

Abstract
Oncolytic viruses induce antitumor immunity following direct viral oncolysis. However, their therapeutic effects are limited in distant untreated tumors because their antitumor function depends on indirect antitumor immunity. Here, we generated a novel fusogenic oncolytic vaccinia virus (FUVAC) and compared its antitumor activity with that of its parental non-fusogenic virus. Compared with the parent, FUVAC exerted the cytopathic effect and induced immunogenic cell death in human and murine cancer cells more efficiently. In a bilateral tumor-bearing syngeneic mouse model, FUVAC administration significantly inhibited tumor growth in both treated and untreated tumors. However, its antitumor effects were completely suppressed by CD8+ T cell depletion. Notably, FUVAC reduced the number of tumor-associated immune-suppressive cells in treated tumors, but not in untreated tumors. Mice treated with FUVAC before an immune checkpoint inhibitor (ICI) treatment achieved complete response (CR) in both treated and untreated tumors, whereas ICI alone did not show antitumor activity. Mice achieving CR rejected rechallenge with the same tumor cells, suggesting establishment of a long-term tumor-specific immune memory. Thus, FUVAC improves the tumor immune microenvironment and enhances systemic antitumor immunity, suggesting that, alone and in combination with ICI, it is a novel immune modulator for overcoming oncolytic virus-resistant tumors.
Keywords: vaccinia virus, oncolytic function, immune checkpoint blockade, antitumor immunity, cell membrane fusion
Graphical abstract
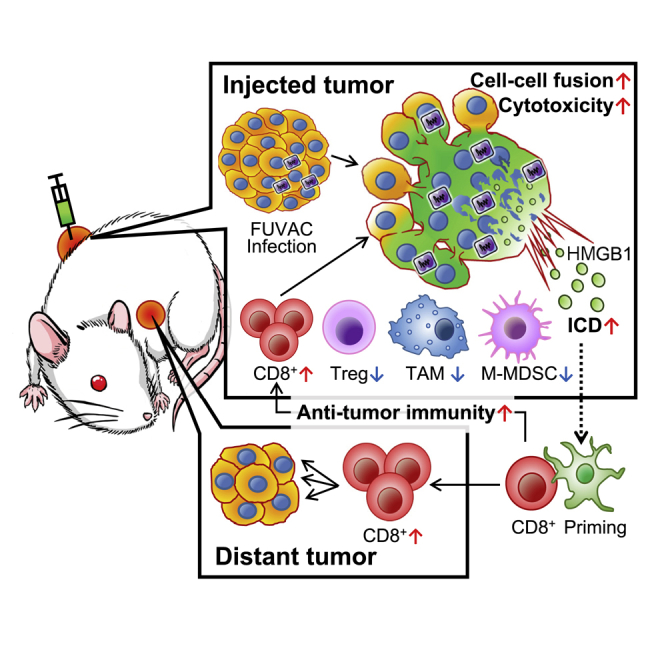
Fusogenic vaccinia virus modulates the tumor immune microenvironment by reducing tumor-associated immune suppressive factors locally and increasing cytotoxic CD8+ T cells systemically. Thus, this study presents a novel approach that maximizes the anti-cancer immunotherapeutic potential by treatment starting with oncolytic virus-induced cancer cell fusion and ending with immune checkpoint blockade.
Introduction
Oncolytic virotherapy is a novel cancer treatment strategy that utilizes tumor-specific replication selected virus as an anticancer medicine. Various oncolytic viruses have shown significant clinical benefits1,2 represented by the FDA approval of talimogene laherparepvec. Oncolytic viruses exert two types of therapeutic functions, namely, direct tumor lysis (oncolysis) and indirect antitumor immunity.1 Currently, oncolytic viruses are most often administered intratumorally. This approach provides rapid and powerful therapeutic effects against the injected tumors via antitumor immune responses following viral oncolysis. However, in non-injected tumors, such as the disseminated or metastatic tumor region, the therapeutic response is delayed and reduced because it depends on indirect antitumor immunity elicited after lysis of the injected tumor cells.3, 4, 5 Therefore, enhancement of the antitumor immunity response to the effect of oncolytic viruses, especially for the treatment of the non-injected tumors, is needed.
Some oncolytic viruses, such as the measles, mumps, Sendai, and Newcastle disease virus, have a fusogenic phenotype. These viruses cause syncytia formation in infected tumor cells, which induces rapid oncolysis and releases several tumor antigens, thereby resulting in immunogenic cell death (ICD).6, 7, 8 There are also some oncolytic viruses armed with exogenous fusion proteins that enhance their therapeutic functions through conferring these characteristics.6,8
The vaccinia virus (VV) is a major oncolytic agent and is known as a non-fusogenic virus. Our laboratories have isolated its mutant clone having the fusion phenotype, named the fusogenic oncolytic vaccinia virus (FUVAC). FUVAC was discovered during plaque purification of the mitogen-activated protein kinase-dependent recombinant vaccinia virus (MDRVV). MDRVV achieved tumor-specific virus replication through the deletion of two viral growth factors, namely the VV growth factor (VGF) and O1L.9,10 FUVAC has a nonsense mutation in the viral gene K2L, whereas other viral genomes are maintained in the MDRVV. K2L encodes the serine protease inhibitor (SPI-3), which conjugates with the A56 polypeptide and blocks the viral entry-fusion complex (EFC).11, 12, 13, 14, 15, 16, 17 EFC, a complex of 11 viral proteins associated with post-attachment membrane fusion,12 presents on the surface of the virus or the infected cellular membrane and has critical roles in virus entry and cell-cell fusion. K2/A56 complex binds with the G9/A16 of EFC to block its membrane fusion initiation,17 thus distinguishing the infected cells from non-infected cells by blocking virus re-entry. However, this inhibition system also limits cellular fusion between infected cells. FUVAC promoted VV fusion by impairing the fusion inhibition of K2L since it has a nonsense mutation in the K2L gene.
It is unknown whether vaccinia virus-mediated cell fusion enhances oncolytic activity and antitumor immunity. By using FUVAC, we evaluate how cell fusion is involved in oncolysis and induces antitumor immune response. We also showed that the novel fusogenic vaccinia virus conferred greater therapeutic potential to treat both the virus injected and non-injected tumors. Its fusogenic phenotype enhanced not only the tumor lysis but also the immune modulation through reducing immune-suppressive cells locally and increasing CD8+ T cells systemically. These anticancer functions were synergistically enhanced by the combination with ICI, especially in the non-injected tumor treatment. FUVAC opened the possibility for oncolytic VV to exert its oncolytic activity by enhancing its fusion function.
Results
Fusogenic vaccinia virus has a natural K2L mutation
FUVAC was isolated from MDRVV having double deletions of VGF and O1L. Whole-genome sequencing identified that FUVAC has a nonsense mutation (254 Trp→stop) in the viral fusion inhibitor K2L (Figure S1), although the VGF and O1 deletions were maintained. The MDRVV was inserted into the gene cassettes expressing luciferase fused with enhanced green fluorescent protein (EGFP) and DsRed into the VGF and O1 gene loci, respectively, and named MDRVV-luciferase-EGFP (LG)/DsRed. Therefore, the fusogenic strain derived from the MDRVV-LG/DsRed was called FUVAC-LG/DsRed. Furthermore, the inserted genes, luciferase-EGFP and DsRed, were replaced with luciferase and LacZ for immune staining and in vivo experiments, resulting in MDRVV-Luc/LacZ and FUVAC-Luc/LacZ, respectively (Figure 1A).
Figure 1.

Viral replication and oncolytic effect in MDRVV and FUVAC
(A) Schematic representation of MDRVV and FUVAC. (B) A549 cells were infected with MDRVV- or FUVAC-LG/DsRed at an MOI of 0.01, 0.1, or 1, and the images were taken 3 days after infection. Scale bar, 500 μm. (C) Viability of the A549 cells 72 h after virus infection as described in (B) was examined with CellTiter 96 Aqueous Nonradioactive Cell Proliferation Assay (Promega). Data are represented as the percent survival of mock-infected cells. (D) A549 cells infected with MDRVV- or FUVAC-LG/DsRed at an MOI of 0.1 were harvested after infection for 24, 48, or 72 h, and the extracted intracellular viruses were titrated in RK13 cells. Data in (C) and (D) are presented as the means ± SEM (n = 3). ∗∗∗p < 0.001 (two-tailed unpaired t test).
FUVAC showed fusogenic cytopathic effect in cancer cell lines
FUVAC induced syncytium formation in human lung carcinoma A549 cells (Figure 1B). The cell viability following FUVAC infection was more reduced than that following MDRVV (Figure 1C), whereas their virus yield was comparable (Figure 1D). The fusion phenotype and oncolytic activity were also examined in several human and murine cancer cell lines. FUVAC induced cell-cell fusion in various tumor types, such as lung, ovarian, pancreatic, colon, mammary, prostate, and epithelial (Figure 2A; Videos S1 and S2). Moreover, the viability in these cell lines was decreased by FUVAC infection, compared with MDRVV (Figure 2B).
Figure 2.

Cytolytic fusion of FUVAC against several cancer cell lines
(A) Several cancer cell lines were infected with MDRVV- or FUVAC-LG/DsRed at each MOI: Panc1, CaCO2, MDA-MB-231, A549, and A431, 0.1; SKOV3 and PC3, 1; and B16-F10, CT26, and TC1, 5. Cells were photographed 3 days after virus infection, except for TC1 cells that were photographed after 2 days. Scale bar, 500 μm. (B) Viability of tumor cell lines described in (A). Data are presented as the percent survival of mock-infected cells and represented as the means ± SEM (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed unpaired t test).
Oncolytic VV induced several types of tumor cell death. MDRVV infection induced apoptosis, necrosis, and ICD against the human A549 and murine CT26 cell lines, whereas FUVAC enhanced the induction of these cell deaths, especially that of ICD as detected by HMGB1 release (Figures 3A and 3B). Another ICD marker, ATP release, was examined using VGF and O1L intact viruses, VGF+/O1+ VV and FUVAC VGF+/O1+ (Figure S2A). FUVAC VGF+/O1+ showed higher ATP release than non-fusogenic VGF+/O1+ VV (Figure S2B).
Figure 3.

Three types of cell death efficiently induced by FUVAC
(A) A549 cells were infected with MDRVV- or FUVAC-Luc/LacZ at an MOI of 1, and the apoptotic or necrotic cells were detected by staining with annexin V or ethidium homodimer, respectively, 30 h after infection. HMGB1 release was detected using ELISA of the culture supernatant of the infected cells 60 h after infection. (B) CT26 cells were infected with MDRVV or FUVAC at an MOI of 10, and apoptosis or necrosis was detected as described in (A) 22 h after infection. Meanwhile, the culture supernatant of CT26 cells infected with MDRVV or FUVAC at an MOI of 5 for 60 h was used for HMGB1 detection as described in (A). Data in (A) and (B) are presented as the means ± SEM (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed unpaired t test).
Furthermore, FUVAC was compared with a K2L-deleted virus (ΔK2L-LG/DsRed, Figure S2A) to confirm the K2L function in VV fusion. FUVAC and ΔK2L showed equal cell-cell fusion, enhanced cytotoxicity, and HMGB1 release in A549 and CT26 cells (Figures S2C–S2E). Therefore, the K2L mutation is causative for the FUVAC phenotype.
FUVAC showed enhanced therapeutic efficacy in injected and non-injected tumor regions
The in vivo oncolytic effect was determined using the syngeneic mice model bearing the bilateral CT26 murine colon cancer cells (Figure 4A). CT26 cells were chosen because they formed the largest syncytia during the FUVAC infection in murine tumor cell lines (Figure 2A). MDRVV or FUVAC was injected into the larger flank, and their therapeutic effects were examined in both injected and non-injected tumors. Viral replication was detected using the firefly luciferase (Fluc) expression of each virus. FUVAC showed higher viral replication than MDRVV in the injected tumor regions (Figures 4B and 4C). Both viruses did not express Fluc luminescence in the non-injected tumors (Figure 4C). Nevertheless, FUVAC showed stronger anticancer effect than MDRVV, not only in the injected tumors, but also in the non-injected tumors (Figure 4D).
Figure 4.

In vivo viral replication and oncolytic effect of FUVAC
(A) Schedule of MDRVV or FUVAC treatment against the bilateral CT26 tumor model. CT26 cells were bilaterally transplanted on BALB/c mice through subcutaneous injection. When the average tumor volume reached 60 mm3, 5 × 107 PFUs of MDRVV or FUVAC-Luc/LacZ were intratumorally injected three times every 2 days. (B) In vivo bioluminescence imaging of the viral replication. Viral Fluc luminescence was detected by VivoGlo Luciferin (3 mg/mouse; Promega) on day 3. (C) Quantification of the viral luminescence on days 1, 3, 5, and 7 after the first virus injection. Fluc signals were separately quantified in the injection and non-injection sites. Mean luminescence (ph/s) ± SEM are shown. ∗p < 0.05 (two-way ANOVA). (D) Tumor growth was separately monitored in the injection and non-injection sites. Results are representative of two independent experiments comprising 6–8 mice/group (n = 12, 14, or 15 for PBS, MDRVV, or FUVAC, respectively). Mean tumor volume (mm3) ± SEM are shown. ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA).
Moreover, immunohistochemical (IHC) analysis (Figure 5) showed that viral GFP expression, for both viruses, was detected only on injected tumors (Figure 5C). FUVAC treatment resulted in a larger GFP region than MDRVV, according to their Fluc intensity (Figures 5B and 5C). Virus infected and lysed regions showed eosin dominance, and FUVAC-treated tumors showed the fusogenic phenotype at the boundary between the infected and non-infected regions (Figure 5D). In comparison with MDRVV, all FUVAC-treated tumors showed similar cell-cell fusion in the infected region (n = 3, Figure S3).
Figure 5.

Fusogenic findings in in vivo tumor tissues
(A) Schedule of the tumor tissue collection for IHC. Mice bearing bilateral CT26 tumors were treated with 5 × 107 PFUs of MDRVV- or FUVAC-LG/DsRed on days 0, 2, and 4, then tumors were collected on day 5. (B) Bioluminescence imaging before tumor collection on day 5. (C) Whole pictures of virus-treated or -untreated tumors. Upper images show the hematoxylin and eosin staining, and lower images show the anti-GFP antibody staining. Scale bar, 1,000 μm. (D) High-power images of the area marked in (C). Arrows show the fusogenic phenotype. Representative images are shown in (C) and (D). Pictures from the other two mice are shown in Figure S3. Scale bar, 50 μm.
On the other hand, MDRVV tended to reduce oncolytic efficacy, owing to the larger tumor volume. FUVAC had a stable oncolytic effect regardless of the base tumor sizes (Figure S4). Even when the initial tumor volume reached more than 100 mm3, FUVAC enhanced viral replication and therapeutic efficacy more than MDRVV (Figures S5A–S5C).
FUVAC enhanced the infiltration of CD8+ T cells and the inhibition of tumor-associated immune suppressive cells
To identify the therapeutic factor of FUVAC, we examined the tumor immune microenvironment. Both virus-treated and -untreated tumors were collected 5 days after virus injection, and then the tumor infiltrating lymphocytes (TILs) were analyzed using flow cytometry (Figure 6A). The total percentage of CD45+ immune cells increased in the injected tumors, but not in non-injected tumors after virus treatment (Figure 6B). The total CD3+ T cell levels remained unchanged. However, FUVAC significantly enhanced the infiltration of CD8+ T cells in the non-injected tumor region (Figure 6C). In the injected tumors, the ratio of CD8+ T cells did not change significantly, but FUVAC decreased the number of CD4+ T cells, especially in the CD25+-FoxP3+ (regulatory T cell, Treg) fraction. Moreover, other tumor-associated immune suppressive cells, tumor-associated macrophages (TAM: CD11b+ F4/80+), and monocytic-myeloid derived suppressor cells (M-MDSC: CD11b+ Ly6C+) were also decreased by FUVAC (Figure 6D), except for granulocytic-myeloid derived suppressor cells (G-MDSC: CD11b+ Ly6C– Ly6G+). The level of these cells did not fluctuate in the non-injected tumors. Overall, FUVAC enhanced the inhibition of tumor-associated immune suppressive cells in the injected tumors and the infiltration of cytotoxic CD8+ T cells in the non-injected tumors.
Figure 6.

Changing tumor immune microenvironment by FUVAC infection
(A) Schedule of the immune microenvironment characterization after MDRVV or FUVAC treatment. Bilateral CT26-bearing mice were treated with MDRVV or FUVAC using the same method as Figure 4. 5 days after the first virus injection (1 day after the third injection), the virus-treated or -untreated tumors were recovered and processed for flow cytometry analysis. (B) The ratio of CD45+ total immune cells and total tumor cells, and the ratio of CD3+ T lymphocyte in CD45+ cells. (C) The ratios of CD3+/CD8+ and CD3+/CD4+ cells in CD45+ T lymphocyte, and the ratio of Treg in CD3+ T cell as determined by the expression levels of CD4, CD25, and FoxP3. (D) The ratio of TAM (CD11b+ F4/80+), M-MDSC (CD11b+ Ly6C+), or G-MDSC (CD11b+ Ly6C– Ly6G+) in CD45+ cells. Data in (B)–(D) are presented as the means ± SEM (n = 7). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-tailed unpaired t test).
7 days after viral injection, CD8+ T cells had infiltrated injected tumors to the same level as non-injected tumors (Figure S6A), which was accompanied by programmed death-ligand 1 (PD-L1) expression (Figure S6B). Meanwhile, both viruses led to an increase in antigen-presenting dendritic cells (DCs) in spleens (Figure S6C). FUVAC particularly increased the splenocyte CD8+ T cell count (Figure S6D) and tumor-specific interferon-γ (IFN-γ) secretion (Figure S6E).
The role of TILs in the FUVAC treatment was also confirmed by immune depletion. Viruses and inhibitor antibodies targeting CD4 or CD8 were alternately injected into the mice bearing bi-flank CT26 tumors (Figure 7A). Both inhibitors did not show any significant effect against the Fluc luminescence of MDRVV (Figure S7). However, that of FUVAC was significantly increased by the CD8 inhibitor, but not by the CD4 inhibitor (Figure S7). CD4 depletion partially reduced the therapeutic efficacy of FUVAC to the same level as that of MDRVV (Figure 7C), although MDRVV was not affected (Figure 7B). However, CD8 depletion completely abolished their anticancer effects not only in the injected tumors, but also in the non-injected tumors. These findings suggest that CD8+ T cells have a critical role in the therapeutic mechanism of oncolytic VV.
Figure 7.

Effect of immune depletion against FUVAC treatment
(A) Schedule of the CD4 or CD8 depletion. CT26-bearing mice were administrated with MDRVV or FUVAC on days 0, 2, and 4, and 200 μg of Isotype ctrl, anti-CD4, or anti-CD8 antibody was injected on days 3, 5, and 7. (B and C) Tumor growth curves of mice after the treatment of each inhibitor with MDRVV (B) or FUVAC (C). Mean tumor volume (mm3) ± SEM are shown (n = 6–7). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (two-way ANOVA).
FUVAC showed synergistic effects with programmed cell death (PD)-1 blockade
In contrast with immune depletion, CD8+ T cell activity was promoted when combined with the PD-1 blockade (Figure 8A). In comparison with the mock (PBS)-treated groups (Figure 8B), anti-PD-1 antibody strongly enhanced the viral oncolytic effects in both viruses. PD-1 blockade by itself had little anticancer effect, but its combination with FUVAC disappeared all the injected tumors (Figure 8C). Notably, FUVAC combined with the PD-1 blockade showed a remarkable regression in non-injected tumors. i.e., 3/6 mice treated with FUVAC prior to the PD-1 blockade achieved complete response (CR), although other mice could not eliminate the non-injected tumors (Table S1). FUVAC and anti-PD-1 antibody treatment significantly prolonged the survival of mice compared with other single or combination treatment groups (Figure 8D). Furthermore, CR mice completely rejected the CT26 tumor re-implantation (Figure 8E). Thus, FUVAC efficiently recruits antitumor CD8+ T cells against the distant tumors and maximizes the systemic antitumor immunity when combined with immune checkpoint blockade.
Figure 8.

Effect of PD-1 blockade under FUVAC treatment
(A) Schedule of the combination of virus with PD-1 blockade. (B and C) Tumor growths in mice treated with PBS, MDRVV, or FUVAC + mock (B) or + anti-PD-1 antibodies (C). Data are presented as the mean tumor volume (mm3) ± SEM (n = 5–6). ∗∗∗p < 0.001 (two-way ANOVA). (D) Survival curves of mice associated with (B) and (C) generated using Kaplan-Meier analysis. FUVAC alone prolonged the survival of mice compared with the PBS treatment (∗p = 0.0112). FUVAC + anti-PD-1 prolonged the survival longer than anti-PD-1 antibody alone or MDRVV + anti-PD-1 (∗∗∗p = 0.0007 or ∗∗p = 0.0066, respectively, using log-rank test). (E) Tumor growths in CR mice and control mice after tumor rechallenge (n = 3).
Discussion
Fusogenic oncolytic viruses showed promising therapeutic benefits through the reinforcement of their oncolytic activity and immune inducement. Various oncolytic viruses armed with exogenous fusion proteins, such as GALV envelope,18, 19, 20, 21 FAST protein,22,23 HIV envelope,24,25 MV-F protein,26 NDV-F protein,27,28 and SV5-F protein,29,30 have shown improved oncolytic functions. Fusion phenotype enhances virus yield, spreading, and cytopathic effect, as well as induces antitumor immunity.6,8 Although the viral fusion phenotype seems to be a reasonable way to evolve various types of oncolytic viruses, there have been no reports regarding the effects of vaccinia virus-mediated cell fusion on therapeutic responses.
In this study, we first demonstrated the therapeutic potential of the novel FUVAC. VV can infrequently produce its progeny virion, having a fusogenic phenotype in an environment where replication is difficult, such as in the plaque separation process. Its fusogenic phenotype is regulated by the viral fusion inhibitor, K2L, which limits virus re-entry and cell-cell fusion by blocking the EFC.13, 14, 15, 16, 17 This function may be a mechanism for the prevention of virus congestion in one place. However, causing impairing of the fusion inhibition would be advantageous when VVs are administered via the intratumoral route. Directly injected viruses stayed in the injected tumor region, as shown in Figures 4C and 5C. Therefore, their oncolytic effects against distant tumors, such as metastatic tumors, strongly depend on their antitumor immune function. Rapid and immunogenic tumor cell death causes efficient antitumor immunity, expanding locally to systemically. To maximize the systemic anticancer immune response following local oncolysis, the suppression of VV fusion activity should be investigated.
FUVAC induces syncytia formation in various types of tumor cells due to fusion deregulation. FUVAC efficiently decreased the viability of infected cells, whereas its progeny virus production was unchanged. Larger syncytium formation tended to increase viral oncolysis. Murine cells tended to form smaller syncytia than human cancer cell lines because VV more efficiently replicated in human cells than in mouse cells. Nevertheless, FUVAC more significantly enhanced tumor cell death, such as ICD, than its parental non-fusogenic virus MDRVV in both human A549 and murine CT26 cells (Figure 3). This implies that, like other fusogenic viruses,18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 the VV fusion function improves oncolytic activity. FUVAC also showed strong oncolytic activity and cell-cell fusion against injected CT26 tumors in vivo (Figures 4D and 5D), regardless of baseline tumor volume. Interestingly, the viral replication of FUVAC in vivo was higher than that of MDRVV (Figures 4C and 5C), whereas their replication efficacies were comparable in vitro. Overall, FUVAC increased viral dissemination in tumor tissues due to the fusogenic spread. This spreading property might support the stable antitumor effect and immune modulation in injected tumors.
FUVAC caused the inhibition of tumor-associated immune suppressive cells, such as Treg, TAM, and M-MDSC in directly treated tumors. Only G-MDSCs increased, but those cells were induced by the VV infection to decrease viral toxicity.31 As a result of the immune remodeling in injected tumors, FUVAC further increased the number of CD8+ T cells and suppressed the growth of non-injected tumors (Figures 4D and 6C). The CD8+ T cell ratio remained unchanged in the injected tumors (Figure 6C), regardless of the higher CD45+ levels on day 5 (Figure 6B). The increase in the CD45+ ratio might be caused by the decrease in the live tumor cell ratio due to the viral tumor lysis, as shown in Figure 5C. On the other hand, both MDRVV and FUVAC induced CD8+ T cell infiltration in injected tumors to the same level as in the non-injected region on day 7 (Figure S6A). At the same time, both viruses increased antigen-presenting DCs, and FUVAC significantly induced tumor-specific T lymphocytes, in the spleen (Figures S6C and S6E). These results suggest that FUVAC efficiently elicited an antitumor immune response through remodeling of the tumor immune microenvironment. This antitumor immune function was completely suppressed by CD8 depletion (Figures 7B and 7C). Despite the direct virus influence, viral therapeutic effects against the injected tumors were abolished, as well as in non-injected tumors. This attenuation of the injected tumor treatment could be caused by the lower VV tropism in murine cells, as described in Figure 2. Therefore, in this model, the viral therapeutic function in injected tumors strongly depended on immune modulation, rather than direct tumor lysis.
In contrast, CD4+ T cells were not increased in both tumor regions, although the base CD4+ levels were larger than the number of CD8+ T cells in CT26 tumors.32, 33, 34 FUVAC rather decreased CD4+ T cells accompanied with decreased Treg in the injected tumors (Figure 6C). Similar reductions in the number of CD4+ T cells and Treg together have been reported in the MCA205- or RenCa-bearing mice model.35,36 In another study, the fusogenic VSV-p14 reduced the CD4 ratio in virus-injected 4T1 tumors, but it increased the CD4+ T cells in the spleen and drained lymph node.23 In addition, CD4 depletion partially suppressed the FUVAC function (Figure 7C), whereas MDRVV was not affected by CD4 depletion (Figure 7B), suggesting that FUVAC utilizes non-tumor associated CD4+ T cells to promote systemic antitumor immunity, and their activity caused the difference in their antitumor immune responses.
FUVAC modulated the immune suppressive environment in the virus-injected tumors, but PD-L1 expression only increased upon CD8+ T cell infiltration (Figures S6A and S6B). In addition, immune suppressive cells remained in the non-injected tumors (Figures 6C and 6D) due to the lack of direct viral activity. The existence of these cells would have become a barrier against anticancer effect, but the combination of FUVAC and PD-1 blockade overcame its prevention (Figure 8). Half of the mice treated with FUVAC and anti-PD-1 antibody showed a complete response (CR) (Figure 8D; Table S1), whereas CR mice had immunological memory confirmed by tumor rejection (Figure 8E). MDRVV also enhanced their anticancer potential in combination with PD-1 blockade; however, not one mouse showed regression in non-injected tumor and survival benefit (Figures 8C and 8D). When a single treatment of anti-PD-1 antibody did not work, FUVAC dramatically triggered its immune checkpoint inhibition. Thus, FUVAC clearly enhanced the anticancer function even in murine CT26 tumors, regardless of lower fusion activity, compared with human tumor cell lines. These indicate the strong therapeutic benefits of FUVAC that can be expected in human cancer treatment.
Interestingly, anti-PD-1 antibody treatment decreased the viral replication of FUVAC (Figure S8). This is in contrast with the induction of CD8 depletion (Figure S7). The former strongly supported the viral oncolytic activity, but the latter completely disabled it. These Fluc fluctuations would reflect the immune response. In other words, viral replication was prevented by enhanced immunity but promoted by the suppressed immunity. Therefore, an increase in viral replication will not yield proportional therapeutic benefits. The necessity of viral replication in cancer immunotherapy is often discussed.37,38 For example, the inactivated non-replicating modified vaccinia Ankara (MVA) had higher immune induction than replicating MVA.39 The cancer vaccine specializes in promoting antitumor immune response without relying on viral tumor lysis. In contrast, FUVAC enhanced the viral replication and lytic efficacy while promoting antitumor immunity. Therefore, it would be a convenient vector for loading other exogenous therapeutic genes, such as cell suicide genes, cytokines, chemokines, T cell costimulatory molecules, and immune checkpoint inhibitors.
In conclusion, we established a novel fusogenic vaccinia virus with improved direct oncolytic activity and indirect antitumor immunity. FUVAC modulates the tumor immune microenvironment by reducing the tumor-associated immune suppressive factors locally and increasing cytotoxic CD8+ T cells systemically. It would be an effective initiator for immune checkpoint blockade. Moreover, FUVAC can evolve other therapeutic functions through the loading of various therapeutic genes because of its high gene expression level. FUVAC would be a next generation platform for oncolytic immunotherapy.
Materials and methods
Virus preparation
Recombinant viruses were developed as previously described.9,10 When both VGF and O1L-deleted viruses9 were cloned during plaque purification, we found unusual viral plaques having fusogenic cell degeneration. The plaques were isolated, and the full genome sequence was analyzed using PacBio RSII (Pacific Bioscience, CA, USA). For in vivo usage, CV1 cells were infected with MDRVV-LG/DsRed or FUVAC-LG/DsRed and transfected with pTNshuttle/VGF-SP-Luc. The recombinant viruses, MDRVV-VGF-SP-Luc/O1L-p7.5K-DsRed or FUVAC-VGF-SP-Luc/O1L-p7.5K-DsRed, were isolated based on the loss of GFP expression and were used to infect the CV1 cells for recombination with pTNshuttle/O1L-SP-LacZ. The resultant viruses, MDRVV- or FUVAC-VGF-SP-Luc/O1L-SP-LacZ (Luc/LacZ), were isolated based on the loss of DsRed expression. Construction of VGF/O1L intact virus and K2L-deleted virus are described in the Supplemental Materials and Methods and Table S2. All viruses were propagated in A549 cells and titrated in RK13 cells. Luc/LacZ viruses were then purified before in vivo usage. Briefly, the viruses were propagated in A549 cells. After that, the infected cells were harvested and mixed with 200 U/mL benzonase (Merck, MA, USA), followed by incubation at 37°C for 1 h. Cell lysates were purified and dialyzed using OptiPrep (Axis-Shield, Oslo, Norway) and Slide-A-Lyzer Dialysis Cassette (Thermo Fisher, MA, USA), according to the manufacturer’s protocol.
Cytotoxicity assay and virus titration
To follow the virus replication, we infected A549 lung carcinoma cells with MDRVV- or FUVAC-LG/DsRed at an MOI of 0, 0.01, 0.1, or 1 Videos S1S2. After 72 h, the cells were photographed under a fluorescence microscope and viability was assessed using the CellTiter 96 Aqueous Nonradioactive Cell Proliferation Assay (Promega, WI, USA). Virus replication was also examined by the titration of cell-associated viruses. Cell lysates were harvested 24, 48, or 72 h after infection, and cell-associated virions were extracted through freeze-thawing, sonication, and centrifugation. The extracted viruses were titrated in RK13 cells, and viral plaques were counted 3 days after infection.
To confirm the cell-cell fusion in various tumor cell lines, we harvested 2.0 × 104 cells of human ovarian SKOV3, pancreatic Panc1, colon CaCO2, breast MDA-MB-231, epithelial A431 cells, 1.0 × 104 cells of human lung A549, murine colon CT26, 2.5 × 104 cells of human prostate PC-3, 1.5 × 104 cells of murine melanoma B16-F10, and 4.0 × 103 cells of murine lung TC1 cells in 96-well plates. After 24 h, the cells were infected with MDRVV- or FUVAC-LG/DsRed at the following conditions: MOI of 0.1 for Panc1, CaCO2, MDA-MB-231, A549, and A431; MOI of 1 for SKOV3 and PC-3; and MOI of 5 for B16-F10, CT26, and TC1. Cells were photographed, and their viability was examined 72 h after infection (for TC1 cells, 48 h) as described above.
Apoptosis, necrosis, and immunogenic cell death assay
Human A549 cells or murine CT26 cells were infected with MDRVV- or FUVAC-Luc/LacZ at an MOI of 0.1 or 10, respectively. After 30 h or 22 h, the cell death was examined using an Apoptotic/Necrotic/Healthy Cells Detection Kit (Takara Bio, Otsu, Japan) and photographed under the fluorescence microscope (BZ-X700; Keyence, Osaka, Japan). Fluorescence intensity was quantified using the Hybrid Cell Count software (Keyence) according to the manufacturer’s protocol. ICD was detected through the ELISA analysis of High Mobility Group Box 1 (HMGB1) release. MDRVV- or FUVAC-Luc/LacZ virus was infected into A549 or CT26 cells at an MOI of 1 or 5, respectively. After 60 h, the infected cellular supernatant was cultured and was analyzed using a HMGB1 ELISA Kit II (Shino-Test, Tokyo, Japan).
In vivo bioimaging and therapeutic effect
CT26.WT (CRL-2638: American Type Culture Collection, VA, USA) tumors (5 × 105 cells) were subcutaneously injected into the bilateral flank of 6-week-old female BALB/c mice. Then, the tumors were grown until an average tumor volume of 60 mm3. The larger flank tumors were directly injected with 5 × 107 plaque-forming units (PFUs) in 40 μL PBS of MDRVV- or FUVAC-Luc/LacZ on days 0, 2, and 4. Tumor volumes were measured using calipers, and the viral replication was monitored using an injection of VivoGlo Luciferin, In Vivo Grade (3 mg/mouse; Promega) on days 1, 3, 5, and 7. The mice were anesthetized using isoflurane during bioimaging and was visualized using NightSHADE LB985 (Berthold Technologies, Bad Wildbad, Germany). Luminescence intensity was quantified according to the manufacturer’s protocol.
IHC analysis
BALB/c mice were bilaterally transplanted with CT26 tumors as described above. After tumor growth, 5 × 107 PFUs of MDRVV- or FUVAC-LG/DsRed were unilaterally injected on days 0, 2, and 4. After viral Fluc confirmation, tumors were harvested on day 5. Formalin-fixed and paraffin-embedded tissues were sliced and stained with hematoxylin and eosin or anti-GFP rabbit antibody (D5.1: Cell Signaling Technology, MA, USA). Anti-GFP antibody reaction was detected with the SignalStain Boost IHC detection reagent and SignalStain DAB Substrate Kit (CST). GFP-detected slides were counterstained with hematoxylin and a coverslip was added with softmount (Wako, Tokyo, Japan). Tissue sections were photographed by BZ-X700 (Keyence).
Fluorescence-activated cell sorting (FACS) analysis
Mice-bearing bilateral CT26 tumors were treated with Luc/LacZ viruses as described above. After 5 days from the first virus injection, the tumors were harvested and homogenized using a Tumor Dissociation Kit, mouse and gentleMACS Octo Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s protocol. After Fc blocking (2.4G2: BD Bioscience, CA, USA), the cells were stained with each antibody (0.5 μg/5 × 105 cells) as described below. The following cell surface staining antibodies were purchased from BioLegend (CA, USA): CD45 (30-F11), CD3 (145-2C11), CD8 (53-6.7), CD11b (M1/70), F4/80 (BM8), and Ly6G (1A8); CD4 (GK1.5) and CD25 (PC61.5) were purchased from Thermo Fisher; and Ly6C (AL21) was purchased from BD Bioscience. 7-AAD (Beckman Coulter, CA, USA) was used for dead cell differentiation. Cells were incubated at 4°C for 25 min, and some of them were intracellularly stained using anti-FoxP3 antibody (FJK-16 s) and FoxP3 staining buffer set (Thermo Fisher) according to the manufacturer’s protocol. Samples were suspended in FACS buffer (PBS/2% FBS/0.02% sodium azide) and were analyzed using CytoFLEX and CytExpert Ver 2.0 (Beckman). FACS analysis performed on day 7 was detailed in the Supplemental Materials and Methods.
Immune depletion and immune checkpoint blockade
For immune depletion, bilateral CT26-implanted mice were intratumorally injected with 2.5 × 107 PFUs of viruses on days 0, 2, and 4. Then, the mice were intraperitoneally treated with 200 μg of inhibitors, InVivo Plus rat IgG2b isotype control (LTF-2), anti-mouse CD4 antibody (GK1.5), or anti-mouse CD8a antibody (2.43: BioXcell, NH, USA) on days 3, 5, and 7. For the immune checkpoint blockade, 5 × 107 PFUs of viruses were directly injected in the larger side of the bilateral CT26 tumors on days 0, 2, and 4. Anti-mouse PD-1 antibodies (200 μg, RMP1-14: InVivo Plus Mab, BioXcell) or control PBS was administered via the intraperitoneal route on days 3, 5, 7, 9, and 11. The tumor volume and viral luciferase expression were measured as described above. For the tumor rechallenge, CR mice and control naive mice were bilaterally transplanted with 5 × 105 cells of CT26 tumors 101 days after the virus treatment. All animal experiments were approved by the Animal Experiment Committee of the Tottori University.
Statistical analysis
Differences in cell viability, immune staining, and FACS analysis among the groups were evaluated using the two-tailed unpaired t test. In vivo bioluminescence and tumor volumes were analyzed using two-way analysis of variance (ANOVA), followed by the Bonferroni test when the ANOVA showed an overall significance. Survival curves were generated using the Kaplan-Meier method and were analyzed using the log-rank test. p <0.05 was considered statistically significant. All statistical analyses were performed using Prism v.5 (GraphPad, CA, USA).
Acknowledgments
This work was funded by The Japan Agency for Medical Research and Development (AMED) (grant numbers JP20am0401017 and JP20ae0201008 to T.N.) and Japan Society for the Promotion of Science (JSPS)-KAKENHI (grant numbers 19H03515 and 15H04310 to T.N.). This work was also supported by the Center for Clinical and Translational Research of Kyushu University.
Author contributions
T.N. supervised the entire project. T.N., M.N., and N.K. designed the study and interpreted the results. M.N. and N.K. performed most of the experiments. H.K. and E.K. generated the recombinant vaccinia viruses and conducted the in vivo experiments. T.N. and M.N. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.024.
Supplemental information
References
- 1.Russell S.J., Peng K.W., Bell J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fountzilas C., Patel S., Mahalingam D. Review: Oncolytic virotherapy, updates and future directions. Oncotarget. 2017;8:102617–102639. doi: 10.18632/oncotarget.18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andtbacka R.H., Ross M., Puzanov I., Milhem M., Collichio F., Delman K.A., Amatruda T., Zager J.S., Cranmer L., Hsueh E. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM Phase III clinical trial. Ann. Surg. Oncol. 2016;23:4169–4177. doi: 10.1245/s10434-016-5286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufman H.L., Amatruda T., Reid T., Gonzalez R., Glaspy J., Whitman E., Harrington K., Nemunaitis J., Zloza A., Wolf M., Senzer N.N. Systemic versus local responses in melanoma patients treated with talimogene laherparepvec from a multi-institutional phase II study. J. Immunother. Cancer. 2016;4:12. doi: 10.1186/s40425-016-0116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park B.H., Hwang T., Liu T.C., Sze D.Y., Kim J.S., Kwon H.C., Oh S.Y., Han S.Y., Yoon J.H., Hong S.H. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 6.Burton C., Bartee E. Syncytia formation in oncolytic virotherapy. Mol. Ther. Oncolytics. 2019;15:131–139. doi: 10.1016/j.omto.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higuchi H., Bronk S.F., Bateman A., Harrington K., Vile R.G., Gores G.J. Viral fusogenic membrane glycoprotein expression causes syncytia formation with bioenergetic cell death: implications for gene therapy. Cancer Res. 2000;60:6396–6402. [PubMed] [Google Scholar]
- 8.Krabbe T., Altomonte J. Fusogenic viruses in oncolytic immunotherapy. Cancers (Basel) 2018;10:216. doi: 10.3390/cancers10070216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura T. 2017. Mitogen-activated protein kinase-dependent recombinant vaccinia virus (MD-RVV) and use thereof. US patent US-9809803-B2, filed November 20, 2014, and granted November 7, 2017. [Google Scholar]
- 10.Nakatake M., Kurosaki H., Kuwano N., Horita K., Ito M., Kono H., Okamura T., Hasegawa K., Yasutomi Y., Nakamura T. Partial deletion of glycoprotein B5R enhances vaccinia virus neutralization escape while preserving oncolytic function. Mol. Ther. Oncolytics. 2019;14:159–171. doi: 10.1016/j.omto.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeHaven B.C., Gupta K., Isaacs S.N. The vaccinia virus A56 protein: a multifunctional transmembrane glycoprotein that anchors two secreted viral proteins. J. Gen. Virol. 2011;92:1971–1980. doi: 10.1099/vir.0.030460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moss B. Membrane fusion during poxvirus entry. Semin. Cell Dev. Biol. 2016;60:89–96. doi: 10.1016/j.semcdb.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turner P.C., Moyer R.W. Orthopoxvirus fusion inhibitor glycoprotein SPI-3 (open reading frame K2L) contains motifs characteristic of serine proteinase inhibitors that are not required for control of cell fusion. J. Virol. 1995;69:5978–5987. doi: 10.1128/jvi.69.10.5978-5987.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner P.C., Moyer R.W. The vaccinia virus fusion inhibitor proteins SPI-3 (K2) and HA (A56) expressed by infected cells reduce the entry of superinfecting virus. Virology. 2008;380:226–233. doi: 10.1016/j.virol.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagenaar T.R., Moss B. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J. Virol. 2007;81:6286–6293. doi: 10.1128/JVI.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagenaar T.R., Moss B. Expression of the A56 and K2 proteins is sufficient to inhibit vaccinia virus entry and cell fusion. J. Virol. 2009;83:1546–1554. doi: 10.1128/JVI.01684-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagenaar T.R., Ojeda S., Moss B. Vaccinia virus A56/K2 fusion regulatory protein interacts with the A16 and G9 subunits of the entry fusion complex. J. Virol. 2008;82:5153–5160. doi: 10.1128/JVI.00162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H.H., Cawood R., El-Sherbini Y., Purdie L., Bazan-Peregrino M., Seymour L.W., Carlisle R.C. Active adenoviral vascular penetration by targeted formation of heterocellular endothelial-epithelial syncytia. Mol. Ther. 2011;19:67–75. doi: 10.1038/mt.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guedan S., Grases D., Rojas J.J., Gros A., Vilardell F., Vile R., Mercade E., Cascallo M., Alemany R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012;19:1048–1057. doi: 10.1038/gt.2011.184. [DOI] [PubMed] [Google Scholar]
- 20.Simpson G.R., Han Z., Liu B., Wang Y., Campbell G., Coffin R.S. Combination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor control. Cancer Res. 2006;66:4835–4842. doi: 10.1158/0008-5472.CAN-05-4352. [DOI] [PubMed] [Google Scholar]
- 21.Thomas S., Kuncheria L., Roulstone V., Kyula J.N., Mansfield D., Bommareddy P.K., Smith H., Kaufman H.L., Harrington K.J., Coffin R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer. 2019;7:214. doi: 10.1186/s40425-019-0682-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Del Papa J., Petryk J., Bell J.C., Parks R.J. An oncolytic adenovirus vector expressing p14 FAST protein induces widespread syncytium formation and reduces tumor growth rate in vivo. Mol. Ther. Oncolytics. 2019;14:107–120. doi: 10.1016/j.omto.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Boeuf F., Gebremeskel S., McMullen N., He H., Greenshields A.L., Hoskin D.W., Bell J.C., Johnston B., Pan C., Duncan R. Reovirus FAST protein enhances vesicular stomatitis virus oncolytic virotherapy in primary and metastatic tumor models. Mol. Ther. Oncolytics. 2017;6:80–89. doi: 10.1016/j.omto.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Betancourt D., Ramos J.C., Barber G.N. Retargeting oncolytic vesicular stomatitis virus to human T-cell lymphotropic virus type 1-associated adult T-cell leukemia. J. Virol. 2015;89:11786–11800. doi: 10.1128/JVI.01356-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., Haviv Y.S., Derdeyn C.A., Lam J., Coolidge C., Hunter E., Curiel D.T., Blackwell J.L. Human immunodeficiency virus type 1-mediated syncytium formation is compatible with adenovirus replication and facilitates efficient dispersion of viral gene products and de novo-synthesized virus particles. Hum. Gene Ther. 2001;12:2155–2165. doi: 10.1089/10430340152710504. [DOI] [PubMed] [Google Scholar]
- 26.Ayala-Breton C., Russell L.O., Russell S.J., Peng K.W. Faster replication and higher expression levels of viral glycoproteins give the vesicular stomatitis virus/measles virus hybrid VSV-FH a growth advantage over measles virus. J. Virol. 2014;88:8332–8339. doi: 10.1128/JVI.03823-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdullahi S., Jäkel M., Behrend S.J., Steiger K., Topping G., Krabbe T., Colombo A., Sandig V., Schiergens T.S., Thasler W.E. A novel chimeric oncolytic virus vector for improved safety and efficacy as a platform for the treatment of hepatocellular carcinoma. J. Virol. 2018;92 doi: 10.1128/JVI.01386-18. e01386–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ebert O., Shinozaki K., Kournioti C., Park M.S., García-Sastre A., Woo S.L. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res. 2004;64:3265–3270. doi: 10.1158/0008-5472.can-03-3753. [DOI] [PubMed] [Google Scholar]
- 29.Chang G., Xu S., Watanabe M., Jayakar H.R., Whitt M.A., Gingrich J.R. Enhanced oncolytic activity of vesicular stomatitis virus encoding SV5-F protein against prostate cancer. J. Urol. 2010;183:1611–1618. doi: 10.1016/j.juro.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Gómez-Treviño A., Castel S., López-Iglesias C., Cortadellas N., Comas-Riu J., Mercadé E. Effects of adenovirus-mediated SV5 fusogenic glycoprotein expression on tumor cells. J. Gene Med. 2003;5:483–492. doi: 10.1002/jgm.371. [DOI] [PubMed] [Google Scholar]
- 31.Fortin C., Huang X., Yang Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J. Immunol. 2012;189:1843–1849. doi: 10.4049/jimmunol.1200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leyland R., Watkins A., Mulgrew K.A., Holoweckyj N., Bamber L., Tigue N.J., Offer E., Andrews J., Yan L., Mullins S. A novel murine GITR ligand fusion protein induces antitumor activity as a monotherapy that is further enhanced in combination with an OX40 agonist. Clin. Cancer Res. 2017;23:3416–3427. doi: 10.1158/1078-0432.CCR-16-2000. [DOI] [PubMed] [Google Scholar]
- 33.McLeod R.L., Angagaw M.H., Baral T.N., Liu L., Moniz R.J., Laskey J., Hsieh S., Lee M., Han J.H., Issafras H. Characterization of murine CEACAM1 in vivo reveals low expression on CD8+ T cells and no tumor growth modulating activity by anti-CEACAM1 mAb CC1. Oncotarget. 2018;9:34459–34470. doi: 10.18632/oncotarget.26108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosely S.I., Prime J.E., Sainson R.C., Koopmann J.O., Wang D.Y., Greenawalt D.M., Ahdesmaki M.J., Leyland R., Mullins S., Pacelli L. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res. 2017;5:29–41. doi: 10.1158/2326-6066.CIR-16-0114. [DOI] [PubMed] [Google Scholar]
- 35.Fend L., Remy-Ziller C., Foloppe J., Kempf J., Cochin S., Barraud L., Accart N., Erbs P., Fournel S., Préville X. Oncolytic virotherapy with an armed vaccinia virus in an orthotopic model of renal carcinoma is associated with modification of the tumor microenvironment. OncoImmunology. 2015;5:e1080414. doi: 10.1080/2162402X.2015.1080414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fend L., Yamazaki T., Remy C., Fahrner C., Gantzer M., Nourtier V., Préville X., Quéméneur E., Kepp O., Adam J. Immune checkpoint blockade, immunogenic chemotherapy or IFN-alpha blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. 2017;77:4146–4157. doi: 10.1158/0008-5472.CAN-16-2165. [DOI] [PubMed] [Google Scholar]
- 37.Davola M.E., Mossman K.L. Oncolytic viruses: how “lytic” must they be for therapeutic efficacy? OncoImmunology. 2019;8:e1581528. doi: 10.1080/2162402X.2019.1596006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Z.S., Lu B., Guo Z., Giehl E., Feist M., Dai E., Liu W., Storkus W.J., He Y., Liu Z., Bartlett D.L. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J. Immunother. Cancer. 2019;7:6. doi: 10.1186/s40425-018-0495-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai P., Wang W., Yang N., Serna-Tamayo C., Ricca J.M., Zamarin D., Shuman S., Merghoub T., Wolchok J.D., Deng L. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci. Immunol. 2017;2:eaal1713. doi: 10.1126/sciimmunol.aal1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.