

Abstract
Dopamine (DA) transporter (DAT) is dynamically regulated by several protein kinases and the Thr53 phosphorylation of DAT (pT53-DAT) is documented in heterologous cell models and in rat brain. However, the role of endogenous pT53-DAT in living animals has never been addressed. Here we generated and studied the pT53-lacking DAT mouse model (DAT-Ala53) by CRISPR/Cas9 technology. DAT-Ala53 mice showed normal growth, body weight, body temperature, grip strength, and sucrose preference while pT53-DAT was completely absent. However, DAT-Ala53 mice showed hyperlocomotion, pronounced vertical exploratory behavior, and stereotypy in a novel environment compared to wild-type littermates (WT). DAT-Ala53 mice displayed unaltered levels of monoamines, glutamate, and GABA in the striatum compared to WT. There were also no significant differences between DAT-Ala53 mice and WT in tyrosine hydroxylase (TH) and phospho-TH levels, or in total and surface DAT levels, or in DA-transport kinetic parameters Vmax and Km. Immunohistochemical and colocalization analyses of TH and DAT in caudate-putamen and nucleus accumbens revealed no significant differences between DAT-Ala53 and WT mice. Interestingly, cocaine’s potency to inhibit striatal DA transport and cocaine-induced locomotor activation were significantly reduced in the DAT-Ala53 mice. Also, ERK1/2 inhibitors completely failed to inhibit striatal DA uptake in DAT-Ala53 mice. Collectively, our findings reveal that the mice lacking pT53-DAT display novelty-induced hyperactive phenotype despite having normal transporter protein expression, DA-transport kinetics and DA-linked markers. The results also reveal that the lack of endogenous pT53-DAT renders DAT resistant to ERK1/2 inhibition and also less susceptible to cocaine inhibition and cocaine-evoked locomotor stimulation.
Keywords: DAT-Ala53 knock-in, ERK1/2, phosphorylation, cocaine, affinity, behavior
1. Introduction
The role of dopamine (DA) transporter (DAT) function in DA signaling, DA-mediated behaviors, psychostimulant actions, and DA pathology in neurological disorders has been demonstrated using animal models of complete DAT gene knock-out [1] or DAT overexpression [2–4] as well as knock-in mouse lines expressing the cocaine-insensitive DAT mutant [5, 6] and the rare human DAT coding variants [7–9]. Psychostimulants including cocaine, amphetamine (AMPH), methamphetamine (METH) and therapeutic stimulant methylphenidate bind to DAT and increase extracellular DA and potentiate DAergic neurotransmission [1, 10–13]. Studies using animal models and postmortem human brains as well as brain imaging studies revealed altered DAT expression in addiction [14–27]. Notably, pharmacological interaction and inhibition of DAT activity by cocaine are required for the manifestation of cocaine behavioral effects [5, 6].
The surface expression, and functional properties of DAT (DA transport and DA efflux) are regulated by several protein kinases (PKC, CaMKII, Cdk5, tyrosine kinase and ERK1/2), protein phosphatases (PP1/PP2Ac), and receptors involving post-translational modifications such as phosphorylation, ubiquitylation, palmitoylation, protein-protein interactions and proteolysis [Reviewed in and see references therein 28, 29-31]. DAT is a known phosphoprotein and possesses several phosphorylation sites in the intracellular cytoplasmic domains [Reviewed in 31]. Truncation of DAT N-terminal region leads to abrogation of PKC-mediated DAT phosphorylation and reduced AMPH-induced DAT-mediated DA efflux [32, 33]. DAT phosphorylation at the N-tail by CaMKII is shown to regulate AMPH-mediated DA efflux [34]. Threonine 53 (Thr53), a membrane proximal proline directed kinase site is phosphorylated by ERK1, JNK, p38 MAPK [35]. PKC activation and protein phosphatase inhibition regulate Thr53 phosphorylation of DAT (pT53-DAT) in LLCPK1 cells expressing rat DAT and in rat striatal synaptosomes [36, 37]. It is known that AMPH and METH stimulate pT53-DAT in vitro and in vivo, and altered DAT functional expression, AMPH-induced efflux and cocaine binding affinity are evident in LLPKC1 cells expressing the Thr53Ala DAT mutant [36, 37]. While these in vitro findings indicated the importance of Thr53 phosphorylation in modulating DAT expression, function and affinity for cocaine, whether endogenous Thr53 phosphorylation is involved in ERK-mediated DAT regulation and also the role of in vivo pT53-DAT in animal behavior and psychostimulant drug mechanisms remain unknown. Therefore, we generated a knock-in mouse model lacking pT53-DAT (DAT-Ala53 mice) using CRISPR/Cas9 technology by replacing Thr53 with non-phosphorylatable Ala53 in mDAT and studied basal motor functions and brain neurochemical levels. We also conducted immunohistochemical analysis of DA terminals and studied DAT biochemical properties in parallel with wild-type littermates. Furthermore, we examined the impact of in vivo Thr53 mutation on ERK1/2 mediated DAT regulation and cocaine evoked locomotor stimulation. We found that the DAT-Ala53 mice exhibit hyperactive phenotype in novel open-field locomotor test while displaying no changes in their body weight, rectal temperature, grip strength, or sucrose preference. There were also no changes in striatal DA transport or total and surface DAT levels or total tyrosine hydroxylase (t-TH) and phospho-TH (p-TH) levels or in the distribution and colocalization of TH and DAT. In addition, ERK1/2 inhibition had no effect on DAT activity in DAT-Als53 mice. Cocaine was less potent in inhibiting striatal DA uptake in DAT-Ala53 mice and DAT-Ala53 mice displayed reduced cocaine-induced locomotor activation. These results suggest that anomalies in cocaine binding, ERK1/2 mediated DAT regulation and cocaine-evoked locomotion exist when DAT-T53 phosphorylation is absent.
2. Materials and methods:
2.1. Animals and housing
All animal studies and care were performed under the guidelines of the Virginia Commonwealth University Institutional Animal Care and Use Committee (IACUC), in accordance with the principles and procedures outlined in the National Research Council “Guide for the Care and Use of Laboratory Animals”. Mice were group (same sex of mixed genotypes up to five) housed in individually ventilated cages in a barrier vivarium which excludes all known mouse viruses and parasites and most bacteria (including helicobacter), under a 12 hr light/12 hr dark light schedule, and were fed standard mouse chow (irradiated Teklad LM-485 diet) and autoclaved water. WT and DAT-Ala53 homozygous male mice were 8-10 weeks old at the start of both behavioral and molecular experiments.
2.2. Generation of DAT-Ala53 knock-in mice
A CRISPR/Cas9 approach was employed to generate DAT (SLC6A3, MGI: 94863) Ala53-knock-in mice, using methods modified from Wefers et al [38]. A guide RNA protospacer sequence was selected using a combination of the Broad Institute sgRNA design tool developed by Doench et al [39] (http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design), and Breaking-Cas, developed by Oliveros et al [40] (see Fig.1A for the protospacer sequence). This sequence was used to generate an Alt-R crRNA, which was then annealed to the Alt-R tracrRNA (both from Integrated DNA Technologies, IDT) to generate the functional guide RNA. Homology-directed repair at the cleavage site utilized a 200-base anti-sense single-stranded oligodeoxynucleotide (ssODN) spanning most of exon 2, and containing two mutations: an A-to-G substitution to create the threonine-to-alanine mutation at codon 53 (and a unique Ban I restriction site), and a G-to-C substitution to eliminate the Cas9 PAM sequence to prevent retargeting. A mix containing 20 ng/ul Cas9 protein (IDT Alt-R S.p. Cas9 Nuclease 3NLS), 20 ng/ul annealed cr/tracrRNA (preincubated with the Cas9 protein at R.T. for 10 min), and 20 ng/μl ssODN (IDT) was microinjected into the pronuclei of fertilized C57BL/6J mouse eggs by the VCU Transgenic/Knockout Mouse Core using standard methods, and the eggs were then implanted into the oviducts of pseudopregnant ICR females (~20 per recipient).
Fig. 1. Generation and characterization of DAT-Ala53 mice.
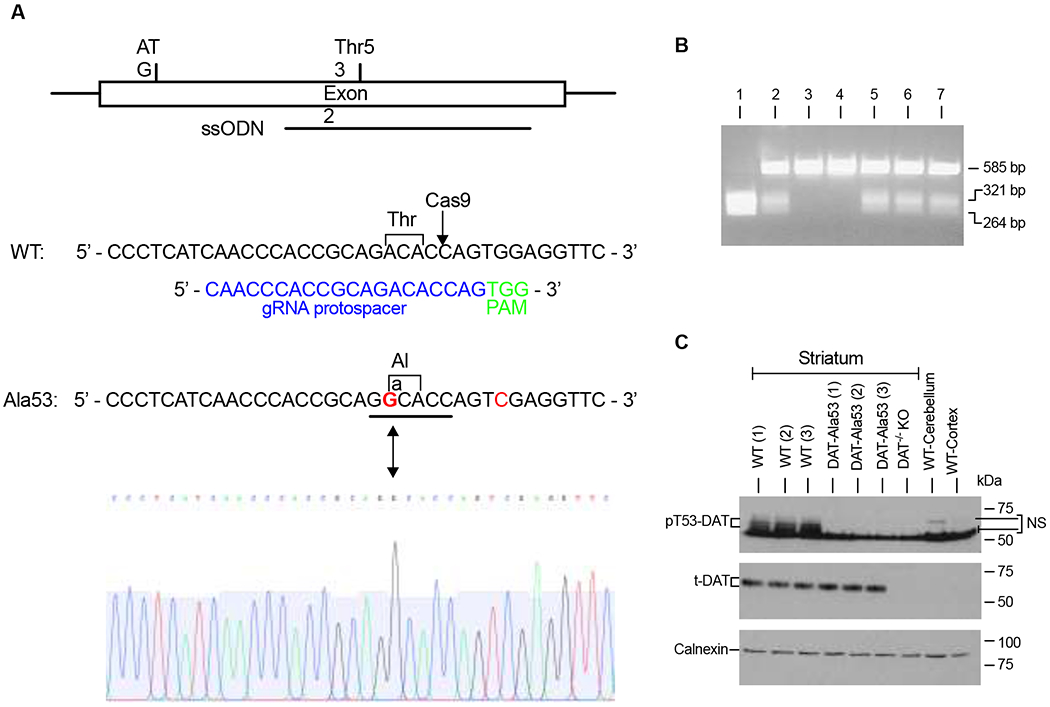
(A) DAT-Thr53Ala knock-in mouse targeting strategy using CRISPR/Cas9. Diagram of DAT exon 2, indicating the locations of the translational start codon, codon Thr53, and 200-base ssODN (anti-sense) repair template carrying two mutations. Sequences of the WT and Thr53Ala DAT alleles. Indicated in the WT sequence is the ACA codon for threonine at amino acid 53 and the Cas9 cleavage site (arrow). Below the WT sequence is the gRNA protospacer sequence (blue) and the adjacent PAM motif (green). Indicated in the Thr53Ala sequence is the A-to-G mutation that creates the GCA codon for alanine at amino acid 53 (bold red), which also creates a unique Ban I restriction site (underlined) that is used for genotyping these mice. An additional (silent) G-to-C mutation (red) was introduced to eliminate the Cas9 PAM sequence to prevent retargeting. Partial sequencing chromatogram from a homozygous DAT-Ala53 mouse corresponding to the same region shown for the Thr53Ala allele. (B) Genotyping of DAT-Ala53 knock-in mice by PCR. Seven pups from a DAT-Ala53 heterozygote x heterozygote mating were genotyped using primers that flank the targeted region to generate a 585-bp PCR product from both the WT and Thr53Ala alleles. The PCR products were then digested with Ban I, which cuts only the Ala53 product into two fragments of 321 bp and 264 bp. Mouse 1 from this litter is homozygous for the Thr53Ala knock-in allele, mice 2 and 5-7 are heterozygotes, and mice 3 and 4 are WT. The PCR product from mouse 1 was sequenced (see A.) (C) Genotyping of DAT-Ala53 knock-in mice by a pT53-DAT antibody. Representative immunoblots show the lack of pT53-DAT (~60-65 kDa) protein band in the striatum of DAT-Ala53 mice with no changes in the level of t-DAT (~ 65 kDa) and calnexin (90 kDa). Protein extracts from DAT-KO (striatum) and lysates from the cerebellum and cortex (without the frontal cortex) of WT mice were used to validate the specificity of pT53-DAT and t-DAT bands. Uncropped immunoblots were provided in supplementary figure 1. NS-nonspecific band.
2.3. Genotyping of DAT-Ala53 knock-in mice
At weaning, DNA was purified from a 5 mm tail snip from each pup, and genotyped by PCR amplification across the targeted region using the forward primer 5’-GGAATTTCAGGTGCTTGGACTACC-3’ and reverse primer 5’-CTCAGCCTTTGCTGTGAGCATTTC-3’. Two potential founder mice were selected for breeding and further characterization, including DNA sequencing. For sequence verification, DNAs from both potential founders and a subset of their offspring, as well as one homozygous DAT-Ala53 mouse generated from each line, were amplified using the primers used for Ban I genotyping, followed by sequencing using the forward primer (performed by Eurofins). Genotyping using tail clippings was done by Transnetyx, Inc. (Cordov, TN).
2.4. Measurement of locomotor activity, stereotypy and vertical exploratory behaviors
Locomotor activity of WT and DAT-Ala53 mice was recorded using open filed activity boxes (Med Associates, St. Albans, VT; Model ENV-510; 10.75 X 10.75 inches) enclosed in a sound-proof outer shell with artificial ventilation. The apparatus consists of 16 equally spaced infrared sources (1 cm above the floor) to track the movement and stereotypic behaviors of a mouse. Additionally, 16 similarly spaced infrared sources are 4 cm elevated above the activity chamber floor to capture vertical, jumping behaviors. Briefly mice were handled gently for three days by an experimenter. Prior to start of experiment, mice were adapted in the testing room for one hour. Naive mice (n = 24 each of WT and DAT-Ala53) were placed in the locomotor boxes for three consecutive days and total distance travelled, and counts of horizontal, vertical and stereotypic activity were recorded in 4 mins intervals for 60 min. On day four, mice were habituated in locomotor boxes for 60 min following a single saline (sterile 0.9% NaCl) injection (1ml/kg; i.p.). On testing day (day 5), same set of mice were divided into four independent groups (n = 6 per group for each genotype). Following a 15 min initial habituation to the boxes, one group from each genotype received saline and other group a single dose of cocaine (2.5 or 7.5 or 10 mg/kg; i.p.) (National Institute on Drug Abuse Supply Program, Bethesda, MD) and their activity was recorded for 60 min post-injection. The software analyzes and provides the total activity traveled in cm, stereotypy, horizontal and vertical counts. The data collected were analyzed. The chamber was cleaned with 70% ethanol and dried with paper towels between animals [41].
2.5. Body temperature measurement
Basal body temperature was measured using a standard rectal thermometer (Fischer Scientific, Pittsburg, PA). The probe was rectally inserted approximately 24 mm and temperature was recorded. The laboratory room temperature varied from 22-24°C from day to day.
2.6. Grip strength behavior
The forelimb grip strength was assessed by Ugo Basile Grip strength Meter. To assess forelimb grip strength measurement, the mouse was held gently by the base of its tail over the top of the grid so that only its front paws were able to grip platform/T-bar. With its torso in a horizontal position the mouse was pulled back steadily until the grip was released down the complete length of the grid/bar. The propensity is that the mouse will cling onto the grid/bar until it can no longer resist the increasing force, before it is released. The grip strength meter digitally displays the maximum force applied as the peak tension (in grams) once the grasp is released. The mean of five consecutive trails was taken as an index of forelimb strength. The mouse was given inter trial gap period of 15 seconds.
2.7. Sucrose preference test
Sucrose preference test in mice is used to measure normal natural reward preference for sucrose solution over a tap water. Male mice WT (n = 5) and DAT-Ala53 (n = 5) were individually housed and had free access ad libitum throughout the whole experiment. Before basal sucrose preference, mice were acclimatized to drink tap water from two bottles for one week. After one week of adaptation, individually housed animals were provided with a free choice of water or increasing order of sucrose solution (0.3, 1.0, 3.0, 10 and 30 %) and each concentration was tested for 48 h. In order to minimize the side preference bias, both sucrose solution and water containing bottles were switched every 24 hours and the intake of sucrose solution and water was measured. Percentage of sucrose consumption was calculated by (volume of sucrose consumed/total volume of liquid × 100) [42].
2.8. Quantification of monoamines and other neurotransmitters by HPLC
Whole striatum was dissected out from WT and DAT-Ala53 mice and flash-frozen on dry ice. The monoamine and other neurotransmitters were quantified by using HPLC with electrochemical detection and the measures of monoamine were obtained by the Neurochemistry Core Facility at Vanderbilt University (Nashville, TN). Frozen tissues were homogenized by using a tissue dismembrator, in 100-750 ul of 0.1 M trichloroacetic acid, which contains 10 mM sodium acetate, 0.1 mM EDTA, and 10.5 % methanol (pH 3.8). Ten microliters of homogenate were used for protein assay and the protein concentration was quantified by BSA Protein Assay Kit (Thermo Scientific, Waltham, MA). Then samples were spun in a microcentrifuge at 10,000 g for 20 min and the supernatant was used for biogenic amines analysis. Concentrations of monoamines were determined by using an Antec Decade II (Oxidation: 0.65) electrochemical detector operated at 33°C. Isoproterenol was used as an internal standard to quantify the specific interest of monoamines and other neurotransmitters. Samples of the supernatant were injected on to a Phenomenex Kintex C18 HPLC column by using a water 2707 autosampler. Using an HPLC solvent monoamines and other neurotransmitters were eluted and data acquisition was done by Empower software (Water Corp).
2.9. Crude synaptosomes preparation and DAT mediated DA uptake assay
Crude synaptosomes were prepared as described [43]. Briefly, WT and DAT-Ala53 male mice were decapitated and quickly brains were collected in ice cold Petri dishes. Dorsal (caudate putamen) and ventral (nucleus accumbens) striatum were dissected and collected in 2 ml of ice cod sucrose buffer (0.32 M sucrose in 5 mM HEPES, pH 7.4). The tissue was homogenized by using a Teflon-glass homogenizer and the homogenized samples were centrifuged at 1000 g for 10 min at 4°C. After centrifugation supernatant was collected and again centrifuged at 12,000 g for 20 min and the resulting pellet was resuspended in ice cold sucrose buffer. The concentration of protein was quantified by DC method using BSA as a standard. Synaptosomes (30 μg of protein) were incubated in a total volume of 0.3 ml of KRH-assay buffer, pH 7.4 (120 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4 and 10 mM Hepes and 10 mM D-glucose) containing 0.1 mM ascorbic acid and 0.1 mM pargyline. Uptake was initiated by the addition of 5 nM [3H] DA (18.32 Ci/mmol: Perkin Elmer, Santa Clara, CA). For DAT kinetic analysis, [3H] DA (20 nM) was mixed with unlabeled DA so that total DA concentration ranges from 25 nM to 1000 nM. DAT mediated DA uptake was assayed in the presence of NET specific inhibitor nisoxetine (50 nM) to block NET mediated DA uptake. To test the effect of MAPK kinase (MEK) inhibitors on DA uptake experiment, synaptosomes (30 μg) were preincubated with vehicle or varying concentrations (0.1, 1 or 5μM) of U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto) butadiene] or PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one] for 30 min at 37°C prior to initiating DA uptake. The concentrations of MEK inhibitors were selected based on published study [44]. To determine the nonspecific DA uptake, synaptosomes were preincubated with the DAT specific inhibitor GBR-12909 (50 nM) and NET specific inhibitor nisoxetine (50 nM) at 37°C for 10 min followed by the addition [3H] DA. Uptake was terminated with the addition of GBR-12909 followed by rapid filtration over GF-B filters on a Brandel Cell Harvester (Brandel Inc., Gaitherburg, MD). Filters were counted by liquid scintillation counter to determine the radioactivity bound to the filters. DA uptake measured in the presence of GBR-12909 and nisoxetine was subtracted from the total accumulation of [3H] DA to yield DAT-specific DA uptake. [3H] DA uptake assays were conducted in duplicates or triplicates from one independent mouse, and each data point is the average of duplicates or triplicates.
2.10. Biotinylation of DAT using striatal synaptosomes
Procedure for surface biotinylation was adapted from our previous study [43]. Striatal synaptosomes were subjected to surface protein biotinylation by incubating with membrane impermeable EZ link NHS-Sulfo-SS-biotin (Pierce, Rockford, IL, USA) (1 mg/1 mg protein) for 30 min at 4°C in ice-cold PBS/Ca/Mg buffer (138 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 9.6 mM Na2HPO4, 1 mM MgCl2, 0.1 mM CaCl2, pH 7.3). At end of the biotinylation, the same buffer containing 100 mM glycine was used to quench excess NHS-Sulfo-SS-biotin. The biotinylated synaptosomes were suspended in ice cold radioimmunoprecipitation assay RIPA buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, and 1% sodium deoxycholate) supplemented with protease (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μM pepstatin, and 250 μM phenylmethylsulfonyl fluoride) and phosphatase inhibitors (10 mM sodium fluoride, 50 mM sodium pyrophosphate, 5 mM sodium orthovanadate, and 1 μM okadaic acid) cocktails and solubilized by passing through a 26-gauge needle six times. The solubilizates were centrifuged at 25,000 g for 30 min and supernatants were incubated with NeutrAvidin Agarose resins (Pierce, Rockford, IL) to isolate biotinylated proteins. The NeutrAvidin Agarose resins were washed three times with RIPA buffer and biotinylated proteins (bound to the resins) were eluted by incubating the resins with 50 μl Laemmli sample buffer (62.5 mM Tris-HCl pH 6.8, 20 % glycerol, 2 % SDS and 5 % ß-mercaptoethanol). Proteins from total, bound and unbound were separated by 7.5 % SDS-PAGE and transferred to PVDF membrane (Bio-Rad, Hercules, CA). The membrane blots were blocked with 5% BSA-TBS-Tween20 buffer for 1 hour at room temperature and immunoprobed with one of the primary antibodies for total DAT (t-DAT) (mouse anti-dopamine transporter monoclonal, clone mAb16, Cat.no. MABN669, 1:2000 dilution, MilliporeSigma, Burlington, MA) or pT53-DAT (rabbit anti-phospho-Thr53 dopamine transporter polyclonal, Cat.no. p435-53, 1:2000 dilution, PhosphoSolutions Inc., Aurora, CO). Further the total protein blot was stripped and reprobed with one of the primary antibodies, anti-TH (mouse anti-tyrosine hydroxylase monoclonal, Cat.no. MAB318, 1:2000 Millipore), or anti-p-TH (rabbit anti-tyrosine hydroxylase, phospho-Ser40 polyclonal, Cat.no. AB5935, 1:2000, MilliporeSigma, Burlington, MA) or anti-calnexin (rabbit anti-calnexin polyclonal Cat.no. SPA 860, 1:2000, Enzo Life Sciences, Inc. Farmingdale, NY) antibody to validate equal protein loading. ECL reagents (Amersham Biosciences, GE Healthcare, Pittsburgh, USA) were used to visualize the protein bands. The protein bands were quantified by using NIH Image J software (1.48v) to ensure that results were within the linear range of the film (HyBlot CL, Thomas Scientific, Swedesboro, NJ) exposure. The density of intracellular endoplasmic reticulum protein calnexin was determined by stripping the membranes and reprobing with anti-calnexin antibody in order to validate that only plasma membrane surface proteins were biotinylated and also to ensure that equal protein was loaded and transferred. Expression levels of arbitrary protein band densities of pT53-DAT, t-DAT and calnexin were quantified using NIH Image J software (version 1.48j).
2.11. Tissue processing and Immunofluorescence
The methodology for immunofluorescence was followed as described previously [45, 46]. WT and DAT-Ala53 male animals were anesthetized with pentobarbital and mice were transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde in PBS. After perfusion, whole brain was excised and post-fixed overnight and were cryoprotected in graded sucrose solutions (10, 20 and 30% in PBS). Coronal sections of 30 μM size containing both dorsal and ventral striatum were obtained with a cryostat (Leica CM1850). The whole immunofluorescence protocol was performed in floating sections. The sections containing dorsal and ventral striatum (bregma 1.18mm to 0.86mm, Paxinos and Franklin, 2001) were rinsed in 1X PBS buffer for three times for 10 min. The sections were blocked with 4% normal goat serum containing 1% Triton X-100 in PBS for 1 h at room temperature. Sections were incubated overnight at 4°C with the following primary antibodies, Anti-dopamine transporter (1:1000, Millipore MAB369) or rabbit anti-tyrosine hydroxylase antibody (1:1000, Millipore AB152). After post-washing, the primary antibody was detected with Alexa Fluor 488 goat anti-rat antibody (1:500, A-11006; Thermo Fisher Scientific, Waltham, MA) or Alexa Fluor 568 goat anti-rabbit antibody (1:500, A-11011; Thermo Fisher Scientific, Waltham, MA) respectively. The sections were mounted with Vectashield H-1000 (Vector Laboratories, Burlingame, CA) and images were taken using Zeiss LSM710 meta confocal microscopy (Zeiss, Germany). The fluorescence intensity of DAT and TH were determined and analyzed as described [47] using NIH Image J software.
2.12. Measurement of inhibition constants (IC50) of psychostimulants
Striatal synaptosomes (30 μg) from WT and DAT-Ala53 were used for transport assays at 37°C in a volume of 300 μl in KRH-assay buffer (120 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4 and 10 mM Hepes and 10 mM D-glucose) containing 0.1 mM ascorbic acid and 0.1 mM pargyline. Inhibitory assays were initiated in synaptosomes with an uptake time of 4 min by addition of mixed 20 nM [3H] DA label with or without varying concentrations of cocaine (National Institute on Drug Abuse Supply Program, Bethesda, MD) or AMPH or METH (Sigma-Aldrich (St. Louis, MO). Uptake was terminated with the addition of ice cold KRH assay buffer followed by rapid filtration over GF-B filters on a Brandel Cell Harvester. Filter was counted by liquid scintillation counter to determine the radioactivity bound to filter. Data were analyzed by using a nonlinear regression to determine the IC50 values of competitors (GraphPad Prism 9).
2.13. Statistical analyses
Statistical analysis and graphical presentation were done by using GraphPad Prism 9 (GraphPad, San Diego, CA). One-way or two-way analysis of variance (ANOVA) was used where appropriate. Following ANOVA, Bonferroni post hoc testing was used for pair-wise comparisons. Groups of two were compared by unpaired two-tailed Student’s t-test. A value of P < 0.05 was considered statistically significant. All values are presented as Mean ± S.D, and individual values from each animal were shown in figures.
3. Results
3.1. Validation of DAT-Ala53 mice.
DAT-Ala53 knock-in mouse line in the C57BL/6 background was created using the CRISPR/Cas9 technique. Design strategy of the generation of DAT-Ala53 mice was shown in Fig. 1A and discussed in detail in the methods section. Genotyping using specific primers (details given in the methods section) yielded a product of 585 bp for both the WT (Thr53) and DAT-Ala53 alleles. Subsequent digestion of the PCR products with Ban I yielded 321-bp and 264-bp fragments for the Thr53Ala allele, while the WT allele remained uncut (Fig. 1B). Additionally, Thr53-phosphospecific DAT antibody (pT53-DAT ab) was used in immunoblot analysis to validate the absence of mouse DAT phosphorylation at Thr53 site in DAT-Ala53 homozygous mice. As shown in Fig. 1C, pT53-DAT ab detected Thr53 phosphorylated DAT (pT53-DAT) protein (~60-65 kDa) in the striatal tissue of the WT mice; however, pT53-DAT bands were completely absent in the DAT-Ala53 mice. Further, the blots were stripped and probed with total DAT antibody (t-DAT ab) and calnexin antibody. When compared with WT, expression of t-DAT (~65 kDa) and calnexin (~90 kDa) were not altered in the striatum of DAT-A53 mice (Fig. 1C). To validate the authenticity of DAT proteins detected by the pT53-DAT ab and t-DAT ab, striatal lysates from DAT knock-out mice, and lysates from WT cerebellum and cortex (without the frontal cortex) were used in parallel immunoblot analysis. Cerebellum and cortex (without the frontal cortex) are devoid of DAT expression. pT53-DAT ab and t-DAT ab detected neither t-DAT (~65 kDa) nor pT53-DAT (~60-65 kDa) from DAT knock-out striatum, and WT cerebellum and cortex (without the frontal cortex), confirming the authenticity of DAT proteins detected by pT53-DAT ab and t-DAT antibody (Fig. 1C). pT53-DAT ab showed additional non-specific bands ~50-54 kDa and~ 68-70 kDa just above the pT53-DAT as evidenced by their presence in DAT-KO striatum as well as in WT cerebellum and cortex. Thus, DAT-Ala53 knock-in mice represent the first construct-valid animal model lacking the Thr53 phosphorylation. Both male and female homozygous DAT-Ala53 mice are viable and exhibit normal health and breeding. Male WT and DAT-Ala53 homozygous mice were used in this study.
3.2. Body weight, body temperature, grip strength, and sucrose preference are not altered developmentally in DAT-Ala53 mice.
As show in Fig. 2A, two-way ANOVA revealed there is no effect between genotype in body weight across different ages [F (3,152) = 3.567, P = 0.6616] or genotype and age interactions [F (3, 152) = 0.6616, P = 5769], however there was a significant aging effect seen in different months of age [F (2.774, 140.6) = 577.8, P < 0.0001]. Unpaired two-tailed Student’s t-test revealed no difference in the body temperature between WT and DAT-Ala53 mice [t = 0.8071, P = 0.4384] (Fig. 2B). Forelimb grip strength behavior showed no significant change in DAT-Ala53 mice compared to WT [t = 1.664, P = 0.1156] (Fig. 2C), suggesting that DAT-Ala53 mice exhibit no developmental defects. We examined the known established natural reward behavior, the sucrose intake and preference involving the activation of dopamine neurons [48–51]. The statistical two-way ANOVA analysis revealed a significant effect across different concentrations of sucrose [F (4, 32) = 122.5, P < 0.0001], but there was no main effect between the genotypes [F (1, 8) = 0.03137, P = 0.8638]. The interaction between genotype and different concentrations of sucrose was not statistically significant [F (4, 32) = 0.07154, P =0.9902] (Fig. 2D). Taken together, these data suggest that the natural reward process was not altered in DAT-Ala53 mice.
Fig. 2. Phenotypic characterization and sucrose preference of DAT-Ala53 mice.
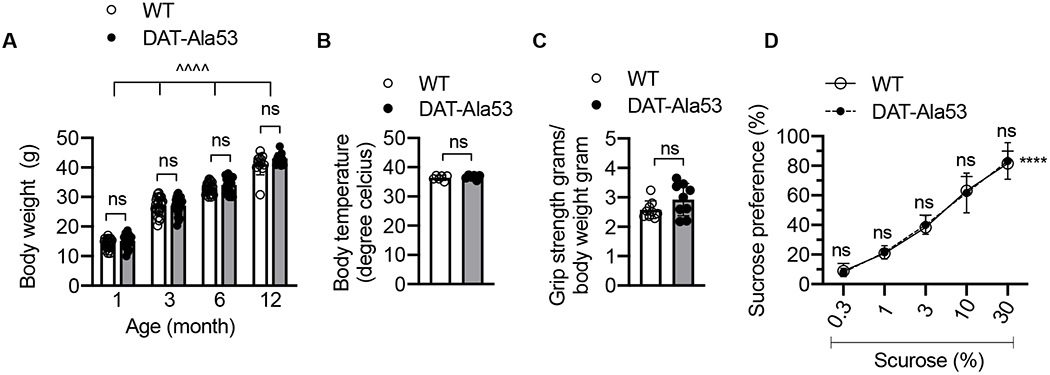
(A) Mice were weighed from 4 weeks until 12 months old age (n = 24 for one, three, and six months; n = 12 for twelve months) for each genotype. ΛΛΛΛ P < 0.0001, mixed two-way ANOVA of aging effect across different months of ages, ns: nonsignificant (Bonferroni’s multiple comparison test). (B) The rectal temperature of WT (n = 6) and DAT-Ala53 (n = 6) mice was measured and revealed no significant differences (P = 0.4382) between genotypes. (C) Both WT (n = 9) and DAT-Ala53 (n = 9) mice showed no deficits in grip strength behavior and revealed no significant differences between genotypes (P = 0.1156). Data are presented as Mean ± S.D. ns: nonsignificant (unpaired two-tailed Student’s t-test). (D). Sucrose preference for each concentration was presented as a percentage of the volume of sucrose solution consumed to the total volume of solution intake WT (n = 5) and DAT-Ala53 (n = 5). ΛΛΛΛ P < 0.0001, two-way ANOVA for effect of sucrose preference across different concentration of sucrose, ns: nonsignificant effect (Bonferroni’s multiple comparison test).
3.3. DAT-Ala53 mice display hyperlocomotion in a novel environment.
While DAT-Ala53 animals displayed no developmental defects or deficits in motor strength behavior, DAT-Ala53 mice showed hyperlocomotion, and enhanced vertical behaviors and stereotypy in unhabituated/novel open field environment on day one. However, on subsequent days (day 2 and day 3) when open field environment becomes familiar environment, there were no differences in the locomotor activity, vertical behaviors, and stereotypy counts between DAT-Ala53 mice and WT littermates. The locomotor activity, vertical behaviors, and stereotypy counts on day 2 were similar to those on day 3, and therefore, data collected on day 3 are shown in Figure 3. Representative locomotor activity path graphs are shown in Fig. 3A and B. The distance traveled, vertical behaviors, and stereotypy counts measured in 4 min bins over a 60 min time period on day 1 are shown in Fig. 3C, D and E and on day 3 are shown in Fig. 3F, G and H. Comparative analyses of the data for day 1 and day 3 are given in Fig. 3I, J and K. Two-way ANOVA analysis of the locomotor activity measured over a 60 min on day 1 and day 3 revealed significant effect of days and genotype for total distance [F(1, 46) = 237.6, P < 0.0001; F(1, 46) = 42.06, P < 0.0001], vertical counts [F(1, 46) = 113.9, P < 0.0001; F(1, 46) = 26.49, P < 0.0001], and stereotypy counts [F(1, 46) = 430.6, P < 0.0001; F(1, 46) = 52.88, P < 0.0001] respectively. There was also a significant genotype x days interaction in total distance [F(1, 46) = 27.41, P < 0.0001, vertical counts [F(1, 46) = 25.69, P < 0.0001] and stereotypy counts [F(1, 46) = 14.82, P = 0.0004]. Together, these data indicate that the novel environment causes motor activation in DAT-Ala53 mice indicating the influence of DAT-Thr53 phosphorylation on novelty induced hyperactivity.
Fig. 3. DAT-Ala53 mice exhibit hyperlocomotion to novel environment.
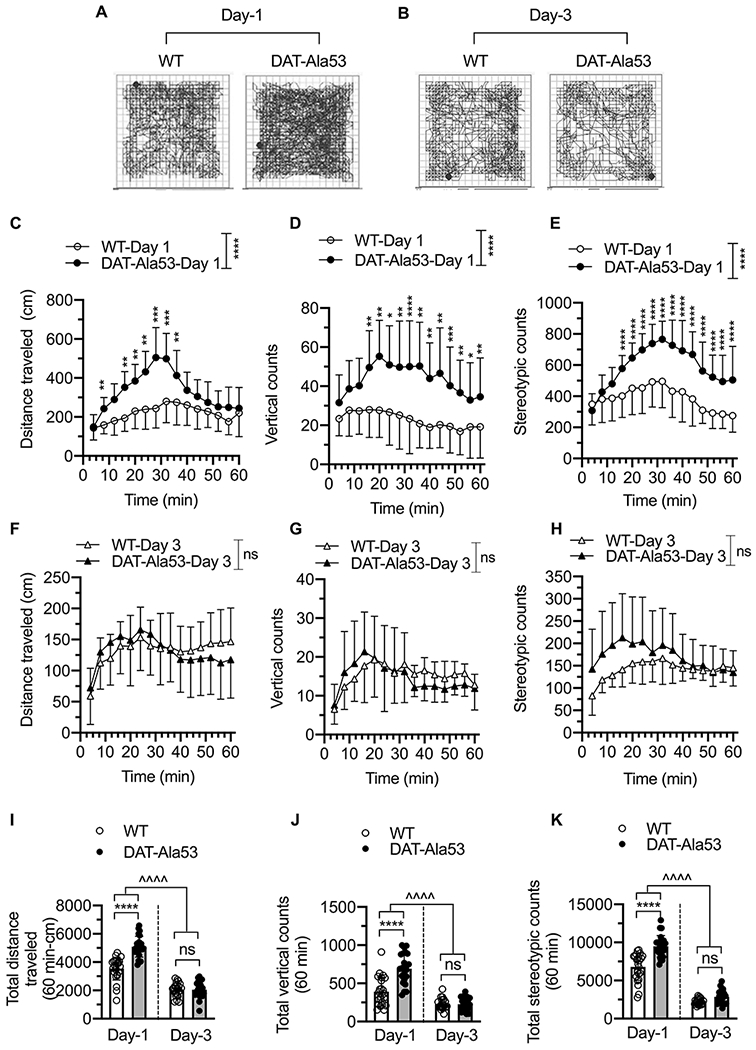
Naive male WT (n = 24) and DAT-Ala53 (n = 24) animals were placed in the locomotor boxes on day 1 without prior habituation and then on subsequent days 2 and 3, and motor activity were monitored for 60 min. Representative activity traces show the locomotor activity of WT and DAT-Ala53 mice on day 1 (A) and day 3 (B). Time course plots show distance traveled, vertical counts, and stereotypy counts measured in 4 min bins over 60 min period on day one (C, D, E) and on day three (F, G, H). Comparative analysis (DAT-Ala53 vs. WT) of total distance traveled (I), vertical counts (J) and stereotypy counts (K) are shown. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 (Day 1), ns: nonsignificant effect (P > 0.9999) compared to WT (Day 3). ΛΛΛΛ P <0.0001, compared between specified pairs. Data are presented as Means ± S.D. Two-way ANOVA followed by Bonferroni’s multiple comparison test
3.4. Neurochemical levels and TH expression in Striatum are unaltered in DAT-Ala53 mice.
Quantification of neurotransmitter levels and their metabolites in striatum revealed no significant genotypic differences between DAT-Ala53 and WT mice in the levels of DA [t = 0.993, df = 19, P = 0.333], DA metabolites DOAPC [t = 1.506, df = 22, P = 0.1464], 3-MT [t = 1.337, df = 22, P = 0.1949] and HVA [t = 0.4684, df = 22, P = 0.6441] (Fig. 4A). There were also no significant differences in the levels of 5-HT [t = 1.538, df = 22, P = 0.1383] and NE [t = 0.7341, df = 22, P = 0.4706] between the genotypes (Fig. 4A). However, there is a significant difference in the level of 5-HT metabolite 5-HIAA [t = 2.569, df = 22, P = 0.0175]. There were no significant genotypic differences in the levels of glutamate [t = 0.0433, df = 22, P = 0.9658] and GABA [t = 0.7587, df = 10, P = 0.4655] (Fig. 4A). To check whether there is any genotypic effect on the expression of TH, the rate-limiting enzyme in the synthesis of DA, we analyzed total TH (t-TH) and phospho-TH (p-TH) levels in the striatum by immunoblot analysis and a representative immunoblot is shown in Fig. 4B. Quantified immunoblot analysis revealed no significant genotypic difference in the p-TH/t-TH ratio when p-TH is normalized to t-TH [t = 0.8296, df = 4, P = 0.4534] (Fig. 4C) or in t-TH/calnexin ratio when t-TH is normalized to calnexin [t = 0.6565, df = 4, P = 0.5474) (Fig. 4D).
Fig. 4. Neurotransmitter levels and expression of t-TH andp-TH in DAT-Ala53 striatum.
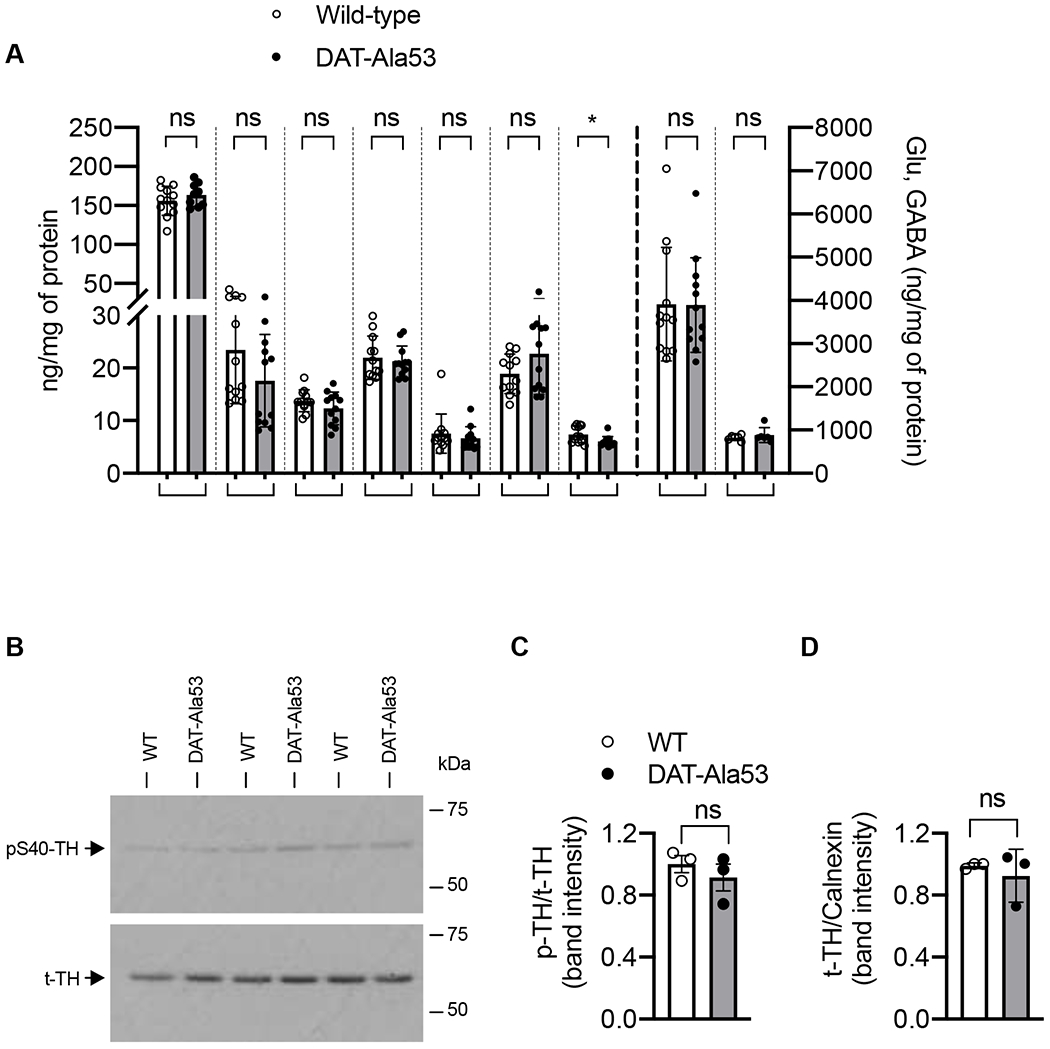
(A) The concentrations of DA, DOPAC, 3-MT, HVA, NE, 5-HT, 5-HIAA, Glu and GABA in the WT and DAT-Ala53 striatal tissues. Values are presented as Mean ± S.D. DAT-Ala53 (n = 12) and WT (n = 12). * P = 0.0175 and ns: nonsignificant compared to WT (unpaired two-tailed Student’s t-test). (B) Representative immunoblots show expression of p-TH (~60 kDa) and t-TH (~60 kDa). (C) Quantified p-TH normalized to t-TH and (D) quantified t-TH normalized to total calnexin (~90 kDa). Values are presented as Mean ± S.D. DAT-Ala53 (n = 3) and WT (n = 3) ns: nonsignificant (unpaired two-tailed Student’s t-test). DA: dopamine, DOPAC: 3,4-dihydroxyphenylacetic acid, 3-MT: 3-methoxytyramine, HVA: homovanillic acid, NE: norepinephrine, 5-HT: 5-hydroxytryptamine (serotonin), 5-HIAA: 5-hydroxyindoleacetic acid, Glu: glutamate, GABA: gamma aminobutyric acid.
3.5. DAT-Ala53 display unaltered striatal TH and DAT immunofluorescence.
We performed immunofluorescence study on dorsal and ventral striatum to examine whether there are any alterations in the specific expression of TH and DAT in DAT-Ala53 mice (Fig. 5A, B and F, G). Immunofluorescence intensities of TH or DAT in the dorsal striatum showed no significant difference between DAT-Ala53 and WT mice [TH: t = 0.4194, df = 6, P = 0.6895; DAT: t = 1.819, df = 6, P = 0.1187] (Fig. 5C and D). Similarly, there was also no significant difference in TH or DAT immunofluorescence in the ventral striatum between DAT-Ala53 and WT mice [TH: t = 0.1872, df = 6, P = 0.8577; DAT: t = 0.4565, df = 6, P = 0.6641] (Fig. 5H and I). Furthermore, when normalized to TH, DAT immunofluorescence in the dorsal or ventral striatum showed no significant difference between the genotypes [dorsal: t = 2.210, df = 6, P = 0.0692; ventral: t = 0.1837, df = 6, P = 0.8603] (Fig. 5E and J).
Fig. 5. Dopamine specific neuronal markers (DAT and TH) in the dorsal and ventral striatum of DAT-Ala53.
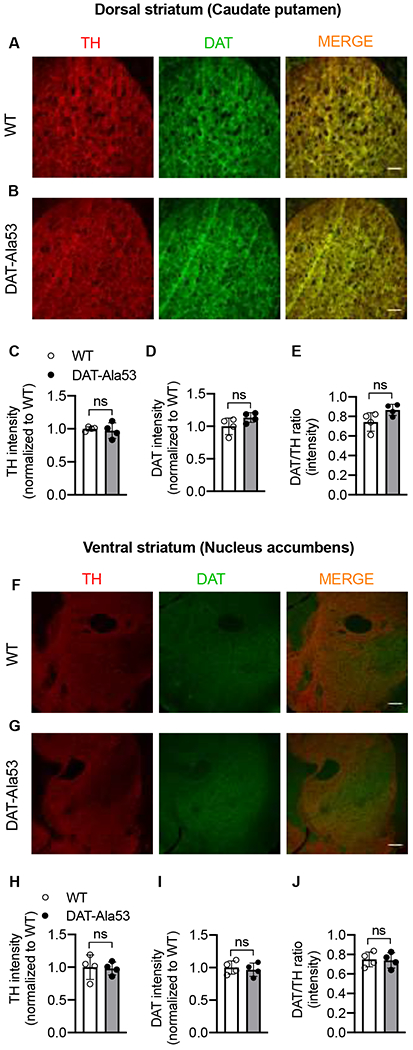
Immunofluorescence intensity and distribution of TH (red), DAT (green) in dorsal and ventral striatum of WT (n = 4) and DAT-Ala53 (n = 4) mice show no significant changes. Representative images of TH and DAT immunofluorescence in the dorsal striatum of WT and DAT-Ala53 along with the superimposed images of TH and DAT immunofluorescence are shown in (A) and (B). Quantified intensities of TH and DAT immunofluorescence in the dorsal striatum and the corresponding DAT/TH ratio are shown in (C, D, E). Representative images of TH and DAT immunofluorescence in the ventral striatum of WT and DAT-Ala53 along with the superimposed images of TH and DAT immunofluorescence are shown in (F) and (G). Quantified intensities of TH and DAT immunofluorescence in the ventral striatum, and the corresponding DAT/TH ratio are shown in (H, I, J). Scale bar represents 100 μM. Values are presented as Mean ± S.D. ns: nonsignificant effect (unpaired two-tailed Student’s t-test).
3.6. DAT-Ala53 mice display unaltered DA uptake, DA transport Kinetics, and total and surface DAT expression.
There were no significant differences in DAT-mediated DA uptake between WT and DAT-Ala53 mice when dorsal or ventral striatal synaptosomes were used [dorsal: t = 0.0672, df = 6, P = 0.9486; ventral: t = 0.9694, df = 6, P = 0.3698] (Fig. 6A). The kinetics of DAT-mediated DA transport activity are important factors in the regulation of DA neurotransmission. The kinetic parameters (maximal velocity, Vmax and DA substrate apparent affinity, Km) were determined using whole striatal synaptosomes from WT and DAT-Ala53 mice. Notably, there were no significant differences in the maximal velocity and apparent affinity between DAT-Ala53 and WT mice when DA uptake was measured over a concentration range of 25-1000 nM using striatal synaptosomes (Fig. 6B). Vmax for DAT-Ala53 was 21.26 ± 2.522 pmol/mg protein/min and for WT was 20.15 ± 1.65 pmol/mg protein/min [t = 0.6345, df = 4, P = 0.5602]. Km for DAT-Ala53 was 71.17 ± 6.766 nM and for WT was 77.80 ± 3.009 nM [t = 1.551, df = 4, P = 0.1957]. We then performed and analyzed the levels of t-DAT and pT53-DAT expression and the distribution between plasma membrane and intracellular pools using biotinylation and immunoblot approach. In parallel, biotinylation experiments were conducted using synaptosomal preparations from WT cerebellum and cortex (without the frontal cortex) to validate t-DAT and pT53-DAT antibodies. Striatal extract from DAT-knock-out was also included as another control to authenticate DAT immunoreactive bands in immunoblot analyses. Figures 7A, B and C show representative immunoblots of striatal t-DAT and pT53-DAT, and their distribution in plasma membrane and intracellular pools. t-DAT protein band was evident in the total extract, biotinylated surface fraction, and non-biotinylated intracellular faction of WT and DAT-A53 mice. However, pT53-DAT bands were completely absent in the DAT-Ala53 mice (Fig. 7A, B and C). Both t-DAT and pT53-DAT bands were undetectable in our control samples (cerebellum, cortex (without the frontal cortex) and striatum from DAT knock-out). Quantification of t-DAT immunoreactive band densities showed no significant differences between DAT-Ala53 and WT mice in the total expression level of t-DAT normalized to total calnexin (Fig. 7D, t = 0.1885, df = 4, P = 0.8597), surface biotinylated t-DAT normalized to total t-DAT (Fig. 7E, t = 1.240, df = 4, P = 0.2828) and intracellular non-biotinylated t-DAT normalized to total t-DAT (Fig. 7F, t = 0.3975, df = 4, P = 0.7113). pT53-DAT immunoreactive bands were detectable in the WT mice and distributed in the total, surface membrane (biotinylated), and intracellular (non-biotinylated) fractions, but completely absent in the DAT-Ala53 mice (Fig. 7G, H and I). There were no differences between DAT-Ala53 and WT mice in the density of calnexin (an intracellular endoplasmic reticulum protein which was used as an internal control) in the total and non-biotinylated intracellular fractions (Fig. 7A and C, quantified data not shown). No detectable levels of calnexin were found in the biotinylated surface membrane fractions (Fig. 7B) evidently showing that the synaptosomes were intact and only the surface proteins were biotinylated during the procedure.
Fig. 6. DAT activity and DA-uptake kinetics in the striatal synaptosomes of DAT-Ala53 mice.
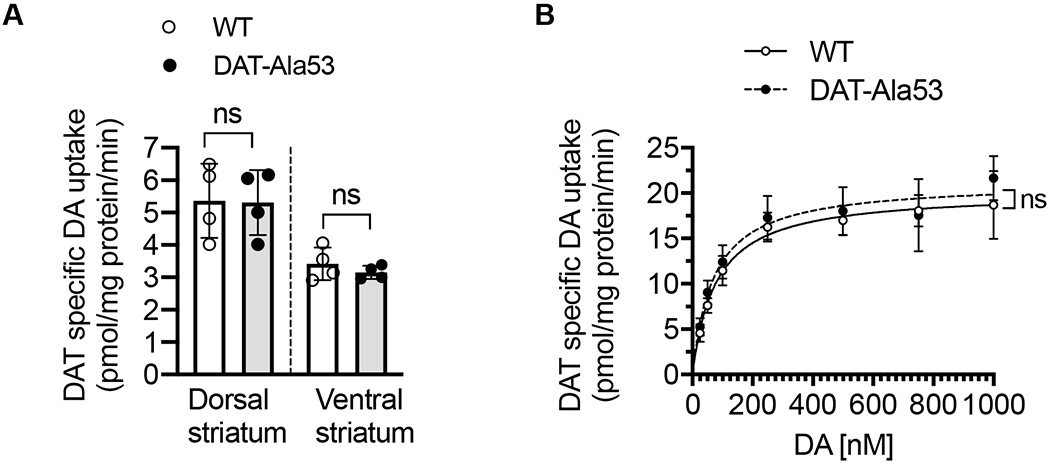
(A) [3H] DA (5 nM) uptake assays were performed using crude synaptosomes from dorsal and ventral striatum of WT and DAT-Ala53 mice as described in the material and methods section 2.8. Values (pmol/mg protein/min) are presented as Mean ± S.D. WT (n = 4) and DAT-Ala53 (n = 4) mice. ns: nonsignificant (unpaired two-tailed Student’s t-test). (B) DAT kinetic analysis was performed in whole striatal synaptosomes using DA concentrations over a range of 25-1000 nM and DAT specific activity was determined as described in the material and methods. Substrate (DA) apparent affinity (Km) and maximal velocity (Vmax) of DAT were determined by using the Michaelis-Menten equation (Prism). Values represented as Mean ± S.D. WT (n = 4) and DAT-Ala53 (n = 4) mice. ns: nonsignificant (unpaired two-tailed Student’s t-test).
Fig. 7. Similar total and cell surface expression of DAT in the striatal synaptosomes of DAT-Ala53 mice.
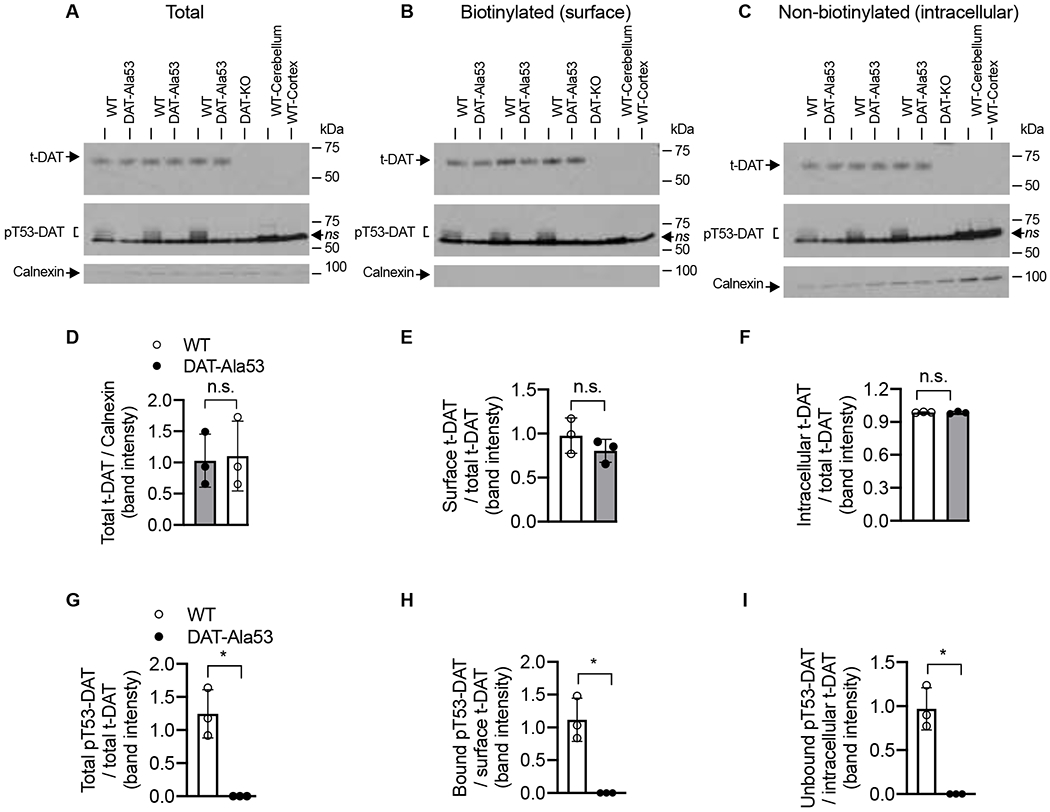
Surface protein biotinylation, immunoblotting, and quantification were done as described in material and methods section 2.9. Representative immunoblots (A, B, C) show total, biotinylated and non-biotinylated t-DAT (~65 kDa) and pT53-DAT (~60-55 kDa). The intracellular calnexin bands (~90 kDa) were shown under each respective blot. In parallel, protein extracts from DAT-KO striatum or lysates of cerebellum and cortex (without the frontal cortex) from WT were used to validate the specificity of t-DAT and pT53-DAT bands. Quantified t-DAT band intensities are as shown. (D) total extract normalized to total calnexin, (E) biotinylated surface fraction normalized to total t-DAT, and (F) non-biotinylated intracellular fraction normalized to total t-DAT. Quantified pT53-DAT band intensities are as shown. (G) total extract normalized to total t-DAT. (H) biotinylated surface fraction normalized to biotinylated t-DAT, and (I) non-biotinylated intracellular fraction normalized to intracellular t-DAT. Values are presented as Mean ± S.D. WT (n = 3) and DAT-Ala53 (n = 3) mice. * P < 0.05 and ns: nonsignificant (unpaired two-tailed Student’s t-test).
3.7. Striatal DA uptake in DAT-Ala53 is not altered by ERK1/2 inhibition.
Given the fact that the DAT-Thr53 is a canonical ERK phosphorylation site [35] and ERK1/2 inhibition by MEK inhibitors U0126 and PD 98059 reduces DA uptake [44], we sought to determine whether endogenous DAT-Thr53 phosphorylation is involved in ERK mediated DAT regulation. We examined the effect of ERK1/2 inhibition by MEK inhibitors, U0126 and PD 98059 on DAT-specific DA uptake in DAT-Ala53 striatal synaptosomes and compared with that in WT. Inhibition of DAT-mediated DA uptake by U0126 or PD98059 at different doses was completely absent in DAT-Ala53 (Fig. 8 A and B). In agreement with previous findings [44], pretreatment with 0.1, 1.0 and 5.0 μM U0126 or PD98059 resulted in a significant dose dependent inhibition of DA uptake in WT [U0126: F(3, 8) = 12.21, P = 0.0023; PD98059: F(3, 8) = 7.279, P = 0.0113]. In contrast to WT, there was no inhibitory effect of U0126 or PD98059 on DAT activity in DAT-Ala53 [U0126: F(3, 8) = 1.789, P = 0.2271; F(3, 8) = 1.023, P = 0.9564]. These results indicate that the downregulation of DAT by ERK1/2 inhibition is dependent on endogenous phosphorylation of Thr53 site in mouse DAT.
Figure 8. Effect of ERK inhibitors on DAT mediated DA uptake in WT and DAT-Ala53 striatal synaptosomes.
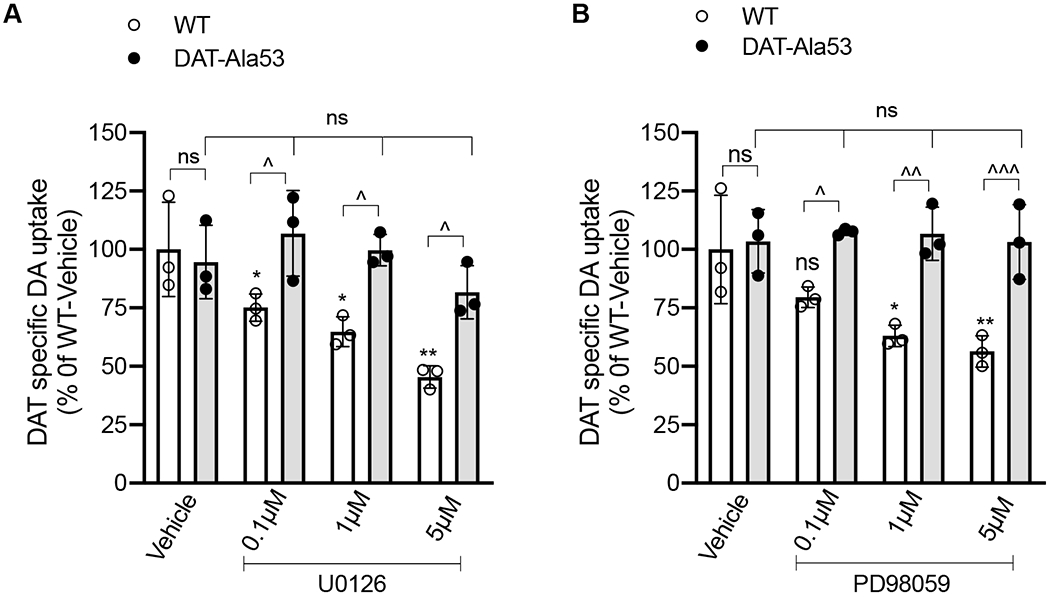
Crude striatal synaptosomes were preincubated with vehicle or indicated concentrations of U0126 or PD98059 for 30 min at 37°C and [3H] DA (5 nM) uptake assays were performed following the treatments as described in material and methods section 2.9. Values are presented as Mean ± S.D. WT (n = 3) and DAT-Ala53 (n = 3) mice. Bonferroni’s multiple comparison test followed by one-way ANOVA: * P <0.05, ** P <0.01 (WT: U0126 or PD98059 compared with WT-Vehicle), ns: nonsignificant effect (DAT-A53: U0126 or PD98059 versus DAT-A53 Vehicle or WT-Vehicle vs DAT-Ala53-Vehicle) and Λ P <0.05, ΛΛ P <0.01, ΛΛΛ P <0.001 (WT versus DAT-Ala53 pretreated with different doses of U0126 or PD98059). DAT activity (DA transport: 0.90559 ± 0.1852 pmol/mg protein/ min for WT-Vehicle; 0.8946 ± 0.1206 pmol/mg protein/ min for DAT-Ala53 Vehicle)
3.8. Striatal DA uptake in DAT-Ala53 is less sensitive to cocaine inhibition and DAT-Ala53 mice exhibit reduced cocaine-induced hyperlocomotion.
Inhibitor sensitivity of cocaine on DA uptake was measured using striatal synaptosomes from DAT-Ala53 mice expressing DAT-Ala53 and WT mice expressing DAT-Thr53 (Fig. 9 A). DA uptake in DAT-Ala53 showed significantly reduced (4.7 fold) inhibition by cocaine than that in WT (t = 5.010, df = 4, P = 0.0074). DA uptake in DAT-Ala53 was inhibited by cocaine with IC50 of 1.268 ± 0.348 μM, while that in WT was inhibited by cocaine with IC50 of 0.268 ± 0.058 μM. DA uptake in DAT-Ala53 and WT showed similar sensitivity to inhibition by amphetamine with IC50 of 0.198 ± 0.068 μM for DAT-Ala53 and 0.183 ± 0.023 μM for WT (t = 0.367, df = 4, P = 0.7320) or by METH with IC50 of 0.262 ± 0.025 μM for DAT-Ala53 and 0.280 ± 0.040 μM for WT (t = 0.669, df = 4, P = 0.5403) (Supplementary figure 2). These results indicate the potential impact of DAT-Thr53 phosphorylation on cocaine potency to inhibit DAT activity. Therefore, we investigated whether DAT-Ala53 mice display reduced cocaine sensitivity to elicit locomotor stimulation.
Fig. 9. Reduced cocaine potency and suppression of cocaine induced locomotor activity in DAT-Ala53 mice.
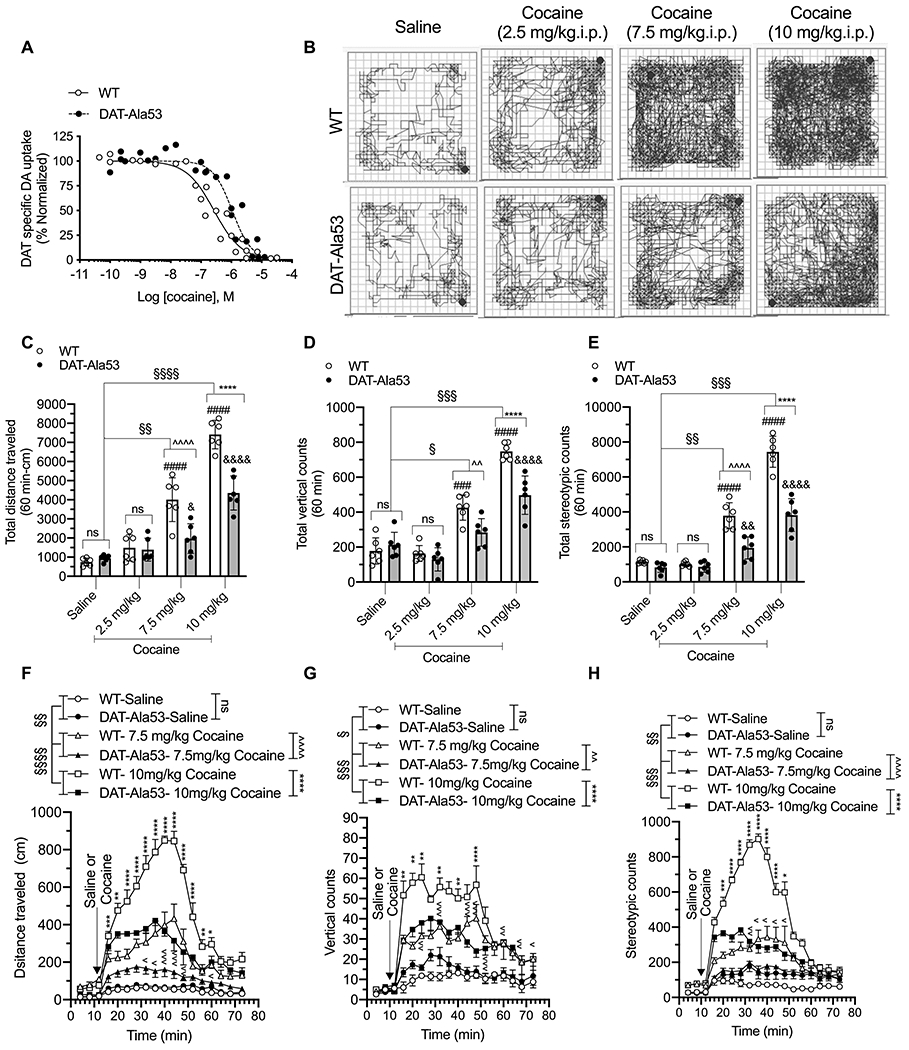
(A) Competition DA uptake assays with cocaine were performed using crude synaptosomes from the striatum of WT and DAT-Ala53 mice. Striatal synaptosomes were incubated with a fixed concentration of [3H] DA (20 nM) and increasing concentrations of cocaine for 4 min as described under material and methods section 2.12. DAT-specific DA uptake was normalized with control uptake in the absence of test compound. Figure shows the normalized data plotted against the log of the molar concentrations of test the compound. IC50 values were determined using nonlinear regression fit. Cocaine IC50 in DAT-Ala53 is significantly shifted to the right compared to WT (P = 0.007 unpaired two-tailed Student’s t-test). WT (n = 3) and DAT-Ala53 (n = 3) mice for each compound. (B - H) Habituated male WT (n = 6 for all groups) and DAT-Ala53 (n = 6 for all groups) animals were injected with saline or cocaine with different doses (2.5 or 7.5 or 10 mg/kg. i.p.) and placed in the locomotor boxes and monitored motor activities for 60 min. Representative activity traces show the locomotor activity of WT and DAT-Ala53 mice post-injection of vehicle or cocaine (B). Comparative analysis (DAT-Ala53 vs. WT) of total distance traveled (C), vertical counts (D), and stereotypy counts (E) are shown. Time course plots show distance traveled, vertical counts, and stereotypy counts measured in 4 min bins over 60 min period post-injection (F, G, H). Bonferroni’s multiple comparison test followed by two-way ANOVA: § P <0.05, §§ P <0.01, §§§ P <0.001, §§§§ P <0.0001, ns: nonsignificant effect (P > 0.9999) compared between specified pairs, Λ P <0.05, ΛΛ P <0.01, ΛΛΛ P <0.001, ΛΛΛ P <0.0001, (WT-cocaine (7.5 mg/kg) versus DAT-Ala53-cocaine (7.5 mg/kg), * P <0.05, ** P <0.01, *** P <0.001, **** P<0.0001, (WT-cocaine (10 mg/kg) versus DAT-Ala53-cocaine (10 mg/kg), ### P <0.001, #### P <0.0001, (WT-saline vs WT-cocaine), & P <0.05, && P <0.001, &&&& P <0.0001(DAT-Ala53-saline vs DAT-Ala53-cocaine). Data are presented as Means ± S.D.
We examined the behavioral effect of i.p. cocaine at 2.5, 7.5 and 10 mg/kg doses in WT and DAT-Ala53 mice by assessing the cocaine-induced hyperlocomotion. Representative locomotor activity path graphs are shown in Fig. 9 B. The total distance traveled, vertical counts, and stereotypy counts along with comparative analyses of the data are shown in Fig. 9 C, D and E and respective 4 min bins over a 60 min time period of post injection are shown in Fig. 9 F, G and H. Two-way ANOVA analysis over a 60 min locomotor activity revealed there was no significant effect on total distance travelled at cocaine dose (2.5 mg/kg) and no significant effect of on total distance travelled [F(1, 10) = 0.05674, P = 0.8165; F(1, 10) = 1.090, P = 0.4472], vertical counts [F(1, 10) = 2.067, P = 0.1811; F(1, 10) = 0.0011, P = 0.9734], and stereotypy counts [F(1, 10) = 4.545, P = 0.0588; F(1, 10) = 4.710, P = 0.0551] and no significant effect of cocaine x genotype interaction [distance travelled; F(1, 10) = 0.5970, P = 0.4576; vertical counts F(1, 10) = 1.020, P = 0.3363; and stereotypy counts F(1, 10) = 1.676, P = 0.2246 respectively]. Interestingly, cocaine-stimulated locomotion at 7.5 and 10 mg/kg doses was significantly reduced in DAT-Ala53 mice compared to that in WT mice. Two-way ANOVA analysis of saline and 7.5mg/kg cocaine groups revealed significant effect of cocaine and genotype on total distance [F(1, 10) = 63.56, P < 0.0001; F(1, 10) = 9.216, P = 0.0126], vertical counts [F(1, 10) = 22.75, P < 0.0001; F(1, 10) = 9.926, P = 0.0103], and stereotypy counts [F(1, 10) = 82.61, P < 0.0001; F(1, 10) = 26.91, P = 0.0004] respectively. In addition, there was significant effect of genotype x cocaine interaction on total distance [F(1, 10) = 17.15, P = 0.0020, vertical counts [F(1, 10) = 7.003, P = 0.0245] and stereotypy counts [F(1, 10) = 13.59, P = 0.0042]. Similar trends were observed with 10 mg/kg cocaine dose. As shown in Figure.9 C – H, two-way ANOVA analysis of saline and 10 mg/kg cocaine groups revealed significant effect of cocaine and genotype on total distance [F(1, 10) = 426.1, P < 0.0001; F(1, 10) = 34.90, P = 0.0001], vertical counts [F(1, 10) = 227.9, P < 0.0001; F(1, 10) = 9.080, P = 0.0130], and stereotypy counts [F(1, 10) = 291.7, P < 0.0001; F(1, 10) = 53.60, P < 0.0001]. There was also a significant effect of genotype x drug interaction on total distance [F(1, 10) = 44.36, P < 0.0001 , vertical counts [F(1, 10) = 25.50, P = 0.0005] and stereotypy counts [F(1, 10) = 36.75, P = 0.0001]. Taken together, these data indicate that the cocaine-induced motor stimulation is suppressed in DAT-Ala53 mice in parallel with reduced DAT sensitivity to cocaine inhibition.
4. Discussion
DAT exists in constitutively phosphorylated form and the phosphorylation of DAT is a dynamically regulated phenomenon. It is known that DAT functional regulation by presynaptic DA auto- and hetero- receptors, protein kinases, and phosphatases is tightly coupled to DAT phosphorylation [Reviewed in and see references therein 28, 29–31]. Phosphorylation of DAT on Thr53 has been identified in rodent striatum and demonstrated using heterologous expression models [35–37, 52]. However, the impact of endogenous DAT-Thr53 phosphorylation on DAT regulation, synaptic DA dynamics and animal behavior has not been investigated. The generation of a mouse line with a targeted mutation of DAT-Thr53 is the logical step toward understanding the in vivo role of DAT-Thr53 phosphorylation. In the present study, we generated a knock-in mouse model lacking Thr53 phosphorylation in DAT protein (DAT-Ala53) and we report comprehensive biochemical, neurochemical, immunochemical, and behavioral characterization of the DAT-Ala53 mice. Immunoblot analysis demonstrated that the DAT-Thr53 phosphorylation was successfully eliminated in DAT-Ala53 knock-in mice. We show that the DAT-Ala53 mice do not display any developmental deficits or any changes in the body weight, temperature, forelimb coordination strength or food (sucrose) preference. Both male and female DAT-Ala53 mice were fertile and produced normal sized litters with no abnormalities in body weight or body temperature (measured from 4 weeks until 12 months).
The DAT-Ala53 mice displayed hyperactivity/hyperlocomotion, enhanced vertical exploratory behavior, and stereotypy in novel open-field environment. Studies on DAT-KO mice showed hyperlocomotion and stereotypy [1, 53–55]. DAT knock-down mice with reduced DAT expression and mice expressing a cocaine-insensitive DAT mutant also exhibit spontaneous hyperactivity, stereotypic/rearing activity, and impaired habituation to a novel environment [6, 56, 57]. However, these changes cannot be accounted for the display of novelty-induced hyperlocomotion observed because the activity and expression of DAT in DAT-Ala53 are equivalent to WT. Previous reports state that the presence or activation of hyperlocomotor behavior in rodents is due to their attraction and excitement to the novel environment [58, 59], and this type of behavior can be reversed by habituation to the novel stimulus or by use of pharmacological agents such as haloperidol or clozapine or serotonin selective reuptake inhibitors (SSRI) [53, 54, 60]. Consistent with this notion, hyperactive behaviors (hyperlocomotion, enhanced vertical exploration, and stereotypy) of DAT-Ala53 mice disappeared following habituation. Thus, the hyperactive behaviors observed in the DAT-Ala53 mice prior to habituation to locomotor boxes appear to be novelty-induced behaviors and therefore reversed by prolonged habituations. A higher extracellular synaptic DA can also trigger higher baseline locomotor activity [1, 6]. DAT ligands, including amphetamine, cocaine, and DAT inhibitor GBR 12909 trigger stereotypy in rodents [57, 61–63]. Interestingly, cocaine failed to produce stereotypic behavior in mice expressing cocaine-insensitive DAT mutant [57]. These observations are pointing out the role of DAT in locomotor behavior, stereotypy, and habituation. However, we found unaltered striatal total DA and DA metabolites in the DAT-Ala53 mice. Future studies investigating DAT-Thr53 phosphorylation, extracellular DA levels, and DA dynamics (release and clearance) during the experience of novel environment and during complex exploratory behaviors using additional behavioral paradigms may provide more insight into the impact of DAT-Thr53 phosphorylation on novelty induced hyperactivity.
Findings from our functional and biochemical studies on the striatum of DAT-Ala53 mice showed no significant alterations in the DA-transport kinetics or in the total or surface DAT expression. Furthermore, pTH and t-TH levels were not altered in DAT-Ala53 mice striatum as evidenced by immunoblot and immunofluorescence studies. These results suggest that there are no basal functional abnormalities in DA synthesis or DAT functional expression arising due to constitutive elimination of DAT-Thr53 phosphorylation. This is different from the reported observations in heterologous expression cell models. While one study reported that mutant rDAT-Ala53 expressing cells exhibit lower DAT expression and Vmax without any alteration in Km values [36], another study reported higher Vmax in mutant rDAT-Ala53 expressing cells [37]. One can anticipate that methodological differences (DAT activity measured by [3H]DA uptake versus rotating disc electrode voltammetry technique) could contribute to conflicting observations. We conclude that the lack of endogenous DAT-Thr53 phosphorylation does not affect the expression and distribution of DAT protein or DA-transport kinetics. Thr53 phosphorylation site in rodent DAT is located within the consensus motif (P-P-X-S.T-P) for proline-directed kinases and it was demonstrated that ERK kinase phosphorylates N-tail fusion protein of rDAT in vitro [35]. Also, ERK1/2 inhibition reduces DA uptake in heterologous EM4-cell models and rat-striatal synaptosomes [44]. However, whether endogenous Thr53 phosphorylation of DAT is involved in ERK-mediated DAT regulation is unknown. We found that the inhibitory effect of ERK1/2 inhibition on DAT activity is completely absent in DAT-Ala53 mice providing the first insight into the molecular link between ERK-dependent Thr53 phosphorylation and DAT activity in the brain.
We previously demonstrated that the activation of dopamine autoreceptors (D2 and D3) and heteroreceptor kappa-opioid receptors regulate DAT function via ERK1/2 dependent mechanisms [64–66]. D2-autoreceptor activation increases dorsal but not ventral striatal DAT-Thr53 phosphorylation in slice preparations [52]. Further studies are needed to identify signaling pathways downstream of receptor activation and upstream of ERK activation, and other kinases that may be involved in the phosphorylation of Thr53 either directly or indirectly in conjunction with other signaling cascades to regulate DAT functional regulations.
We observed an interesting finding that the potency of cocaine to inhibit DA transport in DAT-Ala53 mice was significantly lower while the potencies of amphetamine and methamphetamine to inhibit DA transport in DAT-Ala53 were not affected. An in vitro study showed reduced cocaine binding affinity in LLPKC1 cells transfected with Thr53Ala mutant of rDAT [37]. Furthermore, enhanced cocaine potency has been reported to occur in parallel with enhanced Thr53 phosphorylation of DAT in females during oestrus cycle [67]. Further studies investigating the functional regulation of DAT, DA dynamics, and behaviors in DAT-Ala53 females during the cyclical hormonal phase will reveal the causal link between DAT-Thr53 phosphorylation and sex hormones. DA neuronal activity modulates DAT-T53 phosphorylation, cocaine potency, and cocaine consumption [68]. However, the causal link between DAT-Thr53 phosphorylation and behavioral effects of cocaine is yet to be established. Based on the present findings from DAT-Ala53 mice and previous supportive findings [37, 67], we propose that the reduced cocaine potency coupled to DAT-Ala53 could impact cocaine-induced behaviors. As expected, DAT-Ala53 mice exhibit reduced hyperlocomotion in response to cocaine at 7.5 and 10 mg/kg doses compared to WT. Compared to saline, while both doses of cocaine were capable of increasing locomotor activity in both genotypes, DAT-Ala53 mice showed comparatively less locomotor activation in response to cocaine than WT mice. The reason for this could uniquely be due to reduced cocaine potency to inhibit DAT transport in DAT-Ala53 mice. However, it is important to test whether DAT-Ala53 mice have generalized deficits in psychostimulant-induced locomotor and or specific to cocaine due to reduced potency to inhibit DAT in the absence of DAT-Thr53 phosphorylation. Even though potencies of AMPH and METH to inhibit DA transport were not affected in DAT-Ala53 mice, it is still needed to be validated at behavioral level. Despite these reservations, our data would indicate that DAT-Thr53 phosphorylation affects cocaine potency by modifying DAT’s affinity for cocaine and consequently the behavioral effects of cocaine.
As far as we know, no studies have been reported on how DAT-phosphorylation impacts cocaine binding. Engagement of residues in transmembrane 1, 3, 6, and 8 has been implicated in DA, AMPH, and cocaine binding [69]. Several residues in DAT have been proposed to modulate cocaine interactions with DA transport capacity through allosteric mechanisms [70, 71]. There are many possible interpretations to explain the decreased cocaine potency to inhibit DA transport in DAT-Ala53 mice. DAT-Thr-53 phosphorylation may have direct effects on the stabilization of the outward-facing DAT confirmation subsequently modifying the cocaine binding potency, or DAT-Thr53 phosphorylation allosterically modulates conformational changes of cocaine-DAT binding sites/residues affecting the cocaine potency. Also, DAT-Thr-53 phosphorylation may affect DAT-interacting partners, which might affect cocaine binding sites. While the molecular mechanisms underlying the causal interaction of Thr53 phosphorylation with cocaine binding sites/residues need to be resolved in future studies, based on our findings, we speculate that Thr53 phosphorylation of DAT enhances cocaine potency by retaining normal DAT expression, DA-transport capacity, and apparent DA affinity. Thus, our findings that DAT-Ala53 affects cocaine potency without altering DA-transport kinetics suggest a clear dissociation of cocaine binding and DA transport in vivo. However, the influence of Ala substitution at Thr53 on cocaine potency cannot be ruled out. Cocaine binding and consequent DAT inhibition is a prerequisite for initiating cocaine behavioral effects and cocaine-induced adaptive neuronal plasticity [5, 6, 72]. We previously demonstrated that DAT kinetics, phosphorylation, surface expression, and association with phosphatase 2Ac are differentially altered in the nucleus accumbens and caudate putamen of cocaine self-administered rats [73, 74]. Considering the fact that receptor triggered kinases and phosphatases can modulate the stoichiometry of DAT- Thr53 phosphorylation, potential physiological regulation of cocaine’s affinity/potency to inhibit DA transport in the central nervous system can consequently modulate cocaine linked behaviors.
5. Conclusions
DAT-Ala53 knock-in mouse model has translational values at different levels to fill in the existing gaps in our understanding of the role of DAT phosphorylation in vivo. For example, DAT-Ala53 mice will allow validation of in vitro findings to in vivo settings and provide the first opportunity to study the role of in vivo DAT-Thr53 phosphorylation in DAT expression and regulation, DA-transport kinetics, DA dynamics, and DA-circuit specific molecular changes. Utilizing DAT-Ala53 mice, the causal linkage of DAT-Thr53 phosphorylation can be made to auto- and hetero- receptor mediated DAT regulations, and the influential engagement of DAT-Thr53 phosphorylation on cellular and neurochemical linkages of cocaine reward and other substance-use-disorders can be explored. Thus, our rodent model offers a valuable tool for initiating such translational studies. More significantly, our findings reveal cocaine antagonistic site in DAT regulatory motif that can be used to develop new treatment strategies for cocaine-use-disorder without affecting the normal DA-linked behaviors.
Highlights.
DAT-Ala53 knock-in mice are viable and display no developmental deficits.
DA-transport and the expression of DAT and TH are unaltered in the DAT-Ala53 mice.
DAT-Ala53 mice exhibit hyperactivity in novel environment.
ERK1/2 mediated DAT regulation is blunted in the DAT-Ala53 knock-in mice.
DAT-Ala53 mice exhibit reduced sensitivity to cocaine inhibition and hyperlocomotion.
Acknowledgments
The work was supported by National Institute of Health (MH112731, DA041673) to SR. We thank the Vanderbilt Neurochemistry Core supported by the Vanderbilt brain institute and the Vanderbilt Kennedy Center for neurochemical analysis of our mouse brain samples. We thank Drs. William Dewey and Imad Damaj (Dep. Pharmacology and Toxicology at VCU) for providing the apparatus and guidance in measuring locomotor and grip strength behaviors. We also thank the VCU Massey Cancer Transgenic/Knockout Mouse Core Facility supported by P30 CA016059 for the generation of DAT-Ala53 knock-in mice and the VCU Department of Anatomy and Neurobiology Microscopy Facility supported by P30 NS047463 and P30 CA016059 for immunofluorescence analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: All authors declare that there is no conflict of interest.
References:
- [1].Giros B, Jaber M, Jones SR, Wightman RM, Caron MG, Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter, Nature 379 (1996) 606–612 [DOI] [PubMed] [Google Scholar]
- [2].Donovan DM, Miner LL, Perry MP, Revay RS, Sharpe LG, Przedborski S, Kostic V, Philpot RM, Kirstein CL, Rothman RB, Schindler CW, Uhl GR, Cocaine reward and MPTP toxicity: alteration by regional variant dopamine transporter overexpression, Brain Res Mol Brain Res 73(1-2) (1999) 37–49. 10.1016/s0169-328x(99)00235-1 [DOI] [PubMed] [Google Scholar]
- [3].Salahpour A, Ramsey AJ, Medvedev IO, Kile B, Sotnikova TD, Holmstrand E, Ghisi V, Nicholls PJ, Wong L, Murphy K, Sesack SR, Wightman RM, Gainetdinov RR, Caron MG, Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter, Proc Natl Acad Sci U S A 105(11) (2008) 4405–10. 10.1073/pnas.0707646105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hadar R, Edemann-Callesen H, Reinel C, Wieske F, Voget M, Popova E, Sohr R, Avchalumov Y, Priller J, van Riesen C, Puls I, Bader M, Winter C, Rats overexpressing the dopamine transporter display behavioral and neurobiological abnormalities with relevance to repetitive disorders, Sci Rep 6 (2016) 39145. 10.1038/srep39145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Thomsen M, Han DD, Gu HH, Caine SB, Lack of cocaine self-administration in mice expressing a cocaine-insensitive dopamine transporter, J Pharmacol Exp Ther 331(1) (2009) 204–11. 10.1124/jpet.109.156265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S, Quan N, Stephens RL, Hill ER, Nottoli T, Han DD, Gu HH, Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter, Proc Natl Acad Sci U S A 103(24) (2006) 9333–8. 10.1073/pnas.0600905103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mergy MA, Gowrishankar R, Gresch PJ, Gantz SC, Williams J, Davis GL, Wheeler CA, Stanwood GD, Hahn MK, Blakely RD, The rare DAT coding variant Val559 perturbs DA neuron function, changes behavior, and alters in vivo responses to psychostimulants, Proc Natl Acad Sci U S A 111(44) (2014) E4779–88. 10.1073/pnas.1417294111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].DiCarlo GE, Aguilar JI, Matthies HJ, Harrison FE, Bundschuh KE, West A, Hashemi P, Herborg F, Rickhag M, Chen H, Gether U, Wallace MT, Galli A, Autism-linked dopamine transporter mutation alters striatal dopamine neurotransmission and dopamine-dependent behaviors, J Clin Invest 129(8) (2019) 3407–3419. 10.1172/JCI127411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Davis GL, Stewart A, Stanwood GD, Gowrishankar R, Hahn MK, Blakely RD, Functional coding variation in the presynaptic dopamine transporter associated with neuropsychiatric disorders drives enhanced motivation and context-dependent impulsivity in mice, Behav Brain Res 337 (2018) 61–69. 10.1016/j.bbr.2017.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Giros B, Caron MG, Molecular characterization of the dopamine transporter, Trends Pharmacol Sci 14(2) (1993) 43–9 [DOI] [PubMed] [Google Scholar]
- [11].Wayment HK, Deutsch H, Schweri MM, Schenk JO, Effects of methylphenidate analogues on phenethylamine substrates for the striatal dopamine transporter: potential as amphetamine antagonists?, J Neurochem 72(3) (1999) 1266–1274 [DOI] [PubMed] [Google Scholar]
- [12].Sulzer D, Sonders MS, Poulsen NW, Galli A, Mechanisms of neurotransmitter release by amphetamines: a review, Prog Neurobiol 75(6) (2005) 406–33. 10.1016/j.pneurobio.2005.04.003 [DOI] [PubMed] [Google Scholar]
- [13].Appell M, Berfield JL, Wang LC, Dunn WJ 3rd, Chen N, Reith ME, Structure-activity relationships for substrate recognition by the human dopamine transporter, Biochem Pharmacol 67(2) (2004) 293–302 [DOI] [PubMed] [Google Scholar]
- [14].Dubol M, Trichard C, Leroy C, Sandu AL, Rahim M, Granger B, Tzavara ET, Karila L, Martinot JL, Artiges E, Dopamine Transporter and Reward Anticipation in a Dimensional Perspective: A Multimodal Brain Imaging Study, Neuropsychopharmacology 43(4) (2018) 820–827. 10.1038/npp.2017.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, Hitzemann R, Shea CE, Relationship between subjective effects of cocaine and dopamine transporter occupancy, Nature 386(6627) (1997) 827–30. 10.1038/386827a0 [DOI] [PubMed] [Google Scholar]
- [16].Narendran R, Martinez D, Cocaine abuse and sensitization of striatal dopamine transmission: a critical review of the preclinical and clinical imaging literature, Synapse 62(11) (2008) 851–69. 10.1002/syn.20566 [DOI] [PubMed] [Google Scholar]
- [17].Leroy C, Karila L, Martinot JL, Lukasiewicz M, Duchesnay E, Comtat C, Dolle F, Benyamina A, Artiges E, Ribeiro MJ, Reynaud M, Trichard C, Striatal and extrastriatal dopamine transporter in cannabis and tobacco addiction: a high-resolution PET study, Addict Biol 17(6) (2012) 981–90. 10.1111/j.1369-1600.2011.00356.x [DOI] [PubMed] [Google Scholar]
- [18].Mash DC, Pablo J, Ouyang Q, Hearn WL, Izenwasser S, Dopamine transport function is elevated in cocaine users, J Neurochem 81(2) (2002) 292–300 [DOI] [PubMed] [Google Scholar]
- [19].Little KY, Carroll FI, Butts JD, Striatal [125I]RTI-55 binding sites in cocaine-abusing humans, Prog Neuropsychopharmacol Biol Psychiatry 22(3) (1998) 455–66 [DOI] [PubMed] [Google Scholar]
- [20].Little KY, Zhang L, Desmond T, Frey KA, Dalack GW, Cassin BJ, Striatal dopaminergic abnormalities in human cocaine users, Am J Psychiatry 156(2) (1999) 238–45 [DOI] [PubMed] [Google Scholar]
- [21].Malison RT, Best SE, van Dyck CH, McCance EF, Wallace EA, Laruelle M, Baldwin RM, Seibyl JP, Price LH, Kosten TR, Innis RB, Elevated striatal dopamine transporters during acute cocaine abstinence as measured by [123I] beta-CIT SPECT, Am J Psychiatry 155(6) (1998) 832–4. [DOI] [PubMed] [Google Scholar]
- [22].Staley JK, Hearn WL, Ruttenber AJ, Wetli CV, Mash DC, High affinity cocaine recognition sites on the dopamine transporter are elevated in fatal cocaine overdose victims, The Journal of Pharmacology and Experimental Therapeutics 271 (1994) 1678–1685 [PubMed] [Google Scholar]
- [23].Mash DC, Staley JK, The dopamine transporter in human brain: Characterization and effect of cocaine exposure, in: Reith MEA (Ed.), Neurotransmitter Transporters: Structure, Function, and Regulation, Humana Press, Totowa, NJ, 1997, pp. 315–344. [Google Scholar]
- [24].Hurd YL, Herkenham M, Influence of a single injection of cocaine, amphetamine or GBR 12909 on mRNA expression of striatal neuropeptides, Brain Res Mol Brain Res 16(1-2) (1992) 97–104. [DOI] [PubMed] [Google Scholar]
- [25].Wilson JM, Levey AI, Bergeron C, Kalasinsky K, Ang L, Peretti F, Adams VI, Smialek J, Anderson WR, Shannak K, Deck J, Niznik HB, Kish SJ, Striatal dopamine, dopamine transporter, and vesicular monoamine transporter in chronic cocaine users, Annual Neurology 40 (1996) 428–439 [DOI] [PubMed] [Google Scholar]
- [26].Arroyo M, Baker WA, Everitt BJ, Cocaine self-administration in rats differentially alters mRNA levels of the monoamine transporters and striatal neuropeptides, Brain Res Mol Brain Res 83(1-2) (2000) 107–20. [DOI] [PubMed] [Google Scholar]
- [27].Xia Y, Goebel DJ, Kapatos G, Bannon MJ, Quantitation of rat dopamine transporter mRNA: effects of cocaine treatment and withdrawal, The Journal of Neurochemistry 59 (1992) 1179–1182 [DOI] [PubMed] [Google Scholar]
- [28].Ramamoorthy S, Shippenberg TS, Jayanthi LD, Regulation of monoamine transporters: Role of transporter phosphorylation, Pharmacol Ther 129(2) (2011) 220–38. 10.1016/j.pharmthera.2010.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Foster JD, Cervinski MA, Gorentla BK, Vaughan RA, Regulation of the dopamine transporter by phosphorylation, Handb Exp Pharmacol (175) (2006) 197–214 [DOI] [PubMed] [Google Scholar]
- [30].Eriksen J, Jorgensen TN, Gether U, Regulation of dopamine transporter function by protein-protein interactions: new discoveries and methodological challenges, J Neurochem 113(1) (2010) 27–41. 10.1111/j.1471-4159.2010.06599.x [DOI] [PubMed] [Google Scholar]
- [31].Bermingham DP, Blakely RD, Kinase-dependent Regulation of Monoamine Neurotransmitter Transporters, Pharmacol Rev 68(4) (2016) 888–953. 10.1124/pr.115.012260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Granas C, Ferrer J, Loland CJ, Javitch JA, Gether U, N-terminal truncation of the dopamine transporter abolishes phorbol ester- and substance P receptor-stimulated phosphorylation without impairing transporter internalization, J Biol Chem 278(7) (2003) 4990–5000. 10.1074/jbc.M205058200 [DOI] [PubMed] [Google Scholar]
- [33].Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA, N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux, PLoS Biol 2(3) (2004) 0387–0393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N, Nikandrova Y, Bowton E, McMahon DG, Colbran RJ, Daws LC, Sitte HH, Javitch JA, Galli A, Gether U, Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport, Neuron 51(4) (2006) 417–29 [DOI] [PubMed] [Google Scholar]
- [35].Gorentla BK, Moritz AE, Foster JD, Vaughan RA, Proline-directed phosphorylation of the dopamine transporter N-terminal domain, Biochemistry 48(5) (2009) 1067–76. 10.1021/bi801696n [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Foster JD, Yang JW, Moritz AE, Challasivakanaka S, Smith MA, Holy M, Wilebski K, Sitte HH, Vaughan RA, Dopamine transporter phosphorylation site threonine 53 regulates substrate reuptake and amphetamine-stimulated efflux, J Biol Chem 287(35) (2012) 29702–12. 10.1074/jbc.M112.367706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Challasivakanaka S, Zhen J, Smith ME, Reith MEA, Foster JD, Vaughan RA, Dopamine transporter phosphorylation site threonine 53 is stimulated by amphetamines and regulates dopamine transport, efflux, and cocaine analog binding, J Biol Chem 292(46) (2017) 19066–19075. 10.1074/jbc.M117.787002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wefers B, Bashir S, Rossius J, Wurst W, Kuhn R, Gene editing in mouse zygotes using the CRISPR/Cas9 system, Methods 121-122 (2017) 55–67. 10.1016/j.ymeth.2017.02.008 [DOI] [PubMed] [Google Scholar]
- [39].Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE, Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9, Nat Biotechnol 34(2) (2016) 184–191. 10.1038/nbt.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Oliveros JC, Franch M, Tabas-Madrid D, San-Leon D, Montoliu L, Cubas P, Pazos F, Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes, Nucleic Acids Res 44(W1) (2016) W267–71. 10.1093/nar/gkw407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mannangatti P, NarasimhaNaidu K, Damaj MI, Ramamoorthy S, Jayanthi LD, A Role for p38 Mitogen-activated Protein Kinase-mediated Threonine 30-dependent Norepinephrine Transporter Regulation in Cocaine Sensitization and Conditioned Place Preference, J Biol Chem 290(17) (2015) 10814–27. 10.1074/jbc.M114.612192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Alsio J, Nordenankar K, Arvidsson E, Birgner C, Mahmoudi S, Halbout B, Smith C, Fortin GM, Olson L, Descarries L, Trudeau LE, Kullander K, Levesque D, Wallen-Mackenzie A, Enhanced sucrose and cocaine self-administration and cue-induced drug seeking after loss of VGLUT2 in midbrain dopamine neurons in mice, J Neurosci 31(35) (2011) 12593–603. 10.1523/JNEUROSCI.2397-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ragu Varman D, Jayanthi LD, Ramamoorthy S, Glycogen synthase kinase-3ss supports serotonin transporter function and trafficking in a phosphorylation-dependent manner, J Neurochem (2020). 10.1111/jnc.15152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Moron JA, Zakharova I, Ferrer JV, Merrill GA, Hope B, Lafer EM, Lin ZC, Wang JB, Javitch JA, Galli A, Shippenberg TS, Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity, J Neurosci 23(24) (2003) 8480–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ruan QT, Yazdani N, Blum BC, Beierle JA, Lin W, Coelho MA, Fultz EK, Healy AF, Shahin JR, Kandola AK, Luttik KP, Zheng K, Smith NJ, Cheung J, Mortazavi F, Apicco DJ, Ragu Varman D, Ramamoorthy S, Ash PEA, Rosene DL, Emili A, Wolozin B, Szumlinski KK, Bryant CD, A Mutation in Hnrnph1 That Decreases Methamphetamine-Induced Reinforcement, Reward, and Dopamine Release and Increases Synaptosomal hnRNP H and Mitochondrial Proteins, J Neurosci 40(1) (2020) 107–130. 10.1523/JNEUROSCI.1808-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ragu-Varman D, Macedo-Mendoza M, Labrada-Moncada FE, Reyes-Ortega P, Morales T, Martinez-Torres A, Reyes-Haro D, Anorexia increases microglial density and cytokine expression in the hippocampus of young female rats, Behav Brain Res 363 (2019) 118–125. 10.1016/j.bbr.2019.01.042 [DOI] [PubMed] [Google Scholar]
- [47].Boger HA, Mannangatti P, Samuvel DJ, Saylor AJ, Bender TS, McGinty JF, Fortress AM, Zaman V, Huang P, Middaugh LD, Randall PK, Jayanthi LD, Rohrer B, Helke KL, Granholm AC, Ramamoorthy S, Effects of brain-derived neurotrophic factor on dopaminergic function and motor behavior during aging, Genes Brain Behav 10(2) (2011) 186–98. 10.1111/j.1601-183X.2010.00654.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hajnal A, Norgren R, Repeated access to sucrose augments dopamine turnover in the nucleus accumbens, Neuroreport 13(17) (2002) 2213–6. 10.1097/00001756-200212030-00010 [DOI] [PubMed] [Google Scholar]
- [49].Bello NT, Lucas LR, Hajnal A, Repeated sucrose access influences dopamine D2 receptor density in the striatum, Neuroreport 13(12) (2002) 1575–8. 10.1097/00001756-200208270-00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hajnal A, Norgren R, Accumbens dopamine mechanisms in sucrose intake, Brain Res 904(1) (2001) 76–84. 10.1016/s0006-8993(01)02451-9 [DOI] [PubMed] [Google Scholar]
- [51].Liu MY, Yin CY, Zhu LJ, Zhu XH, Xu C, Luo CX, Chen H, Zhu DY, Zhou QG, Sucrose preference test for measurement of stress-induced anhedonia in mice, Nat Protoc 13(7) (2018) 1686–1698. 10.1038/s41596-018-0011-z [DOI] [PubMed] [Google Scholar]
- [52].Gowrishankar R, Gresch PJ, Davis GL, Katamish RM, Riele JR, Stewart AM, Vaughan RA, Hahn MK, Blakely RD, Region-Specific Regulation of Presynaptic Dopamine Homeostasis by D2 Autoreceptors Shapes the In Vivo Impact of the Neuropsychiatric Disease-Associated DAT Variant Val559, J Neurosci 38(23) (2018) 5302–5312. 10.1523/JNEUROSCI.0055-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG, Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity, Science 283(5400) (1999) 397–401. 10.1126/science.283.5400.397 [DOI] [PubMed] [Google Scholar]
- [54].Spielewoy C, Roubert C, Hamon M, Nosten-Bertrand M, Betancur C, Giros B, Behavioural disturbances associated with hyperdopaminergia in dopamine-transporter knockout mice, Behav Pharmacol 11(3-4) (2000) 279–90. 10.1097/00008877-200006000-00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pogorelov VM, Rodriguiz RM, Insco ML, Caron MG, Wetsel WC, Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene, Neuropsychopharmacology 30(10) (2005) 1818–31. 10.1038/sj.npp.1300724 [DOI] [PubMed] [Google Scholar]
- [56].Zhuang X, Oosting RS, Jones SR, Gainetdinov RR, Miller GW, Caron MG, Hen R, Hyperactivity and impaired response habituation in hyperdopaminergic mice, Proc Natl Acad Sci U S A 98(4) (2001) 1982–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tilley MR, Gu HH, Dopamine transporter inhibition is required for cocaine-induced stereotypy, Neuroreport 19(11) (2008) 1137–40. 10.1097/WNR.0b013e3283063183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hughes RN, Intrinsic exploration in animals: motives and measurement, Behav Processes 41(3) (1997) 213–26. 10.1016/s0376-6357(97)00055-7 [DOI] [PubMed] [Google Scholar]
- [59].Berlyne DE, Koenig ID, Hirota T, Novelty, arousal, and the reinforcement of diversive exploration in the rat, J Comp Physiol Psychol 62(2) (1966) 222–6. 10.1037/h0023681 [DOI] [PubMed] [Google Scholar]
- [60].Hooks MS, Kalivas PW, The role of mesoaccumbens--pallidal circuitry in novelty-induced behavioral activation, Neuroscience 64(3) (1995) 587–97. 10.1016/0306-4522(94)00409-x [DOI] [PubMed] [Google Scholar]
- [61].Creese I, Iversen SD, Blockage of amphetamine induced motor stimulation and stereotypy in the adult rat following neonatal treatment with 6-hydroxydopamine, Brain Res 55(2) (1973) 369–82. 10.1016/0006-8993(73)90302-8 [DOI] [PubMed] [Google Scholar]
- [62].Budygin EA, Kilpatrick MR, Gainetdinov RR, Wightman RM, Correlation between behavior and extracellular dopamine levels in rat striatum: comparison of microdialysis and fast-scan cyclic voltammetry, Neurosci Lett 281(1) (2000) 9–12. 10.1016/s0304-3940(00)00813-2 [DOI] [PubMed] [Google Scholar]
- [63].Aliane V, Perez S, Nieoullon A, Deniau JM, Kemel ML, Cocaine-induced stereotypy is linked to an imbalance between the medial prefrontal and sensorimotor circuits of the basal ganglia, Eur J Neurosci 30(7) (2009) 1269–79. 10.1111/j.1460-9568.2009.06907.x [DOI] [PubMed] [Google Scholar]
- [64].Kivell B, Uzelac Z, Sundaramurthy S, Rajamanickam J, Ewald A, Chefer V, Jaligam V, Bolan E, Simonson B, Annamalai B, Mannangatti P, Prisinzano TE, Gomes I, Devi LA, Jayanthi LD, Sitte HH, Ramamoorthy S, Shippenberg TS, Salvinorin A regulates dopamine transporter function via a kappa opioid receptor and ERK1/2-dependent mechanism, Neuropharmacology 86 (2014) 228–40. 10.1016/j.neuropharm.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zapata A, Kivell B, Han Y, Javitch JA, Bolan EA, Kuraguntla D, Jaligam V, Oz M, Jayanthi LD, Samuvel DJ, Ramamoorthy S, Shippenberg TS, Regulation of dopamine transporter function and cell surface expression by D3 dopamine receptors, J Biol Chem 282(49) (2007) 35842–54 [DOI] [PubMed] [Google Scholar]
- [66].Bolan EA, Kivell B, Jaligam V, Oz M, Jayanthi LD, Han Y, Sen N, Urizar E, Gomes I, Devi LA, Ramamoorthy S, Javitch JA, Zapata A, Shippenberg TS, D2 receptors regulate dopamine transporter function via an extracellular signal-regulated kinases 1 and 2-dependent and phosphoinositide 3 kinase-independent mechanism, Mol Pharmacol 71(5) (2007) 1222–32 [DOI] [PubMed] [Google Scholar]
- [67].Calipari ES, Juarez B, Morel C, Walker DM, Cahill ME, Ribeiro E, Roman-Ortiz C, Ramakrishnan C, Deisseroth K, Han MH, Nestler EJ, Dopaminergic dynamics underlying sex-specific cocaine reward, Nat Commun 8 (2017) 13877. 10.1038/ncomms13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Brodnik ZD, Xu W, Batra A, Lewandowski SI, Ruiz CM, Mortensen OV, Kortagere S, Mahler SV, Espana RA, Chemogenetic Manipulation of Dopamine Neurons Dictates Cocaine Potency at Distal Dopamine Transporters, J Neurosci 40(45) (2020) 8767–8779. 10.1523/JNEUROSCI.0894-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ, The binding sites for cocaine and dopamine in the dopamine transporter overlap, Nat Neurosci 11(7) (2008) 780–9. 10.1038/nn.2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lin Z, Uhl GR, Dopamine transporter mutants with cocaine resistance and normal dopamine uptake provide targets for cocaine antagonism, Mol Pharmacol 61(4) (2002) 885–91. 10.1124/mol.61.4.885 [DOI] [PubMed] [Google Scholar]
- [71].Chen R, Han DD, Gu HH, A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate, J Neurochem 94(2) (2005) 352–9. 10.1111/j.1471-4159.2005.03199.x [DOI] [PubMed] [Google Scholar]
- [72].Martin BJ, Naughton BJ, Thirtamara-Rajamani K, Yoon DJ, Han DD, Devries AC, Gu HH, Dopamine transporter inhibition is necessary for cocaine-induced increases in dendritic spine density in the nucleus accumbens, Synapse 65(6) (2011) 490–6. 10.1002/syn.20865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Samuvel DJ, Jayanthi LD, Manohar S, Kaliyaperumal K, See RE, Ramamoorthy S, Dysregulation of dopamine transporter trafficking and function after abstinence from cocaine self-administration in rats: evidence for differential regulation in caudate putamen and nucleus accumbens, J Pharmacol Exp Ther 325(1) (2008) 293–301 [DOI] [PubMed] [Google Scholar]
- [74].Ramamoorthy S, Samuvel DJ, Balasubramaniam A, See RE, Jayanthi LD, Altered dopamine transporter function and phosphorylation following chronic cocaine self-administration and extinction in rats, Biochem Biophys Res Commun 391(3) (2010) 1517–21. 10.1016/j.bbrc.2009.12.110 [DOI] [PMC free article] [PubMed] [Google Scholar]