

Abstract
Extracellular vesicles (EVs) are an important intercellular communication system facilitating the transfer of macromolecules between cells. Delivery of exogenous cargo tethered to the EV surface or packaged inside the lumen are key strategies for generating therapeutic EVs. We identified two “scaffold” proteins, PTGFRN and BASP1, that are preferentially sorted into EVs and enable high-density surface display and luminal loading of a wide range of molecules, including cytokines, antibody fragments, RNA binding proteins, vaccine antigens, Cas9, and members of the TNF superfamily. Molecules were loaded into EVs at high density and exhibited potent in vitro activity when fused to full-length or truncated forms of PTGFRN or BASP1. Furthermore, these engineered EVs retained pharmacodynamic activity in a variety of animal models. This engineering platform provides a simple approach to functionalize EVs with topologically diverse macromolecules and represents a significant advance toward unlocking the therapeutic potential of EVs.
Keywords: extracellular vesicles, exosomes, EV engineering, exosome engineering, PTGFRN, BASP1, IL-12, vaccine, sacffold
Graphical abstract
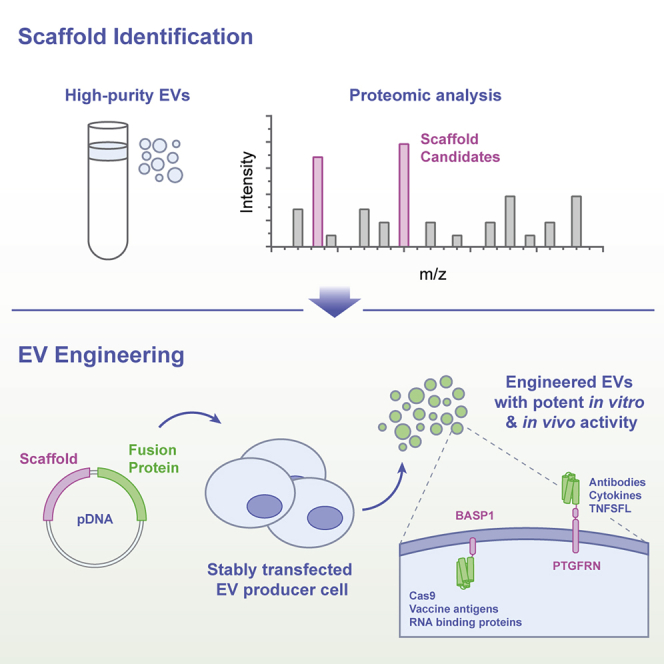
Extracellular vesicles (EVs) are natural mediators of cell-cell communication and thus potential vehicles for drug delivery. Dooley et al. describe an EV engineering platform based on two scaffold proteins, PTGFRN and BASP1, which enable high-density EV surface display and luminal loading of therapeutic cargo, leading to potent, pharmacodynamic activity in vitro and in vivo.
Introduction
Extracellular vesicles (EVs) are nanoscale, lipid membrane delimited particles released by cells across all kingdoms of life with roles in normal physiology and disease pathology. The diverse functions of EVs are mediated by molecules displayed on the surface or inside the vesicle lumen.1 The observation that EVs can transfer RNA and protein cargoes to recipient cells has spurred translational research focused on delivering therapeutic payloads with EVs.2,3 In addition, ligands displayed on the EV surface can engage cell receptors to activate cytosolic signaling pathways. Several studies have reported cargo-specific effects when these two strategies are used alone or in combination in cancer,4,5 muscular dystrophy,6 cardiovascular disease,7 regenerative medicine,8 and neurodegenerative disorders.9,10
While EVs have the potential to deliver therapeutic payloads, robust methods for EV engineering are lacking. Molecules appended to the EV surface after purification by chemical coupling11 or non-covalent attachment can disrupt the EV surface composition or are prone to dissociation from the EV in vivo.6,12 Genetically engineering EVs by fusing cargo to general membrane-targeting sequences (e.g., glycosylphosphatidylinositol [GPI] motifs,13 lipid anchors,14 or pDisplay [pD]5) or to membrane proteins enriched in EVs (e.g., LAMP2B,15 CD9,3 CD63,16 or MFGE817) can result in low-abundance, heterogeneous expression with commensurately low levels of biological activity. There is a clear need for robust, high-density EV engineering strategies to ultimately unlock the therapeutic potential of EVs.
We hypothesized that proteins preferentially packaged into EVs with high efficiency can enable the efficient loading of cargo molecules. Using stringently purified EVs, proteomic analyses identified two protein families previously unexplored as EV scaffolds: the EWI immunoglobulin superfamily (IGSF8 and PTGFRN) and the MARCKS protein family (MARCKS, MARKCSL1, and BASP1). Members of both protein families were found to be abundant in EVs derived from a variety of cell types and were selected for further investigation as scaffold proteins for EV loading.
We find that the overexpression of PTGFRN and MARCKS family proteins results in multi-log enrichment in EVs compared to previously described scaffolds. We delineate the minimal sequences required for EV enrichment and demonstrate the versatility of this EV engineering platform by appending a diverse array of biomolecules to these scaffolds, including protein reporters, enzymes, antibody fragments, cytokines, tumor necrosis factor superfamily (TNFSF) ligands, RNA binding proteins, Cas9, and vaccine antigens. We also describe engineered, multi-functional EVs that simultaneously deliver a small-molecule adjuvant and an antigen loaded luminally with a truncated version of BASP1, inducing adaptive, antigen-specific T cell responses in mice.
Results
Identification of high-density EV scaffold proteins
We reasoned that the detailed characterization of highly purified EVs would allow us to identify proteins we could use as scaffolds to load EVs with cargo. To this end, we developed a methodology to stringently and reproducibly purify a population of EVs from large volumes of cell culture supernatant, similar to those recently reported by Jeppesen et al.18 Cell culture medium from suspension-adapted HEK293 cells was processed by successive filtration and centrifugation steps to prepare a crude EV pellet that was separated on a discontinuous iodixanol gradient (Figures 1A and 1B). We split this gradient into four fractions (F1–F4), collecting material that migrated to the interface between adjacent density layers (Figures 1B and 1C), and analyzed the fractions for the presence of EVs using ultrastructural and biochemical techniques.
Figure 1.

EV purification and characterization
(A) Isolation of fractions F1–F4 from conditioned media. (B) Gradient image after 150,000 × g centrifugation highlighting F1–F4. (C) Density profile of blank iodixanol gradient after 150,000 × g centrifugation. (D) Representative transmission electron micrographs of F1–F4. (E) Cholesterol, DNA, protein, and particle concentrations in F1–F4. Data from 3 EV isolations are plotted as mean ± SD (F) Representative protein normalized SDS-PAGE of F1–F4, producer cell lysate (CL), and crude ultracentrifuged (UC) pellet with immunoblots for individual markers. Molecular weight markers for all immunoblots and SDS-PAGE gels are given in kDa. (G) Heatmap plotting quantitative peptide spectrum matches for proteins indicated. Proteins highly enriched in F1 (top) and F4 (bottom) are shown. Data are averaged from triplicate measurements from a representative EV isolation.
Negative stain transmission electron microscopy (TEM) analysis revealed the presence of 30- to 200-nm diameter vesicles predominantly in the lower-density fractions (F1 and F2), while the higher-density fractions (F3 and F4) appeared to contain mostly proteinaceous, non-vesicular material (Figure 1D). Cryoelectron microscopy (cryo-EM) analysis of F1 revealed the presence of non-vesicular, web-like material that was not apparent by negative stain TEM (Figure S1A). An additional 20,000 × g centrifugation step following density gradient fractionation removed these higher-order complexes, which were composed primarily of actin and actin-binding proteins (Figure S1B). Following the removal of this material, cryo-EM analysis confirmed the presence of lipid membrane delimited vesicles, free of cellular debris (Figure S1A).
We found that cholesterol, a major lipid component of exosomes,19 was predominantly found in F1, while the majority of DNA and protein were found in higher-density fractions (Figure 1E). Particle concentrations determined by nanoparticle tracking analysis (NTA) were similar for F1, F3, and F4 despite substantial differences in the prevalence of vesicles observed by TEM (Figures 1D and 1E). Proteinase K treatment of these fractions resulted in significant changes in NTA profiles of the high-density fractions, whereas F1 was minimally affected (Figures S1C and S1D). These data suggest that detailed biochemical characterization is needed to distinguish EVs from non-EV particles that co-purify with less stringent isolation methods.
SDS-PAGE and immunoblot analysis showed the enrichment of frequently reported EV markers CD9, CD63, CD81, and SDCBP in low-density fractions F1 and F2, consistent with the vesicle content observed by TEM (Figures 1D, 1F, and S1E). Single-particle flow cytometry analysis for tetraspanin markers indicated the presence of vesicle subpopulations within F1, which is consistent with previous reports (Figure S1F).20 Proteins that form large oligomeric complexes, such as LGALS3BP21 and chromatin-associated HMGB1 and HIST3H3,22 were enriched in F4 and absent from low-density EV-enriched fractions. The endoplasmic reticulum protein CANX was only found in cell lysate (CL), suggesting that the majority of this EV contamination marker was removed before ultracentrifugation (Figure 1F).
By liquid chromatography-tandem mass spectrometry (LC-MS/MS), F1 was enriched in known EV proteins such as PDCD6IP, MFGE8, and integrins β1 and α4, which were largely absent from the higher-density fractions F3 and F4 (Figure 1G). F4 was predominantly composed of secreted extracellular matrix (ECM) proteins (LAMA5/B1/C1, NID1, TNC, AGRN, and HSPG2) and histones (HIST-2H2AB, 3H2BB, 1H3A, and 2H2AC) and was depleted of EV-related proteins. Several members of the immunoglobulin superfamily (IgSF)-EWI and MARCKS protein families were identified as highly abundant in F1 and depleted or absent in F3 and F4: PTGFRN, IGSF8, MARCKS, MARCKSL1, and BASP1 (Figure 1G, underlined). These proteins were selected for further evaluation as EV-specific scaffold candidates.
Together with the EM and biochemical analyses, these data demonstrated that F1 was highly enriched in vesicles containing canonical EV markers and free of non-vesicular material that migrated to F2–F4. All of the subsequent experiments were performed using F1 EVs.
EV enrichment of engineered proteins
We next assessed the relative ability of these proteins to direct fusion partners into EVs using FLAG-tagged GFP as a surrogate cargo molecule. The five EV proteins identified by LC-MS/MS were compared to commonly used EV-localizing proteins: the tetraspanins CD9, CD63, and CD81; a palmitoylation tag (Palm); pD; and LAMP2B (Figure 2A). Stable suspension-adapted HEK293 cell lines were established through antibiotic selection using plasmids encoding GFP fusions to each scaffold. All of the engineered EVs described in this report were generated from stably selected cell pools unless otherwise stated.
Figure 2.

Characterization of scaffolds for EV engineering
(A) EV membrane lipid bilayer showing different classes of scaffold proteins with luminal GFP fusions. (B) MFI of EV producer cells stably expressing GFP fusions to the indicated scaffold. Data from 3 biological replicates are plotted as mean ± SD (C) Average number of GFP molecules per EV determined by NTA and GFP ELISA. GFP standard curve used for quantitation shown in inset. Data from 3 biological replicates are plotted as mean ± SD. All statistical comparisons are given in Figure S2D. (D) Representative histograms from flow cytometry measurements of engineered EVs with GFP fused to the indicated scaffold. Data are normalized to the highest count within each sample.
The efficiency by which GFP fusion proteins were packaged into EVs was determined by comparing cellular versus EV GFP levels. While most targeting constructs had similar cellular GFP expression measured by flow cytometry, there was a wide range of EV-associated GFP measured quantitatively by ELISA, with MARCKSL1, BASP1, MARCKS, and PTGFRN showing the highest levels of GFP per EV (Figures 2B, 2C, and S2D). Despite exhibiting 10-fold higher cellular expression than other constructs, untargeted cytoplasmic GFP (cGFP) showed only weak EV localization, suggesting that abundant cellular expression of GFP alone is insufficient for significant stochastic packaging into EVs (Figures 2B and 2C).
We next sought to determine whether overexpressed scaffold proteins were uniformly distributed among EVs or enriched in subsets. A flow nanoanalyzer (NanoFCM), a cytometer specifically designed to analyze particles smaller than the wavelength of visible light, was used to measure GFP fluorescence of individual vesicles.23,24 While EVs from cells expressing GFP fused to previously reported scaffolds showed low (LAMP2B, pD, Palm: 10%–35%) to moderate (CD9, CD63, CD81: 50%–65%) frequencies of GFP+ vesicles, fusions to MARKCSL1, BASP1, MARCKS, and PTGFRN resulted in well-defined, GFP+ peaks, with ∼95% of analyzed particles showing detectable GFP fluorescence (Figure 2D). Taken together with the ELISA measurements (Figure 2C), the data suggest that the overexpression of MARCKSL1, BASP1, MARCKS, or PTGFRN results in abundant, uniform distribution across EVs present in F1.
Characterization of EV scaffold candidates
PTGFRN
PTGFRN and IGSF8 are IgSF-EWI proteins, a family of type I transmembrane glycoproteins structurally composed of tandem IgV domains and short, cytoplasmic tails (Figure 3A).25 Despite their structural similarities, cellular expression of IGSF8-GFP resulted in only modest enrichment in EVs, while PTGFRN-GFP expression resulted in exceptionally high levels (Figures 2C and 2D).
Figure 3.

Determination of scaffold requirements for EV engineering
(A) EV membrane lipid bilayer showing GFP fusions to IgSF-EWI and MARCKS family members. Numbers for PTGFRN truncations indicate the first amino acid included from the PTGFRN protein sequence. “+++++,” polybasic region. (B) Average number of GFP molecules per EV determined by NTA and GFP ELISA. Data from 3 biological replicates are plotted as mean ± SD. B, BASP1; L, MARCKSL1; M, MARCKS. (C) EV normalized anti-GFP immunoblot of EVs with GFP fused to the indicated truncation. The 55-kDa membrane-bound cleaved product and putative ADAM10 cleavage site are shown. (D) EV normalized anti-GFP immunoblot following transient transfection of FL-GFP and Δ395-GFP into WT or ADAM10 knockout (−/−) producer cell lines with anti-ADAM10 immunoblot to confirm deletion. (E) EV-normalized SDS-PAGE and anti-GFP immunoblot of EVs with C-terminal GFP fusions to incremental truncations of BASP1 with amino acid positions and point mutations labeled. (F) EV-normalized anti-GFP immunoblot blot of EVs with C-terminal GFP fusions to single amino acid truncations of BASP1, with minimal sequence required for EV localization highlighted.
To better understand the features of PTGFRN that enable EV localization, a series of PTGFRN-GFP truncations were generated at predicted IgV domain boundaries (Figure 3A). Most truncations showed similar levels of cellular GFP expression as full-length PTGFRN (FL); however, truncations Δ149–Δ537 showed marked reduction in EV localization (Figures 3B and S2A). Immunoblot analysis of purified EVs using an antibody directed against the luminal GFP tag revealed an unexpected 55-kDa membrane-bound product in truncated forms of PTGFRN (Figure 3C). This product was also detected with the FL scaffold, although at much lower levels. PTGFRN-GFP fusion proteins were largely intact in cell lysate, suggesting that proteolysis occurs during EV biogenesis or after release (Figure S2B). Characterization of this proteolytic event and preventing its occurrence may therefore be beneficial for developing stabilized, high-density surface engineered EVs.
We explored the possibility that ADAM10, an abundant EV protease,20 may be responsible for the cleavage of PTGFRN truncations (Figure 3C). An ADAM10 knockout cell line was generated and used to produce EVs following transient transfection of FL-GFP and Δ395-GFP. EVs purified from ADAM10 KO cells did not contain the 55-kDa product found in EVs from wild-type (WT) producer cells transfected with Δ395-GFP (Figure 3D), suggesting that truncated forms of PTGFRN are susceptible to site-specific ADAM10 cleavage. We identified a PTGFRN truncation that was resistant to ADAM10 proteolysis by removing the putative cleavage site (Figure S2C), which resulted in more GFP per EV than any other truncation tested (Figure 3B, Δ687). Both FL and the optimized Δ687 truncated form of PTGFRN were further evaluated as scaffolds for displaying proteins of interest on the EV surface.
BASP1
Despite lacking significant sequence homology, MARCKS, MARCKSL1, and BASP1 associate with the inner leaflet of cellular membranes through N-terminal myristoylation of the glycine residue in position 2 and a polybasic effector domain.26 The importance of the polybasic region was determined when C-terminal truncations of MARCKS and MARCKSL1 (containing only the first 30 amino acids, removing the polybasic region; Figure 3A) reduced GFP loading by 40%–80% (Figure 3B). A similar truncation for BASP1 (containing only the first 30 amino acids but including the polybasic region) resulted in comparable levels to the full-length scaffold by ELISA (Figure 3B). To further define the minimal sequence within the polybasic region required for EV localization, we made a series of incremental BASP1 truncations fused to GFP and observed robust loading of all constructs containing at least the first 8 residues (Figures 3E and 3F). This minimal peptide sequence contains only 3 of the 10 lysine residues present in the BASP1 polybasic domain. While a portion of the polybasic domain is required for EV localization, it is not sufficient, as mutation of the myristoylation site (G2A) completely ablates the loading of a construct containing the entire polybasic domain (Figure 3E). The data suggest that the MARCKS family proteins require lipid modification and a polybasic sequence for high-density loading into EVs, both of which can be accommodated in an 8-amino acid peptide (Figure 3F, bottom).
PTGFRN enables EV surface display of broad classes of proteins
PTGFRN was evaluated as a surface display scaffold by fusing an array of structurally and biologically diverse proteins to the surface-exposed N terminus (Figure 4A). To compare specific protein activity of engineered EVs, in vitro half-maximal effective/inhibitory concentrations (EC50/IC50) data are presented in terms of EV concentration and absolute fusion protein concentration (Figures 4B–4E and S3A–S3D). FL PTGFRN and the truncated Δ687 scaffold were effective in displaying active interleukin-7 (IL-7), a secreted cytokine, on the surface of EVs, as measured by downregulation of the IL-7 receptor (IL-7R) on CD8+ T cells.27 PTGFRN constructs were up to 250-fold more potent per particle than those engineered with IL-7 fused to pD, a commonly used scaffold in EV engineering (IC50 = 2.79E+8 [FL], 3.09E+8 [Δ687] versus 7.10E+10 ([pD] p/mL). All EV constructs exhibited similar IC50 values when normalized to the total amount of IL-7 (Figures 4B and S3A).
Figure 4.

PTGFRN enables EV surface display of protein cargo
(A) EV membrane highlighting the classes of proteins displayed by fusion to PTGFRN. (B) In vitro activity of EVs displaying IL-7. Mean values from 4 donors using 2 EV isolations were compared by 1-way ANOVA and a Tukey post hoc test. ∗p = 0.0108; Rec., recombinant IL-7. (C) In vitro activity of EVs displaying CD40L. Mean values from 4 donors using 2 EV isolations were compared by 1-way ANOVA and a Tukey post hoc test. ∗p = 0.0247; ∗∗∗∗p < 0.0001; Rec., recombinant CD40L ECD. (D) In vitro activity of EVs displaying αCD3 scFab. Mean values are shown from individual mouse spleens using 2 EV isolations. Rec., recombinant αCD3. (E) In vitro activity of EVs displaying IL-12. Mean values from 4 donors using 2 EV isolations were compared by 1-way ANOVA and a Tukey post hoc test (ng/mL) or an unpaired t test (p/mL). Rec., recombinant IL-12; ns, not significant. (F) In vivo activity of EVs displaying murine IL-12 (FL) compared to treatment with recombinant murine IL-12 (Rec.) at 100 and 200 ng doses. Percentages of reduction in tumor volume compared to PBS treatment are given. Independent EV isolations were used for each study and data were compared by 1-way ANOVA and a Tukey post hoc test. ∗p < 0.05; ∗∗∗p < 0.0005; ∗∗∗∗p < 0.0001; ns, not significant. (G) Survival of EV-treated animals compared to recombinant murine IL-12. 100 ng, p = 0.0209; 200 ng, p = 0.0421 by Gehan-Breslow-Wilcoxon test.
TNFSF member CD40 ligand (CD40L) is a type II transmembrane protein transiently expressed on activated T cells, which induces signaling by engaging its cognate receptor, CD40, in a homotrimeric conformation.28,29 We fused a single-chain trimeric form of CD40L extracellular domain (ECD) to PTGFRN and measured potency in a primary B cell activation assay (Figures 4C and S3B). These EVs were >50-fold more potent than those isolated from CD40L overexpressing cells, demonstrating the importance of appending therapeutic molecules to EV-specific scaffolds (EC50 = 1.12E+9 [FL], 1.89E+9 [Δ687] versus 6.09E+10 [CD40L] p/mL). In addition, CD40L-FL EVs were 20-fold more potent than recombinant CD40L ECD alone, suggesting that the presentation of these molecules within a biological membrane may enhance signaling (EC50 = 1.02 [FL] versus 19.7 [Rec.] ng/mL). The same molecular design was used to functionally display the ECD of another TNFSF member, LIGHT, on the EV surface (Figure S3E), demonstrating the versatility of PTGFRN as a scaffold for EV display of TNFSF ligands.
To explore the feasibility of displaying targeting ligands such as antibody fragments on EVs, we appended single-chain antigen binding (scFab) and variable (scFv) fragments targeting immune cells to both PTGFRN scaffolds, including the murine 145-2C11 anti-CD3ε antibody. Anti-CD3 antibodies are potent immunosuppressants and tolerance induction agents used to mitigate symptoms of organ allograft rejection via CD3ε binding.30 EVs decorated with anti-CD3 scFab by fusion to FL and Δ687 PTGFRN similarly downregulated T cell receptor (TCR) expression on primary murine CD4+ T cells compared to soluble anti-CD3 antibody (Figures 4D and S3C). The versatility of this system was further demonstrated using scFv, scFab, or single domain VHH formats displayed on the EV surface by PTGFRN fusion (Figures S3H and S3I).
IL-12 is naturally produced by antigen-presenting cells (APCs) as a stably associated heterodimer and confers anti-tumor immunity in part by stimulating interferon gamma (IFNγ) secretion by immune cells in the tumor microenvironment (TME).31 We constructed PTGFRN fusions to a single-chain version of human IL-12 consisting of the p35 and p40 subunits connected by a flexible linker. Fusion to either FL or Δ687 PTGFRN resulted in similar potency as measured by the IFNγ response in peripheral blood mononuclear cells (PBMCs; EC50 = 4.23E+8 [FL] versus 6.70E+8 [Δ687] p/mL). When normalized to IL-12 concentration, IL-12-FL EVs and recombinant IL-12 exhibited similar potency (EC50 = 0.277 [FL] versus 0.269 [Rec.] ng/mL), ∼6-fold more potent than Δ687-IL-12 (EC50 = 1.77 ng/mL; Figures 4E and S3D). While we observed significant donor-to-donor variability, the relative EC50 values between treatment groups remained consistent within each donor. Murine versions of these IL-12 constructs were generated (Figure S3F), and anti-tumor activity was evaluated in vivo in a syngeneic B16F10 melanoma model at 100 and 200 ng doses of recombinant murine IL-12 or the EV equivalent IL-12 quantity. Intratumoral administration of murine IL-12-FL EVs improved both tumor growth inhibition (Figures 4F and S3G) and survival (Figure 4G) compared to recombinant IL-12.
BASP1 enables EV luminal loading of broad classes of proteins
The ability of BASP1 to drive high levels of GFP into the lumen of EVs (Figure 3B) prompted us to investigate the range of cargo types that we could load into EVs. We successfully loaded proteins of different sizes and complexities by fusion to an N-terminal fragment of BASP1 (Figures 5A and S4A), including the RNA-binding MS2 bacteriophage major coat protein (MCP, 14 kDa), chicken ovalbumin (OVA, 43 kDa), and Cas9 (168 kDa).
Figure 5.

BASP1 enables EV loading with protein cargo
(A) EV membrane lipid bilayer showing proteins loaded with BASP1 along with vaccine formulations of OVA and adjuvant. Vaccination experiments in (B)–(D) were performed with independent EV isolations. (B) OVA and CDN combinations were administered i.n. as free compounds or associated with EVs. Lung and spleen effector memory T cells reactive to the dominant MHC class I epitope in OVA (SIINFEKL) were measured by flow cytometry (means ± SDs; n = 3 per group). ∗p < 0.0182; ∗∗∗∗p < 0.0001; for exoVACC compared to all groups by 1-way ANOVA. (C) OVA and CDN combinations were administered i.n., and ELISpot analysis on viable splenocytes is shown for reactivity against dominant CD4 and CD8 epitopes. Data are presented as background subtracted IFNγ spot-forming units (SFUs) per 100,000 splenocytes (means ± SDs; n = 5 per group). ∗p < 0.0168; ∗∗p < 0.0014; for exoVACC compared to all groups by 1-way ANOVA. (D) OVA-reactive splenic effector memory T cells measured following a single dose of exoVACC administered i.v., i.n., or s.c. alongside s.c. dose-matched free OVA formulated with AddaVax (mean ± SD; n = 5 per group). ∗p = 0.0162; ∗∗p = 0.0015; compared to OVA + AddaVax group by Welch’s 1-way ANOVA.
We examined whether luminal loading of EVs with antigenic proteins would be effective in inducing robust cellular immune responses due to the particulate delivery of EV-associated antigen into APCs.32 In a pilot experiment, we compared the cellular immune response elicited by soluble OVA delivered to mice intranasally (i.n.) as either free protein or loaded into the lumen of EVs as a BASP1 peptide fusion (Figure 5A). In the absence of adjuvant, neither soluble OVA protein nor BASP1-OVA EVs (exoOVA) elicited a significant T cell response in the lung or spleen (Figures 5B and S4B). Associating a cyclic dinucleotide (CDN) stimulator of IFN genes (STING) agonist adjuvant33 with exoOVA (exoVACC) induced local and systemic T cell responses, which were substantially greater than soluble OVA + CDN, or when exoOVA was co-administered with free CDN (Figures 5B and S4B). These results were confirmed in a subsequent study using ELISpot to evaluate systemic CD8+ and CD4+ T cell responses (Figure 5C).
We also assessed the immunogenicity of exoVACC when delivered via different routes of administration. Superior OVA-specific CD8+ effector memory T cell responses were observed in the spleen after a single vaccination by intravenous (i.v.), i.n., or subcutaneous (s.c.) routes compared to s.c. administration of soluble OVA protein with AddaVax (MF59), a standard adjuvant in influenza vaccines (Figures 5D and S4C). These data demonstrate the potential of exoVACC to drive robust T cell responses against ovalbumin and suggest that coincident delivery of adjuvant and antigen to APCs by EVs may improve antigen-specific immune responses. These data also highlight the capacity of BASP1 to localize proteins to the lumen of EVs to generate defined pharmacological effects in vivo.
Discussion
Given their role in intercellular communication, EVs have enormous potential as a unique therapeutic modality. Despite their discovery 35 years ago34 and their first use in human clinical trials over a decade ago,35 no EV-based therapies have been approved. This is due in part to the inability to rationally engineer EVs with potent, predictable therapeutic activity. We describe the identification and development of protein scaffolds that can efficiently load a variety of biologically active cargoes, including fluorescent reporters, cytokines, antibody fragments, enzymes, vaccine antigens, and TNFSF ligands onto the surface or into the lumen of EVs at therapeutically relevant levels.
The identification of these scaffold proteins was enabled by the proteomic analysis of high-purity EVs, free from other contaminants present in conditioned media. A challenge in interpreting the results of published reports on EVs lies in the range of isolation methods commonly used, including ultracentrifugation, precipitation, and size exclusion chromatography. We found that these methods, used alone or in combination, led to high levels of contaminating proteins and nucleic acids (Figures 1E and S5A). A density gradient was necessary to separate EVs from contaminants present in the ultracentrifuged (UC) pellet, including oligomeric protein complexes, ECM constituents, and chromatin-associated proteins, thereby enabling the identification of IgSF-EWI and MARCKS protein families in the low-density F1 vesicle population.
Several reports characterize PTGFRN and its interaction with CD9 and CD81, but little is known about its biological function.36, 37, 38 Deletion or overexpression of PTGFRN in clonal HEK293 producer cell lines did not dramatically alter the EV protein profile by SDS-PAGE analysis. Furthermore, these cell lines had EV production capacities similar to those of the parental cell line, suggesting that PTGFRN is not required for EV biogenesis in HEK293 (Figure S5B). We demonstrated that the cytoplasmic tail, transmembrane region, and the membrane proximal IgV domain of PTGFRN are sufficient for loading proteins onto the surface of EVs (Figures 4B–4E). In addition to exhibiting high levels of EV enrichment, the topology of PTGFRN makes it an attractive scaffold for displaying proteins on the EV surface through fusion to the exposed N terminus. In contrast, both N and C termini of tetraspanins are sequestered in the EV lumen, making the surface display of fusion proteins challenging.16,39
While MARCKS proteins have been implicated in a wide variety of cellular processes, including vesicle trafficking and bridging membrane-anchored signaling systems with the actin cytoskeleton, their function inside EVs is not clear.40 We determined that both the myristoylation site and the polybasic region of MARCKS, MARCKSL1, and BASP1 are important for EV localization. Labeling the inner leaflet of the cellular plasma membrane with a palmitoylation anchor fused to GFP resulted in lower levels of EV enrichment, suggesting that active sorting into EVs is critical for achieving high-efficiency loading (Figure 2, “Palm” sequence).
We validated our EV engineering platform by loading molecules across a spectrum of protein families and complexities to both full-length and truncated forms of PTGFRN and BASP1. PTGFRN enabled the functional display of all proteins tested, ranging from 12 kDa single-domain antibodies to a 170-kDa multi-domain enzymatic clotting factor (Figure S3). Truncated forms of BASP1 showed the capacity to luminally load a wide range of proteins as well, including Cas9. Concomitant increases in activity were shown for EVs engineered with these scaffolds compared to other methods (Figures 4B and 4C). Furthermore, transient transfection of Chinese hamster ovary (CHO) cells with GFP fusions to PTGFRN or BASP1 resulted in EV loading, suggesting that these scaffolds can be used to engineer EVs derived from cells besides HEK293 (Figures S5C and S5D). The concept of broad applicability in using PTGFRN and BASP1 for EV engineering is further bolstered by the detection of endogenous PTGFRN and BASP1 in EVs derived from diverse cellular sources (Figure S5E).
Displaying therapeutic molecules on the EV surface with PTGFRN resulted in enhanced (CD40L), similar (αCD3, IL-12), or attenuated (IL-7) in vitro potency compared to soluble protein (Figures 4B–4E). Target receptor density, requirements for receptor signaling, and multivalent presentation of molecules on the EV surface may contribute to this variability. For instance, TNF receptor clustering is required to initiate signal transduction pathways and cellular responses.41 Multivalent presentation of cognate TNFSF ligands, including CD40L, within a dynamic biological membrane may be important for potently activating these signaling cascades (Figure 4C). Conversely, locally concentrating naturally secreted proteins, such as IL-7, to the EV surface may result in limited access to target receptors when compared to an equivalent dose of free protein in vitro (Figure 4B). It is important to note that these model systems do not capture some potential advantages of EV-mediated delivery, including cell targeting, expanded therapeutic index, and delivery of target molecules across biological membranes,42 all of which are better assessed in vivo.
Despite the similar potency of recombinant IL-12 and IL-12-FL EVs in vitro, EV delivery slightly enhanced in vivo efficacy in the B16F10 tumor model (Figures 4F and 4G). A possible explanation for this is prolonged retention of IL-12-FL EVs in the TME following intratumoral administration,43 whereas recombinant IL-12 readily extravasates into systemic circulation within minutes after injection.44 EV delivery may therefore allow for a more favorable safety profile by limiting systemic exposure, which has hampered the clinical development of IL-12 therapies.45 The vaccine platform demonstrates another example of the potential advantage of EV delivery of cargo protein. Administration of soluble OVA with adjuvant did not result in meaningful T cell responses in any of the in vivo models tested. Antigen packaged into the EV lumen also did not generate strong cellular or humoral immune responses in the absence of adjuvant (exoOVA). Adjuventing with CDN (exoVACC), however, induced strong humoral IgG/IgA and CD4+/CD8+ T cell responses (Figures 5B, 5C, and S4D). It should be noted that the IgG titers observed here are comparable to those obtained with traditional vaccine adjuvants. Compartmentalizing adjuvant and antigen via BASP1 fusion in the same EV may allow for coincident delivery to APCs and elicit more robust effector memory T cell responses than conventional vaccination strategies (Figure 5). While these examples demonstrate defined pharmacological effects of the engineered EVs and confirm the integrity of the displayed/packaged proteins in vivo, further investigation into both therapeutic strategies is required to fully elucidate the biological mechanisms underpinning these observations.
EV engineering enables the presentation of complex, post-translationally modified transmembrane and luminal cargoes in their native conformation with a level of precision not readily attainable using synthetic nanoparticle-based approaches. We have also demonstrated the combinatorial potential of both surface display and luminal loading approaches by the co-expression of multiple cargoes fused to PTGFRN or BASP1 in a single producer cell (Figure S5F). Combining this protein engineering framework with other strategies for ex vivo loading of small molecules46 or nucleic acids47 further broadens the utility and modular approach to designing custom EVs for the targeted delivery of payloads to specific cells.
Materials and methods
Transfection and stable cell line selection
Suspension-adapted HEK293 cells were grown in CDM4PERMab media (GE Healthcare) supplemented with 4 mM l-glutamine. Genes of interest were cloned downstream of a cytomegalovirus (CMV) promoter in a pIRES vector and transfected into HEK293 cells via Neon electroporation (ThermoFisher) or Transporter 5 transfection reagent (Polysciences). Stable cell lines were selected by supplementing puromycin or neomycin with periodic passaging until the cell lines returned to >90% viability, at which point they were cryopreserved.
A PTGFRN knockout HEK293 cell pool was generated by CRISPR/Cas9 editing with commercially available guide RNAs targeting regions in exons 2 and 9 (ThermoFisher). A single clone was selected by limited dilution, and PTGFRN knockout was confirmed by genotyping and immunoblot analysis. The same approach was used to generate the ADAM10 knockout cell pool.
EV isolation from cell culture medium
For small-volume production (<1.2 L), suspension-adapted HEK293 cells were grown in Thompson shake flasks at 37°C in 8.0% CO2, 80% humidity at 150 rpm in working volumes from 200 to 1,200 mL. For larger-scale production, cells were grown in WAVE bioreactors (GE Healthcare) maintained at similar temperature, CO2, and agitation in 10- or 25-L working volumes. Chemically defined CDM4PERMAb media was inoculated with 0.3E+6 viable cells per milliliter and cells were grown for 9 days. Cell density and viability were measured on a Vi-CELL XR cell counter (Beckman Coulter). Upon culture termination, cells were removed by centrifugation at 6,000 × g for 20 min at room temperature. Small-scale harvests (≤1.2 L) were subjected to a 16,000 × g centrifugation step for 30 min to remove large cell debris, whereas larger harvests were filtered using Sartopore 0.8/0.45 μm capsules (Sartorius). Clarified medium was supplemented with 1 mM MgCl2 and benzonase (20 U/mL, Millipore) and incubated at 37°C for 3 h or overnight at room temperature to digest extravesicular nucleic acids. Harvests larger than 1.2 L were concentrated 10× by tangential flow filtration using Pellicon 2 mini 1,000 NMWL modified polyethersulfone (PES) ultrafiltration cassettes (Millipore) on a SARTOFLOW benchtop system. Nuclease treated media was next transferred to 100-mL Quick-Seal Ultra-Clear tubes (Beckman Coulter) and centrifuged for 60 min at 133,900 × g at 4°C in a 45-Ti fixed-angle titanium rotor. The media was discarded, and the crude pellet was resuspended in a minimal volume of PBS.
For large-scale preparations, crude pellets were diluted to 3 mL total volume with PBS, mixed with 9 mL of 60% iodixanol solution (OptiPrep, Sigma), and layered into the bottom of a 38-mL Ultra-Clear tube (Beckman Coulter). Lower-density solutions were prepared by diluting with homogenization buffer (250 mM sucrose, 10 mM Tris-HCl, 1 mM EDTA, pH 7.4) to yield final iodixanol concentrations (vol/vol) of 30%, 23%, and 18%. Successive layers of 30% (9 mL), 23% (6 mL), and 18% (6 mL) iodixanol solutions were carefully pipetted on top. PBS at 3 mL was added to the top of the gradient. The gradient was ultracentrifuged in a swinging-bucket SW 32 Ti rotor (Beckman Coulter) for 16 h at 150,000 × g and 4°C to separate EVs from other cell culture supernatant contaminants. The EVs that migrated to the interface between the PBS and 18% iodixanol layers were carefully extracted and transferred to a new 38-mL Ultra-Clear tube. One-milliliter fractions from an identical empty gradient (no crude pellet loaded) were analyzed by refractometry to calculate the density of the EV samples. The isolated EV fraction was diluted to 35 mL with PBS and centrifuged for 30 min at 20,000 × g at 4°C to pellet contaminating species, including actin and actin-binding proteins. The supernatant was filtered through a Steriflip 0.22-μm PES filter (Millipore) and transferred to a new 38-mL Ultra-Clear tube. The purified EVs were concentrated by ultracentrifugation in a SW 32 Ti swinging bucket rotor for 3 h at 135,000 × g and 4°C. The EV pellet was resuspended in PBS, analyzed, aliquoted at 1E+13 p/mL, and frozen at −80°C. Repeated freeze-thaw cycles were avoided for all EV preparations. Small-scale preparations were processed identically, except that iodixanol gradients were prepared in 13-mL Ultra-Clear tubes with 1/3 all indicated volumes and centrifuged in a swinging bucket SW 41 Ti rotor (Beckman Coulter). Subsequent ultracentrifugation steps were also carried out in an SW 41 Ti rotor.
Material requirements for the analysis described in the report varied depending on the assay and the productivity of each engineered cell pool. Typically, 200–1,200 mL of cell culture at ∼10E+6 vc/mL at cell culture termination was required for all biochemical experiments. Cell culture at 1–10 L was required for all in vitro and in vivo models.
TEM and cryo-EM
For TEM, EV samples at ∼2.0E+11 p/mL were transferred to a 200-mesh Formvar and carbon-coated copper grid (Ted Pella) and incubated for 1 min. After adsorption, the grids were rinsed with water and the excess solution was wicked away. The grids were then stained with a 1% solution of uranyl acetate for ∼30 s. After staining, the excess solution was wicked away and the grids were allowed to dry. The negatively stained samples were analyzed with a FEI Morgagni (Hillsboro) electron microscope operating at 80 kV.
For cryo-TEM, EV samples at 2.0E+12 p/mL were transferred to a glow-discharged R2/2 Quantifoil Holey carbon film grid and incubated for 1 min. Grids were blotted and immediately plunge frozen into nitrogen-cooled liquid ethane using a Vitrobot Mk IV (ThermoFisher). The samples were imaged at 13,000× magnification using a Phillips CM12 electron microscope operated at 100 keV and captured with a TVIPS 1kx1k CCD camera.
EV biochemical characterization
Purified EV stocks were diluted 100–1,000× in PBS and analyzed for total protein, double-stranded DNA, and cholesterol using the micro BCA, Quant-iT PicoGreen, and Amplex Red assay kits (ThermoFisher), respectively. All of the assays were performed according to the manufacturer’s protocols. Particle concentrations and size distributions were measured by nanoparticle tracking analysis on a NanoSight NS300 (Malvern). Samples were freshly diluted 1,000–10,000× in 0.22-μm filtered PBS and analyzed immediately. Three 30-s acquisitions were made for each sample under continuous flow operation and analyzed using NanoSight NTA software version 3.4. Identical samples were treated with a final concentration of 0.5 mg/mL thermolabile Proteinase K (New England Biolabs), incubated for 1 h at ambient temperature, and analyzed as described above. Proteinase K was heat inactivated at 55°C for 10 min before SDS-PAGE and western blotting analysis.
SDS-PAGE samples were normalized to total protein or EV input (10 μg or 1.0–3.0E+10 EVs, respectively) in reducing or non-reducing Laemmli buffer (Bio-Rad), depending on the primary antibody (Table S1). Next, samples were denatured at 95°C for 10 min before loading into 4%–20% TGX stain-free precast gels (Bio-Rad). Separated protein was imaged on a ChemiDoc gel imaging system (Bio-Rad), transferred to a polyvinylidene fluoride (PVDF) membrane using a Trans-Blot Turbo transfer system (Bio-Rad), and blocked in 1% casein for 1 h. Primary antibodies were diluted in blocking buffer and incubated with membranes for 1–3 h; proteins of interest were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies and chemiluminescent substrate.
For Simple Western analysis, samples were prepared according to the manufacturer’s protocol in reducing or non-reducing sample buffer, depending on the primary antibody (Table S1). EV and cell lysates were separated on 12–230 kDa Wes separation modules with 1-h incubation times in primary and secondary antibodies (ProteinSimple). HRP-conjugated secondary antibodies and chemiluminescent substrate were used for detection. The high dynamic range exposure was used for all of the analyses. Molecular weights were approximated by peak integration using Compass software (ProteinSimple).
EV flow cytometry
We adhered to MIFlowCyt-EV guidelines when analyzing vesicles by flow cytometry.48 Cytometer excitation and emission light paths were aligned using 100-nm silica nanoparticles (nanoFCM) immediately before analyzing vesicle samples. A 488-nm laser diode was used to illuminate the sample; side-scattered light and GFP fluorescence were collected in separate channels using avalanche photodiode detectors. Distilled water was used as sheath fluid. We empirically determined acquisition parameters (laser power, detection threshold, detector gain) to minimize background using buffer-only controls to yield a background event rate of 2–4 events per second.24 Sample injection pressure was maintained at 1.5 kPa, yielding well-separated particle detection events. All of the samples were analyzed using identical parameters on the same day. Samples were stored at 4°C in PBS and analyzed within 1 week of purification. Each sample was serially diluted, and the dilution that yielded 100–150 detection events per second was used to analyze single vesicles (∼1.0E+9 p/mL as determined by NTA).
AlphaLISA and GFP ELISA
All AlphaLISA and ELISA measurements were made according to the manufacturers’ protocols using dose titrations of solubilized EV samples (Table S1). For the GFP ELISA, EVs purified from 200-mL production cultures were diluted 500-fold in 1× cell extraction buffer (CEB, Abcam ab171581). EV stock concentrations ranged from 5.5E+11–5.0E+12 p/mL before initial dilution. EV samples were serially diluted 5-fold in 1× CEB to create 8-point dose titration curves with concentrations ranging from 1.0E+10–1.5E+4 p/mL. Diluted EV samples were transferred to pre-coated 96-well microtiter plates for quantitative assessment of GFP concentration by SimpleStep ELISA. The assay sensitivity is 1.8 pg/mL with a detection range between 2.7 and 2,000 pg/mL. Titration points falling within the linear range of the GFP standard were used for further analysis.
MS and data analysis
EV samples were diluted to 125 μg/mL in PBS buffer and lysed at 55°C for 60 min in an equal volume of lysis buffer (50 μL; 60 mM Tris pH 8.5, 400 mM GdmCl, 100 mM EDTA, 20 mM tris(2-carboxyethyl)phosphine [TCEP], 1.0% Triton X-100). Protein precipitation was performed by adding 1.25 mL of ethanol at −20°C. To improve efficiency, samples were vigorously vortexed for ∼10 min and then incubated at −20°C for 60 min. After incubation, samples were sonicated in a water bath for 5 min. Precipitated material was pelleted by centrifuging for 5 min at 15,000 × g at 4°C. The supernatant was decanted, and the pelleted material was thoroughly dried using nitrogen gas. Pellets were resuspended in 30 μL digestion buffer (30 mM Tris pH 8.5, 1.0 M GdmCl, 100 mM EDTA, 50 mM TCEP), which also reduced disulfide bonds. Free cysteine residues were alkylated by adding 5 μL alkylation solution (375 mM iodoacetamide, 50 mM Tris pH 8.5) and incubating the resulting solution at room temperature in the dark for at least 30 min. Next, each sample was diluted using 30 μL 50 mM Tris pH 8.5, and proteolytic digestion was initiated by adding 2 μg trypsin. All of the samples were mixed and then incubated overnight at 37°C. After the incubation, trypsin activity was quenched by adding 5 μL 10% formic acid. Before analysis by LC-MS/MS, each sample was desalted using Pierce C18 spin columns, after which each sample was dried and reconstituted in 50 μL water with 0.1% formic acid and transferred to a high-performance liquid chromatography (HPLC) vial for analysis.
Samples were injected into an UltiMate 3000 RSCLnano (ThermoFisher) low flow chromatography system, and tryptic peptides were loaded onto an Acclaim PepMap 100 C18 trapping column (75 μm × 2 cm, 3 μm particle size, 100 Å pore size; ThermoFisher) using loading mobile phase (MPL: water, 0.1% formic acid) at a flow rate of 1 μL/min. Peptides were eluted and separated with a gradient of mobile phase A (MPA: water, 0.1% formic acid) and mobile phase B (MPB: acetonitrile, 0.1% formic acid) at a flow rate of 300 nL/min across an EASY-Spray C18 analytical column (75 μm – 25 cm, 2 μm particle size, 100 Å pore size; ThermoFisher). The stepwise gradient used for elution began at 2% MPB, where it was held for 8 min during loading. The percentage MPB then increased from 2%–17% over 35 min, again from 17%–25% over 45 min, and finally from 25%–40% over 10 min. The most hydrophobic species were removed by increasing to 98% MPB over 5 min and holding for 10 min.
Mass analyses were performed with a Q Exactive Basic mass spectrometer (ThermoFisher). Precursor ion mass spectra were measured across an m/z range of 400–1,600 Da at a resolution of 70,000. The 10 most intense precursor ions were selected and fragmented in the HCD cell using a collision energy of 27, and MS/MS spectra were measured across an m/z range of 200–2,000 Da at a resolution of 35,000. Ions with charge states from 2 to 4 were selected for fragmentation, and the dynamic exclusion time was set to 30 s. An exclusion list containing 14 common polysiloxanes was used to minimize the misidentification of known contaminants. Proteins were first identified and quantified (label-free) using Proteome Discoverer software (version 2.1.1.21, ThermoFisher) and the Sequest HT algorithm combined with the Target Decoy PSM Validator. Searches were performed against the full Swiss-Prot Homo sapiens reference database.
In vitro activity assays
For all in vitro activity assays, data are presented in terms of EV concentration (p/mL) and absolute fusion protein concentration (ng/mL). EV doses ranged from ∼5.0E+11–1.0E+6 p/mL for each assay. Dose ranges for EVs and recombinant proteins were selected to ensure that full dose-response curves were achieved. WT EV controls were also included for each assay to account for any impact of endogenous EV cargo. The gating strategies for flow cytometry analysis are provided in Figure S6.
IL-7 in vitro activity assay
PBMCs were isolated from whole blood using SepMate tubes (STEMCELL Technologies). T cells were isolated using the negative-selection, human T cell enrichment kit on the RoboSep-S automated cell separator (STEMCELL Technologies) and diluted in complete RPMI media (ThermoFisher) with 10% fetal bovine serum (FBS). A total of 50,000 cells were added per well in 96-well plates. EV samples or recombinant IL-7 were added to the wells in a final volume of 200 μL and incubated overnight at 37°C and 5% CO2. The next day, the cells were pelleted, washed, and stained to assess IL-7 receptor levels on CD8+ T cells by flow cytometry using a SA3800 Spectral Cell Analyzer (Sony). Data were normalized to compare data from multiple donors.
CD40L in vitro activity assay
B cells were isolated from multiple donors using the RosetteSep human B cell negative selection cocktail (STEMCELL Technologies). Isolated B cells were resuspended in RPMI supplemented with 10% FBS and 200,000 cells were added per well in round-bottom 96-well plates. EV samples or recombinant CD40L were added to the wells in a final volume of 200 μL and incubated at 37°C and 5% CO2. The next day, the cells were pelleted, washed, and stained for flow cytometry. Expression of the activation marker CD69 was assessed. Flow cytometry analysis was completed on a SA3800 Spectral Cell Analyzer (Sony). Data were normalized to compare data from multiple donors.
Anti-CD3 in vitro activity assay
Total splenocytes from C57BL/6 mice were isolated by mechanical disruption of the spleen followed by red blood cell lysis with ACK buffer. Cells were plated in round-bottom 96-well plates at 200,000 cells per well in RPMI supplemented with 10% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, and 50 μL M 2-mercaptoethanol. EVs or recombinant anti-mouse CD3 (clone 145-2C11) were added at the specified concentrations in a final volume of 200 μL. The amount of anti-CD3 expressed on EVs was calculated by quantitative Simple Western using a recombinant protein standard curve. Cells were incubated at 37°C and 5% CO2 for 18 h, and the expression of TCRβ was analyzed by flow cytometry. Cells were stained with the following anti-mouse antibodies from Biolegend: anti-CD4 (clone RM4-5), anti-TCRβ (clone H57-597), anti-CD8 (clone 53-6.7), anti-CD19 (clone 1D3), and anti-CD3 (clone 17A2). Samples were analyzed using a Cytoflex LX analyzer (Beckman Coulter).
IL-12 in vitro activity assay
PBMCs were isolated from whole blood using SepMate tubes (STEMCELL Technologies). Cells were plated in round-bottom 96-well plates at 200,000 cells per well in RPMI supplemented with 10% FBS. Cells were stimulated with anti-human CD3 (clone OKT3) at a final concentration of 1 ng/mL. EV samples or recombinant IL-12 were added to the wells in a final volume of 200 μL and incubated at 37°C and 5% CO2. After 4 days, the supernatant was harvested and analyzed for human IFNγ using an AlphaLISA kit according to the manufacturer’s protocol (PerkinElmer). Murine orthologs were tested identically using total splenocytes from C57BL/6 mice, anti-mouse CD3 (clone 145-2C11), and a mouse IFNγ AlphaLISA detection kit.
IL-12 in vivo activity assessment
Codiak’s internal Institutional Animal Care and Use Committee (IACUC) and the Cambridge Animal Commissioner approved the study protocols and provided guidance on the care and ethical treatment of all study animals. Female C57BL/6 mice (Taconic), aged 5–6 weeks, were maintained in accordance with the regulations and guidelines of the IACUC (CB2017-001). B16F10 cells (1E+6 cells per animal) were implanted subcutaneously in the right flank of mice. At day 5 post-implantation, when tumor volumes reached an average volume of 50–100 mm3, mice were randomized into 3 groups with 5 mice per group. Mice were given 3 intratumoral injections (100 ng: d5, d7, d9; 200 ng: d5, d6, d7) of PBS, recombinant murine IL-12, or EVs displaying murine IL-12 (IL-12-FL). Tumor volume was calculated as tumor (width)2 × (length) × 0.5. Mice were euthanized when tumor volume reached ≥2,000 mm3.
Vaccination
Female C57BL/6 mice (Taconic), aged 6–10 weeks, were immunized i.n., s.c., or i.v. with vaccine formulations, all of which included a total of 1 μg OVA in PBS. Vaccine formulations groups were free OVA (OVA); EVs engineered to express luminal OVA as a fusion to amino acids 1–10 of BASP1 (exoOVA); free OVA mixed with 0.2 μg STING agonist (OVA + CDN); EVs engineered with luminal OVA mixed with 0.2 μg STING agonist immediately before injection (exoOVA + CDN); or EVs engineered with luminal OVA and loaded with 0.2 μg STING agonist (exoVACC). The STING loading procedure for the exoVACC group is described in detail below. OVA levels in EVs were assessed by quantitative western blot. The STING agonist used in all of the groups was cAIM(PS)2 Difluor (Rp/Sp) (InvivoGen). In the first study comparing vaccine formulations (Figure 5B), all of the groups received a dose on days 0 and 7 and were sacrificed on day 14. In the follow-up experiment (Figure 5C), mice received doses on days 0 and 14 and were sacrificed on day 28. In the study comparing routes of administration (Figure 5D), animals received a single dose on day 0 and were sacrificed on day 7.
Lungs or spleens were harvested by enzymatic dissociation at 37°C using a gentleMACS and dissociation kits, per manufacturer instructions (Miltenyi Biotec). Single-cell suspensions were enriched for live leukocytes by density centrifugation with Lympholyte M (CedarLane Labs) and the interface collected and washed twice with PBS. For flow cytometry analysis, cells were stained for viability with 2 ng/mL iFluor 860 maleimide (AAT BioQuest) in PBS at room temperature for 15 min, washed twice with 0.5% BSA in PBS, and then stained with APC- and PE-labeled H2Kb-SIINFEKL tetramers at room temperature for 20 min (MBL Bio). Without washing, cell surface markers were stained with fluorescent antibodies against CD45 (30-F11), CD19 (1D3), CD4 (RM4-5, BD Bioscience), or CD8 (KT15, MBL Bio). OVA reactive CD8+ effector/memory T cells were enumerated by gating on live CD45+ CD4− CD19− CD8+ CD62l−CD44+ cells double positive for PE and APC tetramers. ELISpot analysis was performed using the murine IFNγ ELISpotPLUS kit according to the manufacturer’s protocol (Mabtech). Splenocytes were stimulated with peptides corresponding to the dominant CD4+ (ISQAVHAAHAEINEAGR) and CD8+ (SIINFEKL) T cell epitopes (AnaSpec). OVA reactive mouse serum antibodies were quantified according to the manufacturer’s protocols (Chondrex). All of the vaccination experiments were conducted with independent EV isolations.
STING agonist association and quantitation
A total of 1 mM STING agonist was incubated with 1E+12 purified EVs in 300 μL PBS at 37°C overnight. EVs were diluted to 1 mL in PBS and ultracentrifuged in a TLA-120.2 fixed angle rotor at 100,000 × g for 20 min to separate the unincorporated STING agonist. The supernatant was removed and discarded. The EV pellet was resuspended in 1 mL PBS and ultracentrifuged again at 100,000 × g for 20 min. The supernatant was discarded and STING agonist-associated EV samples were resuspended in PBS and analyzed by NTA. Approximately 1%–2% of input STING agonist was incorporated into EV samples after cleanup.
STING agonist concentration was determined by LC-MS/MS. STING agonist standard curves were prepared by serial dilution in phosphate buffer containing 1.0E+11 p/mL EVs such that all standards contained an equal concentration of EVs. All of the samples were appropriately diluted so the final concentration of EVs was equal to that of the standards, 1.0E+11 p/mL. All of the standards and samples were then transferred to HPLC vials and diluted 3:1 with EV lysis buffer (60 mM Tris, 400 mM GdmCl, 100 mM EDTA, 20 mM TCEP, 1.0% Triton X-100), followed by the addition of 2.0 μg Proteinase K enzyme (Dako, reference S3004). All of the vials were then capped, vortexed to mix, and incubated at 55°C for 60 min. Following incubation, all HPLC vials were allowed to cool to room temperature and were held at 4°C–8°C until analysis.
Standards and samples at 5.0 μL were injected neat into an ACQUITY UPLC I-Class System (Waters Corporation) without cleanup. The separation of analytes was performed using an ACQUITY UPLC HSS T3 analytical column (2.1 × 50 mm, 1.8 μm particle size, 100 Å pore size; Waters Corporation) and a gradient of MPA (water, 0.1% formic acid) and MPB (acetonitrile, 0.1% formic acid) at a flow rate of 500 μL/min. The gradient began at 0% MPB, which was held for 1 min to load and desalt the STING agonist analyte. The percentage MPB then increased from 0% to 95% over 1.5 min to elute the analyte. The percentage of MPB was held at 95% for 1.25 min, decreased from 95% to 0% over 0.25 min, and then held at 0% for 1 min to re-equilibrate the column. The total runtime for the method was 5 min, and LC flow was only directed into the MS between 1.0 and 2.5 min. Samples were typically injected in duplicate, with blank injections performed between unique analytical samples.
Mass analyses were performed with a Xevo G2-XS QTof (Waters Corporation) quadrupole time-of-flight (Tof) mass spectrometer with an electrospray ionization (ESI) probe, and source parameters were optimized for the LC flow rate of 500 μL/min. Analyses were performed using Tof-multiple reaction monitoring (MRM) mode, negative polarity, and sensitivity analyzer mode. Tof data (continuum format) were acquired across the m/z range from 100 to 600 Da, with a scan time of 0.1 s. MRM data used for quantitation were acquired using a precursor m/z of 346.5 Da (−2 charge), a fragment m/z of 557.97 Da (−1 charge), a collision energy of 13, a cone voltage of 40, and target enhancement at m/z 557.97 Da. The concentration of STING agonist in a given sample was determined by comparing the STING agonist peak area in that sample to STING agonist peak areas generated by standards.
Quantification and statistical analysis
Experimental replicates, including the number of EV isolations used, are defined in the figure legends for each experiment. Numbers of animals/cells analyzed for specific experiments are reported in the figure legends as well. Statistical analyses were performed in GraphPad Prism 7 using one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test for experiments with three or more groups; for experiments with two groups, comparisons were made using Fisher’s t test. Values are expressed as mean ± standard deviation (SD) or as mean ± standard error of the mean (SEM), as indicated in the figure legends. Box and whisker plots span the maximum and minimum values, with the box showing 25th–75th percentiles. The bar marks the median value. Differences with a p <0.05 were considered significant.
Data and materials availability
The data and biological materials that support the findings of this study are available from the corresponding author upon request. The parental HEK293 cell line used in this work was obtained under license and is not available for distribution, including all subsequent engineered cell lines made from this parent clone.
Acknowledgments
The authors wish to thank Eric S. Lander, Timothy J. Harris, and Tim Soos for important editorial suggestions on the manuscript. Some graphics were created with BioRender.com. This study was funded by Codiak BioSciences.
Author contributions
Conceptualization, K.D., R.E.M., K.X., and J.D.K.; methodology, K.D., R.E.M., K.X., N.D.L., S.M., R.J.M., S.C.J., R.A.H., D.H., B.C., D.B., C.L., and J.D.K.; investigation, K.D., R.E.M., K.X., N.D.L., S.H., M.R.Y., S.M., C.L.S., C.M., R.J.M., O.B., J.S.-S., S.C.J., B.C., R.A.H., D.B., C.L., K.K., N.L.R., L.G., and S.E.; writing – original draft, K.D. and R.E.M.; writing – review & editing, K.D., R.E.M., J.D.F., J.E.T., S.S., and D.E.W.; supervision, K.D., R.E.M., K.X., K.D.E., J.D.K., S.S., and D.E.W.
Declaration of interests
All of the authors are current or former employees and shareholders of Codiak BioSciences. D.E.W. currently serves on the Board of Directors at Ovid Pharmaceuticals, AC Immune, and Cygnal Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.01.020.
Supplemental information
References
- 1.van Niel G., D’Angelo G., Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018;19:213–228. doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- 2.Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 3.Yim N., Ryu S.-W., Choi K., Lee K.R., Lee S., Choi H., Kim J., Shaker M.R., Sun W., Park J.-H. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016;7:12277. doi: 10.1038/ncomms12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pi F., Binzel D.W., Lee T.J., Li Z., Sun M., Rychahou P., Li H., Haque F., Wang S., Croce C.M. Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nat. Nanotechnol. 2018;13:82–89. doi: 10.1038/s41565-017-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao X., Ran N., Dong X., Zuo B., Yang R., Zhou Q., Moulton H.M., Seow Y., Yin H. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018;10:eaat0195. doi: 10.1126/scitranslmed.aat0195. [DOI] [PubMed] [Google Scholar]
- 7.Ibrahim A., Marbán E. Exosomes: Fundamental Biology and Roles in Cardiovascular Physiology. Annu. Rev. Physiol. 2016;78:67–83. doi: 10.1146/annurev-physiol-021115-104929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Jong O.G., Van Balkom B.W.M., Schiffelers R.M., Bouten C.V.C., Verhaar M.C. Extracellular vesicles: potential roles in regenerative medicine. Front. Immunol. 2014;5:608. doi: 10.3389/fimmu.2014.00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Didiot M.-C., Hall L.M., Coles A.H., Haraszti R.A., Godinho B.M., Chase K., Sapp E., Ly S., Alterman J.F., Hassler M.R. Exosome-mediated Delivery of Hydrophobically Modified siRNA for Huntingtin mRNA Silencing. Mol. Ther. 2016;24:1836–1847. doi: 10.1038/mt.2016.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haney M.J., Klyachko N.L., Zhao Y., Gupta R., Plotnikova E.G., He Z., Patel T., Piroyan A., Sokolsky M., Kabanov A.V., Batrakova E.V. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release. 2015;207:18–30. doi: 10.1016/j.jconrel.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smyth T., Petrova K., Payton N.M., Persaud I., Redzic J.S., Graner M.W., Smith-Jones P., Anchordoquy T.J. Surface functionalization of exosomes using click chemistry. Bioconjug. Chem. 2014;25:1777–1784. doi: 10.1021/bc500291r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haraszti R.A., Miller R., Didiot M.C., Biscans A., Alterman J.F., Hassler M.R., Roux L., Echeverria D., Sapp E., DiFiglia M. Optimized Cholesterol-siRNA Chemistry Improves Productive Loading onto Extracellular Vesicles. Mol. Ther. 2018;26:1973–1982. doi: 10.1016/j.ymthe.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kooijmans S.A.A., Aleza C.G., Roffler S.R., van Solinge W.W., Vader P., Schiffelers R.M. Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. J. Extracell. Vesicles. 2016;5:31053. doi: 10.3402/jev.v5.31053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai C.P., Kim E.Y., Badr C.E., Weissleder R., Mempel T.R., Tannous B.A., Breakefield X.O. Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun. 2015;6:7029. doi: 10.1038/ncomms8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 16.Stickney Z., Losacco J., McDevitt S., Zhang Z., Lu B. Development of exosome surface display technology in living human cells. Biochem. Biophys. Res. Commun. 2016;472:53–59. doi: 10.1016/j.bbrc.2016.02.058. [DOI] [PubMed] [Google Scholar]
- 17.Zeelenberg I.S., Ostrowski M., Krumeich S., Bobrie A., Jancic C., Boissonnas A., Delcayre A., Le Pecq J.B., Combadière B., Amigorena S., Théry C. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68:1228–1235. doi: 10.1158/0008-5472.CAN-07-3163. [DOI] [PubMed] [Google Scholar]
- 18.Jeppesen D.K., Fenix A.M., Franklin J.L., Higginbotham J.N., Zhang Q., Zimmerman L.J., Liebler D.C., Ping J., Liu Q., Evans R. Reassessment of Exosome Composition. Cell. 2019;177:428–445.e18. doi: 10.1016/j.cell.2019.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skotland T., Sandvig K., Llorente A. Lipids in exosomes: current knowledge and the way forward. Prog. Lipid Res. 2017;66:30–41. doi: 10.1016/j.plipres.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Kowal J., Arras G., Colombo M., Jouve M., Morath J.P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasaki T., Brakebusch C., Engel J., Timpl R. Mac-2 binding protein is a cell-adhesive protein of the extracellular matrix which self-assembles into ring-like structures and binds beta1 integrins, collagens and fibronectin. EMBO J. 1998;17:1606–1613. doi: 10.1093/emboj/17.6.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watson M., Stott K., Fischl H., Cato L., Thomas J.O. Characterization of the interaction between HMGB1 and H3-a possible means of positioning HMGB1 in chromatin. Nucleic Acids Res. 2014;42:848–859. doi: 10.1093/nar/gkt950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu S., Ma L., Wang S., Chen C., Zhang W., Yang L., Hang W., Nolan J.P., Wu L., Yan X. Light-scattering detection below the level of single fluorescent molecules for high-resolution characterization of functional nanoparticles. ACS Nano. 2014;8:10998–11006. doi: 10.1021/nn505162u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian Y., Ma L., Gong M., Su G., Zhu S., Zhang W., Wang S., Li Z., Chen C., Li L. Protein Profiling and Sizing of Extracellular Vesicles from Colorectal Cancer Patients via Flow Cytometry. ACS Nano. 2018;12:671–680. doi: 10.1021/acsnano.7b07782. [DOI] [PubMed] [Google Scholar]
- 25.André M., Morelle W., Planchon S., Milhiet P.E., Rubinstein E., Mollicone R., Chamot-Rooke J., Le Naour F. Glycosylation status of the membrane protein CD9P-1. Proteomics. 2007;7:3880–3895. doi: 10.1002/pmic.200700355. [DOI] [PubMed] [Google Scholar]
- 26.Murray D., Hermida-Matsumoto L., Buser C.A., Tsang J., Sigal C.T., Ben-Tal N., Honig B., Resh M.D., McLaughlin S. Electrostatics and the membrane association of Src: theory and experiment. Biochemistry. 1998;37:2145–2159. doi: 10.1021/bi972012b. [DOI] [PubMed] [Google Scholar]
- 27.Vranjkovic A., Crawley A.M., Gee K., Kumar A., Angel J.B. IL-7 decreases IL-7 receptor α (CD127) expression and induces the shedding of CD127 by human CD8+ T cells. Int. Immunol. 2007;19:1329–1339. doi: 10.1093/intimm/dxm102. [DOI] [PubMed] [Google Scholar]
- 28.Elgueta R., Benson M.J., de Vries V.C., Wasiuk A., Guo Y., Noelle R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.An H.J., Kim Y.J., Song D.H., Park B.S., Kim H.M., Lee J.D., Paik S.G., Lee J.O., Lee H. Crystallographic and mutational analysis of the CD40-CD154 complex and its implications for receptor activation. J. Biol. Chem. 2011;286:11226–11235. doi: 10.1074/jbc.M110.208215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuhn C., Weiner H.L. Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside. Immunotherapy. 2016;8:889–906. doi: 10.2217/imt-2016-0049. [DOI] [PubMed] [Google Scholar]
- 31.Tugues S., Burkhard S.H., Ohs I., Vrohlings M., Nussbaum K., Vom Berg J., Kulig P., Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22:237–246. doi: 10.1038/cdd.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uto T., Wang X., Sato K., Haraguchi M., Akagi T., Akashi M., Baba M. Targeting of antigen to dendritic cells with poly(γ-glutamic acid) nanoparticles induces antigen-specific humoral and cellular immunity. J. Immunol. 2007;178:2979–2986. doi: 10.4049/jimmunol.178.5.2979. [DOI] [PubMed] [Google Scholar]
- 33.Van Dis E., Sogi K.M., Rae C.S., Sivick K.E., Surh N.H., Leong M.L., Kanne D.B., Metchette K., Leong J.J., Bruml J.R. STING-Activating Adjuvants Elicit a Th17 Immune Response and Protect against Mycobacterium tuberculosis Infection. Cell Rep. 2018;23:1435–1447. doi: 10.1016/j.celrep.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan B.T., Johnstone R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: selective externalization of the receptor. Cell. 1983;33:967–978. doi: 10.1016/0092-8674(83)90040-5. [DOI] [PubMed] [Google Scholar]
- 35.Escudier B., Dorval T., Chaput N., André F., Caby M.P., Novault S., Flament C., Leboulaire C., Borg C., Amigorena S. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J. Transl. Med. 2005;3:10. doi: 10.1186/1479-5876-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charrin S., Le Naour F., Oualid M., Billard M., Faure G., Hanash S.M., Boucheix C., Rubinstein E. The major CD9 and CD81 molecular partner. Identification and characterization of the complexes. J. Biol. Chem. 2001;276:14329–14337. doi: 10.1074/jbc.M011297200. [DOI] [PubMed] [Google Scholar]
- 37.Chambrion C., Le Naour F. The tetraspanins CD9 and CD81 regulate CD9P1-induced effects on cell migration. PLoS ONE. 2010;5:e11219. doi: 10.1371/journal.pone.0011219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charrin S., Latil M., Soave S., Polesskaya A., Chrétien F., Boucheix C., Rubinstein E. Normal muscle regeneration requires tight control of muscle cell fusion by tetraspanins CD9 and CD81. Nat. Commun. 2013;4:1674. doi: 10.1038/ncomms2675. [DOI] [PubMed] [Google Scholar]
- 39.Brown M., Stafford L.J., Onisk D., Joaquim T., Tobb A., Goldman L., Fancy D., Stave J., Chambers R. Snorkel: an epitope tagging system for measuring the surface expression of membrane proteins. PLoS ONE. 2013;8:e73255. doi: 10.1371/journal.pone.0073255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El Amri M., Fitzgerald U., Schlosser G. MARCKS and MARCKS-like proteins in development and regeneration. J. Biomed. Sci. 2018;25:43. doi: 10.1186/s12929-018-0445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward-Kavanagh L.K., Lin W.W., Šedý J.R., Ware C.F. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity. 2016;44:1005–1019. doi: 10.1016/j.immuni.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiklander O.P.B., Brennan M.Á., Lötvall J., Breakefield X.O., El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019;11:eaav8521. doi: 10.1126/scitranslmed.aav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smyth T., Kullberg M., Malik N., Smith-Jones P., Graner M.W., Anchordoquy T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control. Release. 2015;199:145–155. doi: 10.1016/j.jconrel.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Herpen C.M., Huijbens R., Looman M., De Vries J., Marres H., Van De Ven J., Hermsen R., Adema G.J., De Mulder P.H. Pharmacokinetics and immunological aspects of a phase Ib study with intratumoral administration of recombinant human interleukin-12 in patients with head and neck squamous cell carcinoma: a decrease of T-bet in peripheral blood mononuclear cells. Clin. Cancer Res. 2003;9:2950–2956. [PubMed] [Google Scholar]
- 45.Lasek W., Zagożdżon R., Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014;63:419–435. doi: 10.1007/s00262-014-1523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jang S.C., Moniz R.J., Sia C.L., Harrison R.A., Houde D., Ross N., Xu K., Lewis N., Bourdeau R., McCoy C. exoSTING: an engineered exosome therapeutic that selectively delivers STING agonist to the tumor resident antigen-presenting cells resulting in improved tumor antigen-specific adaptive immune response. Cancer Res. 2019;79:944. [Google Scholar]
- 47.Kamerkar S., LeBleu V.S., Sugimoto H., Yang S., Ruivo C.F., Melo S.A., Lee J.J., Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Welsh J.A., Van Der Pol E., Arkesteijn G.J.A., Bremer M., Brisson A., Coumans F., Dignat-George F., Duggan E., Ghiran I., Giebel B. MIFlowCyt-EV: a framework for standardized reporting of extracellular vesicle flow cytometry experiments. J. Extracell. Vesicles. 2020;9:1713526. doi: 10.1080/20013078.2020.1713526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.