

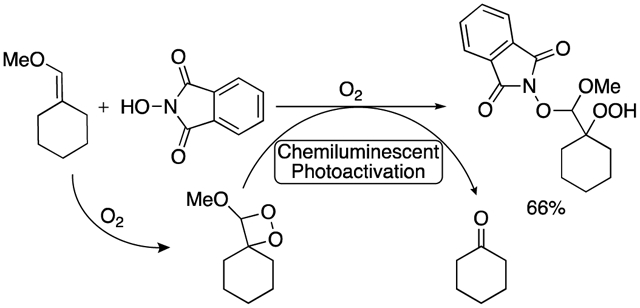
Abstract
The hydroperoxidation of alkyl enol ethers using N-hydroxyphthalimide and molecular oxygen occurred in the absence of catalyst, initiator, or light. The reaction proceeds through a radical mechanism that is initiated by N-hydroxyphthalimide-promoted autoxidation of the enol ether substrate. The resulting dioxetane products decompose in a chemiluminescent reaction that allows for photochemical activation of N-hydroxyphthalimide in the absence of other light sources.
Keywords: Enol ether, Oxidation, Radical, NHPI, Dark, Autoxidation, Chemiluminescence
Graphical Abstract
1. Introduction
Enol ethers are versatile building blocks in organic synthesis that have been utilized in oxidation reactions,1,2 cyclizations,3 carbon-carbon bond forming reactions,4–6 and protection of hydroxyl groups,7 among other transformations. While the reactivity of silyl enol ethers, as well as more highly reactive boron enolates and metal enolates, have been extensively researched over the past several decades,8,9 transformations involving less reactive alkyl enol ethers have remained relatively underdeveloped.10 Oxidation reactions involving alkyl enol ethers, which are far less prevalent than those involving silyl enol ethers,11–17 predominantly rely on the use of metal catalysts.18–20 While less reactive than their silyl counterparts, alkyl enol ethers may be expected to undergo similar transformations, that, when paired with their relative stability, make them uniquely useful.21–23 The identification of reactions unique to alkyl enol ethers, that proceed under mild conditions, is thus desirable.
Herein, we report the oxidation of enol ethers with molecular oxygen and NHPI under mild conditions and in the absence of catalyst (Scheme 1). The reaction proceeds with aryl- and alkyl-substituted enol ethers to generate β-hydroperoxy-N-alkoxyamine acetal products, and it exhibits chemoselectivity in the presence of other alkenes or alkynes. The oxidation reaction occurs through a radical mechanism that does not require heat or light, and is instead initiated by NHPI-promoted autoxidation of the enol ether. The resulting chemiluminescent autoxidation pathway provides both chemical and photochemical means by which the radical addition reaction can be initiated.
Scheme 1:

NHPI-mediated oxidation of alkenes
2. Results/discussion
We recently reported a strain-promoted oxidation of alkylidenecyclopropanes with N-hydroxyphthalimide (NHPI) under mild conditions (Scheme 1).24 The reaction proceeds through a radical pathway that does not require stabilized intermediates such as those involved in oxidations of styrene and enyne derivatives,25–31 but is rather driven by strain-induced destabilization of the starting alkene. We hypothesized that unstrained alkenes with suitable electronic activation could undergo oxidation in the presence of NHPI, whether through destabilization of the starting material or stabilization of the radical intermediate.
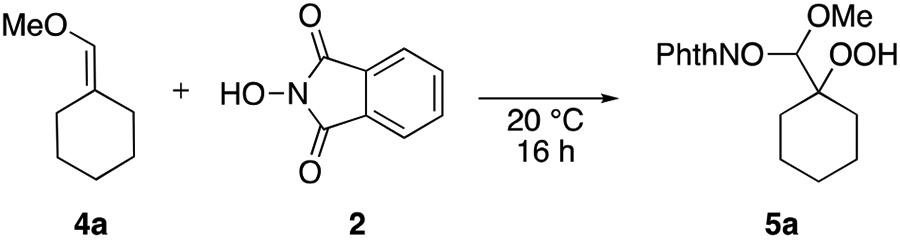
Enol ethers were promising candidates for reaction with the electrophilic phthalimide-N-oxyl (PINO) radical due to their electron-rich character and the potentially stabilizing effect of the oxygen atom on a radical intermediate.32 Treatment of (methoxymethylene)cyclohexane (4a) with NHPI (2) in an oxygen atmosphere resulted in the formation β-hydroperoxy-N-alkoxyamine acetal 5a (Table 1). The yield of 5a was substantially reduced at a higher reaction concentration (Table 1, entry 3), while smaller changes to yield were observed when more dilute concentrations were employed (Table 1, entries 4,5). This sensitivity to concentration is indicative of a radical pathway that can undergo self-termination through dimerization or other undesired side reactions.33 The reaction yield was also highly dependent upon the use of an oxygen atmosphere (Table 1, entry 6), and it proceeded most efficiently in solvents that could dissolve oxygen best, such as acetonitrile and acetone (Table 1, entries 8,9).34 The reaction proceeded largely independently of any light source, and the absence of any light resulted in the highest yield (Table 1, entries 4,7,8).
Table 1:
Optimization of reaction conditions
 | |||||
|---|---|---|---|---|---|
| Entry | Conc. (M) | Solvent | Light | Oxidant | Yield (%)a |
| 1 | 0.1 | MeCN | Blue LED | O2 | 50 |
| 2 | 0.1 | Acetone | Blue LED | O2 | 55 |
| 3 | 0.3 | Acetone | Blue LED | O2 | 16 |
| 4 | 0.05 | Acetone | Blue LED | O2 | 58 |
| 5 | 0.01 | Acetone | Blue LED | O2 | 52 |
| 6 | 0.05 | Acetone | Blue LED | Air | 31 |
| 7 | 0.05 | Acetone | Ambient | O2 | 59 |
| 8 | 0.05 | Acetone | Dark | O2 | 69 |
| 9 | 0.05 | MeCN | Dark | O2 | 53 |
| 10 | 0.05 | 1,4-Dioxetane | Dark | O2 | 41 |
| 11 | 0.05 | DCE | Dark | O2 | 20 |
yields determined by 1H NMR spectroscopy of the unpurified reaction mixture using mesitylene as an internal standard
The oxidation reaction was general for a number of different enol ethers (Scheme 2). Trisubstituted enol ethers 4a–4h all reacted under the present conditions to give their respective β-hydroperoxy-N-alkoxyamine acetals. Carbonyl groups, carbon–carbon double bonds, and carbon-carbon triple bonds were all tolerated. Ketone 5g’, observed in addition to peroxide 5g following treatment of enol ether 4g under the present conditions, likely arises through a Criegee rearrangement of 5g.35 The involvement of an aryl-stabilized radical intermediate was not necessary in the case of trisubstituted enol ethers, as was evidenced by the comparable yields of alkyl- and aryl-substituted substrates 4a–d and 4e–h, respectively. Stabilization with an aryl group was necessary for reaction with disubstituted enol ether 3i, however, as treatment of non-stabilized disubstituted enol ether 4n with NHPI and oxygen resulted in formation of both regioisomers 5n and 5n’ in similar amounts (Scheme 3). Regioselectively was restored in the case of monosubstituted enol ether 4j to give peroxyacetal 5j in moderate yields, although exposure to blue LED light was necessary for the reaction to proceed. Exposure to blue light was also necessary for the oxidation of silyl enol ether 4k. The α,β-unsaturated enol ether 4l did not react to give peroxide 5l, nor was enamine 4m reactive under the present conditions.
Scheme 2:

Scope of NHPI-mediated oxidation of enol ethersa
a Reaction conditions: 4 (0.30 mmol, 1.0 equiv) and 2 (1.2 equiv) in acetone (6 mL) for 16 h. Isolated yields.
b Reaction mixture was subjected to blue LED light
Scheme 3:

Oxidation of disubstituted enol ether 4n
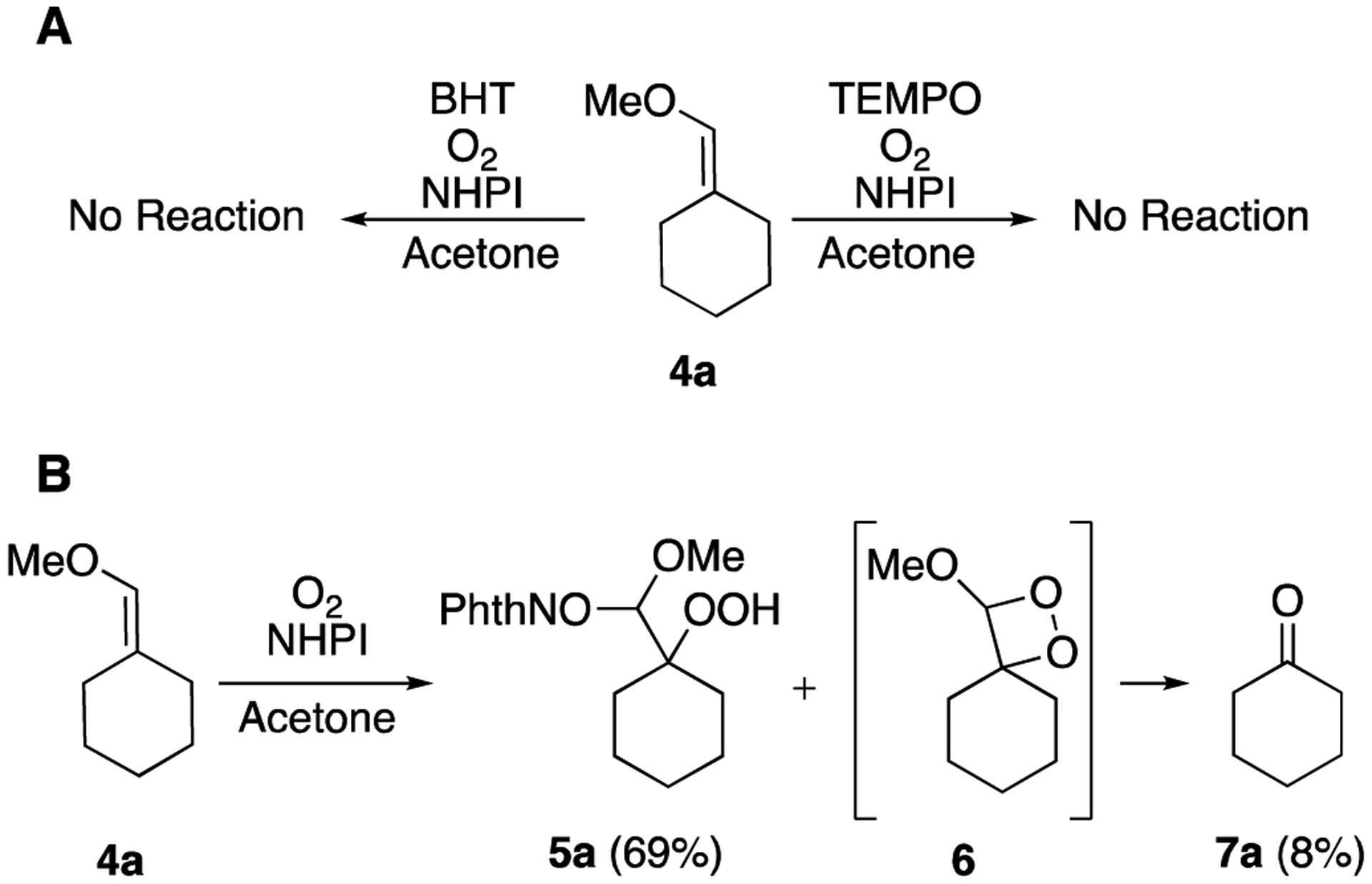
NHPI-mediated oxidations generally proceed through radical pathways that require conversion of NHPI to the phthalimide-N-oxyl (PINO) radical through homolytic cleavage of the hydrogen-oxygen bond.32 The present reaction also appears to proceed through a radical pathway, as is evidenced by the complete inhibition of the reaction in the presence of the radical traps (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and butylated hydroxytoluene (BHT) (Scheme 4A). While initiation to form the PINO radical generally depends upon light,27,36–38 heat,39–42 or catalyst25,26,28,29,32,43–45 as an external source of activation, we recently reported formation of the PINO radical through interaction with a minor autoxidation side reaction in the oxidation of alkylidenecyclopropanes.24 The presence of cyclohexanone 7a in the unpurified reaction mixture following treatment of 4a with NHPI suggests that a similar autoxidation reaction is occurring in the case of oxidations of enol ethers, and that the formation and decomposition of a dioxetane intermediate is likely (Scheme 4B).
Scheme 4:
Mechanistic experiments
The generation of cyclohexanone 7a appears to involve a radical mechanism characteristic of triplet oxygen, as evidenced by the susceptibility of the reaction to radical trapping and the ability of the reaction to proceed in the dark. Although the addition of molecular oxygen to alkenes more commonly involves the excited singlet state of oxygen,46 triplet oxygen can add to alkynes,47 alkenes,48–51 and enols.52–59
The rate of autoxidation of the alkyl enol ethers was enhanced in the presence of NHPI. While cyclohexanone and acetophenone were observed in samples of 4a and 4e, respectively, following 1 week of storage at 4 °C, exposure of either of these enol ethers to an oxygen atmosphere in acetone for 16 h did not result in the formation of autoxidation products at a level detectable by TLC or NMR spectroscopy. The addition of NHPI to 4e resulted in the rapid and concurrent formation of acetophenone and peroxide product, as detected by TLC.
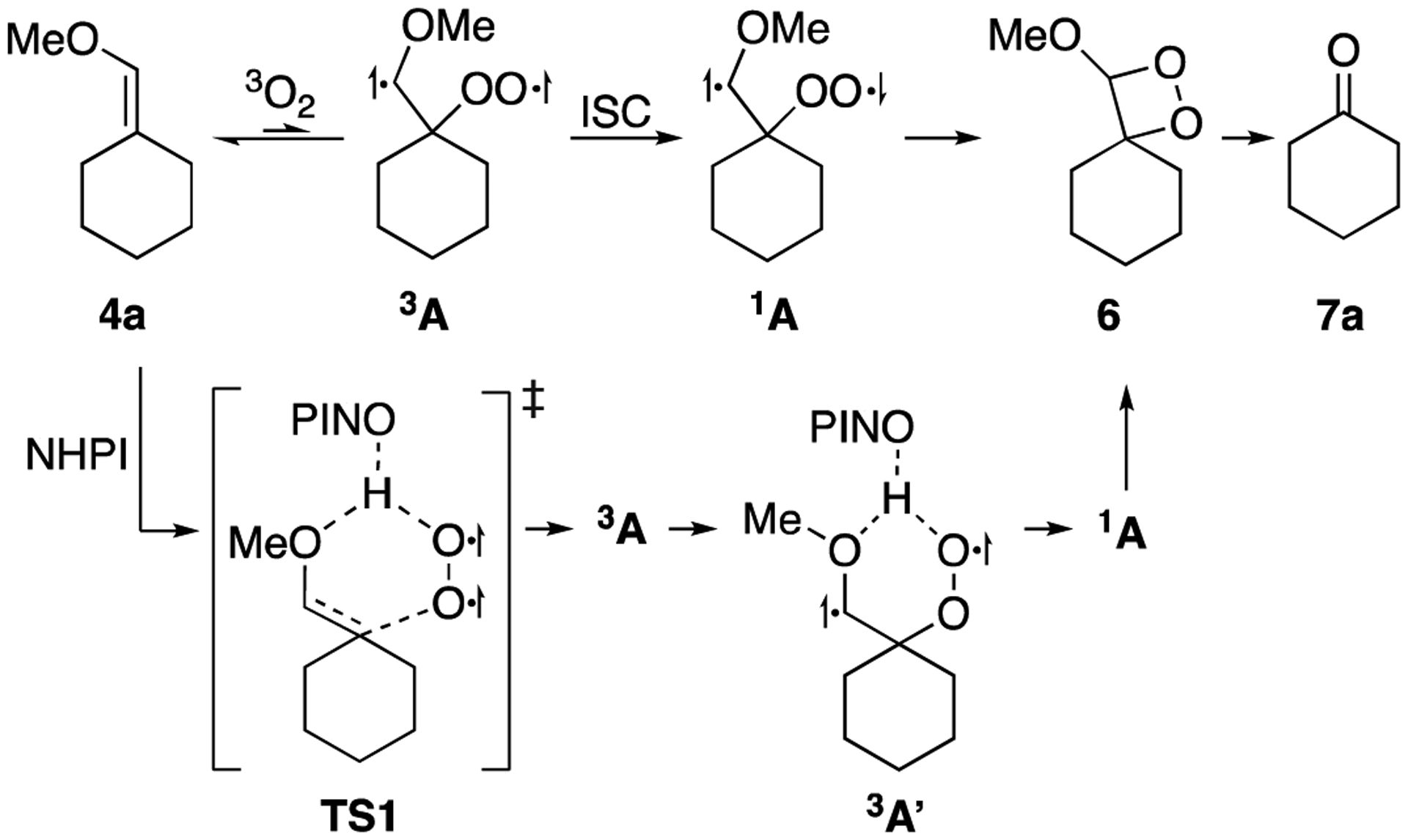
Autoxidation of alkenes with triplet oxygen likely involves the direct addition of triplet oxygen to the alkene, rather than formation of an initial charge-transfer complex.60 Direct addition would form the triplet diradical 3A (Scheme 5).53,60 Formation of dioxetane 4 through radical recombination of 3A is a spin-forbidden process that can only occur following intersystem crossing (ISC) to form the singlet diradical 1A.61 The slow rate of autoxidation in the absence of NHPI suggests that intersystem crossing is largely outcompeted by the dissociation of dioxygen to reform enol ether 4a and triplet oxygen.
Scheme 5:
NHPI-promoted autoxidation of 4a
NHPI may promote autoxidation through stabilization of triplet diradical 3A, thereby allowing time for the formation of the singlet species 1A through ISC. In the absence of hydrogen bonding, the difference in energy between the transition state of addition and the resulting triplet diradical should be small (0.3 kcal/mol).53 NHPI, however, may provide stabilization through hydrogen bonding, both within the transition state TS1 of the addition of triplet oxygen to 3a, as well as in the resulting triplet diradical through complex 3A’ (Scheme 5). Such hydrogen bonding between the methoxy group of the enol ether and the peroxyl radical should result in a larger energy difference between the transition state and the diradical intermediate (0.7 kcal/mol),53 thereby allowing more time for the conversion of 3A to 1A.
Autoxidation of enol ethers is also promoted in the presence of other highly polarized hydrogen bond donors. Treatment of enol ether 4e with N-hydroxysuccinimide (NHS) or acetic acid under an oxygen atmosphere both resulted in the formation of acetophenone 7e (Scheme 7). The higher yields of 7e observed in these reactions compared to that of treatment with NHPI is likely due to the absence of any competing radical addition pathway, as no addition products were observed in either reaction. The absence of any subsequent addition reaction in these hydrogen bond-promoted autoxidation reactions of 4e suggests that these two processes occur discretely in the case of the NHPI-promoted oxidations of enol ethers.
Scheme 7:

Proposed reaction mechanism
The decomposition of dioxetane 6 to form cyclohexanone 7a and methylformate is a chemiluminescent reaction that is accompanied by the emission of blue light (λmax = 420 nm).62–64,65 Given the use of blue light to induce formation of the PINO radical in previous reports,27,66,67 we explored the possibility that this chemiluminescence could drive formation of the PINO radical in the absence of other light sources.
Inhibition of the oxidation reaction of enol ethers by luminescence quenching indicates that light plays a role in the reaction even in the dark. Treatment of enol ether 4a with NHPI in the presence of methyl orange 8, a dye with a peak absorption at 460 nm,68 in the dark resulted in a concentration-dependent inhibition of the oxidation reaction (Table 2, entries 2,3). Reactivity in the presence of methyl orange was restored with exposure to blue light, suggesting that the inhibitory effect is light-dependent (Table 2, entries 4,5).
Table 2:
Inhibition of peroxidation reaction with methyl orange
 | |||
|---|---|---|---|
| Entry | Light Source | Methyl Orange | Yield |
| 1 | Dark | None | 69% |
| 2 | Dark | 0.3 equiv | 33% |
| 3 | Dark | 3.0 equiv | 4% |
| 4 | Blue LED | 0.3 equiv | 66% |
| 5 | Blue LED | 3.0 equiv | 26% |
Photometric analysis of the oxidation reaction of 4a over time revealed a small but detectable emission of light from the reaction mixture (Figure 1). By the second hour of the reaction, chemiluminescence was detected in the reaction mixture, and this chemiluminescence persisted over the next several hours.
Figure 1:
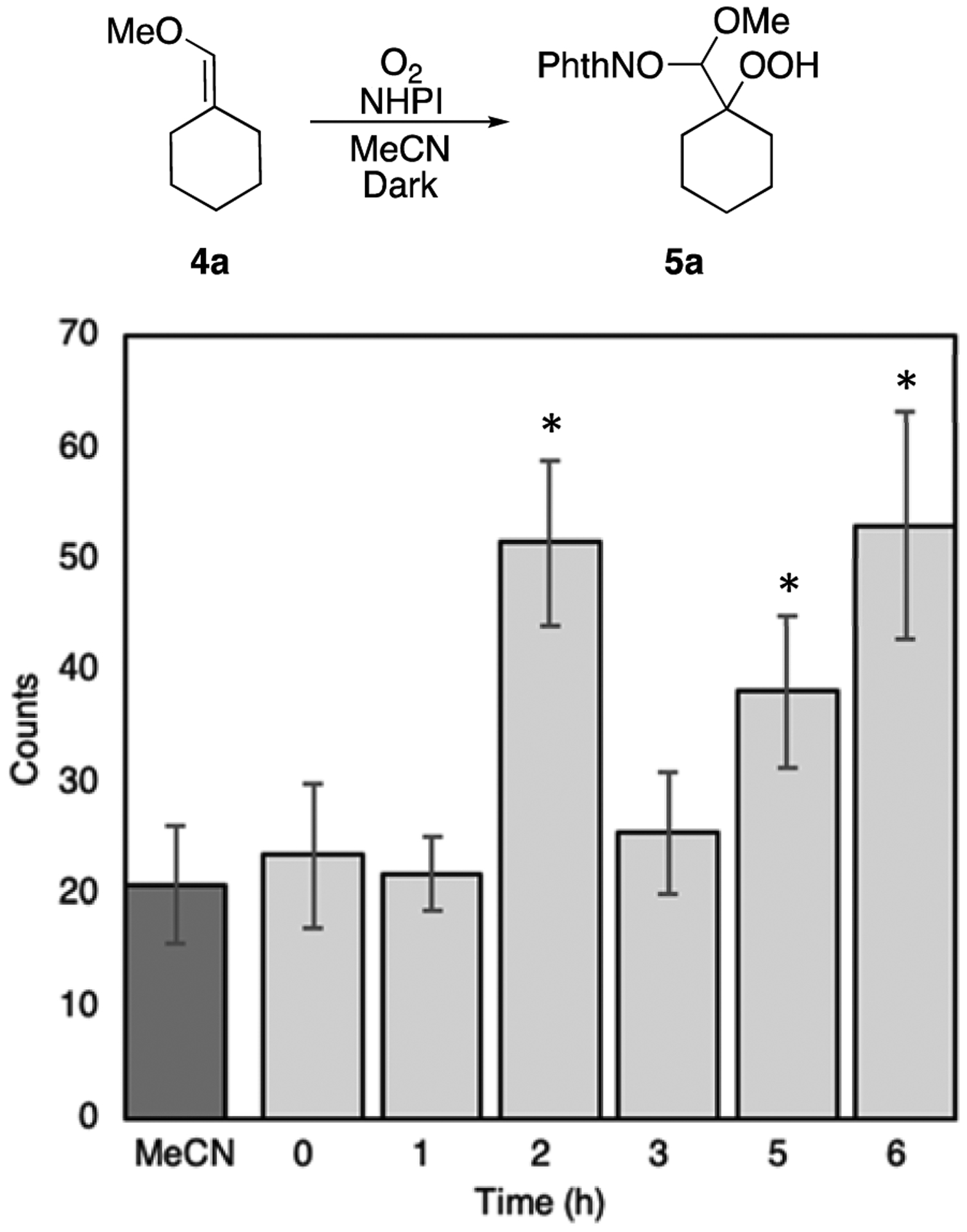
Chemiluminescence during reaction of 4a with NHPIa
aError bars represent standard deviation (n = 10). asterisks represent statistically significant differences in the number of counts compared to measurements of MeCN (p < 0.01).
Based on the above experiments, a reaction mechanism is proposed in Scheme 7. NHPI-promoted autoxidation of enol ether 4a results in formation of dioxetane 6. Decomposition of 6 yields cyclohexanone (7a) and methyl formate (9) in a chemiluminescent reaction that induces homolytic cleavage of the hydrogen-oxygen bond in NHPI to form the PINO radical. While the PINO radical may also form through the interaction of NHPI with the diradical intermediates 3A or 1A (Scheme 6), the susceptibility of the reaction to inhibition through luminescence quenching suggests a light-based pathway for PINO radical initiation (Table 2). Once the PINO radical is formed, the reaction likely proceeds similarly to previously reported oxidations of alkenes with NHPI.25,27–29,69 The PINO radical adds across the alkene to generate alkyl radical B,70 and subsequent addition of molecular oxygen yields peroxyl radical C. Abstraction of hydrogen from another molecule of NHPI yields β-hydroperoxy-N-alkoxyamine acetal 5a and regenerates the PINO radical.
Scheme 6:

Hydrogen bond-promoted autoxidation of 4e
aYields determined by 1H NMR spectroscopy of the unpurified reaction mixture using mesitylene as an internal standard
The regioselectivity observed in the products of the oxidation reaction indicates that, for trisubstituted alkenes, steric interactions are a greater determinant of regiochemistry than the stability of the resulting radical intermediates. Although an alkyl radical α to the methoxy group should be favored over the tertiary β alkyl radical (by 1.9 kcal/mol),71 no products resulting from this intermediate were observed from reaction with trisubstituted enol ethers. The expected HOMO-SOMO interaction between the electron-rich enol ether and the electrophilic PINO radical should also favor addition to the β carbon, where the molecular orbital coefficient is greater.70,72,73 Steric effects are nevertheless a strong determinant of regioselectivity in the addition of even relatively small radicals to alkenes,70,74,75 and preferential addition to the α position has been observed in radical additions to enol ethers.76 The large PINO radical may be expected to be highly sensitive to steric effects within the enol ether substrate.
Generation of the less stable radical intermediate following addition of the PINO radical may also be rationalized through consideration of the possible configurations of the three valence electrons directly involved in the transition state of the reaction.77,78 A polarized electron configuration, in which electron density is localized onto the oxygen atom of the PINO radical, may be expected to be a significant contributor to the transition state due to the electrophilic nature of the PINO radical (Scheme 8). In the case of α-addition, the resulting carbocation is stabilized through resonance with the methoxy group (Scheme 8, TS5a). No such resonance stabilization is possible within the transition state of β-addition, making this electron configuration relatively unfavorable (Scheme 8, TS5a’).
Scheme 8:

Transition states in the addition of NHPI to 4a
The enol ether substrates that required blue light in order to undergo reaction with NHPI are capable of reacting with the PINO radical, but incapable of inducing its formation through reaction with triplet oxygen (Scheme 7). Consequently, when silyl enol ether 4k was treated with NHPI in the dark in the presence of alkyl enol ether 4a, which can induce formation of the PINO radical through autooxidation, peroxide product 5k was formed in yields comparable to those observed with the use of blue light (Table 3).
Table 3:
Alkyl enol ether-activated oxidation of silyl enol ether 4k

Yields determined by 1H NMR spectroscopy of the unpurified reaction mixture using mesitylene as an internal standard
The degree of electron density in the alkene is expected to be an important determinant of success, both for the addition of the enol ether to the PINO radical as well as for the autoxidation reaction, as both of these events involve reaction with an electrophilic radical.32,79 Silyl enol ethers are less nucleophilic than their alkyl enol ether counterparts,80 and the inability of silyl enol ether 4k to undergo reaction with triplet oxygen may reflect this decreased reactivity. The relatively electron-deficient monosubstituted alkyl enol ether 4j also appears to lack a sufficient degree of electron density to undergo reaction with triplet oxygen, requiring blue light for addition to NHPI. The carbonyl group in enol ether 4l renders the alkene too electron deficient to react with either triplet oxygen or the PINO radical, so no reaction was observed. The observed trends in reactivity cannot be entirely be explained by consideration of nucleophilicity, however, because the highly nucleophilic enamine 4m failed to react under the present conditions.81 The presence of an oxygen atom may be necessary for the formation of the hydrogen bonds between the enol ether, NHPI, and molecular oxygen that allow autoxidation to occur (Scheme 6). Even the bulky trimethylsilyl group may interfere with these interactions, as suggested by the failure of silyl enol ether 4k to undergo autoxidation. It is less clear, however, why an α-oxygen substituent, as opposed to a nitrogen atom, is necessary for the reaction of the alkene with the PINO radical.
3. Conclusion
The NHPI-mediated oxidation of enol ethers with molecular oxygen expands the scope of alkene oxidations and suggests a novel means by which NHPI can be converted into the reactive PINO radical. As previously demonstrated with the use of ring strain,24 the identification of substrates with suitable electronic activation allows for oxidation of alkenes in the absence of catalyst, initiator, heat, or light. The utilization of chemiluminescence for photochemical activation in the dark has been implicated in a number of biological processes,82 and this work suggests that there is potential use within synthetic chemistry as well.
4. Experimental Section
4.1. General experimental
1H and 13C NMR spectra were obtained at room temperature using Bruker AV-400 and 100 MHz, Bruker AV-500 and 125 MHz, and Bruker AVIII-600 and 150 MHz spectrometers, respectively, as indicated. The data are reported as follows: chemical shift in ppm referenced to residual solvent (1H NMR: CDCl3 δ 7.26; 13C NMR: CDCl3 δ 77.2, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sept = septet, dd = doublet of doublets, dt = doublet of triplets, dq = doublet of quartets, m = multiplet), coupling constants (Hz), and integration. Structures were determined using COSY, HSQC, and HMBC experiments. Overlapping carbon peaks were determined using HSQC experiments. High resolution mass spectra (HRMS) were acquired on an Agilent 6224 Accurate-Mass time-of-flight LC/MS spectrometer with atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI) sources and were obtained by peak matching. Infrared (IR) spectra were obtained on a Nicolet 6700 FT-IR spectrometer using attenuated total reflectance (ATR). Melting points are reported uncorrected. Analytical thin layer chromatography was performed on silica get 60 Å F254 plates. Liquid chromatography was performed using forced flow (flash chromatography) of the indicated solvent system on Silicycle silica gel (SiO2) 60 (230–400 mesh). All reactions were performed under an atmosphere of nitrogen in glassware that had been flame-dried under vacuum unless otherwise stated. Non-deuterated solvents were purified via the Pure Solv-MD Standard Design Solvent Purification System before use. Aqueous solutions were prepared from nanopure water with a resistivity over 18 MΩ-cm. For reactions conducted under blue light, a Feit Electric 7W blue LED lamp (SKU: PAR38/B/10KLED/BX) was positioned approximately 10 cm away from the reaction vessel. All experiments were conducted in borosilicate glass vessels, which filters frequencies of light below approximately 280 nm.83 Chemiluminescence studies were conducted using a Tecan Spark microplate reader. Unless otherwise stated, all reagents and substrates were commercially available.
4.2. Reaction procedures and characterization data
4.2.1. Representative procedure for the synthesis of enol ethers ((methoxymethylene)cyclohexane (4a)
According to the procedure of Chepiga et al.,84 to a suspension of (methoxymethyl)triphenylphosphonium chloride (8.23 g, 24.0 mmol) in dry THF (100 mL) stirring at −78 °C was added potassium tert-butoxide (2.69 g, 24.0 mmol) in portions. The reaction mixture was allowed to warm to 0 °C over 20 min, then cooled to −78 °C, after which cyclohexanone (2.08 mL, 20.0 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was then washed with saturated aqueous ammonium chloride (2 × 40 mL) and brine (2 × 40 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The resulting oil was purified by flash column chromatography (5:95 CH2Cl2:pentane) followed by distillation at reduced pressure to give (methoxymethylene)cyclohexane (4a) as a colorless oil (1.75 g, 70%). The NMR spectroscopic data are in agreement with those previously reported:84 1H NMR (400 MHz, CDCl3) δ 5.74 (s, 1H), 3.52 (s, 3H), 2.17 (t, J = 5.5 Hz, 2H), 1.93 (t, J = 5.5 Hz, 2H), 1.55–1.44 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 138.9 (CH), 118.6 (C), 59.4 (CH3), 30.7 (CH2), 28.5 (CH2), 27.2 (CH2), 27.0 (CH2), 25.6 (CH2); HRMS (ESI) m / z calcd for C8H15O (M + H)+ 127.1117, found 127.1116.
4.2.1.1. (Methoxymethylene)heptane (4b).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (8.23 g, 24.0 mmol), potassium tert-butoxide (2.69 g, 24.0 mmol), and 4-heptanone (2.79 mL, 20.0 mmol) were combined in THF (100 mL) to give (methoxymethylene)heptane (4b) as a colorless oil (1.82 g, 64%): 1H NMR (400 MHz, CDCl3) δ 5.76 (s, 1H), 3.52 (s, 3H), 2.01 (t, J = 7.6 Hz, 2H), 1.83 (t, J = 7.6 Hz, 2H), 1.43–1.32 (m, 4H), 0.92–0.84 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 142.3 (CH), 118.6 (C), 59.4 (CH3) 33.8 (CH2), 29.0 (CH2), 21.4 (CH2), 21.1 (CH2), 14.2 (CH3), 13.9 (CH3); IR (ATR) 2957, 1740, 1676, 1453, 1137, 1096, 841 cm−1; HRMS (ESI) m / z calcd for C9H16Na (M + Na – H2O)+ 147.1144, found 147.1142.
4.2.1.2. 4-(Methoxymethylene)cyclohexan-1-one (4c).
To a solution of 8-(methoxymethylene)-1,4-dioxaspiro[4.5]decane (S1, 0.737 g, 4.00 mmol) in acetone/H2O (132 mL/28 mL, 5:1) was added pyridinium p-toluenesulfonate (0.202 g, 0.800 mmol). The reaction mixture was heated to 70 °C for 18 h, then poured into saturated aqueous sodium bicarbonate (150 mL). The solution was extracted with diethyl ether (3 × 50 mL), dried over MgSO4, and concentrated in vacuo. The resulting oil was purified by flash column chromatography (20:80 EtOAc:hexanes) to yield 4-(methoxymethylene)cyclohexan-1-one (4c) as a colorless oil (0.306 g, 55%): 1H NMR (400 MHz, CDCl3) δ 5.94 (s, 1H), 3.60 (s, 3H), 2.57–2.51 (m, 2H), 2.41–2.30 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 212.1 (C), 141.6 (CH), 112.6 (C), 59.7 (CH3), 42.3 (CH2), 40.9 (CH2), 28.9 (CH2), 23.9 (CH2); IR (ATR) 2936, 2842, 1709, 1685, 1224, 1122 cm−1; HRMS (ESI) m / z calcd for C8H13O2 (M + H)+ 141.0910, found 141.0908.
4.2.1.3. 1-Methoxy-2-methylhepta-1,6-diene (4d).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (2.06 g, 6.00 mmol), potassium tert-butoxide (0.673 g, 6.00 mmol), and hept-6-en-2-one (S3, 0.561 g, 5.00 mmol) were combined in THF (25 mL) to give 1-methoxy-2-methylhepta-1,6-diene (4d) as a mixture of diastereomers in a 1.00:0.85 ratio as a colorless oil (0.253 g, 36%): 1H NMR (400 MHz, CDCl3) δ 5.90–5.73 (m, 3.7H), 5.05–4.90 (m, 3.7H), 3.57–3.48 (m, 5.55H), 2.10–1.98 (m, 5.55H), 1.88 (t, J = 7.5 Hz, 2H), 1.59–1.56 (m, 3H), 1.53–1.51 (m, 2.55H), 1.50–1.42 (m, 3.55H); 13C NMR (100 MHz, CDCl3) δ 142.1 (CH, major), 141.9 (CH, minor), 139.3 (CH, minor), 139.1 (CH, major), 114.5 (CH2, major), 114.32 (CH2, minor), 114.31 (C, minor), 113.9 (C, major), 59.4 (CH3, major), 59.3 (CH3, minor), 33.7 (CH2, minor), 33.5 (CH2, major), 33.4 (CH2, major), 28.5 (CH2, minor), 27.4 (CH2, major), 26.9 (CH2, minor), 17.3 (CH3, minor), 12.8 (CH3, major); IR (ATR) 2927, 2856, 1684, 1456, 1205, 1134, 909 cm−1; HRMS (ESI) m / z calcd for C9H20NO (M + NH4)+ 158.1539, found 158.1547.
4.2.1.4. (1-Methoxyprop-1-en-2-yl)benzene (4e).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (4.11 g, 12.0 mmol), potassium tert-butoxide (0.673 g, 12.0 mmol), and acetophenone (1.17 mL, 10.0 mmol) were combined in THF (50 mL) to give (1-methoxyprop-1-en-2-yl)benzene (4e) as a mixture of diastereomers in a 1.00:0.58 ratio as a colorless oil (1.14 g, 77%): 1H NMR (400 MHz, CDCl3) δ 7.63–7.58 (m, 1.16H), 7.36–7.26 (m, 5.16H), 7.22–7.14 (m, 1.58H), 6.42 (s, 1H), 6.13 (s, 0.58H), 3.72 (s, 3H), 3.68 (s, 1.74H), 2.00 (s, 3H), 1.93 (1.74H); 13C NMR (100 MHz, CDCl3) δ 145.3 (CH, major), 144.7 (CH, minor), 140.8 (C, major), 138.5 (C, minor), 128.5 (CH, major), 128.1 (CH, minor), 127.6 (CH, major), 126.2 (CH, minor), 126.0 (CH, minor), 125.1 (CH, major), 114.6 (C, major), 111.0 (C, minor), 60.3 (CH3, minor), 60.0 (CH3, major), 18.4 (CH3, minor), 12.7 (CH3, major); IR (ATR) 2932, 2835, 1652, 1442, 1222, 1131, 1075 cm−1; HRMS (ESI) m / z calcd for C10H11 (M + H – H2O)+ 131.0855, found 131.0852.
4.2.1.5. (1-Methoxyhepta-1,6-dien-2-yl)benzene (4f).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (2.06 g, 6.00 mmol), potassium tert-butoxide (0.673 g, 6.00 mmol), and 1-phenylhex-5-en-1-one (S4, 0.871 g, 5.00 mmol) were combined in THF (25 mL) to give (1-methoxyhepta-1,6-dien-2-yl)benzene (4f) as a mixture of diastereomers in a 1.00:0.77 ratio as a colorless oil (0.643 g, 64%): 1H NMR (400 MHz, CDCl3) δ 7.50–7.44 (m, 1.54H), 7.35–7.26 (m, 5.54H), 7.22–7.15 (m, 1.77 H), 6.30 (s, 1H), 6.07 (s, 0.77H), 5.92–5.67 (m, 1.77H), 5.03–4.88 (m, 3.54H), 3.69 (s, 3H), 3.63 (s, 2.31H), 2.55–2.50 (m, 2H), 2.33–2.28 (m, 1.54H), 2.10–2.01 (m, 3.54H), 1.52–1.40 (m, 3.54H); 13C NMR (100 MHz, CDCl3) δ 145.5 (CH, major), 144.4 (CH, minor), 139.9 (C, major), 139.1 (CH, major), 138.9 (CH, minor), 137.5 (C, minor), 128.5 (CH, major), 128.3 (CH, minor), 128.1 (CH, minor), 126.2 (CH, minor), 126.1 (CH, major), 126.0 (CH, major), 119.9 (C, major), 116.5 (C, minor), 114.7 (CH2, minor), 114.4 (CH2, major), 60.2 (CH3, minor), 60.0 (CH3, major), 33.8 (CH2, major), 33.4 (CH2, minor), 32.1 (CH2, minor), 28.2 (CH2, minor), 27.6 (CH2, major), 26.6 (CH2, major); IR (ATR) 2929, 1642, 1440, 1221, 1131, 908, 760 cm−1; HRMS (ESI) m / z calcd for C14 H19O (M + H)+ 203.1430, found 203.1429.
4.2.1.6. (1-Methoxy-3-methylbut-1-en-2-yl)benzene (4g).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (2.06 g, 6.00 mmol), potassium tert-butoxide (0.673 g, 6.00 mmol), and isobutyrophenone (0.750 mL, 5.00 mmol) were combined in THF (25 mL) to give (1-methoxy-3-methylbut-1-en-2-yl)benzene (4g) as a mixture of diastereomers in a 1.00:0.40 ratio as a colorless oil (0.290 g, 33%): 1H NMR (400 MHz, CDCl3) δ 7.35–7.17 (m, 7H), 5.97 (s, 0.4H), 5.93 (s, 1H), 3.64 (s, 3H), 3.56 (s, 1.2H), 3.04 (sept, J = 7.1, 1H), 2.70–2.56 (m, 0.4H), 1.12 (d, J = 7.1, 6H), 1.03 (d, J = 6.8, 2.4H); 13C NMR (100 MHz, CDCl3) δ 145.1 (CH, major), 142.7 (CH, minor), 140.1 (C, major), 138.4 (C, minor), 129.1 (CH, minor), 128.6 (CH, major), 127.99 (CH, minor), 127.95 (CH, major), 126.4 (C, major), 126.3 (CH, minor), 126.2 (CH, major), 124.7 (C, minor), 60.0 (CH3, minor), 59.9 (CH3, major), 31.5 (CH, minor), 28.6 (CH, major), 22.4 (CH3, minor), 21.5 (CH3, major); IR (ATR) 2958, 2929, 1646, 1220, 1129, 1103, 766 cm−1; HRMS (ESI) m / z calcd for C12H17O (M + H)+ 177.1274, found 177.1274.
4.2.1.7. (1-Methoxyhept-1-en-6-yn-2-yl)benzene (4h).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (1.25 g, 3.65 mmol), potassium tert-butoxide (0.410 g, 3.65 mmol), and 1-phenylhex-5-yn-1-one (S6, 0.524 g, 3.04 mmol) were combined in THF (15 mL) to give (1-methoxyhept-1-en-6-yn-2-yl)benzene (4h) as a mixture of diastereomers in 1.00:0.83 ratio as a colorless oil (0.423 g, 69%): 1H NMR (400 MHz, CDCl3) δ 7.49–7.45 (m, 1.66H), 7.36–7.26 (m, 5.66H), 7.22–7.15 (m, 1.83H), 6.33 (s, 1H), 6.12 (s, 0.83H), 3.69 (s, 3H), 3.64 (s, 2.49H), 2.61 (t, J = 7.7 Hz, 2H), 2.43 (t, J = 7.3 Hz, 1.66H), 2.21–2.13 (m, 3.66H), 1.97–1.92 (m, 1.83H), 1.68–1.54 (m, 3.66H); 13C NMR (100 MHz, CDCl3) δ 145.9 (CH, major), 144.8 (CH, minor), 139.4 (C, major), 137.0 (C, minor), 128.5 (CH, major), 128.22 (CH, minor), 128.16 (CH, minor), 126.3 (CH, minor), 126.2 (CH, major), 125.8 (CH, major), 118.8 (C, major), 115.2 (C, minor), 84.8 (C, major), 84.5 (C, minor), 68.6 (CH, minor), 68.3 (CH, major), 60.3 (CH3, minor), 60.1 (CH3, major), 31.3 (CH2, minor), 27.3 (CH2, minor), 27.2 (CH2, major), 26.2 (CH2, major), 18.4 (CH2, major), 17.7 (CH2, minor); IR (ATR) 3293, 2931, 1646, 1455, 1221, 1130, 760 cm−1; HRMS (ESI) m / z calcd for C14H17O (M + H)+ 201.1274, found 201.1272.
4.2.1.8. (2-Methoxyvinyl)benzene (4i).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (4.11 g, 12.0 mmol), potassium tert-butoxide (0.673 g, 12.0 mmol), and benzaldehyde (1.02 mL, 10.0 mmol) were combined in THF (50 mL) to give (2-methoxyvinyl)benzene (4i) as a mixture of diastereomers in a 1.00:0.59 ratio as a colorless oil (1.11 g, 83%): 1H NMR (400 MHz, CDCl3) δ 7.61–7.52 (m, 1.18H), 7.31–7.19 (m, 5.18H), 7.17–7.10 (m, 1.59H), 7.04 (d, J = 13.0 Hz, 1H), 6.13 (d, J = 7.1 Hz, 0.59H), 5.81 (d, J = 13.0 Hz, 1H), 5.22 (d, J = 7.1 Hz, 0.59H), 3.77 (s, 1.77H), 3.68 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 149.0 (CH, major), 148.1 (CH, minor), 136.5 (C, major), 136.0 (C, minor), 128.7 (CH, major), 128.3 (CH, minor), 128.3 (CH, minor ), 125.9 (CH, minor), 125.8 (CH, major), 125.2 (CH, major), 105.8 (CH, minor), 105.2 (CH, major), 60.8 (CH3, minor), 56.6 (CH3, major); IR (ATR) 2933, 1639, 1455, 1235, 1150, 1095, 934 cm−1; HRMS (ESI) m / z calcd for C9H14NO (M + NH4)+ 152.107, found 152.1065.
4.2.1.9. (Cyclohexylidenemethoxy)trimethylsilane (4k).
To a solution of cyclohexanecarboxaldehyde (0.489 mL, 4.04 mmol) in acetonitrile (6 mL) was added sodium iodide (0.719 g, 4.80 mmol). After 5 m, triethylamine (0.836 mL, 6.00 mmol) and chlorotrimethylsilane (0.609 mL, 4.80 mmol) were added and the solution was stirred overnight. The reaction mixture was then cooled to 0 °C and hexanes (10 mL) and saturated aqueous ammonium chloride (10 mL) were added. The organic layer was separated, and the aqueous layer was extracted with hexanes (2 × 10 mL). The combined organic layers were washed with ice water (20 mL) and saturated aqueous ammonium chloride (20 mL), dried over MgSO4, filtered, and concentrated in vacuo. Purification by flash column chromatography (pentane) yielded (cyclohexylidenemethoxy)trimethylsilane (4k) as a colorless oil (0.625 g, 85%). The NMR spectroscopic data are in agreement with those previously reported:85 1H NMR (400 MHz, CDCl3) δ 5.99 (s, 1H), 2.17 (t, J = 5.5 Hz), 1.93 (t, J = 5.5 Hz), 5.55–5.42 (m, 6H), 0.16 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 130.2 (CH), 122.7 (C), 30.7 (CH2), 28.6 (CH2), 27.2 (CH2), 27.1 (CH2), 25.5 (CH2), 0.41 (CH3); HRMS (ESI) m / z calcd for C10H20KOSi (M + K)+ 223.0915, found 223.0917.
4.2.1.10. 1-Methoxyhept-1-ene (4n).
Following the representative procedure for the synthesis of enol ethers, (methoxymethyl)triphenylphosphonium chloride (2.06 g, 6.00 mmol), potassium tert-butoxide (0.673 g, 6.00 mmol), and hexanal (0.614 mL, 5.00 mmol) were combined in THF (25 mL) to give 1-methoxyhept-1-ene (4n) as a mixture of diastereomers in a 1.00:0.53 ratio as a colorless oil (0.457 g, 71%): 1H NMR (400 MHz, CDCl3) δ 6.26 (d, J = 12.6 Hz, 1H), 5.86 (d, J = 6.2 Hz, 0.53 H), 4.73 (dt, J = 7.3, 12.6 Hz, 1H), 4.37–4.30 (m, 0.53H), 3.57 (s, 1.59H), 3.50 (s, 3H), 2.09–2.01 (m, 1.06 H), 1.95–1.87 (m, 2H), 1.37–1.23 (m, 10.71H), 0.91–0.86 (m, 4.59H); 13C NMR (100 MHz, CDCl3) δ 147.0 (CH, major), 146.1 (CH, minor), 107.3 (CH, minor), 103.4 (CH, major), 59.6 (CH3, minor), 56.0 (CH3, major), 31.7 (CH2, minor), 31.4 (CH2, major), 30.6 (CH2, major), 29.7 (CH2, minor), 27.8 (CH2, major), 24.0 (CH2, minor), 22.7 (CH2, major), 22.5 (CH2, minor), 14.24 (CH3, minor), 14.23 (CH3, major); IR (ATR) 2924, 2855, 1655, 1464, 1209, 1107, 931 cm−1; HRMS (ESI) m / z calcd for C8H15K (M + K – H2O)+ 150.0805, found 150.0804.
4.2.2. Representative procedure for peroxidation of enol ethers (Peroxide 5a)
To a foil-covered flask containing acetone (6 mL, sparged with oxygen for 5 min) was added N-hydroxyphthalimide (NHPI, 0.059 g, 0.36 mmol). The flask was then sealed, and the (methoxymethylene)cyclohexane (4a, 0.038 g, 0.30 mmol) was injected by syringe. The reaction mixture was stirred under a balloon filled with O2 in the dark for 16 h, then concentrated in vacuo. The resulting solid was suspended in dichloromethane (6 mL) and the suspension was filtered through a 2-cm plug of Celite. The filtered solid was washed with dichloromethane (12 mL), and the combined filtrates were concentrated in vacuo. Purification of the resulting solid by flash column chromatography (30:70 EtOAc:hexanes) afforded peroxide 5a as a white solid (0.063 g, 66%). No melting point was obtained because peroxide 5a decomposed at a temperature of 150–156 °C: 1H NMR (400 MHz, CDCl3) δ 9.86 (s, 1H), 7.92–7.77 (m, 4H), 4.97 (s, 1H), 3.61 (s, 3H), 2.10–2.01 (m, 1H), 1.95–1.86 (m, 1H), 1.81–1.65 (m, 3H), 1.62–1.45 (m, 4H), 1.30–1.15 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 164.7 (C), 135.2 (CH), 128.8 (C), 124.2 (CH), 114.1 (CH), 84.1 (C), 60.4 (CH3), 28.1 (CH2), 26.1 (CH2), 25.7 (CH2), 20.9 (CH2), 20.6 (CH2); IR (ATR) 3303, 2944, 1786, 1721, 1370, 1205, 968 cm−1; HRMS (ESI) m / z calcd for C16H19NNaO6 (M + Na)+ 344.1105, found 344.1106.
4.2.2.1. Peroxide 5b.
Following the representative procedure for the peroxidation of enol ethers, (methoxymethylene)-heptane (4b, 0.047, 0.33 mmol), NHPI (0.065 g, 0.40 mmol), and acetone (7 mL) were combined to afford peroxide 5b as a white solid (0.055 g, 49%): mp = 98–100 °C; 1H NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.93–7.77 (m, 4H), 5.08 (m, 1H), 3.61 (s, 3H), 1.81–1.62 (m, 4H), 1.57–1.41 (m, 4H), 0.98–0.88 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 164.8 (C), 135.2 (CH), 128.9 (C), 124.2 (CH), 115.1 (CH), 86.6 (C), 60.6 (CH3), 35.4 (CH2), 32.2 (CH2), 16.7 (CH2), 16.5 (CH2), 15.1 (CH3), 14.9 (CH3); IR (ATR) 3349, 2961, 1719, 1376, 1188, 1113, 968 cm−1; HRMS (ESI) m / z calcd for C17H23NNaO6 (M + Na)+ 360.1418, found 360.1425.
4.2.2.2. Peroxide 5c.
Following the representative procedure for the peroxidation of enol ethers, 4-(methoxymethylene)cyclohexan-1-one (4c, 0.048 g, 0.34 mmol), NHPI (0.066 g, 0.41 mmol), and acetone (7 mL) were combined to afford peroxide 5c as a white solid (0.075 g, 66%): mp = 69–75 °C; 1H NMR (400 MHz, CDCl3) δ 10.3 (s, 1H), 7.95–7.80 (m, 4H), 5.13 (s, 1H), 3.64 (s, 3H), 2.77 (dt, J = 6.1, 14.3 Hz, 1H), 2.65–2.53 (m, 1H), 2.48–2.39 (m, 1H), 2.37–2.19 (m, 4H), 2.05–1.95 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 211.0 (C), 164.8 (C), 135.4 (CH), 128.7 (C), 124.4 (CH), 113.2 (CH), 82.8 (C), 60.2 (CH3), 36.2 (CH2), 35.9 (CH2), 28.2 (CH2), 25.6 (CH2); IR (ATR) 3340, 2942, 1724, 1374, 1187, 971, 896 cm−1; HRMS (ESI) m / z calcd for C16H17NNaO7 (M + Na)+ 358.0897, found 358.0900.
4.2.2.3. Peroxide 5d.
Following the representative procedure for the peroxidation of enol ethers, 1-methoxy-2-methylhepta-1,6-diene (4d, 0.043 g, 0.307 mmol), NHPI (0.060 g, 0.368 mmol), and acetone (6 mL) were combined to afford peroxide 5d as a 1.00:0.85 mixture of diastereomers as a white solid (0.046 g, 45%): mp = 146–151 °C; 1H NMR (400 MHz, CDCl3) δ 9.99 (s, 1H), 9.88 (s, 0.85H), 7.92–7.79 (m, 7.4H), 5.90–5.76 (m, 1.85H), 5.09–4.92 (m, 5.55H), 3.63–3.61 (m, 5.55H), 2.15–2.01 (m, 3.7H), 1.91–1.45 (m, 7.4H), 1.37 (s, 2.55H), 1.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.8 (C, major), 164.7 (C, minor), 138.9 (CH, major), 138.8 (CH, minor), 135.2 (CH, major), 135.2 (CH, minor), 128.8 (C, major), 128.8 (C, minor), 124.21 (CH, major), 124.20 (CH, minor), 114.8 (2CH2, major, minor), 114.5 (CH, minor), 114.0 (CH, major), 85.11 (C, minor), 85.07 (C, major), 60.4 (CH3, minor), 60.2 (CH3, major), 34.3 (CH2, major), 34.1 (CH2, minor), 33.2 (CH2, major), 30.5 (CH2, minor), 22.1 (CH2, major), 21.7 (CH2, minor), 18.6 (CH3, minor), 14.8 (CH3, major); IR (ATR) 3345, 2948, 1719, 1377, 1116, 972, 877 cm−1; HRMS (ESI) m / z calcd for C17H21NNaO6 (M + Na)+ 358.1261, found 358.1259.
4.2.2.4. Peroxide 5e.
Following the representative procedure for the peroxidation of enol ethers, (1-methoxyprop-1-en-2-yl)benzene (4e, 0.044 g, 0.30 mmol), NHPI (0.059 g, 0.36 mmol), and acetone (6 mL) were combined to afford peroxide 5e as a 1.00:0.39 mixture of diastereomers as a white solid (0.064 g, 63%). The peroxide was purified by flash column chromatography and characterized as a 1.0:0.11 mixture of diastereomers: mp = 126–130 °C; 1H NMR (400 MHz, CDCl3) δ 9.99 (s, 0.11H), 9.88 (s, 1H), 7.92–7.77 (m, 4.44H), 7.71–7.67 (m, 2H), 7.63–7.69 (m, 0.22H), 7.41–7.27 (m, 3.33H), 5.42 (s, 1H), 5.37 (s, 0.11H), 3.66–3.60 (m, 3.33H), 1.80 (s, 3H), 1.71 (s, 0.33H); ); 13C NMR (100 MHz, CDCl3) δ 164.7 (C, minor), 164.5 (C, major), 141.2 (C, minor), 137.9 (C, major), 135.2 (CH, minor), 135.1 (CH, major), 128.9 (C, minor), 128.8 (C, major), 128.4 (CH, minor), 128.3 (CH, major), 128.1 (CH, major), 127.9 (CH, minor), 127.6 (CH, major), 126.6 (CH, minor), 124.2 (CH, minor), 124.1 (CH, major), 113.4 (CH, major), 112.5 (CH, minor), 86.7 (C, minor), 86.4 (C, major), 60.7 (CH3, major), 60.2 (CH3, minor), 21.2 (CH3, major); 18.7 (CH3, minor); IR (ATR) 3489, 3333, 1785, 1717, 1378, 1124, 877 cm−1; HRMS (ESI) m / z calcd for C18H17NNaO6 (M + Na)+ 366.0948, found 366.0949.
4.2.2.5. Peroxide 5f.
Following the representative procedure for the peroxidation of enol ethers, (1-methoxyhepta-1,6-dien-2-yl)benzene (4f, 0.060 g, 0.30 mmol), NHPI (0.059 g, 0.360 mmol), and acetonitrile (6 mL) were combined to afford peroxide 5f as a 1.00:0.12 mixture of diastereoemers as a white solid (0.047 g, 39%): mp = 146–151 °C; 1H NMR (400 MHz, CDCl3) δ 10.49 (s, 0.12 H), 10.24 (s, 1H), 7.94–7.75 (m, 4.48H), 7.69–7.64 (m, 0.24 H), 7.59–7.53 (m, 2H), 7.41–7.26 (m, 3.36H), 5.84–5.63 (m, 1.12H), 5.36 (s, 1H), 5.30 (s, 0.12H), 5.05–4.84 (m, 2.24H), 3.63–3.57 (m, 3.36H), 2.27–2.00 (m, 4.48H), 1.64–1.51 (m, 1.12H), 1.28–1.16 (m, 1.12H); 13C NMR (100 MHz, CDCl3) δ 164.9 (C, minor), 164.7 (C, major), 139.3 (C, minor), 138.8 (CH, major), 138.6 (CH, minor), 135.9 (C, major), 135.3 (CH, minor), 135.2 (CH, major), 128.8 (C, minor), 128.7 (C, major), 128.2 (CH, minor), 127.9 (CH, major), 127.8 (CH, major), 127.6 (CH, minor), 127.4 (CH, major), 127.2 (CH, minor), 124.3 (CH, minor), 124.2 (CH, major), 114.79 (CH2, major), 114.77 (CH2, minor), 113.9 (CH, major), 113.8 (CH, minor), 88.5 (C, minor), 87.7 (C, major), 61.1 (CH3, minor), 60.8 (CH3, major), 34.0 (CH2, minor), 33.9 (CH2, major), 32.9 (CH2, major), 32.0 (CH2, minor), 22.0 (CH2, minor), 21.8 (CH2, major); IR (ATR) 3335, 2936, 1716, 1377, 1116, 906, 877 cm−1; HRMS (ESI) m / z calcd for C22H23NNaO6 (M + Na)+ 420.1418, found 420.1414.
4.2.2.6. Peroxide 5g and ketone 5g’.
Following the representative procedure for the peroxidation of enol ethers, (1-methoxy-3-methylbut-1-en-2-yl)benzene (4g, 0.053 g, 0.30 mmol), NHPI (0.059 g, 0.36 mmol), and acetonitrile (6 mL) were combined to afford peroxide 5g as a mixture of a pair of diastereomers and ketone 5g’ in a 1.00:0.79:0.50 ratio as a white solid (0.076 g, 68%): mp = 51–55 °C; 1H NMR (400 MHz, CDCl3) δ 10.32–10.25 (m, 1.79H), 8.26–8.20 (m, 1H), 7.92–7.74 (m, 9.16H), 7.69–7.60 (m, 4.08H), 7.55–7.49 (m, 1H), 7.42–7.24 (m, 5.37H), 5.80 (s, 0.5H), 5.69 (s, 0.79H), 5.52 (s, 1H), 3.86 (s, 1.5H), 3.69 (s, 3H), 3.66 (s, 2.37), 2.59–2.43 (m, 1.79H), 1.12 (d, J = 7.0 Hz, 3H), 1.07 (d, J = 6.9 Hz, 2.37H), 0.92 (d, J = 6.9 Hz, 2.37H), 0.79 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 190.4 (5g’ C), 164.8 (5g C, major), 164.7 (5g C, minor), 163.4 (5g’ C), 139.4 (5g C, major), 136.9 (5g C, minor), 135.22 (5g CH, major), 135.17 (5g CH, minor), 134.8 (5g’ CH), 134.5 (5g’ C), 134.2 (5g’ CH), 133.7 (5g’ C), 129.9 (5g’ CH, major), 129.00 (5g’ C), 128.97 (5g C, major), 128.8 (5g C, minor), 128.7 (5g’ CH), 127.9 (5g CH, major), 127.61 (5g CH, minor), 127.59 (5g CH, minor), 127.42 (5g CH, minor), 127.37 (5g CH, major), 127.36 (5g CH, major), 124.2 (5g CH, major), 124.1 (5g CH, minor), 123.9 (5g’ CH), 114.0 (5g CH, minor), 113.9 (5g CH, major), 107.8 (5g’ CH), 90.4 (5g C, major), 89.9 (5g C, minor), 61.7 (5g CH3, major), 61.1 (5g CH3, minor), 57.7 (5g’ CH3), 35.2 (5g CH, minor), 33.9 (5g CH, major), 18.2 (5g CH3, major), 18.0 (5g CH3, major), 17.9 (5g CH3, minor), 17.6 (5g CH3, minor); IR (ATR) 3344, 1723, 1372, 1186, 966, 876, 747 cm−1; 5g HRMS (ESI) m / z calcd for C20H21NNaO6 (M + Na)+ 394.1261, found 394.1260; 5g’ HRMS (ESI) m / z calcd for C17H13NNaO5 (M + Na)+ 335.0719, found 335.0721.
4.2.2.7. Peroxide 5h.
Following the representative procedure for the peroxidation of enol ethers, (1-methoxyhept-1-en-6-yn-2-yl)benzene (4h, 0.061 g, 0.31 mmol), NHPI (0.059 g, 0.36 mmol), and acetonitrile (6 mL) were combined to afford peroxide 5h as a 1.00:0.42 mixture of diastereomers as a white solid (0.066 g, 55%): mp = 149–153 °C; 1H NMR (400 MHz, CDCl3) δ 10.94 (s, 0.42H), 10.28 (s, 1H), 7.93–7.78 (m, 5.68H), 7.68–7.64 (m, 0.84H), 7.59–7.55 (m, 2H), 2.41–2.27 (m, 4.26H), 5.37 (s, 1H), 5.30 (s, 0.42H), 3.62 (s, 3H), 3.58 (s, 1.26H), 2.38–2.07 (m, 5.68H), 1.96 (t, J = 2.6 Hz, 1H), 1.88 (t, J = 2.6 Hz, 0.42H), 1.77–1.57 (m, 1.42H), 1.44–1.32 (m, 1H), 1.30–1.18 (m, 0.42H); 13C NMR (100 MHz, CDCl3) δ 164.8 (C, minor), 164.6 (C, major), 139.0 (C, minor), 135.7 (C, major), 135.3 (CH, minor), 135.2 (CH, major), 128.9 (C, minor), 128.8 (C, major), 128.3 (CH, minor), 127.90 (CH, major), 127.87 (CH, major), 127.7 (CH, minor), 127.5 (CH, major), 127.2 (CH, minor), 124.3 (CH, minor), 124.1 (CH, major), 113.73 (CH, minor), 113.70 (CH, major), 88.4 (C, minor), 87.6 (C, major), 84.5 (C, major), 84.3 (C, minor), 68.62 (CH, major), 68.56 (CH, minor), 61.1 (CH3, minor), 60.7 (CH3, major), 32.4 (CH2, major), 31.6 (CH2, minor), 22.1 (CH2, minor), 21.8 (CH2, major), 18.8 (CH2, minor), 18.6 (CH2, major); IR (ATR) 3338, 3298, 1716, 1378, 1132, 973, 877 cm−1; HRMS (ESI) m / z calcd for C22H25N2O6 (M + NH4)+ 413.1707, found 413.1701.
4.2.2.8. Peroxide 5i.
Following the representative procedure for the peroxidation of enol ethers, (2-methoxyvinyl)benzene (4i, 0.073 g, 0.5455 mmol), NHPI (0.107 g, 0.6546 mmol), and acetone (11 mL) were combined to afford peroxide 5i as a 1.00:0.77 mixture of diastereomers as a white solid (0.100 g, 56%). The peroxide was purified by flash column chromatography and characterized as a 1.0:1.0 mixture of diastereomers: mp = 123–130 °C; 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 9.60 (s, 1H), 7.92–7.76 (m, 8H), 7.55–7.51 (m, 2H), 7.48–7.44 (m, 2H), 7.42–7.34 (m, 6H), 5.45 (d, J = 3.8 Hz, 1H), 5.31 (d, J = 3.8 Hz, 1H), 5.29 (d, J = 7.1 Hz, 1H), 5.17 (d, J = 7.1 Hz, 1H), 3.71 (s, 3H), 3.60 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.7 (C), 164.4 (C), 135.5 (C), 135.1 (CH), 135.0 (CH), 134.4 (C), 129.2 (CH), 128.98 (C), 128.97 (C), 128.9 (CH), 128.8 (CH), 128.56 (CH), 128.55 (CH), 128.4 (CH), 124.1 (CH), 124.0 (CH), 110.9 (CH), 110.0 (CH), 86.7 (CH), 86.5 (CH), 58.7 (CH3), 57.7 (CH3); IR (ATR) 3308, 1716, 1376, 1188, 1156, 981, 877 cm−1; HRMS (ESI) m / z calcd for C17H15NNaO6 (M + Na)+ 352.0792, found 352.0799
4.2.2.9. Peroxide 5j.
To a solution of NHPI (0.111 g, 0.683 mmol) in acetone (10 mL, sparged with O2 for 5 min) was added ethyl vinyl ether (0.041 g, 0.569 mmol). The reaction mixture was stirred under an O2 balloon in front of blue light for 16 h, then concentrated in vacuo. The resulting solid was resuspended in dichloromethane (10 mL) and the suspension was filtered through a celite plug. The filtered solid was washed with dichloromethane (20 mL), and the combined filtrates were concentrated in vacuo. Purification of the resulting solid by flash column chromatography (30% EtOAc/hexanes) afforded peroxide 5j as a colorless oil (0.073 g, 48%): 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 7.89–7.75 (m, 4H), 5.21 (dd, J = 4.1, 7.1 Hz, 1H), 4.41 (dd, J = 4.1, 11.4 Hz, 1H), 4.30 (dd, J = 7.1, 11.4 Hz, 1H), 3.97 (dq, J = 7.1, 9.6 Hz, 1H), 3.67 (dq, J = 7.1, 9.6 Hz, 1H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 163.9 (C), 135.0 (CH), 128.9 (C), 124.0 (CH), 103.8 (CH), 75.6 (CH2), 65.0 (CH2), 15.2 (CH3); IR (ATR) 2978, 1720, 1374, 1186, 1116, 1020, 877 cm−1; HRMS (ESI) m / z calcd for C12H13NNaO6 (M + Na)+ 290.0635, found 290.0640.
4.2.2.10. Peroxide 5k.
Following the representative procedure for the peroxidation of enol ethers, (cyclohexylidenemethoxy)trimethylsilane (4k, 0.038 g, 0.181 mmol), NHPI (0.035 g, 0.217 mmol) and acetonitrile (3.6 mL) were combined to afford peroxide 5k as a white solid (0.037 g, 54%). No melting point was obtained because peroxide 5k decomposed at a temperature of 158– 164 °C; 1H NMR (400 MHz, CDCl3) δ 9.49 (s, 1H), 7.92–7.76 (m, 4H), 5.34 (s, 1H), 2.06–1.92 (m, 2H), 1.82–1.50 (m, 6H), 1.39 (dt, J = 4.4, 13.3 Hz, 1H), 1.28–1.15 (m, 1H), 0.076 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 164.6 (C), 135.1 (CH), 128.9 (C), 124.0 (CH), 104.9 (CH), 84.2 (C), 28.3 (CH2), 26.0 (CH2), 25.8 (CH2), 20.9 (CH2), 20.7 (CH2), 0.36 (CH3); IR (ATR) 3363, 1718, 1376, 1247, 1111, 871, 839 cm−1; HRMS (ESI) m / z calcd for C18H25NNaO6Si (M + Na)+ 402.1343, found 402.1351.
4.2.2.11. Peroxides 5n and 5n’.
Following the representative procedure for the peroxidation of enol ethers, 1-methoxyhept-1-ene (4n, 0.040 g, 0.31 mmol), NHPI (0.059 g, 0.36 mmol), and acetonitrile (6 mL) were combined to afford peroxides 5n and 5n’ as a mixture of regoisomers and diastereomers in a 1.0:0.92:0.59:0.43 ratio as a colorless oil (0.037 g, 36%). The identity of the regioisomers was assigned by comparison of the acetal carbon chemical shifts to other peroxide products: 1H NMR (400 MHz, CDCl3) δ 9.97–9.83 (m, 2.35H), 9.07–9.00 (s, 0.59H), 7.90–7.75 (m, 11.76H), 5.21 (d, J = 4.2 Hz, 0.92H), 5.05 (d, J = 7.0 Hz, 1H), 4.94 (d, J = 4.7 Hz, 0.59H), 4.89 (d, J = 7.6 Hz, 0.43H), 4.36–4.25 (m, 1.02H), 4.18–4.10 (m, 1.92H), 3.73–3.68 (m, 5.76H), 3.59 (s, 1.29H), 3.50 (s, 1.77H), 1.97–1.26 (m, 23.52H), 0.95–0.87 (8.82H); 13C NMR (100 MHz, CDCl3) δ 164.8 (C, 5n’ minor), 164.6 (C, 5n major), 164.5 (C, 5n minor), 164.2 (C, 5n’ major), 135.12 (CH, 5n major), 135.11 (CH, 5n minor), 135.0 (CH, 5n’ minor), 134.8 (CH, 5n’ major), 129.0 (C, 5n’ major), 128.84 (C, 5n major), 128.82 (C, 5n minor), 128.78 (C, 5n’ minor), 124.11 (CH, 5n major), 124.07 (CH, 5n minor), 124.0 (CH, 5n’ minor), 123.8 (CH, 5n’ major), 111.7 (CH, 5n major), 111.0 (CH, 5n minor), 108.4 (CH, 5n’ major), 107.7 (CH, 5n’ minor), 86.6 (CH, 5n’ major), 85.5 (CH, 5n’ minor), 84.2 (CH, 5n minor), 83.6 (CH, 5n major), 58.9 (CH3, 5n minor), 57.4 (CH3, 5n’ major), 56.8 (CH3, 5n’ minor), 56.7 (CH3, 5n major), 32.1 (CH2, 5n’ minor), 31.81 (CH2, 5n minor), 31.78 (CH2, 5n’ major), 31.76 (CH2, 5n major), 30.0 (CH2, 5n’ minor), 28.0 (CH2, 5n major), 27.7 (CH2, 5n’ major), 26.8 (CH2, 5n minor), 25.7 (CH2, 5n minor), 25.2 (CH2, 5n major), 25.0 (CH2, 5n’ major), 23.7 (CH2, 5n’ minor), 22.7 (CH2, 5n major), 22.62 (CH2, 5n minor, 5n’ minor), 22.60 (CH2, 5n’ major), 14.23 (CH3, 5n’ major), 14.21 (CH3, 5n’ minor), 14.19 (CH3, 5n major), 14.19 (CH3, 5n minor); IR (ATR) 3384, 2930, 1724, 1374, 1187, 971, 876 cm−1; HRMS (ESI) m / z calcd for C16H22NO6 (M*)+ 324.1442, found 324.1439.
Supplementary Material
Acknowledgements
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health (1R01GM118730). NMR spectra acquired with the Bruker Avance 400 and 500 NMR spectrometers were supported by the National Science Foundation (CHE-01162222). The authors thank Dr. Chin Lin (NYU) for his assistance with NMR spectroscopy and mass spectrometry, as well as the NYU Genomics Core Facility for use of their Tecan Spark Microplate Reader.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- (1).Rubottom GM; Vazquez MA; Pelegrina DR Tetrahedron Lett. 1974, 15, 4319. [Google Scholar]
- (2).Zlotorzynska M; Zhai H; Sammis GM Org. Lett 2008, 10, 5083. [DOI] [PubMed] [Google Scholar]
- (3).Lempenauer L; Lemière G; Duñach E Adv. Synth. Catal 2019, 361, 5284. [Google Scholar]
- (4).Ohmatsu K; Nakashima T; Sato M; Ooi T Nat Commun 2019, 10, 2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Matsuo J.-i.; Murakami M Angew. Chem. Int. Ed 2013, 52, 9109. [DOI] [PubMed] [Google Scholar]
- (6).Mukaiyama T; Banno K; Narasaka K J. Am. Chem. Soc 1974, 96, 7503. [Google Scholar]
- (7).Manchand PS Ethyl Vinyl Ether. Encyclopedia of Reagents for Organic Synthesis; Wiley: Hoboken, 2001. [Google Scholar]
- (8).Poirier J-M Org. Prep. Proced. Int 1988, 20, 317. [Google Scholar]
- (9).Mukaiyama T Org. React 1982, 28, 203. [Google Scholar]
- (10).Effenberger F Angew. Chem., Int. Ed. Engl 1969, 8, 295. [Google Scholar]
- (11).Jung ME; Pan Y-G; Rathke MW; Sullivan DF; Woodbury RP J. Org. Chem 1977, 42, 3961. [Google Scholar]
- (12).Hintz S; Fröhlich R; Mattay J Tetrahedron Lett. 1996, 37, 7349. [Google Scholar]
- (13).Moriarty RM; Prakash O; Duncan MP; Vaid RK; Musallam HA J. Org. Chem 1987, 52, 150. [Google Scholar]
- (14).Magnus P; Lacour J; Evans PA; Roe MB; Hulme CJ Am. Chem. Soc 1996, 118, 3406. [Google Scholar]
- (15).Nicolaou K; Gray DL; Montagnon T; Harrison ST Angew. Chem. Int. Ed 2002, 41, 996. [DOI] [PubMed] [Google Scholar]
- (16).Snider BB; Kwon T J. Org. Chem 1990, 55, 4786. [Google Scholar]
- (17).Vil’ VA; Gorlov ES; Bityukov OV; Barsegyan YA; Romanova YE; Merkulova VM; Terent’ev AO Adv. Synth. Catal 2019, 361, 3173. [Google Scholar]
- (18).Yamamoto H; Tsuda M; Sakaguchi S; Ishii Y J. Org. Chem 1997, 62, 7174. [DOI] [PubMed] [Google Scholar]
- (19).Boyce RS; Kennedy RM Tetrahedron Lett. 1994, 35, 5133. [Google Scholar]
- (20).Lauer MG; Henderson WH; Awad A; Stambuli JP Org. Lett 2012, 14, 6000. [DOI] [PubMed] [Google Scholar]
- (21).Ciufolini MA; Deaton MV; Zhu S; Chen M Tetrahedron 1997, 53, 16299. [Google Scholar]
- (22).Inokuchi T; Takagishi S; Ogawa K; Kurokawa Y; Torii S Chem. Lett 1988, 17, 1347. [Google Scholar]
- (23).Hachem M; Schneider C; Hoarau C Eur. J. Org. Chem 2020. [Google Scholar]
- (24).Anderson TE; Woerpel KA Org. Lett 2020, 22, 5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bag R; Sar D; Punniyamurthy T Org. Lett 2015, 17, 2010. [DOI] [PubMed] [Google Scholar]
- (26).Andia AA; Miner MR; Woerpel KA Org. Lett 2015, 17, 2704. [DOI] [PubMed] [Google Scholar]
- (27).Samanta S; Ravi C; Joshi A; Pappula V; Adimurthy S Tetrahedron Lett. 2017, 58, 721. [Google Scholar]
- (28).Bag R; Sar D; Punniyamurthy T Org. Biomol. Chem 2016, 14, 3246. [DOI] [PubMed] [Google Scholar]
- (29).Bag R; Punniyamurthy T Chemistryselect 2018, 3, 6152. [Google Scholar]
- (30).Tang S-Q; Wang A-P; Schmitt M; Bihel F Tetrahedron Lett. 2018, 59, 1465. [Google Scholar]
- (31).Xia X-F; Gu Z; Liu W; Wang H; Xia Y; Gao H; Liu X; Liang Y-M J. Org. Chem 2014, 80, 290. [DOI] [PubMed] [Google Scholar]
- (32).Coseri S J. Phys. Org. Chem 2009, 22, 397. [Google Scholar]
- (33).Kothe T; Marque S; Martschke R; Popov M; Fischer H J. Chem. Soc., Perkin Trans 2 1998, 1553. [Google Scholar]
- (34).Quaranta M; Murkovic M; Klimant I Analyst 2013, 138, 6243. [DOI] [PubMed] [Google Scholar]
- (35).Renz M; Meunier B Eur. J. Org. Chem 1999, 1999, 737. [Google Scholar]
- (36).Luo J; Zhang J J Org Chem 2016, 81, 9131. [DOI] [PubMed] [Google Scholar]
- (37).Melone L; Franchi P; Lucarini M; Punta C Adv. Synth. Catal 2013, 355, 3210. [Google Scholar]
- (38).Yadav AK; Yadav LDS ChemComm 2016, 52, 10621. [Google Scholar]
- (39).Lin R; Chen F; Jiao N Org. Lett 2012, 14, 4158. [DOI] [PubMed] [Google Scholar]
- (40).Hirabayashi T; Sakaguchi S; Ishii Y Angew. Chem. Int. Ed 2004, 43, 1120. [DOI] [PubMed] [Google Scholar]
- (41).Ishii Y; Nakayama K; Takeno M; Sakaguchi S; Iwahama T; Nishiyama YJ Org. Chem 1995, 60, 3934. [Google Scholar]
- (42).Yan Y; Feng P; Zheng QZ; Liang YF; Lu JF; Cui Y; Jiao N Angew. Chem. Int. Ed 2013, 52, 5827. [DOI] [PubMed] [Google Scholar]
- (43).Ishii Y; Iwahama T; Sakaguchi S; Nakayama K; Nishiyama Y J Org Chem 1996, 61, 4520. [DOI] [PubMed] [Google Scholar]
- (44).Aguadero A; Falcon H; Campos-Martin J; Al-Zahrani S; Fierro J; Alonso J Angew. Chem. Int. Ed 2011, 50, 6557. [DOI] [PubMed] [Google Scholar]
- (45).Gaster E; Kozuch S; Pappo D Angew. Chem. Int. Ed 2017, 56, 5912. [DOI] [PubMed] [Google Scholar]
- (46).Frimer AA Chem. Rev 1979, 79, 359. [Google Scholar]
- (47).Turro NJ; Ramamurthy V; Liu K-C; Krebs A; Kemper RJ Am. Chem. Soc 1976, 98, 6758. [Google Scholar]
- (48).Petterson R Angew. Chem., Int. Ed. Engl 1970, 9, 644. [Google Scholar]
- (49).Turro NJ; Chow M-F; Ito YJ Am. Chem. Soc 1978, 100, 5580. [Google Scholar]
- (50).Bartlett PD; McCluney RE J. Org. Chem 1983, 48, 4165. [Google Scholar]
- (51).Huang C-S; Peng C-C; Chou C-H Tetrahedron Lett. 1994, 35, 4175. [Google Scholar]
- (52).Lévesque F; Seeberger PH Angew. Chem. Int. Ed 2012, 51, 1706. [DOI] [PubMed] [Google Scholar]
- (53).Riahi A; Muzart J; Abe M; Hoffmann N New J. Chem 2013, 37, 2245. [Google Scholar]
- (54).Attenburrow J; Connett J; Graham W; Oughton J; Ritchie A; Wilkinson PJ Chem. Soc 1961, 4547. [Google Scholar]
- (55).Enslin P Tetrahedron 1971, 27, 1909. [Google Scholar]
- (56).Ghosh P ACS Omega 2019, 4, 8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Chou C-H; Trahanovsky WS J. Org. Chem 1995, 60, 5449. [Google Scholar]
- (58).Adam W; Cueto O; Rebollo H Angew. Chem., Int. Ed. Engl 1982, 21, 75. [Google Scholar]
- (59).Hashmi ASK; Blanco Jaimes MC; Schuster AM; Rominger FJ Org. Chem 2012, 77, 6394. [DOI] [PubMed] [Google Scholar]
- (60).Warmuth R; Marvel MA Chem. Eur. J 2001, 7, 1209. [DOI] [PubMed] [Google Scholar]
- (61).Minaev BJ Struct. Chem 1982, 23, 170. [Google Scholar]
- (62).De Vico L; Liu YJ; Krogh JW; Lindh R J Phys Chem A 2007, 111, 8013. [DOI] [PubMed] [Google Scholar]
- (63).Adam W Adv. Heterocycl. Chem 1977, 21, 437. [Google Scholar]
- (64).Meijer E; Wynberg H Tetrahedron Lett. 1981, 22, 785. [Google Scholar]
- (65).Methyl formate was observed in addition to 4-heptanone when the oxidation reaction of 1b was conducted in deuterated acetonitrile and the reaction mixture was analyzed by 1H NMR spectroscopy
- (66).Zhang C; Huang Z; Lu J; Luo N; Wang FJ Am. Chem. Soc 2018, 140, 2032. [DOI] [PubMed] [Google Scholar]
- (67).Zhang X; Rakesh K; Ravindar L; Qin H-L Green Chem. 2018, 20, 4790. [Google Scholar]
- (68).Taniguchi M; Lindsey JS Photochem. Photobiol 2018, 94, 290. [DOI] [PubMed] [Google Scholar]
- (69).Bag R; De PB; Pradhan S; Punniyamurthy T Eur. J. Org. Chem 2017, 5424. [Google Scholar]
- (70).Delbecq F; Ilavsky D; Anh NT; Lefour J J. Am. Chem. Soc 1985, 107, 1623. [Google Scholar]
- (71).Zipse H Radical stability—a theoretical perspective. Radicals in Synthesis I. Topics in Current Chemistry; Springer: Berlin, 2006; Vol. 263, p 163. [Google Scholar]
- (72).Fleming I Radical Reactions. Frontier Orbitals and Organic Chemical Reactions; Wilely: Chichester, 1976; Vol. 5, p 394. [Google Scholar]
- (73).Bonacic-Koutecky V; Koutecky J; Salem LJ Am. Chem. Soc 1977, 99, 842. [Google Scholar]
- (74).Tedder JM Angew. Chem., Int. Ed. Engl 1982, 21, 401. [Google Scholar]
- (75).Giese B Angew. Chem., Int. Ed. Engl 1983, 22, 753. [Google Scholar]
- (76).Caron G; Lessard J Tetrahedron 1993, 49, 8039. [Google Scholar]
- (77).Wong MW; Pross A; Radom L Isr. J. Chem 1993, 33, 415. [Google Scholar]
- (78).Shaik SS J. Am. Chem. Soc 1981, 103, 3692. [Google Scholar]
- (79).Cvetanović RJ Can. J. Chem 1960, 38, 1678. [Google Scholar]
- (80).Burfeindt J; Patz M; Müller M; Mayr HJ Am. Chem. Soc 1998, 120, 3629. [Google Scholar]
- (81).Leonov AI; Timofeeva DS; Ofial AR; Mayr H Synthesis 2019, 51, 1157. [Google Scholar]
- (82).Baader WJ; Stevani CV; Bechara EJ J. Braz. Chem. Soc 2015, 26, 2430. [Google Scholar]
- (83).Hook BDA; Dohle W; Hirst PR; Pickworth M; Berry MB; Booker-Milburn KI J. Org. Chem 2005, 70, 7558. [DOI] [PubMed] [Google Scholar]
- (84).Chepiga KM; Feng Y; Brunelli NA; Jones CW; Davies HML Org. Lett 2013, 15, 6136. [DOI] [PubMed] [Google Scholar]
- (85).Smietana M; Mioskowski C Org. Lett 2001, 3, 1037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.