

Abstract
Mucopolysaccharidosis II (MPS II), a lysosomal storage disease caused by mutations in iduronate-2-sulfatase (IDS), is characterized by a wide variety of somatic and neurologic symptoms. The currently approved intravenous enzyme replacement therapy with recombinant IDS (idursulfase) is ineffective for CNS manifestations due to its inability to cross the blood-brain barrier (BBB). Here, we demonstrate that the clearance of heparan sulfate (HS) deposited in the brain by a BBB-penetrable antibody-enzyme fusion protein prevents neurodegeneration and neurocognitive dysfunctions in MPS II mice. The fusion protein pabinafusp alfa was chronically administered intravenously to MPS II mice. The drug reduced HS and attenuated histopathological changes in the brain, as well as in peripheral tissues. The loss of spatial learning abilities was completely suppressed by pabinafusp alfa, but not by idursulfase, indicating an association between HS deposition in the brain, neurodegeneration, and CNS manifestations in these mice. Furthermore, HS concentrations in the brain and reduction thereof by pabinafusp alpha correlated with those in the cerebrospinal fluid (CSF). Thus, repeated intravenous administration of pabinafusp alfa to MPS II mice decreased HS deposition in the brain, leading to prevention of neurodegeneration and maintenance of neurocognitive function, which may be predicted from HS concentrations in CSF.
Keywords: mucopolysaccharidosis II, enzyme-replacement therapy, blood-brain barrier, heparan sulfate, neurodegeneration, neurocognitive impairment
Graphical abstract
A blood-brain-barrier-penetrable enzyme clears heparan sulfate (HS) in the brain, prevents neurodegeneration, and maintains neurocognitive function in mucopolysaccharidosis II mice. Brain HS deposition is the primary cause of CNS manifestations. The CNS disease progression and drug efficacy are shown to be predictable based on HS concentrations in cerebrospinal fluid.
Introduction
Mucopolysaccharidoses (MPSs) include several types of diseases caused by mutations in genes encoding lysosomal enzymes involved in the catabolism of glycosaminoglycans (GAGs).1 Defects in iduronate-2-sulfatase (IDS) cause MPS II (also called Hunter syndrome), which is characterized by the pathological accumulation of GAGs such as heparan sulfate (HS) and dermatan sulfate (DS) throughout the body; this leads to various symptoms, including coarse facial features, hepatosplenomegaly, joint stiffness, skeletal deformities, and cardiac failure.1, 2, 3 The clinical features and severity of the disease depend on the mutations in the IDS gene.4,5 Attenuated patients with MPS II exhibit similar somatic manifestations without intellectual disability while severe patients progressively develop neurocognitive impairments.1, 2, 3 Intravenous enzyme replacement therapy (ERT) with recombinant human IDS (idursulfase: Elaprase, Takeda Pharmaceuticals) has been available for the treatment of patients with MPS II and exerts beneficial effects on somatic symptoms along with the reduction of GAG concentrations in peripheral tissues.6,7 However, such a conventional ERT is not effective for neurologic abnormalities because the enzyme cannot cross the blood-brain barrier (BBB) and access the CNS.
BBB-crossing technology utilizing the endogenous protein transport system mediated by receptors expressed on brain capillary endothelial cells has been pioneered by Pardridge and his colleagues.8, 9, 10, 11, 12, 13 The receptors include the insulin receptor and transferrin receptor (TfR).8, 9, 10, 11, 12, 13 We have developed a BBB-penetrable IDS designated pabinafusp alfa (investigational code name, JR-141), which is a genetically engineered fusion protein consisting of an anti-human TfR (hTfR) antibody and human IDS, fused to the C terminus of the immunoglobulin G (IgG) heavy chain.14, 15, 16, 17 Pabinafusp alfa can cross the BBB to reach the brain parenchyma, probably by taking advantage of the mechanism of the receptor-mediated transcytosis of transferrin.14 As a result, pabinafusp alfa reduces elevated GAG levels in the CNS, as well as in peripheral tissues in a mouse model of MPS II.14, 15, 16, 17
The major GAG species accumulated in the body of patients with MPS II are HS and DS.18 Although the precise roles of HS and DS in CNS pathogenesis of the disease are not fully understood, cells in the CNS seem to be more susceptible to HS than to DS. An evidence for this is the fact that, among MPSs, intellectual disability is seen only in patients with MPS type I, II, III, and VII, in which HS is the predominant accumulated substance.19, 20, 21, 22 The fact that patients with MPS VI, in which DS is exclusively accumulated, do not develop intellectual disability23 supports this notion. HS deposited in the brain is thought to initiate a series of pathogenic processes leading to the clinical symptoms of MPS.22 The accumulation of undigested substrates induces functional and morphological changes in the neurons, including impaired autophagy, dysfunction of organelles, induction of ectopic dendrites, altered axonal transport, and altered calcium storage.22, 23, 24, 25, 26 Glial cells also suffer from pathological lysosomal storage. Astroglia and microglia are activated, leading to glial degeneration and neuroinflammation, which are eventually followed by neurodegeneration and neuronal cell death.22,27,28 Clinical CNS symptoms are manifested in patients with MPS II as a result of these pathogenic processes.
In this study, we evaluated the efficacy of the BBB-penetrable IDS fusion protein pabinafusp alfa against neurodegeneration and neurocognitive impairment, as well as HS deposition in MPS II mice. We also sought to identify a practical surrogate biomarker that can predict disease severity and drug efficacy in neuronopathic MPS II.
Results
Dose assessment study in MPS II mice
Since the anti-hTfR antibody moiety used in pabinafusp alfa does not recognize mouse TfR, hTfR-knockin mice lacking Ids gene (TFRC-KI/Ids-KO) were established as an animal model of MPS II to evaluate the drug efficacy.14 We first conducted a 12-week repeated intravenous administration study in 12-week-old TFRC-KI/Ids-KO mice to find the appropriate doses of pabinafusp alfa for the following longitudinal study. Pabinafusp alfa was administered intravenously to MPS II mice at doses of 0.5, 1, 2, 5, or 10 mg/kg once every week for 12 weeks, while idursulfase was administered at the clinically approved dose of 0.5 mg/kg once every week. Due to the higher molecular weight but presence of two IDS enzymes per antibody in pabinafusp alfa, 1 mg/kg equals 0.5 mg/kg idursulfase on a molar dose. Concentrations of HS and DS in all tissues tested were markedly elevated in untreated MPS II (disease control) mice compared with those in the wild-type mice (Figures S1–S3). Pabinafusp alfa dose-dependently reduced HS and DS concentrations in the urine, serum, heart, liver, and spleen (Figures S1 and S2). The efficacy of pabinafusp alfa in the reduction of HS and DS in these peripheral tissues at 1 mg/kg or higher was similar to or greater than that of idursulfase at a dose of 0.5 mg/kg/week (Figures S1 and S2). For the brain and cerebrospinal fluid (CSF), pabinafusp alfa decreased HS levels in a dose-dependent manner, in contrast with idursulfase, which failed to affect HS levels in the CNS (Figure S3). Based on these data, we determined the doses used in the next chronic experiment to be 2 mg/kg for every week and 4 mg/kg for every other week.
Reduction in HS concentrations in MPS II mice by chronic treatment of pabinafusp alfa
To understand the effect of long-term reduction of HS in the CNS on neurodegeneration and cognitive function such as spatial learning ability, we performed a 36-week repeated intravenous dose study of pabinafusp alfa in MPS II mice. Pabinafusp alfa was administered intravenously to the mice at doses of 2 mg/kg once every week or 4 mg/kg once every other week for 36 weeks. The administration begun at 10 weeks of age, at which point the mice exhibit no overt CNS manifestations. As expected, the chronic treatment of pabinafusp alfa decreased HS concentrations in the serum, heart, liver, and spleen to near-normal levels (Figure 1A) and drastically reduced HS levels in the brain and CSF (Figure 1B). Idursulfase reduced HS depositions only in peripheral tissues, but not in the CNS (Figures 1A and 1B). Although the total dose amount was the same, the weekly dosing regimen seemed slightly superior in reducing HS concentrations than the regimen based on dosages administered every other week.
Figure 1.

Reduction in HS by chronic treatment of pabinafusp alfa
Drugs were intravenously administered to 10-week-old MPS II mice once every week (EW) or once every other week (EoW) for 36 weeks. (A) HS concentrations in the serum and peripheral tissues. (B) HS concentrations in the brain and cerebrospinal fluid (CSF). Tissues and body fluids were obtained after the last dosing and subjected to HS measurement. Values are presented as the mean with SD for each group (n = 4–5). Tukey-Kramer test, ∗∗p < 0.01 (versus disease control group). WT, wild-type; IDS, idursulfase (0.5 mg/kg EW).
Histopathological changes in peripheral organs
A histopathological analysis by H&E staining was performed in organs in the same long-term dosing study. Age-related findings, such as fatty liver and inflammatory cell infiltration in the liver, were detected in all animal groups, including wild-type animals (Figure S4; Table S1). As for the MPS II disease control mice, enlarged macrophages in the liver and spleen were observed in all animals (Figure S4; Table S1). In the heart, the vacuolization of atrial and ventricular cardiomyocytes, foamy cell infiltration, and inflammatory cell infiltration were observed in all animals, and fibrosis was observed in 3/8 animals of the disease control group. In addition, an enlargement of interstitial cells in the aortic, pulmonary, tricuspid, or mitral valve was observed in all disease control animals (Figure S4; Table S1). These histopathological changes observed in peripheral organs were almost entirely suppressed by chronic treatment of either pabinafusp alfa or idursulfase (Figure S4; Table S1).
Histopathological changes in the brain
Lamp1 is a lysosomal membrane protein that has been reported to be increased in lysosomal storage disease model animals, as well as in human patients.29, 30, 31 An increase in Lamp1 immunoreactivity in cells is thus likely a hallmark of lysosomal hypertrophy and storage of undigested materials, which leads to the loss of cellular function. We found that the intensity of Lamp1-staining increased throughout the brain in untreated MPS II mice (Figure 2A; Table S2). The recombinant enzyme idursulfase exerted little or no effect on the intensity of Lamp1 staining (Figure 2A; Table S2). In contrast, a chronic treatment with pabinafusp alfa, irrespective of the dosing regimen, decreased the intensity of Lamp1 staining in most regions of the brain including the cerebral cortex, hippocampus, interbrain, medulla oblongata, and cerebellum (Figure 2A; Table S2). This is indicative of an amelioration of lysosomal dysfunction, which is expected to lead to restoration of neuronal cell function.
Figure 2.

Histopathological changes in the brain of MPS II mice observed 1 week after the last dosing
(A) Lamp-1 staining. Representative photomicrographs of sections from the cerebellum, hippocampus, and medulla oblongata are shown. Brown stains indicate positive signals for Lamp-1. (B) H&E staining. Representative photomicrographs of the cerebellum sections are shown. Bottom panels are a high power view of white dashed rectangles shown in the upper panels. Arrows indicate vacuoles in the Purkinje cells. Scale bars, 50 μm.
H&E staining of the brain sections revealed the occurrence of vacuolation of the Purkinje cells in the cerebellum of untreated MPS II mice (Figure 2B; Table S1), which are rarely found in young diseased mice (Table S3). This change was not affected at all by idursulfase. Chronic treatment with pabinafusp alfa effectively prevented the neuronal cell damage obviated by the Purkinje cell vacuolation (Figure 2B; Table S1).
Maintenance of spatial learning abilities in MPS II mice by chronic treatment of pabinafusp alfa
The spatial learning ability of MPS II mice assessed by the Morris water maze test32 deteriorates with age. In the test, goal latency (the time to reach the platform) was shortened day by day in wild-type mice, but not in the MPS II disease control mice at the end of the dosing period (Figure 3), thereby confirming the validity of this experimental system for the evaluation of spatial learning and memory abilities. In the pabinafusp alfa treatment groups, the goal latency decreased to a comparable level to that in the normal control group (Figure 3), indicating that the chronic treatment of pabinafusp alfa maintained the spatial learning ability in MPS II mice. Idursulfase, which did not decrease HS levels in the brain, exerted no effect on the loss of spatial learning abilities (Figure 3).
Figure 3.

Maintenance of spatial learning abilities in MPS II mice upon chronic treatment with pabinafusp alfa
After 36 weeks of treatment, the spatial learning ability was assessed by the Morris water maze test. The time to reach the platform (goal latency) was measured 3 times per day and the means were calculated within each day for individual animals. Values are presented as the mean with SE for each group (n = 12–15). Paired t test, ∗∗p < 0.01 (day 1 versus day 5).
Correlation of HS concentrations between the brain and the CSF
It seems reasonable that HS depositions in the brain are associated with neuronal cell damage, leading to neurologic symptoms observed in MPS II. Therefore, measurement of HS concentration in the brain represents a useful way to examine the drug efficacy in neuronopathic MPS II. However, biopsy of the brain is not practical in a clinical setting. Thus, it is worth assessing whether HS concentrations in body fluids can be a potential and practical surrogate biomarker for HS accumulation in the brain. To this end, individual values of HS concentration in the body fluids measured in the 12-week and 36-week pharmacological studies were integrated and plotted into graphs to identify a potential relationship with HS concentration in the brain of MPS II mice. In our analyses, we found HS concentration in the CSF, but not in the serum, to correlate well with HS in the brain (Figures 4A and 4B). In addition, HS concentrations in the CSF did not correlate with that in the serum (Figure 4C), indicating that CSF HS reflects HS in the CNS tissues, but not in the peripheral tissues of MPS II mice. Therefore, we conclude that HS concentrations in the CSF can serve as a good predictor of HS depositions in the brain when BBB-penetrable enzymes are administered intravenously.
Figure 4.

Correlations in HS concentrations among the brain, CSF, and serum
(A–C) The correlation between HS concentrations in the brain and CSF (A), in the brain and serum (B), and in the serum and CSF (C) are shown. Values are from results obtained in the 12-week and 36-week repeated dose studies as indicated. The linear regression equations and correlation coefficients (R2) are shown in corresponding panels.
Discussion
This study demonstrates that chronic intravenous treatment with pabinafusp alfa, the BBB-penetrating anti-hTfR antibody-fused human IDS, reduced HS concentrations in both peripheral and CNS tissues of MPS II mice, leading to the prevention of histopathological changes in all tissues tested, including the brain. In contrast, the intact recombinant enzyme idursulfase decreased HS concentrations in peripheral tissues but failed to affect HS deposition and neurodegeneration observed in the brain. In addition, only pabinafusp alfa, which cleared HS deposited in the brain, prevented the loss of spatial learning ability in the mice. These findings point to an association between HS deposition in the brain, neurodegeneration, and neurocognitive impairment in MPS II mice.
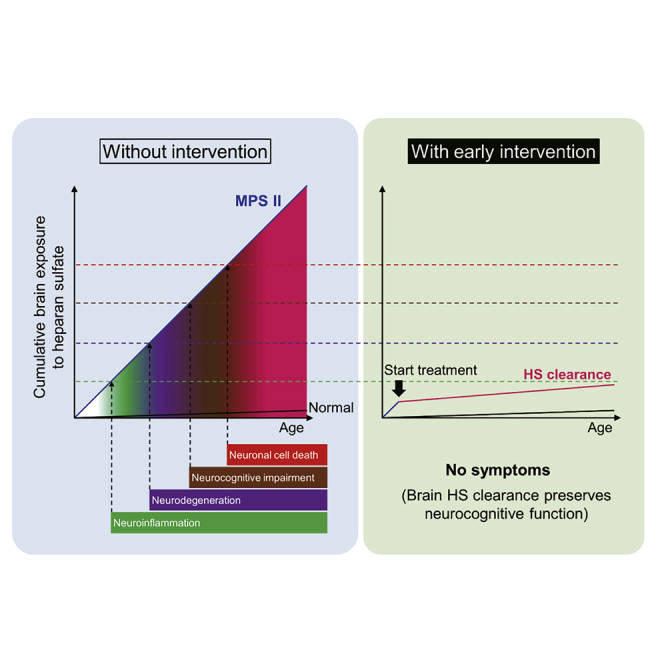
Although HS concentrations in the brain of MPS II mice are markedly increased as early as 10 weeks of age and do not change substantially after 20 weeks of age (Figure S5), disease symptoms continue to progress thereafter. For example, MPS II mice exhibit near-normal spatial learning ability as assessed by the Morris water maze test at 24 weeks of age (Figure S6), deteriorating thereafter at 8 months (approximately 34 weeks) of age33 or older (Figure 3). In addition, histopathological changes in the brain also progress with age even after 30 weeks (Table S3). Thus, a temporal factor in addition to HS concentrations at specific time points is likely to also be responsible for the onset and progression of these neurological events. It seems reasonable to consider that the cumulative exposure of the brain to HS is associated with neurodegeneration and cognitive abnormalities: a sustained elevation in HS levels may increase the risk of neuronal damage (Figure 5). It was reported that lysosomal storage induced glial cell activation (neuroinflammation) preceding neuronal degeneration in MPS II.27,28 Such a glial activation, in addition to the longitudinal exposure of HS to neurons themselves, could gradually drive neurodegeneration and neural cell death.27,28 Conversely, the continuous reduction in HS deposited in the brain may prevent or at least delay the onset of neuroinflammation, neurodegeneration, and CNS symptoms observed in MPS II (Figure 5). The symptoms may also even be reversed by the reduction of the brain HS level below a certain threshold value before the occurrence of irreversible damage to neuronal cells. Applying our model into a clinical setting, the intensity or degree of therapeutic effect would depend on the timing of intervention: the earlier the treatment starts, the better the chance to prevent CNS symptoms of MPS II (Figure 5).
Figure 5.

Conceptual picture representing the association of cumulative exposure of the brain to HS with neurodegeneration and neurocognitive impairments
The cumulative exposure levels (a blue curve for MPS II mice and a black curve for wild-type mice) were estimated by the actual brain HS concentrations presented in Figure S5. Vertical arrows indicate the presumed (not actual) timing of the onset of neurological events occurring in MPS II mice. Horizontal arrows indicate the postulated levels of cumulative exposure of the brain to HS when corresponding neuronal events occur. Values on y-intercepts are tentative. Pink dashed curves indicate presumed cumulative exposure to HS if HS-decreasing treatment for the brain starts at 10, 15, and 20 weeks of age. Note that this graph is simplified to help understand the role of cumulative exposure of the brain to HS over time. Other factors associated with aging may be involved in the development of CNS disorders, but these are not under consideration here. In addition, the treatment not only prevents but also may reverse symptoms before irreversible CNS damage occurs.
In this study, the accumulation of HS in MPS II mice induced an increase in Lamp1 positivity, a hallmark of lysosomal storage and cellular dysfunction, throughout the brain, including the hippocampus, the brain region responsible for spatial learning and memory,34,35 resulting in the loss of learning ability assessed by the Morris water maze test. When HS concentration in the brain decreased by chronic treatment of pabinafusp alfa, from the age of 10 weeks on, Lamp1 positivity in the hippocampus was attenuated, leading to prevention of the loss of spatial learning ability in MPS II mice (Figure 3). These results provide evidence for the prevention of CNS symptoms by the continuous reduction of HS in the brain and further support the concept of a sequential link between HS deposition, neurodegeneration, and neurocognitive impairment.
In order to predict disease progression and drug efficacy in MPS II patients with CNS symptoms, monitoring HS levels in the brain would be ideal, as discussed above. However, it is not practical to determine HS concentrations in the brain in a clinical setting as a biopsy of the brain is highly invasive and incurs the risk of bleeding, seizures, brain swelling, infection, stroke, blood clots, and reactions to anesthesia for the patients. Our previous and present studies clearly indicate that HS concentrations between the CSF and the brain are strongly correlated,15 and the collection of CSF by lumbar puncture is far less invasive than a brain biopsy. Therefore, HS in the CSF appears to be a practical surrogate marker reflecting that in the brain and can thus be used to predict the disease progression and drug efficacy on neurodegeneration leading to various CNS manifestations in patients with MPS II. However, the general applicability of this biomarker to other therapeutic modalities or routes of administration needs to be considered cautiously; although the intrathecal (IT) administration of IDS led to a marked decrease of GAG in the CSF of patients with MPS II,36 subsequent clinical studies were unable to demonstrate a clinically meaningful neurological benefit of IT-administered IDS to patients (ClinicalTrials.gov: NCT02055118). Preceding animal studies did not demonstrate a quantitative reduction of HS or GAGs in the CNS of monkeys, dogs, or MPS II mice.37 We therefore recommend that this biomarker should only be applied clinically for a given therapeutic or route of administration if validating animal studies unanimously demonstrate a linear correlation between the reduction of the brain and CSF HS.
The limitations of this study include the fact that a definite quantitative relationship between the reduction in HS concentrations in the brain and suppression of the loss of spatial learning abilities was not demonstrated. Since substrates are degraded depending on the amount and activity of their corresponding enzymes, it is reasonable to conclude that the degree of reduction of HS concentrations in the brain and CSF correlates with the dose of pabinafusp alfa (Figure S3). However, CNS symptoms of MPS II, such as the loss of learning and memory abilities, represent disorders in higher brain functions that result from a variety of pathological conditions in the CNS at different levels.22 Although these pathologic changes in the CNS are initiated by the deposition of HS in neurons and glial cells, their relationship is complex and interdependent. Nevertheless, HS deposited in the brain represents the hallmark of neurodegeneration leading to CNS symptoms observed in MPS II (Figure 5). As such, we reason that the reduction of HS concentrations in the brain is linked to the prevention of the neurocognitive impairments.
In conclusion, this study shows that the BBB-penetrable antibody-IDS fusion protein, pabinafusp alfa, decreases HS concentration in the brain and prevents neurodegeneration, which in turn maintains spatial learning ability in MPS II mice. Importantly, the disease progression and drug efficacy in MPS II patients may be predictable based on the HS concentration in the CSF, as it reflects the HS level in the brain.
Materials and methods
Recombinant proteins
Pabinafusp alfa (JR-141), a recombinant fusion protein consisting of humanized anti-hTfR antibody and human IDS, was produced as described previously.14 Idursulfase (Elaprase, Shire) was purchased from Shire Pharmaceuticals (now acquired by Takeda Pharmaceuticals).
Animals
All animal experiments were conducted under the approval of Animal Care and Use Committees of Nihon Bioresearch and JCR Pharmaceuticals. TFRC-KI/Ids-KO, a mouse model of MPS II, was established as described previously.14,38 C57BL/6 mice (Charles River, Japan) were used as a normal control.
For the 12-week intravenous repeated dose study, pabinafusp alfa was administered to TFRC-KI/Ids-KO mice through the tail vein at a dose of 0.5, 1, 2, 5, or 10 mg/kg once every week (n = 5/group). Idursulfase was administered similarly at a dose of 0.5 mg/kg for 12 weeks (n = 5). The administration began at 12 weeks of age. Urine samples were collected pre-dose and at weeks 4, 8, and 12 of the dosing. 1 week after the last dosing, tissues (namely the brain, heart, liver, and spleen), CSF, and serum were collected. CSF was taken from the cisterna magna using a glass capillary tube under anesthesia. Subsequently, blood samples were collected from the caudal vena cava, and sera were isolated by centrifugation. Next, the tissues were collected after euthanasia by exsanguination from the abdominal aorta. HS and DS were quantified by liquid chromatography-tandem mass spectrometry.15 Their concentrations in urine were normalized by creatinine.
For the 36-week intravenous repeated dose study, pabinafusp alfa was administered to TFRC-KI/Ids-KO mice through the tail vein once every week at 2 mg/kg or once every other week at 4 mg/kg. Idursulfase was administered to the mice at a dose of 0.5 mg/kg once every week. Administration was begun at 10 weeks of age. The number of animals used was 15 per group, including 5 animals for substrate measurements and 10 animals for histopathological analysis. After 36 weeks of dosing, all mice were subjected to a spatial learning ability test by the Morris water maze test,32 in which a circular pool of 120 cm in diameter and 18 cm in depth equipped with a transparent acrylic resin platform (2 cm below the water surface) was used. Each mouse was placed with its head facing the wall of the pool and the time to reach the platform (goal latency) was measured. If mice were not able to reach the platform within 90 s, the goal latency was counted as 90 s. Trials were performed three times per day for 5 consecutive days. 1 week (for every week dosing groups) or 2 weeks (for every other week dosing group) after the last dosing, tissues, CSF, and serum were obtained as described above, and HS concentrations were measured.
Histopathology
1 week (for every week dosing groups) or 2 weeks (for every other week dosing group) after the last dosing, animals were euthanized by exsanguination from the abdominal aorta and subjected to whole-body perfusion with physiological saline. The brain, liver, spleen, and heart were collected to be immersed in 10% neutral-buffered formalin. Next, the tissues were embedded in paraffin and stained with H&E for histopathological evaluation. The specimens were analyzed by optical microscopy and findings were graded as negative (−), minimal (+/−), mild (+), moderate (++), and severe (+++).
Immunostaining
Lamp1 staining was performed with the paraffin-embedded sections. The sections were deparaffinized with xylene and rehydrated with a graded series of ethanol before staining. The deparaffinized sections were treated with 3% H2O2/methanol to quench endogenous peroxidase and blocked with 1% BSA. The primary antibody used was anti-LAMP1 antibody (Abcam, Cambridge, UK: ab24170, 1:700 dilution) and the secondary antibody used was EnVision+ System-HRP Labeled Polymer Anti-Rabbit (Dako, Glostrup, Denmark), and the reaction was visualized with 3,3'- diaminobenzidine. The specimens were analyzed by optical microscopy and the intensity of immunoreactivity was graded as negative (−), minimal (+/−), mild (+), moderate (++), and severe (+++).
Statistics
The statistical analysis was conducted with SPSS Statistics software (IBM, Armonk, NY, USA). For substrate reduction efficacy, the Tukey-Kramer test was used to compare the differences between groups. A paired t test was applied to assess the shortening of the goal latency in the Morris water maze test. p values <0.05 were considered to indicate statistical significance. No statistical method was employed for immunohistochemistry.
Data availability
The authors confirm that all data supporting the findings of this study are available within the article and its Supplemental information.
Acknowledgments
We thank Miho Okumura for her technical assistance and Mathias Schmidt for critical reading of the manuscript. We would like to thank Editage (https://www.editage.com/) for English language editing.
Author contributions
H.M., K.M., T.H., K.T., and H.S. conceptualized and designed the research; H.M., S.K., E.Y., M.K., N.T., Y.K., and H.T. performed the research; H.M., S.K., E.Y., N.T., R.Y., K.T., T.H., K.M., and H.S. analyzed the data; K.M. wrote the paper.
Declaration of interests
H.M., S.K., E.Y., M.K., N.T., R.Y., Y.K., H.T., K.T., T.H., K.M., and H.S. received compensation as employees of JCR Pharmaceuticals. This study was sponsored by JCR Pharmaceuticals.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.01.027.
Contributor Information
Kohtaro Minami, Email: minami-k@jcrpharm.co.jp.
Hiroyuki Sonoda, Email: sonoda-h@jcrpharm.co.jp.
Supplemental information
References
- 1.Neufeld E.F., Muenzer J. The Mucopolysaccharidoses. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw Hill; 2001. pp. 3421–3452. [Google Scholar]
- 2.Tylki-Szymańska A. Mucopolysaccharidosis type II, Hunter’s syndrome. Pediatr. Endocrinol. Rev. 2014;12(Suppl 1):107–113. [PubMed] [Google Scholar]
- 3.Wilson P.J., Morris C.P., Anson D.S., Occhiodoro T., Bielicki J., Clements P.R., Hopwood J.J. Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc. Natl. Acad. Sci. USA. 1990;87:8531–8535. doi: 10.1073/pnas.87.21.8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dvorakova L., Vlaskova H., Sarajlija A., Ramadza D.P., Poupetova H., Hruba E., Hlavata A., Bzduch V., Peskova K., Storkanova G. Genotype-phenotype correlation in 44 Czech, Slovak, Croatian and Serbian patients with mucopolysaccharidosis type II. Clin. Genet. 2017;91:787–796. doi: 10.1111/cge.12927. [DOI] [PubMed] [Google Scholar]
- 5.Vollebregt A.A.M., Hoogeveen-Westerveld M., Kroos M.A., Oussoren E., Plug I., Ruijter G.J., van der Ploeg A.T., Pijnappel W.W.M.P. Genotype-phenotype relationship in mucopolysaccharidosis II: predictive power of IDS variants for the neuronopathic phenotype. Dev. Med. Child Neurol. 2017;59:1063–1070. doi: 10.1111/dmcn.13467. [DOI] [PubMed] [Google Scholar]
- 6.Muenzer J., Beck M., Giugliani R., Suzuki Y., Tylki-Szymanska A., Valayannopoulos V., Vellodi A., Wraith J.E. Idursulfase treatment of Hunter syndrome in children younger than 6 years: results from the Hunter Outcome Survey. Genet. Med. 2011;13:102–109. doi: 10.1097/GIM.0b013e318206786f. [DOI] [PubMed] [Google Scholar]
- 7.Muenzer J., Beck M., Eng C.M., Giugliani R., Harmatz P., Martin R., Ramaswami U., Vellodi A., Wraith J.E., Cleary M. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet. Med. 2011;13:95–101. doi: 10.1097/GIM.0b013e3181fea459. [DOI] [PubMed] [Google Scholar]
- 8.Pardridge W.M. Receptor-mediated peptide transport through the blood-brain barrier. Endocr. Rev. 1986;7:314–330. doi: 10.1210/edrv-7-3-314. [DOI] [PubMed] [Google Scholar]
- 9.Pardridge W.M., Eisenberg J., Yang J. Human blood-brain barrier transferrin receptor. Metabolism. 1987;36:892–895. doi: 10.1016/0026-0495(87)90099-0. [DOI] [PubMed] [Google Scholar]
- 10.Bickel U., Yoshikawa T., Pardridge W.M. Delivery of peptides and proteins through the blood-brain barrier. Adv. Drug Deliv. Rev. 2001;46:247–279. doi: 10.1016/s0169-409x(00)00139-3. [DOI] [PubMed] [Google Scholar]
- 11.Pardridge W.M. Blood-brain barrier drug delivery of IgG fusion proteins with a transferrin receptor monoclonal antibody. Expert Opin. Drug Deliv. 2015;12:207–222. doi: 10.1517/17425247.2014.952627. [DOI] [PubMed] [Google Scholar]
- 12.Osborn M.J., McElmurry R.T., Peacock B., Tolar J., Blazar B.R. Targeting of the CNS in MPS-IH using a nonviral transferrin-alpha-L-iduronidase fusion gene product. Mol. Ther. 2008;16:1459–1466. doi: 10.1038/mt.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Q.H., Boado R.J., Lu J.Z., Hui E.K., Pardridge W.M. Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab. Dispos. 2012;40:329–335. doi: 10.1124/dmd.111.042903. [DOI] [PubMed] [Google Scholar]
- 14.Sonoda H., Morimoto H., Yoden E., Koshimura Y., Kinoshita M., Golovina G., Takagi H., Yamamoto R., Minami K., Mizoguchi A. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018;26:1366–1374. doi: 10.1016/j.ymthe.2018.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka N., Kida S., Kinoshita M., Morimoto H., Shibasaki T., Tachibana K., Yamamoto R. Evaluation of cerebrospinal fluid heparan sulfate as a biomarker of neuropathology in a murine model of mucopolysaccharidosis type II using high-sensitivity LC/MS/MS. Mol. Genet. Metab. 2018;125:53–58. doi: 10.1016/j.ymgme.2018.07.013. [DOI] [PubMed] [Google Scholar]
- 16.Okuyama T., Eto Y., Sakai N., Minami K., Yamamoto T., Sonoda H., Yamaoka M., Tachibana K., Hirato T., Sato Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019;27:456–464. doi: 10.1016/j.ymthe.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okuyama T., Eto Y., Sakai N., Nakamura K., Yamamoto T., Yamaoka M., Ikeda T., So S., Tanizawa K., Sonoda H., Sato Y. A Phase 2/3 Trial of Pabinafusp Alfa, IDS Fused with Anti-Human Transferrin Receptor Antibody, Targeting Neurodegeneration in MPS-II. Mol. Ther. 2020 doi: 10.1016/j.ymthe.2020.09.039. Published online September 30, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujitsuka H., Sawamoto K., Peracha H., Mason R.W., Mackenzie W., Kobayashi H., Yamaguchi S., Suzuki Y., Orii K., Orii T. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019;19:100455. doi: 10.1016/j.ymgmr.2019.100455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bigger B.W., Begley D.J., Virgintino D., Pshezhetsky A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018;125:322–331. doi: 10.1016/j.ymgme.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Tomatsu S., Shimada T., Mason R.W., Montaño A.M., Kelly J., LaMarr W.A., Kubaski F., Giugliani R., Guha A., Yasuda E. Establishment of glycosaminoglycan assays for mucopolysaccharidoses. Metabolites. 2014;4:655–679. doi: 10.3390/metabo4030655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Węgrzyn G., Jakóbkiewicz-Banecka J., Narajczyk M., Wiśniewski A., Piotrowska E., Gabig-Cimińska M., Kloska A., Słomińska-Wojewódzka M., Korzon-Burakowska A., Węgrzyn A. Why are behaviors of children suffering from various neuronopathic types of mucopolysaccharidoses different? Med. Hypotheses. 2010;75:605–609. doi: 10.1016/j.mehy.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 22.Sato Y., Okuyama T. Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. Int. J. Mol. Sci. 2020;21:400. doi: 10.3390/ijms21020400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valayannopoulos V., Nicely H., Harmatz P., Turbeville S. Mucopolysaccharidosis VI. Orphanet J. Rare Dis. 2010;5:5. doi: 10.1186/1750-1172-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shapiro E.G., Jones S.A., Escolar M.L. Developmental and behavioral aspects of mucopolysaccharidoses with brain manifestations - Neurological signs and symptoms. Mol. Genet. Metab. 2017;122S:1–7. doi: 10.1016/j.ymgme.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Węgrzyn G., Jakóbkiewicz-Banecka J., Gabig-Cimińska M., Kloska A., Malinowska M., Moskot M., Piotrowska E., Narajczyk M., Banecka-Majkutewicz Z., Banecki B. Mechanisms of neurodegeneration in mucopolysaccharidoses. In: Tomatsu S., Lavery C., Giugliani R., Harmatz P.R., Scarpa M., Wegrzyn G., Orii T., editors. Mucopolysaccharidoses Update. Nova Science Publishers; 2018. pp. 87–113. [Google Scholar]
- 26.Sharma J., di Ronza A., Lotfi P., Sardiello M. Lysosomes and Brain Health. Annu. Rev. Neurosci. 2018;41:255–276. doi: 10.1146/annurev-neuro-080317-061804. [DOI] [PubMed] [Google Scholar]
- 27.Fusar Poli E., Zalfa C., D’Avanzo F., Tomanin R., Carlessi L., Bossi M., Nodari L.R., Binda E., Marmiroli P., Scarpa M. Murine neural stem cells model Hunter disease in vitro: glial cell-mediated neurodegeneration as a possible mechanism involved. Cell Death Dis. 2013;4:e906. doi: 10.1038/cddis.2013.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zalfa C., Verpelli C., D’Avanzo F., Tomanin R., Vicidomini C., Cajola L., Manara R., Sala C., Scarpa M., Vescovi A.L., De Filippis L. Glial degeneration with oxidative damage drives neuronal demise in MPSII disease. Cell Death Dis. 2016;7:e2331. doi: 10.1038/cddis.2016.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meikle P.J., Brooks D.A., Ravenscroft E.M., Yan M., Williams R.E., Jaunzems A.E., Chataway T.K., Karageorgos L.E., Davey R.C., Boulter C.D. Diagnosis of lysosomal storage disorders: evaluation of lysosome-associated membrane protein LAMP-1 as a diagnostic marker. Clin. Chem. 1997;43:1325–1335. [PubMed] [Google Scholar]
- 30.Kondagari G.S., Fletcher J.L., Cruz R., Williamson P., Hopwood J.J., Taylor R.M. The effects of intracisternal enzyme replacement versus sham treatment on central neuropathology in preclinical canine fucosidosis. Orphanet J. Rare Dis. 2015;10:143. doi: 10.1186/s13023-015-0357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf H., Damme M., Stroobants S., D’Hooge R., Beck H.C., Hermans-Borgmeyer I., Lüllmann-Rauch R., Dierks T., Lübke T. A mouse model for fucosidosis recapitulates storage pathology and neurological features of the milder form of the human disease. Dis. Model. Mech. 2016;9:1015–1028. doi: 10.1242/dmm.025122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Hooge R., De Deyn P.P. Applications of the Morris water maze in the study of learning and memory. Brain Res. Brain Res. Rev. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- 33.Fu H., Zaraspe K., Murakami N., Meadows A.S., Pineda R.J., McCarty D.M., Muenzer J. Targeting Root Cause by Systemic scAAV9-hIDS Gene Delivery: Functional Correction and Reversal of Severe MPS II in Mice. Mol. Ther. Methods Clin. Dev. 2018;10:327–340. doi: 10.1016/j.omtm.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma S., Rakoczy S., Brown-Borg H. Assessment of spatial memory in mice. Life Sci. 2010;87:521–536. doi: 10.1016/j.lfs.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vorhees C.V., Williams M.T. Assessing spatial learning and memory in rodents. ILAR J. 2014;55:310–332. doi: 10.1093/ilar/ilu013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muenzer J., Hendriksz C.J., Fan Z., Vijayaraghavan S., Perry V., Santra S., Solanki G.A., Mascelli M.A., Pan L., Wang N. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet. Med. 2016;18:73–81. doi: 10.1038/gim.2015.36. [DOI] [PubMed] [Google Scholar]
- 37.Calias P., Papisov M., Pan J., Savioli N., Belov V., Huang Y., Lotterhand J., Alessandrini M., Liu N., Fischman A.J. CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: implications for neurological outcomes of lysosomal storage disorder. PLoS ONE. 2012;7:e30341. doi: 10.1371/journal.pone.0030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Higuchi T., Shimizu H., Fukuda T., Kawagoe S., Matsumoto J., Shimada Y., Kobayashi H., Ida H., Ohashi T., Morimoto H. Enzyme replacement therapy (ERT) procedure for mucopolysaccharidosis type II (MPS II) by intraventricular administration (IVA) in murine MPS II. Mol. Genet. Metab. 2012;107:122–128. doi: 10.1016/j.ymgme.2012.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that all data supporting the findings of this study are available within the article and its Supplemental information.