

1. Introduction
The developing fetus undergoes a dynamic period of rapid growth that is responsive to changes in the in utero environment that may contribute to the development of fetal growth restriction (FGR) or fetal overgrowth resulting in the delivery of a large for gestational age (LGA) infant1. Pregnancies complicated by abnormal fetal growth have a major impact on public health due to the increased perinatal morbidity and mortality associated with FGR and LGA and because abnormal fetal growth is strongly associated with long-term health consequences for offspring including the development of cardiovascular disease, metabolic dysfunction, and obesity later in life2–4. An array of conditions, including changes in maternal nutrient, endocrine, and metabolic status and impaired uteroplacental blood flow are associated with altered fetal growth.5 However, the precise mechanisms causing changes in the growth trajectory of the fetus remain to be fully established. Accumulating evidence suggests that changes in placental function may contribute to, or mediate, both FGR and fetal overgrowth.
Early clinical studies of fetal growth were instrumental in establishing a link between impaired utero-placental blood flow and reduced fetal weight as a result of placental insufficiency6,7. While impaired utero-placental blood flow is an established risk factor for restricted fetal growth, placental insufficiency is more than reduced blood flow and may involve decreased transplacental nutrient transport capacity, altered activity in placental growth factor and inflammatory signaling pathways, and changes in the release of placental extracellular vesicles and their cargo. Placental cell signaling pathways, such as mechanistic target of rapamycin (mTOR), insulin-like growth factor (IGF), adipokine signaling and nutrient transport are regulated by oxygen and nutrient levels as well as maternal metabolic hormones. Changes in placental function therefore link the availability of oxygen and nutrients for fetal growth to the fetal growth trajectory, in some cases resulting in FGR or at the opposite end of the growth spectrum, LGA8,9.
In this review, we will discuss recent work demonstrating compelling associations between changes in placental function and fetal growth. In particular, we will focus on distinct differences in transplacental nutrient transport, cellular signaling pathways, inflammatory markers, and extracellular vesicle regulatory functions that are unique to placentas of LGA and FGR infants. Furthermore, we will discuss novel clinical interventions specifically targeting placental function as an avenue to rescue disordered fetal growth patterns. Lastly, we will speculate on future research and intervention priorities to prevent adverse infant outcomes in pregnancies complicated by altered placental function and fetal growth.
2. Human Placenta
The human placenta is derived from the fetal trophectoderm, the outermost layer of the blastocyst. After implantation, the trophectoderm differentiates into mononuclear villous cytotrophoblasts that can further differentiate into either extravillous trophoblast or they can fuse to form the multinucleated syncytiotrophoblast (STB). The extravillous trophoblast proliferate and invade the myometrium to reach the spiral arteries where they eventually replace smooth muscle cells of the arterial media with eosinophilic materials resulting in decreased vasoreactivity, allowing for the marked increase in uteroplacental blood flow throughout gestation that characterizes a normal pregnancy. During the first weeks after implantation, cytotrophoblasts rapidly proliferate to form the functional unit of the placenta, the trophoblast villous tree, which is lined with a continuous outer layer of STB. Following the onset of the uteroplacental circulation in late first trimester, the STB are directly exposed to maternal blood in the intervillous space, and serve as the primary barrier between maternal and fetal blood supply as well as the site of placental hormone production and maternal-fetal oxygen, nutrient, and ion transfer10,11.
Maternal-fetal exchange occurs across two largely continuous cell layers, the fetal capillary endothelium and STB, which separate maternal and fetal blood supplies. Fetal capillary endothelial cells allow largely unrestricted transfer of small molecules such as glucose, amino acids, and ions through intercellular junctions12, making STB the limiting factor for maternal-fetal solute exchange. In contrast, although the mechanisms involved remain to be fully established, transplacental transfer of lipids likely requires transport across both STB and fetal capillary endothelium13. The STB consists of two polarized plasma membranes, the apical or maternal facing microvillous plasma membrane (MVM), and the fetal facing basal plasma membrane (BM). The MVM and BM express an array of different transporter proteins critical for mediating vectorial maternal-to-fetal transfer of nutrients14 and fetal-to-maternal transfer of waste products (Figure 1).
Figure 1. Some key placental signaling pathways and nutrient transporters.
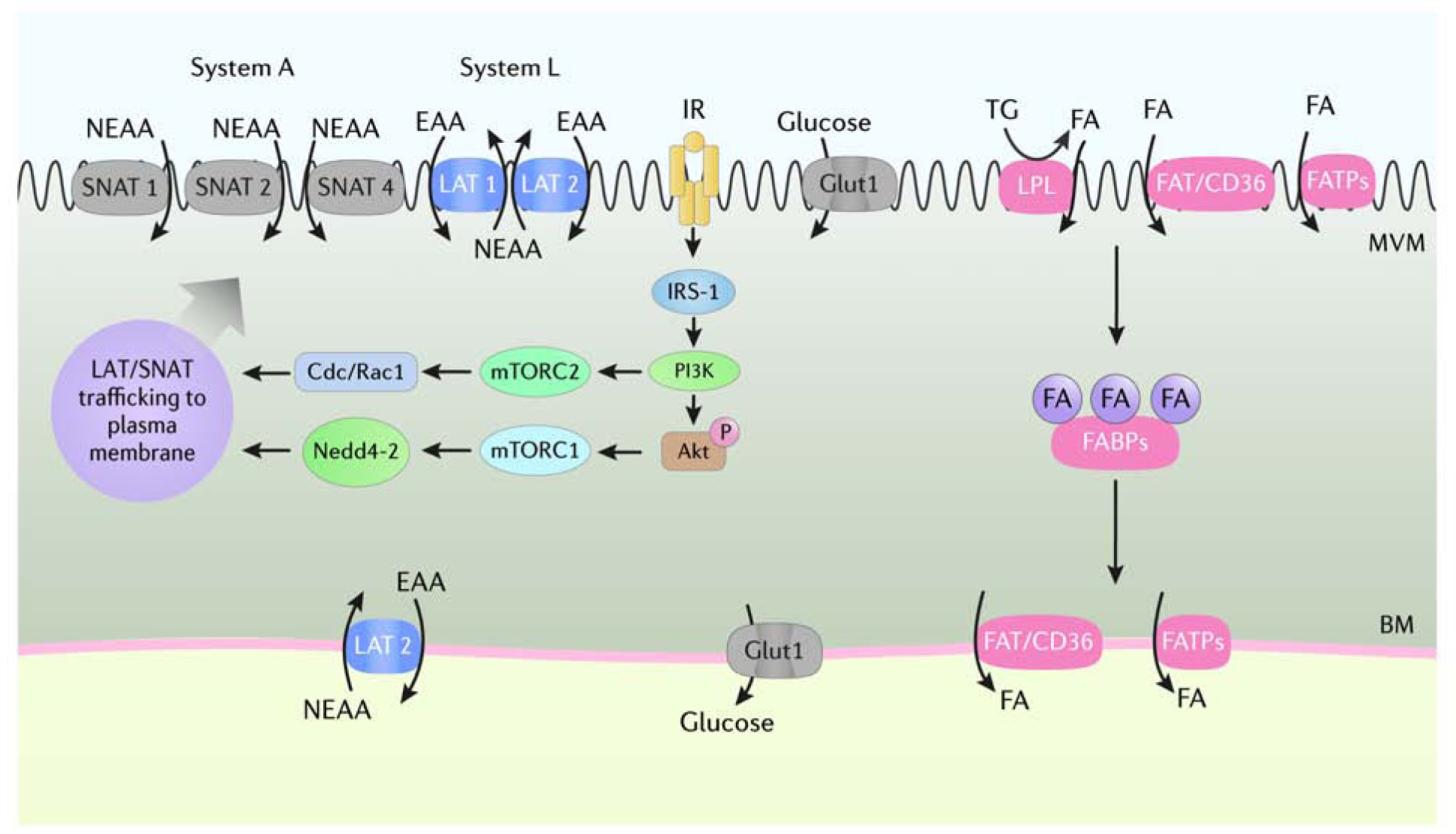
The syncytiotrophoblast consists of two polarized plasma membranes, microvillous plasma membrane (MVM) and basal membrane (BM), that express an array of transport proteins that mediate maternal-to-fetal transfer of amino acids, glucose, and fatty acids. The uptake of nonessential and essential amino acids from maternal circulation across the MVM is mediated by System A (SNAT1, 2, 4) and System L (LAT1, 2) transport systems that are trafficked to the plasma membrane as a result of activation of insulin/IGF-1 and mTOR signaling. GLUT-1 is highly expressed in the MVM and BM of the syncytiotrophoblast and is considered the primary glucose transporter in the human placenta at term. Maternal triglycerides are hydrolyzed into FFA by membrane-bound lipases and transferred across the MVM by FAT/CD36 and FATPs. Internalized FFA are transferred to the BM by FABPs for export into fetal circulation. Akt, Protein Kinase B; Cdc/Rac1, cell division control protein/ras-related C3 botulinum toxin substrate 1; EAA, essential amino acids; FA, fatty acids; FABPs, fatty acid binding proteins; FAT/CD36, fatty acid translocase/cluster of differentiation 36; FATPs, fatty acid transport proteins; GLUT1, glucose transporter 1; IGF-1, insulin-like growth factor; IR, insulin receptor; IRS-1, insulin receptor substrate 1; LAT 1, L-amino acid transporter 1, 2; LPL, lipoprotein lipase, mTORC1, mechanistic target of rapamycin complex 1; mTORC2, mechanistic target of rapamycin complex 2; Nedd4–2, neuronal precursor cell-expressed, developmentally down-regulated gene 4 isoform 2; NEAA, non-essential amino acids; SNAT 1, Sodium-coupled neutral amino acid transporter 1, 2, 4; TG, triglycerides. Courtesy of KIMEN Design4Research, with permission.
3. Placental Signaling
Placental receptors for many metabolic hormones and growth factors, including receptors for insulin15, IGF-116, and adiponectin17 are localized on the MVM of syncytiotrophoblast, mediating regulation of placental function by maternal circulating factors. The coordinated actions of insulin/IGF-1and adiponectin through downstream mTOR signaling act to regulate mitochondrial function, protein synthesis and the flux of glucose, amino acids, lipids, and folate across the placental barrier. Importantly, these signaling pathways are differentially regulated in pregnancies complicated by fetal overgrowth and FGR (Figure 2).
Figure 2. Placental signaling in fetal overgrowth and FGR.
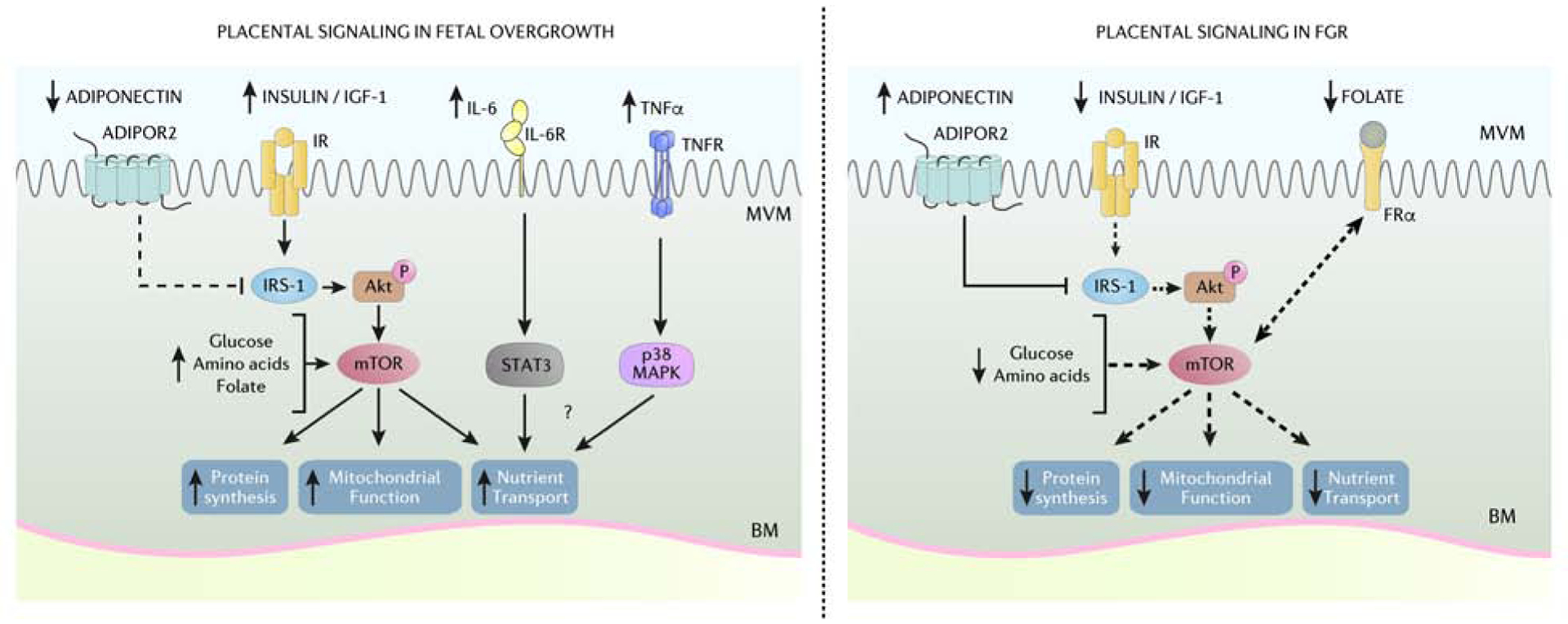
The coordinated actions of placental insulin/IGF-1, mTOR, adiponectin, and inflammatory cytokine signaling pathways act to regulate mitochondrial function, protein synthesis and the flux of glucose, amino acids, lipids, and folate across the placental barrier. Placental insulin/IGF-1 and mTOR signaling is activated in women with obesity delivering LGA infants likely due to elevated circulating maternal insulin/IGF-1 and increased availably of nutrients. Moreover, low maternal levels of circulating adiponectin in pregnancies complicated by maternal obesity contribute to enhance placental insulin signaling due to decreased inhibition of IRS-1. Increased maternal circulating proinflammatory cytokines IL-6 and TNF-α are increased in pregnancies with obesity and may contribute to fetal overgrowth by activating STAT3 and p38 MAPK signaling pathways. Conversely, lower maternal circulating insulin/IGF-1, folate, and increased adiponectin contribute to reduced placental insulin/IGF-1 and mTOR signaling in FGR pregnancies. Thus, differential regulation of placental signaling pathways and subsequent impact on placental function and nutrient transfer likely contribute to fetal overgrowth and FGR. ADIPOR2, adiponectin receptor 2; Akt, Protein Kinase B; BM, basal membrane; FRα, folate receptor-α; IGF-1, insulin-like growth factor 1; IR, insulin receptor; IRS-1, insulin receptor substrate 1; IL-6, interleukin-6; IL-6R, interleukin-6 receptor; FGR, fetal growth restriction; mTORC, mechanistic target of rapamycin complex; MVM, microvillous membrane; p38 MAPK, p38 mitogen-activated protein kinases; STAT3, Signal transducer and activator of transcription 3; TNF-α, tumor necrosis factor-α; TNFR, tumor necrosis factor-α receptor. Courtesy of KIMEN Design4Research, with permission.
3.1. Insulin/IGF-1 and mTOR signaling
The insulin/IGF-1 signaling pathway is primarily comprised of a system of ligands (IGF-1, IGF-2, and insulin), tyrosine kinase receptors [IGF-1 receptor and insulin receptor isoforms (INSR) A and B], and downstream activation of target proteins insulin receptor substrate 1 (IRS-1), protein kinase B (Akt), and mTOR. Activation of placental insulin/IGF-1 signaling is crucial for normal trophoblast function and fetal growth and development by promoting hormone synthesis, protein synthesis, and nutrient transfer, in part as a result of mTOR activation18,19. During pregnancy, IGF-1 availability in the maternal circulation and at the maternal-fetal interface is primarily regulated by IGF binding proteins such as IGFBP-1 synthesized by the decidua20. Phosphorylation of IGFBP-1 at serine residues (Ser101, 119, and 169) markedly increases IGFBP-1 binding affinity for IGF-1, effectively reducing IGF-1 availability and function21. FGR pregnancies are associated with reduced maternal serum IGF-1 concentrations and increased abundance22 and phosphorylation23,24 of IGFBP-1. In addition, decreased placental IGF-1 expression has been reported in human FGR25, which may contribute to the inhibition of placental insulin/IGF-1 signaling pathway in this pregnancy complication26–28. Conversely, placental insulin/IGF-1 signaling is enhanced in women with obesity29 or gestational diabetes (GDM)30 delivering a large infant. There are likely multiple factors underlying the activation of placental insulin/IGF-1 signaling in fetal overgrowth, including elevated maternal IGF-122 and lower circulating levels of adiponectin in the mother31, as discussed below.
Activation of mTOR complex 1 (mTORC1) occurs by several mechanisms including insulin/IGF-1 ligand binding and increased availability of ATP, amino acids, fatty acids, folate, and glucose, whereas mTOR complex 2 (mTORC2) primarily responds to insulin/PI3K (Akt) signaling32. In the placenta, mTORC1 serves as a positive regulator of amino acid33 and folate transport34, and mitochondrial biogenesis35. On the other hand, mTORC2 promotes cell proliferation by phosphorylation of Akt, PKCα, and SGK1, which regulate cytoskeletal remodeling and cell migration32. Placental mTORC1 activity has been found to be closely related to fetal growth. A consistent decrease in placental mTORC1 activity has been reported in humans27,36,37 and animal models38,39 of FGR whereas human GDM40 and obesity29 and rodent models of fetal overgrowth41,42 are often associated with placental mTORC1 activation. Further, inhibition of mTOR in decidual cells increased the release of hyperphosphorylated IGFBP-1, which decreases IGF-1 bioavailability and is associated with restricted fetal growth43. Likewise, mTORC1 is inhibited by AMPK, a critical nutrient sensor that is activated when ATP levels are depleted44. Placental mTORC1 activity is positively correlated to infant birthweight whereas placental AMPK activity shows an inverse relationship, suggesting that placental mTORC1 signaling pathway constitutes an important link between placental nutrient status and fetal growth29.
3.2. Adiponectin signaling
As in non-pregnant individuals, circulating levels of adiponectin are inversely correlated to BMI in pregnant women. Moreover, maternal serum adiponectin is negatively associated with birth weight45. In agreement with these associations, low adiponectin increases the risk of fetal overgrowth31,46–48, whereas maternal adiponectin tends to be increased in FGR45. Adiponectin is the most abundant protein secreted from adipose tissue and has well-documented insulinsensitizing effects in adipose, liver, and skeletal muscle tissues49. Adiponectin binds to adiponectin receptor (AdipoR) 1 and AdipoR2 that activate downstream AMPK, p38 MAPK, and/or PPARα50. Surprisingly, adiponectin blunts insulin signaling in primary human trophoblast cells51 by PPARα mediated synthesis of ceramide, which phosphorylates IRS-1 at an inhibitory site52. These findings are consistent with the possibility that low circulating adiponectin, a feature of maternal obesity and GDM, is mechanistically linked to activation of placental insulin/IGF-1/mTOR signaling, stimulating fetal growth. In support of this model, adiponectin supplementation prevented the adverse effects of maternal obesity on placental function and fetal growth in mice41. Moreover, normalization of maternal circulating adiponectin levels in obese pregnant mice largely prevented the development of cardiac53 and metabolic disease54 in adult progeny, implicating low maternal adiponectin levels as a mechanism underpinning fetal overgrowth and developmental programming of cardiovascular and metabolic disease.
4. Inflammation
Pregnancy is characterized by a tightly regulated balance between pro- and anti-inflammatory cytokines necessary for implantation and placentation. Although results of studies exploring the effect of maternal obesity and GDM on maternal cytokine levels are inconsistent55, women with obesity56, GDM57, or preeclampsia58 generally are thought to have increased circulating pro-inflammatory cytokines, such as IL-6 and TNF-α. Placental cytokine production, which is critical for the maintenance of pregnancy from implantation to parturition59, is altered in pregnancies complicated with obesity and GDM55. Maternal BMI is positively associated with activation of distinct placental inflammatory pathways including p38 MAPK and STAT3 signaling without changes in classical inflammatory pathways or fetal cytokine profile60. These findings suggest that maternal and placental inflammation in maternal obesity and GDM may impact fetal development by altering placental function rather than direct fetal exposure to elevated levels of proinflammatory cytokines.
The transcription of placental IL-6 and TNF-α mRNA is increased in maternal obesity61, which may promote placental lipid and amino acid transfer. In cultured primary human trophoblasts, IL-6 was shown to upregulate STAT3 dependent System A amino acid transport activity through increased SNAT2 expression62, whereas TNF-α stimulated System A amino acid transport through activation of p38 MAPK signaling63. On the other hand, IL-1β down regulates insulin-stimulated System A transport but activates System L activity in primary trophoblasts64. Collectively, data from in vitro experiments demonstrate a mechanistic link between placental response to inflammation and altered nutrient transport. However, the cumulative effects of inflammation on placental nutrient transport are complex and may vary depending on the degree of inflammation and specific cytokines that are elevated.
5. Nutrient Transport
5.1. Glucose
Glucose is the primary energy substrate for the placenta and the fetus. Fetoplacental glucose needs are met entirely by placental uptake from the maternal circulation via facilitated diffusion and in response to increased postprandial maternal glucose levels. Glucose transporter (GLUT)-1 is highly expressed in the MVM and BM of the STB, however, GLUT-1 localized in the BM is considered the primary glucose transporter in the human placenta at term65. BM GLUT-1 expression is associated with birthweight and is increased in pregnancies complicated by obesity and fetal overgrowth66. Similarly, GLUT-4 expression and translocation to the BM is upregulated by insulin, which may enhance glucose transport in response to postprandial hyperinsulinemia15. MVM and BM GLUT-1 protein expression and activity appear not to be affected in FGR65, however, reduced MVM GLUT-1 protein, activity, and glucose transfer have been reported in preeclampsia67. Some FGR fetuses are hypoglycemic in utero, this may becaused by impaired uteroplacental blood flow, increased placental glucose consumption or altered glycolytic pathways21,68.
5.2. Amino acids
Placental uptake of amino acids is mediated by an array of distinct transporter systems. Of these, System A and System L are believed to be some of the most important, in part because these transporters are subjected to regulation. System A is a sodium-dependent transporter mediating the uptake of non-essential neutral amino acids into the cell. System A is comprised of three isoforms, which are all expressed in the human placenta: SNAT 1 (SLC38A1), SNAT 2 (SLC38A2), and SNAT 4 (SLC38A4)69. The System L amino acid transporter is a heterodimer, consisting of a light chain, typically LAT1 (SLC7A5) or LAT2 (SLC7A8), and a heavy chain, 4F2hc/CD98 (SLC3A2). System L is a sodium-independent exchanger mediating cellular uptake of essential amino acids, including leucine, in exchange for non-essential System A substrates such as glycine. As a result, the coordinated activity of both System A and System L activity is required for placental transport of non-essential and essential amino acids across pregnancy70. Activation of trophoblast System A and System L amino acid transport occurs as a direct result of upstream mTORC1 and mTORC2 activation33. mTORC1 modulates SNAT2 and LAT1 trafficking by Nedd4–2-regulated ubiquitination in normal term pregnancies69, whereas mTORC2 regulates amino acid transporter trafficking through interactions with Cdc42 and Rac1 which are down regulated in FGR71. Placental amino acid transport capacity has been consistently shown to be reduced in human FGR37,72–77. We have reported that down-regulation of key placental amino acid transport systems precedes the development of FGR in rodents38,78 and non-human primates39,79,80, supporting the concept that down-regulation of placental nutrient transport is a primary event, which directly contributes to FGR, rather than a response to a changing fetal demand. Conversely, as previously discussed, activation of placental amino acid transport systems due to upstream activation of mTOR signaling by elevated maternal hormones (insulin/IGF, leptin) low adiponectin, excess nutrients, and a mild proinflammatory activation may contribute fetal overgrowth in pregnancies complicated by obesity29,42,52,62.
5.3. Lipids
Placental uptake of lipids from the maternal circulation is mediated by a number of membrane-bound transport proteins and lipases. Triglycerides (TG) packaged in circulating lipoproteins (VLDL, LDL, chylomicrons) are hydrolyzed into non-esterified fatty acids (NEFA) and glycerol by lipoprotein lipase (LPL) and endothelial lipase (EL) prior to entering the syncytiotrophoblast. Long chain fatty acids are transported across the MVM by fatty acid transport proteins (FATP) and FAT/CD3681. Once internalized, free fatty acids are esterified with coenzyme A (CoA) producing acyl-CoAs that are trafficked by cytosolic fatty acid binding proteins for use in placental mitochondrial respiration, incorporation into lipid droplets for placental storage, or transferred to the BM for export82. Maternal obesity is generally believed to increase lipid transport possibly contributing to greater fetal adipogenesis and birthweight61. While the relationship between maternal obesity, placental lipid transport, and fetal growth remains incompletely understood, maternal obesity is associated with decreased placental total FATP1 and 4 mRNA and increased FATP6 and FAT/CD36 protein content83. FATP2 and 4 expression is greater on the BM compared to the MVM in the human placenta with BM FATP2 expression positively correlated to maternal BMI84. Conversely, FAT/CD36 expression is higher in the MVM compared to the BM, but was not associated with maternal BMI84. Increased BM FATP2 expression may reflect increased transplacental lipid transfer, however direct evidence demonstrating an association between increased BM FATP expression and fetal overgrowth is currently lacking. Maternal obesity is also associated with reduced placental EL expression and increased placental storage of long chain polyunsaturated fatty acids (LCPUFA), which may limit transplacental transport of LCPUFA fatty acid species in maternal obesity83.
Pregnancies complicated by FGR are also associated with changes in placental lipid transport mechanisms. Placental EL mRNA and MVM LPL activity have been reported to be decreased, yet placental LPL gene expression is elevated85,86, in FGR. The divergence between LPL activity and mRNA expression may reflect that regulation of LPL mediated lipid transfer does not occur at the transcriptional level21. MVM FATP6 and CD36 protein levels are increased in FGR, and LCPUFA appear to be preferentially routed to storage in placental TG suggesting a possible defect in intracellular LCPUFA trafficking and export87. Further, fetal and placental weights and placental expression of genes related to fatty acid mobilization and TG content were not altered in mice deficient in FATP2 and 488. Interestingly, protein expression of placental FATP1, 2, 4, 5, and 6 is increased across late gestation in a baboon model of FGR, suggesting a coordinated placental adaptation to facilitate fatty acid transport89. However, the potential overlap and redundancy of FATPs involved in placental fatty acid trafficking throughout gestation remains unresolved and warrants further investigation. The expression of low-density lipoprotein protein receptor (LDLr) mRNA and scavenger receptor class B type I (SR-B1) protein, which are membrane bound proteins responsible for the uptake of LDL and HDL from maternal circulation, are reduced in FGR placentas. However, whether changes in placental cholesterol transport in FGR influence fetal development is not fully elicited90.
5.4. Folate
Folate has a well characterized role in DNA replication, amino acid metabolism, and as a methyl donor. Folate deficiency during pregnancy is associated with FGR and congenital abnormalities such as neural tube defects. Interestingly, mTOR functions as a folate sensor in the trophoblast and other cell types34,91 and maternal folate deficiency in mice inhibits placental mTORC1 and 2 signaling and placental amino acid transport contributing to FGR91. Furthermore, both mTORC1 and 2 are positive regulators of trophoblast folate uptake by modulating the cell surface expression of folate receptor-α (FR-α) and the reduced folate carrier (RFC)34,92. Indeed, placental folate transport capacity is decreased in human FGR93, which may contribute to the restricted fetal growth and intrauterine programming of childhood and adult disease.
6. Small Extracellular Vesicles & microRNA
Extracellular vesicles are membrane-bound particles containing bioactive proteins, lipids, DNA, mRNA, and microRNA (miR) that are secreted from most cells and participate in cell to cell communication. Small extracellular vesicles (sEV), commonly referred to as exosomes, are small (<150 nm in diameter) nanovesicles produced from the endosomal pathway and released upon fusion of multivesicular bodies with the plasma membrane. Due to a lack of specific subtypes and mixed origins, the International Society of Extracellular Vesicles has recommended the use of size to categorize extracellular vesicles; therefore, we will refer to this heterogenous population as sEV94.
Currently little is known about the relationship between sEVs, placental or originating from other tissues, and fetal growth. However, mice infused with human total and/or placental sEVs from women with preeclampsia developed hypertension95 and mice infused with sEVs from GDM women became glucose intolerant and insulin resistant96. Therefore, it is highly plausible that sEVs regulate fetal growth, at least secondarily to changes in maternal physiology (Figure 3). Indirect support of this hypothesis is found in the differential expression of sEV miR isolated from maternal serum in the second trimester being correlated with birth weight97. Further, the fraction of total circulating sEVs that are of placental origin is reduced in FGR, likely due to the fact that the levels of circulating placental sEVs was correlated with placental weight, suggesting that decreased release of placental sEV may be a result of reduced placental size98. The functions of placental sEVs remain to be fully established but are believed to be involved in immune response, angiogenesis, and the transfer of nucleic acids and proteins important in normal and complicated pregnancies99. Placental sEVs containing miR-520c-3p have been reported to inhibit CD44/HA-mediated extravillous trophoblast invasion, suggesting a link between placental sEV and placentation100. sEVs isolated from second trimester cytotrophoblasts contain a significant amount of TNF-α and increase decidual stromal cell transcription and secretion of NF-κB targets, including IL-8101. Lastly, placental sEVs from GDM women promote endothelial cytokine release when compared to placenta derived sEVs from normal pregnancies102, in agreement with previous studies demonstrating that placental sEV mediate monocyte recruitment and induce IL-1beta production103. A direct link between sEVs of maternal or placental origin and abnormal fetal growth remains to be established.
Figure 3. The influence of small extracellular vesicles on placental function.
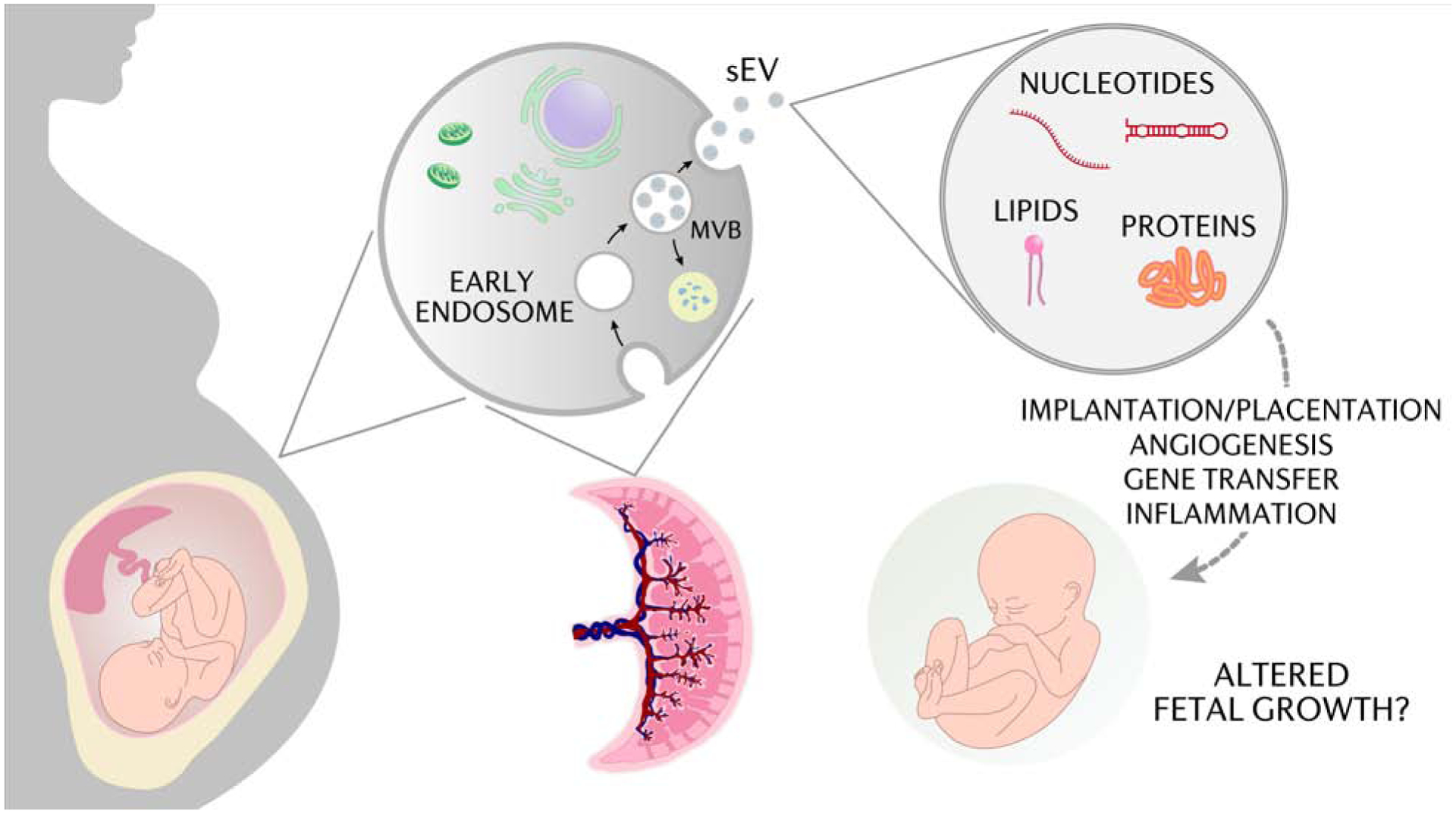
Small extracellular vesicles (sEV) are membrane-bound particles containing bioactive proteins, lipids, DNA, mRNA, and miR that are secreted from most cells and participate in cell to cell communication. The functions of maternal and placental sEVs remain to be fully established but are believed to be involved in immune response, angiogenesis, placentation, and the transfer of nucleic acids and proteins important in normal and complicated pregnancies that may influence fetal growth. miR, microRNA; MVB, multivesicular bodies. Courtesy of KIMEN Design4Research, with permission.
7. Interventions targeting placental function in FGR and fetal overgrowth
7.1. Restoring utero-placental blood flow in FGR
Numerous studies have tested the hypothesis that systemic administration of vasodilators increases utero-placental blood flow and promote fetal growth in FGR (Figure 4). Aspirin is a cyclo-oxygenase inhibitor that suppresses the production of thromboxane, which promotes platelet aggregation and functions as a vasoconstrictor. Thus, aspirin has the potential to increase utero-placental blood flow. Although the evidence on the efficacy of initiating aspirin therapy in early gestation to prevent fetal growth restriction is conflicting, several meta-analyses suggest that low dose aspirin (75–100 mg) has a modest effect on reducing the risk for developing preeclampsia and FGR104–106. Challenges remain in determining the appropriate timing to initiate treatment (before or after 16 weeks of gestation), defining the optimal dose, and delineating the benefits of aspirin in women with chronic hypertension versus those who develop preeclampsia during late gestation. Further, the majority of the studies included in the meta-analyses were designed with preeclampsia prevention as a primary endpoint with effects on fetal growth as a secondary outcome. In an effort to address these concerns, the Chronic Hypertension and Acetyl Salicylic Acid in Pregnancy (CHASP) trial will compare the efficacy of aspirin (150mg/day) introduced before 15 weeks of gestation in the prevention of maternal and fetal-morbidity and mortality, including FGR, in women with chronic hypertension107.
Figure 4. Clinical interventions targeting placental function to restore fetal growth.
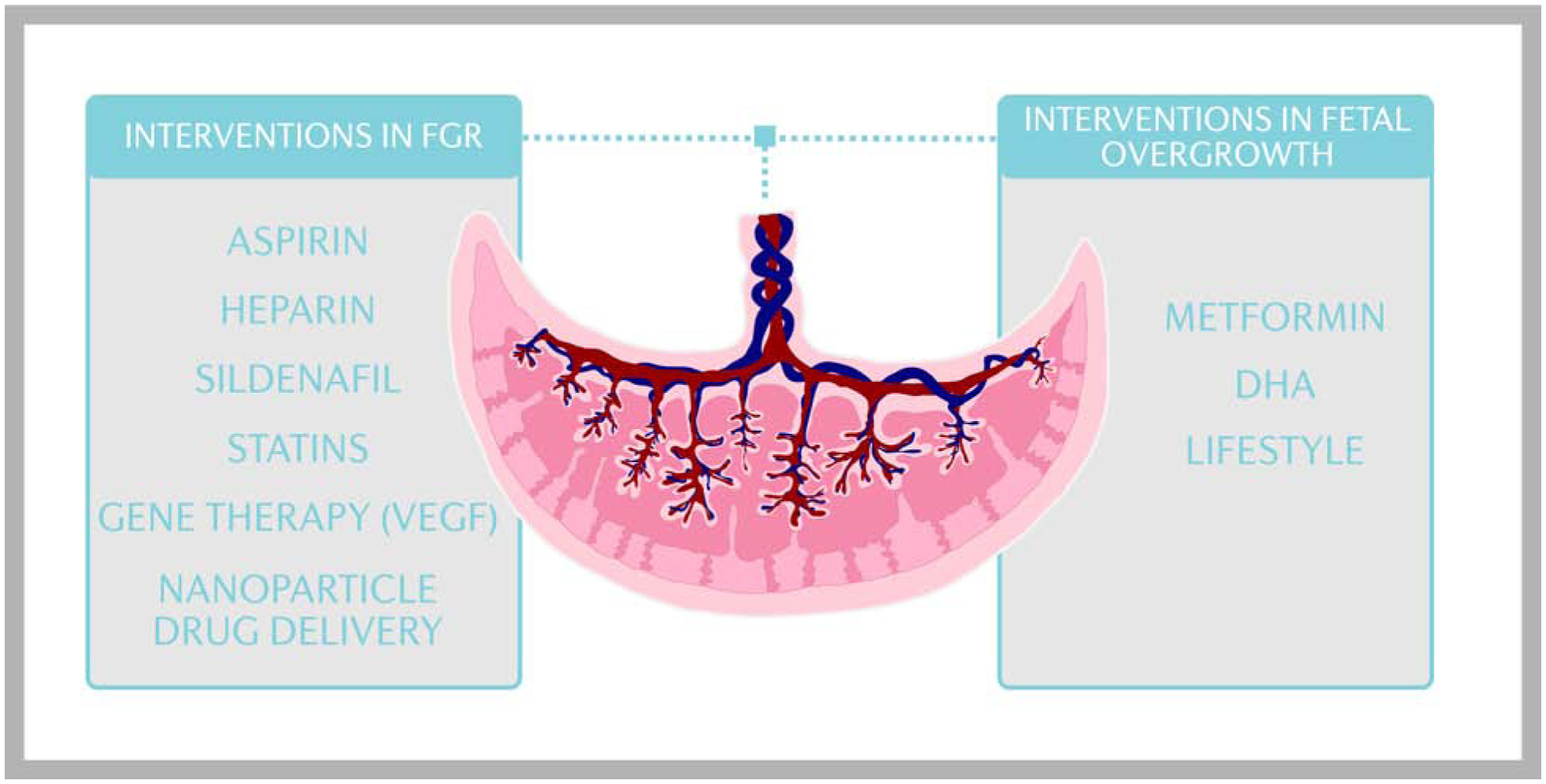
Numerous studies have tested the hypothesis that systemic administration of vasodilators such as aspirin, low molecular weight heparin, and sildenafil increases utero-placental blood flow and promote fetal growth in FGR, however the results have not been encouraging. Clinical trials to improve fetal growth in FGR pregnancies using statins have been initiated. In addition, gene therapy and nanoparticle drug delivery designed to alter the expression of genes in the uteroplacental circulation and the placenta, including VEGF, is an area of active research. Limited clinical approaches to prevent fetal overgrowth currently exist, however, treatment of GDM and/or maternal obesity with metformin, DHA supplementation, or lifestyle interventions may mitigate fetal overgrowth in high risk pregnancies. DHA, docosahexaenoic acid; GDM, gestational diabetes mullites; FGR, fetal growth restriction; VEGF, vascular endothelial growth factor. Courtesy of KIMEN Design4Research, with permission.
Low molecular weight heparin is commonly used in pregnancy as a thromboprophylaxis and for the treatment of venous thromboembolism. Heparin therapy during pregnancy is associated with elevated circulating levels of placental growth factor and decreased risk of recurrence of placental-mediated complications in women without thrombophilia108. Randomized controlled trials initially showed a potential reduction of the incidence of preeclampsia and FGR, however, recent meta-analyses suggest no benefit109,110. Despite early promise in small trials, a recent large trial (STIDER) of the phosphodiesterase type 5 inhibitor, sildenafil, was terminated prior to completion after several fetal deaths were reported111, possibly due to fetal hypotension as a result of transplacental transfer of sildenafil into fetal circulation112. Conversely, a phase II trial (TADAFER) of tadalafil, a phosphodiesterase 5 inhibitor that does not cross the placenta, decreased fetal and infant deaths associated with fetal growth restriction, however, fetal growth velocity and birthweight were unchanged113.
7.3. Clinical interventions targeting placental function in fetal overgrowth
Significant challenges exist in designing interventions targeting placental function in pregnancies complicated by maternal obesity. As described in this review, placental dysfunction as a result of increased nutrient availability and transport in response to elevated insulin, low adiponectin and enhanced mTOR placental signaling may contribute to fetal overgrowth. However, concerns over safety with the use of pharmaceuticals to reduce circulating maternal nutrients has limited the clinical approaches to prevent fetal overgrowth (Figure 4). Using metformin for glucose control in GDM pregnancies decreased the risk of fetal overgrowth as compared to women receiving insulin or glyburide114 but was recently shown to increase the proportion of small for gestation age infants in women the type 2 diabetes and obesity115. While the cause of reduced infant birthweights is unknown, it is possible those infants did not experience accelerated growth highlighting the need for early detection of fetal overgrowth to guide clinical decision making. In contrast, metformin did not prevent fetal overgrowth in obese pregnant women116. Because metformin is transported across the placenta into fetal circulation concerns have been raised that metformin may program the fetus for adverse outcomes, including obesity, later in life117. However, a recent report showed children of obese mothers exposed prenatally to metformin had improved cardiovascular profiles compared to placebo controlled offspring118.
The powerful impact of hormonal regulation on placental function and fetal growth was demonstrated by experimentally increasing adiponectin levels in obese mice compared to those observed in normal weight pregnant mice. Not only did this treatment improve placental function and prevent fetal overgrowth41, the long-term programming effects on metabolism, weight gain, glucose intolerance and cardiac dysfunction were also corrected53,54. In human pregnancy, nutrition and lifestyle interventions and dietary supplements have been explored as treatments to reduce fetal overgrowth in pregnancies complicated by maternal obesity or GDM. The UK Pregnancies Better Eating and Activity Trial (UPBEAT) recently reported that a comprehensive intervention targeting improvements in nutrition and physical activity did not reduce the incidence of LGA infants in pregnant women with obesity119, albeit placental storage of fatty acids in droplets was reported to be modestly decreased in women who received the lifestyle intervention120. Lastly, supplementation with docosahexaenoic acid (DHA) in pregnancy decreases placental inflammation and amino acid transporter expression in obese pregnancies which may mitigate fetal overgrowth121. However, trials assessing DHA on clinical outcomes related to fetal growth are lacking.
7.3. Future interventional approaches and priorities
Targeting the placenta with pharmaceutical interventions and approaches designed to alter placental gene expression is an area of active research. Injection of adenoviral vectors containing vascular endothelial growth factor (VEGF), a potent angiogenic factor, in the uterine artery of sheep was shown to improve uterine blood flow122. Gene therapy targeting maternal VEGF, is currently being investigated as a therapeutic intervention in early onset fetal growth restriction (EVERREST trial)123. The use of statins, particularly lipophilic statins that readily cross plasma membranes, is contraindicated during pregnancy due to the risk of congenital malformations124. Despite recent interest in the use of pravastatin to improve outcomes in preeclampsia and FGR mediated by inhibition of placental sFLT1 secretion125, the evidence that pravastatin is beneficial in these two pregnancy complications is conflicting126,127. Large randomized controlled trials are needed to assess the efficacy of statins in preeclampsia and FGR pregnancies. Lastly, innovative approaches using nanoparticles for drug delivery or gene targeting to the placenta represents an emerging area of clinical interest and warrants further investigation128.
8. Summary
Placental regulation of fetal growth involves the integration of multiple hormonal, inflammatory, nutrient, oxygen and energy-sensing signaling pathways, which modulate an array of placental functions including nutrient transport. As a result, the flux of oxygen and nutrients to the fetus is altered leading to changes in fetal growth. Placental insulin/IGF-1 and mTOR signaling and transport capacity of certain nutrients are inhibited in FGR and activated in some cases of fetal overgrowth, implicating these placental functions in driving fetal growth. Emerging evidence suggests that circulating total and placenta derived sEVs and miR may modulate placental function, however it is currently unknown how these novel signaling systems affect fetal growth. Future research priorities include establishing the role of sEVs in regulating fetal growth and determining the mechanistic role of maternal versus placental sEVs in the development of FGR and fetal overgrowth. Despite a considerable body of evidence linking placental function and fetal growth, clinical interventions specifically designed to restore normal placental function in high risk pregnancies is lacking. While novel interventions utilizing placental gene targeting and nanoparticle drug delivery are currently being investigated, the development of future clinically useful therapies to alleviate abnormal fetal growth is likely to be dependent on a better understanding of the specific placental molecular pathways that regulate fetal growth.
Key Points.
Placental signaling and nutrient transport are determinants of fetal growth.
In FGR, placental insulin/IGF-1 and mTOR signaling and nutrient transport are typically inhibited, which may contribute to the restricted fetal growth.
Activation of placental insulin/IGF-1 and mTOR signaling and nutrient transporters and reduced placental adiponectin signaling may promote fetal overgrowth in some pregnancies complicated by maternal obesity and GDM.
Therapeutic strategies designed to restore normal placental function and fetal growth in high risk pregnancies are limited and have yielded conflicting results.
Robust approaches to specifically target the placenta rather than the mother and the fetus and a better understanding of the placental molecular pathways driving fetal growth are required for the development of successful interventions to modulate placental function.
Clinical Care Points.
Fetal growth restriction and fetal overgrowth increase the risk of perinatal complications and to develop obesity, diabetes, and cardiovascular disease in childhood and later in life.
Changes in placental function contribute to abnormal fetal growth.
The understanding of the causes of abnormal fetal growth is limited and no effective treatments are available.
Therapeutic strategies designed to restore normal placental function in women with an FGR or LGA fetus have largely been unsuccessful and may potentially harm the fetus.
Novel interventions utilizing placental gene targeting and nanoparticle drug delivery may be effective therapeutic strategies to restore normal fetal growth in high risk pregnancies but require rigorous research to determine their clinical usefulness.
Synopsis.
Placental regulation of fetal growth involves the integration of multiple hormonal, inflammatory and nutrient, oxygen and energy-sensing signaling pathways, which modulate an array of placental functions including nutrient transport. As a result, the flux of oxygen and nutrients to the fetus is altered leading to changes in placental and fetal growth. Placental insulin/IGF-1 and mTOR signaling and amino acid transport capacity are inhibited in FGR and activated in fetal overgrowth, implicating these placental functions in driving fetal growth. With novel approaches to specifically target the placenta, clinical interventions to modulate placental function in high risk pregnancies can be developed.
Acknowledgements:
We thank KIMEN Design4Research (kimendesign4research.com) for the graphic design of the figures in this manuscript.
Funding: Supported by grants from NIH [grant numbers R01DK089989, HD089980, HD093950 HD065007, HD068370, HD078376, and T32HD007186].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interests: The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Goldstein RF, Abell SK, Ranasinha S, et al. Association of Gestational Weight Gain With Maternal and Infant Outcomes: A Systematic Review and Meta-analysis. JAMA. 2017;317(21):2207–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359(1):61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reynolds RM, Allan KM, Raja EA, et al. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: follow-up of 1 323 275 person years. BMJ. 2013;347:f4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Catalano PM, Ehrenberg HM. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG. 2006;113(10):1126–1133. [DOI] [PubMed] [Google Scholar]
- 5.Gluckman PD, Hanson MA, Pinal C. The developmental origins of adult disease. Matern Child Nutr. 2005;1(3):130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lao TT, Wong W. The neonatal implications of a high placental ratio in small-for-gestational age infants. Placenta. 1999;20(8):723–726. [DOI] [PubMed] [Google Scholar]
- 7.Karsdorp VH, van Vugt JM, van Geijn HP, et al. Clinical significance of absent or reversed end diastolic velocity waveforms in umbilical artery. Lancet. 1994;344(8938):1664–1668. [DOI] [PubMed] [Google Scholar]
- 8.Vaughan OR, Rosario FJ, Powell TL, Jansson T. Regulation of Placental Amino Acid Transport and Fetal Growth. Prog Mol Biol Transl Sci. 2017;145:217–251. [DOI] [PubMed] [Google Scholar]
- 9.Jansson T, Powell TL. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol. 2013;56(3):591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turco MY, Moffett A. Development of the human placenta. Development. 2019;146(22). [DOI] [PubMed] [Google Scholar]
- 11.Pollheimer J, Vondra S, Baltayeva J, Beristain AG, Knofler M. Regulation of Placental Extravillous Trophoblasts by the Maternal Uterine Environment. Front Immunol. 2018;9:2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leach L, Firth JA. Fine structure of the paracellular junctions of terminal villous capillaries in the perfused human placenta. Cell Tissue Res. 1992;268(3):447–452. [DOI] [PubMed] [Google Scholar]
- 13.Elad D, Levkovitz R, Jaffa AJ, Desoye G, Hod M. Have we neglected the role of fetal endothelium in transplacental transport? Traffic. 2014;15(1):122–126. [DOI] [PubMed] [Google Scholar]
- 14.Burton GJ, Fowden AL. The placenta: a multifaceted, transient organ. Philos Trans R Soc Lond B Biol Sci. 2015;370(1663):20140066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James-Allan LB, Arbet J, Teal SB, Powell TL, Jansson T. Insulin stimulates GLUT4 trafficking to the syncytiotrophoblast basal plasma membrane in the human placenta. J Clin Endocrinol Metab. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang J, Furesz TC, Lurent RS, Smith CH, Fant ME. Spatial polarization of insulin-like growth factor receptors on the human syncytiotrophoblast. Pediatr Res. 1997;41(2):258–265. [DOI] [PubMed] [Google Scholar]
- 17.Aye IL, Powell TL, Jansson T. Review: Adiponectin--the missing link between maternal adiposity, placental transport and fetal growth? Placenta. 2013;34 Suppl:S40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowman CJ, Streck RD, Chapin RE. Maternal-placental insulin-like growth factor (IGF) signaling and its importance to normal embryo-fetal development. Birth Defects Res B Dev Reprod Toxicol. 2010;89(4):339–349. [DOI] [PubMed] [Google Scholar]
- 19.Roos S, Kanai Y, Prasad PD, Powell TL, Jansson T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am J Physiol Cell Physiol. 2009;296(1):C142–150. [DOI] [PubMed] [Google Scholar]
- 20.Martina NA, Kim E, Chitkara U, Wathen NC, Chard T, Giudice LC. Gestational age-dependent expression of insulin-like growth factor-binding protein-1 (IGFBP-1) phosphoisoforms in human extraembryonic cavities, maternal serum, and decidua suggests decidua as the primary source of IGFBP-1 in these fluids during early pregnancy. J Clin Endocrinol Metab. 1997;82(6):1894–1898. [DOI] [PubMed] [Google Scholar]
- 21.Chassen S, Jansson T. Complex, coordinated and highly regulated changes in placental signaling and nutrient transport capacity in IUGR. Biochim Biophys Acta Mol Basis Dis. 2020;1866(2):165373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olausson H, Lof M, Brismar K, Forsum E, Sohlstrom A. Maternal serum concentrations of insulin-like growth factor (IGF)-I and IGF binding protein-1 before and during pregnancy in relation to maternal body weight and composition and infant birth weight. Br J Nutr. 2010;104(6):842–848. [DOI] [PubMed] [Google Scholar]
- 23.Gupta MB, Abu Shehab M, Nygard K, et al. IUGR Is Associated With Marked Hyperphosphorylation of Decidual and Maternal Plasma IGFBP-1. J Clin Endocrinol Metab. 2019;104(2):408–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singal SS, Nygard K, Gratton R, Jansson T, Gupta MB. Increased Insulin-like Growth Factor Binding Protein-1 Phosphorylation in Decidualized Stromal Mesenchymal Cells in Human Intrauterine Growth Restriction Placentas. J Histochem Cytochem. 2018;66(9):617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calvo MT, Romo A, Gutierrez JJ, Relano E, Barrio E, Ferrandez Longas A. Study of genetic expression of intrauterine growth factors IGF-I and EGFR in placental tissue from pregnancies with intrauterine growth retardation. J Pediatr Endocrinol Metab. 2004;17 Suppl 3:445–450. [PubMed] [Google Scholar]
- 26.Laviola L, Perrini S, Belsanti G, et al. Intrauterine growth restriction in humans is associated with abnormalities in placental insulin-like growth factor signaling. Endocrinology. 2005;146(3):1498–1505. [DOI] [PubMed] [Google Scholar]
- 27.Yung HW, Calabrese S, Hynx D, et al. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol. 2008;173(2):451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Street ME, Viani I, Ziveri MA, Volta C, Smerieri A, Bernasconi S. Impairment of insulin receptor signal transduction in placentas of intra-uterine growth-restricted newborns and its relationship with fetal growth. Eur J Endocrinol. 2011;164(1):45–52. [DOI] [PubMed] [Google Scholar]
- 29.Jansson N, Rosario FJ, Gaccioli F, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2013;98(1):105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shang M, Wen Z. Increased placental IGF-1/mTOR activity in macrosomia born to women with gestational diabetes. Diabetes Res Clin Pract. 2018;146:211–219. [DOI] [PubMed] [Google Scholar]
- 31.Jansson N, Nilsfelt A, Gellerstedt M, et al. Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am J Clin Nutr. 2008;87(6):1743–1749. [DOI] [PubMed] [Google Scholar]
- 32.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosario FJ, Kanai Y, Powell TL, Jansson T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol. 2013;591(3):609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosario FJ, Powell TL, Jansson T. mTOR folate sensing links folate availability to trophoblast cell function. J Physiol. 2017;595(13):4189–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosario FJ, Gupta MB, Myatt L, et al. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci Rep. 2019;9(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roos S, Jansson N, Palmberg I, Saljo K, Powell TL, Jansson T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol. 2007;582(Pt 1):449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen YY, Rosario FJ, Shehab MA, Powell TL, Gupta MB, Jansson T. Increased ubiquitination and reduced plasma membrane trafficking of placental amino acid transporter SNAT-2 in human IUGR. Clin Sci (Lond). 2015;129(12):1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosario FJ, Jansson N, Kanai Y, Prasad PD, Powell TL, Jansson T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology. 2011;152(3):1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kavitha JV, Rosario FJ, Nijland MJ, et al. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 2014;28(3):1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sati L, Soygur B, Celik-Ozenci C. Expression of Mammalian Target of Rapamycin and Downstream Targets in Normal and Gestational Diabetic Human Term Placenta. Reprod Sci. 2016;23(3):324–332. [DOI] [PubMed] [Google Scholar]
- 41.Aye IL, Rosario FJ, Powell TL, Jansson T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc Natl Acad Sci U S A. 2015;112(41):12858–12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosario FJ, Powell TL, Jansson T. Activation of placental insulin and mTOR signaling in a mouse model of maternal obesity associated with fetal overgrowth. Am J Physiol Regul Integr Comp Physiol. 2016;310(1):R87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta MB, Jansson T. Novel roles of mechanistic target of rapamycin signaling in regulating fetal growthdagger. Biol Reprod. 2019;100(4):872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kola B, Grossman AB, Korbonits M. The role of AMP-activated protein kinase in obesity. Front Horm Res. 2008;36:198–211. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Shang LX, Dong X, et al. Relationship of adiponectin and resistin levels in umbilical serum, maternal serum and placenta with neonatal birth weight. Aust N Z J Obstet Gynaecol. 2010;50(5):432–438. [DOI] [PubMed] [Google Scholar]
- 46.Haghiac M, Basu S, Presley L, Serre D, Catalano PM, Hauguel-de Mouzon S. Patterns of adiponectin expression in term pregnancy: impact of obesity. J Clin Endocrinol Metab. 2014;99(9):3427–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hendler I, Blackwell SC, Mehta SH, et al. The levels of leptin, adiponectin, and resistin in normal weight, overweight, and obese pregnant women with and without preeclampsia. Am J Obstet Gynecol. 2005;193(3 Pt 2):979–983. [DOI] [PubMed] [Google Scholar]
- 48.Vernini JM, Moreli JB, Costa RA, Negrato CA, Rudge MV, Calderon IM. Maternal adipokines and insulin as biomarkers of pregnancies complicated by overweight and obesity. Diabetol Metab Syndr. 2016;8(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423(6941):762–769. [DOI] [PubMed] [Google Scholar]
- 50.Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55(9):2562–2570. [DOI] [PubMed] [Google Scholar]
- 51.Jones HN, Jansson T, Powell TL. Full-length adiponectin attenuates insulin signaling and inhibits insulin-stimulated amino Acid transport in human primary trophoblast cells. Diabetes. 2010;59(5):1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aye IL, Gao X, Weintraub ST, Jansson T, Powell TL. Adiponectin inhibits insulin function in primary trophoblasts by PPARalpha-mediated ceramide synthesis. Mol Endocrinol. 2014;28(4):512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaughan OR, Rosario FJ, Powell TL, Jansson T. Normalisation of circulating adiponectin levels in obese pregnant mice prevents cardiac dysfunction in adult offspring. Int J Obes (Lond). 2020;44(2):488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paulsen ME, Rosario FJ, Wesolowski SR, Powell TL, Jansson T. Normalizing adiponectin levels in obese pregnant mice prevents adverse metabolic outcomes in offspring. FASEB J. 2019;33(2):2899–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pantham P, Aye IL, Powell TL. Inflammation in maternal obesity and gestational diabetes mellitus. Placenta. 2015;36(7):709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.St-Germain LE, Castellana B, Baltayeva J, Beristain AG. Maternal Obesity and the Uterine Immune Cell Landscape: The Shaping Role of Inflammation. Int J Mol Sci. 2020;21(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nguyen-Ngo C, Jayabalan N, Salomon C, Lappas M. Molecular pathways disrupted by gestational diabetes mellitus. J Mol Endocrinol. 2019;63(3):R51–R72. [DOI] [PubMed] [Google Scholar]
- 58.Tenorio MB, Ferreira RC, Moura FA, Bueno NB, de Oliveira ACM, Goulart MOF. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxid Med Cell Longev. 2019;2019:8238727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hauguel-de Mouzon S, Guerre-Millo M. The placenta cytokine network and inflammatory signals. Placenta. 2006;27(8):794–798. [DOI] [PubMed] [Google Scholar]
- 60.Aye IL, Lager S, Ramirez VI, et al. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod. 2014;90(6):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelly AC, Powell TL, Jansson T. Placental function in maternal obesity. Clin Sci (Lond). 2020;134(8):961–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol. 2009;297(5):C1228–1235. [DOI] [PubMed] [Google Scholar]
- 63.Aye IL, Jansson T, Powell TL. TNF-alpha stimulates System A amino acid transport in primary human trophoblast cells mediated by p38 MAPK signaling. Physiol Rep. 2015;3(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aye IL, Jansson T, Powell TL. Interleukin-1beta inhibits insulin signaling and prevents insulin-stimulated system A amino acid transport in primary human trophoblasts. Mol Cell Endocrinol. 2013;381(1–2):46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jansson T, Wennergren M, Illsley NP. Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J Clin Endocrinol Metab. 1993;77(6):1554–1562. [DOI] [PubMed] [Google Scholar]
- 66.Acosta O, Ramirez VI, Lager S, et al. Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am J Obstet Gynecol. 2015;212(2):227 e221–227. [DOI] [PubMed] [Google Scholar]
- 67.Luscher BP, Marini C, Joerger-Messerli MS, et al. Placental glucose transporter (GLUT)-1 is down-regulated in preeclampsia. Placenta. 2017;55:94–99. [DOI] [PubMed] [Google Scholar]
- 68.Sharma D, Shastri S, Sharma P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin Med Insights Pediatr. 2016;10:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosario FJ, Dimasuay KG, Kanai Y, Powell TL, Jansson T. Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4–2. Clin Sci (Lond). 2016;130(7):499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.James-Allan LB, Teal S, Powell TL, Jansson T. Changes in Placental Nutrient Transporter Protein Expression and Activity Across Gestation in Normal and Obese Women. Reprod Sci. 2020. [DOI] [PubMed] [Google Scholar]
- 71.Jansson T, Castillo-Castrejon M, Gupta MB, Powell TL, Rosario FJ. Down-regulation of placental Cdc42 and Rac1 links mTORC2 inhibition to decreased trophoblast amino acid transport in human intrauterine growth restriction. Clin Sci (Lond). 2020;134(1):53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Norberg S, Powell TL, Jansson T. Intrauterine growth restriction is associated with a reduced activity of placental taurine transporters. Pediatr Res. 1998;44(2):233–238. [DOI] [PubMed] [Google Scholar]
- 73.Jansson T, Scholtbach V, Powell TL. Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatr Res. 1998;44(4):532–537. [DOI] [PubMed] [Google Scholar]
- 74.Mahendran D, Donnai P, Glazier JD, D’Souza SW, Boyd RD, Sibley CP. Amino acid (system A) transporter activity in microvillous membrane vesicles from the placentas of appropriate and small for gestational age babies. Pediatr Res. 1993;34(5):661–665. [DOI] [PubMed] [Google Scholar]
- 75.Glazier JD, Cetin I, Perugino G, et al. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr Res. 1997;42(4):514–519. [DOI] [PubMed] [Google Scholar]
- 76.Marconi AM, Paolini CL, Stramare L, et al. Steady state maternal-fetal leucine enrichments in normal and intrauterine growth-restricted pregnancies. Pediatr Res. 1999;46(1):114–119. [DOI] [PubMed] [Google Scholar]
- 77.Paolini CL, Marconi AM, Ronzoni S, et al. Placental transport of leucine, phenylalanine, glycine, and proline in intrauterine growth-restricted pregnancies. J Clin Endocrinol Metab. 2001;86(11):5427–5432. [DOI] [PubMed] [Google Scholar]
- 78.Jansson N, Pettersson J, Haafiz A, et al. Down-regulation of placental transport of amino acids precedes the development of intrauterine growth restriction in rats fed a low protein diet. J Physiol. 2006;576(Pt 3):935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pantham P, Rosario FJ, Nijland M, et al. Reduced placental amino acid transport in response to maternal nutrient restriction in the baboon. Am J Physiol Regul Integr Comp Physiol. 2015;309(7):R740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pantham P, Rosario FJ, Weintraub ST, et al. Down-Regulation of Placental Transport of Amino Acids Precedes the Development of Intrauterine Growth Restriction in Maternal Nutrient Restricted Baboons. Biol Reprod. 2016;95(5):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lewis RM, Childs CE, Calder PC. New perspectives on placental fatty acid transfer. Prostaglandins Leukot Essent Fatty Acids. 2018;138:24–29. [DOI] [PubMed] [Google Scholar]
- 82.Lewis RM, Wadsack C, Desoye G. Placental fatty acid transfer. Curr Opin Clin Nutr Metab Care. 2018;21(2):78–82. [DOI] [PubMed] [Google Scholar]
- 83.Segura MT, Demmelmair H, Krauss-Etschmann S, et al. Maternal BMI and gestational diabetes alter placental lipid transporters and fatty acid composition. Placenta. 2017;57:144–151. [DOI] [PubMed] [Google Scholar]
- 84.Lager S, Ramirez VI, Gaccioli F, Jang B, Jansson T, Powell TL. Protein expression of fatty acid transporter 2 is polarized to the trophoblast basal plasma membrane and increased in placentas from overweight/obese women. Placenta. 2016;40:60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Magnusson AL, Waterman IJ, Wennergren M, Jansson T, Powell TL. Triglyceride hydrolase activities and expression of fatty acid binding proteins in the human placenta in pregnancies complicated by intrauterine growth restriction and diabetes. J Clin Endocrinol Metab. 2004;89(9):4607–4614. [DOI] [PubMed] [Google Scholar]
- 86.Gauster M, Hiden U, Blaschitz A, et al. Dysregulation of placental endothelial lipase and lipoprotein lipase in intrauterine growth-restricted pregnancies. J Clin Endocrinol Metab. 2007;92(6):2256–2263. [DOI] [PubMed] [Google Scholar]
- 87.Chassen SS, Ferchaud-Roucher V, Gupta MB, Jansson T, Powell TL. Alterations in placental long chain polyunsaturated fatty acid metabolism in human intrauterine growth restriction. Clin Sci (Lond). 2018;132(5):595–607. [DOI] [PubMed] [Google Scholar]
- 88.Mishima T, Miner JH, Morizane M, Stahl A, Sadovsky Y. The expression and function of fatty acid transport protein-2 and −4 in the murine placenta. PLoS One. 2011;6(10):e25865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chassen SS, Ferchaud-Roucher V, Palmer C, et al. Placental fatty acid transport across late gestation in a baboon model of intrauterine growth restriction. J Physiol. 2020;598(12):2469–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Welge JA, Warshak CR, Woollett LA. Maternal plasma cholesterol concentration and preterm birth: a meta-analysis and systematic review of literature. J Matern Fetal Neonatal Med. 2020;33(13):2291–2299. [DOI] [PubMed] [Google Scholar]
- 91.Rosario FJ, Nathanielsz PW, Powell TL, Jansson T. Maternal folate deficiency causes inhibition of mTOR signaling, down-regulation of placental amino acid transporters and fetal growth restriction in mice. Sci Rep. 2017;7(1):3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosario FJ, Powell TL, Jansson T. Mechanistic target of rapamycin (mTOR) regulates trophoblast folate uptake by modulating the cell surface expression of FR-alpha and the RFC. Sci Rep. 2016;6:31705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen YY, Gupta MB, Grattton R, Powell TL, Jansson T. Down-regulation of placental folate transporters in intrauterine growth restriction. J Nutr Biochem. 2018;59:136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han C, Wang C, Chen Y, et al. Placenta-derived extracellular vesicles induce preeclampsia in mouse models. Haematologica. 2020;105(6):1686–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.James-Allan LB, Rosario FJ, Barner K, et al. Regulation of glucose homeostasis by small extracellular vesicles in normal pregnancy and in gestational diabetes. FASEB J. 2020;34(4):5724–5739. [DOI] [PubMed] [Google Scholar]
- 97.Rodosthenous RS, Burris HH, Sanders AP, et al. Second trimester extracellular microRNAs in maternal blood and fetal growth: An exploratory study. Epigenetics. 2017;12(9):804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miranda J, Paules C, Nair S, et al. Placental exosomes profile in maternal and fetal circulation in intrauterine growth restriction - Liquid biopsies to monitoring fetal growth. Placenta. 2018;64:34–43. [DOI] [PubMed] [Google Scholar]
- 99.Jin J, Menon R. Placental exosomes: A proxy to understand pregnancy complications. Am J Reprod Immunol. 2018;79(5):e12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Takahashi H, Ohkuchi A, Kuwata T, et al. Endogenous and exogenous miR-520c-3p modulates CD44-mediated extravillous trophoblast invasion. Placenta. 2017;50:25–31. [DOI] [PubMed] [Google Scholar]
- 101.Taylor SK, Houshdaran S, Robinson JF, et al. Cytotrophoblast extracellular vesicles enhance decidual cell secretion of immune modulators via TNF-alpha. Development. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Salomon C, Scholz-Romero K, Sarker S, et al. Gestational Diabetes Mellitus Is Associated With Changes in the Concentration and Bioactivity of Placenta-Derived Exosomes in Maternal Circulation Across Gestation. Diabetes. 2016;65(3):598–609. [DOI] [PubMed] [Google Scholar]
- 103.Atay S, Gercel-Taylor C, Suttles J, Mor G, Taylor DD. Trophoblast-derived exosomes mediate monocyte recruitment and differentiation. Am J Reprod Immunol. 2011;65(1):65–77. [DOI] [PubMed] [Google Scholar]
- 104.Roberge S, Bujold E, Nicolaides KH. Aspirin for the prevention of preterm and term preeclampsia: systematic review and metaanalysis. Am J Obstet Gynecol. 2018;218(3):287–293 e281. [DOI] [PubMed] [Google Scholar]
- 105.Roberge S, Nicolaides K, Demers S, Hyett J, Chaillet N, Bujold E. The role of aspirin dose on the prevention of preeclampsia and fetal growth restriction: systematic review and meta-analysis. Am J Obstet Gynecol. 2017;216(2):110–120 e116. [DOI] [PubMed] [Google Scholar]
- 106.Meher S, Duley L, Hunter K, Askie L. Antiplatelet therapy before or after 16 weeks’ gestation for preventing preeclampsia: an individual participant data meta-analysis. Am J Obstet Gynecol. 2017;216(2):121–128 e122. [DOI] [PubMed] [Google Scholar]
- 107.Lecarpentier E, Haddad B. Aspirin for the Prevention of Placenta-Mediated Complications in Pregnant Women with Chronic Hypertension. J Gynecol Obstet Hum Reprod. 2020:101845. [DOI] [PubMed] [Google Scholar]
- 108.Nawathe A, David AL. Prophylaxis and treatment of foetal growth restriction. Best Pract Res Clin Obstet Gynaecol. 2018;49:66–78. [DOI] [PubMed] [Google Scholar]
- 109.Rodger MA, Gris JC, de Vries JIP, et al. Low-molecular-weight heparin and recurrent placenta-mediated pregnancy complications: a meta-analysis of individual patient data from randomised controlled trials. Lancet. 2016;388(10060):2629–2641. [DOI] [PubMed] [Google Scholar]
- 110.Mastrolia SA, Novack L, Thachil J, et al. LMWH in the prevention of preeclampsia and fetal growth restriction in women without thrombophilia. A systematic review and meta-analysis. Thromb Haemost. 2016;116(5):868–878. [DOI] [PubMed] [Google Scholar]
- 111.Hawkes N Trial of Viagra for fetal growth restriction is halted after baby deaths. BMJ. 2018;362:k3247. [DOI] [PubMed] [Google Scholar]
- 112.Hitzerd E, Broekhuizen M, Mirabito Colafella KM, et al. Placental effects and transfer of sildenafil in healthy and preeclamptic conditions. EBioMedicine. 2019;45:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maki S, Tanaka H, Tsuji M, et al. Safety Evaluation of Tadalafil Treatment for Fetuses with Early-Onset Growth Restriction (TADAFER): Results from the Phase II Trial. J Clin Med. 2019;8(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tarry-Adkins JL, Aiken CE, Ozanne SE. Comparative impact of pharmacological treatments for gestational diabetes on neonatal anthropometry independent of maternal glycaemic control: A systematic review and meta-analysis. PLoS Med. 2020;17(5):e1003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Feig DS, Donovan LE, Zinman B, et al. Metformin in women with type 2 diabetes in pregnancy (MiTy): a multicentre, international, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2020;8(10):834–844. [DOI] [PubMed] [Google Scholar]
- 116.Nascimento IBD, Sales WB, Dienstmann G, Souza MLR, Fleig R, Silva JC. Metformin for prevention of cesarean delivery and large-for-gestational-age newborns in non-diabetic obese pregnant women: a randomized clinical trial. Arch Endocrinol Metab. 2020;64(3):290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tarry-Adkins JL, Aiken CE, Ozanne SE. Neonatal, infant, and childhood growth following metformin versus insulin treatment for gestational diabetes: A systematic review and meta-analysis. PLoS Med. 2019;16(8):e1002848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Panagiotopoulou O, Syngelaki A, Georgiopoulos G, et al. Metformin use in obese mothers is associated with improved cardiovascular profile in the offspring. Am J Obstet Gynecol. 2020;223(2):246 e241–246 e210. [DOI] [PubMed] [Google Scholar]
- 119.Poston L, Bell R, Croker H, et al. Effect of a behavioural intervention in obese pregnant women (the UPBEAT study): a multicentre, randomised controlled trial. Lancet Diabetes Endocrinol. 2015;3(10):767–777. [DOI] [PubMed] [Google Scholar]
- 120.Gazquez A, Uhl O, Ruiz-Palacios M, et al. Placental lipid droplet composition: Effect of a lifestyle intervention (UPBEAT) in obese pregnant women. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863(9):998–1005. [DOI] [PubMed] [Google Scholar]
- 121.Lager S, Ramirez VI, Acosta O, et al. Docosahexaenoic Acid Supplementation in Pregnancy Modulates Placental Cellular Signaling and Nutrient Transport Capacity in Obese Women. J Clin Endocrinol Metab. 2017;102(12):4557–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Carr DJ, Wallace JM, Aitken RP, et al. Uteroplacental adenovirus vascular endothelial growth factor gene therapy increases fetal growth velocity in growth-restricted sheep pregnancies. Hum Gene Ther. 2014;25(4):375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spencer R, Ambler G, Brodszki J, et al. EVERREST prospective study: a 6-year prospective study to define the clinical and biological characteristics of pregnancies affected by severe early onset fetal growth restriction. BMC Pregnancy Childbirth. 2017;17(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Edison RJ, Muenke M. Mechanistic and epidemiologic considerations in the evaluation of adverse birth outcomes following gestational exposure to statins. Am J Med Genet A. 2004;131(3):287–298. [DOI] [PubMed] [Google Scholar]
- 125.Brownfoot FC, Tong S, Hannan NJ, et al. Effects of Pravastatin on Human Placenta, Endothelium, and Women With Severe Preeclampsia. Hypertension. 2015;66(3):687–697; discussion 445. [DOI] [PubMed] [Google Scholar]
- 126.Ahmed A, Williams DJ, Cheed V, et al. Pravastatin for early-onset pre-eclampsia: a randomised, blinded, placebo-controlled trial. BJOG. 2020;127(4):478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lefkou E, Mamopoulos A, Dagklis T, Vosnakis C, Rousso D, Girardi G. Pravastatin improves pregnancy outcomes in obstetric antiphospholipid syndrome refractory to antithrombotic therapy. J Clin Invest. 2016;126(8):2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Irvin-Choy NS, Nelson KM, Gleghorn JP, Day ES. Design of nanomaterials for applications in maternal/fetal medicine. J Mater Chem B. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]