

Abstract
CD39, expressed by tumor-infiltrating lymphocytes (TILs), is a marker to identify tumor-reactive T cells, which is frequently associated with stronger antitumor activity than bystander T cells in a variety of malignancies. Therefore, CD39 could be a promising marker for identifying the active antitumor immune cells used for cellular immunotherapy. To test this possibility, we constructed the hepatitis B virus (HBV) surface protein-specific chimeric antigen receptor T cells (HBVs-CAR-T cells) and generated the personalized tumor-reactive CD8+ T cells. We subsequently assessed their antitumor efficiency mainly with a co-culture system for autologous HBVs+ HCC organoid and T cells. We found that both CD39+ HBVs-CAR-T and CD39+ personalized tumor-reactive CD8+ T cells induced much more apoptosis in HCC organoids. Although the exhaustion status of CAR-T cells increased in CD39+ CAR-T cells, triple knockdown of PD-1, Tim-3, and Lag-3 with shRNAs further enhanced antitumor activity in CD39+ CAR-T cells. Furthermore, these CD39+ CAR-T cells exerted an increased secretion of interferon-γ and stronger antitumor effect in a patient-derived xenograft mouse model. Our findings demonstrated that CD39 could be a promising biomarker to enrich active immune cells and become an indicator marker for evaluating the prognosis of immunotherapy for HCC patients.
Keywords: CD39, CAR-T, HCC
Graphical abstract
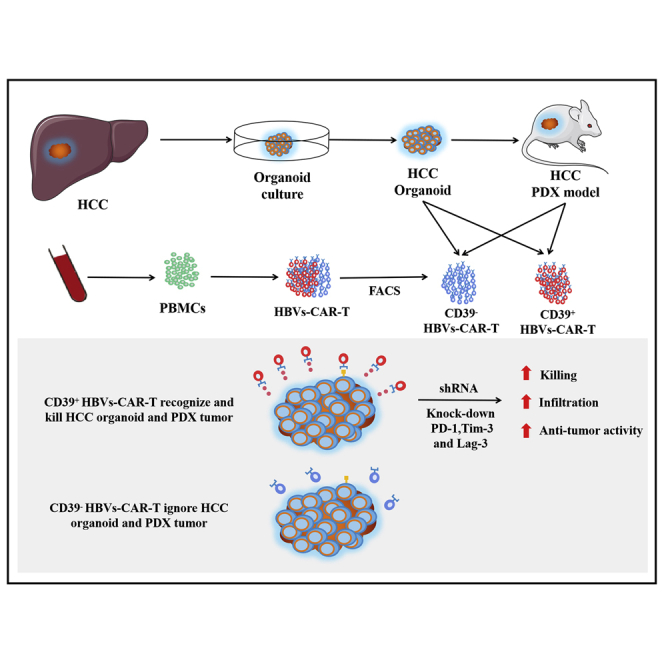
Zou et al. constructed the hepatitis B virus surface protein-specific chimeric antigen receptor T cells (HBVs-CAR-T) and assessed antitumor activity by HCC organoid and PDX model. CD39 may be a promising biomarker to enrich active CAR-T cells, and triple downregulation of inhibitory receptors of CD39+ CAR-T may improve immunotherapy for HCC patients.
Introduction
Hepatocellular carcinoma (HCC) is the fourth most common cause of cancer-related death and ranks sixth in terms of incident cases.1 In recent years, immune checkpoint blockades such as Opdivo (nivolumab) and Keytruda (pembrolizumab) have provided a new class of effective therapeutics for HCC patients.2,3 However, the efficiency of immunotherapy is still uncertain in different patients, and a significant number of patients have shown limited response to checkpoint-blockade immunotherapies. The possible cause may be lack of immunogenic tumor-associated antigen or the overexpression of checkpoint molecules.4,5 For the past few years, a series of novel strategies for HCC cellular immunotherapy has been developed and applied in clinical trials, such as chimeric antigen receptor T (CAR-T) cell and personalized tumor-reactive CD8+ T cell treatments.6, 7, 8 As a promising approach, CAR-T cells can specifically recognize and eliminate cancer cells through direct T cell cytotoxicity in a major histocompatibility complex-I (MHC-I)-independent manner. One of the most popular tumor-associated antigens identified for HCC, glypican-3 (GPC3) was expressed in human embryos and in some adult tissues, such as ovary, mammary gland, mesothelium, lung, and kidney. High-affinity GPC3-specific CAR-T cells may aggravate off-tumor adverse events, with severe or potentially lethal side effects.9, 10, 11 Chronic hepatitis B virus (HBV) infection is responsible for 50%–80% of HCC cases worldwide, considered one of the high-risk factors for human HCC.12 As an attractive immunotherapy strategy, genetically engineered CAR-T cells targeted HBV surface proteins (HBVs) on the surface of infected HCC cells barely induced off-tumor adverse event .13
Given the immunosuppressive microenvironment in HCC and heterogeneity of the patients, 80%–90% of the patients did not have an objective response to the immunotherapy treatments mentioned above.14 It is critical to predict whether they are suitable for immunotherapy.5,15 To accurately assess the sensitivity of individual patients treated with different kinds of immunotherapies, a novel pre-clinical therapy model was developed, which was based on non-small cell lung cancer (NSCLS) and colorectal cancer (CRC) organoid-T cell co-culture systems.16 This system not only preserved the tumor heterogeneity and microenvironment but could also evaluate the efficiency of cellular immunotherapies in vitro. Personalized tumor-reactive CD8+ T cells generated from autologous peripheral blood mononuclear cells (PBMCs) and tumor organoids also exhibited impressive antitumor effects.16,17
CD39 (ENTPD1) is expressed on monocytes, dendritic cells (DCs), B lymphocytes, natural killer (NK), T cells, and neutrophils.18, 19, 20, 21, 22, 23 With the abilities to hydrolyze proinflammatory extracellular ATP (eATP) and promote the synthesis of immunosuppressive adenosine, CD39 plays important roles in limiting inflammation.24 Alternatively, a recent study has shown that CD39+CD8+ tumor infiltrated lymphocytes (TILs) had better effector function and stronger antitumor activity than CD39−CD8+ TILs in multiple malignant tumor types.25 CD39 has been identified as a biomarker for tumor-specific T cells that could be used to distinguish them from bystander CD8+ T cell.26 Therefore, we speculated that CD39 is related to T cell effector function and may influence the cytotoxic efficiency of CAR-T and personalized tumor-reactive CD8+ T cells. Due to CD39+, T cells are frequently co-expressed with inhibitory receptors such as PD-1, Tim-3, or Lag-3, we hypothesized that the combination of CD39+ CAR-T cells with the checkpoint blockage may enhance antitumor activity.
In this study, we generated HBVs+ HCC organoids. Then, we developed HBVs-CAR-T cells and found that CD39 may be associated with the killing effect of CAR-T cells. The HCC organoid-T cell co-culture system was constructed to verify the efficiency of CD39+ HBVs-CAR-T and CD39+ personalized tumor-reactive CD8+ T cells. Based on our previous study, we also showed that they downregulated the expression of PD-1, Tim-3, and Lag-3 simultaneously on CAR-T cells by short hairpin RNA (shRNA)-cluster can significantly improve the cytotoxic and infiltrating ability. Furthermore, the CAR-T cells exhibiting CD39+ plus downregulation of PD-1, Tim-3, and Lag-3 with shRNAs can potently inhibit tumor growth in vivo.
Results
HBVs protein was abundant on HCC organoids, and HBVs-CAR-T cell was constructed for cellular immunotherapy
To evaluate the potent antitumor effector function of anti-HCC CAR-T cells, the organoids derived from three HCC patients’ tumor biopsy samples were constructed. Immunofluorescent analysis revealed that alpha-fetoprotein (AFP) and HBVs, which are the biomarkers of HBV+ HCC, were both highly expressed in tumor organoids in accordance with the expression pattern of the original tumor tissues,27 while GPC3 was not at all expressed (Figures 1A, S1A, and S1B). Flow cytometry analysis revealed the frequency of HBVs protein expression on HCC organoids ranging from 46% to 75% (Figure 1B). To further characterize the HCC organoids, we performed whole-exome sequencing (WES), which showed the similar tumor mutation burdens (TMBs) in both tumor organoids and tissues. Non-synonymous mutations, the top 12 mutations, and the gene functions in these organoids were also listed. Venn diagrams illustrated the number of somatic non-synonymous mutations presented in each HCC tissue and HCC organoid (Figures S1C–S1E). The proportion of coincident mutations ranged from 85.7% to 90.5%, suggesting that the HCC organoid model was highly mimetic of the heterogeneity and microenvironment of tumor tissue and retained the characteristics of original tumor tissues for expansion in vitro.
Figure 1.

HBVs protein was abundant on HCC organoids, and HBVs-CAR-T cells were constructed for cellular immunotherapy
(A) Bright-field microscopy images, H&E staining, and immunofluorescence staining of HCC markers AFP (red) and HBVs (green) for organoids were shown separately. Nuclei were stained with DAPI (blue). Scale bars, 200 μm (bright field), 100 μm (H&E and immunofluorescence). (B) Flow cytometry was used to analyze the frequency of HBVs protein expression on HCC organoids of 3 patients. (C) Schematic representation of the lentiviral vectors carrying a HBVs-specific 3rd-generation CAR moiety. (D) Western blotting and immunofluorescence staining for HBVs in HepG-2.2.15 cells; HepG-2 cells served as the mock cells. (E) Immune response was evaluated by measuring the number of IFN-γ production and spot-forming cells (SFCs) per 104 HBVs-CAR-T cells (E:T = 4:1). CAR-T cells added with PHA served as PC (positive control) and CAR-T cells alone as NC (negative control). (F) Cytotoxic activity of HBVs-CAR-T cells on HepG-2.2.15 targets cells were detected by LDH assay (n = 3, 3 healthy donors). (G) After co-culture with HepG-2.2.15 cells for 24 h (E:T = 4:1), CD107a+ and CD107a− HBVs-CAR-T cells were obtained by flow sorting, and their transactional profiling was analyzed by RNA sequencing (RNA-seq). Heatmap of RNA-seq showed the expression of genes involving in cytotoxicity, adhesion, and chemotaxis in different effector sub-populations, with p < 0.01. RNA expression levels were indicated with a red/blue scale for high and low expression levels, respectively.
Then, a HBVs-specific CAR moiety targeting the pre-S1 domain of HBVs was designed and constructed into the lentiviral vector (Figure 1C). Fluorescence images and western blot assay confirmed the HBVs expression on the surface of HepG-2.2.15 cells (Figure 1D). By evaluating the secreting ability and specific killing efficiency of interferon-γ (IFN-γ), we revealed the potency of CAR-T cells killing HBVs+ target cells (Figures 1E and 1F). To further characterize the effector sub-population of HBVs-CAR-T cells generated from peripheral blood, we compared the RNA profiles between CD107a+ and CD107a− HBVs-CAR-T cells sorted by fluorescence-activated cell sorting (FACS) after co-culture with HepG-2.2.15 cells for 24 h. A heatmap of transcriptional profiling revealed a significant increasing expression of some genes involved in effector molecules such as INF-γ and granzyme B, and cell chemotaxis or adhesion molecules such as CXCL-9, CXCL-10, and CD103 in CD107a+ HBVs-CAR-T cells (Figure 1G). Notably, CD39 was also highly expressed on CD107a+ HBVs-CAR-T cells. The results were validated by quantitative real-time PCR analysis (Figure S2). These data suggested that CD39 may serve as a marker representing effector CAR-T cells.
CD39+ HBVs-CAR-T cells exerted potent antitumor activity in HBV+ HCC organoid-T cell co-culture system
To verify whether CD39 molecule on HBVs-CAR-T cells generated from peripheral blood represent potent antitumor effector function, and to evaluate the possibility of being a particularly suitable biomarker for therapeutic strategy, the organoid-T cell co-culture system was used. Next, the HCC organoid and T cell co-culture system was established to assess the antitumor activity of CD39+ HBVs-CAR-T cells. The CD39+/− HBVs-CAR-T cells generated from PBMCs were first sorted and then co-cultured with HCC tumor organoids at the effector:target ratio E:T = 10:1 for 24 h separately. Sorting efficacy was >95% by FACS (Figure S3). Each group of CAR-T cells was labeled with CellTrace Far Red, and the apoptotic cells were labeled with caspase3/7 probe fluorescence (green). A more significant apoptosis was observed in CD39+ HBVs-CAR-T cell-treated organoids in comparison with that in CD39−HBVs-CAR-T or the control HIV-1 GP120-CAR-T cells (Figures 2A–2C). Meanwhile, we further identified that IFN-γ and CD107a were significantly upregulated on CD39+ HBVs-CAR-T cells (Figures 3A–3C). These data suggested that CD39+ HBVs-CAR-T cells effectively identified and induced apopotosis of HCC organoids. To determine whether CD39 act as a protein with an important pathway in an antitumor response or whether it is only an underlying molecular phenotype of effector T cells, we knocked down CD39 on HBVs-CAR-T cells by shRNA and evaluated the level of cytotoxic activity. Accordingly, inhibiting CD39 expression resulted in the partial dysfunction of HBVs-CAR-T cells when cocultured with HBV+ HCC organoids (Figure S4). However, the expression of multiple checkpoints such as PD-1, Tim-3, and Lag-3 were also increased on CD39+ HBVs-CAR-T cells, which may have detriment effects on the immune response (Figure 3D). In conclusion, we demonstrated in this study that CD39 expressed on HBVs-CAR-T cells induced stronger cytotoxic T lymphocyte activity.
Figure 2.

CD39+ HBVs-CAR-T cells exerted potent antitumor activity in the HBV+ HCC organoid-T cell co-culture system
(A) The CD39+ and CD39− HBVs-CAR T cells were sorted by FACS and co-cultured with autologous tumor organoids for 24 h separately. HBVs-CAR-T cells (red) were labeled with CellTrace Far Red and apoptotic cells (green) were labeled with caspase3/7 probe. Real-time Biotech imaging system was used to take 1 picture per hour for 24 h. GP120-CAR-T cells served as NC. Scale bars, 300 μm. (B) Summaries of quantitative statistics of HBVs-CAR-T cells (red) and apoptotic cells (green). Apoptotic cells (green) and infiltrating CAR-T cells (red) inside the area of organoids were calculated by the Spot function of Imaris software based on the size and intensity threshold. The initial number of infiltrating CAR-T cells at 0 h was defined as 0. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005 (1-way ANOVA). All data are means ± SEMs. Error bars represent SEMs of 3 view fields from each patient. (C) Representative flow cytometry analysis of organoid apoptosis by caspase3/7 probe (FITC).
Figure 3.

The CD39+ HBVs-CAR-T cells exerted an enhanced effector function and a downregulation of inhibitory receptors
(A) Representative flow cytometry plots of CD107a and IFN-γ gated on CD39+/− HBVs-CAR-T cells after being co-cultured with autologous tumor organoids. GP120-CAR-T cells served as NC. (B and C) Relative quantification of IFN-γ and CD107a expression of CD39+/− CAR-T cells. Error bars represent SEMs of 3 biological replicates. (D) Flow cytometry analysis of inhibitory receptor expression of CD39+/− HBVs-CAR T cells.
Downregulation of checkpoints enhanced antitumor activity of CD39+ CAR-T cells
As several checkpoint molecules were highly expressed on CD39+ HBVs-CART cells, we constructed PTL-HBVs-CAR moiety, which included a combination of shRNAs targeting PD-1, Tim-3, and Lag-3 on the basis of HBVs-CAR-T cells for possibly reversing the inhibitory effects on CD39+ HBVs-CAR-T cells28 (Figure 4A). We verified that the shRNA cluster of PD-1, Tim-3, and Lag-3 significantly downregulated the mRNA expression of target genes in CD39+ HBVs-CAR-T cells (Figures S5A and S5B).
Figure 4.

The downregulation of inhibitory receptors enhances antitumor activity of CD39+ HBVs-CAR-T cells
(A) Schematic representation of the lentiviral vectors carrying a HBVs-specific CAR moiety and a cluster of sh-PD-1, sh-Lag-3, and sh-Tim-3. The second structure of this PTL-shRNA cluster was derived from the miR-106b cluster in human genome. (B) CD39+/− HBVs-CAR-T and CD39+/− PTL-HBVs CAR-T were sorted and co-cultured with autologous tumor organoids for 24 h separately. CD39+ PTL-HBVs-CAR-T cells exerted the strongest infiltrating ability compared to CD39+ HBVs-CAR-T cells and CD39-CAR-T cells. CAR-T cells were labeled with CellTrace Far Red (red) and apoptotic cells were labeled with caspase3/7 probe (green). Real-time Biotech imaging system was used to take 1 picture per hour. Scale bar, 300 μm. (C) Representative flow cytometry analysis of organoids apoptosis by caspase3/7 probe (FITC). (D) Summaries of quantitative statistics of (B) were shown. Apoptotic cells (green) and organoid-infiltrated CAR-T cells (red) were calculated by the Spot function of Imaris software based on the size and intensity threshold. The initial number of infiltrating CAR-T cells at 0 h was defined as the base point. ∗∗p < 0.005, ∗∗∗p < 0.0005 (1-way ANOVA). All data are means ± SEMs. Error bars represent SEMs of 3 view fields from each patient.
To validate whether the inhibitory receptors downregulated and CD39+ PTL-HBVs-CAR-T cells exhibited stronger antitumor efficacy, the co-culture system containing CAR-T cells and HCC organoids was applied. The PTL-HBVs-CAR-T and HBVs-CAR-T cells were expanded in vitro for 14 days and CD39+/− cells were sorted by FACS and co-cultured with HCC organoids, respectively (E:T = 10:1). The inhibitory receptor-downregulated CD39+ PTL-HBVs-CAR-T cells showed much stronger antitumor organoid activity than the CD39+/− HBVs-CAR-T cells without the shRNA cluster (Figure 4B). The FACS data further confirmed the stronger efficiency of inducing apoptosis by CD39+ PTL-HBVs-CAR-T cells (Figure 4C). The image analysis by Imaris software also revealed the elevated antitumor activity and tumor-infiltrating ability of CD39+ PTL-HBVs-CAR-T cells (Figure 4D). IFN-γ and CD107a expression in each group of CAR-T cells was tested by flow cytometry. Stronger efficacy was observed in CD39+ PTL-HBVs-CAR-T cells compared to that of CD39+ or CD39− HBVs-CAR-T cells without the shRNA cluster (Figures 5A and 5B).
Figure 5.

CD39+ HBVs-CAR-T cells and CD39+ PTL-HBVs-CAR-T cells exerted potent antitumor activity
(A) Representative flow cytometry analysis of CD107a and IFN-γ gated on CD39+/− CD8+ PTL-HBVs-CAR-T and HBVs-CAR-T cells after being co-cultured with autologous tumor organoids. (B) Relative quantification of IFN-γ production and CD107a expression in CD39+/− PTL-HBVs-CAR-T and HBVs-CAR-T cells. Error bars represent SEMs of 3 biological replicates.
The CD39+ CAR-T cells exerted potent antitumor activity in the patient-derived xenograft (PDX) model
To verify CD39 acted as a key factor contributing to the increased antitumor activity of CAR-T cells, we further constructed the HCC PDX model using HCC organoids. To observe the antitumor activity of CD39+ or CD39− HBVs-CAR-T cells in vivo, 6- to 8-week-old female NSG mice were inoculated subcutaneously (s.c.) into the right flank with HCC organoids with Matrigel. The CD39+/− subset of CAR-T cells and the control CAR-T cells were administered into PDX mice via a single intravenous (i.v.) injection. Much stronger antitumor activities of CD39+ PTL-HBVs-CAR-T cells were observed (Figures 6A and 6B). Notably, the infiltration and IFN-γ secretion of CD39+ CAR-T cells significantly improved in the xenograft tumor tissue compared to CD39− CAR-T cells (Figures 6C–6E). In addition, the infiltration, IFN-γ secretion, and CD39 frequency of sorted CD39+ CAR-T cells were higher than unsorted CAR-T cells in vivo (Figure S6). Given the important role of CD39 in HBVs-CAR-T cells, it was reasonable to assume that CD39 may be used as a marker to identify the active CAR-T cell population.
Figure 6.

CD39+ HBVs-CAR-T cells and CD39+ PTL-HBVs-CAR-T cells exerted potent antitumor activity in the PDX model
(A and B) HCC organoids at 5 × 106 cells were inoculated s.c. into NSG mice. Fifteen days later, various CAR-T cells were infused via tail injection, and GP120-CAR-T cells (NC-CAR-T cells) served as NC. All of the tumors were measured every 5 days (n = 5). (C–E) At day 40, tumors were resected from the mice, followed by digestion and FACS analysis. The percentage of infiltrating CAR-T cells in tumor (C and D) and the percentages of IFN-γ+ cells in tumor-infiltrating CAR-T cells (E) were shown among different groups. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005 (1-way ANOVA). All data are means ± SEMs.
CD39+ personalized tumor-reactive CD8+ T cells exerted potent antitumor activity
To test whether CD39 also identified effector function in tumor-reactive CD8+ T cells, HCC organoids were generated and co-cultured with autologous PBMCs, as previously described.16 Co-stimulation signals were sustained to support T cells proliferation by interleukin-2 (IL-2), anti-CD28, and anti-PD-1 antibodies (Abs). The expanded CD39+ or CD39−CD8+ T cells were sorted by FACS and co-cultured with HCC organoids separately (Figures 7A and 7B). Subsequently, we used the organoid-T cell co-culture platform to assess the tumor reactivity of CD39+/− CD8+ T cells. To determine this, CD39+/− CD8+ T cells after sorting were co-cultured with autologous tumor organoids for 24 h. To visualize the antitumor immune response, each group of CD8+ T cells were labeled by CellTrace Far Red, while the apoptotic cells were labeled with caspase3/7 probe fluorescence (green). Stronger apoptosis was also observed in organoids treated with CD39+ CD8+ T cells rather than in organoids treated with CD39− CD8+ T cells (Figures 7C–7F). Enhanced effector function was also observed in the CD39+ sub-population of tumor-reactive CD8+ T cells (Figures 7G–7I). These data suggested that the CD39 can serve as the biomarker to identify the personalized tumor-reactive CD8+ T cells. These works also showed that it is feasible to induce CD39+ personalized tumor-reactive CD8+ T cells by an organoid-PBMC co-culture platform, which could provide a suitable method to efficiently maintain and expand the tumor-specific T lymphocyte in vitro environment.
Figure 7.

CD39+ personalized tumor-reactive CD8+ T cells generated from organoid-T cell co-culture system exerted stronger antitumor activity
(A) Experimental design. HCC samples were obtained from a needle biopsy of primary tumors. Organoids were established and co-cultured with autologous peripheral blood lymphocytes (PBLs). After 1 week of co-culture, CD39+/− tumor-reactive CD8+ T cells were expanded and sorted by FACS separately. CD39+/− CD8+ T cells were then co-cultured with autologous tumor organoids for 24 h. The antitumor effect was observed by measuring the apoptotic cells within the tumor organoids. (B) Frequency of CD39 gated on tumor-reactive CD8+ T cells after 1 week of co-culture was evaluated by flow cytometry. No organoids added was used as NC. (C) Tumor-reactive CD8+ T cells (red) were labeled with CellTrace Far Red and apoptotic cells (green) were labeled with caspase3/7 probe. Real-time Biotech imaging system was used to take 1 image per hour for 24 h. Representative images for 1 representative patient were shown. Scale bar, 300 μm. (D and E) Summaries of quantitative statistics of (C). Apoptotic cells (green) and infiltrating tumor-reactive CD8+ T cells (red) inside the area of organoids were calculated by the Spot function of Imaris software based on the size and intensity threshold. The initial number of infiltrating T cells at 0 h was defined as 0. ∗∗∗p < 0.0005 (1-way ANOVA). All data are means ± SEMs. Error bars represent SEMs of 3 view fields from each patient. (F) Representative flow cytometry analysis of organoids apoptosis by caspase3/7 probe (FITC). (G–I) Representative flow cytometry analysis and relative quantification of IFN-γ production and CD107a expression of CD39+/− CD8+ T cells. Error bars represent SEMs of 3 biological replicates.
Discussion
Our work developed a novel HCC organoid-T cell co-culture system to assess the antitumor activity of several cellular immunotherapies. Previously, the individualized organoid-specific system was reported to provide a concept that tumor organoids can be used to establish individualized ex vivo systems.29 The organoid-T cell co-culture platform of HCC with high sensitivity and stability was well tested in accordance with previous studies.16,30 With this system, we verified CD39 as a promising marker of effector function for HBVs-CAR-T cells and personalized tumor-reactive CD8+ T cells, which represented stronger antitumor activity.
CD39 is an extracellular ATP hydrolase enzyme expressed by CD8+ T cells. Although some studies suggested that it was an inhibitor receptor like PD-1,31, 32, 33 our data in this report and other related works have demonstrated that CD39 could serve as an effector marker.25,26,30,34 Importantly, Simoni et al.26 found that CD39 was an key molecule to distinguish bystander CD39−CD8+ T cells from tumor-specific CD39+ CD8+ T cells, which were mainly CD45RO+CCR7− T cells and showed a TEM phenotype, while bystander CD39-CD8+ T cells showed a naive and TCM phenotype. According to our previous study, Liu et al.30 found that CD39+ PD-1intCD8+ T cells acted as an effector cytotoxic T lymphocyte in vivo, while CD39+PD-1highCD8+ T cells were an exhaustion phenotype, demonstrating that CD39 was partially close to activation molecules and not the same as PD-1. This results further verified our results that knockdown multiple checkpoints enhanced the antitumor activity of CD39+ CAR-T. In our study, both CD39+ HBVs-CAR-T cells and CD39+ personalized tumor-reactive CD8+ T cells exhibited the effector phenotype, but checkpoints such as PD-1, Tim-3, and Lag-3 were highly expressed on these subsets. According to our previous study, the triple downregulation of inhibitory receptors could enhance the effector function and infiltration of CAR-T cells.28 In the present report, the inhibitory receptors downregulating CD39+ PTL-HBVs-CAR-T cells were developed and exhibited significant stronger antitumor organoid activity compared to CD39+ or CD39− HBVs-CAR-T cells without the shRNA cluster.
To explore the underlying mechanism of the CD39 molecule, we constructed CD39-downregulated HBVs-CAR-T cells. The data showed that knockdown CD39 expression on CD8+ T cells decreased the cytotoxicity activity in vitro, which was in accordance with Li et al.35 They found that the antitumor efficacy of immunotherapy was completely host CD39+CD8+ T lymphocyte dependent. All of these data demonstrated that CD39 was an important molecule to positively regulate the antitumor activity of CD8+ T cells, not simply a molecular phenotype.
Although CD39+ HBVs-CAR-T cells did not display the typical characteristics of the exhausted phenotype in the co-culture system for maintaining the effector function, the downstream molecule CD73 induced the production of adenosine, which can lead to immunosuppression and is regarded as a marker of tumor-reactive T cells.32 Triple inhibitory receptors downregulating CD39+ HBVs-CAR-T cells further enhance the antitumor effect in HCC organoids and PDX models. These results indicate that CD39 is more likely an effector marker than an inhibitory molecule.
Although the organoid model is the best in vitro model to maintain the complex tumor microenvironment, its main defect is that its myeloid cells and stromal elements cannot be preserved long term through passage. We were trying to simulate the tumor microenvironment in this study, so the organoid samples we used were less than five generations, but the limitation of this model still could not be neglected; that is, the importance of the PDX model cannot be replaced by tumor organoids, especially in vivo. The construction of organoids with complex tumor microenvironments, including the myeloid and stromal elements, urgently require future research, which will also improve the patient-derived orthotopic xenograft.
Cellular immunotherapy with CAR-T, T cell receptor-T (TCR-T), antigen-specific T, or cytokine-induced killer (CIK) cell specific for HBV or HBV+ HCC, has provided certain therapeutic effects in several previous clinical trials.6,36, 37, 38, 39, 40, 41 Due to the immunosuppressive microenvironment being potentially important barriers in HCC immune cell therapy, the effects of CAR-T immunotherapy for HCC thus far are unsatisfactory. In our study, T cells derived from PBMCs were constructed to HBVs-CAR-T cells by transducing with CAR, or personalized tumor-reactive CD8+ T cells by co-culturing with autologous tumor organoids, CD39+ subsets in both two modified cells exerted better antitumor activity. Notably, adoptive transferring with CD39+ PTL-HBVs-CAR-T cells showed a potent therapeutic effect in HCC PDX mouse models, suggesting that CD39+ CD8+ T cells may act as a major cellular immunotherapy subset for HCC. Therefore, CD39 can serve as a biomarker to further enrich effective cytotoxic T cells or evaluate the therapeutic effects of cellular immunotherapy with T cells, which could contribute to improving the efficiency of antitumor immunotherapy. Based on our previous study, HCC patients may receive CAR-T cell immunotherapy treatment as soon as possible after peripheral blood collection, which has been shown to be a great clinical application prospect in the cellular immunotherapy of HCC.
Materials and methods
Cell lines
HEK293T, HepG-2.2.15, and HepG2 cell lines were obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Gibco). All cell culture media contained 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. Cell lines were routinely tested for excluding mycoplasma contamination.
Isolation and culture of primary human T lymphocytes
PBMCs were derived from samples obtained from healthy volunteers from anonymous buffy coats of healthy donors (Guangzhou Blood Center, Guangzhou, China) by Ficoll-Hypaque gradient separation. Primary human CD8+ T cells were negatively purified with magnetic beads to a purity of >98% from PBMCs with enrichment set DM (BD-IMag). T lymphocytes were activated by anti-CD3 (R&D Systems, Minneapolis, MN, USA) and anti-CD28 at (R&D Systems) Abs 1 μg mL−1 and infected in retronectin-coated plates (Takara Bio, Shiga, Japan). The transduced T cells were expanded in the conditioned medium containing 90% RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco), 0.1 mM nonessential amino acids (Gibco), 2 mM GlutaMAX (Gibco), and 0.05 mM 2-mercaptoethanol at an initial concentration of 1 × 106 cells mL−1. Cells were fed twice per week with recombinant IL-2 (10 ng mL−1) (R&D Systems).
Construction of 3rd generation CAR-encoding lentiviral vector
The anti-HBV pre-S1 scFv was derived from parental scFv clone 1E4.42 As shown in Figure 1A, the scFv region was fused with the endodomains of CD28 (nucleotides 460–660; GenBank: NM_006139.3), CD137 (nucleotides 640–765; NM_001561.5), and CD3ζ (nucleotides 160–492; NM_198053.2) in tandem with a (GGGS)3 sequence inserted between each signaling domain. GP120-CAR, specifically targeting the gp120 antigen of HIV-1, was used as NC-CAR-T cells. Schematic representations of lentiviral vectors carrying a HBVs-specific CAR moiety and a cluster of shRNAs targeting PD-1, Tim3, and Lag-3, which is derived from the miR-106b cluster, are shown in Figure 4A. Sequences of shRNA are listed in Figures S3 and S4.
Transduction of recombinant lentiviral particles
For the production of pseudotyped lentiviral supernatant, on the day before transduction, HEK293T cells were seeded at 8 × 106 cells per 100-mm dish. Twenty-four hours later, the pseudoviruses were generated by co-transfecting HEK293T cells with plasmids encoding various CAR moieties (13.5 μg), pMD.2G encoding VSV-G envelope (7.5 μg), and a packaging vector psPAX2 (16.5 μg) using the phosphate transfection system following the manufacturer’s instructions. Supernatants were harvested after 48 h and filtered through a 0.45-μm membrane to remove cell debris. Pseudoviruses were concentrated by ultracentrifugation (Optima XE-100, Beckmann Coulter, Indianapolis, IN, USA) at 827,000 × g for 2 h at 4°C. Then, the activated CD8+ T lymphocytes were transduced with lentiviral supernatants using retronectin-coated plates, with polybrene (TR-1003-G, Sigma-Aldrich, St. Louis, MO, USA) at 8 μg mL−1, followed by centrifugation for 90 min at 350 × g and incubation at 37°C. Twelve hours later, the recombinant viruses were removed, and T cells were expanded in the conditioned medium, as described above. The genetically modified T cells were maintained in complete T cell medium in the presence of IL-2 (fed twice per week, 10 ng mL−1) and used for functional assay 14 days after transduction.
Patients and samples collection
Between 2018 and 2020, a cohort of patients with HCC in the First Affiliated Hospital of Sun Yat-sen University underwent needle biopsies and blood collection. The project was approved with the patient’s informed consent and ethical approval (201842). No treatment was given to patients when samples were collected, and patients had received surgery or no therapy previously. Three patients were selected to receive the tumor organoid culture and personalized tumor-reactive T cell generation.
HCC tumor organoid culture
The procedures were developed by following previous procedures with minor modifications.29,43 Briefly, tumor tissues derived from needle biopsies were mechanically dissociated into small pieces using needles and embedded in Matrigel (matrix basement membrane growth factor reduced, Corning #354230, Corning, Corning, NY, USA). After Matrigel was solidified for 20 min at 37°C, cells were overlaid with human HCC organoid medium. Human HCC organoids medium was composed of Advanced DMEM/F12 (Gibco) supplemented with 2 mM Ultra glutamine I (Lonza, Basel, Switzerland), 10 mM HEPES [Gibco], and 100/100 U/mL penicillin/streptomycin [Gibco]), 30% Wnt3a-conditioned medium, 1 × B27 supplement without vitamin A (Gibco), 1 × N2 supplement (Gibco), 2 mM N-acetylcysteine (Sigma-Aldrich), 10 mM nicotinamide (Sigma-Aldrich), 10 nM recombinant human [Leu15]-gastrin I (Sigma-Aldrich), 50 ng/mL human recombinant epidermal growth factor (EGF) (Peprotech, Rocky Hill, NJ, USA), 100 ng/mL human recombinant FGF (fibroblast growth factor)-10 (Peprotech), 50 ng/mL human recombinant human growth factor (HGF) (Peprotech), 50 ng/mL human recombinant R-spondin1 (NovoProtein, Summit, NJ, USA), 5 μM A83-01 (Sigma-Aldrich), and 10 μM MY-27632 (Sigma-Aldrich). Wnt3a-conditioned medium was from L-Wnt3a cells. Organoids were passaged approximately every week by incubation in TrypLE Express (Gibco) for 5–10 min at 37°C to dissociate organoids to single cells and replate in fresh Matrigel. Organoids were cryopreserved in 10% FBS/DMSO and organoids < passage 30 were used in the experiments. All of the organoids were regularly checked for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza).
WES sequence for organoid
Collected tumor/organoids and blood samples were sent to GenomiCare Biotechnology (Shanghai, China), for targeted next-generation sequencing (NGS) analysis. The extracted DNA was sheared using a Covaris L220 sonicator, the exome DNA was captured using the SureSelect Human All Exon V7 kit (cat# 5991-9039EN, Agilent Technologies, Santa Clara, CA, USA), prepared to library using the SureSelectXT Low Input Target Enrichment and Library Preparation system (cat. no. G9703-90000, Agilent), and sequenced on an Illumina NovaSeq-6000 sequencer (Illumina, San Diego, CA, USA) to generate 150 × 150-bp paired end reads. Image analysis and base calling were completed using onboard RT3 software (Illumina). After removing adapters and low-quality reads, the reads were aligned to NCBI human genome reference assembly hg19 using the Burrows-Wheeler Aligner alignment algorithm and further processed using the Genome Analysis Toolkit (GATK, version 3.5), including the GATK Realigner Target Creator to identify regions that needed to be realigned. Somatic single-nucleotide variants (SNVs), Indel, and copy-number variation (CNV) were determined using MuTect/ANNOVAR/dbNSFP31, VarscanIndel, and CNVnator software, respectively, as reported previously.44
RNA isolation and qPCR analysis
Total RNA was isolated with Trizol reagent (Life Technologies, Carlsbad, CA, USA) and served as the template for preparing cDNA using a PrimeScript reverse transcription (RT) reagent kit (TaKaRa, Kusatsu, Japan) according to the manufacturer’s instructions. qPCR was performed with the SYBR Premix ExTaq Kit (TaKaRa) on a CFX96 Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was measured as an endogenous control.
Flow cytometry
To evaluate the proportion of HBVs+ tumor cells in organoids, fluorescein isothiocyanate (FITC)-hepatitis B surface antigen (#ab32914 Abcam, Cambridge, UK) was used to stain and FITC-Rabbit immunoglobulin G (IgG) (#ab37406 Abcam) was regarded as the isotype control. Organoids were isolated from Matrigel and stained in FACS buffer (PBS, supplemented with 1% FBS and 0.01% NaN3). The staining was performed according to the manufacturer’s protocol. Cells were washed twice before flow cytometric recording.
For verifying and sorting CD39+/CD39− CD8 T cells from sorted CD8 by magnetic beads, APC Anti-human CD8 (#344721, Biolegend, San Diego, CA, USA), Brilliant Violet 421 Anti-human CD39 (#328214, Biolegend) were used to stain, and Pacific Blue Mouse IgG1 (#400131, Biolegend) was considered an isotype control. The staining was performed according to the manufacturer’s protocol. Cells were washed twice before flow cytometric detecting and sorting. Sorted CD39+/CD39− T cells were used for further assays.
For the evaluation of the tumor reactivity of T cells, 105 sorted CD39+/CD39− T cells were co-cultured with tumor organoids for 12 h. Golgi-Plug (1:1,000, BD Biosciences, San Jose, CA, USA) and Golgi-Stop (1:1,500, BD Biosciences) was added after 1 h. Cells were washed twice in FACS buffer and stained with the following antibodies: PE Anti-human CD107a (#121624, Biolegend) for 30 min at 4°C. Cells were washed twice in FACS buffer, fixed, and stained for FITC intracellular IFN-γ (#502506, Biolegend) using the Cytofifix/Cytoperm kit (BD Biosciences), according to the manufacturer’s instructions.
For the evaluation of the tumor reactivity of the sub-population of CAR-T cells, 105 CAR-T cells were co-cultured with tumor organoids for 12 h. Golgi-Plug (1:1,000, BD Biosciences) and Golgi-Stop (1:1,500, BD Biosciences) was added after 1 h. Cells were washed twice in FACS buffer and stained with the following antibodies: Brilliant Violet 421 Anti-human CD39 (#328214, Biolegend), PE Anti-human PD-1 (#367403, Biolegend), APC Anti-human Tim-3 (#345011, Biolegend), and PerCP/Cy5.5 Anti-human Lag-3 (#369312, Biolegend) for 30 min at 4°C. Cells were washed twice in FACS buffer, fixed, and stained for FITC intracellular IFN-γ (#502506, Biolegend) using the Cytofifix/Cytoperm kit (BD Biosciences), according to the manufacturer’s instructions.
Enzyme-linked immunosorbent spot (ELISpot)
For the ELISpot assay, CAR-T cells were mixed with target cells (104 cells) at E:T = 4:1 and then added to the anti- IFN-γ antibody-precoated plates from the human IFN-γ ELISpot assay kit (DKW22-1000-096 s; Dakewei, Shenzhen, China), along with a negative control (effector CD8+ T cells alone) or positive control (phytohemagglutinin [PHA] stimulation). Plates were incubated for 20 h in a humidified atmosphere containing 5% CO2 at 37°C. The ELISpot assays were then performed according to the manufacturer’s instructions. The plates were scanned by the S6 ultra immunoscan reader (Cellular Technology, Shaker Heights, OH, USA), and the number of IFN-γ+ T cells was calculated by ImmunoSpot software (version 5.1.34, Cellular Technology).
Cytotoxicity assay
The ability of T cells to kill tumor target cells was measured by lactate dehydrogenase (LDH) assay. Briefly, CAR-T cells were co-cultured with target cells at different ratios (from 16:1 to 1:1) for 24 h in a 96-well plate (V bottom). Then, LDH release was measured by the CytoTox96 nonradioactive cytotoxicity assay (G1781, Promega, Madison, WI, USA) according to the manufacturer’s instructions. Absorbance values of wells containing effector cells alone and target cells alone were detected and subtracted as the background from the values of the co-cultures. Wells containing target cells alone were mixed with a lysis reagent for 30 min at 37°C, and the resulting luminescence was set as 100% lysis. Cytotoxicity was calculated by using the following formula: %Cytotoxicity = (Experimental − Effector spontaneous − Target spontaneous)/(Target maximum − Target spontaneous) × 100%.
Organoid killing assay
Organoids were isolated from Matrigel, and part of the organoids were dissociated into single cells and counted to infer the number of tumor cells per tumor organoid to allow co-culture of organoids and T cells at a 10:1 E:T. Tumor organoids were resuspended in X-Vivo15 medium and seeded in triplicate on a flat-bottomed plate with 1 × 105 autologous CD39+CD8+T cells or CD39−CD8+ T cells obtained by sorting. To facilitate visualization, T cells were previously stained with 1 mM CellTrace Far Red (Invitrogen) in PBS for 20 min at 37°C, followed by blocking with human serum and washing in PBS. At the start of the co-culture, a green fluorescent caspase3/7 probe that binds DNA upon cleavage by caspase3/7 (Invitrogen) was added at a 1:2,000 dilution to visualize cell apoptosis. After 24 h of co-culture, micrographs were taken.
For the quantification of the results, the images of organoids were further analyzed with Imaris software (version 7.4) (Bitplane, Zurich, Switzerland) using the Spot function to locate and enumerate T cells and apoptosis cells inside the organoid threshold. The absolute numbers of T cells and apoptotic cells spot per square millimeter of organoids areas were statistically analyzed with Imaris software (version 7.4) (Bitplane) using the Spot function to locate and enumerate CAR-T cells or apoptotic cells, based on size and intensity threshold. The initial infiltration of T cells inside the organoids was considered to be zero. Finally, organoids were dissociated into single cells with TrypLE Express until the organoids were fully dissociated. Cells were washed in FACS buffer for FACS.
HCC-PDX mouse models
We used the NSG (NOD-PrkdcscidIL2rgtm1/Bcgen, Beijing Biocytogen, Beijing, China) mouse model to build HCC PDX.29 All of the mouse experiments strictly followed the ethical regulations and approved.
To observe the antitumor activity of CAR-T cells in vivo, 6- to 8-week-old female NSG mice were inoculated s.c) in the right flank with 5 × 106 HCC organoid-derived cells in 100 μL f 50:50 Matrigel (Corning) and PBS. On day 15, 1 × 106 transduced CAR-T cells (in 200 μL PBS) were adoptively transferred into PDX mice via the tail vein and all of the mice were intraperitoneally (i.p. injected with IL-2 at 1 μg per mouse every 5 days, and the volume of tumors was measured by two-dimensional measurements (millimeters): volume of tumor = length × width2/2. Tissues were digested with collagenase type IV (2 mg mL−1, Sigma-Aldrich) at 37°C for 30 min, and tumor-infiltrating T cells were separated by centrifugation on a discontinuous Percoll gradient (Haoyang, China).
Genome-wide transcriptional profiling
The CD107a+ HBVs-CAR-T and CD107a− HBVs-CAR-T cells were collected by FACS sorting. Total RNA was extracted from each population of cells using the Trizol reagent kit (Invitrogen), according to the manufacturer’s protocol. RNA quality was assessed on an Agilent 2100 Bioanalyzer (Agilent Technologies) and checked using RNase-free agarose gel for electrophoresis. After total RNA was extracted, eukaryotic mRNA was enriched by oligo(dT) beads, while prokaryotic mRNA was enriched by removing rRNA using the Ribo-Zero Magnetic Kit (Epicenter, Madison, WI, USA). Then, the enriched mRNA was fragmented into short fragments using fragmentation buffer and reverse transcribed into cDNA with random primers. Second-strand cDNA was synthesized by DNA polymerase I, RNase H, dNTP, and buffer. The cDNA fragments were purified with QiaQuick PCR extraction kit (QIAGEN, Venlo, the Netherlands), end repaired, poly(A) added, and ligated to Illumina sequencing adapters. The ligation products were size selected by agarose gel electrophoresis, PCR amplified, and sequenced using Illumina HiSeq2500 by Gene Denovo Biotechnology (Guangzhou, China). Reads obtained from the sequencing machines included raw reads containing adapters or low-quality bases, which will affect the following assembly and analysis. Thus, to obtain high-quality clean reads, reads were further filtered by fastp [1] (version 0.18.0). The parameters removed reads containing >10% of unknown nucleotides (N) and removed low-quality reads containing >50% low-quality (Q value ≤ 20) bases.
H&E and immunofluorescence
Organoid and tissue samples were fixed, processed, and stained according to standard procedures. Briefly, organoid and tissue were fixed with neutrally buffered 4% formaldehyde, and applied to hematoxylin and eosin (H&E) staining conducted by Biopathology Institute (Servicebio, Wuhan, China). For immunofluorescence, the following primary antibodies were used for automated staining on a Benchmark XT device: AFP (#ab3980, Abcam), HBVs-FITC (#ab32914, Abcam), and rabbit IgG (FITC)-Isotype Control (#ab37406, Abcam). Alexa Fluor 594-conjugated goat anti-human IgG Fc (ab97005, Abcam) was used as secondary antibody. DAPI (D1306, Thermo Fisher Scientific, Waltham, MA, USA) was used for the staining of nucleus. Images of both H&E staining samples were obtained using a Leica DM6000B microscope. Fluorescent signals were detected using a laser ZEISS LSM 800 scanning confocal microscope.
Statistical analysis
GraphPad Prism 5.0 (GraphPad, San Diego, CA, USA) was used for all of the statistical analyses. Data samples were compared using a 2-tailed Student’s t test, and a p <0.05 was considered significant.
Study approval
This study was conducted with the approval of the First Affiliated Hospital of Sun Yat-sen University. All of the experimental methods and clinical treatment were carried out in accordance with the approved guidelines. All of the patients signed an informed consent for scientific research statement.
Acknowledgments
The authors thank Qianyang Biomedical Research Institute (Guangdong, China) for their assistance in the bioinformatics sequencing and analysis. This work was supported by grants from the National Natural Science Foundation of China (nos. 81671797, 81971719, and 82003252); the Major Scientific and Technological Project of Guangdong Province (no. 2017B030308006); the Major Program for Tackling Key Problems of Guangzhou City (no. 201704020144); the China Postdoctoral Science Foundation (2019M662853); the Guangdong Key Project in “Development of New Tools for Diagnosis and Treatment of Autism” (2018B030335001); and the Guangzhou Key Project in “Early Diagnosis and Treatment of Autism Spectrum Disorders” (202007030002).
Author contributions
F.Z. and J.T. wrote the manuscript. J.L., F.Z., and J.T. designed the experiments. F.Z., J.T., and T.L. performed the experiments and analyzed the data. J.L. and H.Z. supervised the project.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.01.021.
Contributor Information
Hui Zhang, Email: zhangh92@mail.sysu.edu.cn.
Jiaping Li, Email: lijiap@mail.sysu.edu.cn.
Supplemental information
References
- 1.Villanueva A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 2.Tella S.H., Mahipal A., Kommalapati A., Jin Z. Evaluating the Safety and Efficacy of Nivolumab in Patients with Advanced Hepatocellular Carcinoma: Evidence to Date. OncoTargets Ther. 2019;12:10335–10342. doi: 10.2147/OTT.S214870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng A.L., Hsu C., Chan S.L., Choo S.P., Kudo M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020;72:307–319. doi: 10.1016/j.jhep.2019.09.025. [DOI] [PubMed] [Google Scholar]
- 4.Overman M.J., McDermott R., Leach J.L., Lonardi S., Lenz H.J., Morse M.A., Desai J., Hill A., Axelson M., Moss R.A. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–1191. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel S.A., Minn A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity. 2018;48:417–433. doi: 10.1016/j.immuni.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qasim W., Brunetto M., Gehring A.J., Xue S.A., Schurich A., Khakpoor A., Zhan H., Ciccorossi P., Gilmour K., Cavallone D. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J. Hepatol. 2015;62:486–491. doi: 10.1016/j.jhep.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Fu Y., Urban D.J., Nani R.R., Zhang Y.F., Li N., Fu H., Shah H., Gorka A.P., Guha R., Chen L. Glypican-3-Specific Antibody Drug Conjugates Targeting Hepatocellular Carcinoma. Hepatology. 2019;70:563–576. doi: 10.1002/hep.30326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng M., Mo Y., Wang Y., Wu P., Zhang Y., Xiong F., Guo C., Wu X., Li Y., Li X. Neoantigen vaccine: an emerging tumor immunotherapy. Mol. Cancer. 2019;18:128. doi: 10.1186/s12943-019-1055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gehring A.J., Ho Z.Z., Tan A.T., Aung M.O., Lee K.H., Tan K.C., Lim S.G., Bertoletti A. Profile of tumor antigen-specific CD8 T cells in patients with hepatitis B virus-related hepatocellular carcinoma. Gastroenterology. 2009;137:682–690. doi: 10.1053/j.gastro.2009.04.045. [DOI] [PubMed] [Google Scholar]
- 10.Zhou F., Shang W., Yu X., Tian J. Glypican-3: a promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018;38:741–767. doi: 10.1002/med.21455. [DOI] [PubMed] [Google Scholar]
- 11.Shi D., Shi Y., Kaseb A.O., Qi X., Zhang Y., Chi J., Lu Q., Gao H., Jiang H., Wang H. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020;26:3979–3989. doi: 10.1158/1078-0432.CCR-19-3259. [DOI] [PubMed] [Google Scholar]
- 12.Venook A.P., Papandreou C., Furuse J., de Guevara L.L. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist. 2010;15(Suppl 4):5–13. doi: 10.1634/theoncologist.2010-S4-05. [DOI] [PubMed] [Google Scholar]
- 13.Chu C.M., Liaw Y.F. Membrane staining for hepatitis B surface antigen on hepatocytes: a sensitive and specific marker of active viral replication in hepatitis B. J. Clin. Pathol. 1995;48:470–473. doi: 10.1136/jcp.48.5.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Dika I., Khalil D.N., Abou-Alfa G.K. Immune checkpoint inhibitors for hepatocellular carcinoma. Cancer. 2019;125:3312–3319. doi: 10.1002/cncr.32076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitt J.M., Vétizou M., Daillère R., Roberti M.P., Yamazaki T., Routy B., Lepage P., Boneca I.G., Chamaillard M., Kroemer G., Zitvogel L. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44:1255–1269. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Dijkstra K.K., Cattaneo C.M., Weeber F., Chalabi M., van de Haar J., Fanchi L.F., Slagter M., van der Velden D.L., Kaing S., Kelderman S. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell. 2018;174:1586–1598.e12. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Serag H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273.e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lévesque S.A., Kukulski F., Enjyoji K., Robson S.C., Sévigny J. NTPDase1 governs P2X7-dependent functions in murine macrophages. Eur. J. Immunol. 2010;40:1473–1485. doi: 10.1002/eji.200939741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mascanfroni I.D., Yeste A., Vieira S.M., Burns E.J., Patel B., Sloma I., Wu Y., Mayo L., Ben-Hamo R., Efroni S. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol. 2013;14:1054–1063. doi: 10.1038/ni.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schena F., Volpi S., Faliti C.E., Penco F., Santi S., Proietti M., Schenk U., Damonte G., Salis A., Bellotti M. Dependence of immunoglobulin class switch recombination in B cells on vesicular release of ATP and CD73 ectonucleotidase activity. Cell Rep. 2013;3:1824–1831. doi: 10.1016/j.celrep.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Deaglio S., Dwyer K.M., Gao W., Friedman D., Usheva A., Erat A., Chen J.F., Enjyoji K., Linden J., Oukka M. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mascanfroni I.D., Takenaka M.C., Yeste A., Patel B., Wu Y., Kenison J.E., Siddiqui S., Basso A.S., Otterbein L.E., Pardoll D.M. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med. 2015;21:638–646. doi: 10.1038/nm.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chalmin F., Mignot G., Bruchard M., Chevriaux A., Végran F., Hichami A., Ladoire S., Derangère V., Vincent J., Masson D. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. 2012;36:362–373. doi: 10.1016/j.immuni.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Muller-Haegele S., Muller L., Whiteside T.L. Immunoregulatory activity of adenosine and its role in human cancer progression. Expert Rev. Clin. Immunol. 2014;10:897–914. doi: 10.1586/1744666X.2014.915739. [DOI] [PubMed] [Google Scholar]
- 25.Duhen T., Duhen R., Montler R., Moses J., Moudgil T., de Miranda N.F., Goodall C.P., Blair T.C., Fox B.A., McDermott J.E. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018;9:2724. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 27.Drost J., Clevers H. Organoids in cancer research. Nat. Rev. Cancer. 2018;18:407–418. doi: 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- 28.Zou F., Lu L., Liu J., Xia B., Zhang W., Hu Q., Liu W., Zhang Y., Lin Y., Jing S. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019;10:4109. doi: 10.1038/s41467-019-11893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broutier L., Mastrogiovanni G., Verstegen M.M., Francies H.E., Gavarró L.M., Bradshaw C.R., Allen G.E., Arnes-Benito R., Sidorova O., Gaspersz M.P. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017;23:1424–1435. doi: 10.1038/nm.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu T., Tan J., Wu M., Fan W., Wei J., Zhu B., Guo J., Wang S., Zhou P., Zhang H. High-affinity neoantigens correlate with better prognosis and trigger potent antihepatocellular carcinoma (HCC) activity by activating CD39+CD8+ T cells. Gut. 2020 doi: 10.1136/gutjnl-2020-322196. Published online December 1, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Canale F.P., Ramello M.C., Núñez N., Araujo Furlan C.L., Bossio S.N., Gorosito Serrán M., Tosello Boari J., Del Castillo A., Ledesma M., Sedlik C. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8+ T Cells. Cancer Res. 2018;78:115–128. doi: 10.1158/0008-5472.CAN-16-2684. [DOI] [PubMed] [Google Scholar]
- 32.Gupta P.K., Godec J., Wolski D., Adland E., Yates K., Pauken K.E., Cosgrove C., Ledderose C., Junger W.G., Robson S.C. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015;11:e1005177. doi: 10.1371/journal.ppat.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohme M., Schliffke S., Maire C.L., Rünger A., Glau L., Mende K.C., Matschke J., Gehbauer C., Akyüz N., Zapf S. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-infiltrating Lymphocytes. Clin. Cancer Res. 2018;24:4187–4200. doi: 10.1158/1078-0432.CCR-17-2617. [DOI] [PubMed] [Google Scholar]
- 34.Yang R., Cheng S., Luo N., Gao R., Yu K., Kang B., Wang L., Zhang Q., Fang Q., Zhang L. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019;21:2. doi: 10.1186/s13059-019-1921-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X.Y., Moesta A.K., Xiao C., Nakamura K., Casey M., Zhang H., Madore J., Lepletier A., Aguilera A.R., Sundarrajan A. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019;9:1754–1773. doi: 10.1158/2159-8290.CD-19-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wada Y., Nakashima O., Kutami R., Yamamoto O., Kojiro M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology. 1998;27:407–414. doi: 10.1002/hep.510270214. [DOI] [PubMed] [Google Scholar]
- 37.Takayama T., Sekine T., Makuuchi M., Yamasaki S., Kosuge T., Yamamoto J., Shimada K., Sakamoto M., Hirohashi S., Ohashi Y., Kakizoe T. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet. 2000;356:802–807. doi: 10.1016/S0140-6736(00)02654-4. [DOI] [PubMed] [Google Scholar]
- 38.Yu X., Zhao H., Liu L., Cao S., Ren B., Zhang N., An X., Yu J., Li H., Ren X. A randomized phase II study of autologous cytokine-induced killer cells in treatment of hepatocellular carcinoma. J. Clin. Immunol. 2014;34:194–203. doi: 10.1007/s10875-013-9976-0. [DOI] [PubMed] [Google Scholar]
- 39.Lee J.H., Lee J.H., Lim Y.S., Yeon J.E., Song T.J., Yu S.J., Gwak G.Y., Kim K.M., Kim Y.J., Lee J.W., Yoon J.H. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology. 2015;148:1383. doi: 10.1053/j.gastro.2015.02.055. 91.e6. [DOI] [PubMed] [Google Scholar]
- 40.Ma W., Wu L., Zhou F., Hong Z., Yuan Y., Liu Z. T Cell-Associated Immunotherapy for Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2017;41:609–622. doi: 10.1159/000457883. [DOI] [PubMed] [Google Scholar]
- 41.Saied A., Licata L., Burga R.A., Thorn M., McCormack E., Stainken B.F., Assanah E.O., Khare P.D., Davies R., Espat N.J. Neutrophil:lymphocyte ratios and serum cytokine changes after hepatic artery chimeric antigen receptor-modified T-cell infusions for liver metastases. Cancer Gene Ther. 2014;21:457–462. doi: 10.1038/cgt.2014.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park S.G., Jeong Y.J., Lee Y.Y., Kim I.J., Seo S.K., Kim E.J., Jung H.C., Pan J.G., Park S.J., Lee Y.J. Hepatitis B virus-neutralizing anti-pre-S1 human antibody fragments from large naïve antibody phage library. Antiviral Res. 2005;68:109–115. doi: 10.1016/j.antiviral.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 43.Nuciforo S., Fofana I., Matter M.S., Blumer T., Calabrese D., Boldanova T., Piscuoglio S., Wieland S., Ringnalda F., Schwank G. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018;24:1363–1376. doi: 10.1016/j.celrep.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zang Y.S., Dai C., Xu X., Cai X., Wang G., Wei J., Wu A., Sun W., Jiao S., Xu Q. Comprehensive analysis of potential immunotherapy genomic biomarkers in 1000 Chinese patients with cancer. Cancer Med. 2019;8:4699–4708. doi: 10.1002/cam4.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.