

Abstract
Growing evidence indicates that N6-methyladenosine (m6A) is the most pervasive RNA modification in eukaryotic cells. However, the specific role of METTL3 in papillary thyroid carcinoma (PTC) initiation and development remains elusive. Here we found that downregulation of METTL3 was correlated with malignant progression and poor prognosis in PTC. A variety of gain- and loss-of-function studies clarified the effect of METTL3 on regulation of growth and metastasis of PTC cells in vitro and in vivo. By combining RNA sequencing (RNA-seq) and methylated RNA immunoprecipitation sequencing (meRIP-seq), our mechanistic studies pinpointed c-Rel and RelA as downstream m6A targets of METTL3. Disruption of METTL3 elicited secretion of interleukin-8 (IL-8), and elevated concentrations of IL-8 promoted recruitment of tumor-associated neutrophils (TANs) in chemotaxis assays and mouse models. Administration of the IL-8 antagonist SB225002 substantially retarded tumor growth and abolished TAN accumulation in immunodeficient mice. Our findings revealed that METTL3 played a pivotal tumor-suppressor role in PTC carcinogenesis through c-Rel and RelA inactivation of the nuclear factor κB (NF-κB) pathway by cooperating with YTHDF2 and altered TAN infiltration to regulate tumor growth, which extends our understanding of the relationship between m6A modification and plasticity of the tumor microenvironment.
Keywords: papillary thyroid cancer, N6-methyladenosine, METTL3, c-Rel, YTHDF2, neutrophils
Graphical abstract
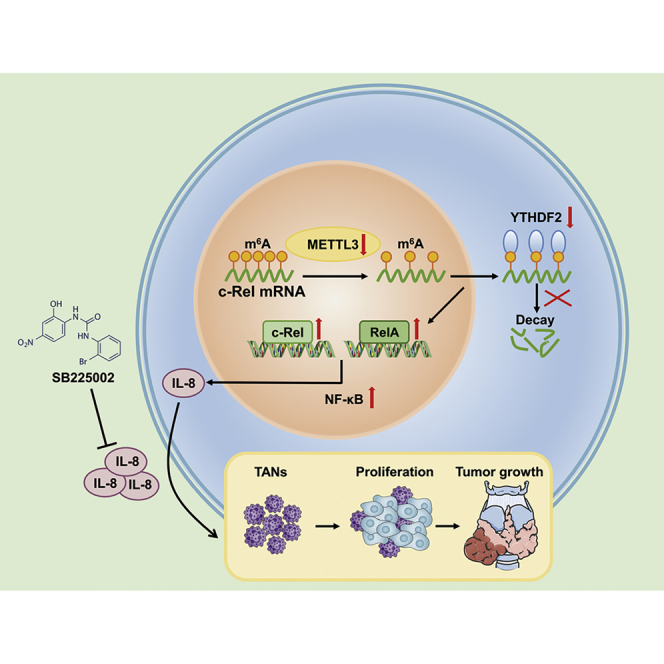
He et al. uncover that METTL3 is downregulated in PTC tissues and cells. METTL3 recognizes c-Rel and RelA m6A modifications and collaborates with YTHDF2 in regulating c-Rel and RelA mRNA homeostasis to silence NF-κB signaling pathway activity, inhibiting chemotaxis of tumor-associated neutrophils and PTC progression.
Introduction
With an estimated 600,000 new cases annually worldwide, thyroid cancer ranks first among endocrine malignancies and is the ninth most common form of cancer in the world.1 Papillary thyroid carcinoma (PTC) comprises about 90% of all cases of thyroid cancer, which is characterized by an increased tendency of capsular invasion and compression of surrounding organs and a high rate of dissemination to local lymph nodes.2 At present, the etiology and molecular pathogenesis of PTC remain mostly unclear. B-type RAF kinase, BRAF (V600E) mutation has been reported to be an initiating factor of PTC carcinogenesis.3 Other potent causes of PTC initiation include v-Ki-ras2 Kirsten rat sarcoma viral oncogene homology gene, KRAS (S65N) mutation, Ret proto-oncogene (RET)/PTC rearrangement, and paired box 8 (PAX8)/perixisome proliferation-activated receptor (PPAR) rearrangement.4 However, many individuals with PTC without these somatic alterations are detected in clinical practice. A better understanding of the molecular mechanisms governing the pathogenesis of PTC is crucial for development of detection biomarkers and therapeutic strategies.
The N6-methyladenosine (m6A) modification is the most universal RNA modification found on the adenosine base of eukaryotic mRNAs, usually occurring on the RRACH consensus sequence (R = A or G and H = A, C or U) of 3′ UTR or stop codon regions.5 The m6A modification is highly reversible because of the tight cooperation of m6A “writers,” “erasers,” and “readers.” The m6A modification can be installed by m6A methyltransferase (a complex consisting of methyltransferase-like 3 [METTL3], methyltransferase-like 14 [METTL14], and WT1 associated protein [WTAP]); removed by m6A demethylases (fat mass and obesity-associated protein [FTO] and AlkB homolog 5 RNA demethylase [ALKBH5]); and recognized by the YT521-B homology (YTH) domain family (YTHDF1-3 and YTHDC1-2), insulin-like growth factor 2 mRNA binding proteins (IGF2BP1–IGF2BP3), or eukaryotic initiation factor 3 (EIF3).6 Accumulating evidence suggests that dysfunctional m6A methylation participates in multiple human diseases, including type 2 diabetes, colitis, cardiac hypertrophy, and malignancies.7, 8, 9, 10, 11 Although many studies have established a close relationship between METTL3 expression and malignant phenotypes of different cancers, the potential role of METTL3 in PTC is largely elusive.
Here we carried out a series of in vivo and in vitro experiments to systemically delineate the biological functions of METTL3 and its underlying molecular mechanisms in human PTC carcinogenesis. We uncovered that the tumor-suppressing effect of METTL3 could be attributed to c-Rel- and RelA-mediated inactivation of the nuclear factor κB (NF-κB) pathway. Silencing of METTL3 increased secretion of interleukin-8 (IL-8) from PTC cells, driving chemotaxis of tumor-associated neutrophils (TANs) to organize the immunosuppressive tumor microenvironment to support PTC progression. Our findings provide novel insights into the molecular mechanism of PTC carcinogenesis epigenetic alterations and indicate that METTL3 and its related c-Rel/IL-8 axis could be pivotal targets for treatment of PTC.
Results
Decreased expression of METTL3 correlates with a poor prognosis of individuals with PTC
To identify the roles of the m6A modification in progression of PTC, we first examined m6A writer (METTL3, METTL14, and WTAP) and m6A eraser (FTO and ALKBH5) expression in 60 pairs of PTC and adjacent normal thyroid tissue in our cohort. RT-PCR results showed that METTL3 and METTL14 were reduced in tumor tissue related to non-tumor samples, whereas WTAP, FTO, and ALKBH5 levels showed no pronounced differences between tumor and normal specimens (Figure 1A). We also screened the expression profiles of these m6A-related genes in the GEPIA platform, and the pattern of METTL3 expression was consistent with our RT-PCR data (Figure S1A). Because METTL3 exhibited the most distinct differential expression, we chose METTL3 for further analysis. Next we detected the protein level of METTL3 in PTC tissue. Western blot revealed that METTL3 was more abundantly expressed in human thyroid tissue (Figure 1B). Immunohistochemistry results from 58 PTC specimens corroborated these findings; immunohistochemistry (IHC) scores confirmed that METTL3 was silenced in human PTC tissue (Figures 1C and 1D). METTL3 was downregulated in a panel of PTC cell lines compared with the normal thyroid follicular cell line Nthy-ori 3-1 at the mRNA and protein levels (Figures 1E and 1F). Immunofluorescence staining showed that METTL3 was mainly localized in the cell nucleus and expressed weakly in tumor cells (Figure 1G). Furthermore, individuals with PTC with low expression of METTL3 had a worse prognosis, and METTL3 expression was inversely correlated with poor recurrence-free survival (RFS) (Figure S1B). We also noticed that higher METTL14 expression was tightly correlated with shorter overall survival (Figure S1C).
Figure 1.

METTL3 is silenced in human PTC samples and cell lines
(A) Violin plots showing METTL3, METTL14, WTAP, FTO, and ALKBH5 expression on the basis of the 60 human PTC tissues and paired normal thyroid tissues. (B) Western blot analysis of METTL3 protein expression in 11 paired human PTC samples and non-tumor thyroid tissues. β-Actin was served as the loading control. (C) Representative IHC staining of METTL3 in thyroid tissue sections from individuals with PTC. Magnification, 200×. Scale bar, 200 μm. (D) Quantification of IHC staining for METTL3 in 58 primary human PTC samples and matched normal tissues. The score was assessed on a scale of 0–3 in staining areas and densities. (E and F) Western blot and RT-PCR analysis showing that METTL3 expression was decreased in PTC cells. (G) Representative images of immunofluorescence staining, showing expression of METTL3 in thyroid follicular epithelial cells and PTC cells. Magnification, 200×. Scale bar, 200 μm. Data represent the mean ± SD (∗p < 0.05, ∗∗∗p < 0.001).
METTL3 deficiency accelerates PTC cell proliferation
We proceeded to dissect the global m6A level in PTC tissue and cell lines using an m6A RNA methylation quantification kit. The m6A RNA levels were remarkably higher in normal thyroid tissues compared with matched tumor tissues and in thyroid follicular cells in comparison with PTC cells (Figure 2A). To clarify the putative role of METTL3 in PTC tumorigenesis, we constructed vectors stably expressing wild-type METTL3 and catalytic mutant (amino acids [aa] 395–398, DPPW–APPA) METTL3 using the pCDH plasmid in KTC-1 cells with the lowest endogenous level of METTL3. Additionally, two specific short hairpin RNAs (shRNAs) targeting METTL3 were transfected into BCPAP and TPC-1 cells. The knockdown and overexpression efficiency was determined by RT-PCR and western blot in these PTC cell lines (Figures 2B and 2C; Figure S2A). We examined the change in m6A RNA levels upon METTL3 overexpression or inhibition in PTC cells. As anticipated, knockdown of METTL3 prominently decreased the m6A level in PTC cells (Figure 2D). In contrast, METTL3 overexpression elevated the level of m6A modification, whereas the catalytic mutant METTL3 abolished this (Figure S2B). EdU incorporation assays were conducted to find out the effects of METTL3 on cell proliferation. These results implied that silencing of METTL3 considerably accelerated the growth of BAPAP and TPC-1 cells (Figure 2E). PTC cell viability was measured by Cell Counting Kit-8 (CCK-8) and CellTiter-Glo assay. Deficiency in METTL3 resulted in enhanced cell viability, whereas ectopic expression of METTL3 evoked opposite effects (Figures 2F and 2G; Figures S2C and S2D). Strikingly, the m6A catalytic activity of METTL3 was indispensable for regulating the growth of PTC cells because mutant METTL3 could not decelerate cell proliferation. Moreover, a robust decrease in colony numbers was observed in METTL3-overexpressing KTC-1 cells, as evidenced by colony formation assays (Figure S2E). On the contrary, METTL3 knockdown promoted the colony formation ability in BCPAP and TPC-1 cells (Figure 2H). These results illustrate the anti-proliferative role of METTL3 in PTC cells.
Figure 2.

Silencing of METTL3 decreases m6A levels and promoted growth of PTC cells
(A) Decreased m6A mRNA methylation levels were confirmed in PTC tumor tissues and PTC cell lines by m6A colorimetry analysis. n = 12 for paired PTC and normal tissues. (B) BCPAP and TPC-1 cells were transfected with different shMETTL3 vectors and a scramble vector (negative control), and the knockdown effect was verified by western blot. (C and D) Two shRNAs targeting METTL3 decreased METTL3 mRNA expression and endogenous m6A levels in BCPAP and TPC-1 cells. (E) The effect of METTL3 on proliferation of BCPAP and TPC-1 cells was determined by EdU assays. Magnification, 200×. Scale bar, 200 μm. (F) Knockdown of METTL3 enhanced cell viability in both cell lines, as shown by CCK-8 assays. (G) Knockdown of METTL3 accelerated PTC cell proliferation, as evidenced by CellTiter-Glo luminescence assays. (H) The number of colonies increased dramatically after knockdown of METTL3 in the BCPAP and TPC-1 cell lines. Data are expressed as mean ± SD (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Loss of METTL3 triggers PTC cell metastasis
Cancer metastasis is the leading cause of death in individuals with PTC. Gain- and loss-of-function studies were utilized to assess the effects of METTL3 on PTC cell mobility. Boyden chamber assays demonstrated that depletion of METTL3 induced, but overexpression of METTL3 weakened, PTC cell migration and invasion in vitro (Figures 3A and 3B; Figures S3A and S3B). These results were further substantiated by in vivo experimental lung metastasis models. The same amounts of METTL3-deficient and control BCPAP cells were injected into immunocompromised nude mice through the tail vein. Ablation of METTL3 efficiently augmented the number of metastatic lung nodules in nude mice after 8 weeks of injection (Figures 3C and 3D). The opposite was observed in wild-type and mutant METTL3 overexpression and corresponding control KTC-1 cells. Exogenous expression of METTL3 suppressed tumor cells seeding, as detected by H&E staining of lung sections of these mice (Figures S3C and S3D). In conclusion, METTL3 has inhibitive effects on PTC metastasis with dependence on its m6A catalytic activity.
Figure 3.

Inhibition of METTL3 drives PTC cell metastasis in vitro and in vivo
(A and B) Transwell migration assays and Matrigel invasion assays were used to determine the cell migration and invasion capabilities of BCPAP and TPC-1 cells transfected with the shMETTL3 and control vectors. Shown is quantification of cell numbers from three independent experiments. Magnification, 200×. Scale bar, 300 μm. (C) Representative images of lung metastasis lesions derived from nude mice 8 weeks after injection of control and METTL3 knockdown BCPAP cells (n = 5 mice per group). (D) H&E staining and quantification of metastatic nodules in the lungs of the indicated groups of nude mice. Magnification, 50×. Scale bar, 200 μm. Data represent the mean ± SD (∗∗p < 0.01, ∗∗∗p < 0.001).
Transcriptome-wide RNA-seq and m6A-seq identify c-Rel as m6A-mediated targets
To better understand the possible mechanism of METTL3 participation in PTC progression, we first performed RNA sequencing (RNA-seq) analysis using METTL3-silenced BCPAP and TPC-1 and their control cells. We identified 486 and 1,119 differentially expressed genes in BCPAP and TPC-1 cells, respectively (Figures S4A and S4B). Among them, 52 genes were coherently altered by METTL3 knockdown in both BCPAP and TPC-1 cells. Gene Ontology (GO) and KEGG enrichment analysis revealed that these genes were mostly linked to cytokine receptor binding, the NF-κB signaling pathway, and transcriptional misregulation in cancer, supporting a regulatory role of METTL3 in carcinogenesis of PTC (Figures S4C and S4D). Next we performed m6A-seq in METTL3-deficient BCPAP cells to detect METTL3-mediated m6A methylated transcripts. A total of 472 and 832 up- and downregulated m6A peaks were defined globally from m6A-seq libraries. m6A signals were specifically abundant in the 3′ UTR, and the consensus motif GAAC was highly enriched in the control and METTL3 knockdown groups (Figures S5A and S5B; Figure 4A). Considering that METTL3 is responsible for catalyzing the m6A modification, we mainly focused on the downregulated m6A peaks caused by METTL3 silencing in PTC cells. These m6A-modified transcripts exhibited apparent associations with the NF-κB signaling pathway, RNA degradation, and cellular nitrogen compound metabolic process (Figures S5C and S6D). Combining the hypomethylated m6A peaks from m6A-seq with differentially expressed genes from RNA-seq, 19 genes were filtered (Figure 4B). Among these overlapping genes, c-Rel has been recognized as an important transcription factor in the NF-κB family. From our m6A-seq data, we confirmed that m6A abundance in the c-Rel mRNA was diminished in response to METTL3 knockdown (Figure 4C).We also found that another NF-κB subunit, RelA/p65, showed decreased m6A methylation in METTL3 knockdown cells with respect to control cells. Hence, these two genes were chosen as candidate targets of METTL3 for subsequent research. Methylated RNA immunoprecipitation (meRIP)-qPCR using an m6A-specific antibody detected that the m6A levels of c-Rel and RelA were decreased in METTL3-silenced BCPAP and TPC-1 cells and increased in METTL3-overexpressing KTC-1 cells. However, mutant METTL3 lost the ability to regulate the levels of m6A modification of c-Rel and RelA (Figure S6A). Additionally, inhibition of METTL3 contributed to the increased mRNA levels of c-Rel and RelA and overexpression of METTL3 showed the opposite trends, as evidenced by RT-PCR (Figures 4D and 4E; Figure S6B). Corroborating the transcriptional levels, immunoblotting confirmed the negative regulation of METTL3 on the protein levels of c-Rel and RelA (Figure 4F; Figure S6C). We then surmised that the m6A modification might affect the stability of the c-Rel and RelA mRNAs. To test this, PTC cells were incubated with the transcription inhibitor actinomycin D for 3 h and 6 h. RT-PCR data showed that METTL3 knockdown prolonged the lifetime of the c-Rel and RelA mRNAs and that forced expression of METTL3 shortened the half-life of c-Rel and RelA in KTC-1 cells (Figures 4G and 4H; Figure S6D). We also treated PTC cells with the global methylation inhibitor 3-deazaadenosine (DAA) to block global m6A activity. We found that DAA treatment stimulated the mRNA levels of c-Rel and RelA in KTC-1 cells stably overexpressing wild-type or mutant METTL3, implying that c-Rel and RelA were modulated by m6A methylation (Figure S6E). Finally, we analyzed the activity of the NF-κB pathway using the pNF-κB-Luc reporter. As expected, ablation of METTL3 led to activation of the NF-κB pathway, whereas catalytic mutant METTL3 abrogated the ability to induce the NF-κB pathway (Figures S6F and S6G). Additionally, c-Rel and RelA deletion in BCPAP or TPC-1 cells prominently weakened NF-κB pathway activity. These results proved the importance of c-Rel and RelA in METTL3-mediated NF-κB signaling (Figure S7A). Collectively, these data establish that METTL3 regulates c-Rel and RelA mRNA homeostasis via m6A methyltransferase activity.
Figure 4.

Identification of potential targets of METTL3-mediated m6A modification in PTC cells
(A) The predominant consensus motif GAAC was identified in m6A-seq results from control and shMETTL3 cells. (B) Venn diagram demonstrating that 19 genes overlap in differentially expressed genes by RNA-seq and downregulated peaks by m6A-seq. (C) m6A-seq identification of m6A modification sites in c-Rel mRNA. (D) meRIP-qPCR analysis of c-Rel and RelA RNA m6A levels in BCPAP (top panel) and TPC-1 (bottom panel) cells with METTL3 knockdown. (E) RT-PCR analysis of alterations of the mRNA levels of c-Rel and RelA in METTL3 knockdown and control PTC cells. (F) Western blot of c-Rel and RelA protein expression upon knockdown of METTL3 in PTC cells. (G and H) The mRNA half-life of c-Rel (G) and RelA (H) was evaluated in METTL3 knockdown cells and their corresponding control PTC cells treated with actinomycin D (5 μg/mL) at the indicated time point by RT-PCR. Data represent the mean ± SD (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
METTL3 regulates c-Rel in an m6A-YTHDF2-dependent manner
To further ascertain the role of METTL3 in controlling m6A modification of c-Rel, we introduced three synonymous mutations at the putative m6A sites in c-Rel, predicted by SRAMP (http://www.cuilab.cn/sramp).12 The c-Rel 3′ UTR containing different point mutations (adenine was replaced with thymine to eradicate the m6A modification) was cloned into the pmirGlo dual-luciferase reporter, and the luciferase activity of the wild-type or mutant c-Rel-fused reporter was measured in control and METTL3 knockdown BCPAP and TPC-1 cells. A 2- to 3-fold increase in luciferase activity was observed in the wild-type 3′ UTR of c-Rel in BCPAP and TPC-1 cells lacking METTL3. Mut2 and mut3 lost METTL3-mediated activation of c-Rel luciferase activity in BCPAP cells (Figure 5A). However, only mut3 abrogated this induction by suppression of METTL3 in TPC-1 cells, suggesting that the AGTCT sequence (mut3) in the 3′ UTR of c-Rel is essential for METTL3-mediated m6A modulation. Because c-Rel was negatively regulated by m6A methylation, we presumed that YTHDF2, the m6A reader protein that facilitates decay of m6A methylated transcripts, may participate in m6A methylation of c-Rel mRNA. Moreover, we screened previous published RIP sequencing (RIP-seq) data (GSE49339) and discovered direct binding between the c-Rel transcript and YTHDF2 in HeLa cells. To validate our hypothesis, small interfering RNA (siRNA) targeting YTHDF2 was integrated into PTC cells with the indicated vectors. We noticed that inhibition of YTHDF2 indeed stimulated c-Rel mRNA expression to different degrees in BCPAP and TPC-1 cells, whereas inhibition of METTL3 impaired regulation of YTHDF2 for stabilizing c-Rel mRNA (Figure 5B). RIP-qPCR was performed using anti-YTHDF2 to confirm the interaction between c-Rel mRNA and YTHDF2 in all three PTC cell lines. Notably, YTHDF2 could also bind to the mRNA of RelA in PTC cells (Figure 5C). Given that YTHDF2 promotes mRNA degradation by selectively binding m6A-modified mRNA, we quantified the mRNA stability of c-Rel and RelA upon knockdown of YTHDF2. YTHDF2 downregulation postponed the decay of m6A-methylated transcripts in YTHDF2-depleted BCPAP and TPC-1 cells (Figures 5D and 5F). Deletion of YTHDF2 partly prolonged the lifespan of c-Rel and RelA mRNA in METTL3-overexpressing KTC-1 cells (Figures 5E and 5G). To further unveil whether the tumor-restraining effects of METTL3 were dependent on YTHDF2, we examined cell proliferation and metastasis by CCK-8 and Transwell assays. These results showed that the anti-tumor phenotypes initiated by METTL3 overexpression were restored by inhibition of YTHDF2 in KTC-1 cells (Figures 5H and 5I). In summary, YTHDF2 recognizes the m6A modification of c-Rel and RelA and assist METTL3 to participate in PTC progression.
Figure 5.

METTL3 regulates m6A modification of c-Rel in a YTHDF2-dependent manner
(A) Putative m6A modification sites in the sequences of c-Rel 3′ UTR mRNA transcripts. Shown is the relative luciferase activity of the c-Rel 3′ UTR luciferase reporter with wild-type or synonymous mutant m6A sites in scramble and shMETTL3 BCPAP (center panel) and TPC-1 (right panel) cells. (B) The mRNA level of c-Rel was quantified by qRT-PCR upon YTHDF2 depletion in BCPAP and TPC-1 cells transfected with control and shMETTL3 vectors. (C) RIP-qPCR analysis using an anti-YTHDF2 antibody and IgG antibody, confirming YTHDF2 binding to c-Rel and RelA mRNA. HPRT1, reported previously to contain no m6A modifications and not to be bound by YTHDF2, was used as a negative control. (D) The decay rate of c-Rel mRNA by RT-PCR analysis at the indicated times after treatment with actinomycin D in BCPAP and TPC-1 cells after YTHDF2 inhibition. (E) RT-PCR analysis of the mRNA stability of c-Rel in KTC-1 cells stably expressing METTL3 with or without YTHDF2 knockdown. (F and G) METTL3-silenced or -overexpressing PTC cells were treated with actinomycin D and harvested at 0, 3, and 6 h. The mRNA level of RelA was normalized to expression at 0 h and measured by RT-PCR. (H and I) CCK-8 (H) and Transwell (I) assays showing the proliferation and migration ability of KTC-1 cells transfected with the indicated plasmids. Data represent the mean ± SD (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant).
Inhibition of c-Rel retards malignant progression of PTC
c-Rel, which is encoded by the REL gene in humans, plays a crucial role in regulation of inflammation and apoptosis.13 Evidence has highlighted a strong link between c-Rel and multiple malignancies. However, the precise role of c-Rel in PTC tumorigenesis has not been documented. Therefore, we first determined the effect of c-Rel expression on PTC tumor metastasis in vitro and in vivo. We observed that the increased metastatic potential associated with METTL3 deficiency could be partially attenuated by depletion of c-Rel, as evidenced by Transwell migration and invasion experiments (Figures 6A and 6B; Figure S7B). In line with the in vitro findings, knockdown of c-Rel appreciably influenced lung metastasis colonization in tail vein injection models (Figure 6C). H&E staining substantiated the reduced lung metastatic burden in the double knockdown METTL3 and c-Rel group versus METTL3-deficient BCPAP cells (Figure 6D). Furthermore, genetic ablation of c-Rel also impaired the effects of METTL3 inhibition on cell growth, as assessed by EdU immunofluorescence assays (Figures 6E and 6F). Similar results were observed in CCK-8 and CellTiter-Glo experiments. Depletion of endogenous c-Rel in METTL3 knockdown BCPAP and TPC-1 cells mitigated the effects of METTL3-induced enhanced cell viability (Figures 6G and 6H). Moreover, we also assessed the potential effects of RelA in METTL3-deficient PTC cells. The data showed that RelA depletion resulted in attenuation of increased proliferation and migration induced by METTL3 silencing, highlighting the essential role of RelA in METTL3-controlled malignant behavior (Figures S7C and S7D). Subsequently, we tested the roles of METTL3 and c-Rel in tumorigenicity using tumor xenograft models. We generated xenografts on a background of BCPAP cells transfected with a control vector, METTL3-knockdown vector, and METTL3 plus c-Rel knockdown vector in BALB/c mice. The average tumor weights and volumes were persistently increased in mouse xenografts injected with BCPAP cells bearing shMETTL3 vectors, whereas inhibition of c-Rel in the METTL3 deletion group caused reduced tumor growth compared with the shMETTL3 groups (Figures 7A–7C). IHC staining of tumor samples showed a dramatic increase in c-Rel and RelA levels upon METTL3 knockdown (Figure 7D). Conversely, KTC-1 cells bearing stable METTL3 expression showed restrained tumor growth compared with control KTC-1 cells and mutant METTL3 groups (Figures 7E–7G). Primary tumor sections from METTL3-overexpressing mice exhibited a lower positive rate of c-Rel and RelA, reaffirming inactivation of the NF-κB pathway upon METTL3 overexpression (Figure 7H). Our results clarify the critical role of c-Rel in METTL3-mediated PTC progression in vitro and in vivo.
Figure 6.

c-Rel is critical for METTL3-regulated PTC cells metastasis and proliferation
(A and B) Effects of c-Rel on migration and invasion of BCPAP (A) and TPC-1 (B) cells with the shMETTL3 vector. Inhibition of c-Rel rescued metastasis in METTL3 knockdown PTC cells. Magnification, 200×. Scale bar, 300 μm. (C) Representative views of lung metastasis foci in the tail vein metastasis model in the tested mice (n = 5 mice per group). (D) Representative microscopic views of lung metastasis foci from H&E staining and statistical analysis of metastatic lung nodules at the endpoint (8 weeks). Magnification, 50×. Scale bar, 200 μm. (E and F) Representative micrographs of EdU incorporation assays with the indicated BCPAP (E) and TPC-1 (F) cells. Magnification, 200×. Scale bar, 200 μm. (G) A CCK-8 assay was performed to measure proliferation of PTC cells transfected with shMETTL3 and shMETTL3 plus shRel compared with those transfected with a scramble vector. (H) Cell viability was also investigated by CellTiter-Glo luminescence assays in BCPAP and TPC-1 cells. Data are presented as the mean ± SD from triplicate experiments (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Figure 7.

Inhibition of METTL3 has a tumor-promoting effect in vivo
(A) Representative pictures of tumors harvested from BCPAP xenografts transfected with scramble negative control, shMETTL3, and shMETTL3 with shRel in BALB/c nude mice. (B and C) Relative tumor weights (B) and volumes (C) of mice bearing BCPAP cells with the indicated vectors (n = 5 mice per group). (D) Representative IHC images of c-Rel and RelA staining in serial sections of tumor tissues isolated from xenograft models and statistical analysis of c-Rel+ and RelA+ cells in the indicated groups of tumors. Magnification, 200×. Scale bar, 200 μm. (E) Primary tumor samples from mice with subcutaneous injection of KTC-1 cells transfected with different vectors. (F) Relative tumor weights were measured at the endpoint. (G) Primary tumor growth curves of the indicated groups monitored every 4 days (n = 5 biologically independent samples). (H) Representative IHC staining images showing expression of c-Rel and RelA in xenograft tumor tissues and quantification of c-Rel+ and RelA+ cells in the indicated subcutaneous xenograft tumors. Magnification, 200×. Scale bar, 200 μm. Data represent the mean ± SD (∗∗p < 0.01, ∗∗∗p < 0.001).
IL-8 recruits neutrophils to facilitate METTL3-deficient tumor cell growth
Given the core contribution of the NF-κB signaling pathway to the inflammatory tumor microenvironment, a proteome profiler human cytokine array was implemented to visualize the cytokine and chemokine profile changes following METTL3 knockdown in PTC cells. We found that inhibition of METTL3 presented with elevated levels of a series of inflammatory cytokines, including CXCL1, CXCL11, IL-6, IL-8, and TREM1. Anti-inflammatory cytokines, especially IL-4 and IL-10, were decreased after knockdown of METTL3 (Figure 8A). IL-8 was shown to be robustly enriched in BCPAP and TPC-1 cells with METTL3 silencing and was selected for further research. We measured the IL-8 levels in culture media of indicated PTC cells and verified that genetic silencing of METTL3 augmented IL-8 production, as evidenced by ELISA. Intriguingly, the NF-κB pathway inhibitor BAY 11-7082 could noticeably reduce IL-8 levels, indicating a critical role of this pathway in regulating IL-8 expression (Figure 8B). In addition, RT-PCR analysis revealed that loss of c-Rel or blockade of the NF-κB pathway attenuated the induction of silencing METTL3 on IL-8. In contrast, ectopic expression of METTL3 decreased the mRNA level of IL-8 (Figures S8A and S8B). We next determined the IL-8 concentration in serum collected from individuals with PTC and healthy donors. ELISAs showed that IL-8 protein levels were consistently elevated in serum from individuals with PTC compared with serum from healthy donors (Figure 8C). Data from TCGA also showed a pronounced increase in IL-8 mRNA levels in PTC samples (Figure 8D). IL-8 has been reported previously to be able to attract neutrophils to inflammatory foci.14 We wanted to ascertain the relevance of IL-8 and the NF-κB pathway to TAN regulation. Subcutaneous tumor tissues from BCPAP cells with the indicated vectors were dissociated and analyzed by flow cytometry. Knockdown of METTL3 in BCPAP cells indeed increased the population of TANs in the tumors, as shown by CD45+CD11b+Ly6G+ numbers, whereas inhibition of c-Rel could block TAN recruitment (Figure 8E). In contrast, overexpression of METTL3 in KTC-1 xenograft tumors showed less TAN infiltration, as depicted in Figure S8C. Next we performed chemotaxis assays using a Transwell co-culture system. Conditional medium from cultures of METTL3 knockdown or control BCPAP or TPC-1 cells was placed in the bottom wells, and freshly isolated TANs from PTC cell-bearing mice were seeded in the upper chambers. The chemotaxis assays results showed that the migration index of TANs was increased markedly following co-culture with METTL3-deleted cells (Figure 8F). In contrast, conditional medium from METTL3-overexpressing cells weakened the migration of TANs compared with that from control KTC-1 cells or METTL3 mutant cells. Notably, these chemotactic effects were apparently blocked with an IL-8-neutralizing antibody or inhibition of c-Rel or RelA (Figures S8D and S8E). Considering that IL-8-related chemo-attraction could be advantageous to tumors, we explored the possibility of therapeutic inhibition targeting IL-8 in PTC cells. Equal amounts of BCPAP cells stably expressing shMETTL3 or a control vector were injected subcutaneously into nude mice (10 mice per group). When palpable tumors formed on day 14, mice in each group were divided randomly into two subgroups, and the IL-8 antagonist SB225002 or its carrier solution, DMSO, was administered intraperitoneally into tumor-bearing mice 3 times per week (Figure 8G). After monitoring for 7 weeks, we found that SB225002 showed satisfactory anti-tumor efficacy in PTC cells (Figures 8H and 8I). Treatment with the IL-8 antagonist SB225002 also suppressed TAN recruitment to tumors in the control and shMETTL3 groups (Figure 8J). We demonstrate that silencing of METTL3 elevates IL-8 expression, promoting TAN recruitment, and that blockade of IL-8 suppresses TAN accumulation and tumorigenesis in PTC cells.
Figure 8.

The METTL3/c-Rel/IL-8 axis regulates neutrophil recruitment in the PTC cell microenvironment
(A) A series of human cytokines and chemokines of cell lysates from BCPAP (top panel) and TPC-1 (bottom panel) cells transfected with the control and shMETTL3 vectors were detected using the Proteome Profiler Human Cytokine Array Kit. The relative level was based on the intensities normalized to the positive controls on the same membrane. (B) Protein expression of IL-8 in BCPAP (top panel) and TPC-1 (bottom panel) cells with the indicated vectors in the presence or absence of the NF-κB inhibitor BAY 11-7082 by ELISA. (C) ELISA of serum IL-8 concentrations from individuals with PTC and healthy donors (n = 8 for each group). (D) The mRNA level of IL-8 in PTC tumor tissues and normal thyroid tissues from the GEPIA database. (E) Tumors samples were collected from subcutaneous models, and the proportions of CD11b+Ly6G+ cells in the CD45+ cell population were determined by flow cytometry. (F) The Transwell system was used to detect neutrophil chemotaxis. TANs were seeded in the upper chamber, and conditioned medium from shMETTL3 or control BCPAP and TPC-1 cells was placed in the lower chamber added IL-8 neutralizing antibody. (G) A schematic of subcutaneous injection of control and shMETTL3 BCPAP cells with SB225002 or DMSO into nude mice. (H) Relative tumor volumes of different groups, monitored every week; treatment started after week 2 (n = 5 mice per group). (I) Tumor weight was measured after sacrifice in the indicated groups. (J) Flow cytometric analyses of tumor cells derived from subcutaneous BCPAP cells treated with SB225002 or DMSO. The percentage of neutrophils relative to CD45+ cell count is plotted. (K) Schematic summarizing the METTL3/c-Rel/IL-8 axis and its role in modulating PTC progression. Data represent the mean ± SD (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
To illustrate the clinical relevance of the METTL3/c-Rel/IL-8 axis in promoting PTC progression, we examined the expression levels of METTL3, c-Rel, IL-8, and MPO (a marker of neutrophil infiltration) in 42 individuals with PTC by RT-PCR. Expression of METTL3 was negatively correlated with the levels of c-Rel and IL-8. We also observed an inverse relationship between METTL3 and MPO in individuals with PTC (Figure S9A). Furthermore, we evaluated the prognostic value of downstream METTL3 targets in clinical samples. Individuals with PTC with increased c-Rel and IL-8 levels had shorter RFS compared with individuals who had low levels of c-Rel and IL-8, as shown via the bioinformatics tool Kaplan-Meier Plotter (Figure S9B). These results underpin the clinical significance of downstream METTL3 targets in individuals with PTC.
Discussion
Many studies have investigated epitranscriptomics over the past decades because of recent technical advances in characterizing RNA modifications.15,16 It is generally assumed that more than 100 RNA modifications exist in mammalian cells. Among these distinct chemical modifications, m6A RNA methylation, the most prevalent RNA modification in eukaryotic cells, performs a great diversity of biological functions in normal development and disease, including initiating translation, stabilizing transcripts, splicing pre-mRNA, and facilitating nuclear export.17,18 In this study, we investigated the expression profiles of m6A RNA methylation-related proteins in PTC tissues and paired normal thyroid tissues. We focused on METTL3, the first documented methyltransferase for m6A modification, which is frequently silenced in individuals with PTC and functions as a tumor suppressor in PTC cells. Gain- and loss-of-function studies uncovered that METTL3 regulates PTC cell proliferation and spread in vitro and in vivo and that these effects rely on the m6A catalytic activity of METTL3.
As a highly dynamic mRNA modification, m6A methylation is catalyzed by methyltransferases (also known as writers), demethylases (erasers), and binding proteins (readers). Lin et al.19 provided the first report about METTL3 in cancer; they uncovered a key role of METTL3 in promoting translation of oncogenes such as epidermal growth factor receptor (EGFR) and Tafazzin (TAZ) and accelerating growth and survival of human lung cancer cells. Since then, the controversial METTL3 has been intensively investigated in multiple malignancies. METTL3 has tumor-promoting effects in gastric cancer, colorectal cancer, liver cancer, and bladder cancer and presents opposite patterns in glioblastoma, endometrial cancer, and renal cell carcinoma.20, 21, 22, 23, 24, 25, 26 Cui et al.24 found that inhibition of METTL3 promotes tumorigenesis and self-renewal of glioblastoma stem cells (GSCs) by elevating the expression of oncogenes (ADAM19, EPHA3, and KLF4) and downregulating tumor suppressors (CDKN2A and BRCA2). Another group discovered that METTL3 was elevated in GSCs and increased SOX2 mRNA stability and expression to maintain neurosphere formation, whereas METTL3 silencing diminished glioblastoma propagation in vivo.27 These contradictory results reflect the complicated role of METTL3 in tumor progression. To date, the biological function and clinical implication of METTL3 in PTC are not fully understood. A new report shows that METTL3 aggravates progression of thyroid carcinoma by regulating m6A methylation of TCF1 to activate the Wnt pathway.28 The researchers concluded that METTL3 is upregulated in thyroid carcinoma by simply examining 24 paracancerous thyroid tissues as a control group without detecting the protein levels of METTL3 or the intrinsic m6A level in thyroid cancer tissues. It is also unclear which subtype of thyroid cancer were used in this study. Here, using high-throughput RNA-seq and MeRIP-seq, we identified c-Rel and RelA as bona fide m6A targets of METTL3. The tumor-suppressive function of METTL3 was mediated by increasing m6A levels and directly destabilizing c-Rel mRNA together with YTHDF2, inactivating the NF-κB pathway. The m6A modification machinery is delicately balanced, relying on cooperation of writers and readers. The YTH family is the largest family of m6A reader proteins, containing YTHDF1–YTHDF3, YTHDC1, and YTHDC2. YTHDF2 is the first characterized m6A reader that mediates mRNA degradation by specifically recognizing m6A-containing transcripts and recruiting them to mRNA decay sites. Chen et al.29 showed that upregulated YTHDF2 in pancreatic tumor cells accelerates cell proliferation but impedes tumor migration and invasion, validating the dual role of YTHDF2 and providing evidence for the “migration-proliferation dichotomy” theory. Another study proved that YTHDF2 might serve as a tumor suppressor by facilitating degradation of EGFR mRNA in hepatocellular carcinoma.30 Here we found that YTHDF2 exercised its suppressing function by binding to the c-Rel transcript and negatively modulating c-Rel mRNA stability, consequently impeding cell proliferation and migration. To our knowledge, this is the first report regarding the physiological function of YTHDF2 and its target mRNA in PTC.
Tumor-related inflammation is considered an important hallmark of cancer.31 Tumor cells communicate with multiple types of immune cells (e.g., lymphocytes, macrophages, neutrophils, and dendritic cells) in the tumor microenvironment, and this crosstalk implies paradoxical roles in promoting and inhibiting cancer progression, depending on the context.32,33 Ample evidence has emphasized that PTC possesses an inflammatory tumor microenvironment.34 Understanding this will be of paramount importance to develop targeting therapy against the components of the tumor microenvironment. Neutrophils comprise around 70% of circulating leukocytes in the human body, and the primary function of neutrophils is defense against extracellular pathogens. TANs play various and conflicting roles in cancer, including sustaining tumor angiogenesis, stimulating tumor cell proliferation, and promoting tumor cell dissemination.35 TANs have also been documented to possess anti-tumor properties, evoking an adaptive immune response and secreting cytotoxic mediators to render tumor cells more susceptible to apoptosis.36 The neutrophil-to-lymphocyte ratio (NLR) has been proposed as an easily visible indicator of the systemic inflammatory response in individuals with tumors. A previous study supports that a higher blood NLR is positively correlated with larger tumor size and a higher risk of recurrence in individuals with thyroid cancer, but the authors could not detect benign or malignant thyroid nodules.37 The functional state of neutrophils in development of PTC remains to be determined. Our data elucidate that METTL3 deficiency was capable of activating the NF-κB pathway and increasing IL-8 production. Inhibition of the NF-κB pathway with BAY 11-7082 could block IL-8 secretion from PTC cells, and an IL-8-neutralizing antibody abolished the enhanced chemotactic effect of METTL3 knockdown PTC cells on TANs. Last, targeting IL-8 with SB225002 achieved satisfactory antineoplastic effects by selectively eliminating neutrophils in the PTC model. Our research provides a scientific basis for the importance of TANs in the tumor microenvironment for progression of PTC.
In summary, we observed reduced METTL3 expression and overall m6A methylation levels in PTC tissues and cell lines. METTL3 recognized c-Rel and RelA m6A modification and collaborated with YTHDF2 in regulating c-Rel and RelA mRNA stability to trigger the NF-κB signaling pathway. METTL3 deficiency elevated IL-8 production of PTC cells to recruit TANs, ultimately promoting PTC progression. Being the first study to establish the link between m6A RNA methylation and TANs, our results underscore the crucial significance of m6A methylation in regulating PTC cell biology and offers a rationale for promising therapeutic targeting of m6A modulators in PTC.
Materials and methods
Human PTC samples
Primary PTC cancerous tissues, their matching adjacent normal thyroid tissues, and healthy control samples were obtained from the First Affiliated Hospital of Zhengzhou University. All individuals with PTC were diagnosed for the first time, did not receive any treatment prior to surgery, and then PTC was confirmed histologically and classified according to the American Joint Committee on Cancer (AJCC) staging system. This study was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University, and written informed consent was obtained from each participant.
Cell culture and chemicals
The human PTC cell lines BCPAP and KTC-1 were cultured in RPMI 1640 medium (HyClone, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and penicillin-streptomycin solution (Gibco, USA). Human immortalized thyroid cells (Nthy-ori 3-1) and PTC cells (TPC-1) were maintained in high-glucose DMEM (HyClone, USA). All cultured cells were tested regularly for mycoplasma contamination and authenticated by short tandem repeat (STR) profiling. The internal m6A inhibitor DAA was obtained from Sigma (St. Louis, MO, USA). The NF-κB inhibitor BAY 11-7082 and the IL-8/CXCR2 inhibitor SB225002 were purchased from Selleckchem (Houston, TX, USA). Recombinant human tumor necrosis factor alpha (TNF-α) protein was obtained from R&D Systems (Minneapolis, MN, USA).
IHC and immunofluorescence staining
Paraffin-embedded PTC tissues were sliced into 4-μm thickness and incubated overnight at 60°C. Next, the slides were deparaffinized, hydrated, and boiled for 30 min in citrate buffer (pH 6.0). The IHC staining index was determined based on the product of the proportion of positively stained tumor cells and the staining intensity score, as described previously.38 For immunofluorescence staining, cells were cultured on coverslips and fixed using 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 for 15 min, and washed with PBS, followed by blocking in 5% bovine serum albumin (BSA) for 1 h and incubation with anti-METTL3 (1:200 dilution) at 4°C overnight. Immunofluorescence was visualized with a fluorescence microscope (Leica, Wetzlar, Germany).
Western blot
Tissues and cells were lysed in ice-cold RIPA lysis buffer (Beyotime Biotechnology, China) supplemented with protease inhibitors. Protein concentrations were measured with the Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific, USA). Equal amounts of proteins were resolved on SDS-PAGE gel and transferred electrophoretically onto polyvinylidene fluoride (PVDF) membranes. The antibodies used in this study were as follows: anti-METTL3 (bs-17609R, BIOSS), anti-c-Rel (4727, Cell Signaling Technology), anti-RelA (8242, Cell Signaling Technology), anti-m6A antibody (ab208577, Abcam), anti-YTHDF2 (ab220163, Abcam), and anti-β-actin (4970, Cell Signaling Technology).
RNA extraction, quantitative real-time PCR, and mRNA stability assays
Total RNA was isolated from tissues and cells with TRIzol reagent (Life Technologies, Carlsbad, CA, USA), and reverse transcription was performed with the PrimeScript RT Reagent Kit with gDNA Eraser (TaKaRa, Dalian, China). Real-time PCR was conducted with TB Green Premix Ex Taq (TaKaRa, Dalian, China). Relative mRNA expression was determined using the 2−ΔΔCt method, and β-actin was applied as the internal control for normalization. Detailed primer sequences are listed in Table S1. To evaluate the mRNA stability of different PTC cells, actinomycin D (Sigma, USA) at 5 μg/mL was added. After incubation for the indicated times, cells were harvested, and total RNA was isolated for real-time PCR to detect stability.
Plasmid construction
The cDNA encoding the METTL3 CDS region was amplified and cloned into the pCDH-CMV-MCS-EF1-Puro lentivirus vector. The catalytic mutant METTL3 (residues 395–398, DPPW–APPA) was synthesized by GENEWIZ using the same vector for as wild-type METTL3. shRNAs targeting human METTL3, c-Rel, or RelA were cloned into pLKO.1-puro. siRNA targeting human YTHDF2 was purchased from GenePharma (Shanghai, China). All constructed vectors were verified by DNA sequencing. The targeting sequences were as follows: shMETTL3-1, 5′-GCAAGTATGTTCACTATGAAA-3′; shMETTL3-2, 5′-GCCAAGGAACAATCCATTGTT-3′; shRel, 5′-GCAGGAATCAATCCATTCAAT-3′; shRelA, 5′-CACCATCAACTATGATGAGTT-3′; siYTHDF2, 5′-AAGGACGTTCCCAATAGCCAA-3′. Plasmid transfection into 293T cells was accomplished using Lipofectamine 3000 (Invitrogen, USA) with Opti-MEM I reduced serum medium (Gibco, USA) and packaging vectors (psPAX2 and pMD2.G). Viral supernatant was harvested 48 h after transfection, passed through a 0.45-μm filter, and then used to infect the target cells at 50% confluence.
Functional assays in vitro
The CCK-8, EdU, colony formation, and Transwell migration and invasion assays were performed as described previously.39 The viability of PTC cells was also measured with a CellTiter-Glo luminescence assay (Promega, USA) according to the manufacturer’s protocols. PTC cells were seeded at 2,000 cells per well in an opaque, flat-bottomed, 96-well plate and cultured continuously for 5 days. Cell viability was confirmed by measuring the luminescence signal of each well using a luminescence plate reader and incubation with CellTiter-Glo reagent for 10 min at room temperature.
RNA-seq and meRIP-seq
For RNA-seq, total RNA was first extracted from BCPAP and TPC-1 cells with stable METTL3 knockdown and vector-transfected cells. Total RNA of each sample was extracted by RNeasy Mini Kit (QIAGEN, USA), and the quality and quantity of the RNA were assessed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Poly(A) mRNA isolation was performed using a poly(A) mRNA magnetic isolation module. RNA libraries were multiplexed and loaded on an Illumina HiSeq platform, and the sequences were processed and analyzed by GENEWIZ (Suzhou, China). Transcripts in fasta format were converted from known gff annotation files and indexed properly. Then, with the file as a reference gene file, HTSeq (version 0.6.1) was employed to estimate gene and isoform expression levels from the paired-end clean data. For meRIP-seq, total RNAs were isolated from METTL3-depleted or control TPC-1 cells using the Arraystar Seq-Star Poly(A) mRNA Isolation Kit. The extracted mRNA was chemically fragmented into approximately 100-nt fragments and immunoprecipitated with anti-m6A antibody. The eluted m6A mRNA fragments were then concentrated for RNA-seq library construction using the KAPA Stranded mRNA-Seq Kit (Illumina) and sequenced on the Illumina HiSeq 4000 platform. The rest of the RNAs were used for meRIP-qPCR to detect the m6A levels of c-Rel and RelA and normalized to the input mRNAs. Differential expression analysis was conducted using the DESeq2 Bioconductor package, and an adjusted p value of less than 0.05 was set as a cutoff criterion. Functional enrichment analysis was performed by R Package clusterProfiler (version 3.10.1).
RIP
The indicated cells were lysed in Polysome lysis buffer containing 10 mM HEPES-NaOH (pH 7.0), 100 mM KCl, 5 mM MgCl2, 0.5% NP40, 1 mM DTT, 200 U/mL SUPERase in RNase inhibitor (Invitrogen, USA), and protease inhibitor cocktails. Then Pierce protein A/G magnetic beads (Thermo Scientific, USA) were washed with NT-2 buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM MgCl2, and 0.05% NP-40), and anti-YTHDF2 or immunoglobulin G (IgG) antibody was added to the beads and incubated with rotation for 2 h. Cellular lysates were immunoprecipitated to each antibody bead tubes on a rocker at 4°C overnight. After washing five times with NT-2 buffer and incubation at 55°C for 30 min with proteinase K buffer to remove the proteins, the co-precipitated RNAs were extracted for detection of c-Rel expression by qRT-PCR.
Luciferase reporter assay
An NF-κB reporter kit (BPS Bioscience, San Diego, CA, USA) was utilized to monitor NF-κB pathway activity. Briefly, the pNF-κB-Luc reporter vector and Renilla luciferase vector were co-transfected into PTC cells using Lipofectamine 3000, and the relative luciferase activity was evaluated with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) 24 h after transfection. Fragments of the c-Rel-3′ UTR containing the wild-type m6A motifs and mutant m6A motifs were constructed by GENEWIZ (Suzhou, China) and fused into the pmirGlo dual-luciferase expression vector (Promega, Madison, WI, USA). The relative level of firefly luciferase activity was normalized to the Renilla luciferase activity levels and represents the effect of m6A modification on c-Rel expression.
Analysis of TANs
Tumor tissues from BALB/c nude mice were excised into small pieces and digested with 1 mg/mL collagenase D (Sigma, USA) and 100 μg/ml DNase I (STEMCELL Technologies, USA) in PBS with 5% FBS for 1 h in a 37°C shaker. Then digested tumor tissues were passed through 70-μm cell strainers, and mononuclear cells were collected on the interface fraction between 40% and 70% Percoll density gradient medium (GE Healthcare). After blocking with rat anti-mouse CD16/CD32 antibody (BD Pharmingen), cells were stained with PerCP-Cy5.5 anti-CD45, anti-PE-Cy7 anti-CD11b, and fluorescein isothiocyanate (FITC) anti-Ly6G (BioLegend) for analysis of proportions of neutrophils in the tumor microenvironment or sorted by FACSAria Cell Sorter (BD Biosciences) for further experiments.
Chemotaxis assay
The in vitro chemotaxis assay was performed using a filter Transwell system (3.0-μm pore size). In brief, TANs were collected from subcutaneous tumors in mice and sorted by flow cytometry. 5 × 104 TANs were suspended in RPMI 1640 medium with 5% FBS and seeded into the upper chamber and then incubated at 37°C for 3 h. Conditioned medium (CM) from PTC cell lines with METTL3, mutant METTL3, or shMETTL3 plasmids, and their corresponding control cells were placed in the bottom chamber. The subsequent steps were identical to those used in the cell migration assay. Specifically, a neutralizing antibody against IL-8 was added to CM from the shMETTL3 group to elucidate the chemotactic effect.
ELISA
RNA m6A quantification in PTC tissue and cell lines was performed by colorimetric ELISA using the EpiQuik m6A RNA Methylation Quantification Kit (Epigentek, USA), following the manufacturer’s protocol. 200 ng of RNA was coated into each well, and the absorbance was read on a microplate reader at 450 nm. The IL-8 levels in CM from different PTC cells and sera from individuals with PTC were detected using the Human IL-8 Quantikine ELISA Kit (R&D Systems, USA).
Animal models
All animal procedures were approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Zhengzhou University. For xenograft models, 5 × 106 BCPAP or KTC-1 cells from each group were injected subcutaneously into the flanks of female BALB/c nude mice (4–6 weeks old, Shanghai SLAC Laboratory Animal, China, n = 5 per group) in a volume of 150 μL PBS. Tumor growth was measured with a digital caliper every 4 days and calculated using the following formula: (length × width2)/2. To study the effect of IL-8 on tumor growth in vivo, scramble or shMETTL3 BCPAP cells were implanted hypodermically into BALB/c nude mice (2 × 106 cells in 150 μL PBS, n = 10 per group). When palpable tumors formed on day 14, mice were treated with DMSO or the IL-8 inhibitor SB225002 (10 mg/kg) by intraperitoneal injection 3 times per week for 3 weeks. Six weeks post-injection, the mice were sacrificed, and the tumors were collected to analyze the frequency of TANs by flow cytometry. For the lung metastasis model, BCPAP and KTC-1 cells (2 × 106 cells in 100 μL PBS) with the corresponding vectors were injected into the tail veins of BALB/c nude mice. Eight weeks after injection, the mice were euthanized, and metastatic lung nodules were analyzed (n = 5 for each group).
Statistical analysis
Experimental data are presented as mean ± standard deviation (SD) and analyzed using GraphPad Prism 8.0 software. All in vitro results are representative of at least three independent trials. Two-group comparison was assessed by Mann-Whitney U test or Student’s t test, and paired t test was employed for paired PTC and corresponding normal thyroid samples. The association between METTL3 and its downstream targets was analyzed by Pearson correlation test. A two-tailed p value of 0.05 was considered statistically significant.
Acknowledgments
We thank all patients enrolled in this study for their contribution to science. This work was supported by the Science and Technology Project of Henan Province of China (grant nos.192102310133 and 202102310112), the National Nature Science Foundation of China (grant no. 81702780), and the Medical Scientific Research Foundation of the Health Department of Henan Province of China (grant no. 201403077).
Author contributions
J.H., M.Z. and F.H. conceived and designed the study. J.H., M.Z., J.Y., and J.W. performed the experiments and analyzed the data. J.C, J.J., J.S., and C.W. helped to collect the tissue samples and conduct animal experiments. J.H. and M.Z. wrote and revised the manuscript. F.H. and H.Y. supervised the study.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.01.019.
Supplemental information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Cabanillas M.E., McFadden D.G., Durante C. Thyroid cancer. Lancet. 2016;388:2783–2795. doi: 10.1016/S0140-6736(16)30172-6. [DOI] [PubMed] [Google Scholar]
- 3.Xing M., Alzahrani A.S., Carson K.A., Viola D., Elisei R., Bendlova B., Yip L., Mian C., Vianello F., Tuttle R.M. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA. 2013;309:1493–1501. doi: 10.1001/jama.2013.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou M., Baitei E.Y., Alzahrani A.S., BinHumaid F.S., Alkhafaji D., Al-Rijjal R.A., Meyer B.F., Shi Y. Concomitant RAS, RET/PTC, or BRAF mutations in advanced stage of papillary thyroid carcinoma. Thyroid. 2014;24:1256–1266. doi: 10.1089/thy.2013.0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao M.D., Jiang Q., Ma Y., Liu C., Zhu C.Y., Sun Y.N., Shan K., Ge H.M., Zhang Q.Y., Zhang H.Y. Role of METTL3-Dependent N6-Methyladenosine mRNA Modification in the Promotion of Angiogenesis. Mol. Ther. 2020;28:2191–2202. doi: 10.1016/j.ymthe.2020.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen X., Xu M., Xu X., Zeng K., Liu X., Sun L., Pan B., He B., Pan Y., Sun H. METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol. Ther. 2020;28:599–612. doi: 10.1016/j.ymthe.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.De Jesus D.F., Zhang Z., Kahraman S., Brown N.K., Chen M., Hu J., Gupta M.K., He C., Kulkarni R.N. m6A mRNA Methylation Regulates Human β-Cell Biology in Physiological States and in Type 2 Diabetes. Nat. Metab. 2019;1:765–774. doi: 10.1038/s42255-019-0089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H.B., Tong J., Zhu S., Batista P.J., Duffy E.E., Zhao J., Bailis W., Cao G., Kroehling L., Chen Y. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–342. doi: 10.1038/nature23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorn L.E., Lasman L., Chen J., Xu X., Hund T.J., Medvedovic M., Hanna J.H., van Berlo J.H., Accornero F. The N6-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation. 2019;139:533–545. doi: 10.1161/CIRCULATIONAHA.118.036146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X., Gonzalez G., Dai X., Miao W., Yuan J., Huang M., Bade D., Li L., Sun Y., Wang Y. Adenylate Kinase 4 Modulates the Resistance of Breast Cancer Cells to Tamoxifen through an m6A-Based Epitranscriptomic Mechanism. Mol. Ther. 2020;28:2593–2604. doi: 10.1016/j.ymthe.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu L., Wu Y., Li Q., Liang J., He Q., Zhao L., Chen J., Cheng M., Huang Z., Ren H. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 m6A Methylation in Oral Squamous Cell Carcinoma. Mol. Ther. 2020;28:2177–2190. doi: 10.1016/j.ymthe.2020.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016;44:e91. doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter J.E., Leslie J., Perkins N.D. c-Rel and its many roles in cancer: an old story with new twists. Br. J. Cancer. 2016;114:1–6. doi: 10.1038/bjc.2015.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baggiolini M., Walz A., Kunkel S.L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Invest. 1989;84:1045–1049. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu P.J., Shi H., He C. Epitranscriptomic influences on development and disease. Genome Biol. 2017;18:197. doi: 10.1186/s13059-017-1336-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nachtergaele S., He C. Chemical Modifications in the Life of an mRNA Transcript. Annu. Rev. Genet. 2018;52:349–372. doi: 10.1146/annurev-genet-120417-031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu S., Zhang S., Wu X., Zhou X. m6A RNA Methylation in Cardiovascular Diseases. Mol. Ther. 2020;28:2111–2119. doi: 10.1016/j.ymthe.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer K.D., Jaffrey S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin S., Choe J., Du P., Triboulet R., Gregory R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell. 2016;62:335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q., Chen C., Ding Q., Zhao Y., Wang Z., Chen J., Jiang Z., Zhang Y., Xu G., Zhang J. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193–1205. doi: 10.1136/gutjnl-2019-319639. [DOI] [PubMed] [Google Scholar]
- 21.Li T., Hu P.S., Zuo Z., Lin J.F., Li X., Wu Q.N., Chen Z.H., Zeng Z.L., Wang F., Zheng J. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer. 2019;18:112. doi: 10.1186/s12943-019-1038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M., Wei L., Law C.T., Tsang F.H., Shen J., Cheng C.L., Tsang L.H., Ho D.W., Chiu D.K., Lee J.M. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 23.Cheng M., Sheng L., Gao Q., Xiong Q., Zhang H., Wu M., Liang Y., Zhu F., Zhang Y., Zhang X. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene. 2019;38:3667–3680. doi: 10.1038/s41388-019-0683-z. [DOI] [PubMed] [Google Scholar]
- 24.Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G., Sun G., Lu Z., Huang Y., Yang C.G. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J., Eckert M.A., Harada B.T., Liu S.M., Lu Z., Yu K., Tienda S.M., Chryplewicz A., Zhu A.C., Yang Y. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018;20:1074–1083. doi: 10.1038/s41556-018-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X., Tang J., Huang W., Wang F., Li P., Qin C., Qin Z., Zou Q., Wei J., Hua L. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8:96103–96116. doi: 10.18632/oncotarget.21726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Visvanathan A., Patil V., Arora A., Hegde A.S., Arivazhagan A., Santosh V., Somasundaram K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–533. doi: 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- 28.Wang K., Jiang L., Zhang Y., Chen C. Progression of Thyroid Carcinoma Is Promoted by the m6A Methyltransferase METTL3 Through Regulating m6A Methylation on TCF1. OncoTargets Ther. 2020;13:1605–1612. doi: 10.2147/OTT.S234751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J., Sun Y., Xu X., Wang D., He J., Zhou H., Lu Y., Zeng J., Du F., Gong A., Xu M. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16:2259–2271. doi: 10.1080/15384101.2017.1380125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong L., Liao D., Zhang M., Zeng C., Li X., Zhang R., Ma H., Kang T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–261. doi: 10.1016/j.canlet.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Wu T., Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68. doi: 10.1016/j.canlet.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 33.Hinshaw D.C., Shevde L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79:4557–4566. doi: 10.1158/0008-5472.CAN-18-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cunha L.L., Marcello M.A., Ward L.S. The role of the inflammatory microenvironment in thyroid carcinogenesis. Endocr. Relat. Cancer. 2014;21:R85–R103. doi: 10.1530/ERC-13-0431. [DOI] [PubMed] [Google Scholar]
- 35.Granot Z. Neutrophils as a Therapeutic Target in Cancer. Front. Immunol. 2019;10:1710. doi: 10.3389/fimmu.2019.01710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granot Z., Henke E., Comen E.A., King T.A., Norton L., Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20:300–314. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu C.L., Lee J.J., Liu T.P., Chang Y.C., Hsu Y.C., Cheng S.P. Blood neutrophil-to-lymphocyte ratio correlates with tumor size in patients with differentiated thyroid cancer. J. Surg. Oncol. 2013;107:493–497. doi: 10.1002/jso.23270. [DOI] [PubMed] [Google Scholar]
- 38.He J., Jin Y., Zhou M., Li X., Chen W., Wang Y., Gu S., Cao Y., Chu C., Liu X., Zou Q. Solute carrier family 35 member F2 is indispensable for papillary thyroid carcinoma progression through activation of transforming growth factor-β type I receptor/apoptosis signal-regulating kinase 1/mitogen-activated protein kinase signaling axis. Cancer Sci. 2018;109:642–655. doi: 10.1111/cas.13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He J., Zhou M., Li X., Gu S., Cao Y., Xing T., Chen W., Chu C., Gu F., Zhou J. SLC34A2 simultaneously promotes papillary thyroid carcinoma growth and invasion through distinct mechanisms. Oncogene. 2020;39:2658–2675. doi: 10.1038/s41388-020-1181-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.