

Abstract
The inclusion of genes that control cell fate (so-called suicide, or kill-switch, genes) into gene therapy vectors is based on a compelling rationale for the safe and selective elimination of aberrant transfected cells. Prodrug-activated systems were developed in the 1980s and 1990s and rely on the enzymatic conversion of non-active prodrugs to active metabolites that lead to cell death. Although considerable effort and ingenuity has gone into vector design for gene therapy, less attention has been directed at the efficacy or associated adverse effects of the prodrug systems employed. In this review, we discuss prodrug systems employed in clinical trials and consider their role in the field of gene therapy. We highlight potential drawbacks associated with the use of specific prodrugs, such as systemic toxicity of the activated compound, the paucity of data on biodistribution of prodrugs, bystander effects, and destruction of genetically modified cells, and how these can inform future advances in cell therapies.
Graphical abstract
Gene therapy to treat monogenic conditions and as a component of cancer and cellular therapies is gaining increasing traction. The inclusion of a conditional gene switch deployed to selectively eliminate aberrant or unwanted cells is an attractive strategy. This review describes and compares prodrug-activated systems in gene therapy and advocates for greater focus on prodrug selection and efficacy of targeted cell killing.
Introduction
The development of gene therapy over the last four decades has been driven by increasingly sophisticated molecular techniques. Defined as the transfer of genetic information in order to modify the phenotype of a cell, gene therapy today has wide therapeutic applications for the treatment of inherited and acquired diseases, including cancer. Consequently, gene therapy can be categorized in a number of ways, such as by cellular target, mode of delivery of the genetic material, and the underlying rationale of treatment.
With regard to mode of delivery, cells can be genetically modified in vivo, where vectors carrying the genetic material are targeted to specific cells or tissues, whereas ex vivo approaches involve the removal of target cells from the patient, followed by genetic modification and re-infusion of the engineered product. Vectors used in gene therapy may be viral or non-viral, including plasmids or plasmids combined with liposomes. At present, the two most commonly used vector systems are adenoviruses, or adeno-associated viruses for in vivo gene transfer to post-mitotic cells, and lentiviral vectors for ex vivo approaches where genetic material is transferred to stem/progenitor cells.1,2
Ultimately, the success of gene therapy relies on two main factors: first, on the ability to safely administer genetic material; and second, to achieve adequate expression of the desired gene product at therapeutic levels in specific cells or tissues. With regard to the latter, much research has centered on the optimization of vectors and mode of vector delivery, a topic that is beyond the scope of this review.
In terms of safety, several issues require consideration: an important challenge is the potential for insertional mutagenesis caused by integration of vectors into the host genome and the resultant development of malignancy.3 Although the use of lentiviral vectors reduces the likelihood of integration near known oncogenes or tumor suppressor genes, other mechanisms of disrupted cellular function remain of concern. Moreover, when modifying stem cells, the transduced cells have the potential to last the lifetime of the host, expand in number, and have a persistent clinical effect that over time may be unwanted or harmful.4 In this context, long-term follow-up of patients receiving gene therapy is crucial to understand its long-term effects and the risks for oncogenesis.2
Prodrug-activated gene therapy
Early gene therapy trials carried out for monogenic diseases, such as severe combined immune deficiency (SCID) and alpha 1-antitypsin deficiency, initially focused on replacing a single deficient or mutated gene with a normal copy. However, the safety concerns inherent in cellular therapy were highlighted when 4 of 10 patients treated with gene therapy for SCID developed acute T cell lymphoblastic leukemia as the result of insertional mutagenesis into the host genome.4
One strategy to mitigate potential adverse effects (AEs) caused by genetically modified cells is to include a cell-fate control system, such as a suicide gene, in the transfer vector; thus, a second transgene accompanies the gene of interest and acts as a suicide switch that allows transduced cells to be destroyed when exposed to a particular signal.1,5 Several mechanisms can be used to control cell fate in this way (Figure 1). Prodrug-activated gene therapy (PAGT) strategies aim to enable the selective, on-demand destruction of transduced cells. A suicide transgene that encodes an enzyme is introduced into a target cell, and the transduced cell then becomes able to convert a specific, non-toxic prodrug into a toxic product.3,6 The activated form of the prodrug is usually a small molecule, and therefore also can diffuse along a concentration gradient into surrounding tissues, such as adjacent tumor tissue.6, 7, 8, 9
Figure 1.

Suicide, or kill switch, gene strategies
The final common pathway is destruction of selected cells “on demand” by apoptosis.
(A) Prodrug-mediated approach. Cells are transduced with the gene for an enzyme that converts an inactive prodrug (in the examples shown, GCV or 5-FC) to an active toxic compound.
(B) Monoclonal antibody mediated. Cells transduced with a targetable cell surface marker (in the example, CD20) are selectively targeted with a monoclonal antibody.
(C) Inducible fusion protein. Cells transfected with the gene for a protein that dimerizes on administration of a small molecule (in the example, AP1903) can be selectively induced to undergo apoptosis.
PAGT strategies were initially devised as a safety switch in the event of excess or off-target toxicity in adoptively transferred cells. However, the same mechanism can also be deployed to directly target and destroy any aberrant cells, such as cancer cells that display a vulnerability to the activated prodrug (Figure 2). For example, in patients with solid cancers, the transfer of a prodrug-activated gene alone into tumor cells can be used to target the disease when conventional treatments have failed. In addition to targeted killing of transduced tumor cells using this mechanism, pre-clinical studies have shown a bystander effect on non-transfected cells that can enhance cytotoxicity.9,10
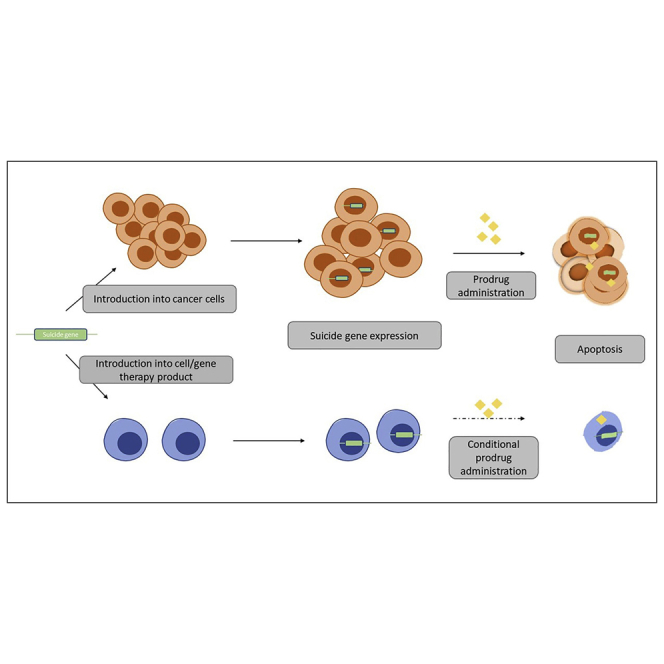
Figure 2.

Schematic of prodrug-activated suicide gene therapy in relevant treatment contexts
In cancer therapy, the introduction of a suicide gene is a primary treatment strategy, in that the suicide gene/prodrug combination targets the tumor directly and to a degree also indirectly through the bystander effect. By contrast, using a PAGT approach with cellular products such as chimeric antigen receptor (CAR) T cells, or alongside the gene of interest in monogenic gene therapy, allows the on-demand/selective destruction of the co-transfected cells in the event of safety concerns or unacceptable side effects/toxicity. In this case, the administration of the prodrug occurs only when the kill switch is to be deployed.
Conversely, in patients receiving allogeneic hematopoietic stem cell transplants (HSCTs), PAGT has been used to attenuate the effects of graft-versus-host disease (GvHD),1, 2, 3, 4, 5 acting as a safety mechanism that can be activated as required when unacceptable toxicity of the cellular product occurs.11
The most common enzymes that have been used in PAGT systems are herpes simplex virus (HSV)-derived thymidine kinase (TK; HSV-TK), cytosine deaminase (CD) from Escherichia coli or yeast, and E. coli-associated nitroreductase (NTR), which render cells sensitive to their respective prodrugs, ganciclovir (GCV), 5-fluorocytosine (5-FC), and CB1954.
Early experience from clinical trials demonstrated the feasibility of PAGT, especially in the setting of cancer therapy. This has served as a model for evaluating PAGT as a strategy in other areas targeted by genetic modification, such as monogenic gene therapy and cellular therapy.
In recent years, inherent problems with the vectors used for PAGT led to the field developing novel approaches to ensure the safety of genetically modified cells, subsequently focusing on methods that do not rely on prodrug metabolism. Table 1 presents an overview of suicide-gene systems, including PAGT, and highlights some of the distinct features associated with each approach.
Table 1.
Selected prodrug-activated gene therapy (PAGT) and alternative “suicide gene” systems, activating/prodrugs used and potential advantages and disadvantages.
| Category | Prodrug(s) (route of administration) | Active metabolite | Time to peak serum concentration of prodrug (estimated) | Duration of prodrug administration | Advantages | Disadvantages | References |
|---|---|---|---|---|---|---|---|
| Prodrug-activated systems | |||||||
| Herpes simplex thymidine kinase (HSV-TK) | Ganciclovir (iv) Valganciclovir (po) Valaciclovir (po) Aciclovir (po/iv) |
Ganciclovir triphosphate (GCV-3P) | 1.5-3 hours (po) Immediate (iv) |
7-21 days | -Widely used in clinical trials; -Effective; -Safe |
-Immunogenicity; -Alternative splice sites; -Toxicity of prodrug (e.g. bone marrow suppression); -Only effective for proliferating cells; -Prolonged prodrug schedule (2-3 weeks); -Need for treating CMV infection with GCV can lead to premature destruction of cells |
1,3,5,12, 13, 14, 15, 16, 17, 18 |
| Cytosine deaminase (CD) | 5-Fluorocytosine (5-FC) (po/iv) | 5-Fluorouracil | 1-2 hours | 7-21 days | -Used in clinical trials; -Safe; -Some efficacy |
-Immunogenicity; -Toxicity of prodrug (mainly hematologic); -Prolonged prodrug schedule (2-3 weeks) |
19, 20, 21, 22, 23 |
| Nitroreductase | CB1954 (iv/ip) | 5-(aziridin-1-yl)-4-N-acetoxy-2-nitrobenzamide | 3-4 hours | One time treatment, repeated 3-weekly for up to 6 cycles | -Safe; -One-time prodrug treatment fast; -Re-treatment with prodrug possible |
-Limited clinical trial experience; -Toxicity of prodrug (lymphopenia, transaminitis) |
24, 25, 26 |
| Cytochrome P450 (CYP450) | Oxazaphosphorines (e.g. cyclophosphamide, ifosfamide) (iv) | 4-hydroxy derivative of oxazaphorine | 24 hours | 14 days every 28 days, repeated up to 7 cycles | -Less immunogenic; -Well tolerated |
-Limited clinical trial experience; -Metabolism through endogenous P450 system causing systemic toxicity |
27 |
| Purine Nucleoside Phosphorylase/ | 6-Methylpurine Deoxyriboside or Fludarabine (iv/po) | 2-deoxyribose-1-phosphate | 0-24 hours | 3 days | -Potent bystander effect; -Well tolerated |
-Limited clinical trial experience | 28 |
| Carboxypeptidase G2 | Nitrogen Mustard (e.g. ZD2767P) (iv) | DNA alkylating agent | minutes | 3 bolus injections on one day | -Causes apoptosis in dividing and non-dividing cells; -Well tolerated |
-Limited clinical trial experience -Immunogenicity -Limited clinical effect |
29 |
| Therapeutic monoclonal antibody-mediated | |||||||
| CD20 tEGFR |
N/A | anti-CD20 antibody (e.g. rituximab) anti-tEGFR antibody (e.g. cetuximab) | hours hours |
2-8 hours iv 60-120 mins (iv) |
-Rapid onset; -Extensive experience with several antibodies (e.g. rituximab, cetuximab) for other clinical indications; -Non-immunogenic |
-Limited clinical data | 30, 31, 32 |
| Dimerization-inducing | |||||||
| iCasp9 | N/A | AP1903 | minutes (iv) | minutes | -Rapid onset; -Non-immunogenic |
-Limited clinical data | 33 |
i.p., intraperitoneally; N/A, not applicable; p.o., per os (orally).
In this review, we describe prodrug-activated suicide systems that have been employed in clinical trials, associated advantages and drawbacks, as well as some of the innovative solutions developed to circumvent respective shortcomings. In particular, we aim to focus on inherent problems of the prodrugs themselves, an area that is often not addressed or reported in clinical studies. In an era of resurgent interest in cellular and genetic therapies, with many hundreds of clinical studies ongoing for a broad range of applications, these insights may inform the design of new cellular safety switches in this constantly evolving field.
Preclinical and clinical experience with the HSV-TK, CD, and NTR prodrug-activating systems reported prodrug activity and toxicities
HSV-TK/GCV PAGT
The HSV-TK gene is arguably the PAGT most extensively studied in human clinical trials. HSV-TK is a cell-cycle-dependent enzyme with high affinity predominantly for GCV, an acyclic nucleoside analog of 2′-deoxyguanosine that is commonly used in the treatment of herpes virus infections. HSV-TK catalyzes the phosphorylation of GCV, leading to the generation of di- and tri-phosphate derivatives, which incorporate into replicating DNA in proliferating cells, thereby inhibiting DNA chain elongation and ultimately causing apoptosis. It has been shown that cells transduced with the HSV-TK gene retain sensitivity to GCV. Importantly, however, GCV kills only those HSV-TK-expressing cells that are actively proliferating and spares resting HSV-TK-transduced cells.12,34,35
The feasibility of inserting HSV-TK into cells to exploit their drug sensitivity after administration of a prodrug was first shown in cancer models in the 1980s. Treatment of in vitro cell lines with the HSV-TK-specific substrate 9-([2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)-guanine ablated their clonogenic potential. Similarly, administration of this prodrug to BALB/c mice bearing HSV-TK-positive tumors could induce complete regression of tumor growth.36
Following these supportive preclinical data, the first published clinical experience that demonstrated the proof of concept that PAGT as a kill switch using cellular therapy was feasible and safe in human subjects was reported in 1997 by Bonini et al.5 in allogeneic bone marrow transplant recipients. Eight patients who relapsed after HSCT or developed post-transplant lymphoproliferative disease (PTLD) were treated with donor lymphocytes that had been transduced with the HSV-TK suicide gene (TK cells). Three patients developed GvHD after receiving donor lymphocytes and were effectively treated with GCV administration at a dose of 10 mg/kg/day until GvHD resolution. Some patients also required steroids to achieve complete remission, but no specific toxicities from administration of the GCV prodrug were reported.
Approximately 150 patients have since been recruited into multiple clinical trials in different countries in order to receive TK cells after HLA-identical or haploidentical HSCT to mitigate the effects of GvHD.3
One study, the TK007 trial, enrolled 50 patients who underwent haploidentical HSCT between 2002 and 2008. Twenty-eight patients received TK cells, of whom 11 developed GvHD. In all patients, GCV administration to mediate prodrug-activated cell destruction led to resolution of GvHD. No adverse events observed in this patient cohort were attributed to the cellular product or indeed the prodrug.13 A long-term follow-up study of patients treated in this way at several European centers reported that of the 11 patients who received TK cells, 2 developed GvHD, which was successfully controlled with GCV administration. HSV-TK gene expression was found to persist until about 2 years after infusion of TK cells. Overall, the use of transduced TK cells was considered to be safe and efficient.13,14,37 Given these promising results, TK PAGT was advanced into a multicenter, randomized phase 3 clinical trial in patients with high-risk leukemia receiving a haploidentical HSCT (ClinicalTrials.gov: NCT00914628). This trial is currently ongoing and expected to complete in 2021.
PAGT strategies with the HSV-TK system have also been studied in solid cancers. Intra-tumoral implantation of HSV-TK-modified cells into experimental rodent brain tumors led to regression of tumors after GCV administration. In this approach to PAGT, introduction of vector-producer cells directly into the tumor allowed HSV-TK to sensitize adjacent tumor cells to the GCV prodrug, and the administration of the prodrug is required to achieve the desired anti-tumor effect. In a cohort of 15 patients, HSV-TK-engineered cells were shown to have anti-tumor activity, although only patients with very small tumors responded to the treatment. This phenomenon was thought to be related to the constraints of transferring large, relatively non-motile cells containing a vector that cannot replicate into a specific tumor site.38
In the single-arm phase 1/2 GLI328 European-Canadian study, 48 patients with relapsed glioblastoma multiforme received HSV-TK-transduced cells by intra-tumoral injection, followed by systemic administration of GCV on days 14–27. The PAGT approach in this setting was found to be safe and feasible, including use of the prodrug, but with limited therapeutic efficacy of the cell product, as only one patient remained disease-free at 24 months.15 Subsequently, a prospective, multicenter, open-label phase 3 trial randomized patients with previously untreated glioblastoma multiforme to receive either standard treatment (surgical resection followed by radiation) or standard therapy plus PAGT. Patients randomized to the PAGT treatment arm received cells derived from a murine fibroblast cell line transfected with a replication-deficient retrovirus containing the HSV-TK gene (G1Tk1SvNa.7); GCV was administered to all PAGT-treated patients at a dose of 5 mg/kg twice daily on days 14–27 after tumor resection. Progression-free and 12-month overall survival rates between the gene therapy group and the control group showed no statistically significant differences. PAGT therefore improved neither time to tumor progression nor overall survival, although the feasibility and favorable biosafety profile of this gene therapy strategy were further supported. No specific toxicities of using GCV in this context were described.16
One of the problems in human trials of HSV-TK PAGT has been the effective delivery of the vector to the tumor site and dissemination throughout the tumor, which is thought to be limited in part by the presence of abnormal gap junctions, especially in solid tumors.39 The main focus in much of the work on PAGT systems with the HSV-TK vector has therefore focused on increasing the efficacy of vector delivery. Conversely, the role of individual prodrugs, their route of administration, distribution, and specific side-effect profiles have not been addressed in equal measure. For example, to our knowledge, dosing strategies of GCV or the management of anticipated AEs of GCV are often not discussed in detail in the HSV-TK PAGT literature, and this makes it difficult to extrapolate and quantify how the prodrug itself may influence the overall efficacy of this PAGT approach.
Use of oral equivalents to GCV: valganciclovir and valacyclovir
Valganciclovir (vGCV) is a prodrug of GCV that can be given orally. vGCV is considered to be as efficacious as intravenous (i.v.) GCV for its licensed indication, the treatment of cytomegalovirus (CMV) disease. Oral valacyclovir (VAV) is another suitable and well-tolerated alternative to i.v. GCV and is often used for the prophylaxis of CMV infection after HSCT. VAV has greater bioavailability than acyclovir and in a randomized comparison with i.v. GCV was shown in HSCT recipients to have less dose-limiting neutropenia but equivalent efficacy in the prevention of CMV disease.40 These data led to the adoption of oral GCV equivalents in clinical trials with PAGT-based systems.
Chiocca et al.17 used VAV as the prodrug in a phase 1b study in which 13 patients were treated with neoadjuvant AdV-TK, an adenoviral vector containing the HSV-TK gene, for newly diagnosed malignant gliomas. Patients received vector particles via tumor bed injection at the time of surgery, followed by 14 days of VAV at a dose of 2 g three times a day. Several AEs observed during the study, including headache, mood alteration, speech impairment, fever (1 patient each), and transient transaminitis (11 patients), all occurring at grade 1–2, were thought to be possibly, but not definitely, related to the combined administration of the AdV-TK vector plus the prodrug, and not exclusively attributed to the prodrug itself.17 A subsequent phase 2 study by the same group recruited 48 patients who were treated post-surgery with AdV-TK and oral VAV, followed by standard-of-care radiation therapy and temozolomide. No dose-limiting toxicities of the vector plus prodrug treatment were described; fever, fatigue, and headache (mainly grade 2) were the most common symptoms possibly attributable to the combination of vector and prodrug, because they occurred during the period of prodrug administration (weeks 1–3 of treatment). Again, none of the AEs were directly attributed to the use of VAV.18
In summary, therefore, oral equivalents of i.v. GCV are well tolerated and safe and may be as efficacious as their i.v. equivalents in activating the prodrug system, although systematic head-to-head comparisons between i.v. and oral GCV in PAGT systems have not been carried out.
CD/5-FC PAGT
CD is an enzyme found in fungi and bacteria, such as Escherichia coli or yeast.41 CD catalyzes the hydrolytic deamination of cytosine into uracil and therefore can convert the non-toxic prodrug 5-FC (flucytosine) to 5-fluorouracil (5-FU), which is then transformed by cellular enzymes into three potent pyrimidine anti-metabolites: 5-FdUTP, 5-FUTP, and 5-FdUMP. Cytotoxicity subsequently ensues as a result of three different pathways: thymidylate synthase inhibition and the formation of (5-FU)-RNA and (5-FU)-DNA complexes.42
Mammalian and plant cells are naturally deficient in CD activity because they lack the enzyme and therefore are resistant to 5-FC. CD-transformed cells acquire CD activity and thus become sensitive to treatment with 5-FC, forming the basis for the use of the E. coli or yeast CD (yCD) genes as a suicide gene.9
5-FC is a synthetic compound developed initially for its antimycotic activity. It does not have intrinsic antifungal capacity but is converted into 5-FU after it is taken up by susceptible fungal cells, and its metabolites lead to inhibition of fungal RNA, DNA, and protein synthesis. With systemic administration, 5-FC can also be deaminated to 5-FU by gut bacteria, leading to potential gastrointestinal (GI) side effects. 5-FU is a widely used cytotoxic chemotherapy agent in the treatment of many solid tumors but requires the administration of high doses to achieve an anti-tumor response. One of the advantages of the CD/5-FC system as a PAGT strategy, therefore, is that it enables a more targeted approach to tumor cells, allowing the use of standard doses of 5-FC and bypassing some of the side effects of systemic 5-FU administration.9 In addition, the CD/5-FC system has a stronger local bystander effect compared with the HSV-TK/GCV system because it does not require cell-to-cell contact and is less dependent on gap junctions.7, 8, 9,43
The anti-cancer effects of the CD/5-FC SGT system in the preclinical setting were demonstrated in human cancer cell lines and multiple xenograft tumor models, including for glioblastoma, and lung and cervical cancer.19,43, 44, 45, 46
In the clinical setting, the CD/5-FC PAGT system has been described mostly in small case series and early-phase clinical trials in patients with different solid cancers. One of the first trials of the E. coli CD/5-FC system in humans involved the CD gene driven by a tumor-specific erbB-2 promotor. In 12 patients with breast cancer, this approach was demonstrated to result in selective tumor-targeted gene expression and met safety endpoints. The prodrug itself was well tolerated without any unexpected AEs or negative impact on treatment efficacy.47
In a study of two patients with malignant pleural effusions as a result of advanced lung cancer relapsing after frontline chemotherapy, the subjects received two doses of an adenoviral CD-containing vector (Ad.CD) administered intra-pleurally. Patients were subsequently treated with 14 days of oral 5-FC at a dose of 500 mg four times a day. Both patients had complete resolution of their pleural effusions but also were reported to have grade 3 neutropenia and anemia attributed to the effects of 5-FC compounding bone marrow suppression as a result of prior systemic chemotherapy.20, 21, 22
A pilot trial recruited three patients with refractory solid cancers for treatment with TAPET-CD, an attenuated Salmonella bacterium strain (VNP20009) with the E. coli CD gene inserted at the DmsbB locus to enhance its effectiveness. TAPET-CD vector particles were injected directly into tumor tissue once every 28 days, and patients then received treatment with 5-FC for 10 days at a dose of 100 mg/kg/day orally in three divided doses. The approach was shown to be safe and feasible with manageable toxicity. Reported AEs, including coronary vasospasm, weakness, fever, and pain, were attributed to the vector, but not the prodrug.23
NTR/CB1954 PAGT
CB1954 is a weak alkylating agent that can be converted by the E. coli bacterial enzyme NTR into the cytotoxic compound 5-(aziridin-1-yl)-4-N-acetoxy-2-nitrobenzamide and induces cell death by forming inter-strand DNA cross-links. In vitro experiments have shown that NTR-expressing cancer cell lines were up to 500- to 2,000-fold more sensitive to CB1954 than parental cell lines. Cell killing by activated CB1954 is cell cycle independent. No human homolog of NTR exists, and CB1954 is a poor substrate for human DT-diaphorase. CB1954 therefore is not readily converted to the activated species in non-NTR-expressing cells. Targeting tumors with NTR delivered through an adenovirus vector, followed by administration of CB1954, may lead to tumor-specific cell killing because only cells transduced with NTR should be directly susceptible to CB1954.48,49 This hypothesis was tested in a number of animal models; in an NTR-expressing pancreatic cancer xenograft model, 80% of mice treated with CB1954 achieved lasting remissions compared with animals with wild-type tumors.49, 50, 51
Importantly, in the clinical setting, the toxicity and pharmacokinetics of CB1954 as a single agent were first assessed in a phase 1 trial prior to administering the vector/prodrug combination, an approach that may allow to distinguish more precisely between effects caused by the prodrug versus the vector. Thirteen patients with a range of solid tumors, including colorectal, gastric, and esophageal cancers, received CB1954 as an i.v. injection every 3 weeks for up to a total of six cycles. Dose-limiting toxicities were diarrhea and hepatic toxicity, at a dose of 37.5 mg/m2. A dose of 24 mg/m2 was advanced into a phase 1/2 trial with an adenovirus-NTR vector.24 In this study, a group of 20 patients with prostate cancer received escalating doses of the replication-defective adenovirus vector CTL102 encoding bacterial NTR, to establish the safety of the vector. Subsequently, 19 patients then received viral vector particles in combination with the CB1954 prodrug (given i.v. over 5 min), and 14 patients were able to be successfully re-treated.25 The only AEs attributed to the PAGT treatment were GI symptoms, pain at the injection site, low-grade fever, lymphopenia (mostly grade 3), and transaminitis (mostly grade 2), all of which were transient and did not require intervention. Although the treatment overall was felt to be safe and deliverable with minimal toxicities, only seven patients achieved >10% reduction in prostate-specific antigen (PSA). Further development of the system may be warranted nonetheless, and it has been debated whether this can be achieved by increasing the catalytic activity of NTR through targeted mutations or by using an alternative prodrug.
Drawbacks of PAGT and alternative strategies
In general, strategies that aim to improve some of the shortcomings of PAGT have focused mainly on modifications in different areas, such as vector design, consideration of bystander effects, or developing alternative suicide approaches that allow gene-modified cells to be destroyed. The contribution of the prodrug itself on the safety and efficacy of a given PAGT approach may be difficult to determine but is also worth considering. We describe some important areas of PAGT development below.
Use and dosing of prodrugs
GCV and its oral derivatives are commonly used to treat viral infections such as CMV, which commonly occur in immunocompromised patients, especially after HSCT. Consequently, administration of the prodrug in the context of active CMV infection would lead to an unwanted prodrug-activated destruction of engineered cells and premature abrogation of their intended function. Some authors have argued that immune reconstitution aided by the administration of TK cells may counterbalance the risk for infection with viruses such as CMV, therefore making the need for GCV administration less likely, and studies in HSCT patients have used alternate agents, such as foscarnet, to substitute GCV (or its equivalent oral form) in the treatment of CMV infections.13,14
Another important consideration relates to prodrug dosing in clinical trials. This is generally in line with standard therapeutic dosing recommendations as per the drug summary of product characteristics. Even in clinical studies that combine suicide genes such as HSV-TK and CD in one vector, the respective prodrugs are administered at full licensed therapeutic dose. Bearing in mind that PAGT strategies are often used in patients with limited or already compromised bone marrow reserve, such as HSCT patients or a heavily pretreated oncology patient population, hematological toxicity especially needs to be anticipated and may limit the administration and effect of the prodrug itself.
Finally, oral administration of prodrugs may be highly desirable and advantageous in terms of time spent in hospital and ease of administration, especially if a prodrug is given over a period of several weeks in the ambulatory care setting. However, monitoring patient adherence with prodrugs in this context may be more difficult, and non-compliance with medication could become an issue affecting the efficacy of the PAGT approach.
Vector design
The most widely studied PAGT approach to date has employed the HSV-TK gene. From the available clinical trials data, several problems with this particular PAGT system are broadly related to the TK gene itself and/or vector design and highlight limitations of the field in general: first, viral epitopes encoded by the TK gene are immunogenic, which can result in the unintended elimination of the transduced cell population.1,14,52,53 Second, alternative splicing sites in the TK gene can lead to non-functional enzymes, and therefore a suboptimal response to the administered prodrug.54 Third, the PAGT strategy is dependent on the cell cycle, and its effectiveness relies on the presence of actively dividing cells. Therefore, the targeted elimination of transduced cells is not immediate, whereas resting or post-mitotic cells harboring the prodrug-activating gene are usually spared.35
Bystander effects
Depending on the PAGT system used, bystander effects can differ and may affect treatment efficacy. Bystander effects, both local and distant, have mostly been described in cancer models, and in this context are defined as evidence of tumor regression when not all tumor cells express the prodrug-activating/suicide gene. Local bystander effects are thought to be precipitated by a number of mechanisms, such as the passive diffusion of the activated drug, transfer through gap junctions, or mediated through apoptotic bodies generated by dying cells, which can be taken up by non-modified adjacent cells.9,55 Distant bystander effects refer to regression of tumor tissue distant from the sites expressing the prodrug-activating gene and are most likely related to activation of immune effector cells, such as T cells and NK cells.
The efficiency of tumor cell transduction with the prodrug-activating gene vector may considerably influence the anti-tumor activity of local bystander effects, as evidenced by experiments in mouse models comparing the efficacy of HSV-TK and CD systems.55,56 Additional preclinical work also has shown that compounds that increased the expression of connexin moieties in gap junctions, such as the polyphenol resveratrol, can enhance the local bystander effect of PAGT systems.56
It is important to consider the contribution, especially of local bystander effects, in the evaluation of individual PAGT systems. Thus, in cancer therapy, a bystander effect may be desirable, and potential additional toxicity arising from this and/or the dose of prodrug required to achieve this could be more acceptable than in the setting of gene therapy for non-malignant diseases where PAGT is employed specifically as a safety switch, and additional tissue damage through bystander effects is an unintended consequence. Bystander effects are often not routinely described in clinical trials in terms of an outcome measure, and therefore it is difficult to quantify the impact on surrounding healthy tissues.
Modifying existing PAGT systems
HSV-TK systems
The generation of mutated HSV-TK variants can avoid cryptic splice sites and therefore the likelihood of the development of GCV-resistant genetically modified cells during clinical trials.57 The HSV-TK platform may also be further improved by expressing HSV-TK under the control of a promoter known to be active in the tumor microenvironment, as reported in a mouse model where a novel STAT3/nuclear factor κB (NF-κB)-based reporter system was used to drive the expression of HSV-TK and was therapeutically active after GCV administration.58
A novel mechanism through which the safety of cell therapy could be optimized to eliminate unwanted proliferating cells is creating a transcriptional link of a gene absolutely essential to cell division, such as CDK-1 with a prodrug-activated gene such as HSV-TK. Using this approach in a mouse model, Liang et al.59 were able to show how potentially tumorigenic proliferating cells contaminating a terminally differentiated cell product derived from induced pluripotent or embryonic stem cells can be successfully eliminated. This study used mathematical modeling to estimate a safe number of cells based on the cell dose and the extent of homozygosity of the genome editing undertaken and is the first to quantify the safety of cell therapy. Finally, mesenchymal stromal cells engineered to express HSV-TK and injected into tumor tissue in murine cancer models have been shown to lead to tumor regression, as well as induce measurable NK cell and cytolytic T cell responses after treatment with GCV, highlighting the potential of advanced cell therapy approaches that may harness a PAGT strategy.60,61
CD systems
In mice, yCD has superior kinetic properties toward 5-FC and somewhat improved efficacy for treating tumors than bacterial CD (bCD).62 However, wild-type yCD is relatively thermolabile as compared with bCD, a property that may limit its performance in therapeutic applications. The half-life of yCD can be stabilized and extended by repacking of its hydrophobic core at several positions distant from the active site. Random mutagenesis of residues selected based on alignment with similar enzymes, followed by selection for enhanced sensitization to 5-FC, can produce enzyme variants with an increased activity half-life. These mutations may significantly improve the enzyme’s performance in PAGT assays, as shown both in cell culture and in animal models.63
Retroviral replicating vectors (RRVs) maintain viral persistence in tumors by non-lytic replication, stable integration into the cancer cell genome, and reduced viral immunogenicity. RRVs can achieve efficient tumor transduction and improve therapeutic benefit in a variety of cancer models. Although not intrinsically cytolytic, RRVs can be engineered to carry prodrug activator genes, such as HSV-TK and yCD, to mediate synchronized cell killing of infected tumor cells upon prodrug administration.
yCD can be delivered into cancer cells via the RRV Toca 511 system. It converts the investigational prodrug Toca-FC (extended-release 5-FC) into 5-FU and showed promising results in a phase 1 clinical trial in patients with recurrent glioblastoma: in a comparison with a matched external control group of patients receiving conventional standard-of-care treatment, patients treated with the RRV Toca 511 system survived for longer, and interestingly, translational studies indicate antitumor immunity mediated by memory T cells.64 Further investigation into the mechanisms underlying the therapeutic efficacy of Toca 511/5-FC in syngeneic models of murine glioma and xenograft models of human glioma demonstrated that long-term tumor control in immunodeficient models could be achieved but was dependent upon continued administration of prodrug using a cyclical dosing schedule; by contrast, in immunocompetent models, the prodrug could be discontinued after a few cycles without further tumor recurrence, suggesting that the induction of anti-cancer immunity may play an important role in achieving long-term disease control.64,65
Also under development are more convenient chromatography-based detection methods that allow the determination of the in vivo conversion rate of 5-FC to 5-FU, which ultimately may allow a tailored approach to the administration of prodrug.66
Combined HSV-TK and CD PAGT approaches
Several studies have been reported in prostate cancer patients treated with an adenovirus vector containing both the CD and TK genes. The initial phase 1 clinical trial experience was published by Freytag et al.67 in 2002. Subjects were treated with a replication-competent adenovirus carrying a CD/HSV-TK fusion gene (Ad5-CD/TK). All patients then received the prodrugs 5-FC and GCV at doses of 150 and 10 mg/kg/day, respectively, for either 7 or 14 days. Three of the 16 patients enrolled showed a decrease in PSA. One patient experienced grade 3 leukopenia attributed to the prodrugs, but this resolved spontaneously after completion of treatment.67
The double-PAGT approach has been shown to be effective and safe, also when combined with additional therapeutic modalities such as radiation. In an initial phase 1 study of this combined approach, patients received escalating doses of two prodrugs, 5-FC and vGCV: 5-FC was administered orally for 1, 2, or 3 weeks (depending on trial cohort), at a total dose of 150 mg/kg/day in four divided doses; vGCV was also given orally for up to a total of 3 weeks at a dose of 1,800 mg/day in two divided doses. The vast majority (94%) of AEs were mild to moderate in nature (grades 1–2). AEs specifically attributed to prodrug therapy were lymphopenia (93%), anemia (67%), diarrhea (53%), and GI discomfort (33%). Hyperglycemia was observed in 47% of patients, especially in elderly patients who were known to be borderline diabetic, and was also thought to be related to prodrug therapy.68 A further phase 1 study went on to evaluate a similar combined radiation/PAGT approach using an improved second-generation adenovirus, Ad5-yCD/mutTKSER39rep ADP. The enzyme products of the yCD/mutTKSER39 vector were both demonstrated to be catalytically more active compared with the first-generation vector, thereby leading to an enhanced chemotherapeutic and radiation-sensitizing effect. Again, the majority of AEs were mild to moderate (grades 1–2), with only five events being grade 3 or above. Anemia, neutropenia, and lymphopenia were attributed to the well-known hematological effects of the prodrugs used and were self-limited. Most of the treatment effect was observed in the intermediate-risk group, with all 12 patients showing no evidence of residual cancer in their latest biopsy.69 In terms of safety, a pooled analysis of the findings of three phase 1 trials in this patient population showed no late side effects of this approach to gene therapy at 5-year follow-up.70
In a phase 2 clinical trial, the same research group reported on the safety and efficacy of combining oncolytic adenovirus-mediated cytotoxic gene therapy with intensity-modulated radiation therapy (IMRT) in 44 patients with intermediate-risk prostate cancer.71 The vector was an adenovirus combining PAGT with CD/5-FC and HSV-TK/vGCV, which was shown to render prostate cancer cells sensitive to pharmacological therapy, as well as ionizing radiation. Patients were treated with intra-prostatic injection of viral particles on day 1, and 2 days later received radiation in addition to commencing oral treatment with 5-FC (4 × 150 mg/kg/day) and vGCV (2 × 1,800 mg/kg/day) for 2 weeks. The majority of AEs (98%) were grade 1–2, with those specifically attributed to the prodrug regimen being neutropenia (7 patients) and thrombocytopenia (10 patients). Sixty-seven percent of patients received the full prescribed doses of prodrug. As per the study protocol, the doses of 5-FC and vGCV were reduced to 50% and 75%, respectively, in six patients, owing to transient thrombocytopenia (n = 2), neutropenia (n = 2), and nausea/vomiting (n = 2). One patient received a partial dose of prodrug because of noncompliance. Overall, the authors concluded that the PAGT approach in conjunction with radiation was well tolerated without any exacerbation of the most common side effects of prostate radiation, with a clinically meaningful reduction in positive biopsy results at 2-year follow-up.71
In a different approach, dual RRV-mediated TK/GCV and CD/5-FC combinatorial gene therapy has been shown to achieve synergistic cytotoxic efficacy compared with single-vector gene therapy, and co-infection of cancer cells by two different RRVs carrying different prodrug activator genes may also avoid drug resistance.72
NTR systems
Analysis of the crystal structure of the NTR enzyme-ligand complex identified nine different amino acid residues within the active catalytic site that may directly influence prodrug binding and catalytic activity. Mutant libraries generated for each of these residues were screened for their ability to sensitize E. coli to CB1954, and amino acid substitutions at six positions were shown to confer a markedly greater sensitivity to CB1954 compared with the wild-type enzyme. Introducing this NTR mutant into human SKOV-3 cells sensitized these cells to CB1954 at clinically relevant prodrug concentrations by approximately 5-fold versus the wild-type enzyme, thereby improving the efficacy of the NTR/CB1954 combination in these in vitro experiments.73 Another modification of this system describes the co-expression of bacterial NTR with the human arylamine N-acetyltransferase NAT2. This approach was also examined in the ovarian cancer SKOV3 cell line and was shown to increase the sensitivity of these cells to CB1954, suggesting that co-expression of these two genes may significantly enhance responsiveness to prodrug treatment. Addition of the acetyltransferase also resulted in a markedly decreased bystander effect, possibly because of a lower concentration of reactive metabolites in the culture medium. These results suggest that a combination of bacterial NTR and human NAT2 may provide a greater clinical response at therapeutic concentrations of CB1954, or even allow dose reduction of the prodrug without loss of efficacy, provided the reduction in bystander effect is not clinically significant.74 Similarly, combining NTR/CB1954 and γ-rays has been shown to lead to a synergistic effect on HeLa cells in vitro, leading to enhanced radiosensitivity of tumor cells.75 Molecular modeling studies may furthermore allow optimization of the prodrug for the NTR system, and cell viability studies with a series of nitro-substituted benzamide prodrugs have led to the discovery of novel prodrug candidates with promising kinetic and molecular docking profiles for NTR-based therapy.76
Other PAGT approaches/systems
Several other prodrug systems are under development (see Table 1). Clinical experience with the cytochrome P450/oxazaphosphorine, carboxypeptidase G2/nitrogen mustard, or purine nucleosidase phosphorylase/fludarabine systems is relatively limited and has been hampered partly by problems with targeted prodrug metabolism, the efficient conversion of the respective prodrug, or the efficacy of the vector/PAGT system.77,78
Cytochrome P450/oxazaphosphorine system
In two phase 1/2 trials, a total of 27 patients with pancreatic cancer were treated with encapsulated genetically modified allogeneic cells expressing a cytochrome P450 enzyme. The cells were delivered into the tumor vasculature by super-selective intra-arterial angiography, with the aim of locally activating systemically administered low-dose ifosfamide. At a dose of 1 g/m2, ifosfamide was well tolerated by all patients in the first part of the trial, with regression of tumors in four patients and stable disease in the other participants. A higher ifosfamide dose of 2 g/m2 was found, however, to be toxic in the majority of patients in the phase 2 portion of the trial, necessitating dose reduction in one patient. Expected side effects attributed to the prodrug included nausea and vomiting, malaise, anorexia, and mild hematuria, but no serious adverse events were reported as a result of the intervention.79,80
Purine nucleoside phosphorylase/fludarabine system
Using E. coli purine nucleoside phosphorylase (PNP) to activate fludarabine was demonstrated to be safe and result in anti-tumor activity in preclinical studies. Specifically, fludarabine monophosphate is converted by E. coli PNP into fluoroadenine, which is phosphorylated into its ATP analog (F-ATP) and incorporated into RNA, a process that disrupts RNA and protein synthesis at a cellular level.
The E. coli/PNP system was advanced into a phase 1 clinical trial in 12 patients with head and neck squamous cell carcinoma. All patients experienced AEs attributed to the study drug, but there were no dose-limiting toxicities. Observed tumor responses were dose dependent, and antitumor activity was confined to lesions that received intratumoral injection, whereas non-target lesions remained stable or progressed. Analysis of patient serum confirmed the lack of systemic exposure to fluoroadenine, indicating that release of the compound into the systemic circulation occurred at levels below the toxic range.81
Alternative approaches for drug-activated gene therapies
Given the limitations inherent in the PAGT approaches described above, alternative drug-activated systems are under development and have been explored in clinical trials (Figure 1).
Monoclonal antibody-mediated systems
In order to overcome the immunogenicity described with some PAGT vectors, cell-based studies have shown that targeting naturally occurring or transduced surface molecules such as CD20, or a truncated human epithelial growth factor receptor (tEGFR) polypeptide, followed by injection of specific targeted monoclonal antibodies after the infusion of the gene-modified cellular product, may be a feasible strategy (Figure 1B).30,31,82
tEGFR is derived from the human full-length EGFR sequence and has been modified to ensure it is physiologically non-functional while retaining its binding site for the antibody cetuximab. tEGFR has been shown to be effective in the tracking, selection, and depletion of engineered T cells. In a murine CAR T cell model, co-expression of tEGFR on the surface of engineered T cells could be targeted with cetuximab, eliminating the CAR T cell product and leading to recovery of functional B cells, without disease relapse.31,32 Using this approach, unwanted CAR T cell-related complications such as cytokine release syndrome (CRS), neurotoxicity, or long-term side effects such as B cell aplasia may be mitigated, provided durable responses can be induced and maintained after elimination of the therapeutic cell product. Several early-phase clinical trials with CAR T cells transduced with tEGFR are ongoing and targeting numerous different tumor antigens such as EGFR itself, HER2, or CD19 in solid or hematological malignancies (e.g., ClinicalTrials.gov: NCT03618381, NCT03500991, and NCT01865617).
The epitope-based selection marker CD34/suicide gene CD20 RQR8 system allows for selective rituximab-mediated antibody-dependent cellular cytotoxicity (ADCC) destruction of adoptively transduced CAR T cells (ClinicalTrials.gov: NCT03590574). To date, no published clinical trials data are available describing the outcome of these approaches in the setting of CAR T cell therapy. It will be of particular interest to determine the rate and efficiency of cell elimination and whether the unique side effects of CRS and neurotoxicity associated with CAR T cell therapy can be mitigated with these strategies, given that CRS, for example, may be perpetuated by immune elements recruited into the tumor microenvironment.83
Dimerization-mediated systems: inducible caspase-9 (iCasp9)/AP1903
iCasp9, an endogenous enzyme in humans that acts as an initiator caspase in the intrinsic apoptosis pathway, has generated interest as an inducible suicide switch. Inactive iCasp9 subunits coupled to the FK-506 binding protein, FKB12, can be expressed in transduced cells, and functional iCasp9 can subsequently be induced through subunit dimerization, leading to DNA fragmentation and apoptosis of engineered cells, including non-replicating cells (Figure 1C).
The otherwise biologically inert dimerizing agent, AP1903, is well tolerated in patients and does not have any effects on endogenous, non-transduced T cells. Pharmacokinetic data show that plasma levels of AP1903 fall rapidly after i.v. infusion, decreasing to 18% and 7% within 30 min and 2 h, respectively. In recipients of haploidentical HSCT who also received genetically modified T cells, the administration of AP1903 led to rapid apoptosis of up to 95% of transduced cells within 30 min and control of GvHD.33,84 In vitro data show that repeat infusions of the dimerizing agent are feasible, and in the study by Di Stasi et al.,33 the trial protocol allowed for the infusion of additional doses of AP1903; however, no patients treated with AP1903 required this because there was no recurrence of GvHD.79 Interestingly, three of four patients treated with AP1903 did go on to have disease relapse, prompting concern that the graft-versus-leukemia effect of alloreactive T cells could be compromised with this strategy.85
In a preclinical model of natural killer CAR (NK-CAR) cells, cord blood-derived NK cells were transduced with three constructs, including the CD19 CAR, an interleukin-15 gene, as well as iCasp9. The authors were able to show that the resultant NK-CAR cells could be eliminated effectively by addition of the AP1903 dimerizer in cell culture and also in an in vivo mouse model, in which NK-CAR cells expressing iCasp9 were shown to undergo apoptosis in the peripheral blood, bone marrow, liver, and spleen.86
Conclusions and future outlook
Results of preclinical and clinical studies, mainly in the field of cancer medicine, show that prodrug-activated gene switches can be incorporated safely into gene therapy strategies. With increasing interest in gene transfer technologies and adoptive cellular therapeutics such as CAR T cells or modified NK cells, it is envisaged that their application can also be extended to non-malignant disorders, potentially benefitting many more patients. The long-term safety of gene therapy strategies will increasingly come into focus with increased clinical success of these treatments, and regulatory agencies are mandating stringent and long-term follow-up of individuals enrolled in gene therapy clinical trials.
One of the major challenges of the PAGT approach is the efficacy of the prodrug system and how this can be optimized. Arguably, much work over the last two decades has focused on improving the delivery and efficiency of viral transfer vectors, which through sophisticated molecular techniques can be rendered more potent and less immunogenic. At the same time, the design of prodrugs has lagged behind. Most prodrugs in use today have effectively been re-purposed for this indication and have well-known safety and side-effect profiles, usually extrapolated from experience with their use for other indications, as, for example, the treatment of CMV disease in the case of GCV. In many of the target patient populations for gene therapy, currently available prodrugs may induce predictable toxicities and limit the effectiveness of a PAGT approach if the drug is slow acting, has to be dose reduced, or if the patient is non-compliant with therapy. Few clinical studies report on AEs specifically attributable to the use of a prodrug, and strategies for mitigating these (such as, for instance, dose-reduction or stopping rules) are often not described in detail. Similarly, accessibility of the prodrug to the cells harboring the prodrug-activating gene should guide the choice of prodrug system. For example, transduced cells may migrate into tissues after infusion or are directly implanted into solid tumors, but few data on the bioavailability and biodistribution of prodrugs have been published.
Future study designs should therefore consider these limitations, and efficient and sustainable future PAGT systems may greatly benefit from innovative prodrug protocols that improve prodrug dosing and delivery.
Acknowledgments
We thank Marieke de Korte for her help with the illustration of Figure 1.
Declaration of interests
S.S. and D.E. have no competing interests. A.K. is a co-founder and holds equity in the biotech startup panCELLa Therapeutics, Inc.
Contributor Information
Semira Sheikh, Email: semira.sheikh@uhn.ca.
Armand Keating, Email: armand.keating@uhn.ca.
References
- 1.Bonini C., Brenner M.K., Heslop H.E., Morgan R.A. Genetic modification of T cells. Biol. Blood Marrow Transplant. 2011;17(Suppl 1):S15–S20. doi: 10.1016/j.bbmt.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagree M.S., López-Vásquez L., Medin J.A. Towards in vivo amplification: Overcoming hurdles in the use of hematopoietic stem cells in transplantation and gene therapy. World J. Stem Cells. 2015;7:1233–1250. doi: 10.4252/wjsc.v7.i11.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greco R., Oliveira G., Stanghellini M.T., Vago L., Bondanza A., Peccatori J., Cieri N., Marktel S., Mastaglio S., Bordignon C. Improving the safety of cell therapy with the TK-suicide gene. Front. Pharmacol. 2015;6:95. doi: 10.3389/fphar.2015.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 5.Bonini C., Ferrari G., Verzeletti S., Servida P., Zappone E., Ruggieri L., Ponzoni M., Rossini S., Mavilio F., Traversari C., Bordignon C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 6.Marais R., Spooner R.A., Light Y., Martin J., Springer C.J. Gene-directed enzyme prodrug therapy with a mustard prodrug/carboxypeptidase G2 combination. Cancer Res. 1996;56:4735–4742. [PubMed] [Google Scholar]
- 7.Touraine R.L., Vahanian N., Ramsey W.J., Blaese R.M. Enhancement of the herpes simplex virus thymidine kinase/ganciclovir bystander effect and its antitumor efficacy in vivo by pharmacologic manipulation of gap junctions. Hum. Gene Ther. 1998;9:2385–2391. doi: 10.1089/hum.1998.9.16-2385. [DOI] [PubMed] [Google Scholar]
- 8.Touraine R.L., Ishii-Morita H., Ramsey W.J., Blaese R.M. The bystander effect in the HSVtk/ganciclovir system and its relationship to gap junctional communication. Gene Ther. 1998;5:1705–1711. doi: 10.1038/sj.gt.3300784. [DOI] [PubMed] [Google Scholar]
- 9.Huber B.E., Austin E.A., Richards C.A., Davis S.T., Good S.S. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc. Natl. Acad. Sci. USA. 1994;91:8302–8306. doi: 10.1073/pnas.91.17.8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duarte S., Carle G., Faneca H., de Lima M.C., Pierrefite-Carle V. Suicide gene therapy in cancer: where do we stand now? Cancer Lett. 2012;324:160–170. doi: 10.1016/j.canlet.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 11.Khalil D.N., Smith E.L., Brentjens R.J., Wolchok J.D. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016;13:273–290. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weissinger E.M., Borchers S., Silvani A., Provasi E., Radrizzani M., Beckmann I.K., Benati C., Schmidtke J., Kuehnau W., Schweier P. Long term follow up of patients after allogeneic stem cell transplantation and transfusion of HSV-TK transduced T-cells. Front. Pharmacol. 2015;6:76. doi: 10.3389/fphar.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciceri F., Bonini C., Stanghellini M.T., Bondanza A., Traversari C., Salomoni M., Turchetto L., Colombi S., Bernardi M., Peccatori J. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009;10:489–500. doi: 10.1016/S1470-2045(09)70074-9. [DOI] [PubMed] [Google Scholar]
- 14.Lupo-Stanghellini M.T., Provasi E., Bondanza A., Ciceri F., Bordignon C., Bonini C. Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum. Gene Ther. 2010;21:241–250. doi: 10.1089/hum.2010.014. [DOI] [PubMed] [Google Scholar]
- 15.Shand N., Weber F., Mariani L., Bernstein M., Gianella-Borradori A., Long Z., Sorensen A.G., Barbier N. A phase 1-2 clinical trial of gene therapy for recurrent glioblastoma multiforme by tumor transduction with the herpes simplex thymidine kinase gene followed by ganciclovir. GLI328 European-Canadian Study Group. Hum. Gene Ther. 1999;10:2325–2335. doi: 10.1089/10430349950016979. [DOI] [PubMed] [Google Scholar]
- 16.Rainov N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000;11:2389–2401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 17.Chiocca E.A., Aguilar L.K., Bell S.D., Kaur B., Hardcastle J., Cavaliere R., McGregor J., Lo S., Ray-Chaudhuri A., Chakravarti A. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 2011;29:3611–3619. doi: 10.1200/JCO.2011.35.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wheeler L.A., Manzanera A.G., Bell S.D., Cavaliere R., McGregor J.M., Grecula J.C., Newton H.B., Lo S.S., Badie B., Portnow J. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro-oncol. 2016;18:1137–1145. doi: 10.1093/neuonc/now002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller C.R., Williams C.R., Buchsbaum D.J., Gillespie G.Y. Intratumoral 5-fluorouracil produced by cytosine deaminase/5-fluorocytosine gene therapy is effective for experimental human glioblastomas. Cancer Res. 2002;62:773–780. [PubMed] [Google Scholar]
- 20.Zarogoulidis P., Chatzaki E., Hohenforst-Schmidt W., Goldberg E.P., Galaktidou G., Kontakiotis T., Karamanos N., Zarogoulidis K. Management of malignant pleural effusion by suicide gene therapy in advanced stage lung cancer: a case series and literature review. Cancer Gene Ther. 2012;19:593–600. doi: 10.1038/cgt.2012.36. [DOI] [PubMed] [Google Scholar]
- 21.Zarogoulidis P., Domvri K., Huang H., Zarogoulidis K. Gene therapy for lung cancer malignant pleural effusion: current and future nano-biotechnology. Transl. Lung Cancer Res. 2012;1:234–237. doi: 10.3978/j.issn.2218-6751.2012.08.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zarogoulidis P., Giraleli C., Karamanos N.K. Inhaled chemotherapy in lung cancer: safety concerns of nanocomplexes delivered. Ther. Deliv. 2012;3:1021–1023. doi: 10.4155/tde.12.77. [DOI] [PubMed] [Google Scholar]
- 23.Nemunaitis J., Cunningham C., Senzer N., Kuhn J., Cramm J., Litz C., Cavagnolo R., Cahill A., Clairmont C., Sznol M. Pilot trial of genetically modified, attenuated Salmonella expressing the E. coli cytosine deaminase gene in refractory cancer patients. Cancer Gene Ther. 2003;10:737–744. doi: 10.1038/sj.cgt.7700634. [DOI] [PubMed] [Google Scholar]
- 24.Chung-Faye G., Palmer D., Anderson D., Clark J., Downes M., Baddeley J., Hussain S., Murray P.I., Searle P., Seymour L. Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: a phase I and pharmacokinetic study of its prodrug, CB1954. Clin. Cancer Res. 2001;7:2662–2668. [PubMed] [Google Scholar]
- 25.Patel P., Young J.G., Mautner V., Ashdown D., Bonney S., Pineda R.G., Collins S.I., Searle P.F., Hull D., Peers E. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954 [correction of CB1984] Mol. Ther. 2009;17:1292–1299. doi: 10.1038/mt.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu L., Yu C., Jiang Y., Han J., Li Z., Browne P., Race P.R., Knox R.J., Searle P.F., Hyde E.I. Nitroaryl phosphoramides as novel prodrugs for E. coli nitroreductase activation in enzyme prodrug therapy. J. Med. Chem. 2003;46:4818–4821. doi: 10.1021/jm034133h. [DOI] [PubMed] [Google Scholar]
- 27.Braybrooke J.P., Slade A., Deplanque G., Harrop R., Madhusudan S., Forster M.D., Gibson R., Makris A., Talbot D.C., Steiner J. Phase I study of MetXia-P450 gene therapy and oral cyclophosphamide for patients with advanced breast cancer or melanoma. Clin. Cancer Res. 2005;11:1512–1520. doi: 10.1158/1078-0432.CCR-04-0155. [DOI] [PubMed] [Google Scholar]
- 28.Sorscher E.J., Hong J.S., Allan P.W., Waud W.R., Parker W.B. In vivo antitumor activity of intratumoral fludarabine phosphate in refractory tumors expressing E. coli purine nucleoside phosphorylase. Cancer Chemother. Pharmacol. 2012;70:321–329. doi: 10.1007/s00280-012-1908-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Francis R.J., Sharma S.K., Springer C., Green A.J., Hope-Stone L.D., Sena L., Martin J., Adamson K.L., Robbins A., Gumbrell L. A phase I trial of antibody directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumours. Br. J. Cancer. 2002;87:600–607. doi: 10.1038/sj.bjc.6600517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffioen M., van Egmond E.H., Kester M.G., Willemze R., Falkenburg J.H., Heemskerk M.H. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. 2009;94:1316–1320. doi: 10.3324/haematol.2008.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X., Chang W.C., Wong C.W., Colcher D., Sherman M., Ostberg J.R., Forman S.J., Riddell S.R., Jensen M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paszkiewicz P.J., Fräßle S.P., Srivastava S., Sommermeyer D., Hudecek M., Drexler I., Sadelain M., Liu L., Jensen M.C., Riddell S.R., Busch D.H. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 2016;126:4262–4272. doi: 10.1172/JCI84813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bondanza A., Valtolina V., Magnani Z., Ponzoni M., Fleischhauer K., Bonyhadi M., Traversari C., Sanvito F., Toma S., Radrizzani M. Suicide gene therapy of graft-versus-host disease induced by central memory human T lymphocytes. Blood. 2006;107:1828–1836. doi: 10.1182/blood-2005-09-3716. [DOI] [PubMed] [Google Scholar]
- 35.Bonini C., Mondino A. Adoptive T-cell therapy for cancer: The era of engineered T cells. Eur. J. Immunol. 2015;45:2457–2469. doi: 10.1002/eji.201545552. [DOI] [PubMed] [Google Scholar]
- 36.Moolten F.L. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46:5276–5281. [PubMed] [Google Scholar]
- 37.Oliveira G., Ruggiero E., Stanghellini M.T., Cieri N., D’Agostino M., Fronza R., Lulay C., Dionisio F., Mastaglio S., Greco R. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci. Transl. Med. 2015;7:317ra198. doi: 10.1126/scitranslmed.aac8265. [DOI] [PubMed] [Google Scholar]
- 38.Ram Z., Culver K.W., Oshiro E.M., Viola J.J., DeVroom H.L., Otto E., Long Z., Chiang Y., McGarrity G.J., Muul L.M. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat. Med. 1997;3:1354–1361. doi: 10.1038/nm1297-1354. [DOI] [PubMed] [Google Scholar]
- 39.Trinh Q.T., Austin E.A., Murray D.M., Knick V.C., Huber B.E. Enzyme/prodrug gene therapy: comparison of cytosine deaminase/5-fluorocytosine versus thymidine kinase/ganciclovir enzyme/prodrug systems in a human colorectal carcinoma cell line. Cancer Res. 1995;55:4808–4812. [PubMed] [Google Scholar]
- 40.Winston D.J., Yeager A.M., Chandrasekar P.H., Snydman D.R., Petersen F.B., Territo M.C., Valacyclovir Cytomegalovirus Study Group Randomized comparison of oral valacyclovir and intravenous ganciclovir for prevention of cytomegalovirus disease after allogeneic bone marrow transplantation. Clin. Infect. Dis. 2003;36:749–758. doi: 10.1086/367836. [DOI] [PubMed] [Google Scholar]
- 41.Kilstrup M., Meng L.M., Neuhard J., Nygaard P. Genetic evidence for a repressor of synthesis of cytosine deaminase and purine biosynthesis enzymes in Escherichia coli. J. Bacteriol. 1989;171:2124–2127. doi: 10.1128/jb.171.4.2124-2127.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Springer C.J., Bavetsias V., Jackman A.L., Boyle F.T., Marshall D., Pedley R.B., Bisset G.M. Prodrugs of thymidylate synthase inhibitors: potential for antibody directed enzyme prodrug therapy (ADEPT) Anticancer Drug Des. 1996;11:625–636. [PubMed] [Google Scholar]
- 43.Zarogoulidis P., Darwiche K., Sakkas A., Yarmus L., Huang H., Li Q., Freitag L., Zarogoulidis K., Malecki M. Suicide Gene Therapy for Cancer - Current Strategies. J. Genet. Syndr. Gene Ther. 2013;4:16849. doi: 10.4172/2157-7412.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim H.S., Yi B.R., Hwang K.A., Kim S.U., Choi K.C. Anticancer effects of the engineered stem cells transduced with therapeutic genes via a selective tumor tropism caused by vascular endothelial growth factor toward HeLa cervical cancer cells. Mol. Cells. 2013;36:347–354. doi: 10.1007/s10059-013-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon S.K., Kim S.U., Song J.J., Cho C.G., Park S.W. Selective delivery of a therapeutic gene for treatment of head and neck squamous cell carcinoma using human neural stem cells. Clin. Exp. Otorhinolaryngol. 2013;6:176–183. doi: 10.3342/ceo.2013.6.3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yi B.R., Hwang K.A., Aboody K.S., Jeung E.B., Kim S.U., Choi K.C. Selective antitumor effect of neural stem cells expressing cytosine deaminase and interferon-beta against ductal breast cancer cells in cellular and xenograft models. Stem Cell Res. (Amst.) 2014;12:36–48. doi: 10.1016/j.scr.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 47.Pandha H.S., Martin L.A., Rigg A., Hurst H.C., Stamp G.W., Sikora K., Lemoine N.R. Genetic prodrug activation therapy for breast cancer: A phase I clinical trial of erbB-2-directed suicide gene expression. J. Clin. Oncol. 1999;17:2180–2189. doi: 10.1200/JCO.1999.17.7.2180. [DOI] [PubMed] [Google Scholar]
- 48.Grove J.I., Searle P.F., Weedon S.J., Green N.K., McNeish I.A., Kerr D.J. Virus-directed enzyme prodrug therapy using CB1954. Anticancer Drug Des. 1999;14:461–472. [PubMed] [Google Scholar]
- 49.Weedon S.J., Green N.K., McNeish I.A., Gilligan M.G., Mautner V., Wrighton C.J., Mountain A., Young L.S., Kerr D.J., Searle P.F. Sensitisation of human carcinoma cells to the prodrug CB1954 by adenovirus vector-mediated expression of E. coli nitroreductase. Int. J. Cancer. 2000;86:848–854. doi: 10.1002/(sici)1097-0215(20000615)86:6<848::aid-ijc14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 50.Bridgewater J.A., Springer C.J., Knox R.J., Minton N.P., Michael N.P., Collins M.K. Expression of the bacterial nitroreductase enzyme in mammalian cells renders them selectively sensitive to killing by the prodrug CB1954. Eur. J. Cancer. 1995;31A:2362–2370. doi: 10.1016/0959-8049(95)00436-x. [DOI] [PubMed] [Google Scholar]
- 51.Green N.K., Youngs D.J., Neoptolemos J.P., Friedlos F., Knox R.J., Springer C.J., Anlezark G.M., Michael N.P., Melton R.G., Ford M.J. Sensitization of colorectal and pancreatic cancer cell lines to the prodrug 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) by retroviral transduction and expression of the E. coli nitroreductase gene. Cancer Gene Ther. 1997;4:229–238. [PubMed] [Google Scholar]
- 52.Berger C., Flowers M.E., Warren E.H., Riddell S.R. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–2302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Traversari C., Marktel S., Magnani Z., Mangia P., Russo V., Ciceri F., Bonini C., Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 54.Garin M.I., Garrett E., Tiberghien P., Apperley J.F., Chalmers D., Melo J.V., Ferrand C. Molecular mechanism for ganciclovir resistance in human T lymphocytes transduced with retroviral vectors carrying the herpes simplex virus thymidine kinase gene. Blood. 2001;97:122–129. doi: 10.1182/blood.v97.1.122. [DOI] [PubMed] [Google Scholar]
- 55.Freeman S.M., Abboud C.N., Whartenby K.A., Packman C.H., Koeplin D.S., Moolten F.L., Abraham G.N. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274–5283. [PubMed] [Google Scholar]
- 56.Chen Y., Li H., Zhang G., Wu Y., Xiao J., Liu J., Qiu P., Liu X., Sun L., Du B., Tan Y. Synergistic inhibitory effect of resveratrol and TK/GCV therapy on melanoma cells. J. Cancer Res. Clin. Oncol. 2020;146:1489–1499. doi: 10.1007/s00432-020-03203-z. [DOI] [PubMed] [Google Scholar]
- 57.Chalmers D., Ferrand C., Apperley J.F., Melo J.V., Ebeling S., Newton I., Duperrier A., Hagenbeek A., Garrett E., Tiberghien P., Garin M. Elimination of the truncated message from the herpes simplex virus thymidine kinase suicide gene. Mol. Ther. 2001;4:146–148. doi: 10.1006/mthe.2001.0433. [DOI] [PubMed] [Google Scholar]
- 58.Kuo W.Y., Hwu L., Wu C.Y., Lee J.S., Chang C.W., Liu R.S. STAT3/NF-κB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer. Theranostics. 2017;7:647–663. doi: 10.7150/thno.16827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang Q., Monetti C., Shutova M.V., Neely E.J., Hacibekiroglu S., Yang H., Kim C., Zhang P., Li C., Nagy K. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature. 2018;563:701–704. doi: 10.1038/s41586-018-0733-7. [DOI] [PubMed] [Google Scholar]
- 60.Jo E.B., Lee H., Lee K.W., Kim S.J., Hong D., Park J.B. Complete regression of metastatic de-differentiated liposarcoma with engineered mesenchymal stromal cells with dTRAIL and HSV-TK. Am. J. Transl. Res. 2020;12:3993–4000. [PMC free article] [PubMed] [Google Scholar]
- 61.Kenarkoohi A., Bamdad T., Soleimani M., Soleimanjahi H., Fallah A., Falahi S. HSV-TK Expressing Mesenchymal Stem Cells Exert Inhibitory Effect on Cervical Cancer Model. Int. J. Mol. Cell. Med. 2020;9:146–154. doi: 10.22088/IJMCM.BUMS.9.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kievit E., Bershad E., Ng E., Sethna P., Dev I., Lawrence T.S., Rehemtulla A. Superiority of yeast over bacterial cytosine deaminase for enzyme/prodrug gene therapy in colon cancer xenografts. Cancer Res. 1999;59:1417–1421. [PubMed] [Google Scholar]
- 63.Stolworthy T.S., Korkegian A.M., Willmon C.L., Ardiani A., Cundiff J., Stoddard B.L., Black M.E. Yeast cytosine deaminase mutants with increased thermostability impart sensitivity to 5-fluorocytosine. J. Mol. Biol. 2008;377:854–869. doi: 10.1016/j.jmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cloughesy T.F., Landolfi J., Hogan D.J., Bloomfield S., Carter B., Chen C.C., Elder J.B., Kalkanis S.N., Kesari S., Lai A. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016;8:341ra75. doi: 10.1126/scitranslmed.aad9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hiraoka K., Inagaki A., Kato Y., Huang T.T., Mitchell L.A., Kamijima S., Takahashi M., Matsumoto H., Hacke K., Kruse C.A. Retroviral replicating vector-mediated gene therapy achieves long-term control of tumor recurrence and leads to durable anticancer immunity. Neuro-oncol. 2017;19:918–929. doi: 10.1093/neuonc/nox038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y., Zhu P., Huang Z., Zhou L., Shi P. Simultaneous detection of 5-fluorocytosine and 5-fluorouracil in human cells carrying CD/5-FC suicide gene system by using capillary zone electrophoresis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018;1076:1–7. doi: 10.1016/j.jchromb.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 67.Freytag S.O., Khil M., Stricker H., Peabody J., Menon M., DePeralta-Venturina M., Nafziger D., Pegg J., Paielli D., Brown S. Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer. Cancer Res. 2002;62:4968–4976. [PubMed] [Google Scholar]
- 68.Freytag S.O., Stricker H., Pegg J., Paielli D., Pradhan D.G., Peabody J., DePeralta-Venturina M., Xia X., Brown S., Lu M., Kim J.H. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003;63:7497–7506. [PubMed] [Google Scholar]
- 69.Freytag S.O., Movsas B., Aref I., Stricker H., Peabody J., Pegg J., Zhang Y., Barton K.N., Brown S.L., Lu M. Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer. Mol. Ther. 2007;15:1016–1023. doi: 10.1038/mt.sj.6300120. [DOI] [PubMed] [Google Scholar]
- 70.Freytag S.O., Stricker H., Peabody J., Pegg J., Paielli D., Movsas B., Barton K.N., Brown S.L., Lu M., Kim J.H. Five-year follow-up of trial of replication-competent adenovirus-mediated suicide gene therapy for treatment of prostate cancer. Mol. Ther. 2007;15:636–642. doi: 10.1038/sj.mt.6300068. [DOI] [PubMed] [Google Scholar]
- 71.Freytag S.O., Stricker H., Lu M., Elshaikh M., Aref I., Pradhan D., Levin K., Kim J.H., Peabody J., Siddiqui F. Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 2014;89:268–276. doi: 10.1016/j.ijrobp.2014.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kubo S., Takagi-Kimura M., Tagawa M., Kasahara N. Dual-vector prodrug activator gene therapy using retroviral replicating vectors. Cancer Gene Ther. 2019;26:128–135. doi: 10.1038/s41417-018-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grove J.I., Lovering A.L., Guise C., Race P.R., Wrighton C.J., White S.A., Hyde E.I., Searle P.F. Generation of Escherichia coli nitroreductase mutants conferring improved cell sensitization to the prodrug CB1954. Cancer Res. 2003;63:5532–5537. [PubMed] [Google Scholar]
- 74.Mitchell D.J., Minchin R.F.E. E. coli nitroreductase/CB1954 gene-directed enzyme prodrug therapy: role of arylamine N-acetlytransferase 2. Cancer Gene Ther. 2008;15:758–764. doi: 10.1038/cgt.2008.47. [DOI] [PubMed] [Google Scholar]
- 75.Teng G., Ju Y., Yang Y., Hua H., Chi J., Mu X. Combined antitumor activity of the nitroreductase/CB1954 suicide gene system and γ-rays in HeLa cells in vitro. Mol. Med. Rep. 2016;14:5164–5170. doi: 10.3892/mmr.2016.5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gungor T., Yetis G., Onder F.C., Tokay E., Tok T.T., Celik A., Ay M., Kockar F. Prodrugs for Nitroreductase Based Cancer Therapy- 1: Metabolite Profile, Cell Cytotoxicity and Molecular Modeling Interactions of Nitro Benzamides with Ssap-NtrB. Med. Chem. 2018;14:495–507. doi: 10.2174/1573406413666171129224424. [DOI] [PubMed] [Google Scholar]
- 77.Malekshah O.M., Chen X., Nomani A., Sarkar S., Hatefi A. Enzyme/Prodrug Systems for Cancer Gene Therapy. Curr. Pharmacol. Rep. 2016;2:299–308. doi: 10.1007/s40495-016-0073-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rama A.R., Zafra I., Burgos M., Prados J. On advances in cancer suicide-genes therapy. SOJ Genet. Sci. 2014;1:1–6. [Google Scholar]
- 79.Löhr M., Hoffmeyer A., Kröger J., Freund M., Hain J., Holle A., Karle P., Knöfel W.T., Liebe S., Müller P. Microencapsulated cell-mediated treatment of inoperable pancreatic carcinoma. Lancet. 2001;357:1591–1592. doi: 10.1016/s0140-6736(00)04749-8. [DOI] [PubMed] [Google Scholar]
- 80.Löhr J.M., Haas S.L., Kröger J.C., Friess H.M., Höft R., Goretzki P.E., Peschel C., Schweigert M., Salmons B., Gunzburg W.H. Encapsulated cells expressing a chemotherapeutic activating enzyme allow the targeting of subtoxic chemotherapy and are safe and efficacious: data from two clinical trials in pancreatic cancer. Pharmaceutics. 2014;6:447–466. doi: 10.3390/pharmaceutics6030447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rosenthal E.L., Chung T.K., Parker W.B., Allan P.W., Clemons L., Lowman D., Hong J., Hunt F.R., Richman J., Conry R.M. Phase I dose-escalating trial of Escherichia coli purine nucleoside phosphorylase and fludarabine gene therapy for advanced solid tumors. Ann. Oncol. 2015;26:1481–1487. doi: 10.1093/annonc/mdv196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Andrea A.E., Chiron A., Bessoles S., Hacein-Bey-Abina S. Engineering Next-Generation CAR-T Cells for Better Toxicity Management. Int. J. Mol. Sci. 2020;21:E8620. doi: 10.3390/ijms21228620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giavridis T., van der Stegen S.J.C., Eyquem J., Hamieh M., Piersigilli A., Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou X., Dotti G., Krance R.A., Martinez C.A., Naik S., Kamble R.T., Durett A.G., Dakhova O., Savoldo B., Di Stasi A. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood. 2015;125:4103–4113. doi: 10.1182/blood-2015-02-628354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou X., Di Stasi A., Tey S.K., Krance R.A., Martinez C., Leung K.S., Durett A.G., Wu M.F., Liu H., Leen A.M. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123:3895–3905. doi: 10.1182/blood-2014-01-551671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu E., Tong Y., Dotti G., Shaim H., Savoldo B., Mukherjee M., Orange J., Wan X., Lu X., Reynolds A. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]