

Abstract
Fifteen water-soluble rhenium compounds of the general formula [Re(CO)3(NN)(PR3)]+, where NN is a diimine ligand and PR3 is either 1,3,5-triaza-7-phosphaadamantane (PTA), tris(hydroxymethyl)phosphine (THP), or 1,4-diacetyl-1,3,7-triaza-5-phosphabicylco[3.3.1]nonane (DAPTA), were synthesized and characterized by multinuclear NMR spectroscopy, IR spectroscopy, and X-ray crystallography. The complexes bearing the THP and DAPTA ligands exhibit triplet-based luminescence in air-equilibrated aqueous solutions with quantum yields ranging from 3.4 to 11.5%. Furthermore, the THP and DAPTA complexes undergo photosubstitution of a CO ligand when irradiated with 365 nm light with quantum yields ranging from 1.1 to 5.5% and sensitize the formation of 1O2 with quantum yields as high as 70%. By contrast, all of the complexes bearing the PTA ligand are non-emissive and do not undergo photosubstitution when irradiated with 365 nm light. These compounds were evaluated as photoactivated anticancer agents in human cervical (HeLa), ovarian (A2780), and cisplatin-resistant ovarian (A2780CP70) cancer cell lines. All of the complexes bearing THP and DAPTA exhibited a cytotoxic response upon irradiation with minimal toxicity in the absence of light. Notably, the complex with DAPTA and 1,10-phenanthroline gave rise to an IC50 value of 6 μM in HeLa cells upon irradiation, rendering it the most phototoxic compound in this library. The nature of the photoinduced cytotoxicity of this compound was explored in further detail. These data indicate that the phototoxic response may result from the release of both CO and the rhenium-containing photoproduct, as well as the production of 1O2.
Graphical Abstract
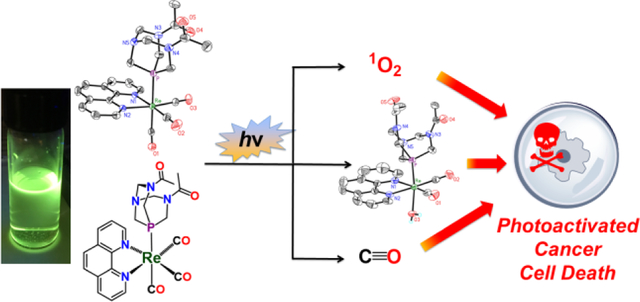
Fifteen rhenium(I) tricarbonyl complexes bearing water-soluble phosphines were synthesized and characterized. Detailed photophysical studies on these compounds reveal that they undergo photosubstitution reactions and are photoluminescent in a manner that depends on the nature of the axial phosphine ligand. Based on their photophysical properties, these complexes were evaluated as photoactivated anticancer agents in cervical and ovarian cancer cell lines.
Introduction
Chemotherapy is a critical component for the treatment of nearly all types of cancer. Conventional chemotherapeutic agents, however, are non-discriminating cytotoxic compounds that induce cell death in both cancerous and healthy tissue, a feature that manifests in the form of undesirable patient side effects. One approach to increase selectivity and minimize toxic side effects is to employ drugs that are activated by light. This concept, known as photodynamic therapy (PDT)1–4 or photoactivated chemotherapy (PACT),5 allows for further spatiotemporal control of the cytotoxic effects to maximize cancer cell death, while minimizing healthy cell damage. An ideal candidate for a PDT or PACT agent should be non-toxic in the absence of light, and therefore only give rise to toxic effects in regions of light exposure.6 The concept of PDT is currently employed in the clinic in the form of the FDA-approved porphyrin derivatives, Photofrin, Foscan, and Verteporfin.7,8 A potential drawback of the currently used PDT agents is that their mechanism of action stems from their abilities to photosensitize the formation of singlet oxygen (1O2).1,9–19 In hypoxic tumors, the efficacy of these PDT agents is substantially diminished due to the lower concentrations of oxygen present.20–23 An additional limitation of these porphyrin analogues is their poor aqueous solubility, which requires clever formulation strategies for in vivo administration.2,24,25 PACT is currently the subject of significant preclinical investigation.5,26 PACT agents generally operate under an oxygen-independent mechanism,5 as irradiation with light triggers release of a cytotoxic chemical compound.
Towards the development of improved PDT27 and PACT5 agents, many research efforts have explored the potential of transition metal complexes. In addition to functioning as 1O2 photosensitizers for PDT applications, many inorganic complexes also undergo photoinduced ligand substitution reactions, which make them useful as PACT agents. Following the concept of PACT, these photosubstitution reactions can be applied to generate cytotoxic complexes from benign precursors in an oxygen-independent manner, rendering them suitable for use in hypoxic conditions.28 Ruthenium PDT29–51 and PACT22,35,40,42,50,52–55 agents have received the majority of focus, because they possess ideal photophysical properties for this application. The diversity of photophysical processes accessible for other metal ion complexes have prompted researchers to expand their search for new PDT or PACT agents.
Complexes of rhenium, for example, possess rich spectroscopic and photophysical properties that can be leveraged for use in PDT or PACT.56 This concept has been demonstrated for several rhenium(I) tricarbonyl complexes, which exhibit potent phototoxic effects in vitro.57–61 All of the potential rhenium-based phototoxic agents to date, however, induce cell death via production of singlet oxygen, and therefore suffer from some of the same limitations as the conventional porphyrin-based PDT agents. The development of rhenium anticancer PACT agents that act via alternative mechanisms of action remains an interesting unmet research objective that could provide access to new therapeutic modalities. In this study, we describe our efforts towards this goal, in which we have developed a series of rhenium(I) tricarbonyl complexes bearing phosphine ligands that exhibit potent UVA (365 nm) light-activated toxicity against several types of cancer cell lines. Although the biological penetration depth of 365 nm light it too shallow to be useful for in vivo applications, this study provides a proof of concept for the future development of rhenium-based PDT and PACT agents.
Results and Discussion
Rationale and Approach.
Diimine rhenium(I) tricarbonyl complexes are usually inert to photosubstitution reactions and exhibit small luminescence quantum yields.62 By contrast, when a phosphine is introduced to the inner coordination sphere, the resulting complexes [Re(CO)3(NN)(PR3)]+, where NN = diimine ligand and PR3 = phosphine ligand, are brightly luminescent 63–66 and photolabile.62,67 Upon irradiation with UVA light, the CO trans to the phosphine dissociates, forming the dicarbonyl complex [Re(CO)2(NN)(PR3)(S)]+, where S is a coordinating solvent molecule.62,68 Complexes like these have been investigated as photo CO-releasing molecules (photoCORMS),68–74 tools to understand the significance of CO as a biological signaling molecule. We envisioned an alternative role of these complexes as photoactivated anticancer agents. The rhenium(I) dicarbonyl photoproduct with a labile coordination site could interact with biological targets, such as DNA, in a manner similar to that reported for rhenium(I) tricarbonyl complexes.75–77 In this context, our lab has recently demonstrated that rhenium(I) tricarbonyl complexes bearing labile aqua ligands exhibit potent in vitro anticancer activity via a mechanism of action that is distinct from that of cisplatin.78 Thus, we anticipated that the structurally similar rhenium(I) dicarbonyl phosphine photoproducts would also be active against cancer cells. Furthermore, the released CO molecule could elicit an additional cytotoxic effect (Scheme 1).79–84 Such “dual action” chemotherapeutic agents may be particularly effective against drug-resistant forms of cancer.
Scheme 1.
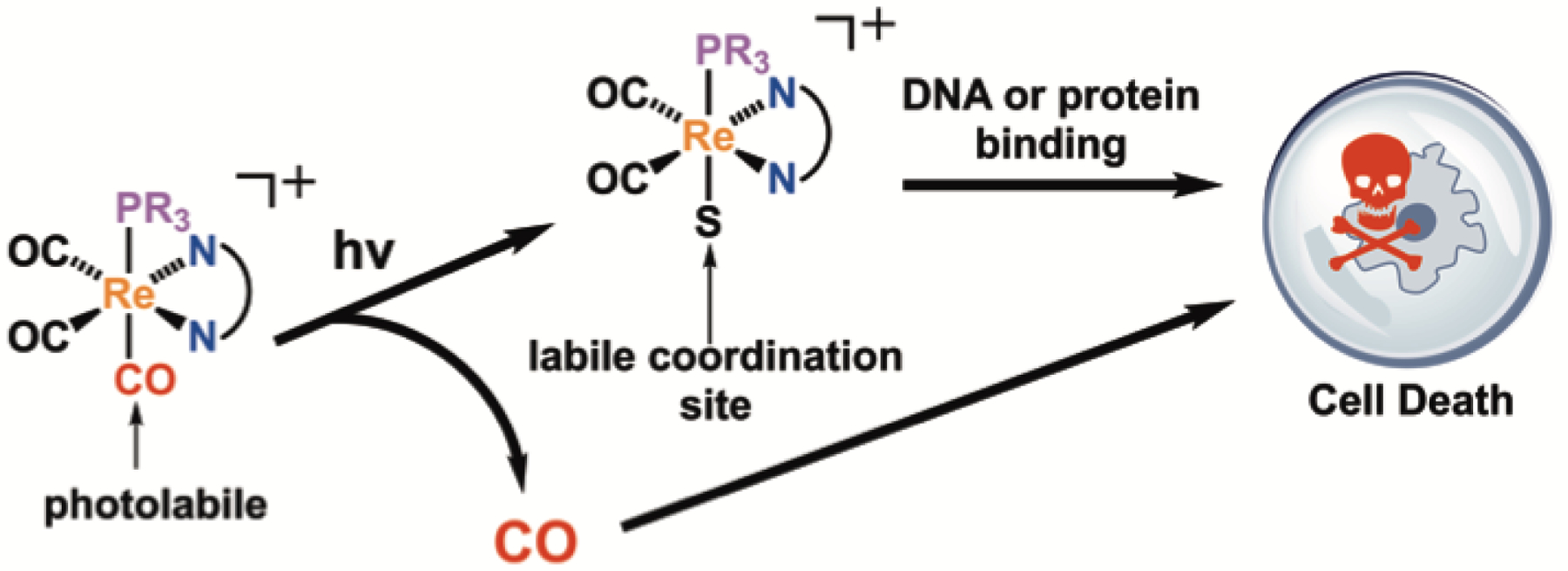
The photoreaction of [Re(CO)3(NN)(PR3)]+ releases an equivalent of CO and a labile rhenium(I) complex, which may both give rise to cytotoxicity in cancer cells. (S is a solvent molecule).
In designing our compound library, we employed the phosphines 1,3,5-triaza-7-phosphaadamantane (PTA), tris(hydroxymethyl)phosphine (THP), and 1,4-diacetyl-1,3,7-triaza-5-phosphabicylco[3.3.1]nonane (DAPTA) because these ligands effectively confer water solubility on their coordination complexes.85–87 To further screen for cytotoxic effects of the rhenium photoproducts, we explored five different diimine ligands, 1,10-phenanthroline (1), 2,9-dimethyl-1,10-phenanthroline (2), 2,2’-bipyridine (3), 4,4’-dimethyl-2,2’-dipyridyl (4), or 4,4’-dimethoxy-2,2’-bipyridine (5). We hypothesized that this diverse set of compounds would possess a range of photophysical and biological properties, some of which would be valuable for use in PACT.
Synthesis and Characterization.
From the three phosphines and five diimine ligands, a total of fifteen rhenium tricarbonyl complexes were synthesized. The synthetic approach followed a previously reported procedure (Scheme 2).88,89 In the first step, Re(CO)5Cl and a diimine ligand were heated to reflux in toluene to afford the yellow complexes, fac-[Re(CO)3(NN)Cl]. These complexes were treated with AgOTf to remove the axial chloride as insoluble AgCl, and then allowed to react with the desired phosphine in refluxing tetrahydrofuran (THF)68 to afford the compounds PTA-1–PTA-5, THP-1–THP-5, and DAPTA-1–DAPTA-5 (Scheme 2). The PTA and DAPTA complexes were isolated via precipitation as the triflate salts. By contrast, all of the THP complexes, except for THP-5, required additional purification by preparative high-performance liquid chromatography (HPLC). Because the HPLC mobile-phase employed 0.1% trifluoroacetic acid, the THP compounds were isolated as trifluoroacetate salts with the exception of THP-5, which was isolated as the triflate salt upon recrystallization. All of the complexes exhibited good water solubility, enabling the preparation of aqueous solutions at concentrations exceeding 1 mM. By contrast, the fac-[Re(CO)3(NN)Cl] starting materials are very poorly soluble in water.78 Among the fifteen complexes prepared in this investigation, PTA-1 and THP-3 have previously been reported and developed as photoCORMs.68,83
Scheme 2.
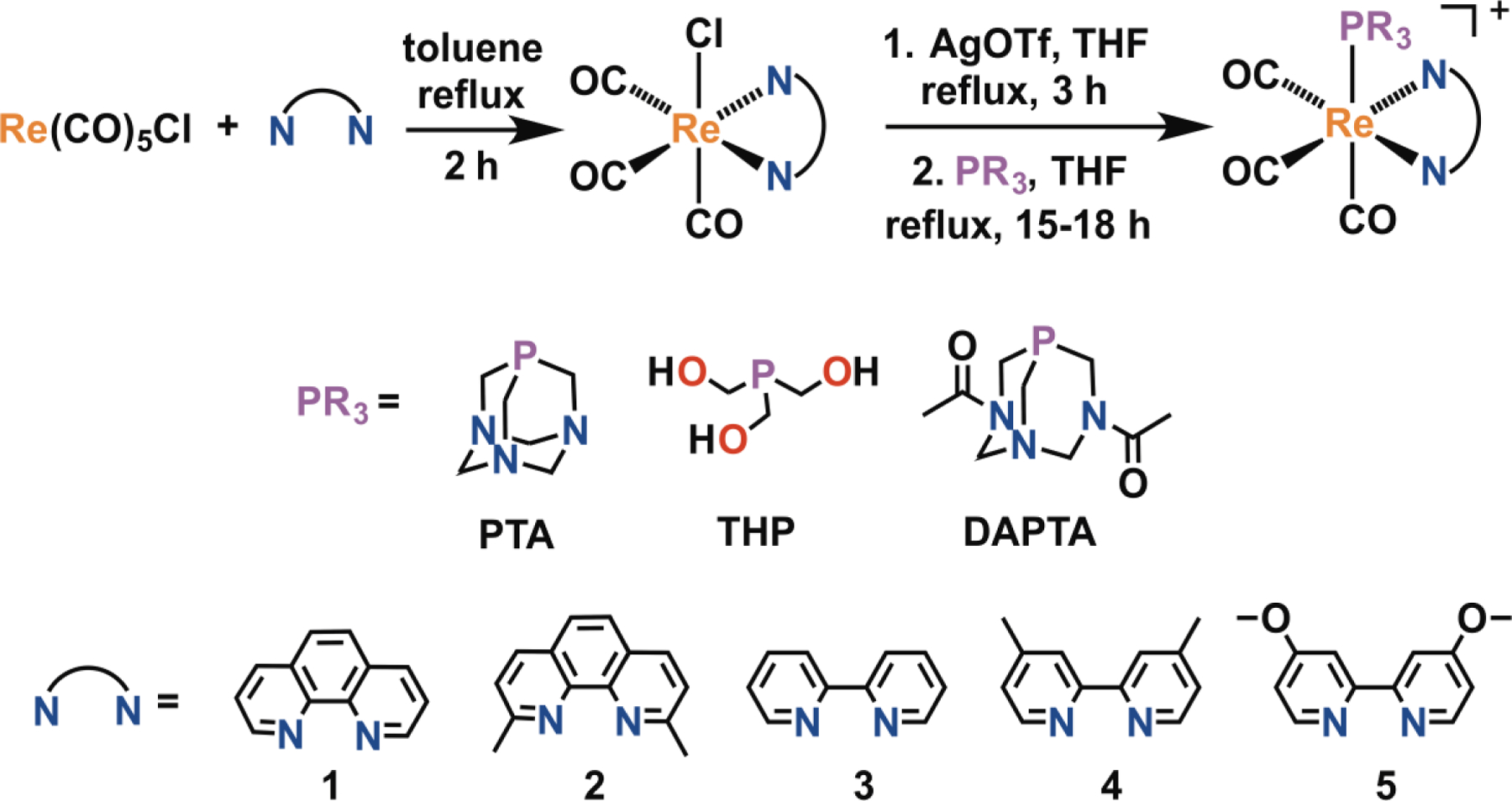
Synthetic scheme for the preparation of the [Re(CO)3(NN)(PR3)]+ complexes.
The complexes were characterized by 1H (Figure S1–S15, Supporting Information), 31P{1H} (Figures S18–S32), and 19F (Figures S35–S49) NMR spectroscopy, IR spectroscopy (Figure S50–S64), and electrospray ionization mass spectrometry (ESI-MS, Figures S84–S98). Purity of the complexes was verified with elemental analysis and HPLC (Figures S67–S81). The IR spectra of the complexes exhibited three distinct CO stretches, as expected for fac-[Re(CO)3(NN)X] (X= H2O, Cl, Br) complexes of Cs symmetry.78,90–93 The energy of these stretching frequencies were invariant with respect to the coordinating diimine ligands. By contrast, a greater dependency on the nature of the axial phosphine ligand was observed. For example, PTA-1, THP-1, and DAPTA-1 exhibited pronounced CO stretches corresponding to the A′′ mode at 2030, 2040, and 2038 cm−1 respectively and two less intense stretches corresponding to the two A′ modes at 1940 and 1920, 1930 and 1910, and 1948 and 1914 cm−1, respectively (Figures S50–S64). These values are consistent with other fac-[Re(CO)3(NN)(PR3)]+ complexes.68,93 The presence of the axial phosphine shifts the CO stretching frequencies to higher energies compared to the precursor complexes fac-[Re(CO)3(NN)Cl], which exhibit stretching frequencies near 2020 and 1880 cm−1.88 The CO stretching energy of the phosphine complexes is similar to cationic complexes, such as fac-[Re(CO)3(NN)(OH2)]+ (νCO = 2030, 1930, and 1900 cm−1)78 and fac-[Re(CO)3(NN)(pyridine)]+ (νCO = 2036 and 1931 cm−1).94 The higher energy CO stretching frequencies of the phosphine complexes may therefore be a result of both the π acidity of the trans phosphine ligand and the overall cationic charge of the complex, features which decrease π-backbonding to CO π* orbitals.
The 31P{1H} NMR spectra of the PTA and THP complexes show a single resonance. This resonance is shifted downfield from the free ligand, signifying coordination to the rhenium center.85,86,95,96 For the DAPTA complexes, two signals in the 31P NMR spectra were observed. We attribute the two signals to arise from conformational isomers of the DAPTA ligand. As shown in Chart 1, the relative orientation of the two acetyl groups gives rise to anti and syn isomers, which will result in distinct NMR signals if their interconversion is slow. The observations of these two isomers in other metal complexes of DAPTA was previously reported.97 The 1H NMR spectra of the DAPTA complexes similarly display resonances of two species that can be attributed to the two conformers. For all of the DAPTA complexes, the anti isomer was the major species in solution. This assignment is based on the 1H NMR resonances of the methyl groups of the DAPTA ligand, which are inequivalent only for the anti isomer. Lastly, the 19F NMR spectra confirm the nature of the counterions of these complexes. As expected, the PTA and DAPTA complexes give rise to a signal at –79 ppm, which corresponds to the outer-sphere triflate ion, and the THP complexes display a signal at –76 ppm, arising from the TFA counterion, except for THP-5, which exhibits a signal corresponding to the triflate counterion at –79 ppm.
Chart 1.
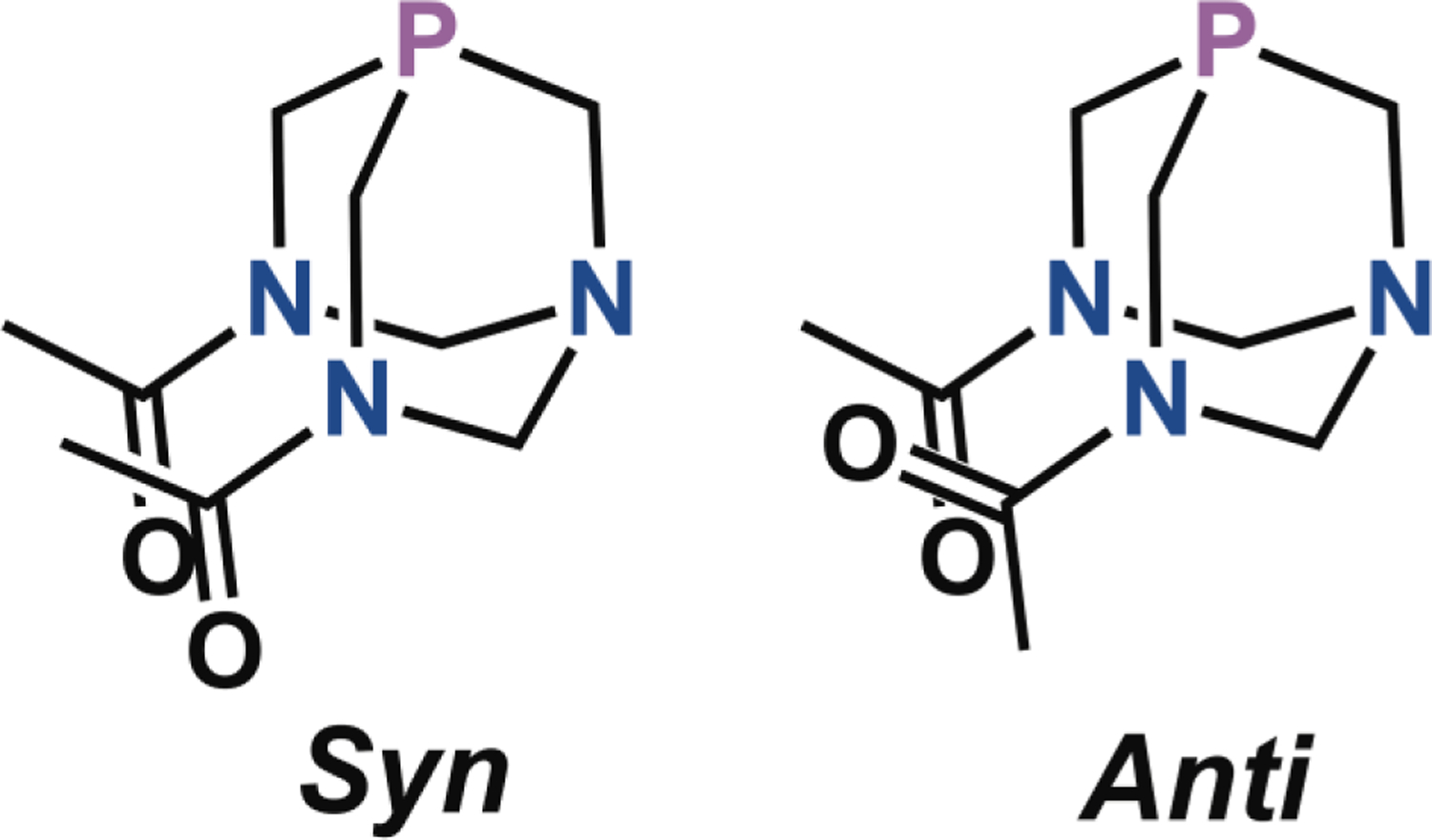
Syn and anti conformational isomers of DAPTA.
X-Ray Crystallography.
Single crystals of all fifteen complexes were obtained, and the crystal structures (Figures 1, S99–S102), with the exception of the previously reported THP-3,68 were determined by X-ray crystallography. Crystal structures for PTA-1, THP-1, and DAPTA-1 are shown in Figure 1, and selected interatomic distances and angles for all fourteen structures are collected in Table S2. We note that the crystal structure of PTA-1 was recently reported, albeit for a different polymorph of this compound;83 a comparison of this previously reported structure and ours reveals no significant differences with respect to the geometry of the rhenium complex.83 All complexes exhibit the expected octahedral geometry for a d6 rhenium(I) center. All DAPTA complexes, except for DAPTA-2, crystallized as the anti isomer, which was the major species exhibited by NMR spectroscopy. No evidence for rotational disorder of the DAPTA acetyl groups was observed in the crystal structures, verifying our conformer assignments. The Re–P bond lengths were relatively invariant among complexes bearing different diimine ligands. A more pronounced dependence was observed as the phosphine ligand was altered. For example, the Re–P distances in PTA-1, THP-1, and DAPTA-1 are 2.4343(6), 2.4602(7), and 2.4449(7) Å, respectively. These values are in the range expected for [Re(CO)3(NN)(PR3)]+ complexes.68,83,98 Among the fifteen complexes, the THP complexes exhibit a slightly longer average Re–P bond distance of 2.4574 ± 0.013 Å (error is the standard deviation over the five THP compounds) compared to the PTA and DAPTA complexes, for which the average Re–P distances are 2.4340 ± 0.0045 and 2.4414 ± 0.0077 Å, respectively. The longer Re–P distances found in the THP complexes are most likely a consequence of the larger cone angle of this phosphine in comparison to PTA and DAPTA.99,100 The Re–CO distances trans to the phosphine vary in a manner that is dependent on the nature of the phosphine as well. In general, this distance is longest for complexes of PTA and DAPTA and shortest for those of THP. For example, the Re–CO distances in PTA-1, THP-1, and DAPTA-1 are 1.962(3), 1.953(3), and 1.963(3) Å.68,83 The longer trans Re–CO distances in the DAPTA and PTA complexes may reflect a greater π-acidity of this ligand relative to THP. For comparison, the axial Re–CO distances of fac-[Re(CO)3(NN)X]+ complexes, where X is a solvent molecule, range from 1.882–1.917 Å, and are therefore approximately 0.036–0.089 Å shorter than the phosphine derivatives.78,101,102
Figure 1.
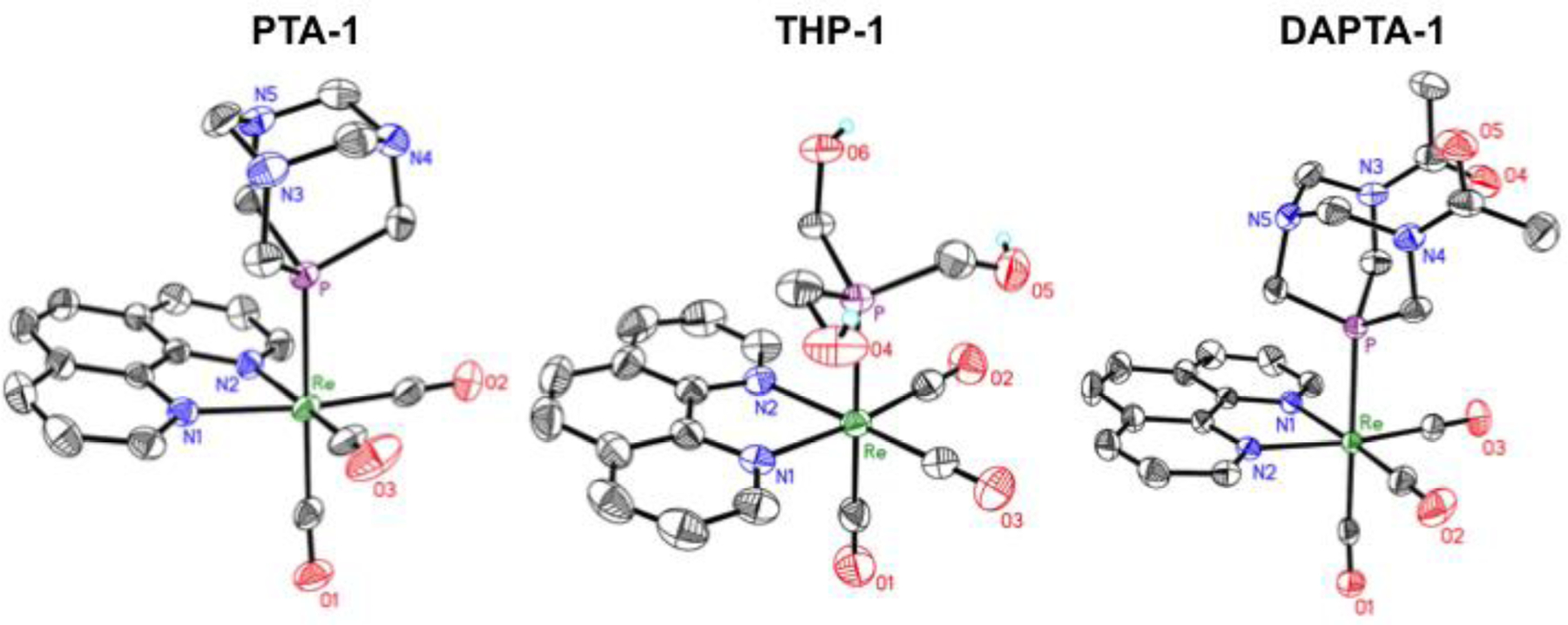
X-ray crystal structures of PTA-1, THP-1, and DAPTA-1. Ellipsoids are drawn at the 50% probability level. H atoms bound to C atoms and counterions are omitted for clarity.
Photophysical Properties.
UV-vis and emission spectra of all 15 complexes were measured in air-equilibrated pH 7.4 phosphate-buffered saline. This data is collected in Table 1, and the UV-vis spectra of these complexes are shown in Figures S103–S107.
Table 1.
Photophysical Propertiesa
| Compound | λmax, nm (ε, M−1cm−1) | Φlum, % (λmax, nm) |
Φrxn, % | τair (μs)b | τN2 (μs)c |
|---|---|---|---|---|---|
| PTA-1 | 226 (35900 ± 2600), 225 (22300 ± 600), 274 (26400 ± 1900), 368 (3200 ± 300) | <0.1 (n.d.) | 0.06 | n.d. | n.d |
| THP-1 | 226 (36800 ± 1300), 275 (26800 ± 900), 322 (6400 ± 200), 366 (3600 ± 100) | 5.1 ± 1.1 (528) | 1.4 ± 0.4 | 1.5 | 4.8 |
| DAPTA-1 | 225 (41600 ± 7300), 275 (27200 ± 4900), 323 (5700 ± 900), 367 (3200 ± 400) | 10.7 ± 0.6 (516) | 1.1 ± 0.1 | 1.9 | 5.6 |
| PTA-2 | 228 (40500 ± 1100), 253 (26100 ± 600), 283 (26200 ± 600), 372 (2500 ± 300) | <0.1 (n.d.) | 0.015 | n.d. | n.d |
| THP-2 | 228 (34600 ± 1200), 286 (23400 ± 800), 308 (11900 ± 400), 372 (2100 ± 70) | 4.5 ± 0.4 (518) | 2.0 ± 0.4 | 1.0 | 3.7 |
| DAPTA-2 | 227 (36800 ± 6800), 285 (23800 ± 700), 309 (11500 ± 300), 373 (2100 ± 50) | 7.2 ± 0.2 (507) | 1.5 ± 0.1 | 2.0 | 7.8 |
| PTA-3 | 248 (24400 ± 800), 307 (11700 ± 500), 319 (12900 ± 500), 349 (3300 ± 300) | <0.1 (538) | 0.0044 | n.d. | n.d. |
| THP-3 | 222 (18900 ± 200), 249 (20100 ± 1900), 308 (10700 ± 400), 318 (12500 ± 400), 343 (3600 ± 700) | 6.1 ± 1.7 (536) | 1.6 ± 0.1 | 0.4 | 0.5 |
| DAPTA-3 | 246 (22600 ± 500), 308 (10700 ± 200), 319 (13000 ± 300), 345 (3400 ± 100) | 9.1 ± 3.0 (528) | 2.3 ± 0.5 | 0.6 | 0.8 |
| PTA-4 | 252 (29200 ± 3600), 303 (13600 ± 1800), 315 (14700 ± 2100), 345 (4300 ± 500) | <0.1 (530) | 0.021 | n.d. | n.d. |
| THP-4 | 252 (24000 ± 3000), 304 (11800 ± 1500), 315 (13400 ± 1700), 338 (4600 ± 500) | 6.5 ± 2.0 (528) | 3.8 ± 0.9 | 0.4 | 0.6 |
| DAPTA-4 | 250 (8200 ± 4300), 305 (12800 ± 2000), 315 (14900 ± 2300), 339 (4000 ± 400) | 11.5 ± 3.9 (518) | 5.5 ± 0.3 | 0.6 | 1.0 |
| PTA-5 | 227 (36100 ± 2600), 249 (32900 ± 2300), 304 (9800 ± 700), 338 (4400 ± 300) | <0.1 (520) | 0.018 | n.d. | n.d. |
| THP-5 | 223 (33900 ± 1000), 251 (30500 ± 900), 303 (8800 ± 300), 337 (4500 ± 80) | 3.4 ± 1.1 (537) | 1.2 ± 0.2 | 0.3 | 0.4 |
| DAPTA-5 | 224 (41400 ± 2000), 251 (34100 ± 1900), 303 (9600 ± 500), 332 (4700 ± 300) | 7.1 ± 3.0 (527) | 1.9 ± 0.1 | 0.4 | 0.6 |
n.d. = not determined due to weak or undetectable luminescence intensity.
Luminescence lifetime measured in air-equilibrated pH 7.4 PBS.
Luminescence lifetime measured in nitrogen-saturated pH 7.4 PBS.
Representative spectra of the phen complexes (PTA, THP, DAPTA)-1 and the bpy complexes (PTA, THP, DAPTA)-3 are given in Figure 2. The overall band shape and structures of the absorbance profiles appear to be dependent predominantly on the nature of the diimine ligand rather than the phosphine. For example, the spectra of PTA-1, THP-1, and DAPTA-1 are nearly superimposable, as are those of PTA-3, THP-3, and DAPTA-3. The high-energy (< 325 nm) features in these spectra most likely correspond to intraligand π–π* transitions, and are therefore expected to be only dependent on the nature of the diimine ligand. The low energy features correspond to the metal-to-ligand charge transfer (MLCT) transition,63,93,103–108 a state that dominates the photochemical properties of Re(CO)3 complexes. Similarly, the energy of the MLCT transition is almost independent of the nature of the phosphine. This result suggests that the phosphine ligands only weakly perturb the energies of metal-based donor orbitals, so that variations in the MLCT bands can be attributed to differences in the ligand π* orbitals. For the phen complexes (1 series), the MLCT band is centered at 367 nm; for the bpy complexes (3 series) this band occurs at 345 nm, consistent with the higher energy π* orbitals of this less conjugated ligand.
Figure 2.
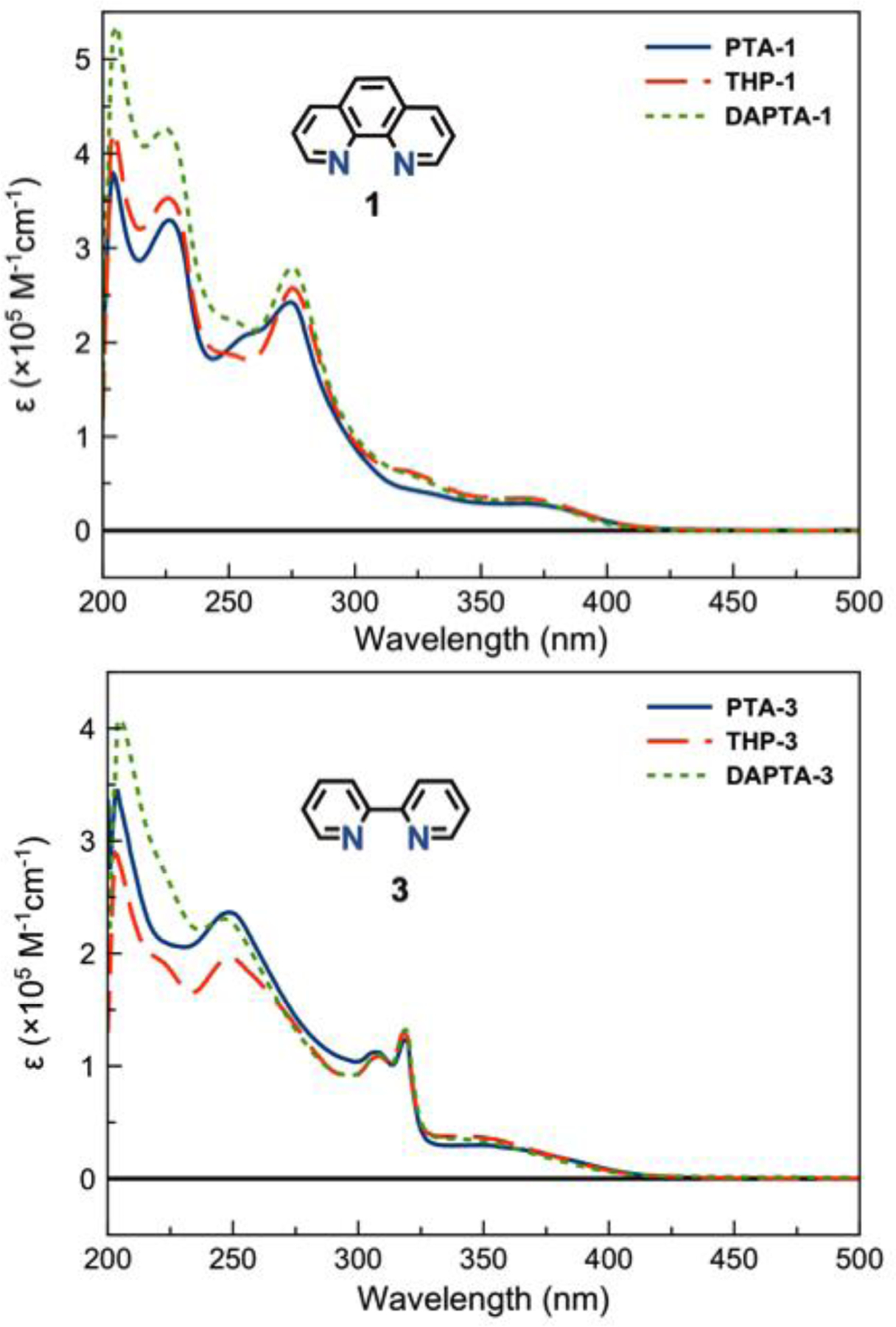
UV-vis spectra in pH 7.4 PBS solutions for complexes (PTA, THP, DAPTA)−1 (top) and (PTA, THP, DAPTA)−3 (bottom).
The luminescence properties of the complexes were also evaluated (Figures S108–S112). Representative emission spectra for complexes (PTA, THP, DAPTA)-1 are shown in Figure 3. All complexes bearing the THP and DAPTA ligands were emissive in air-equilibrated pH 7.4 phosphate-buffered saline (PBS). The quantum yields for emission under these conditions ranged from 3.4–6.1% for the THP complexes, depending on the nature of the diimine ligand. The DAPTA complexes were generally more emissive with quantum yields ranging from 7.1–11.5%. Notably, complexes of the PTA ligand were effectively non-emissive under these conditions. The energy of the emission, which arises from the 3MLCT state,109,110 varies with both the nature of the diimine ligand and the phosphine. For example, DAPTA complexes give rise to higher emission energies compared to the analogous THP complexes (Table 1). This result indicates that the phosphine ligand plays a larger role in the 3MLCT compared to the 1MLCT state that was probed by the absorption spectra, as described above. The excited state lifetimes were also measured in both air-equilibrated (τair) and deoxygenated PBS (τN2) (Table 1, Figures S113–S117), and these range from 0.2 to 2.0 μs for the air-equilibrated PBS solutions, consistent with triplet-based MLCT excited state lifetimes measured for similar rhenium complexes.62,93 Notably, the lifetimes of these complexes are extended under nitrogen atmosphere, suggesting that O2 is an effective quenching agent.
Figure 3.
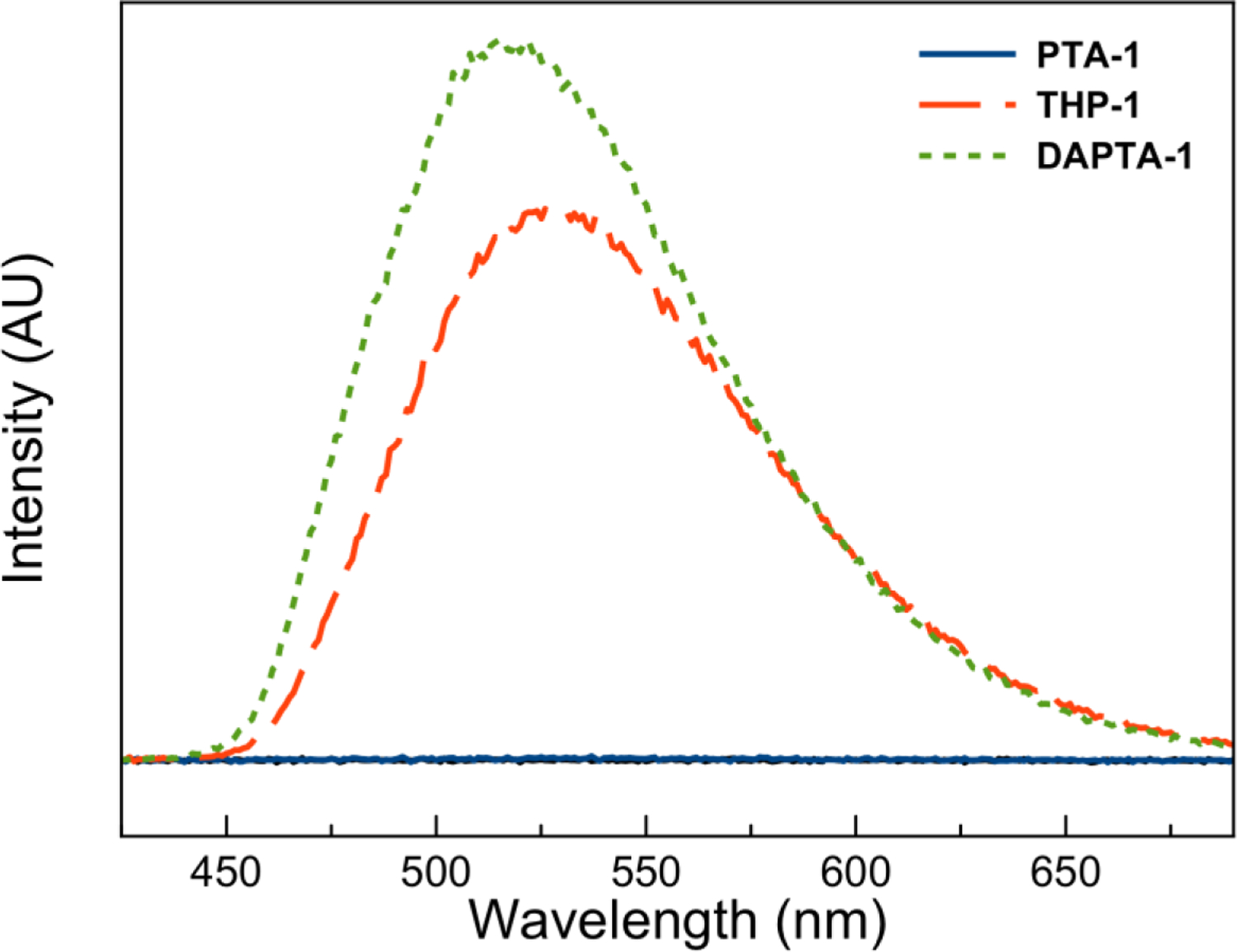
Emission spectra in pH 7.4 PBS (excitation wavelength 350 nm, concentration 30 μM) for complexes PTA-1 (solid blue line), THP-1 (large dash red line), and DAPTA-1 (small dash green line).
Photolysis of the axial CO ligand was investigated by monitoring the UV-vis spectral profiles of the compounds in pH 7.4 PBS as they were irradiated with 365 nm light (Figures S118–S132). Representative spectral changes of PTA-1, THP-1, and DAPTA-1 upon irradiation are shown in Figure 4. Whereas all complexes of PTA were not photoreactive at this excitation wavelength, the THP and DAPTA complexes exhibited a marked change in their UV-vis spectra. We note that, in contrast to our results, PTA-1 was previously reported to undergo CO loss upon excitation at 365 nm.83 We only observed photoreactivity of the PTA complexes when they were irradiated with higher energy UVB light (280–300 nm). Upon irradiation of the THP and DAPTA complexes, the bands near 340–360 nm decayed with the concomitant formation of a new broad feature at lower energies. In all cases, the photoconversion of these complexes proceeded cleanly, as evidenced by the presence of well-defined isosbestic points. Furthermore, gas chromatographic analysis of three representative compounds showed that one equivalent of CO is released per molecule (Table S3).
Figure 4.
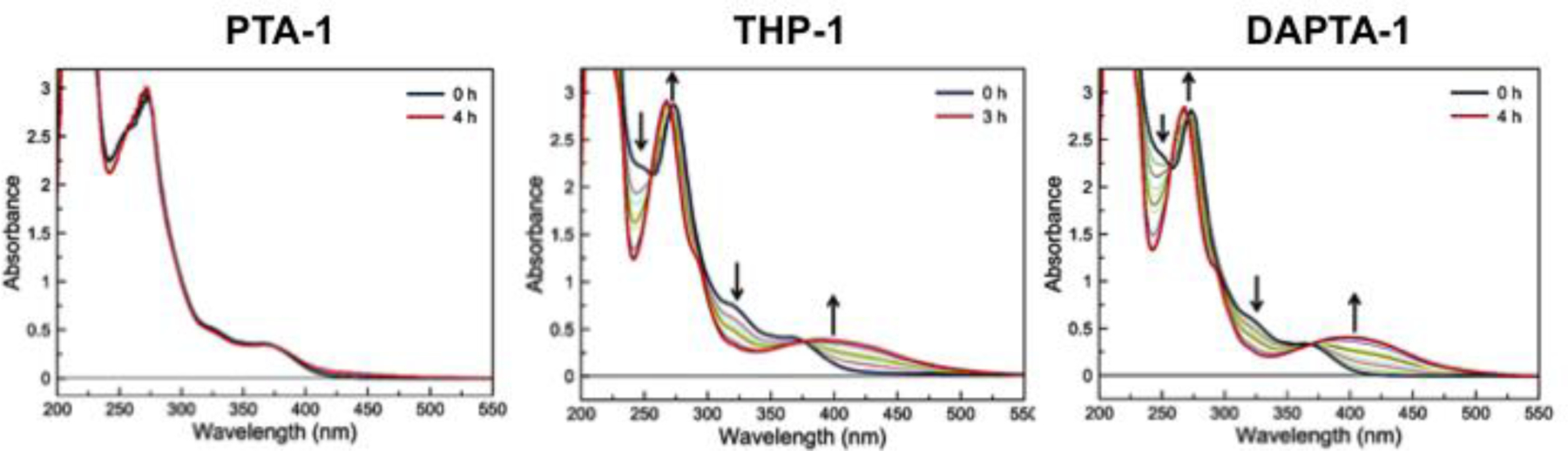
UV-vis spectra of the photoreactions of PTA-1 (110 μM, left), THP-1 (115 μM, middle), and DAPTA-1 (105 μM, right) in pH 7.4 PBS over a 4 h irradiation period with 365 nm light.
Photochemical quantum yields for these compounds were measured via ferrioxalate actinometry and are given in Table 1. The non-luminescent PTA complexes were generally not photoreactive at this excitation wavelength; upper estimates for their photoreaction quantum yields are less than 0.1%. By contrast, the DAPTA and THP complexes exhibited quantum yields ranging from 1.1 to 5.5%. These values are substantially lower than those previously measured for related rhenium phosphine complexes in degassed organic solvents;62,68 the use of aerated aqueous solutions in this case may lead to the lower quantum yields measured for the compounds described here.
Because PDT agents act by generating singlet oxygen, we investigated the 1O2-sensitization quantum yields (ΦΔ) for the fifteen rhenium complexes in air-equilibrated pH 7.4 PBS solutions (Table 2). Upon irradiation with 365 nm light, 1O2 was quantified via the N,N-4-dimethyl-nitrosoaniline/histidine assay following previously reported procedures.59,111 The quantum yields were determined in reference to those of phenalenone (ΦΔ = 0.98). All PTA compounds exhibited no significant production of 1O2 upon irradiation. This result suggests that the excited state quenching of these complexes occurs in an O2-independent manner. The THP and DAPTA complexes produce 1O2 with low to high quantum yields; DAPTA-2 exhibits the largest quantum yield of 70%, whereas both THP-4 and DAPTA-4 do not give rise to detectable quantities of 1O2 upon irradiation.
Table 2.
Singlet Oxygen Quantum Yields
| Compound | ΦΔ, % |
|---|---|
| PTA-1 | <1 |
| THP-1 | 48.5 ± 0.5 |
| DAPTA-1 | 54.0 ± 1 |
| PTA-2 | <1 |
| THP-2 | 45.5 ± 0.5 |
| DAPTA-2 | 70.5 ± 2.5 |
| PTA-3 | <1 |
| THP-3 | 8.7 ± 0.1 |
| DAPTA-3 | 9.1 ± 0.3 |
| PTA-4 | <1 |
| THP-4 | <1 |
| DAPTA-4 | <1 |
| PTA-5 | <1 |
| THP-5 | 15.0 ± 0 |
| DAPTA-5 | 12.5 ± 0.5 |
Reinvestigation of the Photophysical Properties of THP-3.
The photophysical properties of the compound THP-3 were described in one of our previous publications.68 In this earlier study, a luminescence quantum yield of 18% and a photochemical quantum yield of 21% were reported. Additionally, the dicarbonyl photoproduct of THP-3 was found to exhibit photoluminescence with a maximum emission wavelength of 585 nm. Upon reanalysis of THP-3 in this investigation, we have found substantially lower luminescence and photochemical reaction quantum yields of 6.1% and 1.6%, respectively. Furthermore, the dicarbonyl photoproduct, obtained by exhaustive photolysis of THP-3, was found to be nonemissive. These contradictory results prompted further investigations of this compound in both our labs at Cornell and at UC Santa Barbara.
As originally reported, THP-3 was isolated as the triflate salt,68 rather than the TFA salt. The triflate salt of THP-3 was independently prepared as previously described, and characterized by elemental analysis and X-ray crystallography, which gave unit cell parameters that matched the previous report. The photophysical properties were investigated using identical conditions to those employed in the original study with equipment and facilities at UC Santa Barbara. The photoluminescence quantum yield was determined using rhodamine B (Φlum = 65%) as a reference standard. Quantum yields in pH 7.4 PBS in deaerated and air-equilibrated solutions were determined to be 9.7% and 8.0%, respectively (Figure S133). The latter value is in reasonable agreement with that measured at Cornell for the TFA analogue of THP-3 under similar conditions. Furthermore, after exhaustive photolysis, no photoluminescence was detected, demonstrating that the triflate counter ion does not alter this aspect of THP-3 either. Lastly, the photochemical reaction quantum yield of this compound was reanalyzed using a FieldMaxII-TO power meter from Coherent with a thermopile photodetector PM10V1 (calibrated) to measure the light intensities. These studies afford a quantum yield of 2.4% (Figure S134), in reasonable agreement with the value of 1.6% obtained at Cornell. These photoreaction quantum yields are approximately a factor of ten smaller than the value previously reported,68 and we speculate that a simple calculation error may account for the error in the original measurement. This also may be the case for luminescence quantum yield, although another source of such an error would be a partially oxidized rhodamine B standard. The presence of an impurity in the original samples of THP-3 may explain the claimed photoproduct luminescence.68 A likely candidate is fac-[Re(CO)3(bpy)(OH2)]+, as this complex is formed by aquation of the fac-[Re(CO)3(bpy)(THF)]+, the starting material for THP-3. We have independently prepared and characterized fac-[Re(CO)3(bpy)(OH2)](OTf) and shown it to have an absorption spectrum close to that of THP-3 (Figure S135) and to exhibit weak luminescence in the region of ~580 nm (Figure S136). These results underline the importance of confirming the purity of all compounds prior to photophysical measurements due to the sensitivity of these measurements to small quantities of photoactive impurities.
Density Functional Theory.
DFT calculations were employed to gain a deeper understanding of the photophysical properties of these compounds. The phen compounds, PTA-1, THP-1, and DAPTA-1, were explored as a representative subset of all of the complexes. Geometries were optimized at the BP86 level112 of theory in the gas phase, and vertical singlet excited state energies were calculated with time-dependent DFT (TD-DFT) employing a solvent continuum dielectric model for water113 and the hybrid functional PBE0. A map of the lowest energy singlet excited states of these complexes is given in Figure 5. For THP-1 and DAPTA-1, the lowest energy singlet excited states are MLCT in character, as can be discerned from the difference density maps that show a shift of electron density from the metal to the phenanthroline ligand in the excited state. By contrast, the four lowest energy singlet excited states for PTA-1 cannot be reasonably described as MLCT states; the difference density maps of these excited states represent N 2p → L π* transitions, or ligand-to-ligand charge transfers where the N 2p electrons are initially localized on the PTA phosphine. The fifth excited state of PTA-1 is the definitive MLCT, as indicated by the difference density map. Because the MLCT is generally the state which mediates the luminescence and facilitates internal conversion to the photoreactive ligand field excited state, the presence of four lower energy N 2p → L π* states may enhance non-radiative and non-reactive decay pathways to the ground state. One can alternatively think of this as a photoinduced electron transfer (PeT) quenching mechanism, one that commonly employs modulation of nitrogen lone pair orbital energies to elicit a fluorescent response.114 The efficient quenching of the MLCT due to the nitrogen lone pairs of the PTA complexes would, therefore, explain the lack of observed luminescence for this class of compounds. Furthermore, the lack of photosubstitution is also a consequence of the efficient quenching of the 1MLCT state, which must occur more rapidly than intersystem crossing to states that would enable these photoreactivities.62 We note that the DAPTA ligand likewise possesses an N 2p lone pair that may also be available for PeT quenching. We hypothesized that the presence of the electron withdrawing acetyl groups increase the redox potential of the nitrogen lone pair, such that PeT quenching is less thermodynamically viable. This hypothesis was further supported by examining the DFT energies of the nitrogen lone pair orbitals of PTA-1 and DAPTA-1. The energy of this orbital in PTA-1 is higher than that of DAPTA-1 by approximately 0.3 eV. As such, the nitrogen lone pair in DAPTA-1 is less susceptible to oxidation, rendering PeT quenching less thermodynamically favored.
Figure 5.
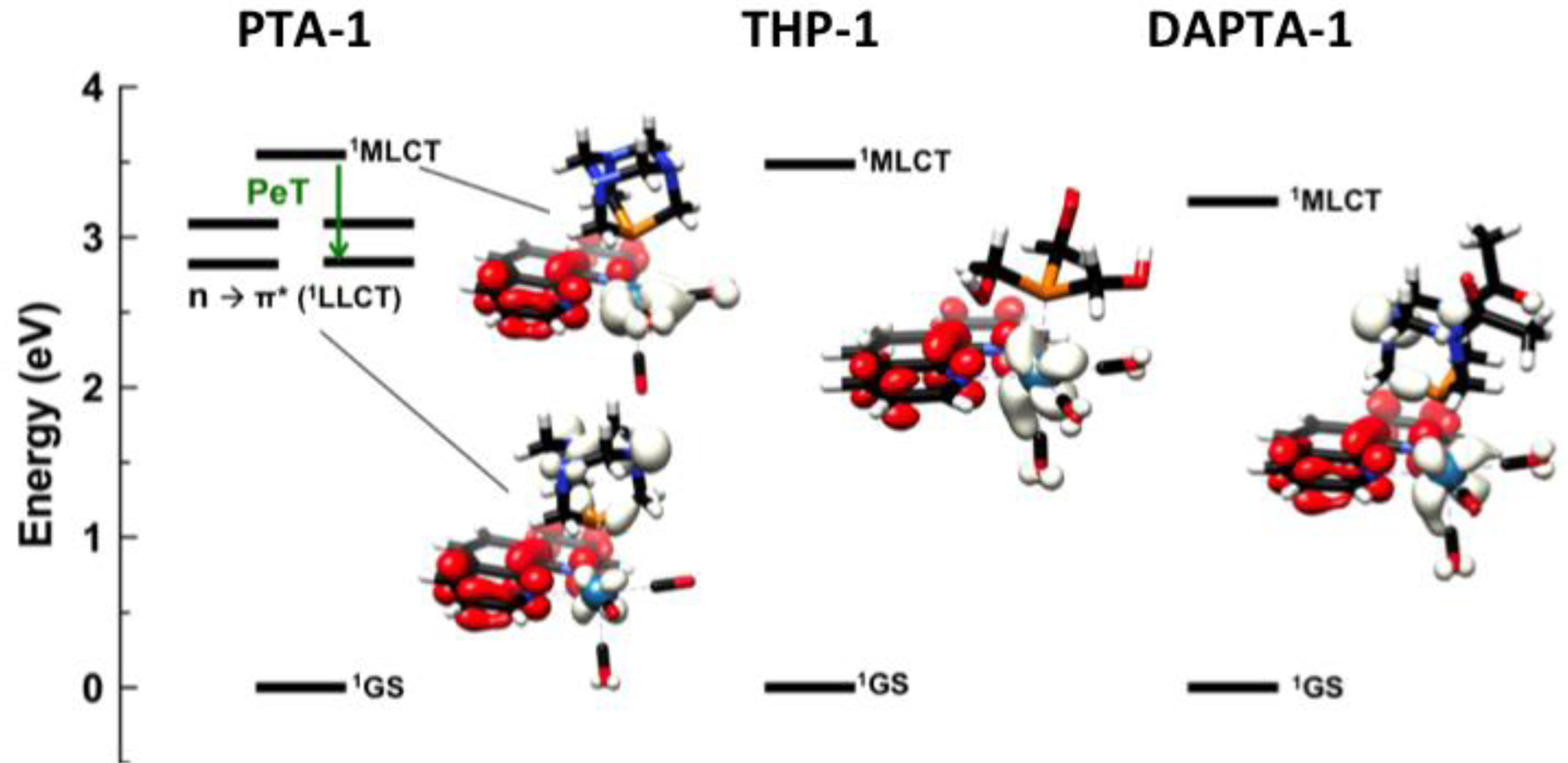
State energy diagram of PTA-1 (left), THP-1 (middle), and DAPTA-1 (right), and the corresponding electron density difference maps for relevant excited states. Negative (white) and positive (red) electron density illustrates the transitions.
Photoinduced Anticancer Activity.
To rapidly screen for photoactivated in vitro anticancer activity, HeLa cells were treated with the fifteen compounds at a single dose of 200 μM. The cells were incubated for 4 h with the rhenium compounds, and then irradiated with 365 nm light for 30 min. Another batch of cells, used as the non-irradiated control, was subjected to the same conditions in the absence of light. The viability of the cells in the presence of the 15 different compounds under the conditions of light irradiation and dark are shown in Figure 6 and Table S10. All compounds at this concentration level, with the exception of THP-2, THP-5, and DAPTA-5 exhibited little or no toxicity in the dark (within experimental uncertainty). Upon irradiation, all of the THP and DAPTA complexes, with the exception of THP-3 for which that effect was much smaller, induced significant cell death, indicating that these compounds are potential PDT or PACT agents. Consistent with their lack of photoreactivity, none of the PTA complexes exhibited significant cytotoxicity when exposed to light.
Figure 6.
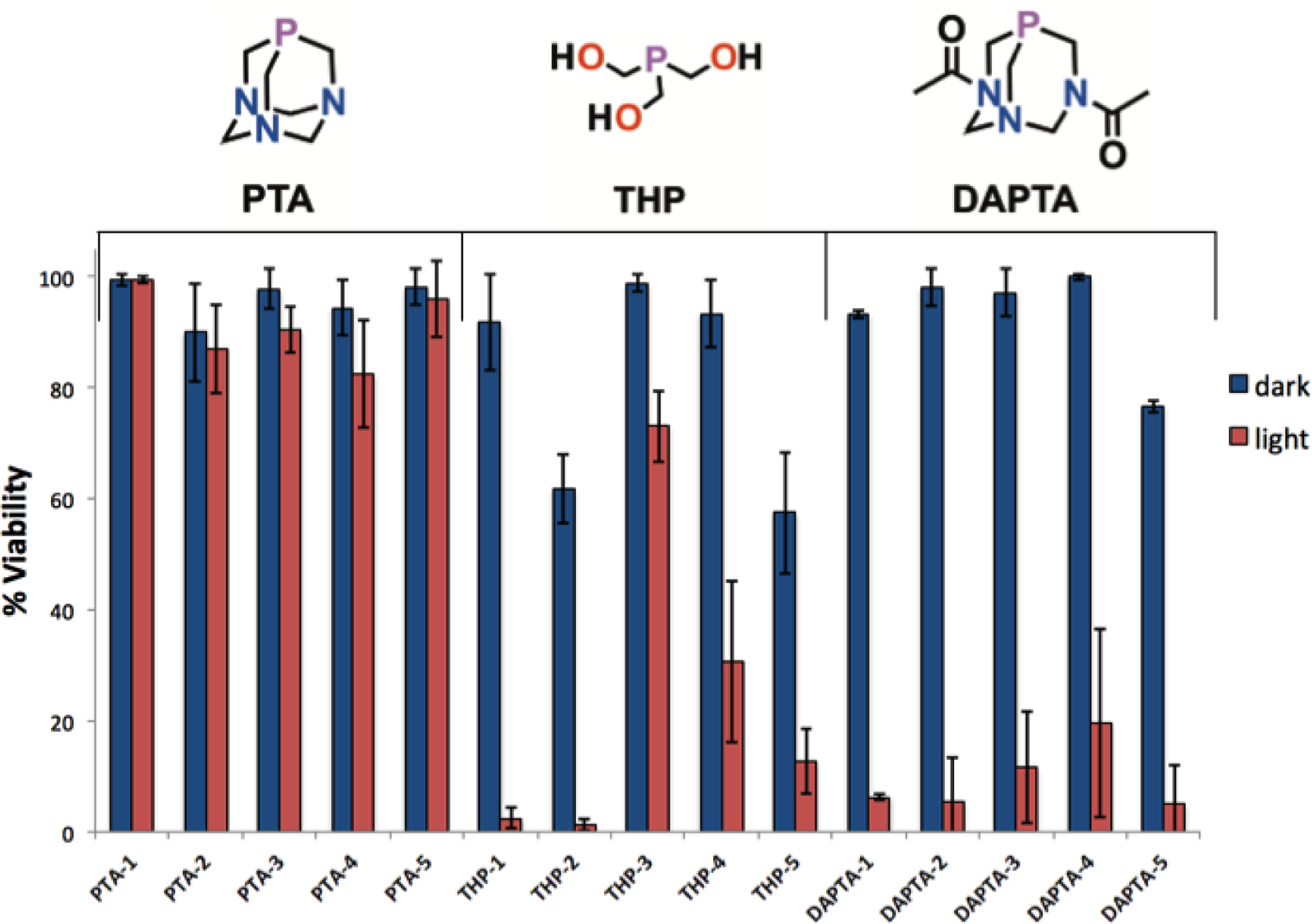
Single-dose (200 μM) cell viability of HeLa cells for all complexes. The compounds were tested in triplicate under both light and dark conditions. For light-irradiated samples, an irradiation time 30 min with 365 nm light at a photon flux (2.38 ± 0.31) × 10−10 Einsteins/s was employed.
Based on this initial screening, the most potent compounds, THP-1, THP-2, THP-5, and all DAPTA complexes, were further evaluated in full dose-response studies to determine 50% growth inhibitory concentration (IC50) values in HeLa cells. These values are collected in Table 3 (Figures S140–S148). Consistent with the single-dose studies, the compounds were all non-toxic in the absence of light. Under conditions of light irradiation at 365 nm, the compounds exhibited enhanced cytotoxicity. Notably, THP-2 and DAPTA-1 possess IC50 values that are less than 10 μM. The ratio of IC50 values in the dark over those determined upon irradiation give phototoxicity indices (PI) that are >34 for DAPTA-1 (Figure 7) and >21 for THP-2. The exact phototoxicity indices could not be determined because the compounds do not induce cell death in the dark at the highest concentration (200 μM) screened.
Table 3.
IC50 values in HeLa (cervical cancer) cells in the absence and presence of 365 nm light.
| Compound | IC50 Value (μM) |
||
|---|---|---|---|
| HeLa |
PIa | ||
| Dark | Light | ||
| THP-1 | >200 | 26.4 ± 9.2 | >7.6 |
| DAPTA-1 | >200 | 5.9 ± 1.4 | >33.9 |
| THP-2 | >200 | 9.6 ± 4.2 | >20.8 |
| DAPTA-2 | >200 | 19.2 ± 2.9 | >10.4 |
| DAPTA-3 | >200 | 14.9 ± 3.2 | >13.4 |
| DAPTA-4 | >200 | 60.3 ± 18.2 | >3.3 |
| THP-5 | 72.6 ± 23.2 | 68.0 ± 4.3 | 1.1 |
| DAPTA-5 | >200 | 24.3 ± 9.1 | >8.2 |
Phototoxic index (PI) is the dark IC50 value divided by the light IC50 value. For light-irradiated samples, an irradiation time 1 h with 365 nm light at a photon flux (2.38 ± 0.31) × 10−10 Einsteins/s was employed.
Figure 7.
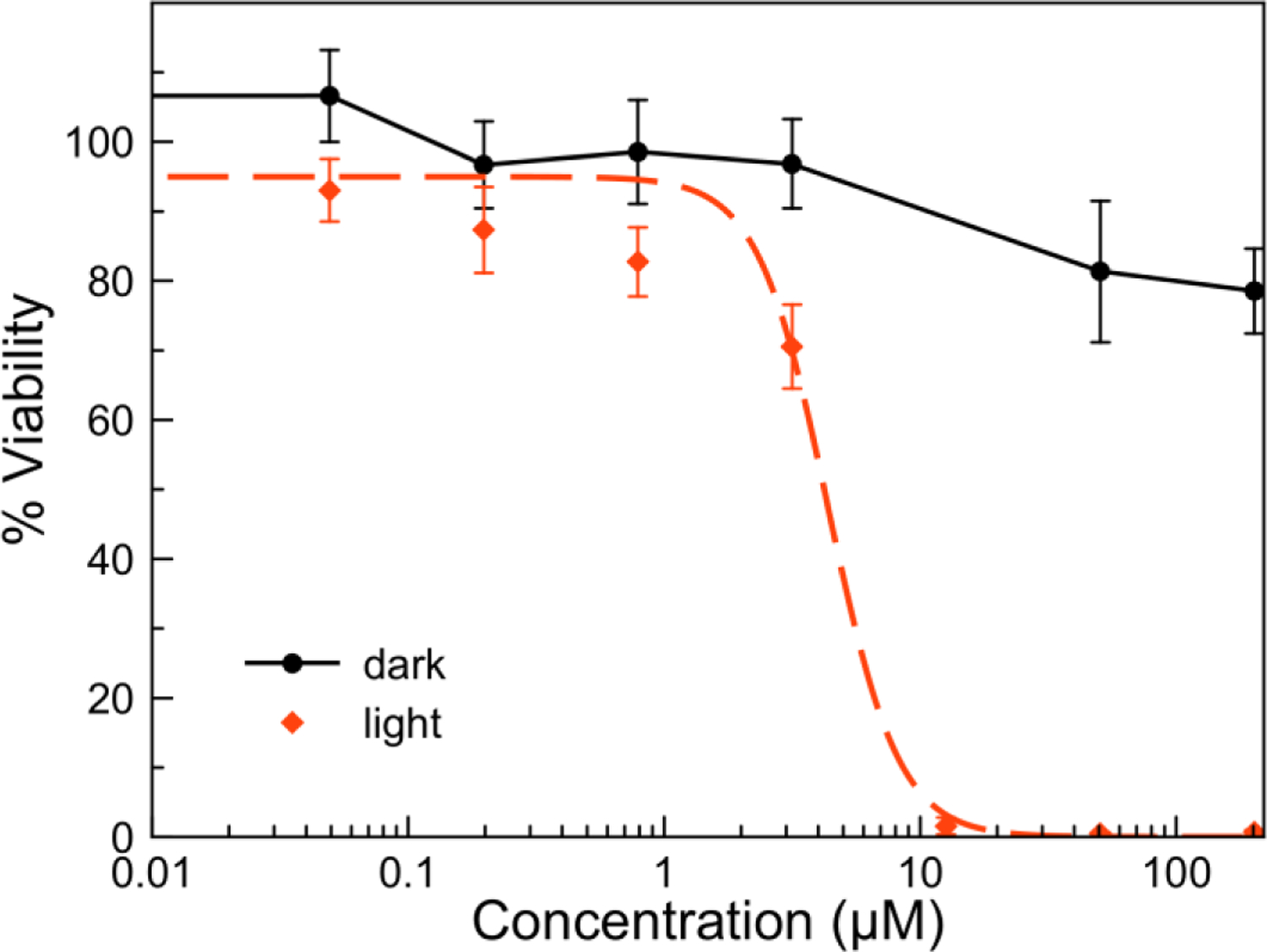
Dose-response curves for DAPTA-1 in HeLa cells in the presence (red dashed) and absence (black solid) 365 nm light irradiation. For light-irradiated samples, an irradiation time 1 h with 365 nm light at a photon flux (2.38 ± 0.31) × 10−10 Einsteins/s was employed.
DAPTA-1 and THP-1 were further evaluated in wild-type (A2780) and cisplatin-resistant ovarian (A2780CP70) cancer cell lines (Table 4, Figures S149–S152). THP-1 exhibited enhanced cytotoxic effects in the presence of light in both A2780 and A2780CP70 cell lines. The IC50 values of the irradiated THP-1, however, are approximately 6-fold higher in the cisplatin-resistant cell line, indicating that this photoactivated product is susceptible to platinum resistance mechanisms. By contrast, DAPTA-1 gave rise to light-induced IC50 values of 2.1 and 3.2 μM in the A2780 and A2780CP70 cell lines, indicating that this compound can circumvent cisplatin resistance.
Table 4.
IC50 values in A2780 (ovarian) and A2780CP70 (ovarian cisplatin-resistant) cells in the absence and presence of 365 nm light.
| Compound | IC50 Value (μM) | |||||
|---|---|---|---|---|---|---|
| A2780 |
PI | A2780CP70 |
PIa | |||
| Dark | Light | Dark | Light | |||
| THP-1 | >200 | 4.6 ± 1.4 | 43.5 | >200 | 29.9 ± 7.7 | 6.7 |
| DAPTA-1 | >200 | 2.2 ± 1.1 | 90.9 | >200 | 3.2 ± 0.7 | 62.5 |
| Cisplatin | 0.18 ± 0.07 | – | – | 5.14 ± 1.1 | – | – |
Phototoxic index (PI) is the dark IC50 value divided by the light IC50 value. For light-irradiated samples, an irradiation time 1 h with 365 nm light at a photon flux (2.38 ± 0.31) × 10−10 Einsteins/s was employed.
Identifying the Cytotoxic Species.
Upon photolysis of this class of compounds in water, both CO and the rhenium dicarbonyl complexes fac-[Re(NN)(PR3)(CO)2(OH2)]+ are released, and singlet oxygen is produced. We hypothesized that the resulting dicarbonyl rhenium photoproduct could induce cytotoxicity by interacting covalently with biomolecules, in a similar manner to the corresponding tricarbonyl aqua species that we have studied.78 To explore this possibility, we synthesized two dicarbonyl analogues, one of DAPTA-1 and the other of PTA-3, for further evaluation of their anticancer activity. The dicarbonyl analogue of DAPTA-1 was pursued because DAPTA-1 is the most potent compound in our library. By contrast, the dicarbonyl analogue of PTA-3 was investigated because this compound exhibits no phototoxicity; this compound would enable us to determine if the dicarbonyl species could elicit a cytotoxic response in the absence of CO release. The target compounds, fac-[Re(phen)(CO)2(DAPTA)Cl] (DAPTA-1A) and fac-[Re(bpy)(CO)2(PTA)Cl] (PTA-3A) were synthesized in two steps from DAPTA-1 or PTA-3 (Scheme 3). Following previously reported procedures,115–118 DAPTA-1 or PTA-3 was treated with trimethylamine-N-oxide (TMAO) and NEt4Cl, and heated to reflux in a mixture of CH2Cl2 and CH3OH. This reaction induces the liberation of the axial CO ligand as CO2, providing the open coordination site that is then occupied by the chloride ions in solution to yield DAPTA-1A or PTA-3A as red solids. The compounds DAPTA-1A (Figures S16, S33, S65, S82) and PTA-3A (Figures S17, S34, S66, S83) were characterized by conventional spectroscopic methods. The chlorido complexes were sufficiently soluble for biological studies after stirring them in water for several hours; their dissolution is hypothesized to occur with concomitant aquation of the chlorido ligand. Photolyzed solutions of DAPTA-1 give rise to a peak with the same retention time on HPLC as DAPTA-1A (Figure S153). Furthermore, solutions of photolyzed DAPTA-1 exhibit the same UV-vis (Figure S154) and 1H NMR spectra (Figure S155) as DAPTA-1A, confirming this compound to be the photoproduct.
Scheme 3.
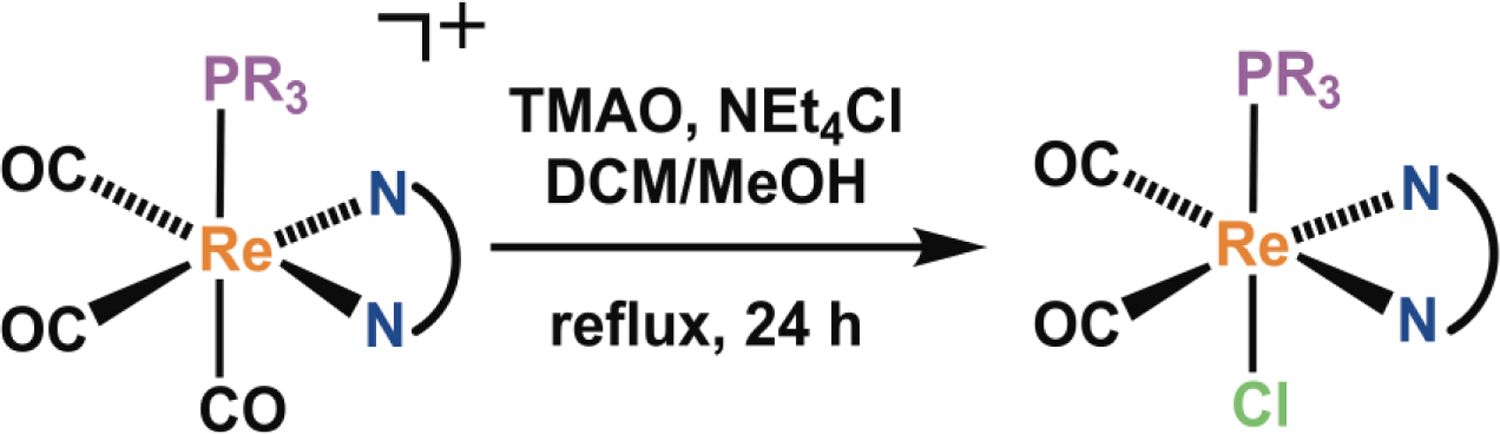
Synthetic scheme for the photoproducts, DAPTA-1A and PTA-3A.
From the photolysis of DAPTA-1 in D2O (Figure S155), single crystals of an aqua analogue of DAPTA-1A, fac-[Re(phen)(DAPTA)(CO)2(OD2)]+ (DAPTA-1A-aqua) precipitated. Crystals of PTA-3A were obtained by vapor diffusion of diethyl ether into N,N-dimethylformamide (DMF). The structures of these complexes were determined by single-crystal X-ray diffraction (Figure 8). The equatorial Re–CO distances are approximately 0.06 Å shorter than those observed in DAPTA-1 and 0.03 Å shorter for PTA-3. The decrease in distance presumably arises from the loss of a π-accepting CO ligand. PTA-3A also exhibits a notably smaller Cl–Re–P angle of 168.77(3)°, significantly deviating from linearity. The O–Re–P distance in DAPTA-1A-aqua is also considerably bent at an angle of 171.70(9)°. This axial ligand bend represents an interesting deviation from the corresponding angles found in DAPTA-1 and PTA-1 and other complexes of the type fac-[Re(CO)3(NN)X], with C–Re–X angles between 174–178°.102,119 A similar rhenium phosphine compound, fac-[Re(CO)2(NN)(PR3)Cl] (PR3 = P(OEt)3), reported exhibits a Cl–Re–P angle of 173°.117
Figure 8.
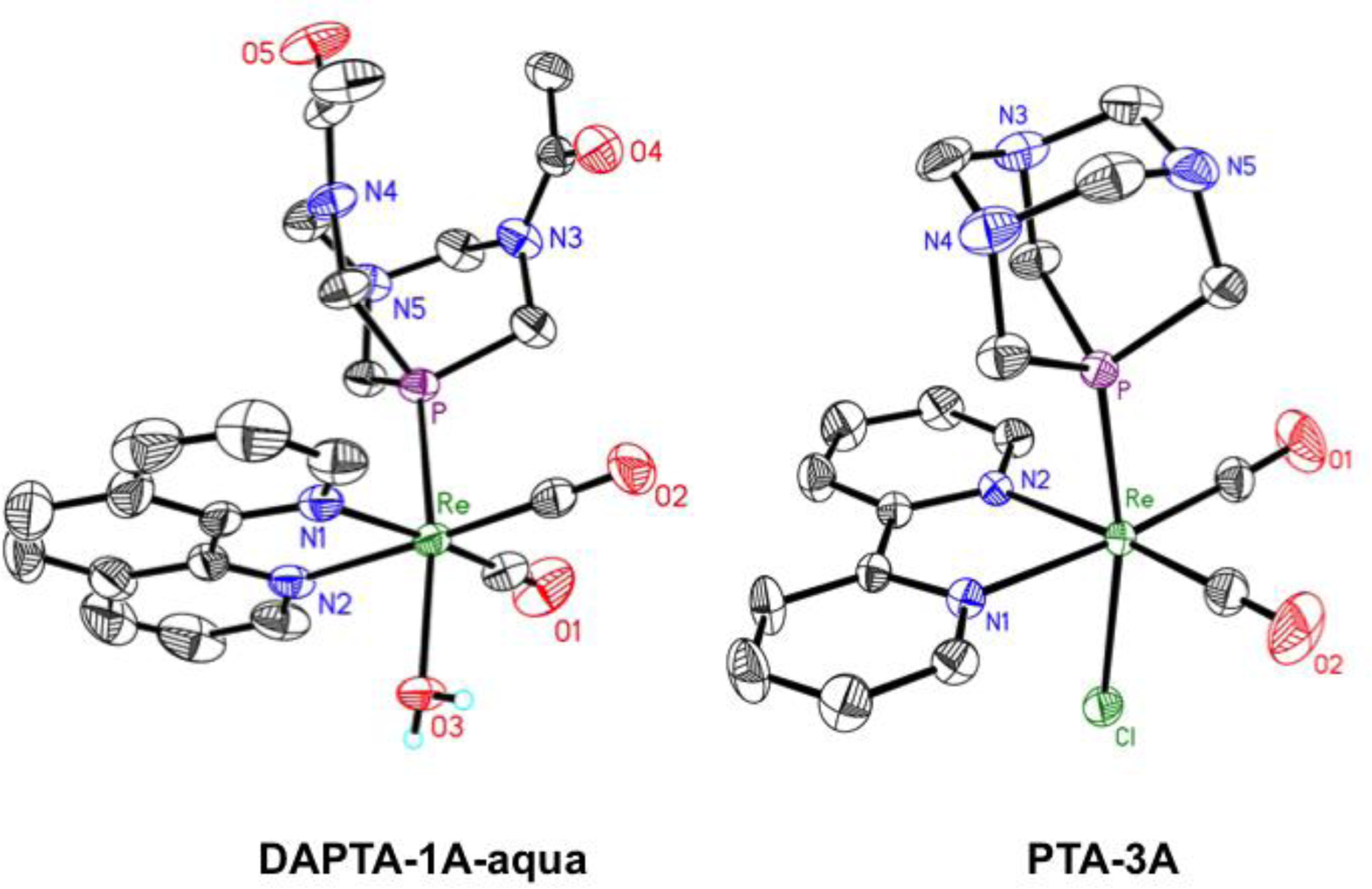
X-ray crystal structure of DAPTA-1A-aqua (left) and PTA-3A (right). Bond lengths and angles are reported in Table S2. Ellipsoids are drawn at the 50% probability level. H atoms bound to C atoms and counterions are omitted for clarity.
With purified DAPTA-1A and PTA-3A in hand, the anticancer activity of these photoproducts was analyzed. PTA-3A (Figure S156) was non-toxic in HeLa cells up to a 200 μM in the dark. DAPTA-1A (Figure 9, Figure S157) exhibited a mild cytotoxic effect at concentrations >50 μM. Additionally, solutions of DAPTA-1 after its exhaustive photolysis gave rise to a similar cytotoxic response as DAPTA-1A (Figure S158), supporting the toxic effects of this dicarbonyl complex. The cytotoxicity of PTA-3A and DAPTA-1A were further investigated under conditions of UVA light irradiation to probe for additional phototoxicity of these complexes. Neither complex, however, induced additional cytotoxic effects in the presence of light. Taken together, these results indicate that the phototoxicity of DAPTA-1 could arise only in part from the dicarbonyl photoproduct and that additional cytotoxicity is elicited through a separate mechanism. The lack of cytotoxicity of PTA-3A is surprising because related rhenium carbonyl complexes with a labile coordination site bind covalently to biomolecules75–77 and induce cell death.78,120–122 Although, we only investigated DAPTA-1A and PTA-3A, we hypothesize that the phototoxicity of the other compounds studied here could arise in part from the dicarbonyl product.
Figure 9.
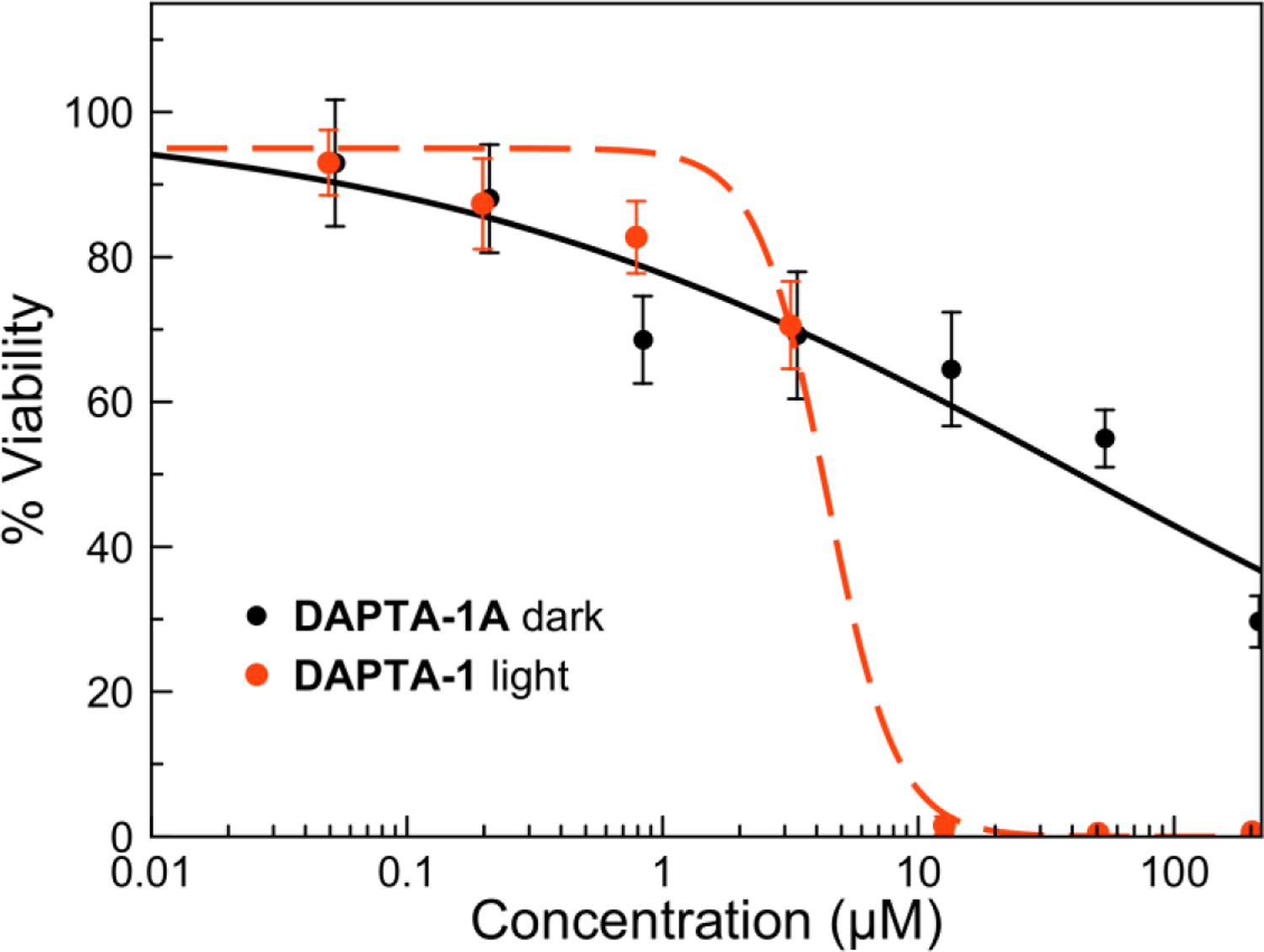
Dose-response curve for DAPTA-1A (black solid) in HeLa cells in the absence of light and DAPTA-1 (red dash) under 365 nm light irradiation.
The limited toxicity of these dicarbonyl complexes could possibly be attributed to their different cellular uptake and intracellular distribution compared to the tricarbonyl phosphine precursors. Upon photolysis in aqueous solution, dicarbonyl aqua complexes, like DAPTA-1A-aqua (Figure 8), are formed. When coordinated to metal centers, the pKa of water decreases substantially. Depending on their pKa values, the photogenerated dicarbonyl complexes could exist either as the charge-neutral hydroxides or as the cationic aqua complexes at physiological pH. On the basis of charge, such species would be expected to have different cellular uptake and localization. To investigate the relative abundances of these species, the pKa values of aquated solutions of DAPTA-1A, PTA-3A, and the photoproduct of THP-1 were determined. These solutions were titrated with base as the pH and UV-vis spectra were monitored. Upon basification, a shift in the absorbance spectra from approximately 370 to 430 nm was observed. This shift arises from the deprotonation of the aqua complex. Based on this data, the pKa values for the dicarbonyl aqua photoproducts of DAPTA-1, THP-1, and PTA-3 are 9.51, 10.07, and 8.75, respectively (Figures S159–S164). These values are similar to those measured for related monoaqua transition metal complexes,123,124 and they indicate that at physiological pH the compounds exist as the cationic aqua complexes.
The low cytotoxicity of the rhenium photoproducts, DAPTA-1A and PTA-3A, prompted us to consider the potential role of 1O2 and CO in mediating cell death. Nearly all of the rhenium complexes in this study sensitize the formation of highly toxic 1O2 (Table 2). No clear direct correlation between ΦΔ and the phototoxicity exist, however, indicating that 1O2 sensitization is not the only mechanism of activity of these complexes. To further test this hypothesis, the phototoxicity of DAPTA-1 was investigated under hypoxia, conditions in which 1O2 sensitizers are less effective. HeLa cells were treated with DAPTA-1 with a similar procedure as described above, except the cells were subjected to hypoxic conditions (5% CO2, 95% N2) before and after exposure to 365 nm light for 1 h. Under hypoxic conditions, DAPTA-1 induced moderate phototoxicity characterized by an IC50 value of 20 ± 1.5 μM (Figure S165). This value is somewhat larger than that measured in normoxic conditions (Table 3), suggesting that 1O2 sensitization does play a role in this compound’s mechanism of action. The lack of a complete suppression of phototoxicity under these conditions, however, does verify that other factors, namely the dicarbonyl photoproduct and CO, may also contribute to the observed activity.
A third possible contributor to the complexes’ phototoxicity is CO. Understanding the biological effects of CO is an active area of research. CO at low concentrations can have therapeutic properties as it elicits anti-inflammatory effects and vasodilation.125–136 The application of CO as a cytotoxic agent for the destruction of malignant cells has also been explored.72,79–81,84,137–141 CO most likely kills cells by binding with high affinity to cytochrome c oxidase,142 thereby inhibiting the mitochondrial respiration pathway. Inhibition of the mitochondrial respiration pathway in this manner with CO reduces cancer cell proliferation by metabolically exhausting cells.143 If the cytotoxic effects of these complexes are mediated solely by CO, then the photoactivated anticancer activity could potentially correlate to the quantum yield for CO release. Across the fifteen compounds tested, however, we do not observe this correlation, suggesting that additional factors are in operation. One possible explanation is that the quantum yields measured in buffer are not representative of those in the complex biological media where binding to proteins such as albumin may alter the values in an unexpected manner. For example, the photophysical properties of the organic dye Rose Bengal and TPPS [meso-tetra(4-sulfanatophenyl)porphyrin] are dramatically altered in the presence of albumin and the cell membrane environment.144 Alternatively, differential cell uptake and intracellular localization of these compounds due to their different lipophilicities may play a large role in mediating the cytotoxic effects of the photoreleased CO and would explain the clear lack of a correlation between CO photorelease quantum yield and cytotoxicity.
Conclusions
The structural and photophysical properties of fifteen rhenium tricarbonyl complexes bearing one of three water-soluble phosphines and five different diimine ligands were investigated. THP and DAPTA complexes exhibited relatively efficient quantum yields for luminescence, photosubstitution of CO, and the production of singlet oxygen. The lack of photoreactivity of the PTA complexes was attributed to quenching from the nitrogen lone pair orbitals, via a photoinduced electron transfer mechanism. The photoreactivities of the THP and DAPTA complexes was leveraged to generate PDT or PACT agents that simultaneously released CO and a labile rhenium complex upon exposure to UV light. The complex DAPTA-1 exhibited the most potent cytotoxic effects upon light exposure in HeLa cells and in wild-type and cisplatin-resistant ovarian cancer cells. The dicarbonyl photoproduct, DAPTA-1A, was only moderately cytotoxic, suggesting that an additional mechanism of cell death may be operable, which could be attributed to 1O2 and the released CO. Because no direct correlations between phototoxicity and both 1O2 sensitization and CO photosubstitution reaction quantum yields could be discerned, we believe that more complicated and subtle factors are in operation for this class of compounds. Regardless, these compounds represent a new type of combined PDT and PACT agent that operates under a multi-action mechanism. They can be easily synthesized, are water-soluble, and exhibit high phototoxic indices. Although this class of compounds is activated only by UVA light, they represent an important proof of principle for the development of new phototoxic rhenium compounds. Ongoing efforts are being pursued to shift the excitation wavelength of these complexes to the red where in vivo tissue penetration would be deeper.
Experimental Section
Methods and Materials.
Rhenium carbonyl was purchased from Pressure Chemicals (Pittsburgh, Pennsylvania, USA). Iodobenzene dichloride was synthesized as previously reported.145 The diimine ligands, 2,2’-bipyridine (bpy), 4,4’-dimethyl-2,2’-dipyridyl (dmbpy), 4,4′-dimethoxy-2,2’-bipyridine (dmobpy), 1,10-phenanthroline (phen), and 2,9-dimethyl-1,10-phenanthroline (dmphen) were purchased from either Sigma Aldrich (St. Louis, Missouri, USA), Alfa Aesar (Heysham, England), or Beantown Chemical (Hudson, New Hampshire, USA) and were used as received. [Re(CO)3(phen)Cl], [Re(CO)3(dmphen)Cl], [Re(CO)3(bpy)Cl], [Re(CO)3(dmbpy)Cl], and [Re(CO)3(dmobpy)Cl] were synthesized using a previously reported procedure.88,89 The phosphine ligands 1,3,5-triaza-7-phosphaadamantane (PTA) and tris(hydroxymethyl)phosphine (THP) were purchased from Alfa Aesar (Heysham, England) and Acros (Geel, Belgium), respectively, and were both used as received. The compound 1,4-diacetyl-1,3,7-triaza-5-phosphabicylco[3.3.1]nonane (DAPTA) was synthesized from PTA using a previously reported procedure.86 All solvents were ACS grade or higher. All reactions were carried out under ambient atmospheric conditions without any effort to exclude water or oxygen.
Physical Measurements.
NMR samples were prepared as solutions using either MeOD-d4, DMSO-d6, DMF-d7, and D2O as the solvents. NMR spectra were acquired on a Varian Inova 400 MHz spectrometer. 1H NMR chemical shifts were referenced to residual solvent peaks versus tetramethylsilane (TMS) at 0 ppm or an external standard, 1,4-dioxane at 3.75 ppm in D2O. 19F and 31P NMR spectra were referenced using an external standard of KPF6 in D2O (19F δ = –72 ppm vs CFCl3 at 0 ppm; 31P δ = –145 ppm vs H3PO4 at 0 ppm). Samples for IR spectroscopy were prepared as KBr pellets and were analyzed on a Nicolet Avatar 370 DTGS (ThermoFisher Scientific, Waltham, MA). The HPLC system (LC-20AP) used for purification of the Re-THP complexes (excluding THP-5) consisted of a SPD-20AV UV-vis detector monitored at 270 and 220 nm (Shimadzu, Japan) and a Epic Polar preparative column, 120 Å, 10 µm, 25 cm × 20 mm (ES Industries, West Berlin, NJ) at a flow rate of 14 mL/min using a binary mobile phase containing 0.1% trifluoroacetic acid (TFA) and H2O. The method for purification consisted of 0–5 min at 10% MeOH, followed by a linear gradient of 100% MeOH for over 20 min, then 10% MeOH for 1 min. Analytical chromatography was carried out on a LC-20AT pump with a SPD-20AV UV-vis detector monitored at 270 and 220 nm (Shimadzu, Japan) using an Ultra Aqueous C18 column (100 Å, 5 µm, 250 mm × 4.6 mm, Restek, Bellefonte, PA) at a flow rate of 1 mL/min with a mobile phase containing 0.1% trifluoroacetic acid (TFA) in H2O or MeOH. The method consisted of 5 min at 10% MeOH, followed by a linear gradient to 100% MeOH over 20 min. High-resolution mass spectra (HRMS) were recorded on an Exactive Orbitrap mass spectrometer in positive ESI mode (ThermoFisher Scientific, Waltham, MA) with samples injected as acetonitrile/water solutions with 1% formic acid. Elemental analyses (C, H, N) were performed by Atlantic Microlab Inc. (Norcross, Georgia, USA). UV-visible spectra were recorded on a Cary 8454 UV-vis (Agilent Technologies, Santa Clara, CA) or a Beckman Coulter DU800 UV-vis using 1-cm quartz cuvettes. Lifetime measurements were collected as described below. Phototoxicity experiments were conducted using a UVA handlamp elevated 8 cm from the cells to give a photon flux of (2.38 ± 0.31) × 10−10 Einsteins/s at 365 nm. Analytical photochemical measurements were performed using a Newport Mercury/Xenon Arc Lamp. The light output was modulated using a combination of a Newport heat absorbing glass filter (50.8 × 50.8 mm) with infrared cut-off (Schott KG5 filter glass), a Newport mercury line bandpass filter (25.4 mm, center wavelength 365.0 ± 2 nm), and a Newport visible absorbing filter (50.8 × 50.8 mm, center wavelength 340 nm) made of dark optical glass to isolate monochromatic 365 nm light. Luminescence quantum yield measurements were carried out on a Beckman Coulter DU800 UV-vis and Varian Eclipse Fluorometer.
X-Ray Crystallography.
Single crystals were grown using vapor diffusion of diethyl ether into solutions of methanol or acetonitrile. For compound DAPTA-1A, the crystals were isolated from a solution of D2O, and those for PTA-3A were collected from vapor diffusion of diethyl ether into DMF. Low-temperature (223 K) X-ray diffraction data for PTA-1, THP-1, DAPTA-1, PTA-2, THP-2, DAPTA-2, PTA-3, DAPTA-3, PTA-4, THP-4, DAPTA-4, PTA-5, THP-5, DAPTA-5, DAPTA-1A, and PTA-3A were collected on a Bruker X8 Kappa diffractometer coupled to an ApexII CCD detector with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were solved through intrinsic phasing using SHELXT146 and refined against F2 on all data by full-matrix least squares with SHELXL147 following established refinement strategies.148 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms bound to carbon were included in the models at geometrically calculated positions and refined using a riding model. Hydrogen atoms bound to oxygen were located in the difference Fourier synthesis and subsequently refined semi-freely with the help of distance restraints. The isotropic displacement parameters of all hydrogen atoms were fixed to 1.2 times the U value of the atoms they are linked to (1.5 times for methyl groups). Details of the data quality and a summary of the residual values of all the refinements are listed in Tables S1 and S2.
Photochemistry.
Photochemical reactions were monitored by UV-vis spectroscopy. Stock solutions of the rhenium compounds at 1–2 mM concentrations in PBS (pH 7.4) were diluted in PBS (pH 7.4) to a final volume of 3 ml at a concentration of approximately 40–150 μM. The absorbance of these samples was between 0.1 and 0.5 at 365 nm. The solutions were stirred in a 1-cm quartz cuvette and irradiated with 365 nm light using the UVA handlamp described above. At different time points, the UV-vis spectra were acquired. The photoreaction was deemed complete when no further changes in the UV-vis spectra were observed. For DAPTA-1, the photoreaction was monitored by HPLC (Figure S153), UV-vis (Figure S154) and 1H NMR spectroscopy (Figure S155).
CO Release Measurements.
These measurements were carried out as previously reported.149 Millimolar stock solutions of DAPTA-1, DAPTA-4, and THP-3 were prepared by dissolving a known amount of the respective complex in 10 mL of pH 7.4 PBS. A custom designed Schlenk cuvette71 (internal volume of 23.6 mL) was loaded with 6 mL of the stock solution, a magnetic stir bar, and sealed. The UV-vis spectrum was measured using a Shimadzu UV2401 (PC) spectrophotometer. The sample was then exposed to 365 nm light using an Oriel 200–500 W/Hg Arc lamp filtered by a 365 nm mercury line interference filter. The power of the incident light (50 mW) was measured using a Coherent FieldMaxII-TO Power Meter. After 1 h, the UV-vis spectrum was measured again. This was repeated until there were no further significant absorbance changes at the λmax (~ 400 nm) of the product, indicating that the photoreaction was complete (~3 h).
A gas tight syringe was used to remove a 100 µL sample from the headspace of the cell through a special screw top fitted with a 3-layer laminated silicone GC septum. This sample was analyzed by gas chromatography with thermal conductivity detection (GC-TCD) using an Agilent 6890N gas chromatograph fitting with a Carboxen 1010 PLOT fused silica capillary column (L × I.D. = 30 m × 0.53 mm, average thickness 30 μm). The GC inlet temperature was 230°C. The oven was held at 35 °C for 14 min, then ramped at 20°C/min to 245°C, and finally held at 245°C for 10 min. The retention time for carbon monoxide (CO) is about 11.2 min and carbon dioxide (CO2) is 21 min. Dioxygen and dinitrogen from ambient air have retention times of about 8.2 and 8.4 min, respectively. The ChemStation auto-integration function was used to calculate the peak area for CO.
The amount of CO in the 100 µL sample was calculated using a calibration curve made by injecting known amounts of CO from a Schlenk flask. The moles of CO in the headspace of the Schlenk cuvette (SC moles of CO) was calculated using the following equation. The volume of the headspace for the Schlenk cuvette (SC headspace) is calculated by subtracting the solution volume from the total volume of the Schlenk cuvette.
| (1) |
For CO, the partition coefficient between the gas phase and water is ~50 at ambient temperature (41 mmoles/liter-atm versus 0.84 mmoles/liter-atm), thus essentially all the CO will be in the gas phase. The stoichiometric ratio (moles CO released/mole of photoreacted complex) was calculated by dividing the SC moles of CO by the moles of complex photolyzed. The average and sample standard deviation of three injections is reported. The results are shown in Table S3.
Emission Quantum Yield.
The luminescence quantum yields were measured relative to the standard quinine sulfate (Φ = 0.52, 0.05 M H2SO4), which was cross-referenced in our lab to harmaline (Φ = 0.32, 0.005 M H2SO4).150 An excitation wavelength of 350 nm was used for the samples and standards. The compounds were measured as solutions in pH 7.4 PBS with the absorbance maintained below 0.1 to prevent inner filter effects.150 At least five different concentrations of the samples and standards were measured by UV-vis and fluorescence spectroscopy, and the absorbance at 350 nm was plotted versus the integrated emission intensity. The slopes of the resulting lines were used in the equation:
| (2) |
where Φref is the quantum yield of the reference, quinine sulfate, and S is the slope of either the sample or the reference, and η is the refractive index.
Photochemical Reaction Quantum Yield.
The photochemical reaction quantum yields were determined via potassium ferrioxalate actinometry.151 All solutions were prepared with minimal light exposure and stored in the dark. To determine the photon flux of our lamp, a 1.5 mL volume of potassium ferrioxalate (6.1 mM, 0.05 M H2SO4) was irradiated with monochromatic 365 nm light for 30 seconds with continuous stirring in a 1-cm quartz cuvette. After irradiation, a 0.1 mL aliquot of the ferrioxalate solution was added to 2.9 mL of 1,10-phenanthroline (5.7 mM, 0.5 mM H2SO4) containing sodium acetate buffer (1.8 M, pH 5). This procedure was repeated for an identical potassium ferrioxalate solution kept in the dark to correct for any thermal reactions. The resulting solution was shaken for 30 s and then incubated for 1 min. The absorbance was then recorded for both the irradiated solution and the dark solution. The equation used to determine photon flux is as follows:
| (3) |
where NA is Avogadro’s number, q is photon flux (photons/s), ΔA is the change in absorbance at 510 nm for the irradiated solution minus the dark solution, V1 (ml) is the total volume after dilution, V2 (ml) is the volume irradiated, and V3 (ml) is the volume of irradiated sample diluted into 1,10-phenanthroline, Φ is the quantum yield at 365 nm, ε is the extinction coefficient (M−1 cm−1), l is path length (cm), and t is time (s). For potassium ferrioxalate at 365 nm, Φ = 1.25 and ε = 11,000 M−1 cm−1.151 Photon flux and quantum yields were cross-referenced to the actinometer, Reinecke’s salt (Φ = 0.38 at 365 nm) to verify the accuracy of our measurements.151
The quantum yields of the rhenium compounds were determined by first measuring the photon flux of the light source with potassium ferrioxalate, as described above. The rhenium compound was irradiated in pH 7.4 PBS with the monochromatic 365 nm light. The concentration of these samples were such that the absorbance was greater than 2 at 365 nm, ensuring that all photons were absorbed by the sample. For some samples, a correction factor (1–10−A) was applied to the photon flux if the solution was below an absorbance of 2, where A is the absorbance of the sample at the irradiation wavelength, 365 nm. This correction factor accounts for the fact that in more optically dilute solutions not all photons may be absorbed. After a given time period of irradiation, the sample was diluted in 2.5 ml PBS and the UV-vis absorbance was measured. This diluted sample was subjected to continued irradiation for 0.5–2 h until the photoreaction was complete, enabling us to determine the extinction coefficient of the photoproduct. To determine the quantum yield of the rhenium compounds, equation 3 was applied to determine Φ, using the photon flux determined from the potassium ferrioxalate actinometry. Absorbance change was monitored at 425–450 nm, depending on the sample.
Lifetime Measurements.
Laser excitation for the phosphorescence lifetime measurements was provided by pulsing the 405 nm laser line from a four-line iChrome MLE laser (Toptica Photonics AG, Munich, Germany). The diode laser in the iChrome was triggered by a DG535 Digital Delay/Pulse Generator (Stanford Research, Sunnyvale, CA) at 100 KHz and delivered 100 ns FWHM 405 nm excitation pulses. The 405 nm pulses were fiber-delivered to a sample-filled cuvette and phosphorescence was collected at 90 degrees through second fiber for delivery to a Bialkali photomultiplier tube (HC125, Hamamatsu, Bridgewater, NJ) through a 470 nm long pass filter (HQ470lp, Chroma Technology, Bellows Falls, VT). The time-resolved photon counts were collected in 40 ns time bins using a SR430 Multi-channel scaler (Stanford Research, Sunnyvale, CA). Data was transferred to a PC via the SR430 GPIB bus and fit to the standard exponential decay model using MagicPlot Pro software. Measurements were collected in PBS solutions at 1000 μM. For deoxygenated measurements, nitrogen gas was bubbled into the PBS solutions for 15 min and then the lifetime was determined.
Singlet Oxygen Measurements.
Indirect singlet oxygen measurements were performed using the N,N-dimethyl-4-nitrosoaniline/histidine assay, where histidine is oxidized by singlet oxygen and the oxidized histidine reacts with N,N-dimethyl-4-nitrosoaniline.111 All measurements were performed in PBS solutions containing N,N-dimethyl-4-nitrosoaniline (25 μM), histidine (10 mM) 59 The reaction was irradiated with monochromatic 365 nm light in 1 cm glass cuvettes at 2 min increments, and the bleaching of N,N-dimethyl-4-nitrosoaniline was monitored at 440 nm. The absorbance at 440 nm was plotted against irradiation time. The singlet oxygen quantum yield (Φsample) of the rhenium complexes was determined relative to the quantum yield of phenalenone (Φref = 0.98)152 using the following formula:
| (4) |
Where S is the slope of plot of absorbance vs. time and I is the overlap of the lamp emission spectra and the absorbance spectra using the formula:
| (5) |
Where I0 is the light-flux intensity of the lamp and A is absorbance of the compound at the irradiation wavelength. The standard, phenalenone was cross-referenced to the complex, fac-[Re(CO)3(NNN)]+ where NNN = N,N-bis(quinolin-2-ylmethyl)amino]-5-valeric acid (Re–COOH), reported by Leonidova et al.59 They reported a singlet oxygen quantum yield of 20% in water. Using our experimental setup, we measured a value of 33–37%.
DFT Studies.
All computational calculations were performed using version 3.0.3 the ORCA computational package.153 The resolution of identity (RI) approximation was used throughout for computational efficiency. Geometry optimizations were performed in the gas phase using the functional BP86,112 the basis set def2-TZVP and its auxiliary def2-TZV/J,154 and the zeroth-order regular approximation for relativistic effects (ZORA).155–157 Optimized geometries were subjected to TD-DFT calculations to determine the nature of the relevant excited states.113 For these calculations, the hybrid functional PBE0 functional was employed,158 using the same basis sets and ZORA as described above. For the TD-DFT calculations, the SMD solvation model for water was employed.159 Twenty singlet excited states were calculated for PTA-1, THP-1, and DAPTA-1. All optimized geometry coordinates are listed in the Supporting Information in Tables S4–S6 the lowest energy singlet excited states are summarized in Tables S7–S9, and the frontier Kohn-Sham orbitals are located Figures S137–S139.
Cell Culture and Cytotoxicity.
HeLa (cervical cancer) cell line was obtained from American Type Culture Collection (ATCC) and cultured using Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). A2780 (ovarian cancer) and A2780CP70 (cisplatin-resistant ovarian cancer) cell lines were provided by the Cell Culture Facility of Fox Chase Cancer Center160 (Philadelphia, PA). These cells were cultured as monolayers with Roswell Park Memorial Institute (RPMI)-1640 culture media supplemented with 10% FBS. All cell lines were grown in a humidified incubator at 37 °C with an atmosphere of 5% CO2. Cells were passed at 80–90% confluence using trypsin/EDTA. Cells were tested monthly for mycoplasma contamination with the PlasmoTest™ mycoplasma detection kit from InvivoGen.
The procedure for determining phototoxicity was modified from the OECD Guidelines, except MTT was replaced with Neutral Red for determining cell viability.161 All compounds, except for DAPTA-1A and PTA-3A were dissolved in PBS at pH 7.4 to prepare a 1–2 mM stock solution. DAPTA-1A and PTA-3A were solubilized in water after exhaustive stirring. For single-dose studies, only HeLa cells were used. For both the single dose and full dose response all cells were grown to 80–90% confluence, detached with trypsin/EDTA, seeded in 96-well plates at 2000 cells/well for HeLa cells and 4000 cells/well for A2780 and A2780CP70 cells in 100 μL of growth media, and incubated for 24 h. The medium was removed and replaced with fresh medium (200 μL) containing a single concentration of either PTA, THP, or DAPTA-1, 2, 3, 4, 5 or media. The cells were then incubated for 4 h and then the cells were irradiated with 365 nm for 30 min (one plate under the same dosing conditions was kept outside the incubator in the dark as a control). After exposure to light, the plates were incubated for an additional 44 h, the medium was removed from the wells, and 3-(4,5-dimethylthiazol-2-yl)-2,5-tetrazolium bromide (MTT) in DMEM or RPMI (200 μL, 1 mg/mL) was added. The additional 44 h incubation was performed to ensure that the cells were in the logarithmic growth phase and that the cells had adequate time to regrow after exposure to the complexes. After 4 h, the MTT/DMEM or RPMI solution was removed, and the formazan crystals were dissolved in 200 μL of an 8:1 mixture of DMSO and pH 10 glycine buffer. The absorbance at 570 nm in each well was measured using a BioTek Synergy HT plate reader. Cell viability was determined by normalizing the absorbance of the treated wells to untreated wells. The % viability data shown is an average of three independent experiments with six replicates per concentration. The same procedure was followed as above for the full dose response but instead after 24 h, varying concentrations of the desired compounds were added and they were incubated for 4 h then irradiated for 1 h. Cell viability was again determined by normalizing the absorbance of the treated wells to untreated wells. The concentrations of the compounds versus % viability were plotted to produce the dose-response curves, which were analyzed using a logistic sigmoid function fit with MagicPlot Pro software. The reported IC50 values are the average of three independent experiments with six replicates per concentration level. For hypoxic conditions, the cells were dosed with the rhenium complex then incubated in a hypoxia chamber (Billips-Rothenburg, Inc., Del Mar, CA, USA), which was purged with an atmosphere of 95% N2 and 5% CO2, for 4 h. The chamber was removed from the incubator, purged with the hypoxic gas mixture again, and then irradiated with 365 nm for 1 h. After this irradiation period, the cells were further incubated under hypoxia at 37 °C for 16 h. The plates were then removed from the hypoxia chamber, and allowed to recover under normoxic (5% CO2, 21% O2) conditions at 37 °C for an additional 24 h prior to analysis of cell viability with MTT, as described above. As an additional control experiment to test the effect of the UVA light on the cells, cells in the absence of rhenium complexes were subjected to the same irradiation conditions. The cell viability of the irradiated cells, relative to control cells that were not exposed to light, was always >85%, indicating that the direct light exposure alone has only a minimal effect on the cells.
pKa Measurements.
Stock solutions of DAPTA-1, THP-1, and PTA-3A were made in water at approximately 2 mM and then diluted to 84–89 μM in water. DAPTA-1 and THP-1 were irradiated with 365 nm light until the photoreaction was complete as evidenced by UV-vis spectroscopy. PTA-3A was dissolved directly for analysis. The solutions containing the rhenium complexes were acidified to an approximate pH of 2.0 with perchloric acid. These solutions were titrated with aqueous KOH, and the pH and UV-vis absorbance spectra were measured at each stage of the titration. The titration apparatus consisted of a 100 mL water-jacketed glass vessel at 25 °C. Nitrogen gas was passed through the solution containing 30% KOH during the experiment to exclude any CO2. The electrode was calibrated using standard buffer solutions prior to the experiment. Carbonate-free solutions of KOH were prepared in freshly boiled Milli-Q water and standardized against potassium hydrogen phthalate.162 The spectral changes resulting from the different pH values were used to determined the pKa values using the HypSpec software package.162,163 Species distribution diagrams were plotted using HySS.164 The titration curves were fit from a wavelength range of 360–405 nm. All absorbance spectra and titration curves are in Figures S159–S164.
Synthesis of Re(CO)5Cl.
A mixture of rhenium carbonyl, Re2(CO)10 (1.90 g, 2.91 mmol) and CH2Cl2 (20 ml) was stirred until Re2(CO)10 was completely dissolved. Then iodobenzene dichloride (0.711 g, 2.59 mmol) was added as a solid to the solution, and the reaction mixture was left to stir for 4 h at room temperature. The desired product, a white solid, was filtered and washed with hexanes (20–30 mL). Yield: 1.268 g (68%). IR (KBr, cm−1): 3435 w, 2462 w, 2157 m, 2035 s, 1958 s, 1002 w, 942 w, 912 w, 590 s, and 555 s.
Synthesis of [Re(CO)3(phen)(PTA)]OTf (PTA-1).
A mixture of [Re(CO)3(phen)Cl] (0.224 g, 0.485 mmol) and AgOTf (0.125 g, 0.485 mmol) in THF (50 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid AgCl was removed via vacuum filtration. To the remaining yellow filtrate, PTA (0.0915 g, 0.582 mmol) was added, and the resulting solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimal amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) precipitated the desired product as a yellow solid, which was isolated by filtration and dried in vacuo. Yield: 0.298 g (78%). 1H NMR (400 MHz, MeOD-d4): δ 9.53 (d, 2H, J = 5.2 Hz), 9.00 (d, 2H, J = 8.3 Hz), 8.34 (s, 2H), 8.15 (dd, 2H, J = 5.2, 8.3 Hz), 4.38 (d, 3H, J = 13.1 Hz), 4.26 (d, 3H, J = 13.1 Hz) 3.64 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.56. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –78.24. IR (KBr, cm−1): 3460 m, 2910 w, 2360 m, 2330 m, 2030 s, 1940 s, 1920 s, 1430 m, 1265 m, 1244 m, 1153 m, 1029 m, 970 w, 950 w, 850 w, 726 w, 637 m, 440 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 608.08745 ([M]+, calcd. 608.08558). Anal. Calcd. for PTA-1·CH3OH (C23H24F3N5O7PReS): C, 35.03; H, 3.07; N, 8.88. Found: C, 34.85; H, 3.02; N, 8.81.
Synthesis of [Re(CO)3(phen)(THP)]TFA (THP-1).
A mixture of [Re(CO)3(phen)Cl] (0.100 g, 0.206 mmol) and AgOTf (0.053 g, 0.206 mmol) in THF (24 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid AgCl was removed via vacuum filtration. To the remaining yellow filtrate, THP (0.038 g, 0.309 mmol) was added and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature and then THF was removed by rotary evaporation. The remaining yellow solid was dissolved in a minimum volume of methanol (5 ml) and filtered through Celite. The crude yellow oil (0.167 g) was purified using preparatory RP-HPLC using a CH3OH/H2O gradient (0–5 min, 10% CH3OH; 5–30 min, 10–100% CH3OH). Purified yield: 0.100 g (69%). 1H NMR (400 MHz, MeOD-d4): δ 9.56 (d, 2H, J = 5.2 Hz), 8.90 (d, 2H, J = 8.3 Hz), 8.25 (s, 2H), 8.07 (dd, 2H, J = 5.2, 8.3 Hz), 3.75 (t, 2H), 3.75 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –76.37. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ 2.43. IR (KBr, cm−1) 3420 m, 3210 m, 3070 m, 2830 w, 2780 w, 2650 w, 2040 s, 1930 s, 1910 w, 1670s, 1520 w, 1430 s, 1200 s, 1130 s, 1050 s, 888 m, 856 s, 800 s, 723 s, 635 w, 618 w, 538 m, 500 m, 483 w, 436 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 575.03961 ([M]+, calcd. 575.03763). Anal. Calcd. for THP-1·H2O (C20H19F3N2O9PRe): C, 34.05; H, 2.71; N, 3.97. Found: C, 34.02; H, 2.61; N, 4.03.
Synthesis of [Re(CO)3(phen)(DAPTA)]OTf (DAPTA-1).
A mixture of [Re(CO)3(phen)Cl] (0.200 g, 0.42 mmol) and AgOTf (0.106 g, 0.42 mmol) in THF (47 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid AgCl was removed via vacuum filtration. To the remaining yellow filtrate, DAPTA (0.113 g, 0.46 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and THF was removed by rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried under in vacuo. Yield: 0.255 g (73%). 1H NMR (400 MHz, MeOD-d4): DAPTA complexes show the syn and anti isomers in a 1:5 ratio; however in the aromatic region the isomers are indistinguishable, δ 9.60 (t, 2H, anti + syn), 9.02 (d, 2H, anti + syn), 8.35 (s, 2H, anti + syn), 8.18 (m, 2H, anti + syn), 5.56 (d, anti), 5.04 (d, syn), 4.75 (d, anti) 4.48 (d, syn), 4.41 (d, anti), 4.12 (m, anti), 3.93 (d, anti), 3.78 (d, anti), 3.64 (s, syn), 3.41 (d, syn), 3.34 (d, anti), 3.04 (d, syn), 2.95 (d, anti) 1.94 (s, syn), 1.91 (s, anti), 1.80 (s, anti).19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.54. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –44.80 ( (syn), –48.75 (anti). IR (KBr, cm−1): 3452 m, 2038 s, 1948 s, 1914 s, 1643 s, 1427 m, 1330 m, 1278 m, 1258 m, 1225 w, 1148 w, 1029 w, 987 w, 888 w, 849 w, 802 w, 720 w, 638 s, 540 m, 498 w, 425 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 680.10846 ([M]+, calcd. 680.10671). Anal. Calcd. for DAPTA-1 (C23H24F3N5O8PReS): C, 36.23; H, 2.92; N, 8.45. Found: C, 36.35; H, 2.79; N, 8.66.
Synthesis of [Re(CO)3(dmphen)(PTA)]OTf (PTA-2).
A mixture of [Re(CO)3(dmphen)Cl] (0.243 g, 0.473 mmol) and AgOTf (0.121 g, 0.473 mmol) in THF (54 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid AgCl was removed via vacuum filtration. To the remaining yellow filtrate, PTA (0.111 g, 0.71 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and then the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried in vacuo. Yield: 0.290 g (78%). 1H NMR (400 MHz, MeOD-d4): δ 8.80 (d, 2H, J = 8.4 Hz), 8.23 (s, 2H), 8.13 (d, 2H, J = 8.4 Hz), 4.33 (d, 3H, J = 13.2 Hz), 4.17 (d, 3H, J = 13.2 Hz), 3.42 (s, 6H), 3.34 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.56. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –79.09. IR (KBr, cm−1): 3448 m, 3052 w, 2913 w, 2033 s, 1958 s, 1934 s, 1445 m, 1384 w, 1266 s, 1222 w, 1150 m, 1031 m, 1013 w, 972 m, 873 m, 802 w, 743 w, 638 s, 582 w, 509 w, 440 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 636.11922 ([M]+, calcd. 636.11743). Anal. Calcd. for PTA-2 (C24H24F3N5O6PReS): C, 36.73; H, 3.08; N, 8.92. Found: C, 36.66; H, 3.09; N, 8.98.
Synthesis of [Re(CO)3(dmphen)(THP)]TFA (THP-2).
A mixture of [Re(CO)3(dmphen)Cl] (0.209 g, 0.407 mmol) and AgOTf (0.104 g, 0.407 mmol) in THF (46 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid AgCl was removed via vacuum filtration. To the remaining yellow filtrate, THP (0.076 g, 0.612 mmol) was added and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and then THF was removed by rotary evaporation. The crude yellow oil was purified using preparatory RP-HPLC using a CH3OH/H2O gradient (0–5 min, 10% CH3OH; 5–50 min, 10–100% CH3OH). Purified yield: 0.158 g (53%). 1H NMR (400 MHz, MeOD-d4): δ 8.70 (d, 2H, J = 8.4 Hz), 8.13 (s, 2H), 8.03 (d, 2H, J = 8.4 Hz), 3.44 (s, 6H), 3.37 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –76.35. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ 6.53. IR (KBr cm−1): 3036 m, 2837 w, 2449 w, 2044 s, 1945 s, 1909 s, 1674 s, 1594 w, 1506 w, 1442 m, 1370 w, 1202 m, 1140 m, 1043 m, 892 w, 859 m, 837 w, 800 w, 781 w, 722 w, 652 w, 619 w, 552 w, 513 w, 499 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 603.07053 ([M]+, calcd. 603.06947). Anal. Calcd. for THP-2·H2O (C22H23F3N2O9PRe): C, 36.02; H, 3.16; N, 3.82. Found: C, 36.03; H, 2.90; N, 3.77.
Synthesis of [Re(CO)3(dmphen)(DAPTA)]OTf (DAPTA-2).
A mixture of [Re(CO)3(dmphen)Cl] (0.232 g, 0.451 mmol) and AgOTf (0.116 g, 0.51 mmol) in THF (54 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid AgCl was removed via vacuum filtration. To the remaining yellow filtrate, DAPTA (0.144 g, 0.59 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature and then the yellow precipitate was isolated by filtration and washed with a small amount of methanol (5 ml) and diethyl ether (20 ml). Yield: 0.319 g (83%). 1H NMR (400 MHz, MeOD-d4): DAPTA complexes show the syn and anti isomers in a 1:5 ratio; however in the aromatic region the isomers are indistinguishable, δ 8.85 (d, 2H, anti + syn), 8.26 (d, 2H, anti + syn), 8.16 (dd, 2H, anti + syn), 5.51 (d, anti), 4.98 (d, syn), 4.44 (d, anti), 3.93 (d, anti), 3.55 (m, anti), 3.37 (d, 3H, anti + syn), 2.98 (m, syn), 2.84 (d, anti), 1.90 (s, syn), 1.84 (s, anti), 1.46 (s, 5H, anti). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.57. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –41.15 (syn), –45.58 (anti). IR (KBr, cm−1): 3440 m, 3070 w, 2040 s, 1970 m, 1930 s, 1640 s, 1510 w, 1420 m, 1380 w, 1340 m, 1270 s, 1150 m, 1030 s, 989 m, 890 m, 867 m, 799 m, 705 w, 638 s, 551 m, 513 m, 469 w, 417 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 708.13962 ([M]+, calcd. 708.13801). Anal. Calcd. for DAPTA-2 (C27H28F3N5O8PReS): C, 37.85; H, 3.29; N, 8.17. Found: C, 38.02; H, 3.24; N, 8.09.
Synthesis of [Re(CO)3(bpy)(PTA)]OTf (PTA-3).
A mixture of [Re(CO)3(bpy)Cl] (0.564 g, 1.22 mmol) and AgOTf (0.314 g, 1.22 mmol) in THF (123 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid, AgCl, was removed via vacuum filtration. To the remaining yellow filtrate, PTA (0.192 g, 1.22 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and then the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried in vacuo. Yield: 0.523 g (59%). 1H NMR (400 MHz, MeOD-d4): δ 9.13 (d, 2H, J = 5.6 Hz), 8.74 (d, 2H, J = 8.3 Hz), 8.40 (t, 2H, J = 8.3 Hz), 7.81 (t, 2H, J = 5.6 Hz), 4.47 (d, 3H, J = 13.0 Hz), 4.38 (d, 3H, J = 13.0 Hz), 3.80 (s, 6H). 19F (376 MHz, MeOD-d4): δ –79.52. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –77.64. IR (KBr cm−1): 3440 m, 2040 s, 1950 s, 1910 s, 1610 m, 1470 m, 1440 m, 1280 s, 1260 s, 1140 m, 1100 m, 1030 s, 1010 m, 969 m, 946 m, 903 w, 801 m, 770 s, 740 m, 631 s, 614 m, 587 m, 573 m, 512 m, 454 m. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 584.08746 ([M]+, calcd. 584.08558). Anal. Calcd. for PTA-3 (C20H20F3N5O6PReS): C, 32.79; H, 2.75; N, 9.56. Found: C, 32.51; H, 2.76; N, 9.38.
Synthesis of [Re(CO)3(bpy)(THP)]TFA (THP-3).
A mixture of [Re(CO)3(bpy)Cl] (0.100 g, 0.217 mmol) and AgOTf (0.056 g, 0.217 mmol) in THF (22 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid, AgCl, was removed via vacuum filtration. To the remaining yellow filtrate, THP (0.040 g, 0.326 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and then THF was removed by rotary evaporation. The remaining yellow oil was then dissolved in a minimal amount of methanol (5 ml) and filtered through Celite. The crude yellow oil (0.170 g) was purified using preparatory RP-HPLC using a CH3OH/H2O gradient (0–5 min, 10% CH3OH; 5–30 min, 10–100% CH3OH). Purified yield: 0.070 g (45%). 1H NMR (400 MHz, MeOD-d4): δ 9.17 (d, 2H, J = 5.8 Hz), 8.64 (d, 2H, J = 8.1 Hz), 8.31 (t, 2H, J = 8.1 Hz), 7.72 (t, 2H, J = 6.4 Hz), 3.90 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.34. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ 2.50. IR (KBr cm−1): 3433 s, 2874 m, 2038 s, 1930 s, 1915 s, 1681 s, 1633 s, 1473 m, 1450 m, 1181 s, 1030 m, 841 w, 769 m, 731 w, 534 w, 497 w, 417 m. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 551.03927 ([M]+, calcd. 551.03763). Anal. Calcd. for THP-3·H2O (C18H19F3N2O9PRe): C, 31.72; H, 2.81; N, 4.11. Found: C, 31.68; H, 2.61; N, 4.09.
Synthesis of [Re(CO)3(bpy)(DAPTA)]OTf (DAPTA-3).
A mixture of [Re(CO)3(bpy)Cl] (0.236 g, 0.51 mmol) and AgOTf (0.131 g, 0.51 mmol) in THF (55 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid, AgCl, precipitate was removed via vacuum filtration. To the remaining yellow filtrate, DAPTA (0.150 g, 0.61 mmol) was added, and the solution was heated to reflux for an additional 24 h. The reaction mixture was allowed to cool to room temperature and then the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried in vacuo. Yield: 0.204 g (48%). 1H NMR (400 MHz, MeOD-d4): DAPTA complexes show the syn and anti isomers in a 1:5 ratio; however in the aromatic region the isomers are indistinguishable, δ 9.21 (t, 2H, anti + syn), 8.77 (d, 2H, anti + syn), 8.43 (t, 2H, anti + syn), 7.84 (t, 2H, anti + syn), 5.65 (d, anti), 5.12 (d, syn), 5.01 (d, anti), 4.92 (m, syn), 4.53 (d, anti), 4.22 (m, anti), 4.03 (d, anti), 3.91 (d, anti), 3.80 (s, syn), 3.58 (d, anti), 3.14 (d, anti), 2.01 (s, syn), 1.98 (s, anti), 1.90 (s, anti). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.56. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): –44.42 (syn), –48.33 (anti). IR (KBr cm−1): 3457 m, 2965 w, 2880 w, 2037 s, 1958 s, 1918 s, 1646 s, 1473 m, 1419 m, 1331 m, 1265 s, 1155 m, 1024 s, 987 m, 892 m, 776 m, 632 s, 515 m, 422 m. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 656.10825([M]+, calcd. 656.10671). Anal. Calcd. for DAPTA-3·0.5(CH3CH2)2O (C25H29F3N5O8.5PReS): C, 35.67; H, 3.47; N, 8.32. Found: C, 35.17; H, 3.49; N, 8.38.
Synthesis of [Re(CO)3(dmbpy)(PTA)]OTf (PTA-4).
A mixture of [Re(CO)3(dmbpy)Cl] (0.200 g, 0.43 mmol) and AgOTf (0.111 g, 0.43 mmol) in THF (47 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid, AgCl, precipitate was removed via vacuum filtration. To the remaining yellow filtrate, PTA (0.102 g, 0.65 mmol) was added and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature and then the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried in vacuo. Yield: 0.254 g (77%). 1H NMR (400 MHz, MeOD-d4): δ 8.91 (d, 2H, J = 5.8 Hz), 8.60 (s, 2H), 7.62 (d, 2H, J = 5.8 Hz), 4.47 (d, 3H, J = 13.2 Hz), 4.37 (d, 3H, J = 13.2 Hz), 3.78 (s, 6H), 2.67 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.53. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –77.22. IR (KBr cm−1): 3452 m, 2940 w, 2033 s, 1951 s, 1921 s, 1626 w, 1266 s, 1139 w, 1031 m, 972 m, 948 m, 834 m, 801 m, 740 m, 637 s, 580 m, 500 m, 436 m. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 612.11845 ([M]+, calcd. 612.11743). Anal. Calcd. for PTA-4 (C22H24F3N5O6PReS): C, 34.74; H, 3.18; N, 9.21. Found: C, 34.97; H, 3.22; N, 9.11.
Synthesis of [Re(CO)3(dmbpy)(THP)]TFA (THP-4).
A mixture of [Re(CO)3(dmbpy)Cl] (0.200 g, 0.43 mmol) and AgOTf (0.111 g, 0.43 mmol) in THF (47 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid, AgCl, precipitate was removed via vacuum filtration. To the remaining yellow filtrate, THP (0.081 g, 0.65 mmol) was added and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature and then THF was removed by rotary evaporation. The crude yellow solid (0.307 g) was purified using preparatory RP-HPLC using a CH3OH/H2O gradient (0–5 min, 10% CH3OH; 5–30 min, 10–100% CH3OH). Purified yield: 0.073 g (25%). 1H NMR (400 MHz, MeOD-d4): δ 8.96 (d, 2H, J = 5.7 Hz), 8.50 (s, 2H), 7.54 (d, 2H, J = 5.7 Hz), 3.88 (s, 6H), 2.63 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –76.36. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ 3.26. IR (KBr cm−1): 3404 m, 2900 w, 2031 s, 1935 s, 1920 s, 1675 m, 1621 w, 1440 w, 1200 m, 1130 m, 1035 m, 905 w, 830 m, 723 w, 637 w, 534 w, 508 m, 444 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 579.07103 ([M]+, calcd. 579.06893). Anal. Calcd. for THP-4 (C20H21F3N2O8PReS): C, 32.79; H, 2.75; N, 9.56. Found: C, 32.51; H, 2.76; N, 9.38.
Synthesis of [Re(CO)3(dmbpy)(DAPTA)]OTf (DAPTA-4).
A mixture of [Re(CO)3(dmbpy)Cl] (0.267 g, 0.57 mmol) and AgOTf (0.146 g, 0.57 mmol) in THF (65 ml) was heated to reflux at 75 °C for 3 h in the dark. The white solid, AgCl, precipitate was removed via vacuum filtration. To the remaining yellow filtrate, DAPTA (0.211 g, 0.86 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature and then the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried in vacuo. Yield: 0.194 g (39%). 1H NMR (400 MHz, MeOD-d4): DAPTA complexes show the syn and anti isomers in a 1:5 ratio; however in the aromatic region the isomers are indistinguishable, δ 8.99 (t, 2H, anti + syn), 8.63 (s, 2H, anti + syn), 7.65 (t, 2H, anti + syn), 5.64 (d, anti), 5.13 (d, syn), 5.00 (d, anti), 4.54 (m, anti + syn), 4.22 (m, anti), 4.02 (d, anti), 3.89 (d, anti), 3.56 (m, syn) 3.12 (d, anti), 2.68 (s, 3H, anti + syn) 2.02 (s, syn), 1.98 (s, anti), 1.91 (s, anti). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.53. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –44.35 (syn), –47.93 (anti). IR (KBr cm−1): 3433 m, 2038 s, 1943 s, 1924 s, 1648 m, 1420 m, 1333 w, 1262 m, 1156 w, 1030 m, 986 w, 890 w, 826 w, 782 w, 638 m, 500 w, 421 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 684.13967 ([M]+, calcd. 684.13801). Anal. Calcd. for DAPTA-4·CH3OH (C26H32F3N5O9PReS): C, 36.11; H, 3.73; N, 8.10. Found: C, 36.31; H, 3.59; N, 8.19.
Synthesis of [Re(CO)3(dmobpy)(PTA)]OTf (PTA-5).
A mixture of [Re(CO)3(dmobpy)Cl] (0.200 g, 0.38 mmol) and AgOTf (0.099 g, 0.38 mmol) in THF (47 ml) was heated to reflux at 75 oC for 3 h in the dark. The white solid, AgCl, precipitate was removed via vacuum filtration. To the remaining yellow filtrate, PTA (0.079 g, 0.50 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and then the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried in vacuo. Yield: 0.158 g (52%). 1H NMR (400 MHz, MeOD-d4): δ 8.84 (d, 2H, J = 6.5 Hz), 8.26 (d, 2H, J = 2.6 Hz), 7.34 (dd, 2H, J = 2.6, 6.5 Hz), 4.49 (d, 3H, J = 13.1 Hz), 4.39 (d, 3H, J = 13.1 Hz), 4.14 (s, 6H), 3.80 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.59. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –75.88. IR (KBr cm−1): 3443 m, 2031 s, 1942 s, 1921 s, 1615 m, 1560 w, 1497 w, 1426 w, 1350 w, 1262 m, 1242 w, 1147 w, 1029 m, 974 w, 948 w, 899 w, 828 w, 801 w, 741 w, 636 m, 581 w, 514 w, 429 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 644.10822 ([M]+, calcd. 644.10671). Anal. Calcd. for PTA-5 (C22H24F3N5O8PReS): C, 33.33; H, 3.05; N, 8.84. Found: C, 33.39; H, 3.10; N, 8.80.
Synthesis of [Re(CO)3(dmobpy)(THP)]OTf (THP-5).
A mixture of [Re(CO)3(dmobpy)Cl] (0.297 g, 0.57 mmol) and AgOTf (0.146 g, 0.57 mmol) in THF (70 ml) was heated to reflux at 75 oC for 3 h in the dark. The white solid, AgCl, precipitate was removed via vacuum filtration. To the remaining yellow filtrate, THP (0.092 g, 0.74 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and then THF was removed by rotary evaporation, then dissolved in minimal methanol (5 ml) and filtered through Celite. The crude yellow oil (0.439 g) was recrystallized using vapor diffusion of diethyl ether into a solution of methanol to afford yellow crystals. Yield: 0.030 g (7%). 1H NMR (400 MHz, MeOD-d4): δ 8.88 (d, 2H, J = 6.6 Hz), 8.16 (d, 2H, J = 2.7 Hz), 7.27 (dd, 2H, J = 2.7, 6.6 Hz), 4.10 (s, 6H), 3.90 (s, 6H). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.57. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ 3.16. IR (KBr cm−1): 3448 m, 2028 s, 1939 w, 1919 s, 1618 s, 1560 m, 1500 m, 1343 w, 1270 m, 1182 w, 1031 w, 905 w, 826 w, 639 w, 513 w, 434 w. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 611.06037 ([M]+, calcd. 611.05875). Anal. Calcd. for THP-5 (C19H21F3N2O11PReS): C, 30.04; H, 2.79; N, 3.69. Found: C, 30.30; H, 2.79; N, 3.89.
Synthesis of [Re(CO)3(dmobpy)(DAPTA)]OTf (DAPTA-5).
A mixture of [Re(CO)3(dmobpy)Cl] (0.210 g, 0.40 mmol) and AgOTf (0.103 g, 0.40 mmol) in THF (45 ml) was heated to reflux at 75 oC for 3 h in the dark. The white solid, AgCl, precipitate was removed via vacuum filtration. To the remaining yellow filtrate, DAPTA (0.123 g, 0.52 mmol) was added, and the solution was heated to reflux for an additional 15 h. The reaction mixture was allowed to cool to room temperature, and then the THF was removed using rotary evaporation. The remaining yellow solid was dissolved in a minimum amount of methanol (5–10 ml) and filtered through Celite. The addition of diethyl ether (20–30 ml) afforded the desired product as a yellow solid, which was isolated by vacuum filtration and dried in vacuo. The solid material (0.343 g) was recrystallized using vapor diffusion of diethyl ether into a solution of methanol to afford pure yellow crystals. Purified yield: 0.028 g (8%). 1H NMR (400 MHz, MeOD-d4): DAPTA complexes show the syn and anti isomers in a 1:5 ratio; however in the aromatic region the isomers are indistinguishable, δ 8.91 (t, 2H, anti + syn), 8.29 (s, 2H, anti + syn), 7.37 (t, 2H, anti + syn), 5.67 (d, anti), 5.15 (d, syn), 5.02 (d, anti), 4.56 (m, anti + syn), 4.22 (m, anti), 4.15 (s, 3H, anti + syn), 4.05 (d, anti), 3.92 (d, anti), 3.54 (m, anti + syn), 3.14 (d, anti + syn), 2.03 (s, syn), 1.99 (s, anti), 1.92 (s, anti). 19F (376 MHz, MeOD-d4, external stnd: KPF6): δ –79.57. 31P{1H} (162 MHz, MeOD-d4, external stnd: KPF6): δ –43.34 (syn), –46.87 (anti). IR (KBr cm−1): 3456 m, 2040 s, 1939 s, 1919 s, 1637 s, 1500 m, 1420 m, 1340 w, 1260 m, 1150 w, 1030 s, 889 w, 796 w, 638 m, 516 w, 419 m. ESI-MS (pos. ion mode, CH3CN:H2O 70:30 and 1% formic acid): m/z 716.12966 ([M]+, calcd. 716.12784). Anal. Calcd. for DAPTA-5 (C25H28F3N5O10PReS): C, 34.72; H, 3.26; N, 8.10. Found: C, 34.44; H, 3.50; N, 8.08.
Synthesis of [Re(CO)2(phen)(DAPTA)Cl] (DAPTA-1A).
Trimethylamine-N-oxide (TMAO, 0.040 g, 0.36 mmol) was dissolved in a mixture of CH2Cl2 (130 ml) and CH3OH (5 ml). A solution of DAPTA-1 (0.262 g, 0.32 mmol) and NEt4Cl (0.919 g, 5 mmol) in CH2Cl2 (105 ml) was heated to reflux at 50 °C, and the TMAO solution was added dropwise over the course of 1 h, changing the color of the solution from light yellow to dark red. The solution was heated under reflux for an additional 24 h. The CH2Cl2/CH3OH was then removed via rotary evaporation, and the remaining red solid was suspended in approximately 20 ml of CH3OH. The solid was isolated by centrifugation and then resuspended in 5–10 ml of CH3OH. This process was repeated three times, yielding the product DAPTA-1A as a red solid. Yield: 0.150 g (94%). 1H NMR (400 MHz, DMF-d7): DAPTA complexes show the syn and anti isomers in a 1:5 ratio; however in the aromatic region the isomers are indistinguishable, δ 9.52 (m, 2H, anti + syn), 8.95 (d, 2H, anti + syn), 8.33 (s, 2H, anti + syn), 8.14 (m, 2H, anti + syn), 5.52 (d, anti), 5.06 (d, anti), 4.89 (d, syn), 4.47 (d, anti), 4.21(m, syn), 3.93 (d, syn), 3.73 (d, anti), 3.32 (m, anti + syn), 3.12 (d, anti), 1.97 (s, syn), 1.90 (s, anti), 1.86 (s, anti). 31P{1H} (162 MHz, DMF-d7, external stnd: KPF6): δ –32.32 (syn), –34.72 (anti). IR (KBr, cm−1): 3458 s, 1927s, 1839 s, 1638 s, 1427 m, 1338 m, 1240 m, 1097 w, 1050 w, 997 w, 890 w, 855 w, 795 w, 727 w, 614 w. Anal. Calcd. for DAPTA-1A·2H2O (C23H28 ClN5O6PRe): C, 38.20; H, 3.90; N, 9.68. Found: C, 38.30; H, 3.94; N, 9.42.
Synthesis of [Re(CO)2(bpy)(PTA)Cl] (PTA-3A).
TMAO (0.052 g, 0.47 mmol) was dissolved in a mixture of CH2Cl2 (150 ml) and CH3OH (6 ml). A mixture of PTA-3 (0.298 g, 0.41 mmol) and NEt4Cl (1.17 g, 6.4 mmol) in CH2Cl2 (120 ml) was heated to reflux at 50 °C, and then the TMAO solution was added dropwise over the course of 1 h, resulting in a color change from yellow to red. The solution was heated to reflux for an additional 24 h. The CH2Cl2/CH3OH was removed via rotary evaporation. To the remaining red solid, approximately 20 ml of water was added. The solid was isolated by centrifugation, and then resuspended in 5–10 ml of water. This process was repeated 3× to obtain the product PTA-3A as a red solid. Yield: 0.112 g (46%). 1H NMR (400 MHz, DMSO-d6): δ 8.88 (d, 2H, J = 5.9 Hz), 8.67 (d, 2H, J = 8.3 Hz), 8.22 (t, 2H, J = 8.3 Hz), 7.65 (t, 2H, J = 5.9 Hz), 4.28 (d, 3H, J = 12.8 Hz), 4.15 (d, 3H, J = 12.8 Hz), 3.49 (s, 6H). 31P{1H} (162 MHz, DMSO-d6, external stnd: KPF6): δ –59.1. IR (KBr, cm−1): 3471 m, 1912 s, 1834 s, 1605 w, 1452 w, 1239 w, 1099 w, 1014 w, 966 w, 948 w, 781 m, 594 w, 580 m, 483 w. Anal. Calcd. for PTA-3A·H2O (C18H22 ClN5O3PRe): C, 35.56; H, 3.64; N, 11.50. Found: C, 35.38; H, 3.66; N, 11.36.
Supplementary Material
Acknowledgements.
This research was supported by Cornell University and by the Office of the Assistant Secretary of Defense for Health Affairs through the Ovarian Cancer Research Program under award no. W81XWH-17-1-0097. This work made use of the NMR facility at Cornell University, which is supported, in part, by the NSF under award number CHE-1531632. Studies at UCSB were supported by the National Science Foundation under award number CHE-1565702. Dr. Nikki Thiele (Cornell University) and Mr. Jacob Barrett (UCSB) are thanked for assistance with the pKa measurements and the CO release quantification, respectively.
Footnotes
Accession Codes
CCDC 1581545–1581560 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Supporting Information. The Supporting Information is available free of charge on the ACS publications website at http://pubs.acs.org.
Complex characterization data, cell viability curves, crystal data tables, DFT-optimized geometries and excited states, UV-vis and emission spectra, and transient emission decay profiles, and species distribution diagrams (PDF)
Crystallographic data (CIF).
The authors declare no competing financial interests.
References:
- (1).Dolmans DEJGJ; Fukumura D; Jain RK Photodynamic Therapy for Cancer. Nat. Rev. Cancer 2003, 3, 380–387. [DOI] [PubMed] [Google Scholar]
- (2).Castano AP; Demidova TN; Hamblin MR Mechanisms in Photodynamic Therapy: Part One—Photosensitizers, Photochemistry and Cellular Localization. Photodiag. Photodyn. Ther 2004, 1, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Castano AP; Mroz P; Hamblin MR Photodynamic Therapy and Anti-Tumour Immunity. Nat. Rev. Cancer 2006, 6, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Agostinis P; Berg K; Cengel KA; Foster TH; Girotti AW; Gollnick SO; Hahn SM; Hamblin MR; Juzeniene A; Kessel D; Korbelik M; Moan J; Mroz P; Nowis D; Piette J; Wilson BC; Golab J Photodynamic Therapy of Cancer: An Update. CA-Cancer J. Clin 2011, 61, 250–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Farrer NJ; Salassa L; Sadler PJ Photoactivated Chemotherapy (PACT): The Potential of Excited-State d-Block Metals in Medicine. Dalton Trans 2009, 10690–10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Taub AF Photodynamic Therapy: Other Uses. Dermatol. Clin 2007, 25, 101–109. [DOI] [PubMed] [Google Scholar]
- (7).DeRosa MC; Crutchley RJ Photosensitized Singlet Oxygen and Its Applications. Coord. Chem. Rev 2002, 233–234, 351–371. [Google Scholar]
- (8).O’Connor AE; Gallagher WM; Byrne AT Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy. Photochem. Photobiol 2009, 85, 1053–1074. [DOI] [PubMed] [Google Scholar]
- (9).Yang JZ; Van Vugt DA; Kennedy JC; Reid RL Intrauterine 5-Aminolevulinic Acid Induces Selective Fluorescence and Photodynamic Ablation of the Rat Endometrium. Photochem. Photobiol 1993, 57, 803–807. [DOI] [PubMed] [Google Scholar]
- (10).Bonnett R Photosensitizers of the Porphyrin and Phthalocyanine Series for Photodynamic Therapy. Chem. Soc. Rev 1995, 19–33. [Google Scholar]
- (11).Ochsner M Photophysical and Photobiological Processes in the Photodynamic Therapy of Tumours. J. Photochem. Photobiol., B 1997, 39, 1–18. [DOI] [PubMed] [Google Scholar]
- (12).Dougherty TJ; Gomer CJ; Henderson BW; Jori G; Kessel D; Korbelik M; Moan J; Peng Q Photodynamic Therapy. J. Natl. Cancer Inst 1998, 90, 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sharman WM; Allen CM; van Lier JE Photodynamic Therapeutics: Basic Principles and Clinical Applications. Drug Discovery Today 1999, 4, 507–517. [DOI] [PubMed] [Google Scholar]
- (14).MacDonald IJ; Dougherty TJ Basic Principles of Photodynamic Therapy. J. Porphyrins Phthalocyanines 2001, 5, 105–129. [Google Scholar]
- (15).Pervaiz S; Olivo M Art and Science of Photodynamic Therapy. Clin. Exp. Pharmacol. Physiol 2006, 33, 551–556. [DOI] [PubMed] [Google Scholar]
- (16).Palumbo G Photodynamic Therapy and Cancer: A Brief Sightseeing Tour. Expert Opin. Drug Delivery 2007, 4, 131–148. [DOI] [PubMed] [Google Scholar]
- (17).Juarranz Á; Jaén P; Sanz-Rodríguez F; Cuevas J; González S Photodynamic Therapy of Cancer. Basic Principles and Applications. Clin. Transl. Oncol 2008, 10, 148–154. [DOI] [PubMed] [Google Scholar]
- (18).Bhuvaneswari R; Gan YY; Soo KC; Olivo M The Effect of Photodynamic Therapy on Tumor Angiogenesis. Cell. Mol. Life Sci 2009, 66, 2275–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Plaetzer K; Krammer B; Berlanda J; Berr F; Kiesslich T Photophysics and Photochemistry of Photodynamic Therapy: Fundamental Aspects. Lasers Med. Sci 2009, 24, 259–268. [DOI] [PubMed] [Google Scholar]
- (20).Harris AL Hypoxia––A Key Regulatory Factor in Tumour Growth. Nat. Rev. Cancer 2002, 2, 38–47. [DOI] [PubMed] [Google Scholar]
- (21).Brown JM Tumor Hypoxia in Cancer Therapy. In Methods in Enzymology; 2007; Vol. 435, pp 296–321. [DOI] [PubMed] [Google Scholar]
- (22).Howerton BS; Heidary DK; Glazer EC Strained Ruthenium Complexes Are Potent Light-Activated Anticancer Agents. J. Am. Chem. Soc 2012, 134, 8324–8327. [DOI] [PubMed] [Google Scholar]
- (23).Bozzini G; Colin P; Betrouni N; Maurage CA; Leroy X; Simonin S; Martin-Schmitt C; Villers A; Mordon S Efficiency of 5-ALA Mediated Photodynamic Therapy on Hypoxic Prostate Cancer: A Preclinical Study on the Dunning R3327-AT2 Rat Tumor Model. Photodiag. Photodyn. Ther 2013, 10, 296–303. [DOI] [PubMed] [Google Scholar]
- (24).Kinsella TJ; Colussi VC; Oleinick NL; Sibata CH Photodynamic Therapy in Oncology. Expert Opin. Pharmacother 2001, 2, 917–927. [DOI] [PubMed] [Google Scholar]
- (25).Baas P; van Mansom I; van Tinteren H; Stewart FA; van Zandwijk N Effect of N-Acetylcysteine on Photofrin-Induced Skin Photosensitivity in Patients. Lasers Surg. Med 1995, 16, 359–367. [DOI] [PubMed] [Google Scholar]
- (26).Farrer NJ; Sadler PJ Photochemotherapy: Targeted Activation of Metal Anticancer Complexes. Aust. J. Chem 2008, 61, 669–674. [Google Scholar]
- (27).Josefsen LB; Boyle RW Photodynamic Therapy and the Development of Metal-Based Photosensitisers. Metal-Based Drugs 2008, 2008, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lameijer LN; Ernst D; Hopkins SL; Meijer MS; Askes SHC; Le Dévédec SE; Bonnet S A Red-Light-Activated Ruthenium-Caged NAMPT Inhibitor Remains Phototoxic in Hypoxic Cancer Cells. Angew. Chem. Int. Ed 2017, 56, 11549–11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Charlesworth P; Truscott TG; Brooks RC; Wilson BC The Photophysical Properties of a Ruthenium-Substituted Phthalocyanine. J. Photochem. Photobiol., B 1994, 26, 277–282. [Google Scholar]
- (30).Zhou J; Liu J; Feng Y; Wei S; Gu X; Wang X; Zhang B Synthesis and Characterization of the Monomer Ruthenium Complex of Hypocrellin B. Bioorg. Med. Chem. Lett 2005, 15, 3067–3070. [DOI] [PubMed] [Google Scholar]
- (31).Davia K; King D; Hong Y; Swavey S A Porphyrin-Ruthenium Photosensitizer as a Potential Photodynamic Therapy Agent. Inorg. Chem. Commun 2008, 11, 584–586. [DOI] [PubMed] [Google Scholar]
- (32).Schmitt F; Govindaswamy P; Süss-Fink G; Ang WH; Dyson PJ; Juillerat-Jeanneret L; Therrien B Ruthenium Porphyrin Compounds for Photodynamic Therapy of Cancer. J. Med. Chem 2008, 51, 1811–1816. [DOI] [PubMed] [Google Scholar]
- (33).Gianferrara T; Bergamo A; Bratsos I; Milani B; Spagnul C; Sava G; Alessio E Ruthenium-Porphyrin Conjugates with Cytotoxic and Phototoxic Antitumor Activity. J. Med. Chem 2010, 53, 4678–4690. [DOI] [PubMed] [Google Scholar]
- (34).Sun Y; Joyce LE; Dickson NM; Turro C Efficient DNA Photocleavage by [Ru(bpy)2(dppn)]2+ with Visible Light. Chem. Commun 2010, 46, 2426–2428. [DOI] [PubMed] [Google Scholar]
- (35).Carneiro ZA; de Moraes JCB; Rodrigues FP; de Lima RG; Curti C; da Rocha ZN; Paulo M; Bendhack LM; Tedesco AC; Formiga ALB; da Silva RS Photocytotoxic Activity of a Nitrosyl Phthalocyanine Ruthenium Complex –– A System Capable of Producing Nitric Oxide and Singlet Oxygen. J. Inorg. Biochem 2011, 105, 1035–1043. [DOI] [PubMed] [Google Scholar]
- (36).Johnpeter JP; Schmitt F; Denoyelle-Di-Muro E; Wagnières G; Juillerat-Jeanneret L; Therrien B Photoactive Sawhorse-Type Diruthenium Tetracarbonyl Complexes. Inorg. Chim. Acta 2012, 393, 246–251. [Google Scholar]
- (37).Pernot M; Bastogne T; Barry NPE; Therrien B; Koellensperger G; Hann S; Reshetov V; Barberi-Heyob M Systems Biology Approach for in vivo Photodynamic Therapy Optimization of Ruthenium-Porphyrin Compounds. J. Photochem. Photobiol., B 2012, 117, 80–89. [DOI] [PubMed] [Google Scholar]
- (38).Zhang J-X; Zhou J-W; Chan C-F; Lau TC-K; Kwong DWJ; Tam H-L; Mak N-K; Wong K-L; Wong W-K Comparative Studies of the Cellular Uptake, Subcellular Localization, and Cytotoxic and Phototoxic Antitumor Properties of Ruthenium(II)–Porphyrin Conjugates with Different Linkers. Bioconjugate Chem 2012, 23, 1623–1638. [DOI] [PubMed] [Google Scholar]
- (39).Lincoln R; Kohler L; Monro S; Yin H; Stephenson M; Zong R; Chouai A; Dorsey C; Hennigar R; Thummel RP; Mcfarland SA Exploitation of Long-Lived 3IL Excited States for Metal–Organic Photodynamic Therapy: Verification in a Metastatic Melanoma Model. J. Am. Chem. Soc 2013, 135, 17161–17175. [DOI] [PubMed] [Google Scholar]
- (40).Albani BA; Peña B; Leed NA; de Paula NABG; Pavani C; Baptista MS; Dunbar KR; Turro C Marked Improvement in Photoinduced Cell Death by a New Tris-Heteroleptic Complex with Dual Action: Singlet Oxygen Sensitization and Ligand Dissociation. J. Am. Chem. Soc 2014, 136, 17095–17101. [DOI] [PubMed] [Google Scholar]
- (41).Frei A; Rubbiani R; Tubafard S; Blacque O; Anstaett P; Felgenträger A; Maisch T; Spiccia L; Gasser G Synthesis, Characterization, and Biological Evaluation of New Ru(II) Polypyridyl Photosensitizers for Photodynamic Therapy. J. Med. Chem 2014, 57, 7280–7292. [DOI] [PubMed] [Google Scholar]
- (42).Joshi T; Pierroz V; Mari C; Gemperle L; Ferrari S; Gasser G A Bis(dipyridophenazine)(2-(2-pyridyl)pyrimidine-4-carboxylic acid)ruthenium(II) Complex with Anticancer Action upon Photodeprotection. Angew. Chem. Int. Ed 2014, 53, 2960–2963. [DOI] [PubMed] [Google Scholar]
- (43).Mari C; Pierroz V; Rubbiani R; Patra M; Hess J; Spingler B; Oehninger L; Schur J; Ott I; Salassa L; Ferrari S; Gasser G DNA Intercalating RuII Polypyridyl Complexes as Effective Photosensitizers in Photodynamic Therapy. Chem. Eur. J 2014, 20, 14421–14436. [DOI] [PubMed] [Google Scholar]
- (44).Fong J; Kasimova K; Arenas Y; Kaspler P; Lazic S; Mandel A; Lilge L A Novel Class of Ruthenium-Based Photosensitizers Effectively Kills in vitro Cancer Cells and in vivo Tumors. Photochem. Photobiol. Sci 2015, 14, 2014–2023. [DOI] [PubMed] [Google Scholar]
- (45).Huang H; Yu B; Zhang P; Huang J; Chen Y; Gasser G; Ji L; Chao H Highly Charged Ruthenium(II) Polypyridyl Complexes as Lysosome-Localized Photosensitizers for Two-Photon Photodynamic Therapy. Angew. Chem. Int. Ed 2015, 54, 14049–14052. [DOI] [PubMed] [Google Scholar]
- (46).Shi G; Monro S; Hennigar R; Colpitts J; Fong J; Kasimova K; Yin H; DeCoste R; Spencer C; Chamberlain L; Mandel A; Lilge L; McFarland SA Ru(II) Dyads Derived from α-Oligothiophenes: A New Class of Potent and Versatile Photosensitizers for PDT. Coord. Chem. Rev 2015, 282–283, 127–138. [Google Scholar]
- (47).Kaspler P; Lazic S; Forward S; Arenas Y; Mandel A; Lilge L A Ruthenium(II) Based Photosensitizer and Transferrin Complexes Enhance Photo-Physical Properties, Cell Uptake, and Photodynamic Therapy Safety and Efficacy. Photochem. Photobiol. Sci 2016, 15, 481–495. [DOI] [PubMed] [Google Scholar]
- (48).Mari C; Huang H; Rubbiani R; Schulze M; Würthner F; Chao H; Gasser G Evaluation of Perylene Bisimide-Based RuII and IrIII Complexes as Photosensitizers for Photodynamic Therapy. Eur. J. Inorg. Chem 2017, 1745–1752. [Google Scholar]
- (49).Pierroz V; Rubbiani R; Gentili C; Patra M; Mari C; Gasser G; Ferrari S Dual Mode of Cell Death upon Photo-Irradiation of a RuII Polypyridyl Complex in Interphase or Mitosis. Chem. Sci 2016, 7, 6115–6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Havrylyuk D; Heidary DK; Nease L; Parkin S; Glazer EC Photochemical Properties and Structure-Activity Relationships of RuII Complexes with Pyridylbenzazole Ligands as Promising Anticancer Agents. Eur. J. Inorg. Chem 2017, 1687–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mari C; Rubbiani R; Gasser G Biological Evaluation of Nitrile Containing Ru(II) Polypyridyl Complexes as Potential Photodynamic Therapy Agents. Inorg. Chim. Acta 2017, 454, 21–26. [Google Scholar]
- (52).Wachter E; Heidary DK; Howerton BS; Parkin S; Glazer EC Light-Activated Ruthenium Complexes Photobind DNA and are Cytotoxic in the Photodynamic Therapy Window. Chem. Commun 2012, 48, 9649–9651. [DOI] [PubMed] [Google Scholar]
- (53).Sgambellone MA; David A; Garner RN; Dunbar KR; Turro C Cellular Toxicity Induced by the Photorelease of a Caged Bioactive Molecule: Design of a Potential Dual-Action Ru(II) Complex. J. Am. Chem. Soc 2013, 135, 11274–11282. [DOI] [PubMed] [Google Scholar]
- (54).Hidayatullah AN; Wachter E; Heidary DK; Parkin S; Glazer EC Photoactive Ru(II) Complexes with Dioxinophenanthroline Ligands Are Potent Cytotoxic Agents. Inorg. Chem 2014, 53, 10030–10032. [DOI] [PubMed] [Google Scholar]
- (55).Mari C; Pierroz V; Leonidova A; Ferrari S; Gasser G Towards Selective Light-Activated RuII-Based Prodrug Candidates. Eur. J. Inorg. Chem 2015, 3879–3891. [Google Scholar]
- (56).Kumar A; Sun S-S; Lees AJ Photophysics and Photochemistry of Organometallic Rhenium Diimine Complexes. In Photophysics of Organometallics; Lees AJ, Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2010; pp 1–36. [Google Scholar]
- (57).Kastl A; Dieckmann S; Wähler K; Völker T; Kastl L; Merkel AL; Vultur A; Shannan B; Harms K; Ocker M; Parak WJ; Herlyn M; Meggers E Rhenium Complexes with Visible-Light-Induced Anticancer Activity. ChemMedChem 2013, 8, 924–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Leonidova A; Gasser G Underestimated Potential of Organometallic Rhenium Complexes as Anticancer Agents. ACS Chem. Biol 2014, 9, 2180–2193. [DOI] [PubMed] [Google Scholar]
- (59).Leonidova A; Pierroz V; Rubbiani R; Heier J; Ferrari S; Gasser G Towards Cancer Cell-Specific Phototoxic Organometallic Rhenium(I) Complexes. Dalton Trans 2014, 43, 4287–4294. [DOI] [PubMed] [Google Scholar]
- (60).Wähler K; Ludewig A; Szabo P; Harms K; Meggers E Rhenium Complexes with Red-Light-Induced Anticancer Activity. Eur. J. Inorg. Chem 2014, 807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Quental L; Raposinho P; Mendes F; Santos I; Navarro-Ranninger C; Alvarez-Valdes A; Huang H; Chao H; Rubbiani R; Gasser G; Quiroga AG; Paulo A Combining Imaging and Anticancer Properties with New Heterobimetallic Pt(II)/M(I) (M = Re 99mTc) Complexes. Dalton Trans 2017, 46, 14523–14536. [DOI] [PubMed] [Google Scholar]
- (62).Koike K; Okoshi N; Hori H; Takeuchi K; Ishitani O; Tsubaki H; Clark IP; George MW; Johnson FPA; Turner JJ Mechanism of the Photochemical Ligand Substitution Reactions of fac-[Re(bpy)(CO)3(PR3)]+ Complexes and the Properties of Their Triplet Ligand-Field Excited States. J. Am. Chem. Soc 2002, 124, 11448–11455. [DOI] [PubMed] [Google Scholar]
- (63).Wrighton M; Morse DL The Nature of the Lowest Excited State in Tricarbonylchloro-1,10-phenanthrolinerhenium(I) and Related Complexes. J. Am. Chem. Soc 1974, 96, 998–1003. [Google Scholar]
- (64).Caspar JV; Meyer TJ Application of the Energy Gap Law to Nonradiative, Excited-State Decay. J. Phys. Chem 1983, 87, 952–957. [Google Scholar]
- (65).Kalyanasundaram K Luminescence and Redox Reactions of the Metal-to-Ligand Charge-Transfer Excited State of Tricarbonylchloro-(polypyridyl)rhenium(I) Complexes. J. Chem. Soc., Faraday Trans. 2 1986, 82, 2401–2415. [Google Scholar]
- (66).Kalyanasundaram K Mononuclear Complexes of Re(I). In Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press Ltd: London, 1992; pp 321–329. [Google Scholar]
- (67).Koike K; Tanabe J; Toyama S; Tsubaki H; Sakamoto K; Westwell JR; Johnson FPA; Hori H; Saitoh H; Ishitani O New Synthetic Routes to Biscarbonylbipyridinerhenium(I) Complexes cis,trans-[Re(X2bpy)(CO)2(PR3)(Y)]n+ (X2bpy = 4,4’-X2-2,2’-bipyridine) via Photochemical Ligand Substitution Reactions, and Their Photophysical and Electrochemical Properties. Inorg. Chem 2000, 39, 2777–2783. [DOI] [PubMed] [Google Scholar]
- (68).Pierri AE; Pallaoro A; Wu G; Ford PC A Luminescent and Biocompatible PhotoCORM. J. Am. Chem. Soc 2012, 134, 18197–18200. [DOI] [PubMed] [Google Scholar]
- (69).Kunz PC; Huber W; Rojas A; Schatzschneider U; Spingler B Tricarbonylmanganese(I) and –Rhenium(I) Complexes of Imidazol-Based Phosphane Ligands: Influence of the Substitution Pattern on the CO Release Properties. Eur. J. Inorg. Chem 2009, 5358–5366. [Google Scholar]
- (70).Pfeiffer H; Rojas A; Niesel J; Schatzschneider U Sonogashira and “Click” Reactions for the N-Terminal and Side-Chain Functionalization of Peptides with [Mn(CO)3(tpm)]+-Based CO Releasing Molecules (tpm = tris(pyrazolyl)methane). Dalton Trans 2009, 0, 4292–4298. [DOI] [PubMed] [Google Scholar]
- (71).Rimmer RD; Richter H; Ford PC A Photochemical Precursor for Carbon Monoxide Release in Aerated Aqueous Media. Inorg. Chem 2010, 49, 1180–1185. [DOI] [PubMed] [Google Scholar]
- (72).Brückmann NE; Wahl M; Reiß GJ; Kohns M; Wätjen W; Kunz PC Polymer Conjugates of Photoinducible CO-Releasing Molecules. Eur. J. Inorg. Chem 2011, 4571–4577. [Google Scholar]
- (73).Kretschmer R; Gessner G; Görls H; Heinemann SH; Westerhausen M Dicarbonyl-bis(cysteamine)iron(II): A Light Induced Carbon Monoxide Releasing Molecule Based on Iron (CORM-S1). J. Inorg. Biochem 2011, 105, 6–9. [DOI] [PubMed] [Google Scholar]
- (74).Schatzschneider U PhotoCORMs: Light-Triggered Release of Carbon Monoxide from the Coordination Sphere of Transition Metal Complexes for Biological Applications. Inorg. Chim. Acta 2011, 374, 19–23. [Google Scholar]
- (75).Zobi F; Spingler B; Fox T; Alberto R Toward Novel DNA Binding Metal Complexes: Structure and Basic Kinetic Data of [M(9MeG)2(CH3OH)(CO)3]+ (M = 99Tc, Re). Inorg. Chem 2003, 42, 2818–2820. [DOI] [PubMed] [Google Scholar]
- (76).Zobi F; Blacque O; Schmalle HW; Spingler B; Alberto R Head-to-Head (HH) and Head-to-Tail (HT) Conformers of cis-Bis Guanine Ligands Bound to the [Re(CO)3]+ Core. Inorg. Chem 2004, 43, 2087–2096. [DOI] [PubMed] [Google Scholar]
- (77).Zobi F; Spingler B; Alberto R Guanine and Plasmid DNA Binding of Mono- and Trinuclear fac-[Re(CO)3]+ Complexes with Amino Acid Ligands. ChemBioChem 2005, 6, 1397–1405. [DOI] [PubMed] [Google Scholar]
- (78).Knopf KM; Murphy BL; MacMillan SN; Baskin JM; Barr MP; Boros E; Wilson JJ In Vitro Anticancer Activity and in Vivo Biodistribution of Rhenium(I) Tricarbonyl Aqua Complexes. J. Am. Chem. Soc 2017, 139, 14302–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Niesel J; Pinto A; Peindy N’Dongo HW; Merz K; Ott I; Gust R; Schatzschneider U Photoinduced CO Release, Cellular Uptake and Cytotoxicity of a Tris(pyrazolyl)methane (tpm) Manganese Tricarbonyl Complex. Chem. Commun 2008, 1798–1800. [DOI] [PubMed] [Google Scholar]
- (80).Carrington SJ; Chakraborty I; Mascharak PK Rapid CO Release from a Mn(I) Carbonyl Complex Derived from Azopyridine upon Exposure to Visible Light and Its Phototoxicity toward Malignant Cells. Chem. Commun 2013, 49, 11254–11256. [DOI] [PubMed] [Google Scholar]
- (81).Carrington SJ; Chakraborty I; Bernard JML; Mascharak PK Synthesis and Characterization of a “Turn-on” photoCORM for Trackable CO Delivery to Biological Targets. ACS Med. Chem. Lett 2014, 5, 1324–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Carrington SJ; Chakraborty I; Bernard JML; Mascharak PK A Theranostic Two-Tone Luminescent PhotoCORM Derived from Re(I) and (2-Pyridyl)-benzothiazole: Trackable CO Delivery to Malignant Cells. Inorg. Chem 2016, 55, 7852–7858. [DOI] [PubMed] [Google Scholar]
- (83).Chakraborty I; Carrington SJ; Roseman G; Mascharak PK Synthesis, Structures, and CO Release Capacity of a Family of Water-Soluble PhotoCORMs: Assessment of the Biocompatibility and Their Phototoxicity toward Human Breast Cancer Cells. Inorg. Chem 2017, 56, 1534–1545. [DOI] [PubMed] [Google Scholar]
- (84).Kawahara B; Moller T; Hu-Moore K; Carrington S; Faull KF; Sen S; Mascharak PK Attenuation of Antioxidant Capacity in Human Breast Cancer Cells by Carbon Monoxide through Inhibition of Cystathionine β-Synthase Activity: Implications in Chemotherapeutic Drug Sensitivity. J. Med. Chem 2017, 60, 8000–8010. [DOI] [PubMed] [Google Scholar]
- (85).Fisher KJ; Alyea EC; Shahnazarian NA 31P NMR Study of the Water Soluble Derivatives of 1,3,5-Triaza-7-Phosphaadamantane (PTA). Phosphorus, Sulfur Silicon Relat. Elem 1990, 48, 37–40. [Google Scholar]
- (86).Darensbourg DJ; Ortiz CG; Kamplain JW A New Water-Soluble Phosphine Derived from 1,3,5-Triaza-7-Phosphaadamantane (PTA), 3,7-Diacetyl-1,3,7-Triaza-5-phosphabicyclo[3.3.1]nonane. Structural, Bonding, and Solubility Properties. Organometallics 2004, 23, 1747–1754. [Google Scholar]
- (87).Gonsalvi L; Peruzzini M 1,3,5-Triaza-7-Phosphaadamantane (PTA). In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd: Chichester, UK, 2010; pp 3–7. [Google Scholar]
- (88).Kurz P; Probst B; Spingler B; Alberto R Ligand Variations in [ReX(diimine)(CO)3] Complexes: Effects on Photocatalytic CO2 Reduction. Eur. J. Inorg. Chem 2006, 2966–2974. [Google Scholar]
- (89).Smieja JM; Kubiak CP Re(bipy-tBu)(CO)3Cl–improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem 2010, 49, 9283–9289. [DOI] [PubMed] [Google Scholar]
- (90).Giordano PJ; Wrighton MS The Nature of the Lowest Excited State in fac-Tricarbonylhalobis(4-phenylpyridine)rhenium(I) and fac-tricarbonylhalobis(4,4’-bipyridine)rhenium(I): Emissive Organometallic Complexes in Fluid Solution. J. Am. Chem. Soc 1979, 101, 2888–2897. [Google Scholar]
- (91).Dattelbaum DM; Martin RL; Schoonover JR; Meyer TJ Molecular and Electronic Structure in the Metal-to-Ligand Charge Transfer Excited States of fac-[Re(4,4’-X2bpy)(CO)3(4-Etpy)]+ (X = CH3, H, Co2Et). Application of Density Functional Theory and Time-Resolved Infrared Spectroscopy. J. Phys. Chem. A 2004, 108, 3518–3526. [Google Scholar]
- (92).Ramos LD; Sampaio RN; de Assis FF; de Oliveira KT; Homem-de-Mello P; Patrocinio AOT; Frin KPM Contrasting Photophysical Properties of Rhenium(I) Tricarbonyl Complexes Having Carbazole Groups Attached to the Polypyridine Ligand. Dalton Trans 2016, 45, 11688–11698. [DOI] [PubMed] [Google Scholar]
- (93).Gonçalves MR; Frin KPM Synthesis, Characterization, Photophysical and Electrochemical Properties of Rhenium(I) Tricarbonyl Diimine Complexes with Triphenylphosphine Ligand. Polyhedron 2017, 132, 20–27. [Google Scholar]
- (94).Fredericks SM; Luong JC; Wrighton MS Multiple Emissions from Rhenium(I) Complexes: Intraligand and Charge-Transfer Emission from Substituted Metal Carbonyl Cations. J. Am. Chem. Soc 1979, 101, 7415–7417. [Google Scholar]
- (95).Darensbourg DJ; Robertson JB; Larkins DL; Reibenspies JH Water-Soluble Organometallic Compounds. 7. Further Studies of 1,3,5-Triaza-7-Phosphaadamantane Derivatives of Group 10 Metals, Including Metal Carbonyls and Hydrides. Inorg. Chem 1999, 38, 2473–2481. [Google Scholar]
- (96).DuBois DL; Miedaner A Mediated Electrochemical Reduction of CO2. Preparation and Comparison of an Isoelectronic Series of Complexes. J. Am. Chem. Soc 1987, 109, 113–117. [Google Scholar]
- (97).Mohr F; Sanz S; Tiekink ERT; Laguna M Water-Soluble and Water-Stable Organometallic Gold(II) Complexes. Organometallics 2006, 25, 3084–3087. [Google Scholar]
- (98).Tsubaki H; Tohyama S; Koike K; Saitoh H; Ishitani O Effect of Intramolecular π–π and CH–π Interactions between Ligands on Structure, Electrochemical and Spectroscopic Properties of fac-[Re(bpy)(CO)3(PR3)]+ (bpy = 2,2’-bipyridine; PR3= trialkyl or triarylphosphines). Dalton Trans 2005, 385–395. [DOI] [PubMed] [Google Scholar]
- (99).Stahl L; Ernst RD Equilibria Studies Involving Ligand Coordination to “Open Titanocenes”: Phosphine and Pentadienyl Cone Angle Influences and the Existence of These Electron-Deficient Molecules. J. Am. Chem. Soc 1987, 109, 5673–5680. [Google Scholar]
- (100).Tolman CA Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev 1977, 77, 313–348. [Google Scholar]
- (101).Connick WB; Di Bilio AJ; Schaeffer WP; Gray HB The Red Form of [Re(phen)(CO)3(H2O)]-CF3SO3·H2O. Acta Crystallogr. Sect., C: Cryst. Struct. Commun 1999, C55, 913–916. [DOI] [PubMed] [Google Scholar]
- (102).Salignac B; Grundler PV; Cayemittes S; Frey U; Scopelliti R; Merbach AE; Hedinger R; Hegetschweiler K; Alberto R; Prinz U; Raabe G; Kölle U; Hall S Reactivity of the Organometallic fac-[(CO)3ReI(H2O)3]+ Aquaion. Kinetic and Thermodynamic Properties of H2O Substitution. Inorg. Chem 2003, 42, 3516–3526. [DOI] [PubMed] [Google Scholar]
- (103).Sacksteder L; Zipp AP; Brown EA; Streich J; Demas JN; DeGraff BA Luminescence Studies of Pyridine α-Diimine Rhenium(I) Tricarbonyl Complexes. Inorg. Chem 1990, 29, 4335–4340. [Google Scholar]
- (104).Tapolsky G; Duesing R; Meyer TJ Synthetic Control of Excited-State Properties in Ligand-Bridged Complexes of Rhenium(I). Intramolecular Energy Transfer by an Electron-Transfer/Energy-Transfer Cascade. Inorg. Chem 1990, 29, 2285–2297. [Google Scholar]
- (105).Worl LA; Duesing R; Chen P; Ciana L Della; Meyer, T. J. Photophysical Properties of Polypyridyl Carbonyl Complexes of Rhenium(I). J. Chem. Soc., Dalton Trans 1991, 849–858. [Google Scholar]
- (106).Wallace L; Rillema DP Photophysical Properties of Rhenium(I) Tricarbonyl Complexes Containing Alkyl- and Aryl-Substituted Phenanthrolines as Ligands. Inorg. Chem 1993, 32, 3836–3843. [Google Scholar]
- (107).Itokazu MK; Polo AS; de Faria DLA; Bignozzi CA; Iha NYM Syntheses and Spectroscopic Characterization of fac-[Re(CO)3(phen)(L)]PF6, L = trans- and cis-1,2-bis(4-pyridyl)ethylene. Inorg. Chim. Acta 2001, 313, 149–155. [Google Scholar]
- (108).Polo AS; Itokazu MK; Frin KM; de Toledo Patrocínio AO; Iha NYM Light Driven Trans-to-Cis Isomerization of Stilbene-like Ligands in fac-[Re(CO)3(NN)(trans-L)]+ and Luminescence of Their Photoproducts. Coord. Chem. Rev 2006, 250, 1669–1680. [Google Scholar]
- (109).Kalyanasundaram K; Grazel M Photosensitization and Photocatalysis Using Inorganic and Organometallic Compounds; Kalyanasundaram K, Grätzel M, Eds.; Catalysis by Metal Complexes; Springer Netherlands: Dordrecht, 1993; Vol. 14. [Google Scholar]
- (110).Balzani V; Juris A; Venturi M; Campagna S; Serroni S Luminescent and Redox-Active Polynuclear Transition Metal Complexes. Chem. Rev 1996, 96, 759–833. [DOI] [PubMed] [Google Scholar]
- (111).Kraljić I; Mohsni S El. A New Method for the Detection of Singlet Oxygen in Aqueous Solutions. Photochem. Photobiol 1978, 28, 577–581. [Google Scholar]
- (112).Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- (113).Adamo C; Jacquemin D The Calculations of Excited-State Properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev 2013, 42, 845–856. [DOI] [PubMed] [Google Scholar]
- (114).de Silva AP; Gunaratne HQN; Gunnlaugsson T; Huxley AJM; McCoy CP; Rademacher JT; Rice TE Signaling Recognition Events with Fluorescent Sensors and Switches. Chem. Rev 1997, 97, 1515–1566. [DOI] [PubMed] [Google Scholar]
- (115).Basolo F Early Kinetic Studies on CO Substitution Reactions of Metal Carbonyls. J. Organomet. Chem 1990, 383, 579–586. [Google Scholar]
- (116).Basolo F Associative Substitution Reactions of Metal Carbonyls. Inorg. Chim. Acta 1985, 100, 33–39. [Google Scholar]
- (117).Kurtz DA; Dhakal B; Donovan ES; Nichol GS; Felton GAN Non-Photochemical Synthesis of Re(diimine)(CO)2(L)Cl (L = Phosphine or Phosphite) Compounds. Inorg. Chem. Commun 2015, 59, 80–83. [Google Scholar]
- (118).Shen J-K; Gaob Y-C; Shib Q-Z; Basoloa F Kinetic Studies of CO Substitution of Metal Carbonyls in the Presence of O-Atom Transfer Reagents. Coord. Chem. Rev 1993, 128, 69–88. [Google Scholar]
- (119).Schutte M; Kemp G; Visser HG; Roodt A Tuning the Reactivity in Classic Low-Spin d6 Rhenium(I) Tricarbonyl Radiopharmaceutical Synthon by Selective Bidentate Ligand Variation (L,L’-bid; L,L’= N,N’, N,O, and O,O’ Donor Atom Sets) in fac-[Re(CO)3(L,L’-bid)(MeOH)]n Complexes. Inorg. Chem 2011, 50, 12486–12498. [DOI] [PubMed] [Google Scholar]
- (120).Kaplanis M; Stamatakis G; Papakonstantinou VD; Paravatou-Petsotas M; Demopoulos CA; Mitsopoulou CA Re(I) Tricarbonyl Complex of 1,10-Phenanthroline-5,6-Dione: DNA Binding, Cytotoxicity, Anti-Inflammatory and Anti-Coagulant Effects towards Platelet Activating Factor. J. Inorg. Biochem 2014, 135, 1–9. [DOI] [PubMed] [Google Scholar]
- (121).Balakrishnan G; Rajendran T; Murugan KS; Kumar MS; Sivasubramanian VK; Ganesan M; Mahesh A; Thirunalasundari T; Rajagopal S Interaction of Rhenium(I) Complex Carrying Long Alkyl Chain with Calf Thymus DNA: Cytotoxic and Cell Imaging Studies. Inorg. Chim. Acta 2015, 434, 51–59. [Google Scholar]
- (122).Medley J; Payne G; Banerjee HN; Giri D; Winstead A; Wachira JM; Krause JA; Shaw R; Pramanik SK; Mandal SK DNA-Binding and Cytotoxic Efficacy Studies of Organorhenium Pentylcarbonate Compounds. Mol. Cell. Biochem 2015, 398, 21–30. [DOI] [PubMed] [Google Scholar]
- (123).Llobet A; Doppelt P; Meyer TJ Redox Properties of Aqua Complexes of Ruthenium(II) Containing the Tridentate Ligands 2,2′:6′,2′′-Terpyridine and Tris(1-pyrazolyl)methane. Inorg. Chem 1988, 27, 514–520. [Google Scholar]
- (124).Dadci L; Elias H; Frey U; Hörnig A; Koelle U; Merbach AE; Paulus H; Schneider JS π-Arene Aqua Complexes of Cobalt, Rhodium, Iridium, and Ruthenium: Preparation, Structure, and Kinetics of Water Exchange and Water Substitution. Inorg. Chem 1995, 34, 306–315. [Google Scholar]
- (125).Motterlini R; Gonzales A; Foresti R; Clark JE; Green CJ; Winslow RM Heme Oxygenase-1 Derived Carbon Monoxide Contributes to the Suppression of Acute Hypertensive Responses In Vivo. Circ. Res 1998, 83, 568–577. [DOI] [PubMed] [Google Scholar]
- (126).Otterbein LE Carbon Monoxide: Innovative Anti-Inflammatory Properties of an Age-Old Gas Molecule. Antioxid. Redox Signaling 2002, 4, 309–319. [DOI] [PubMed] [Google Scholar]
- (127).Otterbein LE; Zuckerbraun BS; Haga M; Liu F; Song R; Usheva A; Stachulak C; Bodyak N; Smith RN; Csizmadia E; Tyagi S; Akamatsu Y; Flavell RJ; Billiar TR; Tzeng E; Bach FH; Choi AMK; Soares MP Carbon Monoxide Suppresses Arteriosclerotic Lesions Associated with Chronic Graft Rejection and with Balloon Injury. Nat. Med 2003, 9, 183–190. [DOI] [PubMed] [Google Scholar]
- (128).Neto JS; Nakao A; Kimizuka K; Romanosky AJ; Stolz DB; Uchiyama T; Nalesnik MA; Otterbein LE; Murase N Protection of Transplant-Induced Renal Ischemia-Reperfusion Injury with Carbon Monoxide. Am. J. Physiol. Renal, Fluid Electrolyte Physiol 2004, 287, F979–F989. [DOI] [PubMed] [Google Scholar]
- (129).Kim HP; Ryter SW; Choi AMK CO as a Cellular Signaling Molecule. Annu. Rev. Pharmacol. Toxicol 2006, 46, 411–449. [DOI] [PubMed] [Google Scholar]
- (130).Bilban M; Haschemi A; Wegiel B; Chin BY; Wagner O; Otterbein LE Heme Oxygenase and Carbon Monoxide Initiate Homeostatic Signaling. J. Mol. Med 2008, 86, 267–279. [DOI] [PubMed] [Google Scholar]
- (131).Nakao A; Kaczorowski DJ; Sugimoto R; Billiar TR; McCurry KR Application of Heme Oxygenase-1, Carbon Monoxide and Biliverdin for the Prevention of Intestinal Ischemia/Reperfusion Injury. J. Clin. Biochem. Nutr 2008, 42, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Halilovic A; Patil KA; Bellner L; Marrazzo G; Castellano K; Cullaro G; Dunn MW; Schwartzman ML Knockdown of Heme Oxygenase-2 Impairs Corneal Epithelial Cell Wound Healing. J. Cell. Physiol 2011, 226, 1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Tavares AFN; Teixeira M; Romão CC; Seixas JD; Nobre LS; Saraiva LM Reactive Oxygen Species Mediate Bactericidal Killing Elicited by Carbon Monoxide-Releasing Molecules. J. Biol. Chem 2011, 286, 26708–26717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Wu M-L; Ho Y-C; Yet S-F A Central Role of Heme Oxygenase-1 in Cardiovascular Protection. Antioxid. Redox Signaling 2011, 15, 1835–1846. [DOI] [PubMed] [Google Scholar]
- (135).Gullotta F; di Masi A; Ascenzi P Carbon Monoxide: An Unusual Drug. IUBMB Life 2012, 64, 378–386. [DOI] [PubMed] [Google Scholar]
- (136).Ling K; Men F; Wang W-C; Zhou Y-Q; Zhang H-W; Ye D-W Carbon Monoxide and Its Controlled Release: Therapeutic Application, Detection, and Development of Carbon Monoxide Releasing Molecules (CORMs). J. Med. Chem 2017, doi: 10.1021/acs.jmedchem.6b01153. [DOI] [PubMed] [Google Scholar]
- (137).Jackson CS; Schmitt S; Dou QP; Kodanko JJ Synthesis, Characterization, and Reactivity of the Stable Iron Carbonyl Complex [Fe(CO)(N4Py)](ClO4)2: Photoactivated Carbon Monoxide Release, Growth Inhibitory Activity, and Peptide Ligation. Inorg. Chem 2011, 50, 5336–5338. [DOI] [PubMed] [Google Scholar]
- (138).Peng P; Wang C; Shi Z; Johns VK; Ma L; Oyer J; Copik A; Igarashi R; Liao Y Visible-Light Activatable Organic CO-Releasing Molecules (PhotoCORMs) That Simultaneously Generate Fluorophores. Org. Biomol. Chem 2013, 11, 6671–6674. [DOI] [PubMed] [Google Scholar]
- (139).Romão CC; Vieira HL A. Metal Carbonyl Prodrugs: CO Delivery and Beyond. In Bioorganometallic Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp 165–202. [Google Scholar]
- (140).Loboda A; Jozkowicz A; Dulak J HO-1/CO System in Tumor Growth, Angiogenesis and Metabolism – Targeting HO-1 as an Anti-Tumor Therapy. Vascul. Pharmacol 2015, 74, 11–22. [DOI] [PubMed] [Google Scholar]
- (141).Üstün E; Özgür A; Coşkun KA; Demir S; Özdemir İ; Tutar Y CO-Releasing Properties and Anticancer Activities of Manganese Complexes with Imidazole/benzimidazole Ligands. J. Coord. Chem 2016, 69, 3384–3394. [Google Scholar]
- (142).Boczkowski J; Poderoso JJ; Motterlini R CO–Metal Interaction: Vital Signaling from a Lethal Gas. Trends Biochem. Sci 2006, 31, 614–621. [DOI] [PubMed] [Google Scholar]
- (143).Wegiel B; Gallo D; Csizmadia E; Harris C; Belcher J; Vercellotti GM; Penacho N; Seth P; Sukhatme V; Ahmed A; Pandolfi PP; Helczynski L; Bjartell A; Persson JL; Otterbein LE Carbon Monoxide Expedites Metabolic Exhaustion to Inhibit Tumor Growth. Cancer Res 2013, 73, 7009–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Rabek JF Photophysics in the Membrane Environment. In Photophysics and Photochemistry, Volume 3; 1991; Vol. 3, pp 157–161. [Google Scholar]
- (145).Hepples C; Murphy GK Synthesis of 3,3-Dichloro-2-Oxindoles from Isatin-3-p-Tosylhydrazones and (Dichloroiodo)benzene. Tetrahedron Lett 2015, 56, 4971–4974. [Google Scholar]
- (146).Sheldrick GM Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect., C: Cryst. Struct. Commun 2015, C71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Sheldrick GM A Short History of SHELX. Acta Crystallogr. Sect., A: Found. Crystallogr 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- (148).Müller P Practical Suggestions for Better Crystal Structures. Crystallogr. Rev 2009, 15, 57–83. [Google Scholar]
- (149).Li Z; Pierri AE; Huang PJ; Wu G; Iretskii AV; Ford PC Dinuclear PhotoCORMs: Dioxygen-Assisted Carbon Monoxide Uncaging from Long-Wavelength-Absorbing Metal–Metal-Bonded Carbonyl Complexes. Inorg. Chem 2017, 56, 6094–6104. [DOI] [PubMed] [Google Scholar]
- (150).Brouwer AM Standards for Photoluminescence Quantum Yield Measurements in Solution (IUPAC Technical Report). Pure Appl. Chem 2011, 83, 2213–2228. [Google Scholar]
- (151).Montalti M; Credi A; Luca P; Gandolfi MT Chemical Actinometry. In Handbook of Photochemistry, Third Edition; CRC Press, 2006; pp 601–616. [Google Scholar]
- (152).Schmidt R; Tanielian C; Dunsbach R; Wolff C Phenalenone, a Universal Reference Compound for the Determination of Quantum Yields of Singlet Oxygen O2 (1Δg) Sensitization. J. Photochem. Photobiol. A Chem 1994, 79, 11–17. [Google Scholar]
- (153).Neese F The ORCA Program System. WIREs Comput. Mol. Sci 2012, 2, 73–78. [Google Scholar]
- (154).Pantazis DA; Chen X-Y; Landis CR; Neese F All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput 2008, 4, 908–919. [DOI] [PubMed] [Google Scholar]
- (155).van Lenthe E; van der Avoird A; Wormer PES Density Functional Calculations of Molecular Hyperfine Interactions in the Zero Order Regular Approximation for Relativistic Effects. J. Chem. Phys 1998, 108, 4783–4796. [Google Scholar]
- (156).van Wüllen C Molecular Density Functional Calculations in the Regular Relativistic Approximation: Method, Application to Coinage Metal Diatomics, Hydrides, Fluorides and Chlorides, and Comparison with First-Order Relativistic Calculations. J. Chem. Phys 1998, 109, 392–399. [Google Scholar]
- (157).Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (158).Adamo C; Barone V Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys 1999, 110, 6158–6170. [Google Scholar]
- (159).Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (160).Godwin AK; Meister A; O’Dwyer PJ; Huang CS; Hamilton TC; Anderson ME High Resistance to Cisplatin in Human Ovarian Cancer Cell Lines is Associated with Marked Increase of Glutathione Synthesis. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 3070–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).OECD Guideline For Testing of Chemicals: In Vitro 3T3 NRU Phototoxicity Test https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecdtg432-508.pdf (accessed Dec 7, 2017).
- (162).Weekes DM; Ramogida CF; Jaraquemada-Peláez MDG; Patrick BO; Apte C; Kostelnik TI; Cawthray JF; Murphy L; Orvig C Dipicolinate Complexes of Gallium(III) and Lanthanum(III). Inorg. Chem 2016, 55, 12544–12558. [DOI] [PubMed] [Google Scholar]
- (163).Equilibrium Constants from Solution Spectrophotometric Data http://www.hyperquad.co.uk/HypSpec.htm (accessed Dec 12, 2017).
- (164).Alderighi L; Gans P; Ienco A; Peters D; Sabatini A; Vacca A Hyperquad Simulation and Speciation (HySS): A Utility Program for the Investigation of Equilibria Involving Soluble and Partially Soluble Species. Coord. Chem. Rev 1999, 184, 311–318. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.