

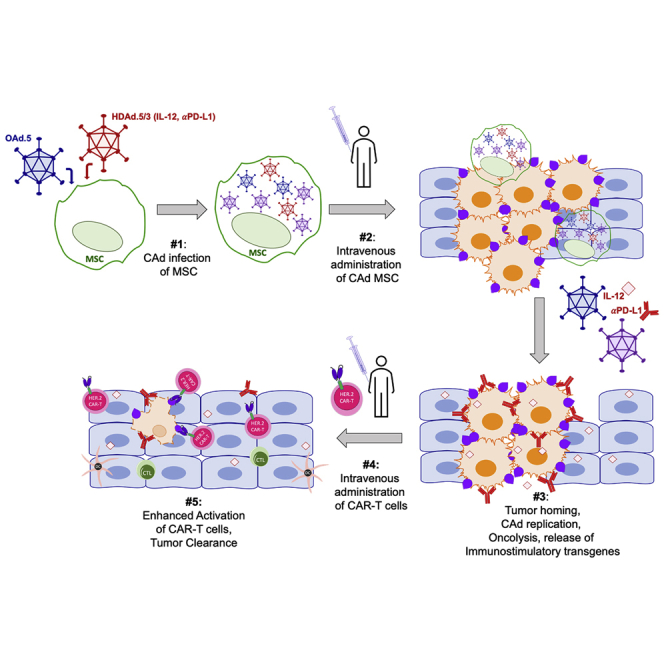
Abstract
The immunosuppressive tumor microenvironment (TME) is a formidable barrier to the success of adoptive cell therapies for solid tumors. Oncolytic immunotherapy with engineered adenoviruses (OAd) may disrupt the TME by infecting tumor cells, as well as surrounding stroma, to improve the functionality of tumor-directed chimeric antigen receptor (CAR)-T cells, yet efficient delivery of OAds to solid tumors has been challenging. Here we describe how mesenchymal stromal cells (MSCs) can be used to systemically deliver a binary vector containing an OAd together with a helper-dependent Ad (HDAd; combinatorial Ad vector [CAd]) that expresses interleukin-12 (IL-12) and checkpoint PD-L1 (programmed death-ligand 1) blocker. CAd-infected MSCs deliver and produce functional virus to infect and lyse lung tumor cells while stimulating CAR-T cell anti-tumor activity by release of IL-12 and PD-L1 blocker. The combination of this approach with administration of HER.2-specific CAR-T cells eliminates 3D tumor spheroids in vitro and suppresses tumor growth in two orthotopic lung cancer models in vivo. Treatment with CAd MSCs increases the overall numbers of human T cells in vivo compared to CAR-T cell only treatment and enhances their polyfunctional cytokine secretion. These studies combine the predictable targeting of CAR-T cells with the advantages of cancer cell lysis and TME disruption by systemic MSC delivery of oncolytic virotherapy: incorporation of immunostimulation by cytokine and checkpoint inhibitor production through the HDAd further enhances anti-tumor activity.
Keywords: oncolytic immunotherapy, CAR-T cells, mesenchymal stromal cells, oncolytic adenovirus, helper dependent adenovirus, systemic delivery, HER.2, lung cancer, solid tumor
Graphical abstract
Oncolytic adenoviruses (OAds) disrupt the tumor microenvironment. Their activity is enhanced by engineering them to release immunostimulatory factors. Here, McKenna and colleagues use mesenchymal stromal cells to systemically deliver oncolytic and helper adenoviruses to the tumor. The OAds further replicate and augment the anti-tumor activity of CAR-T cells.
Introduction
The tumor microenvironment (TME) comprises stromal cells, aberrant vasculature, and an extracellular matrix (ECM) that limit the infiltration and, thus, success of chimeric antigen receptor (CAR-T) cells for the treatment of solid tumors. In addition, the TME produces a potent network of immune-inhibitory ligands, cytokines, and chemokines1, 2, 3 that diminish cytotoxic T cell responses and skew infiltrating myeloid cells to an inhibitory phenotype.4 Due to the complexity of the TME and heterogeneity of solid tumor antigen expression, any approach targeting a single antigen with a single effector will likely fail to eliminate tumors.5,6 Therefore, we are studying therapies in animal models and now in humans (NCT03740256) that both disrupt the TME and favor recruitment of a broad immune response, promoting T cell persistence. Here we investigate the combination of cell-carrier-delivered oncolytic adenovirus (OAd) and CAR-T cells to achieve these prerequisites for the treatment of lung tumors.
Oncolytic virotherapy (OV) can kill tumor cells and stimulate bystander T cells through their T cell receptors (TCRs) to initiate and amplify the host anti-tumor immune response.7 OV can be genetically modified to promote an immune response to the tumor they damage and an oncolytic herpes virus that also encodes human granulocyte-macrophage colony-stimulating factor (GM-CSF) sequence (talimogene laherparepvec [TVEC]) has been approved for the treatment of melanoma.8
Direct intralesional administration of OVs can be problematic for many tumor locations,9 while intravenous administration is often precluded due to innate or adaptive immune responses to the OV.10 Cell carrier systems, such as mesenchymal stromal cells (MSCs), can overcome this limitation through inherent tumor tropism,11 have been successfully used to deliver OAd to tumors,12,13 and have been adapted for human use.14
Here, we use MSCs to deliver a combinatorial Ad vector (termed CAd) consisting of OAd and helper-dependent adenovirus (HDAd), which has a 34 kb cargo capacity and can express a broad array of immunomodulatory transgenes in a single vector.15 The tropism of the HDAd can be engineered to infect non-malignant cells within the TME and produce sustained release of immunostimulatory cytokines, intended to increase the potency and persistence of anti-tumor immune responses.15 To show the ability of MSC-delivered CAd to enhance immune mediated tumor destruction, we co-administered these cells with clinically validated tumor-directed HER.2 CAR-T cells.16 Our results demonstrate the combination of oncolysis and secretion of interleukin-12 (IL-12) and programmed death-ligand 1 (PD-L1) blocking antibody by CAd-infected MSCs augments the anti-tumor activity of HER.2 CAR-T cells9,15,17 in a range of in vitro and in vivo models and tumors.
Results
MSCs are susceptible to CAd infection and produce functional cytotoxic virus
We first assessed whether bone-marrow-derived MSCs from healthy donors were able to replicate both OAds and HDAds and express their transgenes. We infected MSCs with OAd (100 vp/cell) and a HDAd-expressing green fluorescent protein (GFP; 1,000 vp/cell) and compared GFP expression in these MSCs over time with expression by A549 cells directly infected with virus as a standard18 (Figure 1A). MSCs expressed HDAd GFP more slowly than A549 cells but reached peak intensity after 72 h post infection (Figure 1B). By contrast, GFP intensity in A549 began to decline by 40 h, indicating oncolysis (Figure 1A). We confirmed expression of both oncolytic and helper-dependent viral genes in MSCs by qPCR at 24 and 72 h post infection (Figure S1A). MSCs are defined by co-expression of CD73, CD90, and CD105 while remaining negative for the following hematopoetic lineage markers: CD34, CD45, CD14, CD19, and CD3.19 We found no shift in phenotype after co-infection of OAd and HDAd encoding IL-12 and anti-PD-L1 (Figure S1B).
Figure 1.

MSCs are susceptible to combinatorial Ad vector (CAd) infection and produce functional cytotoxic virus
(A) MSCs (left) and A549 lung tumor cells (right) were infected with 100 vp OAd.5 and 1,000 vp HDAd.5/3-expressing GFP. GFP expression was detected by Incucyte live image analysis over time indicated by GCU, green object intensity. (B) MSCs were imaged 72 h post infection with 1,000 vp HDAd.5/3-expressing GFP. Cells were collected and percent GFP was measured by flow cytometry. (C) MSCs were infected with 100 vp OAd.5-expressing RFP and 1,000 vp HDAd.5/3-expressing GFP. Supernatant was collected 72 h post infection and applied to A549 and H1650 cells at 100%, 50%, and 25% dilutions. The number of cells expressing both RFP and GFP were measured by Incucyte indicated by Overlap Object count/well. (D) Viability was measured at 6 days post supernatant addition through 7-AAD and Annexin V staining analyzed by flow cytometry. Significance was determined by Student’s t test for each dilution compared to the no-treatment (No-tmt) group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Error bars represent standard deviation. (E) Infected MSCs co-cultured with different ratios of tumor cells 24 h post MSC infection. Tumor cell viability was determined by gating on CD90-A549 cells and measured 7-AAD and Annexin V staining (left). The percentage of MSCs remaining in co-culture after 5 days is shown to the right as determined by CD90 positivity through fluorescence-activated cell sorting (FACS) analysis. (F) Viability of MSCs post infection in vitro over time. MSC cell death determined by Annexin V+ 7-AAD+ staining and analyzed by flow cytometry. n = 2 MSC donors.
To determine whether MSCs produced functional virus, we infected MSCs and 72 h later transferred the infected-cell supernatant onto two non-small cell lung cancer (NSCLC) cell lines (A549, H1650). We measured infection of our target cells by OAd red fluorescent protein (RFP) and HDAd GFP expression (Video S1) and quantified co-expressing target cells over time. Serial dilutions of the supernatant resulted in dose-dependent expression of fluorescent protein transgenes, which translated into dose-dependent cell death of NSCLC cells (Figures 1C and 1D).
MSCs were infected with 100 vp/cell OAd-expressing RFP and 1,000 vp/cell HDAd-expressing GFP. Supernatant was collected 72 h later and applied to A549 (left) and H1650 (right) cell lines. Images were acquired every 2 h by Incucyte Live Cell image analysis for 3 days.
After confirming that MSCs replicated and expressed all components of the CAd system, we co-cultured CAd MSCs with tumor cells and measured target cell death. We replaced the RFP- and GFP-expressing proteins with an OAd- and HDAd-expressing IL-12 and anti-PDL1 (CAd12_PD-L1) as previously described.20 MSCs infected with OAd or CAd system killed A549 cells at tumor to MSC ratios ranging from 1:1 to 50:1 (Figure 1E). We found reduced frequency of MSCs after 5 days culture, correlating with their death post infection. Figure 1F shows that 90% of MSCs are apoptotic on day 6 post infection by Annexin V and 7-AAD staining unlike uninfected MSCs or MSCs infected by non-replicating HDAd that remain viable.
We confirmed that MSCs produced functional transgenes encoded by the HDAd, IL-12, and anti-PDL1, by measuring IL-12 secreted from MSCs (Figure S2A). Additional IL-12 was produced when MSC supernatant was applied to tumor cells, indicating the potential for continued infection of tumor cells over this period and consequent replication of MSC-produced CAd (Figure S2B). The IL-12 produced by tumor cells after infection with MSC-derived CAd supernatant was functionally active, inducing phospho-STAT4 signaling in activated T cells (Figure S2C). Similarly, PD-L1 minibody was produced from the MSC of multiple donors in sufficient quantity to block PD-L1 detection on interferon-γ (IFN-γ) stimulated NSCLC cell lines, in contrast to the lack of effect with media or supernatant from uninfected MSC supernatant (p = 0.002 [A549] and 0.0001 [H1650]; Figure S2D).
To determine the specificity and cytotoxicity of the CAd system in the lung tumor microenvironment, we infected MSCs with both OAd- and HDAd-expressing GFP and then transferred the supernatant 24 h later to either A549 and H1650 tumor cells or multiple fibroblast lines (primary normal human lung fibroblasts [NHLFs], MRC5, HS-5, and Tig320) and monitored cell viability by Annexin V and 7-AAD staining. We confirmed that fibroblasts expressed HDAd transgenes by detection of transgenic GFP expression, but we observed no significant increase in cell death compared to uninfected fibroblasts (Figure S3), compared to a doubling of cell death observed in A549 tumor cells at 48 h after the application of supernatant. These data support the tumor-specific replication of CAd.
Thus, MSCs can simultaneously replicate both OAd and HDAd to produce infectious and functional particles that destroy tumor cells and the MSCs themselves and induce expression of the desired immunomodulatory transgenes.
CAd12_PD-L1 MSCs enhance antitumor activity of HER.2 CAR-T cells in vitro
HER.2-specific CAR-T cells with CD28ζ signaling domain have been safely used to treat NSCLC (NCT00902044) and currently are being evaluated in patients with glioblastoma, osteosarcoma, and head and neck cancers.9,21,22 We tested the efficacy of these HER.2 CAR-T cells against two NSCLC cell lines in a 96 h co-culture assay. The cells were able to control A549 growth but not completely eliminate tumor cells (Figure 2A); their activity against the H1650 lung cancer cell line was greater, despite similar levels of Her2/neu antigen expression (Figure 2B).
Figure 2.

CAd12_PD-L1 MSCs enhance antitumor activity of HER.2 CAR-T cells in vitro
(A and B) Co-culture of HER.2-specific CAR-T cells with target NSCLC cell lines (A) A549 and (B) H1650 at a ratio of 4 targets to 1 effector T cell overtime and respective Her2/neu surface antigen expression on each tumor line. Error bars represent standard deviation. (C and D) CAd12_PD-L1 or uninfected MSCs were co-cultured 24 h after infection at a ratio of 1 MSC to 10 (C) A549 cells or (D) H1650 cells. 48 h after co-culture, HER.2-specific CAR-T cells or non-transduced (NTR) T cells were added to the culture at a 1:4 effector to target ratio. Residual tumor cells were collected 48, 72, and 96 h later and quantified by flow cytometry. p values determined by Student’s t test.
To test the combinatorial treatment of CAd MSCs with CAR-T cells, we first infected MSCs with our CAd system and then set up a co-culture of MSCs at a ratio of 1:10 with GFP-labeled tumor cell lines. We added either non-transduced (NTR) or CAR-T cells 48 h later at a ratio of 1:4 with tumor cells. Residual tumor was quantified 48, 72, and 96 h post T cell addition by flow cytometry (Figures 3C and 3D). Compared to culture with uninfected MSCs and no treatment, the addition of CAd MSCs enhanced CAR-T cell killing and significantly reduced viable tumor cells in both lung cancer lines (A549: 96 h p = 0.06; H1650: 48 h p = 0.002, 72 h p = 0.007, 96 h p = 0.011, determined by Student’s t test). These measures of cytotoxicity were replicated in an MTS (viability) assay using the H1650 cell line in which we observed significant reduction in tumor cell viability in the presence of CAd MSCs and HER.2 CAR-T cells in combination, compared to HER.2 CAR-T alone (p = 0.001; Figure S4).
Figure 3.

CAdIL-12_PD-L1 MSCs enhance HER.2 CAR-T cell polyfunctionality
Uninfected MSCs or CAd IL-12_PD-L1 infected MSCs were co-cultured at a 1:10 ratio to A549 tumor cells for 48 h. NTR or CAR-T cells were then added to the culture or added to tumor only. We isolated T cells 20 h later and selected for CD4 and CD8 populations. We measured single cell cytokine secretion on the Isoplexis platform for 32 different cytokines. (A and D) Umap analysis shows clusters of CD4 (A) and CD8 (D) cells color coded by each condition. We determined polyfunctionality by the number of cytokines secreted from each cell and represented as a fraction in selected population. (B) Polyfunctionality and secreted cytokines for inset #1, uninfected MSC, and NTR CD4 T cells. (C) Polyfunctionality and secreted cytokines for inset #2, CAd MSC, and HER.2 CD4 T cells. (E) Polyfunctionality and secreted cytokines for inset #3, CAd MSC and HER.2 CD8 T cells. (F) Polyfunctionality and secreted cytokines for inset #4, non-MSC, and HER.2 CD8 T cells. (G) Overall secretion frequency of CD4 and CD8 populations. Percent of population secreting is represented by color intensity for each detected cytokine. NTR, n = 2 donors; HER2, n = 3 donors for each MSC condition.
CAd12_PD-L1 MSCs enhance effector function of CD4+ HER.2 CAR-T cells
To investigate the mechanisms through which CAd-infected MSCs enhance the efficacy of HER.2 CAR-T cells, we measured the cytokine polyfunctionality of individual T cells, a characteristic linked in pre-clinical and clinical studies to increased anti-tumor activity.23 No Her2/neu expression was detected on MSCs from healthy donors (Figure S5) so that the CAR-T cells recognize Her2/neu on the malignant population and not the MSC. CD4 HER.2 CAR-T cells showed increased cytokine secretion frequency when co-cultured with CAd MSCs (36.8%) compared to no MSCs (18.5%) or uninfected MSCs (25.8%; Figure S6). When stimulated with antigen-expressing tumor cells, 18% CD4 and 30% CD8 HER.2 CAR-T cells secrete cytokine, but after CAd MSC addition these numbers increased to 36% and 38%, respectively. Of cells that secrete cytokine, 34% of the CD4 and 40% of CD8 CAR-T cells secreted more than one cytokine in the CAd MSC treatment group compared to 5% CD4 and 16% CD8 CAR-T cells in the HER.2 only group (Figure S7). These results support the ability of combination therapy to improve the activity of HER.2 CAR-T cells by increasing the number of cells secreting multiple cytokines from CD4 T cells. Further analysis of T cells 72 h after exposure to tumor and CAd MSCs showed an increase in the proportion of cells expressing PD-1 in both CD4 and CD8 populations compared to cells exposed to uninfected MSCs alone (Figure S8).
UMap analysis segregated clusters of distinct CD4 (Figure 3A) and CD8 populations (Figure 3D) dependent on the number and types of cytokines secreted (Figure S9). Figure 3B shows a subset of NTR CD4 cells co-cultured with uninfected MSCs that primarily secrete IL-8. Another cluster shown in inset #2 of Figure 3A correlates to CD4 HER.2 CAR-T cells cultured with CAd MSC. These cells secreted up to 9 cytokines per cell, with the majority simultaneously secreting IFN-γ, granzyme B, and CCL11 (Figure 3C). Figure 3D shows the UMap depiction of CD8 T cells where particular populations emerged based on treatment group. Inset #3 highlights CD8 HER.2 CAR-T cells stimulated with tumor without MSCs that express 4–6 cytokines per cell. These cells secreted effector cytokines IFN-γ, granzyme b, tumor necrosis factor alpha (TNF-α) and low levels of perforin but also high levels of macrophage inflammatory protein 1α (MIP1α) and MIP1β (Figure 3E). MIP1 is primarily secreted by macrophages and monocytes, but upon activation, T cells also can secrete it.24 CD8 T cells secreting MIP1α and MIP1β have been associated with a non-cytolytic phenotype to control HIV viral replication.25 Inset #4 draws attention to CD8 HER.2 CAR-T cells cultured with CAd MSCs secreting between 2–7 cytokines per cell. These cells still secreted IFN-γ, granzyme b, TNF-α, and perforin, but lost the ability to secrete MIP1α and MIP1β (Figure 3F). This CAd-MSC associated shift in CD8 CAR-T cell phenotype is consistent with induction of greater cytolytic activity26,27 although the consequences of reduced MIP1α/β secreted from CAR-T cells are less clear.
We also found that CAd MSCs enhanced CD4 and CD8 CAR-T cell secretion of CCL11, a chemokine that has been associated with increased adhesion molecules and T cell chemotaxis28 (Figures 3C and 3F).
Unmodified T cells and uninfected MSCs produced low levels of cytokines other than IL-8 (Figure 3G). While CD8 HER.2 CAR-T cells generally expressed pro-inflammatory cytokines, as well as perforin and granzyme B following exposure to tumor, this pattern was uninfluenced by MSCs. In contrast, CD4 HER.2 CAR-T cells increased secretion of IFN-γ, granzyme B, perforin, and TNF-α when cultured with CAd MSCs. Thus, CAd MSCs increase production of pro-inflammatory mediators by CD4 CAR-T cells upon exposure to cognate tumor.
CAd12_PD-L1 MSCs increase cytotoxicity of HER.2 CAR-T cells in 3D tumor spheroids
To understand how the TME structure responds to combination therapy, we established a 3D tumor model to better recapitulate the physical structure of solid tumors. We generated multicellular tumor spheroids with both A549 and H1650 tumor cell lines by forced suspension of 2 x 103 tumor cells followed by the addition of MSCs 24–48 h later. We labeled MSCs with cell trace dye to visualize their integration into the spheroids (Figure 4; Video S2). We followed viral spread from MSCs infected with OAd expressing RFP within the tumor spheroids with the Incucyte live imaging system. The level of RFP expression was dependent on the initial number of MSCs added to the tumor spheroids (Figure 4B).
Figure 4.

CAd12_PD-L1 MSC improves HER.2 CAR-T cytotoxicity in 3D tumor spheroids
(A) 2 x 103 tumor cells were added to 96-well agarose-coated plates. 24 h after spheroid generation, 400 eflor 450 labeled uninfected of CAd_RFP infected MSCs were added to culture. Fluorescent images were acquired 24 h after MSC addition. Phase images are shown for tumor-only control. Scale bar represents 100 μM. (B) RFP virus detected in H1650 GFP tumor spheroids on day 2 and day 5 post MSC addition by confocal microscopy post 400 MSC addition. Scale bar represents 100 μM. RFP expression was measured overtime by Incucyte imaging system indicated by total red object integrated intensity (RCU/image). (C) 2 x 103 cell H1650 tumor spheroids were generated. 24 h later, 200 CAd-infected MSCs were added followed by 12 NTR or 12 HER2 CAR-T cells 48 h later. Tumor cell cytotoxicity was measured by Annexin V AF594 (Red) staining overtime detected by Incucyte imaging analysis. p = 0.0186 determined by linear regression analysis, n = 4 T cell donors.
2,000 GFP+ H1650 or A549 lung tumor cells were seeded to an agarose coated 96-well plate. 200 uninfected or CAd infected were stained with efluor 670 and added 48 h after spheroid generation. CAd-infected MSCs were infected with 100 vp/cell OAd-expressing RFP and 1,000 vp/cell HDAd-expressing IL-12 and PD-L1 blocker. Images were collected at time of MSC addition and then every 24 h for 4 days on a GE Healthcare DeltaVision LIVE High Resolution Deconvolution Microscope.
To evaluate the anti-tumor effect of both CAd MSCs with CAR-T cells in our multicellular tumor spheroids, we generated the spheroids and then added either 200 uninfected or CAd infected MSCs 24 h later. Consistent with our 2D studies, we kept the 1:10 MSC to tumor ratio. After 48 h of co-culture, 50 HER.2 CAR-T cells were added per spheroid, and we measured tumor cell death by Annexin V staining. Video S3 shows the destruction of the tumor spheroid by HER.2 CAR-T cells, HER.2 CAR-T cells, and uninfected MSCs or HER.2 CAR-T cells with CAd MSCs compared to respective NTR T cell and MSC combinations. We further stressed the system by reducing the number of CAR-T cells from 50 to 12 (T cell to tumor ratio of 1:167). Even in this condition, we observed greater cytotoxicity with the combination of CAd MSCs and CAR-T cells compared to CAR-T cells only (p = 0.0186). Thus, CAd MSCs increase the tumor cell killing of HER.2 CAR-T cells compared to either monotherapy alone (Figure 4C).
2,000 GFP+ H1650 lung tumor cells were seeded to an agarose coated 96-well plate. 200 MSCs were added 48 h later followed by 50 NTR or HER2-specific CAR-T cells. GFP fluorescence indicated viable tumor with RFP Annexin V+ staining used to show tumor cell apoptosis. Images were acquired every 2 h for 5 days on the Incucyte.
Intravenous infusion of CAd MSCs produces virus at primary tumor site
To discover whether these in vitro results could be replicated in vivo, we first analyzed the biodistribution of MSCs and the viruses they delivered. We tracked MSC homing by administering firefly luciferase-expressing MSCs via tail vein injection into mice with and without lung tumors at a ratio of 3 MSCs:1 tumor cell. We detected MSCs in the lung by IVIS bioluminescence imaging up to 5 days post infusion (Figure S10).29 To demonstrate MSC production of OAd in vivo, we measured RFP virus expression in relation to GFP-labeled A549 tumor cells. In tissue sections, we detected OAd co-expression in GFP-positive tumor cells, while surrounding stromal cells were GFP negative (Figure S11), confirming our in vitro observation that OAd produced by MSCs can infect both cell types (Figure S3) and demonstrating that stromal cells can be transduced with CAd to broaden the cellular distribution of the immunomodulatory transgenes within primary tumors.
CAd12_PD-L1 MSCs improve HER.2 CAR-T cell anti-tumor activity in vivo
We assessed the in vivo anti-tumor activity of each treatment alone and in combination in an orthotopic NSCLC xenograft model using A549 lung cancer cells. The combination of CAd MSCs and HER.2 CAR-T cells markedly reduced tumor growth compared to either treatment alone (Figure S12A). HER.2 CAR-T treatment alone reduced tumor growth initially but could not eradicate tumors, and uninfected MSCs marginally increased tumor growth, likely due to their anti-inflammatory properties.30 However, though CAd MSCs alone produced no control of tumor expansion (Figure S13), we observed that tumor growth was prevented from day 10 to day 22 in the CAd MSC and NTR T cell group (Figure S12B) suggesting that the immunostimulation of the HDAd encoded transgenes promoted T cell-mediated anti-tumor activity, albeit of limited potency and duration. Because we observed limited tumor control even with non-modified T cells, we re-challenged the mice with the same A549 tumor line. Only animals who received CAd MSCs and HER.2 CAR-T cells could prevent tumor growth upon second challenge (Figure S14). Furthermore, analysis of T cells in the blood, spleen, and lung tissue showed CD8+ T cells in the CAd MSC + CAR-T cell group after re-challenge exhibited reduced expression of exhaustion marker Tim3 compared to CAR-T cell only group, supporting the ability of CAd MSCs to promote functional T cell persistence.
Studies in the H1650 xenograft lung tumor model produced results consistent with the A549 model. Combination treatment cleared tumors within 2 weeks of HER.2 CAR-T infusion (Figure 5A) and appeared to be well tolerated: mice showed no weight loss or degradation of body condition in any combination (Figure S15). Furthermore, we could not detect residual H1650 tumor in bioluminescence imaging of mice treated with CAd MSC and HER.2 CAR-T cells, confirming superior anti-tumor activity with combination treatment. In the H1650 lung tumor model, we evaluated T cell infiltration in the lungs of animals on day 18 post tumor engraftment (Figure S16). Immunohistochemistry revealed increased T cell infiltration in lungs of mice treated with CAd MSCs and HER.2 CAR-T cells compared to other groups. These results are congruent with the improved T cell activity shown in tumor spheroid models and the detection of T cells in the peripheral tissues (Figure S16B). Similar to our in vitro observations (Figure 1C), H1650 was minimally susceptible to CAd MSC treatment alone in vivo.
Figure 5.

CAd12_PD-L1 MSC improves anti-tumor activity of HER2 CAR-T cells
(A) NSG mice were engrafted with GFP-Ffluc-labeled H1650 cells intravenously followed by intravenous infusion of 2 × 106 uninfected or CAd12_PD-L1 MSCs on day 3. On day 6, mice were infused with 5 × 105 NTR or HER2-specific CAR-T cells. Fold change was determined from baseline imaging on day 3 post tumor engraftment. p values were determined by Mann-Whitney U test. (B) Tumor growth was monitored by in vivo imaging to measure bioluminescence ∗p = 0.01 as determined by Student’s t test, n = 4–5 per group. Bioluminescent animal images are shown for the last day of measurement. (C) 5 × 105 NTR or HER2-specific CAR-T cells labeled with GFP-Ffluc were injected 48 h post MSC infusion and detected by bioluminescence imaging. p = value determined by non-linear regression analysis between HER2 CAR-T cell only control and CAd MSC + HER.2 CAR-T cells. (D) T cells were isolated from tissues on days 6, 13, and 28 post infusion. PD-1 expression for one representative mouse is shown in the histogram measured by flow cytometric analysis. The average percent of PD-1 expression for CD4 and CD8 T cells is shown in the line graph. (E) Residual tumor was stained for Her2/neu surface expression. p value is determined by Student’s t test. Error bars represent standard deviation.
To measure the influence of CAd MSC on T cell activity in vivo, we labeled the infused HER.2 CAR-T cells with GFP Ffluc to track their homing and persistence. To confirm the presence of CAd MSC produced transgenes, we measured IL-12 in the plasma of mice on days 6, 13, and 28 post T cell infusion and detected IL-12 only in animals receiving CAd MSC (Figure S17). We saw no difference in the strength or duration of the overall HER.2 CAR-T cell signal between animals receiving CAd MSC versus uninfected MSC groups (Figure 5C). Isolated lung tissue, however, had greater T cell signal intensity and persistence, as reflected in an increased area under the curve when plotting time versus signal intensity (Figure S18). There was no significant difference in the CD4 and CD8 ratios between different treatment groups (data not shown) nor in their pattern of CCR7/CD45RA expression (Figure S19). Analysis of in vivo T cell activation-associated markers, CD69, CD25, 41BB, and PD-1 revealed higher expression on day 6 when HER.2 CAR-T cells were combined with CAd MSC compared to other conditions (Figure S20). Higher T cell activation was also reflected in increased IFN-γ detected in the serum of mice treated with CAd MSCs and HER.2 CAR-T cells on day 6 compared to other groups (Figure S21). PD-1 expression was primarily elevated on CD8 T cells (Figure 5D) at multiple time points (Figure S22).
Discussion
We showed that MSC co-delivery of OAd and HDAd encoding IL-12 and anti-PD-L1 can both directly disrupt tumor spheroid structures in vitro and augment CAR-T cell responses against orthotopic tumors in vivo in two distinct NSCLC models. Systemic delivery of CAd by MSCs enhanced virus expression at the primary tumor site and improved CAR-T cell infiltration and expansion. Mechanistically, combination treatment of CAd MSCs with CAR-T cells improved T cell infiltration into bulky tumors, enhanced effector cell function, and improved IFN-γ, granzyme B, and perforin production.
MSCs have been used to deliver Ad-based treatments in multiple tumor models.31, 32, 33, 34 MSCs are successful allogeneic cell carriers because of low MHC expression while maintaining the ability to shelter viruses from immune destruction. MSCs administered intravenously, however, are primarily trapped in the lungs due to the pulmonary first pass effect.29 We previously used MSCs to deliver an OAd coupled with a suicide gene therapy in an orthotopic NSCLC model.12,13 These studies found that MSCs successfully traveled to the lung and expressed OAd exclusively at the tumor site with no evidence of Ad infection in other tissues including heart, kidney, and spleen. Allogeneic MSC produce little alloreactivity, and banked MSCs from unrelated donors have been widely and safely used clinically.35 As anticipated, therefore, we observed no evidence for alloreactivity between MSCs and therapeutic T cells. In clinical studies, the risk of alloreactivity could be further reduced by staggering administration of CAR-T cells until the MSCs have completed delivery of their CAd and undergone lysis.
In this study, we targeted lung cancer36,37 and altered the MSC payload by incorporating HDAd, which has the capacity to encode 34 kb of transgenes. This CAd allows expression of multiple immunomodulatory molecules that can be readily varied. Moreover, since the HDAds are non-lytic, modifications to their serotype permit entry and transgene expression in non-malignant cells of the TME with sustained release of immunomodulatory agents.
MSCs may exert a pro- or anti-tumor effect when delivered therapeutically.38,39 Although we observed a pro-tumorigenic effect with uninfected MSCs, CAd infection eliminated this activity likely due to a combination of oncolysis and immune stimulation. For example, uninfected MSCs promote IL-8 secretion from NTR T cells (Figure 3B), which is associated with induction of angiogenesis by endothelial cells and cancer cell proliferation.40 In contrast, CAd MSC infection eliminated IL-8 secretion from T cells, suggesting that CAd MSCs will beneficially modulate the TME. Additionally, we observed that CAd MSCs enhanced CCL11 from HER2 CAR-Ts. CCL11, or Eotaxin-1, is a chemokine produced by a variety of cells that is implicated in the recruitment of eosinophils to inflammatory sites.41 CCL11 also has been shown to play a role in eosinophil immunosurveillance, as CCL11 knockout mice have increased fibrosarcoma growth associated with poor recruitment of eosinophils to the tumor.42 As eosinophils can infiltrate tumors and are associated with improved prognosis,43 increased secretion of CCL11 may be one mechanism through which CAd MSCs improve CAR-T cell antitumor activity. While we did not measure other factors produced from CAd MSCs that may influence the polyfunctionality of T cells, we are able to confirm that the transgenes IL-12 and anti-PD-L1 provided by CAd enhance T cell effector cytokine production.
3D cultures of tumor cells can recapitulate the biophysical characteristics of solid tumors better than conventional 2D tissue culture44 by incorporating cell-cell and cell-matrix interactions and maintaining cell polarity.45,46 These considerations are particularly important in studies of OAd spread, for which 2D cultures poorly predict in vivo performance.47,48 Previous studies have characterized cancer cells located at the perimeter of tumors that interact with the surrounding environment exhibit distinct gene-expression patterns compared to interior cells, of which affects OAd infectivity.49,50 Here, we test our combination therapy in 3D tumor spheroids in which only 23% of cells are exposed to the surface to mimic the structure of solid tumors. MSCs were absorbed into tumor spheroids allowing for CAd spread over time, expanding and disrupting the spheroid structure. The addition of T cells then allowed perimeter targeting of the spheroid and complete destruction. With similar cellular structure and heterogeneity, this 3D modeling system may more predictively measure the cytotoxic kinetics of both OAd and CAR-T cells for clinical translation.47,51 Together, both therapies improved the kinetics of tumor killing, which could be attributed to the enhanced CD4 HER.2 CAR-T cell cytolytic function when cultured with CAd MSCs.
Polyfunctional T cells that secrete more than one cytokine have superior anti-tumor activity compared to single secretors.23,52 Our studies show that CAd MSCs enhance CD4 HER.2 CAR-T cell effector cytokine secretion in the presence of tumor (Figure 3G). Although both CD4 and CD8 CAR-expressing T cells can target and kill tumor cells, CD4 T cell killing is often delayed due to less intracellular granzyme and perforin expression.53, 54, 55 CD4 HER.2 CAR-T cells cultured with CAd MSCs overcame this delay and their pattern of cytotoxic effector molecule expression more closely resembled CD8 CAR-T cell cytokine and perforin secretion. CAd expressing IL-12 could improve CD4 cytotoxicity, as the cytokine has been shown to alter TCR signaling to stimulate a range of cytokine secretion.56,57 Additionally, adenovirus infection itself has been shown to stimulate IFN-γ secretion from CD4 virus-specific T cells.58
Importantly, oncolysis of tumor cells by OVs releases tumor antigens that may recruit additional T cells through epitope spreading and skew the TME to a more pro-inflammatory setting.59 Although our current models did not allow us to directly interrogate epitope spreading, we observed an increase in the frequency of NTR CD8 T cells secreting pro-inflammatory cytokines after exposure to CAd MSCs and tumor cells (Figure S6) and mice treated with the combination therapy were now resistant to tumor re-challenge. These data are all consistent with improved T cell recruitment, and it will be of interest to develop models that can discern whether the increased T cell recruitment and associated anti-tumor effects are in part attributable to epitope spreading to additional tumor-associated antigens.
We demonstrated a multi-pronged approach that overcomes the limitations of OAd and CAR-T cell monotherapies by bypassing pre-existing immune OAd neutralization with our MSC carrier system. Overall, this combinatorial treatment enhances CAR-T cell activity through release of HDAd-encoded immunostimulatory transgenes and, despite the immunosuppressive TME, improves T cell tumor infiltration. As both HER.2 CAR-T cells and MSCs have been proven safe in the clinic (NCT00902044, NCT02530047) and a current clinical trial combining locally injected CAd and HER.2 CAR-T cells (NCT03740256) is approved to accrue patients, the proposed system should be suited for clinical translation.
Materials and methods
Cells and culture condition
A549 and H1650 cell lines were obtained from ATCC (Manassas, VA, USA). GFP Ffluc-expressing cell lines were generated by gamma retroviral transduction with PG13 vector packaging cell line. MSCs were isolated from healthy donors as previously described and used between the 3rd and 6th passages.12 Tumor cell lines and MSCs were maintained in Dulbecco’s modified Eagle’s medium (DMEM; supplemented with 10% fetal bovine serum; GE Healthcare Life Sciences, Marlborough, MA, USA) and 2 mmol/L L-glutamine. All cell lines were routinely tested for mycoplasma using the Mycoalert detection kit (Lonza, Basal, Switzerland).
Adenoviral vectors and transduction
Both OAd and HDAd expressing IL-12p70 and PD-L1 blocker were produced as previously described.9,15,20 For adenoviral transduction, MSCs or tumor cells were plated in 6-well plate at 2 × 106 cells per well and cultured for 24 h in DMEM media. Media was then aspirated and replaced with 350 μL incomplete Iscove’s Modified Dulbecco’s Media (IMDM). 100 vp/cell of OAd and 1,000 vp/cell of HDAd were then added to each well. Plates were incubated at 37°C and rocked every 10 min for 1 h followed by every 20 min for another 2 h. 650 μL of complete IMDM was then added to each well. 24 h later, MSCs or tumor cells were then washed three times with PBS and either used for assays or replaced with 2 mL of DMEM media. To further transduce tumor cells with MSC-produced virus, we harvested media from infected MSCs 72 h post infection and applied it to tumor cell lines.
Quantitative PCR for viral gene expression
We infected MSCs with 100 vp/cell OAd and 1,000 vp/cell HDAd and collected them at the time points indicated (24 h and 72 h post infection). Total DNA was extracted from cells and quantified with primer sets as previously described (Table 1).17
Table 1.
Primer sets
| Primer name | Sequence |
|---|---|
| OAd fwd | 5′-TCCGGTTTCTATGCCAAACCT-3′ |
| OAd rev | 5′-TCCTCCGGTGATAATGACAAGA-3′ |
| HDAd fwd | 5′-TCTGAATAATTTTGTGTTACTCATAGCGCG-3′ |
| HDAd rev | 5′-CCCATAAGCTCCTTTTAACTTGTTAAAGTC-3′ |
| GAPDH fwd | 5′-CATGCCTTCTTGCCTCTTGTCTCTTAGAT-3′ |
| GAPDH rev | 5′-CCATGGGTGGAATCATATTGGAACATGTAA-3′ |
Flow cytometry
We collected MSCs after adenoviral infection and stained for CD90, CD105, and CD73 and a cocktail of CD34, CD45, CD14, CD3, CD19, and HLA-DR (BD Biosciences, San Jose, CA, USA). T cells were stained intracellularly for phoso-Stat4 according to the manufacturer’s protocol (BD Biosciences, San Jose, CA, USA), and tumor cells were stained for PD-L1. Data were acquired on a BD CANTO II flow cytometer for all assays, with the exception of Figure 5, measuring T cell phenotype and activation that used Cytek Aurora Spectral Flow Cytometer and analyzed by FlowJo software.
IL-12 and IFN-γ ELISA
For IL-12 detection in vitro, we infected MSCs with 100 vp/cell OAd and 1,000 vp/cell HDAd IL-12_PD-L1 according to protocol.12 Supernatant was collected 72 h later and analyzed for IL-12 secretion (R&D Systems, Minneapolis, MN, USA). We applied MSC supernatant to A549 tumor cells and sampled at various time points to measure IL-12 continually produced from tumor cells. For in vivo experiments, plasma was isolated from mice at indicated time points and then analyzed for both IFN-γ and IL-12 by ELISA according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN, USA).
HER.2 CAR-T cell generation
We obtained healthy donor peripheral blood mononuclear cells (PBMCs) through an institutional review board (IRB)-approved protocol at Baylor College of Medicine. PBMCs were isolated using Lymphoprep according to the manufacturer’s instructions (Axis-Shield PoC AS, Dundee, Scotland). T cells were activated in 24-well non-tissue culture-treated plates coated with OKT3 (1 mg/mL; Ortho Biotech, Bridgewater, NJ, USA) and NA/LE (no azide/low endotoxin) anti-human CD28 antibodies (1 mg/mL; BD Biosciences, San Jose, CA, USA) at a density of 1 × 106 cells per well. Primary human T cells were cultured in complete CTL media containing 45% RPMI-1640 media (Hyclone Laboratories, Marlborough, MA, USA), 45% Click’s medium (Irvine Scientific, Santa Ana, CA, USA), 10% heat-inactivated FBS (Hyclone Laboratories, Marlborough, MA, USA), 100 U/mL Pen Strep, and 2 mmol/L glutaMAX (GIBCO by Life Technologies, Carlsbad, CA, USA) supplemented with recombinant human IL-7 (10 ng/mL) and IL-15 (10 ng/mL).60
The vector encoding the HER.2 directed CAR (2nd generation HER2.28z; clone FRP5) was a kind gift from Dr. Stephen Gottshalk. We completed transfection for retroviral supernatant with 293T cells as described here.61 Retroviral transduction was performed as previously described.21 Transduction efficiency was measured by flow cytometry using a chimeric Erb2-Fc fusion protein (R&D Systems, Minneapolis, MN, USA) with AF-647 anti-Fc antibody (Southern Biotech, Birmingham, AL, USA).
Apoptosis assays
MSCs and GFP+ tumor cells were collected after infection at indicated time points. All media were collected for dead cells and remaining cells were trypsinized and pelleted by centrifugation. Cells were stained for Annexin V conjugated to Pacific Blue fluorochrome according to manufacturer’s protocol (BioLegend, San Diego, CA, USA) and then washed in Annexin V binding buffer and stained with 7-AAD (7-amino-actinomycin D). Annexin V/7-AAD-positive cells were quantified by flow cytometry on a BD CANTO II and analyzed by FlowJo software.
Incucyte assays
For tumor transduction assays with OAd.5-expressing RFP and HDAd.5/3-expressing GFP, unlabeled tumor cells were plated in a 24-well plate at 1 × 105 cells per well. Transduced MSC supernatant was applied at various dilution, and RFP and GFP cells were quantified by fluorescent expression measured using the Incucyte Live Imaging system. Green object intensity (GCU) and overlap expression parameters (RFP+GFP+ cells) were used for analysis.
For spheroid assays, tumor cells were labeled with GFP and AF594 Annexin V (Invitrogen by Thermo Fisher Scientific, Carlsbad, CA, USA) stain was used at 1:2,000 dilution to indicate cell death. Multispheroid protocol was used for image acquisition and quantified by total red object integrated intensity parameter that represents the summed pixel intensity in calibrated units.
Co-culture and tri-culture assays
HER.2 CAR-T and NTR T cells were co-cultured with A549 or H1650 cell lines expressing GFP at 1:4 (effector to target) ratio. Residual tumor was measured by GFP+7-AAD– staining quantified by flow cytometry and CountBright Absolute Counting Beads (Invitrogen by Thermo Fisher Scientific, Carlsbad, CA, USA).
For tri-culture experiments, MSCs were infected with adenovirus and 24 h later co-cultured with tumor cells at a 1:10 (MSC to tumor) ratio. 48 h later T cells were added at 1:4 effector to target ratio and residual tumor was measured by flow cytometry and CountBright Absolute Counting Beads (Invitrogen by Thermo Fisher Scientific, Carlsbad, CA, USA).
Single cell cytokine secretion
We infected 2 × 105 MSCs with CAd and 24 h later co-cultured them with A549 tumor cells at a 1:10 (MSC to tumor) ratio in a 6-well plate. We added T cells 48 h later at 1:4 effector to target ratio and then collected cells 20 h later. CD4 and CD8 T cells were then isolated by microBeads enrichment and medium selection (MS) columns (Miltenyi Biotech, Bergisch Gladbach, Germany). We then loaded 3 × 104 T cells onto Isocode chips according to the manufacturer’s protocol (Isoplexis, Branford, CT, USA) and analyzed on 32-plex cytokine secretion. Isospeak software was used to quantify number of polyfunctional T cells and measure relative cytokine secretions.62 We used a similar protocol for uninfected MSC conditions with HER.2 CAR-T cells and NTR T cells. For no MSC control, HER.2 CAR-T or NTR T cells were cultured with tumor only.
UMap analysis
Single cell cytokine secretion data was exported using the Isospeak software and transcribed into MATLAB and Simulink software. Data was analyzed by input file run_umap.m and implemented to produce lower-dimensional representation of the data for visualization to cluster T cells according to cytokine secretion and polyfunctionality.
3D spheroid assays
We coated 96-well tissue culture plates with 1% agarose to create a low-attachment binding surface to allow forced suspension of tumor cells as described previously.48,63 We seeded 2 × 103 tumor cells in 100 μL and culture for 24–48 h until spheroids formed spontaneously and confirmed by microscope visualization. MSCs were then added in 50 μL volume and cultured for 48 h followed by T cell addition. Spheroids were monitored by fluorescent microscopy, confocal imaging, and Incucyte Live cell image analysis. We used NIH ImageJ software for image processing.
Xenograft mouse models
Breeder pairs of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (NSG, Stock No. 005557) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in a Baylor College of Medicine animal facility. Female and male (8- to 16-week-old) were used for experiments. All animal experiments were conducted in compliance with the Baylor College of Medicine IACUC (Protocol #AN-4758).
We injected 2 × 106 A549 or H1650 cells expressing firefly luciferase (Ffluc) or unlabeled H1650 cells via tail vein and confirmed tumor engraftment 3 days later by IVIS bioluminescent imaging. We delivered 2 × 106 uninfected or CAd infected MSCs intravenously by tail vein injection 4 days after tumor establishment. NTR or HER.2 CAR-T cells were then infused (5 × 105 or 1 × 106) and tumor was monitored by Xenogen-IVIS (In Vivo Imaging System) (Caliper Life Sciences, Hopkinton, MA, USA). Mice were injected intraperitoneally twice a week with D-luciferin (150 mg/kg), and signal intensity was measured as total photon/sec/cm2/sr (p/s/cm2/sr) after imaging for both 10 s and 1-min exposures.12 For GFP Ffluc-labeled T cell experiments, mice were imaged post T cell infusion on day 0 and then twice a week for 3 min exposures for the remainder of the experiment. We collected and processed tissues (blood, lung, spleen, and liver) for flow cytometry to measure circulating T cells. Lung tissue sections were processed and stained for CD3 T cell infiltration by Baylor Pathology Core.
Statistics
All statistical analyses used GraphPad Prism software. We used paired two-tailed Student’s t test to determine the statistical significance of differences between samples. Linear regression analysis was used for time course studies. In vivo tumor fold change was determined by Mann-Whitney U test. All numerical data are represented as mean with standard deviation. Results were considered statistically significant when p < 0.05.
Acknowledgments
Imaging for this project was supported by the Integrated Microscopy Core at Baylor College of Medicine with funding from NIH (DK56338 and CA125123), CPRIT (RP150578 and RP170719), the Dan L. Duncan Comprehensive Cancer Center, and the John S. Dunn Gulf Coast Consortium for Chemical Genomics. The authors extend special thanks to Ms. Hannah Johnson with the Integrated Microscopy Core. The authors thank the Baylor College of Medicine Pathology Core (HTAP) and Dr. Patricia Castro for performing immunohistochemistry and H&E staining for tissue microarray slides supported by NCI award P30 CA125123. The authors thank Dr. Reid Powell with Texas A&M Center for Advanced Imaging for image analysis. The authors are grateful to Ms. Catherine Gillespie for editing the manuscript. This project was supported by the National Heart, Lung, and Blood Institute of The National Institutes of Health under award number 5T32HL092332-17 to Principal Investigator: Dr. Helen Heslop and The National Cancer Institute under the award 5PO1CA094237-15.
Author contributions
Conceptualization, M.K.M and M.K.B.; investigation, M.K.M., A.E., and T.S., formal analysis, M.K.M., A.E., software, B.B. writing – original draft, M.K.M.; writing – review & editing, M.K.M. and M.K.B.; supervision, M.K.B.; funding acquisition, M.K.B.
Declaration of interests
M.S. is a consultant for Tessa Therapeutic. M.K.B. is a co-founder of the following with equity: Tessa Therapeutics and Marker Therapeutics. Scientific Advisory Boards: Bluebird Bio, Turnstone, Tessa Therapeutics, Marker Therapeutics, Allogene, Walking Fish, Memgen, KUUR, Bellicum Pharmaceuticals, Tscan, Poseida, and Abintus. Research funding: Tessa Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.02.004.
Supplemental information
References
- 1.Labani-Motlagh A., Ashja-Mahdavi M., Loskog A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020;11:940. doi: 10.3389/fimmu.2020.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henke E., Nandigama R., Ergün S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020;6:160. doi: 10.3389/fmolb.2019.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaaf M.B., Garg A.D., Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9:115. doi: 10.1038/s41419-017-0061-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzo-Sanz L., Muñoz P. Tumor-Infiltrating Immunosuppressive Cells in Cancer-Cell Plasticity, Tumor Progression and Therapy Response. Cancer Microenviron. 2019;12:119–132. doi: 10.1007/s12307-019-00232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J., Wang Y., Shi J., Liu J., Li Q., Chen L. Combination therapy: A feasibility strategy for CAR-T cell therapy in the treatment of solid tumors. Oncol. Lett. 2018;16:2063–2070. doi: 10.3892/ol.2018.8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guedan S., Alemany R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 2018;9:2460. doi: 10.3389/fimmu.2018.02460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gujar S., Pol J.G., Kim Y., Lee P.W., Kroemer G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018;39:209–221. doi: 10.1016/j.it.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Harrington K.J., Puzanov I., Hecht J.R., Hodi F.S., Szabo Z., Murugappan S., Kaufman H.L. Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev. Anticancer Ther. 2015;15:1389–1403. doi: 10.1586/14737140.2015.1115725. [DOI] [PubMed] [Google Scholar]
- 9.Rosewell Shaw A., Porter C.E., Watanabe N., Tanoue K., Sikora A., Gottschalk S., Brenner M.K., Suzuki M. Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 2017;25:2440–2451. doi: 10.1016/j.ymthe.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy D.G., Bell J.C. Cell carriers for oncolytic viruses: current challenges and future directions. Oncolytic Virother. 2013;2:47–56. doi: 10.2147/OV.S36623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hadryś A., Sochanik A., McFadden G., Jazowiecka-Rakus J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020;874:172991. doi: 10.1016/j.ejphar.2020.172991. [DOI] [PubMed] [Google Scholar]
- 12.Hoyos V., Del Bufalo F., Yagyu S., Ando M., Dotti G., Suzuki M., Bouchier-Hayes L., Alemany R., Brenner M.K. Mesenchymal Stromal Cells for Linked Delivery of Oncolytic and Apoptotic Adenoviruses to Non-small-cell Lung Cancers. Mol. Ther. 2015;23:1497–1506. doi: 10.1038/mt.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ando M., Hoyos V., Yagyu S., Tao W., Ramos C.A., Dotti G., Brenner M.K., Bouchier-Hayes L. Bortezomib sensitizes non-small cell lung cancer to mesenchymal stromal cell-delivered inducible caspase-9-mediated cytotoxicity. Cancer Gene Ther. 2014;21:472–482. doi: 10.1038/cgt.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruano D., López-Martín J.A., Moreno L., Lassaletta Á., Bautista F., Andión M., Hernández C., González-Murillo Á., Melen G., Alemany R. First-in-Human, First-in-Child Trial of Autologous MSCs Carrying the Oncolytic Virus Icovir-5 in Patients with Advanced Tumors. Mol. Ther. 2020;28:1033–1042. doi: 10.1016/j.ymthe.2020.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farzad L., Cerullo V., Yagyu S., Bertin T., Hemminki A., Rooney C., Lee B., Suzuki M. Combinatorial treatment with oncolytic adenovirus and helper-dependent adenovirus augments adenoviral cancer gene therapy. Mol. Ther. Oncolytics. 2014;1:14008. doi: 10.1038/mto.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hegde M., Joseph S.K., Pashankar F., DeRenzo C., Sanber K., Navai S., Byrd T.T., Hicks J., Xu M.L., Gerken C. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020;11:3549. doi: 10.1038/s41467-020-17175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanoue K., Rosewell Shaw A., Watanabe N., Porter C., Rana B., Gottschalk S., Brenner M., Suzuki M. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017;77:2040–2051. doi: 10.1158/0008-5472.CAN-16-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao K., Bi H., Ding Y.X., Li Y.H., Han C.M., Guo Y., Rao C.M. [Quality control of recombinant oncolytic adenovirus/p53] Yao Xue Xue Bao. 2011;46:1476–1482. [PubMed] [Google Scholar]
- 19.Keating A. Mesenchymal stromal cells. Curr. Opin. Hematol. 2006;13:419–425. doi: 10.1097/01.moh.0000245697.54887.6f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porter C.E., Rosewell Shaw A., Jung Y., Yip T., Castro P.D., Sandulache V.C., Sikora A., Gottschalk S., Ittman M.M., Brenner M.K., Suzuki M. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020;28:1251–1262. doi: 10.1016/j.ymthe.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phan-Lai V., Dang Y., Gad E., Childs J., Disis M.L. The Antitumor Efficacy of IL2/IL21-Cultured Polyfunctional Neu-Specific T Cells Is TNFα/IL17 Dependent. Clin. Cancer Res. 2016;22:2207–2216. doi: 10.1158/1078-0432.CCR-15-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menten P., Wuyts A., Van Damme J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002;13:455–481. doi: 10.1016/s1359-6101(02)00045-x. [DOI] [PubMed] [Google Scholar]
- 25.Saunders K.O., Ward-Caviness C., Schutte R.J., Freel S.A., Overman R.G., Thielman N.M., Cunningham C.K., Kepler T.B., Tomaras G.D. Secretion of MIP-1β and MIP-1α by CD8(+) T-lymphocytes correlates with HIV-1 inhibition independent of coreceptor usage. Cell. Immunol. 2011;266:154–164. doi: 10.1016/j.cellimm.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durgeau A., Virk Y., Corgnac S., Mami-Chouaib F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018;9:14. doi: 10.3389/fimmu.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J., Pemberton A., Morrison W.I., Connelley T. Granzyme B Is an Essential Mediator in CD8(+) T Cell Killing of Theileria parva-Infected Cells. Infect. Immun. 2019;87:e00386–e00418. doi: 10.1128/IAI.00386-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jinquan T., Quan S., Feili G., Larsen C.G., Thestrup-Pedersen K. Eotaxin activates T cells to chemotaxis and adhesion only if induced to express CCR3 by IL-2 together with IL-4. J. Immunol. 1999;162:4285–4292. [PubMed] [Google Scholar]
- 29.Fischer U.M., Harting M.T., Jimenez F., Monzon-Posadas W.O., Xue H., Savitz S.I., Laine G.A., Cox C.S., Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18:683–692. doi: 10.1089/scd.2008.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee M.W., Ryu S., Kim D.S., Lee J.W., Sung K.W., Koo H.H., Yoo K.H. Mesenchymal stem cells in suppression or progression of hematologic malignancy: current status and challenges. Leukemia. 2019;33:597–611. doi: 10.1038/s41375-018-0373-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franco-Luzón L., González-Murillo Á., Alcántara-Sánchez C., García-García L., Tabasi M., Huertas A.L., Chesler L., Ramírez M. Systemic oncolytic adenovirus delivered in mesenchymal carrier cells modulate tumor infiltrating immune cells and tumor microenvironment in mice with neuroblastoma. Oncotarget. 2020;11:347–361. doi: 10.18632/oncotarget.27401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon A.R., Hong J., Li Y., Shin H.C., Lee H., Kim H.S., Yun C.O. Mesenchymal Stem Cell-Mediated Delivery of an Oncolytic Adenovirus Enhances Antitumor Efficacy in Hepatocellular Carcinoma. Cancer Res. 2019;79:4503–4514. doi: 10.1158/0008-5472.CAN-18-3900. [DOI] [PubMed] [Google Scholar]
- 33.Muhammad T., Sakhawat A., Khan A.A., Ma L., Gjerset R.A., Huang Y. Mesenchymal stem cell-mediated delivery of therapeutic adenoviral vectors to prostate cancer. Stem Cell Res. Ther. 2019;10:190. doi: 10.1186/s13287-019-1268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marofi F., Vahedi G., Biglari A., Esmaeilzadeh A., Athari S.S. Mesenchymal Stromal/Stem Cells: A New Era in the Cell-Based Targeted Gene Therapy of Cancer. Front. Immunol. 2017;8:1770. doi: 10.3389/fimmu.2017.01770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J., Huang X., Wang H., Liu X., Zhang T., Wang Y., Hu D. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res. Ther. 2015;6:234. doi: 10.1186/s13287-015-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie C., Yang Z., Suo Y., Chen Q., Wei D., Weng X., Gu Z., Wei X. Systemically Infused Mesenchymal Stem Cells Show Different Homing Profiles in Healthy and Tumor Mouse Models. Stem Cells Transl. Med. 2017;6:1120–1131. doi: 10.1002/sctm.16-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leibacher J., Henschler R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res. Ther. 2016;7:7. doi: 10.1186/s13287-015-0271-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poggi A., Varesano S., Zocchi M.R. How to Hit Mesenchymal Stromal Cells and Make the Tumor Microenvironment Immunostimulant Rather Than Immunosuppressive. Front. Immunol. 2018;9:262. doi: 10.3389/fimmu.2018.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klopp A.H., Gupta A., Spaeth E., Andreeff M., Marini F., 3rd Concise review: Dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells. 2011;29:11–19. doi: 10.1002/stem.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waugh D.J., Wilson C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 41.Kindstedt E., Holm C.K., Sulniute R., Martinez-Carrasco I., Lundmark R., Lundberg P. CCL11, a novel mediator of inflammatory bone resorption. Sci. Rep. 2017;7:5334. doi: 10.1038/s41598-017-05654-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simson L., Ellyard J.I., Dent L.A., Matthaei K.I., Rothenberg M.E., Foster P.S., Smyth M.J., Parish C.R. Regulation of carcinogenesis by IL-5 and CCL11: a potential role for eosinophils in tumor immune surveillance. J. Immunol. 2007;178:4222–4229. doi: 10.4049/jimmunol.178.7.4222. [DOI] [PubMed] [Google Scholar]
- 43.Davis B.P., Rothenberg M.E. Eosinophils and cancer. Cancer Immunol. Res. 2014;2:1–8. doi: 10.1158/2326-6066.CIR-13-0196. [DOI] [PubMed] [Google Scholar]
- 44.Del Bufalo F., Manzo T., Hoyos V., Yagyu S., Caruana I., Jacot J., Benavides O., Rosen D., Brenner M.K. 3D modeling of human cancer: A PEG-fibrin hydrogel system to study the role of tumor microenvironment and recapitulate the in vivo effect of oncolytic adenovirus. Biomaterials. 2016;84:76–85. doi: 10.1016/j.biomaterials.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 45.Katt M.E., Placone A.L., Wong A.D., Xu Z.S., Searson P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016;4:12. doi: 10.3389/fbioe.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenna M.K., Rosewell-Shaw A., Suzuki M. Modeling the Efficacy of Oncolytic Adenoviruses In Vitro and In Vivo: Current and Future Perspectives. Cancers (Basel) 2020;12:619. doi: 10.3390/cancers12030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grill J., Lamfers M.L., van Beusechem V.W., Dirven C.M., Pherai D.S., Kater M., Van der Valk P., Vogels R., Vandertop W.P., Pinedo H.M. The organotypic multicellular spheroid is a relevant three-dimensional model to study adenovirus replication and penetration in human tumors in vitro. Mol. Ther. 2002;6:609–614. [PubMed] [Google Scholar]
- 48.Lv D., Hu Z., Lu L., Lu H., Xu X. Three-dimensional cell culture: A powerful tool in tumor research and drug discovery. Oncol. Lett. 2017;14:6999–7010. doi: 10.3892/ol.2017.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lloyd M.C., Cunningham J.J., Bui M.M., Gillies R.J., Brown J.S., Gatenby R.A. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer Res. 2016;76:3136–3144. doi: 10.1158/0008-5472.CAN-15-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam J.T., Bauerschmitz G.J., Kanerva A., Barker S.D., Straughn J.M., Wang M., Barnes M.N., Blackwell J.L., Siegal G.P., Alvarez R.D. Replication of an integrin targeted conditionally replicating adenovirus on primary ovarian cancer spheroids. Cancer Gene Ther. 2003;10:377–387. doi: 10.1038/sj.cgt.7700578. [DOI] [PubMed] [Google Scholar]
- 51.Hoarau-Véchot J., Rafii A., Touboul C., Pasquier J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? Int. J. Mol. Sci. 2018;19:181. doi: 10.3390/ijms19010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Groot R., Van Loenen M.M., Guislain A., Nicolet B.P., Freen-Van Heeren J.J., Verhagen O.J.H.M., Van Den Heuvel M.M., De Jong J., Burger P., Van Der Schoot C.E. Polyfunctional tumor-reactive T cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged T cell profile. OncoImmunology. 2019;8:e1648170. doi: 10.1080/2162402X.2019.1648170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hombach A., Köhler H., Rappl G., Abken H. Human CD4+ T cells lyse target cells via granzyme/perforin upon circumvention of MHC class II restriction by an antibody-like immunoreceptor. J. Immunol. 2006;177:5668–5675. doi: 10.4049/jimmunol.177.8.5668. [DOI] [PubMed] [Google Scholar]
- 54.Benmebarek M.R., Karches C.H., Cadilha B.L., Lesch S., Endres S., Kobold S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019;20:1283. doi: 10.3390/ijms20061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liadi I., Singh H., Romain G., Rey-Villamizar N., Merouane A., Adolacion J.R., Kebriaei P., Huls H., Qiu P., Roysam B. Individual Motile CD4(+) T Cells Can Participate in Efficient Multikilling through Conjugation to Multiple Tumor Cells. Cancer Immunol. Res. 2015;3:473–482. doi: 10.1158/2326-6066.CIR-14-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vacaflores A., Chapman N.M., Harty J.T., Richer M.J., Houtman J.C. Exposure of Human CD4 T Cells to IL-12 Results in Enhanced TCR-Induced Cytokine Production, Altered TCR Signaling, and Increased Oxidative Metabolism. PLoS ONE. 2016;11:e0157175. doi: 10.1371/journal.pone.0157175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.King I.L., Segal B.M. Cutting edge: IL-12 induces CD4+CD25- T cell activation in the presence of T regulatory cells. J. Immunol. 2005;175:641–645. doi: 10.4049/jimmunol.175.2.641. [DOI] [PubMed] [Google Scholar]
- 58.Hutnick N.A., Carnathan D., Demers K., Makedonas G., Ertl H.C., Betts M.R. Adenovirus-specific human T cells are pervasive, polyfunctional, and cross-reactive. Vaccine. 2010;28:1932–1941. doi: 10.1016/j.vaccine.2009.10.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Melcher A., Parato K., Rooney C.M., Bell J.C. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol. Ther. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mo F., Watanabe N., McKenna M.K., Hicks M.J., Srinivasan M., Gomes-Silva D. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat. Biotechnol. 2020;39:56–63. doi: 10.1038/s41587-020-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mamonkin M., Rouce R.H., Tashiro H., Brenner M.K. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–992. doi: 10.1182/blood-2015-02-629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xue Q., Bettini E., Paczkowski P., Ng C., Kaiser A., McConnell T., Kodrasi O., Quigley M.F., Heath J., Fan R. Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. J. Immunother. Cancer. 2017;5:85. doi: 10.1186/s40425-017-0293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomsen A.R., Aldrian C., Bronsert P., Thomann Y., Nanko N., Melin N., Rücker G., Follo M., Grosu A.L., Niedermann G. A deep conical agarose microwell array for adhesion independent three-dimensional cell culture and dynamic volume measurement. Lab Chip. 2017;18:179–189. doi: 10.1039/c7lc00832e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MSCs were infected with 100 vp/cell OAd-expressing RFP and 1,000 vp/cell HDAd-expressing GFP. Supernatant was collected 72 h later and applied to A549 (left) and H1650 (right) cell lines. Images were acquired every 2 h by Incucyte Live Cell image analysis for 3 days.
2,000 GFP+ H1650 or A549 lung tumor cells were seeded to an agarose coated 96-well plate. 200 uninfected or CAd infected were stained with efluor 670 and added 48 h after spheroid generation. CAd-infected MSCs were infected with 100 vp/cell OAd-expressing RFP and 1,000 vp/cell HDAd-expressing IL-12 and PD-L1 blocker. Images were collected at time of MSC addition and then every 24 h for 4 days on a GE Healthcare DeltaVision LIVE High Resolution Deconvolution Microscope.
2,000 GFP+ H1650 lung tumor cells were seeded to an agarose coated 96-well plate. 200 MSCs were added 48 h later followed by 50 NTR or HER2-specific CAR-T cells. GFP fluorescence indicated viable tumor with RFP Annexin V+ staining used to show tumor cell apoptosis. Images were acquired every 2 h for 5 days on the Incucyte.