bedding was replaced to account for olfactory signals from previous mice.
Immunoblotting
Mice were injected with either eFT508 or vehicle intraperitoneally. Hippocampal, striatal, and cortical tissue were collected 1 h after injection and homogenized in ice-cold RIPA buffer containing protease and phosphatase inhibitors [42]. After 20-min centrifugation at 12,000 r.c.f., protein lysates were collected and total protein concentration was determined by BCA assay. 30 μg of tissue lysate was denatured with heat and 2-mercaptoethanol and resolved on a 12% SDS-PAGE gel. Proteins were transferred to a nitrocellulose membrane for 1 h at 100 V. Membranes were washed with 1x TBS-T and blocked in 5% milk. All primary antibodies were diluted in 5% BSA at a 1:1000 dilution. Immunoblotting for the following antibodies was performed overnight at 4 °C: phospho-eIF4E S209 (Abcam, ab76256, Cambridge, UK), eIF4E (CST, 9742S), phospho-MNK (CST, 2111S), MNK (CST, 2195S), and beta-Actin (CST, 4970S). After primary antibody incubation and 3x washes with 1X TBS-T, membranes were incubated with the following secondary antibody (HRP a-Rb) diluted in 5% BSA at a 1:10,000 dilution for 2 h. Phospho-eIF4E values were normalized to total eIF4E and beta-Actin levels . Immunoreactivity was detected by enhanced chemiluminescence and quantification was performed using ImageLab and GraphPad Prism.
Immunohistochemistry
Mice received injections of either eFT508 or vehicle intraperitoneally. 1 h after injection, they were deeply anesthetized with isoflurane with a single-channel veterinary anesthesia stand and perfused with 4% paraformaldehyde and 1X PBS. The brain was exposed immediately after decapitation and submerged in 4% paraformaldehyde. After 24 h at 4 °C, the tissue was placed into a 30% sucrose solution at 4 °C for 3 days for cryoprotection. The tissue was embedded in O.C.T. and frozen at − 20 °C. Slices (20 μm) were mounted on SuperFrost Plus slides (Thermo Fisher Scientific, Waltham, MA). Immunostaining was then performed as previously described [42]. Antibodies used were NeuN (1:100, EMD Millipore, MAB377, Burlington, MA), p-eIF4E (1:1000, Abcam, ab76256), eIF4E (1:1000, CST, 9742S), AF 488 (1:1000, Invitrogen, A11094, Carlsbad, CA), and Cy3 (1:1000, Invitrogen, A10520). Slices were incubated with DAPI (1 mM) for 15 min. Images were acquired using the Olympus FV3000RS laser confocal (Tokyo, Japan). Quantification was performed using ImageJ. Values were normalized to NeuN expression.
Extracellular Slice Recordings
Animals were treated with eFT508 or 0.9% saline 1 h prior to slice collection. Mice were briefly anesthetized and decapitated. Coronal slices (250 μm) containing the dorsal hippocampus were obtained using a vibrating microtome. Brains were submerged in oxygenated, ice-cold cutting aCSF (206 mM sucrose, 3 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, 7 mM MgCl2, 0.5 mM CaCl2, 1 mM sodium pyruvate, and 0.89 mM sodium l-ascorbate) during slicing. Slices were incubated at 34 °C for at least 1 h in oxygenated recording aCSF (124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, 1.3 mM MgCl2, 2 mM CaCl2, 1 mM sodium pyruvate, and 0.89 mM sodium l-ascorbate) to recover prior to recording. Slices were then placed in the recording chamber and continuously perfused with approximately 400 mL of recirculating oxygenated recording aCSF. A parallel bipolar tungsten stimulating electrode was placed in the stratum radiatum near the CA3 region of the hippocampus. A glass recording electrode (6-7 MΩ) filled with recording aCSF was placed in the stratum radiatum in the CA1 region to record evoked fEPSPs. Baseline fEPSP responses were evoked as previously described, delivering 0.1-ms pulses at 0.033 Hz [15]. Stable baseline recordings were recorded for 20 min prior to LTD induction. LTD was then induced by delivering 0.1 ms biphasic paired pulses at 1 Hz for 15 min. fEPSPs were recorded for at least 40 min after LTD induction using the same stimulation parameters as during the baseline period. Slices were excluded from the final data analysis if baseline fEPSP recording was not stable during the first 20 min of stimulation. Specifically, if the change in fEPSP slope varied more than 20% of the slice average during baseline recording, the slices were excluded. Similarly, slices were excluded if sustained LTD was not observed or if individual recordings did not stabilize during the 40-min recovery period. Recordings from slices from individual animals were compiled to acquire traces and data for each animal. Then, final data for each group (wild-type vehicle n = 5, Fmr1−/− vehicle n = 6, and Fmr1−/− eFT508 n = 4) was compiled by taking the fEPSP slope average from each individual animal. Data was acquired in Matlab using WaveSurfer (Janelia Research Campus) and analyzed using custom Matlab scripts. Statistical analysis and graphs were plotted using GraphPad Prism V8.0. The last 20 min of recording was used to conduct statistical analysis to compare change in fEPSP slope between the 3 groups using 1-way ANOVA Tukey’s multiple comparisons test.
Dendritic Spine Analysis
Mice were injected daily with eFT508 or saline intraperitoneally for 6 days. Afterward, they were anesthetized with isoflurane and decapitated. Brain tissue was collected immediately after decapitation. Golgi–Cox staining was conducted using the FD NeuroTechnologies (Columbia, MD) Golgi–Cox staining kit per the manufacturer’s instructions. Hippocampal pyramidal neurons were analyzed and imaged. Images were acquired using the Olympus FV3000RS laser confocal.
Statistical Analysis
All data are presented as mean value with error calculated from standard error of measurement. For Golgi–Cox dendritic spine analysis, a minimum of 10 neurons per slice were imaged and dendritic spines were counted. An average of 3 slices per mouse were included for quantification. For immunohistochemical analysis, an average of 6 individual slices per mouse were used for quantification. Group sizes are indicated in the figure legend for each experiment. Statistical analysis and graphs were plotted using the GraphPad Prism V8.0 software. Statistical evaluation was performed using 1-way analysis of variance (ANOVA), followed by Tukey’s post hoc test or 2-way ANOVA followed by Bonferroni’s post hoc test. Individual statistical analyses are noted next to each result.
Results
eFT508 Inhibits eIF4E Phosphorylation in the Brain
Exaggerated phosphorylation of eIF4E in the cortex is a key abnormality in Fmr1−/− mice [6]. Genetic elimination of eIF4E phosphorylation through introduction of a single S209A substitution resolves behavioral deficits associated with FXS [6]. To investigate whether eFT508 is capable of inhibiting MNK in the brain, we examined eIF4E phosphorylation in brain regions implicated in the cognitive and behavioral deficits associated with FXS: the hippocampus, prefrontal cortex, and striatum [43–46]. To determine the effect of eFT508 on eIF4E phosphorylation quantitatively, we conducted systemic intraperitoneal (i.p.) injections spanning a range of concentrations between 1 and 8 mg/kg and assayed eIF4E S209 phosphorylation with immunoblots (Fig. 1). The ratio of phosphorylated eIF4E to total eIF4E levels was significantly reduced in the hippocampus following injections of 5 mg/kg and 8 mg/kg (Fig. 1A). Similarly, in the striatum, 3 mg/kg, 5 mg/kg, and 8 mg/kg doses of eFT508 doses led to reduced levels of eIF4E phosphorylation (Fig. 1B). We observe comparable effects on eIF4E phosphorylation after eFT508 treatment in the prefrontal cortex (Fig. 1C). We observe a similar decrease in eIF4E phosphorylation in key brain regions in Fmr1−/− mice after eFT508 treatment (Fig. S1). We conclude that eFT508 attenuates eIF4E phosphorylation in cortical tissue of Fmr1−/− mice.
Fig. 1.
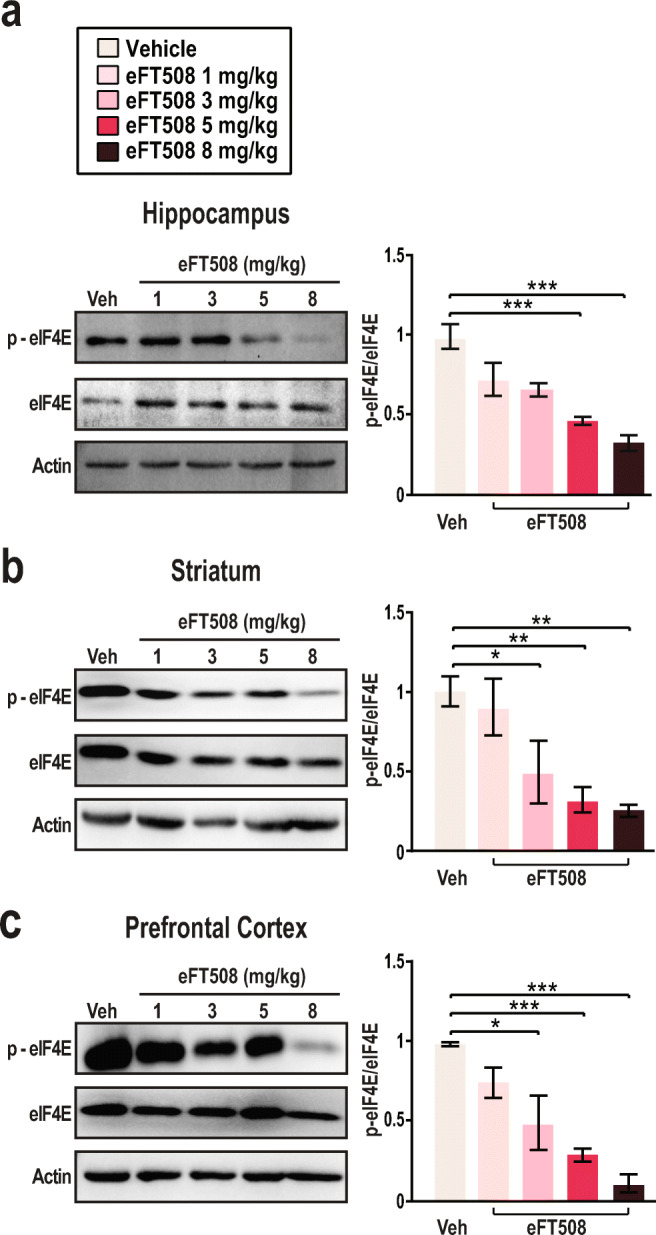
eFT508 attenuates eIF4E phosphorylation in the brain. (A) Representative immunoblot and quantification from hippocampal tissue lysate in vehicle-treated and eFT508-treated WT mice (n = 3 per group). Mice were treated intraperitoneally (i.p.) with vehicle or eFT508 doses 1 h prior to tissue collection (saline 1 ± 0.078, 1 mg/kg 0.729 ± 0.104, 3 mg/kg 0.676 ± 0.042, 5 mg/kg 0.479 ± 0.025, ***p = 0.0009, 8 mg/kg 0.383 ± 0.049, ***p < 0.0001; 1-way ANOVA Tukey’s multiple comparisons test). (B) Immunoblot and quantification of protein lysate from the striatum WT mice (n = 3 per group) were treated with vehicle or varying doses of eFT508. Immunoblots and quantification display levels of phosphorylated eIF4E (denoted as p-eIF4E), total eIF4E, and beta-Actin an hour after the indicated treatment (saline 1 ± 0.093, 1 mg/kg 0.893 ± 0.176, 3 mg/kg 0.489 ± 0.195, *p = 0.0272, 5 mg/kg 0.317 ± 0.079, **p = 0.007, 8 mg/kg 0.262 ± 0.037, ***p < 0.0018; 1-way ANOVA Tukey’s multiple comparisons test). (C) Immunoblot and quantification of p-eIF4E, total eIF4E, and beta-Actin levels in the pre-frontal cortex. Tissue from the pre-frontal cortex was collected 1 h after treatment in wild-type animals (n = 3 per group). Quantification of p-eIF4E signal compared to total eIF4E signal is presented as a ratio relative to vehicle-treated WT p-eIF4E expression (saline 1 ± 0.0131, 1 mg/kg 0.758 ± 0.096, 3 mg/kg 0.492 ± 0.170, *p = 0.005, 5 mg/kg 0.303 ± 0.041, ***p = 0.0009, 8 mg/kg 0.113 ± 0.056, ***p < 0.0001; 1-way ANOVA Tukey’s multiple-comparisons test). Actin was used as a load control. All data are presented as mean ± s.e.m. Statistical analysis was calculated by 1-way ANOVA with Tukey’s post hoc test
eFT508 Treatment Resolves Core Behavioral Abnormalities Associated with FXS
Hyperactivity and anxiety-like behaviors are notable characteristics of FXS [17, 38]. Increased fecal boli in an isolated animal indicates an increase in anxiety and fear [47]. Fmr1−/− mice have significantly more fecal boli in the arena at the end of testing as compared to wild-type animals (Fig. 2A). After eFT508 administration, fecal boli in the arena are significantly reduced in the Fmr1−/− mice in a dose dependent manner (Fig. 2A). As a second test, we made use of the open field assay. This test measures locomotion, exploratory behavior, and hyperactivity in freely moving animals [38]. We scored the total distance traveled by each mouse in the testing arena as a measure of hyperactivity. We observe the significant increase in distance traveled in the Fmr1−/− mice is resolved after a 5-mg/kg dose of eFT508 (Fig. 2B). The Fmr1−/− vehicle-treated group data are consistent with increased locomotion and activity within the arena, indicating hyperactivity. The hyperactive phenotype is resolved after treatment with eFT508. Moreover, wild-type mice treated with 5 mg/kg eFT508 are not negatively affected and retain a similar phenotype to the wild-type vehicle-treated group (Fig. 2B). These data are consistent with increased locomotion and activity within the arena, indicating hyperactivity. However, when treated with eFT508, Fmr1−/− mice display a decrease in distance traveled when treated with a dose of 5 mg/kg (Fig. 2B). Similarly, to measure the lasting effects of persistent eFT508 treatment in Fmr1−/− mice, we conducted the open field assay 72 h after the final treatment of eFT508 (Fig. S2B). We observe a consistent reduction in hyperactive behavior by eFT508-treated Fmr1−/− mice as indicated by a decrease in total distance traveled (Fig. S2B). We failed to detect differences in the amount of time spent in the center of the arena between wild-type and Fmr1−/− mice (Fig. S2A). Based on the similarity between the 5-mg/kg eFT508 treatment group and wild-type controls, this dosage was selected given for subsequent behavioral experiments.
Fig. 2.

eFT508 treatment resolves core behavioral deficits in Fmr1−/− mice. (A) Male mice were treated with vehicle or eFT508 intraperitoneally 1 h prior to the start of the open field assay. Fecal boli were counted at the culmination of the open field test, after 30 min. Boli from WT vehicle-treated mice (n = 12), WT eFT508 (5 mg/kg)-treated mice (n = 8), Fmr1−/− vehicle-treated mice (n = 12), and eFT508 (1 mg/kg, 3 mg/kg, 5 mg/kg, 8 mg/kg)-treated mice (n = 12 per group) were scored (wild-type vehicle 2.667 ± 0.6779, *p = 0.039; wild-type 5 mg/kg 1.875 ± 0.581, **p = 0.007; Fmr1−/− vehicle 4.750 ± 0.6528; Fmr1−/− 1 mg/kg 4.667 ± 0.482; Fmr1−/− 3 mg/kg 2.500 ± 0.417, *p = 0.022; Fmr1−/− 5 mg/kg 2.000 ± 0.603, **p = 0.003; 1-way ANOVA Tukey’s multiple comparisons test). (B) The open field assay was conducted as a measure of hyperactivity and anxiety-like behavior. Total distance traveled in the arena was scored. During the testing phase of the open field assay, mouse behavior was video recorded and analyzed electronically using the Noldus EthoVision XT tracking software. Data from WT vehicle-treated mice (n = 12), WT eFT508 (5 mg/kg)-treated mice (n = 8), Fmr1−/− vehicle-treated mice (n = 12), and eFT508-treated (varying doses) Fmr1−/− mice (n = 12 for each group) were collected and presented as total distance traveled in meters (wild-type vehicle 8.678 ± 1.323, ***p = 0.0006; wild-type 5 mg/kg 7.525 ± 0.620, **p = 0.0015; Fmr1−/− vehicle 17.19 ± 1.817; Fmr1−/− 1 mg/kg 9.972 ± 2.011, *p = 0.0348; Fmr1−/− 3 mg/kg 10.08 ± 1.966, *p = 0.0392; Fmr1−/− 5 mg/kg 5.256 ± 0.615, ****p < 0.0001; 1-way ANOVA Tukey’s multiple comparisons test). (C) Nest-building behavior is a measure of obsessive and repetitive behavior. At the end of the testing period, pictures of the dissociated nestlet were taken to be scored by a blinded observer on a scale of 1 to 5. Data from WT vehicle-treated mice (n = 12), WT eFT508 (5 mg/kg)-treated mice (n = 8), Fmr1−/− vehicle-treated mice (n = 12), and eFT508-treated (5 mg/kg) Fmr1−/− mice (n = 12) were collected (wild-type vehicle 1 ± 0.258, ****p < 0.0001; wild-type 5 mg/kg 0.500 ± 0.189, ****p < 0.0001; Fmr1-/- vehicle 3.833 ± 0.401; Fmr1-/- 5 mg/kg 1.125 ± 0.124, ****p < 0.0001; 1-way ANOVA Tukey’s multiple comparisons test). Representative images depicting WT vehicle-treated (left) and Fmr1−/− vehicle-treated (right) dissociated nests after 30 min of testing are shown. (D) Marble burying serves as another measure of repetitive behavior. At the end of the testing period, pictures of the buried marbles were taken to be scored by a blinded observer. Total marbles buried are counted and scored for WT vehicle-treated mice (n = 12), WT eFT508 (5 mg/kg)-treated mice (n = 8), Fmr1−/− vehicle-treated mice (n = 12), and eFT508-treated (5 mg/kg) Fmr1−/− mice (n = 12) groups (wild-type vehicle 1.667 ± 0.666, ****p < 0.0001; wild-type 5 mg/kg 1.000 ± 0.327, ****p < 0.0001; Fmr1−/− vehicle 9.625 ± 1.179; Fmr1−/− 5 mg/kg 2.714 ± 1.322, ***p = 0.0002; 1-way ANOVA Tukey’s multiple comparisons test). Representative images depicting WT vehicle-treated (left) and Fmr1−/− vehicle-treated (right) buried marbles after 30 min of testing are shown. Values are presented as mean ± s.e.m. Statistical analysis was conducted by 1-way ANOVA with Tukey’s post hoc test
Next, we probed obsessive and repetitive behaviors using 2 behavioral assays: nest building and marble burying [39, 41]. Nestlet shredding is scored on a graded scale of 0 to 5, with 0 indicating no dissociation of the nest and 5 indicating total dissociation of the nest. Fmr1−/− animals have a higher overall nest score (ca. 4) relative to wildtype (ca. 1) (Fig. 2C). Higher nest scores indicate increased engagement in obsessive and repetitive behaviors. After eFT508 administration, nest scores in the Fmr1−/− animals are reduced from 4 to 1 (Fig. 2C). As an additional test, we examined digging and burrowing behavior using a marble burying assay [39]. In these experiments, the total number of marbles buried beneath standard corncob bedding are counted at the end of the 30-min testing phase. Compared to wild-type, Fmr1−/− mice buried more marbles at the end of the testing phase (Fig. 2D). The increase in marbles buried in the FXS background indicate a higher tendency of digging behavior. Engagement in burrowing behaviors is a key marker of anxiety-like behaviors in mice. FXS animals treated with 5 mg/kg eFT508 displayed a significant decrease in marble burying (Fig. 2D). We observe no deviance from wild-type behavior in the wild-type eFT508-treated mice in the nest-building and marble-burying assays (Fig. 2C, D). Our experiments suggest that eFT508 resolves obsessive and repetitive behavioral patterns in 2 distinct assays.
Learning and Social Memory Improvement after eFT508 Treatment
Object recognition and social interaction are impaired in FXS [48]. To test cognitive function and preference for object novelty, we conducted a novel object recognition behavioral assay. During the testing phase, wild-type mice show a clear preference for novel objects, as quantified by total number of interactions (Fig. 3A). In comparison, Fmr1−/− mice do not display a bias for the novel or familiar objects during the testing phase (Fig. 3A). eFT508 treatment rescues this deficit. eFT508-treated Fmr1−/− animals interact more frequently with the novel object during testing (Fig. 3A). Furthermore, wild-type littermates treated with eFT508 displayed no significant change from the wild-type vehicle-treated groups (Fig. 3A). Representative heat maps indicate spatial preference for objects by highlighting the number of interactions with each object (Fig. 3A). These data suggest that eFT508 resolves core cognitive deficits in Fmr1−/− mice.
Fig. 3.

eFT508 treatment restores object recognition and social interactive deficits in fragile X syndrome. a Preference for novelty is assessed through the novel object recognition assay. Total interactions with novel and familiar objects during the 15-min testing phase are scored electronically using Noldus EthoVision XT tracking software. Data from WT vehicle-treated mice (n = 12), WT eFT508 (5 mg/kg)-treated mice (n = 8), Fmr1−/− vehicle-treated mice (n = 12), and eFT508-treated (5 mg/kg,) Fmr1−/− mice (n = 12) were collected (wild-type vehicle familiar 28.0 ± 4.694, wild-type vehicle novel 114.875 ± 11.606, ****p < 0.0001; wild-type 5 mg/kg familiar 21.375 ± 2.872, wild-type 5 mg/kg novel 64.375 ± 6.383, *p = 0.0132; Fmr1−/− vehicle familiar 57.571 ± 12.522, Fmr1−/− vehicle novel 61.0 ± 9.499; Fmr1−/− 5 mg/kg familiar 23.167 ± 2.845, Fmr1−/− 5 mg/kg novel 142.167 ± 23.590, ****p < 0.0001; 2-way ANOVA Sidak’s multiple comparisons test). Representative heat maps providing visual summary of mouse location during the testing phase of the behavioral assay are included for WT vehicle-treated and Fmr1−/− vehicle-treated animals. The white arrows indicate the locations of the novel and familiar objects in the testing arena. b The social approach assay compares total interactions between a novel male mouse and familiar inanimate object over a 15-min testing period. Mice from each testing group; WT vehicle (n = 12), WT eFT508 (5 mg/kg)-treated mice (n = 8), Fmr1−/− vehicle-treated (n = 12), and eFT508-treated (5 mg/kg,) Fmr1−/− (n = 12), are scored based on total interactions with the object or novel male (wild-type vehicle familiar 23.0 ± 2.708, wild-type vehicle novel 58.40 ± 13.430, *p = 0.0215; wild-type 5 mg/kg familiar 23.375 ± 6.032, wild-type 5 mg/kg novel 55.00 ± 5.292, *p = 0.011; Fmr1−/− vehicle familiar 61.917 ± 8.128, Fmr1−/− vehicle novel 20.625 ± 3.417, ***p = 0.0002; Fmr1−/− 5 mg/kg familiar 15.556 ± 4.014, Fmr1−/− 5 mg/kg novel 60.250 ± 16.044, **p = 0.0021; 2-way ANOVA Bonferroni’s multiple comparisons test). Heat maps display data indicating spatial preference of WT vehicle (left)- and Fmr1−/− vehicle (right)-treated mice during this phase of testing. c Social interaction I measures total interactions between a familiar male mouse compared to a novel male mouse. Total interactions between familiar and novel mice are quantified from WT vehicle (n = 12), WT eFT508 (5 mg/kg)-treated mice (n = 8), Fmr1−/− vehicle (n = 12), and eFT508-treated Fmr1−/− (n = 12) groups (wild-type vehicle familiar 27.571 ± 5.485, wild-type vehicle novel 84.833 ± 6.554, *p = 0.0139; wild-type 5 mg/kg familiar 22.625 ± 2.367, wild-type 5 mg/kg novel 73.875 ± 11.698, *p = 0.0144; Fmr1−/− vehicle familiar 58.571 ± 6.924, Fmr1−/− vehicle novel 29.222 ± 4.873; Fmr1−/− 5 mg/kg familiar 52.714 ± 7.963, Fmr1−/− 5 mg/kg novel 118.22 ± 24.458, **p = 0.0012; 2-way ANOVA Bonferroni’s multiple comparisons test). Representative heat maps for both WT vehicle (left) and Fmr1−/− vehicle (right) groups are included as visual summary. d Social interaction II measures total interactions between a familiar male mouse and a novel female mouse. To compare preferences between male and female mice, WT vehicle (n = 12), WT eFT508 (5 mg/kg) (n = 8), Fmr1−/− vehicle (n = 12), and eFT508-treated Fmr1−/− (n = 12) groups were scored (wild-type vehicle familiar 29.333 ± 2.716, wild-type vehicle novel 65.667 ± 16.180, ***p = 0.0006; wild-type 5 mg/kg familiar 37.250 ± 3.895, wild-type 5 mg/kg novel 63.000 ± 6.216, ***p = 0.0006; Fmr1−/− vehicle familiar 36.625 ± 3.973, Fmr1−/− vehicle novel 21.889 ± 3.634; Fmr1−/− 5 mg/kg familiar 13.571 ± 1.888, Fmr1−/− 5 mg/kg novel 40.000 ± 3.813, ***p = 0.0006; 2-way ANOVA Bonferroni’s multiple comparisons test). Heat maps for WT vehicle (left) and Fmr1−/− vehicle (right) groups provide representative data on spatial preference for each group in the arena. Social novelty assays are scored electronically with the Noldus EthoVision XT tracking software. White arrows on all representative heat maps indicate the locations of the novel and familiar animals in the testing arena. Data are presented as mean ± s.e.m. Statistical analysis was calculated by 2-way ANOVA with Bonferroni post hoc test
To examine social behavior and cognitive function, mice were subjected to a social interaction assay consisting of 3 stages. In the first phase of testing, mice are exposed to a familiar inanimate object and a novel mouse. Wild-type mice display a strong bias for the novel mouse, as indicated by the total number of interactions (Fig. 3B). Conversely, Fmr1−/− mice fail to display a significant difference in total number of interactions between novel and familiar stimuli (Fig. 3B). Additionally, no significant change in social approach behavior is observed in wild-type animals after eFT508 administration (Fig. 3B). Normal social approach behavior is restored after eFT508 treatment. Fmr1−/− animals demonstrate significant preference for the novel mouse after eFT508 administration (Fig. 3B). The second phase of the behavioral assay measures social preference between novel and familiar male mice by counting total number of interactions. During testing, wild-type mice interact more frequently with the novel male (Fig. 3C). Fmr1−/− mice do not display a significant difference between interactions with the novel male mouse or familiar male mouse (Fig. 3C). After treatment with eFT508, the deficit is rescued and the Fmr1−/− mice exhibit a social behavioral pattern similar to wild-type (Fig. 3C). Wild-type mice treated with eFT508 display no change in behavior after treatment during phase 2 of the social interactive assay (Fig. 3C). Consistent with the quantification, heat maps indicate spatial preference for the novel mouse in the wild-type background and lack of spatial bias in the Fmr1−/− mice (Fig. 3C). The last phase of social behavior testing measures total interactions of the experimental group between a novel female mouse and a familiar male mouse. As indicated by both the heat map and quantitative measurements of total interactions, wild-type mice have a higher incidence of interactions with the novel female (Fig. 3D). This phenotype is also observed in wild-type mice treated with eFT508, indicating no significant change in behavior in the wild-type littermates due to drug treatment (Fig. 3D). Fmr1−/− mice lack preference for novel social interaction and have a comparable number of interactions between novel and familiar stimuli (Fig. 3D). eFT508-treated Fmr1−/− mice interact with the novel female mouse more frequently during testing (Fig. 3D). Collectively, the data suggest that eFT508 rescues cognitive and social impairments in Fmr1−/− animals.
eFT508 Administration Corrects Physiological Deficits and Neuronal Maturation in the FXS Background
We sought to determine if additional cellular and physiologic features of FXS were reversed by eFT508. We focused specifically on aberrant dendritic spine density formation, exaggerated long-term depression (LTD), and macroorchidism [2, 3, 48]. Dendritic spines are a key morphological marker of plasticity given their involvement in synaptic strengthening. We observe a significant increase in spine density in the Fmr1-deficient mice as compared to wild type, suggesting neuronal immaturity and dendritic abnormalities (Fig. 4A) [6, 15]. Furthermore, an increase in dendritic spine density suggests abnormal dendritic pruning in the cortex of Fmr1−/− mice [49]. Injection of eFT508 over the course of 6 days reverts the abnormal dendritic spine density in mutant animals to levels comparable to that of wild-type mice (Fig. 4A). These results suggest that eFT508 promotes dendritic spine maturation and can correct aberrant spinogenesis associated with loss of Fmr1.
Fig. 4.

eFT508 treatment resolves aberrant spine density formation, macroorchidism, exaggerated LTD, and exaggerated p-eIF4E levels in the hippocampus. a Quantification of dendritic spine density is scored as the total number of spines per 10 μm of the total dendritic length. Spines are measured for each dendrite branching off of the pyramidal cell body (n = 20 for each animal). Data from WT vehicle-treated mice (n = 6), Fmr1−/− vehicle-treated mice (n = 7), and eFT508-treated (5 mg/kg) Fmr1−/− mice (n = 7) were collected (wild-type vehicle 10.031 ± 0.505, ****p < 0.0001, n = 6; Fmr1−/− vehicle 17.250 ± 0.939, n = 7; Fmr1−/− 5 mg/kg 10.843 ± 0.544, ***p < 0.0001, 1-way ANOVA Tukey’s multiple comparisons test). Representative images depicting spine density for WT vehicle-treated, Fmr1−/− vehicle-treated, and Fmr1−/− eFT508-treated groups are included. Scale bar represents 2 μm. b LTD extracellular field potential response is recorded as a function of percent change in baseline rising fEPSP slope. Stable baseline recording is established and recorded for 20 min prior to LTD induction. LTD is induced through low-frequency stimulation of 0.1-ms pulses at 1 Hz for 15 min. fEPSP recording and LTD recovery are recorded for 40 min after LTD induction. Data are presented as an average of 1 to 3 slices per animal in WT vehicle-treated mice (n = 5), Fmr1−/− vehicle-treated mice (n = 9), and eFT508-treated (5 mg/kg) Fmr1−/− mice (n = 6). c Quantification of LTD response during the last 20 min of electrophysiological extracellular field potential response. Change in fEPSP slope for individual slices is quantified for wild-type vehicle (n = 10), Fmr1−/− vehicle (n = 11), and eFT508-treated (5 mg/kg) Fmr1−/− slices (n = 10) (wild-type vehicle 82.540 ± 13.66, *p = 0.016, Fmr1−/− vehicle 32.40 ± 8.93, Fmr1−/− 5 mg/kg 97.20 ± 18.15, *p = 0.01; 1-way ANOVA Tukey’s multiple comparisons test). d Localization of p-eIF4E signal to neurons in the hippocampus of Fmr1−/− mice after vehicle and eFT508 treatment is represented. Hippocampal slices are stained with DAPI, NeuN, and p-eIF4E (left to right). e Quantification and localization of p-eIF4E levels in the hippocampus of Fmr1−/− mice. p-eIF4E florescence staining is normalized to NeuN staining. Scale bar represents 100 μm. Data are presented as an average of 5 slices per animal in Fmr1−/− vehicle-treated (n = 8) and Fmr1−/− eFT508-treated (n = 8) groups (total hippocampal quantification – vehicle, eFT508) (DG vehicle 2.495 ± 0.385, DG eFT508 1.404 ± 0.293, *p = 0.014, CA1 vehicle 1.902 ± 0.225, CA1 eFT508 1.004 ± 0.226, *p = 0.0432, CA3 vehicle 0.934 ± 0.087, CA3 eFT508 1.137 ± 0.230, p = 0.927; 2-way ANOVA Sidak’s multiple comparisons test). Statistical analysis was conducted by 2-way ANOVA with Sidak’s multiple comparisons test. f Phosphorylated eIF4E levels in neurons of the hippocampus of wild-type mice after vehicle and eFT508 treatment are depicted. Slices of the hippocampus are stained with DAPI, NeuN, and phosphorylated eIF4E (left to right). g Normalized phosphorylated eIF4E levels in the hippocampus of wild-type mice in the total hippocampal area, dentate gyrus, CA1 region, and CA3 region. The scale bar indicates 100 μm. Phosphorylated eIF4E florescence signal is normalized to NeuN staining. Data are presented as an average of 6 slices from each animal in the wild-type vehicle-treated group (n = 9) and the wild-type eFT508 5 mg/kg treated group (n = 9) (Total hippocampal quantification – vehicle 1.120 ± 0.047, eFT508 0.879 ± 0.041, **p = 0.002) (DG vehicle 1.255 ± 0.075, DG eFT508 0.967 ± 0.074, *p = 0.01, CA1 vehicle 1.163 ± 0.073, CA1 eFT508 0.905 ± 0.054, **p = 0.008, CA3 vehicle 0.943 ± 0.087, CA3 eFT508 0.728 ± 0.083, p = 0.075; 2-way ANOVA Sidak’s multiple comparisons test). Statistical analysis was conducted by 2-way ANOVA with Sidak’s multiple comparisons test. (H) Mean testicular weight was collected from WT vehicle-treated (n = 4), Fmr1−/− vehicle-treated mice (n = 4), and eFT508-treated (5 mg/kg) Fmr1−/− mice (n = 4) after 6 days of treatment (wild-type vehicle 143.3 ± 2.910, **p = 0.009, Fmr1−/− vehicle 194.7 ± 3.535, Fmr1−/− 5 mg/kg 168.3 ± 10.262, *p = 0.0443; n = 4 per group; 1-way ANOVA Tukey’s multiple comparisons test). Representative images for WT, Fmr1−/− vehicle, and eFT508-treated (5 mg/kg) Fmr1−/− mice are included. Scale bar represents 2.5 mm. All data are presented as mean ± s.e.m. Statistical analysis was calculated by 1-way ANOVA with Tukey’s post hoc test or 2-way ANOVA with Bonferroni post hoc test unless further specified
Fmr1−/− mice display an exaggerated and sustained LTD response in the hippocampus [6, 7]. An altered LTD response interferes with normal synaptic function resulting in cognitive and behavioral deficits [50]. Consistent with previous reports, we found that LTD induced by low-frequency stimulation (LFS-LTD) of the hippocampal Schaffer collateral pathway is amplified in the Fmr1−/− compared to wild-type mice (Fig. 4B). A single eFT508 injection 1 h prior to slice collection eliminated the exaggerated LFS-LTD response in the Schaffer collateral pathway in the FXS background (Fig. 4B). Additional quantification of the electrophysiological response indicates that LFS-LTD in the Fmr1−/− eFT508-treated background is comparable to the wild-type response (Fig. 4C). The data suggest that administration of eFT508 is sufficient to normalize this important form of synaptic plasticity in the CA1 region of the hippocampus in Fmr1−/− mice.
To determine if eFT508 treatment had differential impacts in specific regions of the brain, we examined eIF4E phosphorylation using immunohistochemistry in Fmr1−/− mice (Fig. 4D). A single dose of eFT508 significantly reduced phosphorylation by 55% in the dentate gyrus (DG) and 42% in CA1 (Fig. 4E). We observe similar effects of eFT508 treatment in the hippocampus of wild-type mice (Fig. 4F). Treatment with eFT508 in wild-type mice causes significant reduction of eIF4E phosphorylation in the dentate gyrus and CA1 regions of the hippocampus (Fig. 4G). These results indicate that eFT508 reduces eIF4E phosphorylation in the brain.
A physiological feature of Fmr1−/− mice that mirrors that of FXS patients is macroorchidism [2, 51]. Increased proliferation of Sertoli cells in the FXS background is the cause for increase testicular weight [50–52]. Consistent with previous literature, we observe that the Fmr1−/− mice have a significant increase in testicular weight when compared with wild type (Fig. 4H). Administration of eFT508 over 5 days resolves macroorchidism (Fig. 4H). This suggests that eFT508 acts in the testes in addition to the brain.
Discussion
Our experiments elucidate the potential of MNK inhibition by eFT508 for treatment of FXS. The data support 3 major conclusions. First, eFT508 attenuates eIF4E phosphorylation in the central nervous system after systemic injection. Second, eFT508 resolves core behavioral phenotypes present in Fmr1−/− animals. Finally, eFT508 reverses abnormal neuronal physiology caused by FMRP loss.
Aberrant activation of the MAP kinase pathway, as observed in the Fmr1−/− mice, is also observed in brain samples from FXS patients [6, 14, 53–55]. Specifically, the extracellular regulated kinase 1/2 (ERK1/2) directly upstream of MNK has emerged as a potential target for FXS [56, 57]. Lovastatin, a commonly used cholesterol-reducing agent, attenuates ERK1/2 and is currently being investigated in clinical trials for fragile X [58–62]. One important downstream consequence of increased MAPK and ERK1/2 is increased activity of MNK. The best characterized substrate of MNK is eIF4E; thus, MNK inhibition should provide an effective and potentially more specific way to control eIF4E phosphorylation [24, 25]. Previous MNK inhibitors, including cercosporamide, CGP052088, and CGP57380, have proven to be both less potent and less specific than eFT508 [54–63]. For example, the IC50 values for cercosporamide are 116 nM (MNK1) and 11 nM (MNK2) [64–68], suggesting a preference for inhibiting MNK2. CGP57380, a MNK1-specific inhibitor, has low potency with an IC50 of 3 μM [69–71], while CGP052088, which is derived from the kinase inhibitor staurosporine, has an IC50 value of 70 nM for MNK1 but also inhibits CK1, SGK, BRSK2, and DYRK [72]. In contrast, the IC50 values for eFT508 are 2.4 nM (MNK1) and 1 nM (MNK2) [33]. Thus, eFT508 is both more potent and selective than other MNK inhibitors. To the best of our knowledge, it is the only MNK inhibitor currently in clinical trials. Phase I clinical data indicate that eFT508 is well tolerated in patients with advanced solid tumors [73]. Moreover, eFT508 has shown sufficient inhibition of eIF4E phosphorylation systemically in patients after oral administration [73]. Our experiments illustrate the potency and efficacy of eFT508 administration in a model of FXS. Given that we observed effects of eFT508 outside of the CNS (e.g., macroorchidism), a key consideration for any future clinical studies will be monitoring for signs of off pathway effects. We found that acute eFT508 treatment resolves core deficits in Fmr1−/− mice with systemic intraperitoneal injections. Our experiments further establish that eFT508 crosses the blood–brain barrier and can reduce eIF4E phosphorylation in the brain.
We found that exaggerated eIF4E phosphorylation in the hippocampus, striatum, and prefrontal cortex of Fmr1−/− mice is attenuated by a single dose of eFT508. However, it also targets non-neuronal cell types. For example, we found that eFT508 treatment resolves macroorchidism after repeated dosing. Multiple sites of action are consistent with the fact that eFT508 reverses pain-associated behaviors on mouse models of chemotherapy-induced peripheral neuropathy and nerve injury [35–37]. Nevertheless, systemic administration of eFT508 is advantageous in the context of FXS as it resolves macroorchidism—a cardinal hallmark of the disorder in humans. A potential means to increase the potency of eFT508 in the context of FXS could be through different formulations. For example, polymeric nanoparticles and liposomes could provide a potential vehicle to increase delivery to the CNS [74].
eIF4E has been implicated in multiple functions in the nervous system. For example, eIF4E phosphorylation is required for the chronic action of antidepressants [75]. Nonphosphorylatable eIF4E mutants display behaviors associated with depression and anxiety [6, 75]. We found that behavioral phenotypes caused by deletion of Fmr1 including hyperactivity, anxiety-like behaviors, and cognitive and social recognition deficits are normalized after eFT508 treatment. In particular, we observe a significant increase in marbles buried in the marble burying test and an increase in nesting behavior in the fragile X animals. Our marble burying data are consistent with the current understanding that increased burrowing behavior in this assay is indicative of increased anxiety-like behaviors in Fmr1−/− mice [76–78]. Our nest-building data indicate that FXS animals engage in more nest-building behavior than wild type, contrary to previously reported data [79–81]. We reconcile these differences with 2 explanations. First, all nest-building behavior was conducted in the mouse’s home cage, with the littermates removed, rather than a new environment. This difference in environment might be cause for the difference in phenotype. Second, nest-building behavior in male mice requires involvement in shredding behavior and repetitive actions. Engagement in repetitive behaviors is also a core phenotype of FXS animals [76, 77, 82]. Deviance from wild-type behavior in the FXS animals is resolved after eFT508 administration in both the nest-building and marble-burying tests. This suggests that eFT508 could be broadly applicable to a range of neurological disorders driven by eIF4E.
FXS causes numerous changes in neuroanatomy and physiology. These include increased dendritic spine density and an exaggerated LTD response [2, 3, 7, 9]. FMRP is present in neurons and is localized to dendrites [83–85]. Loss of FMRP causes an increase in immature dendritic spine populations concurrent with exaggerated spine density in cortical neurons [4, 5, 53, 86]. This deficit is likely due to the role of FMRP in local translation. FMRP cofractionates with ribosomes isolated from cortical synaptosomes and regulates translation elongation [10, 14, 85, 87]. After repeated treatment of eFT508, which impacts translation initiation, spine density decreases significantly [6]. In addition to factors associated with recognition of the 5′ end of mRNA, polyadenylation has been linked to spine density. Depletion of factors involved in polyadenylation including the cytoplasmic polyadenylation element binding (CPEB) protein, a poly(A) polymerase (Gld-2), the deadenylase (PARN), or neuroguidin increases dendritic spine density [88, 89]. Thus, multiple mechanisms govern translational control converge on spinogenesis.
What are the downstream targets of translational regulation that are relevant to FXS? There are hundreds of targets of FMRP that have been identified using functional genomics [11, 90–94]. Some of these targets have been proven to be relevant in FXS. For example, Fmr1−/− mice have an enhanced LTD response that is driven by increased levels of metabotropic glutamate receptors (mGluRs) [95–98]. In addition to regulation by FMRP, translation of mGluRs is also controlled by CPEB. Deletion of CPEB in Fmr1−/− mice reduces exaggerated LTD in these animals [99]. Matrix metallopeptidase 9 (Mmp-9), a putative mRNA regulated by eIF4E phosphorylation, has also been suggested as a relevant target in the context of Fmr1 loss. Modulation of Mmp-9, in a mouse model of FXS, resolves social deficits, hyperactivity, spine density formation, and an enhanced LTD response [30, 100]. An obvious next step in understanding the mechanism of eFT508 will be profiling its effects on the landscape of nascent translation in brain tissue.
Our findings suggest that the potent MNK inhibitor, eFT508, reverses several phenotypic abnormalities in the Fmr1−/− mouse model of FXS. Additional tests are needed to determine if the approach is viable in additional animal and patient models. Aberrant translation is a dominant theme in neurobiology beyond FXS and has been reported in Alzheimer’s, ALS, and vanishing white matter disease [101–103]. The identification of a highly specific inhibitor that can access the CNS provides a powerful new tool for attenuating dysregulated cap-dependent translation in the brain.
Electronic Supplementary Material
(DOCX 3.96mb)
(PDF 436 kb)
Acknowledgments
We thank Faruck Morcos and Xianli Jiang for technical assistance with Matlab.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author Contributions
T.S., J.E.P., and Z.T.C. conceived and designed the project. T.S., J.B.D.P., J.M.P., and C.A.T. conducted the experiments and data analysis. C.R.S. and K.R.W. developed and/or supported the development of eFT508. T.S., J.B.D.P., C.R.S. and Z.T.C. prepared the manuscript.
Funding
C.R.S. and K.R.W. are employed by eFFECTOR therapeutics. T.S., J.B.D.P., J.M.P., J.E.P., C.A.T., and Z.T.C. declare no competing interests. This work was supported by NIH grant R01NS100788 (ZTC) and R01NS114018 (ZTC).
Footnotes
eFT508 Attenuates Behavioral Deficits in a Mouse Model of FXS
Summary
The behavioral deficits associated with fragile X syndrome (FXS) are driven by molecular mechanisms that remain largely unknown. Hyperphosphorylation of eIF4E, a key translation initiation factor, is well studied in the context of fragile X syndrome. Here, we reveal that precise control of protein translation with a small molecule inhibitor, eFT508, can resolve key phenotypes present in FXS. Systemic treatment with a potent MNK inhibitor rescues the core phenotypic deficiencies in FXS. This research provides evidence for a potential therapeutic in the context of FXS.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hernandez RN, Feinberg RL, Vaurio R, Passanante NM, Thompson RE, Kaufmann WE. Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Med Genet A [Internet]. 2009 [cited 2019 Aug 16];149A(6):1125–37. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19441123 [DOI] [PMC free article] [PubMed]
- 2.Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet [Internet]. 2008 9 [cited 2019 Sep 26];16(6):666–72. Available from: http://www.nature.com/articles/ejhg200861 [DOI] [PMC free article] [PubMed]
- 3.Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB. Neurodevelopmental effects of the FMR-1 full mutation in humans. Nat Med. 1995;1(2):159–67. doi: 10.1038/nm0295-159. [DOI] [PubMed] [Google Scholar]
- 4.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A [Internet]. 2002 [cited 2019 Sep 27];99(11):7746–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12032354 [DOI] [PMC free article] [PubMed]
- 5.Greenough WT, Klintsova AY, Irwin SA, et al. Synaptic regulation of protein synthesis and the fragile X protein. Proc Natl Acad Sci [Internet]. 2001 [cited 2019 Jun 6];98(13):7101–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11416194 [DOI] [PMC free article] [PubMed]
- 6.Gkogkas CG, Khoutorsky A, Cao R, et al. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep [Internet]. 2014/12/04. 2014 [cited 2018 Jul 17];9(5):1742–55. Available from: 10.1016/j.celrep.2014.10.064. [DOI] [PMC free article] [PubMed]
- 7.Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, et al. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. 2012 [cited 2018 Jul 17]; Available from: 10.1016/j.neuron.2012.07.022 [DOI] [PMC free article] [PubMed]
- 8.Verkerk AJMH, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–14. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 9.Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Vol. 12, Lancet Neurol. 2013. p. 786–98. [DOI] [PMC free article] [PubMed]
- 10.Stefani G, Fraser CE, Darnell JC, Darnell RB. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci [Internet]. 2004 [cited 2019 Aug 16];24(33):7272–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15317853 [DOI] [PMC free article] [PubMed]
- 11.Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell [Internet]. 2011 [cited 2019 Aug 16];146(2):247–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21784246 [DOI] [PMC free article] [PubMed]
- 12.Chen E, Sharma MR, Shi X, Agrawal RK, Joseph S. Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol Cell [Internet]. 2014 [cited 2018 Jul 17];54(3):407–17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24746697 [DOI] [PMC free article] [PubMed]
- 13.Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet [Internet]. 2001 [cited 2018 Aug 27];10(4):329–38. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11157796 [DOI] [PubMed]
- 14.Das Sharma S, Metz JB, Li H, et al. Widespread alterations in translation elongation in the brain of juvenile Fmr1 knockout mice. Cell Rep. 2019;26(12):3313–3322. doi: 10.1016/j.celrep.2019.02.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gantois I, Khoutorsky A, Popic J, et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat Med [Internet]. 2017 15 [cited 2018 Jul 16];23(6):674–7. Available from: http://www.nature.com/doifinder/10.1038/nm.4335 [DOI] [PubMed]
- 16.The Dutch-Belgian Fragile X Consorthium, Bakker CE, Verheij C, et al. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell [Internet]. 1994 [cited 2020 Jan 9];78(1):23–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8033209 [PubMed]
- 17.Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes, Brain Behav [Internet]. 2004 1 [cited 2019 Sep 26];3(6):337–59. Available from: http://doi.wiley.com/10.1111/j.1601-183X.2004.00087.x [DOI] [PubMed]
- 18.Santini E, Huynh TN, Longo F, Koo SY, Mojica E, D'Andrea L, Bagni C, Klann E. Reducing eIF4E-eIF4G interactions restores the balance between protein synthesis and actin dynamics in fragile X syndrome model mice. Sci Signal. 2017 Nov 7;10(504):eaan0665. 10.1126/scisignal.aan0665. [DOI] [PMC free article] [PubMed]
- 19.Sonenberg N, Rupprecht KM, Hecht SM, Shatkin AJ. Eukaryotic mRNA cap binding protein: purification by affinity chromatography on sepharose-coupled m7GDP. Proc Natl Acad Sci U S A. 1979;76(9):4345–9. doi: 10.1073/pnas.76.9.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gingras A-C, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem [Internet]. 1999 [cited 2019 Aug 30];68(1):913–63. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10872469 [DOI] [PubMed]
- 21.Neff CL, Sachs AB. Eukaryotic translation initiation factors 4G and 4A from Saccharomyces cerevisiae interact physically and functionally. Mol Cell Biol [Internet] 1999;19(8):5557–64. doi: 10.1128/mcb.19.8.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galicia-Vázquez G, Cencic R, Robert F, Agenor AQ, Pelletier J. A cellular response linking eIF4AI activity to eIF4AII transcription. RNA. 2012;18(7):1373–84. doi: 10.1261/rna.033209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duncan R, Hershey JW. Identification and quantitation of levels of protein synthesis initiation factors in crude HeLa cell lysates by two-dimensional polyacrylamide gel electrophoresis. J Biol Chem [Internet]. 1983 [cited 2020 Jan 9];258(11):7228–35. Available from: http://www.ncbi.nlm.nih.gov/pubmed/6853516 [PubMed]
- 24.Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999;18(1):270–9. doi: 10.1093/emboj/18.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol. 1999;19(3):1871–80. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Yue P, Deng X, Ueda T, Fukunaga R, Khuri FR, et al. Protein phosphatase 2A negatively regulates eukaryotic initiation factor 4E phosphorylation and eif4F assembly through direct dephosphorylation of mnk and eif4E. Neoplasia [Internet]. 2010 [cited 2020 Aug 26];12(10):848–55. Available from: /pmc/articles/PMC2950334/?report=abstract [DOI] [PMC free article] [PubMed]
- 27.Kolupaeva V. Serine-threonine protein phosphatases: lost in translation [Internet]. Vol. 1866, Biochimica et Biophysica Acta - Molecular Cell Research. Elsevier B.V.; 2019 [cited 2020 Aug 26]. p. 83–9. Available from: /pmc/articles/PMC6430236/?report=abstract [DOI] [PMC free article] [PubMed]
- 28.Bu X, Hagedorn CH. Phosphoprotein phosphatase 2A dephosphorylates eIF-4E and does not alter binding to the mRNA cap. FEBS Lett [Internet]. 1992 13 [cited 2020 Aug 26];301(1):15–8. Available from: https://pubmed.ncbi.nlm.nih.gov/1333409/ [DOI] [PubMed]
- 29.Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51(4):441–54. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 30.Sidhu H, Dansie LE, Hickmott PW, Ethell DW, Ethell IM. Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J Neurosci. 2014;34(30):9867–79. doi: 10.1523/JNEUROSCI.1162-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sahra IB, Regazzetti C, Robert G, Laurent K, Lemarchandbrustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71(13):4366–72. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 32.Reich SH, Sprengeler PA, Chiang GG, et al. Structure-based design of pyridone-aminal eFT508 targeting dysregulated translation by selective mitogen-activated protein kinase interacting kinases 1 and 2 (MNK1/2) inhibition. J Med Chem [Internet]. 2018 26 [cited 2020 Jan 6];61(8):3516–40. Available from: https://pubs.acs.org/doi/10.1021/acs.jmedchem.7b01795 [DOI] [PubMed]
- 33.Webster KR, Goel VK, Staunton J, et al. eFT508, a potent and highly selective inhibitor of MNK 1/2, regulates immune checkpoint and cytokine expression promoting anti-tumor immunity [Internet]. [cited 2019 Aug 16]. Available from: https://effector.com/wp-content/uploads/2018/07/eFFECTOR-eFT508-AACR-2017-poster.pdf
- 34.Xu Y, Poggio M, Jin HY, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25(2):301–11. doi: 10.1038/s41591-018-0321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Megat S, Ray PR, Moy JK, et al. Nociceptor translational profiling reveals the ragulator-rag gtpase complex as a critical generator of neuropathic pain. J Neurosci. 2019;39(3):393–411. doi: 10.1523/JNEUROSCI.2661-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiers S, Mwirigi J, Pradhan G, et al. Reversal of peripheral nerve injury-induced neuropathic pain and cognitive dysfunction via genetic and tomivosertib targeting of MNK. Neuropsychopharmacology. 2019; [DOI] [PMC free article] [PubMed]
- 37.Moy JK, Khoutorsky A, Asiedu MN, et al. The MNK–eIF4E signaling axis contributes to injury-induced nociceptive plasticity and the development of chronic pain. J Neurosci [Internet]. 2017 2 [cited 2019 Aug 28];37(31):7481–99. Available from: https://www.jneurosci.org/content/37/31/7481/tab-figures-data [DOI] [PMC free article] [PubMed]
- 38.Seibenhener ML, Wooten MC. Use of the open field maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp 2015;(96). [DOI] [PMC free article] [PubMed]
- 39.Deacon RMJ. Digging and marble burying in mice: Simple methods for in vivo identification of biological impacts. Nat Protoc. 2006;1(1):122–4. doi: 10.1038/nprot.2006.20. [DOI] [PubMed] [Google Scholar]
- 40.Crawley J, Bailey K. Anxiety-related behaviors in mice. In 2008. p. 77–101. [PubMed]
- 41.Deacon RMJ. Assessing nest building in mice. Nat Protoc. 2006;1(3):1117–9. doi: 10.1038/nprot.2006.170. [DOI] [PubMed] [Google Scholar]
- 42.Barragán-Iglesias P, Lou T-F, Bhat VD, et al. Inhibition of poly(A)-binding protein with a synthetic RNA mimic reduces pain sensitization in mice. Nat Commun [Internet]. 2018 2 [cited 2019 Sep 3];9(1):10. Available from: http://www.nature.com/articles/s41467-017-02449-5 [DOI] [PMC free article] [PubMed]
- 43.Bostrom C, Yau S Y, Majaess N, Vetrici M, Gil-Mohapel J, Christie BR. Hippocampal dysfunction and cognitive impairment in fragile-X syndrome. Vol. 68, Neurosci Biobehav Rev. Elsevier Ltd; 2016. p. 563–74. [DOI] [PubMed]
- 44.Krueger DD, Osterweil EK, Chen SP, Tye LD, Bear MF. Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2011;108(6):2587–92. doi: 10.1073/pnas.1013855108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gothelf D, Furfaro JA, Hoeft F, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP) Ann Neurol. 2008;63(1):40–51. doi: 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoeft F, Hernandez A, Parthasarathy S, Watson CL, Hall SS, Reiss AL. Fronto-striatal dysfunction and potential compensatory mechanisms in male adolescents with fragile X syndrome. Hum Brain Mapp. 2007;28(6):543–54. doi: 10.1002/hbm.20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall CS. Emotional behavior in the rat. I. Defecation and urination as measures of individual differences in emotionality. J Comp Psychol. 1934;18(3):385–403. [Google Scholar]
- 48.Kazdoba TM, Leach PT, Silverman JL, Crawley JN. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable rare Dis Res [Internet]. 2014 Nov [cited 2018 Jul 17];3(4). Available from: www.irdrjournal.com [DOI] [PMC free article] [PubMed]
- 49.He CX, Portera-cailliau C. The trouble with spines in fragile X syndrome: density, maturity and plasticity. Vol. 251, Neuroscience. 2013. p. 120–8. [DOI] [PMC free article] [PubMed]
- 50.Oostra BA, Nelson DL. Animal models of fragile X syndrome: mice and flies. 2006.
- 51.Slegtenhorst-Eegdeman KE, De Rooij DG, Verhoef-Post M, et al. Macroorchidism in FMR1 knockout mice is caused by increased Sertoli cell proliferation during testicular development. 1998 [cited 2018 Jul 16]. Available from: https://academic.oup.com/endo/article-abstract/139/1/156/2987048 [DOI] [PubMed]
- 52.Jégou B, Stéphan JP, Cudicini C, et al. The Sertoli cell-germ cell interactions and the seminiferous tubule interleukin-1 and interleukin-6 system. Vol. 28, Results Probl Cell Differ. 2000. p. 53–68. [DOI] [PubMed]
- 53.Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. 2010 [cited 2018 Aug 9]; Available from: www.jneurosci.org [DOI] [PMC free article] [PubMed]
- 54.Sawicka K, Pyronneau A, Chao M, Bennett MVL, Zukin RS, Purpura DP. Elevated ERK/p90 ribosomal S6 kinase activity underlies audiogenic seizure susceptibility in fragile X mice. [cited 2018 Jul 16]; Available from: www.pnas.org/cgi/doi/10.1073/pnas.1610812113 [DOI] [PMC free article] [PubMed]
- 55.Wang X, Snape M, Klann E, et al. Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J Neurochem. 2012;121(4):672–9. doi: 10.1111/j.1471-4159.2012.07722.x. [DOI] [PubMed] [Google Scholar]
- 56.Asiminas A, Jackson AD, Louros SR, et al. Sustained correction of associative learning deficits after brief, early treatment in a rat model of Fragile X Syndrome. Sci Transl Med 2019;11(494). [DOI] [PMC free article] [PubMed]
- 57.Muscas M, Louros SR, Osterweil EK. Lovastatin, not simvastatin, corrects core phenotypes in the fragile X mouse model. eNeuro 2019 1;6(3). [DOI] [PMC free article] [PubMed]
- 58.Pellerin D, Çaku A, Fradet M, Bouvier P, Dubé J, Corbin F. Lovastatin corrects ERK pathway hyperactivation in fragile X syndrome: potential of platelet’s signaling cascades as new outcome measures in clinical trials. Biomarkers [Internet]. 2016 17 [cited 2020 Mar 10];21(6):497–508. Available from: http://www.tandfonline.com/doi/full/10.3109/1354750X.2016.1160289 [DOI] [PubMed]
- 59.Çaku A, Pellerin D, Bouvier P, Riou E, Corbin F. Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. Am J Med Genet Part A [Internet]. 2014 [cited 2020 Mar 10];164(11):2834–42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25258112 [DOI] [PubMed]
- 60.Berry-Kravis EM, Lindemann L, Jønch AE, et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Vol. 17, Nat Rev Drug Discov. Nature Publishing Group; 2018. p. 280–98. [DOI] [PMC free article] [PubMed]
- 61.Thurman AJ, Potter LA, Kim K, et al. Controlled trial of lovastatin combined with an open-label treatment of a parent-implemented language intervention in youth with fragile X syndrome. J Neurodev Disord [Internet]. 2020 [cited 2020 Aug 26];12(1):12. Available from: https://jneurodevdisorders.biomedcentral.com/articles/10.1186/s11689-020-09315-4 [DOI] [PMC free article] [PubMed]
- 62.Osterweil EK, Chuang SC, Chubykin AA, et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron [Internet]. 2013 [cited 2020 Aug 26];77(2):243–50. Available from: https://pubmed.ncbi.nlm.nih.gov/23352161/ [DOI] [PMC free article] [PubMed]
- 63.Chrestensen CA, Shuman JK, Eschenroeder A, Worthington M, Gram H, Sturgill TW. MNK1 and MNK2 regulation in HER2-overexpressing breast cancer lines. J Biol Chem. 2007;282(7):4243–52. doi: 10.1074/jbc.M607368200. [DOI] [PubMed] [Google Scholar]
- 64.Sugawara F, Strobel S, Strobel G, et al. The structure and biological activity of cercosporamide from Cercosporidium henningsii. J Org Chem [Internet]. 1991 Feb [cited 2020 Jan 12];56(3):909–10. Available from: https://pubs.acs.org/doi/abs/10.1021/jo00003a002
- 65.Sussman A, Huss K, Chio LC, et al. Discovery of cercosporamide, a known antifungal natural product, as a selective Pkc1 kinase inhibitor through high-throughput screening. Eukaryot Cell. 2004;3(4):932–43. doi: 10.1128/EC.3.4.932-943.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Furukawa A, Arita T, Satoh S, Araki K, Kuroha M, Ohsumi J. (-)-Cercosporamide derivatives as novel antihyperglycemic agents. Bioorg Med Chem Lett. 2009;19(3):724–6. doi: 10.1016/j.bmcl.2008.12.035. [DOI] [PubMed] [Google Scholar]
- 67.Konicek BW, Stephens JR, McNulty AM, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res [Internet]. 2011 [cited 2019 Sep 3];71(5):1849–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21233335 [DOI] [PubMed]
- 68.Hou J, Lam F, Proud C, Wang S. Targeting Mnks for cancer therapy. Vol. 3, Oncotarget Impact J LLC; 2012. p. 118–31. [DOI] [PMC free article] [PubMed]
- 69.Knauf U, Tschopp C, Gram H. Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol Cell Biol. 2001;21(16):5500–11. doi: 10.1128/MCB.21.16.5500-5511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishida M, Ishida T, Nakashima H, et al. Mnk1 is required for angiotensin II-induced protein synthesis in vascular smooth muscle cells. Circ Res. 2003;93(12):1218–24. doi: 10.1161/01.RES.0000105570.34585.F2. [DOI] [PubMed] [Google Scholar]
- 71.Andersson K, Sundler R. Posttranscriptional regulation of TNFα expression via eukaryotic initiation factor 4E (eIF4E) phosphorylation in mouse macrophages. Cytokine. 2006;33(1):52–7. doi: 10.1016/j.cyto.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 72.Tschopp C, Knauf U, Brauchle M, et al. Phosphorylation of eIF-4E on Ser 209 in response to mitogenic and inflammatory stimuli is faithfully detected by specific antibodies. Mol Cell Biol Res Commun. 2000;3(4):205–11. doi: 10.1006/mcbr.2000.0217. [DOI] [PubMed] [Google Scholar]
- 73.Falchook GS, Infante JR, Meric-Bernstam F, et al. A phase 1 dose escalation study of eFT508, an inhibitor of mitogen-activated protein kinase-interacting serine/threonine kinase-1 (MNK-1) and MNK-2 in patients with advanced solid tumors. J Clin Oncol. 2017;35(15_suppl):2579–2579. [Google Scholar]
- 74.Khan AR, Yang X, Fu M, Zhai G. Recent progress of drug nanoformulations targeting to brain. Vol. 291, J Control Release. Elsevier B.V.; 2018. p. 37–64. [DOI] [PubMed]
- 75.Amorim IS, Kedia S, Kouloulia S, et al. Loss of eIF4E phosphorylation engenders depression-like behaviors via selective mRNA translation. J Neurosci. 2018;38(8):2118–33. doi: 10.1523/JNEUROSCI.2673-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gross C, Raj N, Molinaro G, et al. Selective role of the catalytic PI3K subunit p110β in impaired higher order cognition in fragile X syndrome. Cell Rep. 2015;11(5):681–8. doi: 10.1016/j.celrep.2015.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gross C, Chang CW, Kelly SM, et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 2015;11(5):727–36. doi: 10.1016/j.celrep.2015.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saré RM, Figueroa C, Lemons A, Loutaev I, Smith CB. Comparative behavioral phenotypes of fmr1 KO, fxr2 het, and fmr1 KO/fxr2 het mice. Brain Sci [Internet]. 2019 [cited 2020 Jul 8];9(1). Available from: /pmc/articles/PMC6356887/?report=abstract [DOI] [PMC free article] [PubMed]
- 79.Carreno-Munoz MI, Martins F, Medrano MC et al. Potential involvement of impaired BK Ca channel function in sensory defensiveness and some behavioral disturbances induced by unfamiliar environment in a mouse model of fragile X syndrome. Neuropsychopharmacology [Internet]. 2018 [cited 2020 Jul 8];43(3):492–502. Available from: /pmc/articles/PMC5770751/?report=abstract [DOI] [PMC free article] [PubMed]
- 80.Gurney ME, Cogram P, Deacon RM, Rex C, Tranfaglia M. Multiple behavior phenotypes of the fragile-X syndrome mouse model respond to chronic inhibition of phosphodiesterase-4D (PDE4D). Sci Rep [Internet]. 2017 [cited 2020 Jul 8];7(1):1–11. Available from: www.nature.com/scientificreports [DOI] [PMC free article] [PubMed]
- 81.Banerjee A, Ifrim MF, Valdez AN, Raj N, Bassell GJ. Aberrant RNA translation in fragile X syndrome: from FMRP mechanisms to emerging therapeutic strategies. Vol. 1693, Brain Res. Elsevier B.V.; 2018. p. 24–36. [DOI] [PMC free article] [PubMed]
- 82.Dahlhaus R. Of men and mice: modeling the fragile X syndrome [Internet]. Vol. 11, Frontiers in Molecular Neuroscience. Frontiers Media S.A.; 2018 [cited 2020 Jul 8]. Available from: /pmc/articles/PMC5862809/?report=abstract [DOI] [PMC free article] [PubMed]
- 83.Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4(4):335–40. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- 84.Verheij C, Bakker CE, De Graaff E, et al. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature. 1993;363(6431):722–4. doi: 10.1038/363722a0. [DOI] [PubMed] [Google Scholar]
- 85.Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17(5):1539–47. doi: 10.1523/JNEUROSCI.17-05-01539.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smidak R, Sialana FJ, Kristofova M, et al. Reduced levels of the synaptic functional regulator FMRP in dentate gyrus of the aging Sprague-Dawley rat. Front Aging Neurosci [Internet]. 2017 23 [cited 2018 Dec 17];9. Available from: http://journal.frontiersin.org/article/10.3389/fnagi.2017.00384/full [DOI] [PMC free article] [PubMed]
- 87.Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–61. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Udagawa T, Swanger SA, Takeuchi K, et al. Bidirectional control of mRNA translation and synaptic plasticity by the cytoplasmic polyadenylation complex. Mol Cell. 2012;47(2):253–66. doi: 10.1016/j.molcel.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McEvoy M, Cao G, Llopis PM, et al. Cytoplasmic polyadenylation element binding protein 1-mediated mRNA translation in Purkinje neurons is required for cerebellar long-term depression and motor coordination. J Neurosci. 2007;27(24):6400–11. doi: 10.1523/JNEUROSCI.5211-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sohani Das Sharma A, Metz JB, Li H, Sulzer D, Tang G, Sims Correspondence PA. Widespread alterations in translation elongation in the brain of juvenile Fmr1 knockout mice. Cell Rep [Internet]. 2019 [cited 2020 Jan 9];26:3313–22. Available from: 10.1016/j.celrep.2019.02.086 [DOI] [PMC free article] [PubMed]
- 91.Ascano M, Mukherjee N, Bandaru P, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature [Internet]. 2012 [cited 2018 Jul 17];492(7429):382–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23235829 [DOI] [PMC free article] [PubMed]
- 92.Brown V, Jin P, Ceman S, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107(4):477–87. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 93.Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell [Internet]. 2001 [cited 2020 Jan 12];107(4):489–99. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11719189 [DOI] [PubMed]
- 94.Satterstrom FK, Kosmicki JA, Wang J, Roeder K, Daly MJ, Buxbaum JD. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism article large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020;1–17. [DOI] [PMC free article] [PubMed]
- 95.Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and Fmr1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–55. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Godfraind J-M, Reyniers E, De Boulle K, et al. Long-term potentiation in the hippocampus of fragile X knockout mice. Am J Med Genet [Internet]. 1996 [cited 2020 Jan 11];64(2):246–51. Available from: http://doi.wiley.com/10.1002/%28SICI%291096-8628%2819960809%2964%3A2%3C246%3A%3AAID-AJMG2%3E3.0.CO%3B2-S [DOI] [PubMed]
- 97.Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST. Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience. 1999;94(1):185–92. doi: 10.1016/s0306-4522(99)00285-7. [DOI] [PubMed] [Google Scholar]
- 98.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci [Internet]. 2004 [cited 2019 Sep 27];27(7):370–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15219735 [DOI] [PubMed]
- 99.Udagawa T, Farny NG, Jakovcevski M, et al. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med [Internet]. 2013 [cited 2019 Sep 26];19(11):1473–7. Available from: http://www.nature.com/articles/nm.3353 [DOI] [PMC free article] [PubMed]
- 100.Janusz A, Miłek J, Perycz M, et al. The fragile X mental retardation protein regulates matrix metalloproteinase 9 mRNA at synapses. J Neurosci. 2013;33(46):18234–41. doi: 10.1523/JNEUROSCI.2207-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ghosh A, Mizuno K, Tiwari SS, et al. Alzheimer’s disease-related dysregulation of protein synthesis causes key pathological features with ageing. bioRxiv. 2019 21;2019.12.20.884783.
- 102.Russo A, Scardigli R, La Regina F, Murray ME, Romano N, Dickson DW, et al. Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum Mol Genet [Internet]. 2017 15 [cited 2020 Jan 12];26(8):1407–18. Available from: https://academic.oup.com/hmg/article/26/8/1407/2965931 [DOI] [PMC free article] [PubMed]
- 103.Scheper GC, Proud CG, Van der Knaap MS. Defective translation initiation causes vanishing of cerebral white matter. Vol. 12, Trends Mol Med. 2006. p. 159–66. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 3.96mb)
(PDF 436 kb)