

Abstract
The nondystrophic myotonias are rare muscle hyperexcitability disorders caused by gain-of-function mutations in the SCN4A gene or loss-of-function mutations in the CLCN1 gene. Clinically, they are characterized by myotonia, defined as delayed muscle relaxation after voluntary contraction, which leads to symptoms of muscle stiffness, pain, fatigue, and weakness. Diagnosis is based on history and examination findings, the presence of electrical myotonia on electromyography, and genetic confirmation. In the absence of genetic confirmation, the diagnosis is supported by detailed electrophysiological testing, exclusion of other related disorders, and analysis of a variant of uncertain significance if present. Symptomatic treatment with a sodium channel blocker, such as mexiletine, is usually the first step in management, as well as educating patients about potential anesthetic complications.
Keywords: management, myotonia congenita, nondystrophic myotonias, paramyotonia congenita, skeletal muscle channelopathies
1 |. INTRODUCTION
The nondystrophic myotonias (NDMs) are a group of rare monogenetic muscle disorders caused by mutations in the voltage-gated skeletal muscle sodium (SCN4A) or chloride ion channel (CLCN1) genes that lead to muscle membrane hyperexcitability.1,2 The clinical correlate of this muscle membrane hyperexcitability is myotonia (from the Greek words “muscle” and “tension”), defined as a delayed relaxation of skeletal muscles after voluntary contraction (as first described by Strümpell in 1881)3 or after percussion (as first described by Erb).4 Nationwide point prevalence estimates for the NDMs have been reported in the United Kingdom (0.75/100,000) and Netherlands (1.70/100,000). The prevalence is reported to be higher in some regions, probably due to founder effects and more geographically isolated populations.5,6 In contrast to myotonic dystrophy (DM) types 1 and 2 (DM1 and DM2), which are multisystemic disorders with progressive muscle wasting, the NDMs are characterized by exclusive skeletal muscle dysfunction in the absence of progressive muscle wasting, and patients usually have a normal life expectancy.7 Key patient-reported symptoms in NDMs are muscle stiffness or cramps, weakness, fatigue, and pain.7–9 Quality of life perception seems to be especially impacted by the presence of pain and fatigue.10 Overall, quality of life measures were similar to that of patients with DM and lower compared with the healthy population,11 which justifies pharmacological treatment. Here, we report guidelines created after a group of experts reviewed the literature, agreed to an outline of key concepts related to the diagnosis and management of NDM, and participated in drafting the review. We describe the pathophysiology, clinical characteristics, current approach to diagnosis, and management of these rare disorders.
2 |. TYPES OF NDMs AND RELATED DISORDERS
NDMs are first divided into two genotype groups (skeletal muscle chloride and sodium channelopathies) depending on whether the disorder is caused by a mutation in the chloride (CLCN1) or sodium channel gene (SCN4A). Historically, these disorders have then been further subdivided based on mode of inheritance, clinical or electrophysiological features (Figure 1).
FIGURE 1.
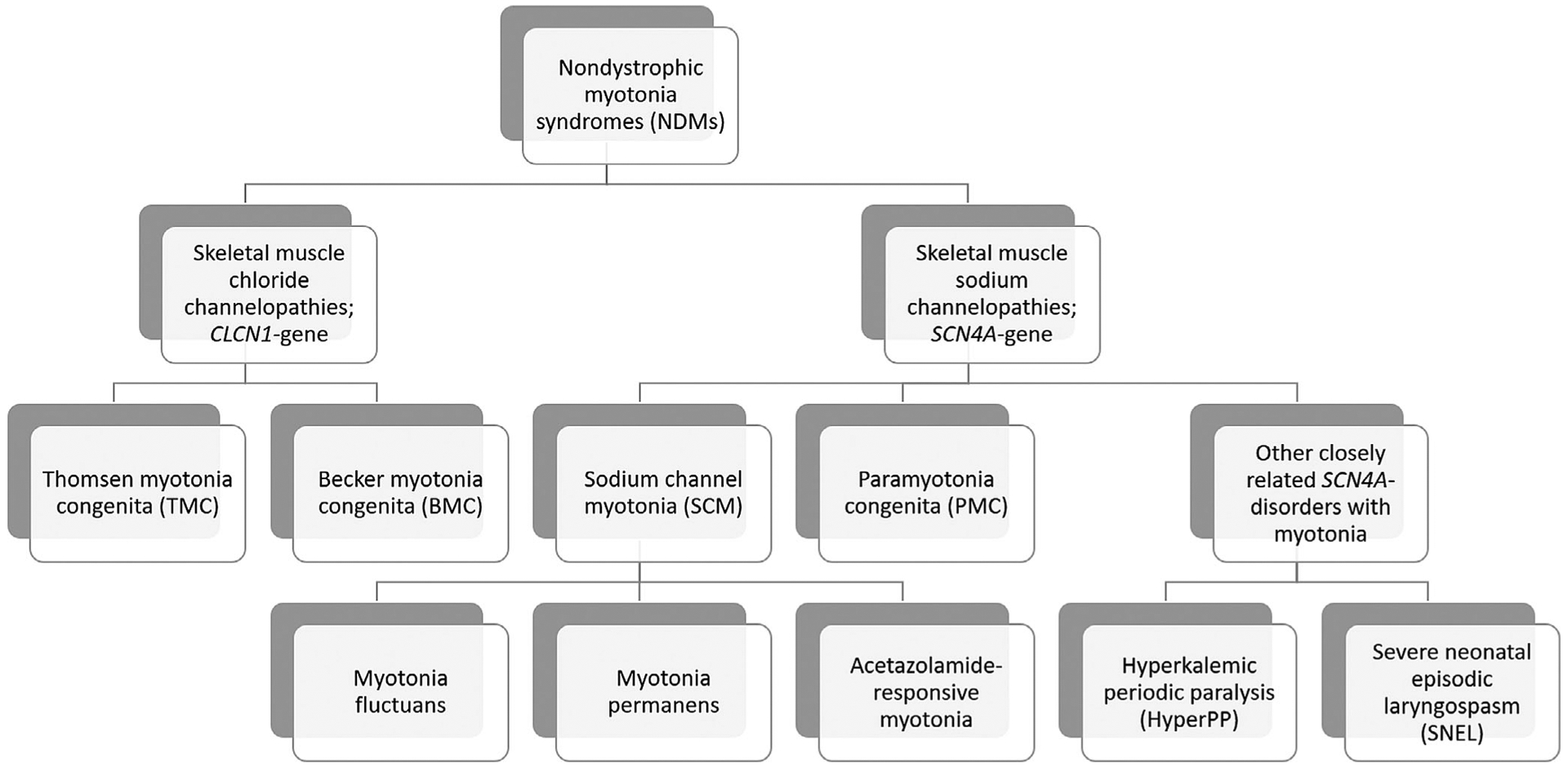
Classification scheme of nondystrophic myotonias and closely related disorders
Skeletal muscle chloride channelopathies, also known as myotonia congenita (MC), are subdivided based on their mode of inheritance: autosomal dominant or Thomsen myotonia congenita (TMC)12 and autosomal recessive or Becker myotonia congenita (BMC).13,14 Both TMC and BMC cause muscle stiffness that can be improved with repetitive muscle activity, called the warm-up phenomenon.15 BMC is thought to have a more severe phenotype with transient weakness that can recover after exercise.15
On the other hand, skeletal muscle sodium channelopathies all have autosomal dominant inheritance and can be subdivided into two major clinical phenotypes: paramyotonia congenita (PMC) and sodium channel myotonia (SCM). PMC, originally described by Eulenberg,16 is characterized by myotonia that worsens, instead of improving, with repeated muscle activity (paradoxical myotonia). In addition, the myotonia is usually cold induced and more prominent in the face compared with that seen in MC.17 Patients with PMC can also experience episodic weakness but it is usually not the most prominent symptom. SCM is clinically characterized as a purely myotonic disorder with occasional additional features such as fluctuations in myotonia (myotonia fluctuans),18 permanent myotonia (myotonia permanens),19 or acetazolamide-responsiveness (acetazolamide-responsive myotonia).20 SCM has historically been referred to as potassium-aggravated myotonia (PAM), but Rudel et al. suggested that it be referred to as SCM in the absence of a potassium loading test21 and not all cases are potassium sensitive.
In the presence of myotonia with episodic weakness, where weakness is the most prominent symptom, the related disorder hyperkalemic periodic paralysis (HyperPP), also caused by mutations in SCN4A, must be considered. HyperPP is characterized by episodes of weakness that can last hours to days, triggered by fasting, exercise, or potassium ingestion.15 Patients with HyperPP can have myotonia, but it only occurs in approximately 50% of cases.7,8,22 Another important disorder to recognize is severe neonatal episodic laryngospasm (SNEL), which occurs in a subset of neonatal sodium channelopathy patients. In these neonatal cases, muscle hypertonia is present along with episodic laryngospasm (especially under circumstances that initiate or aggravate myotonia such as crying or cold exposure) that can lead to life-threatening periods of apnea if not diagnosed in time and treated with anti-myotonic drugs.23
While the focus of this review will be on the NDMs, other allelic disorders caused by mutations in SCN4A are reported. These diseases, caused by loss-of-function, rather than gain-of-function mutations, do not usually cause myotonia. Examples include congenital myopathy with fetal hypokinesia,24 some cases of congenital myasthenic syndrome (CMS),25,26 and hypokalemic periodic paralysis (HypoPP).27
3 |. PATHOPHYSIOLOGY
3.1 |. Skeletal muscle chloride channelopathies
Skeletal muscle action potentials are generated through activation of voltage-gated sodium channels that depolarize the sarcolemmal membrane. Repolarization of the sarcolemma and stability of the resting potential occur through the combined activity of potassium and chloride channels. In contrast to neurons where the resting conductance is dominated by potassium channels, chloride channels contribute the most to the resting membrane conductance in muscle.28 The causal relationship between a reduced chloride conductance and fiber hyperexcitability with after-discharges was discovered in muscle fibers from the myotonic goat by Lipicky and Bryant in 1966. They also showed a two-fold decrease in chloride permeability29 in myotonic fibers from humans in 1971.17,30,31
In the setting of a reduced chloride conductance, the normal activity-dependent increase of K+ concentration in the transverse tubules after muscle contraction produces an anomalously large after-depolarization of the sarcolemma.32–34 These cumulative after-depolarizations give rise to the self-sustained bursts of discharges (seen as electrical myotonia on electromyography [EMG]) that prevent the muscle from relaxing after voluntary contraction.33,34 However, this model does not explain why myotonia stops after a period of seconds, nor does it explain the basis of warm-up whereby with repeated activity of the same muscle the myotonia diminishes in intensity or may even cease. An additional model has been suggested where the increase in K+ causes an initial depolarization and subsequently activates a sodium persistent inward current (NaPIC) that leads to the further depolarization and generation of myotonic action potentials.35
Molecular genetic confirmation of the reduced chloride conductance hypothesis came in 1992 when the first mutations in the CLCN1 gene were identified in patients with dominant and recessive MC.1,17 Functional expression studies36–39 and genetic cohort studies have now identified over 100 CLCN1-mutations.40 The chloride channel is a dimer of ClC-1 subunits, and dominant inheritance for myotonia congenita occurs when a mutant subunit is able to interact with a wild-type subunit to disrupt the function of the overall channel complex (dominant-negative effect). Conversely, mutations associated with recessive inheritance often result in a null allele for which the mutant subunit is not able to interact with a wild-type counterpart (eg, nonsense mutation with a frame-shift and early termination).41,42
A single recessive allele is clinically asymptomatic because, as shown in pharmacologic studies with ClC-1 blockers, the chloride conductance must be reduced to less than 50% of normal to consistently produce myotonia.31 Most of the mutations represent missense mutations (with no specific exon predominance) and around 30% represent small deletions, duplications, insertions, and frameshift mutations.5,6 However, these mutations alone do not entirely explain each person’s phenotype because family members with the same mutation can have varying disease severity.19 Aside from the clinical variability that may occur with a loss of the chloride conductance that is at the threshold for myotonia, another source of variability may be differences in the amount of extracellular Ca2+ and Mg2+. In rat muscle exposed to a chloride channel inhibitor, elevations in these cations have been shown to reduce myotonia through shifts in sodium channel activation.43
3.2 |. Skeletal muscle sodium channelopathies
All NDM sodium channelopathies are due to gain-of-function SCN4A mutations, which either cause an increase of channel activation or a decrease of channel inactivation. These changes lead to inappropriate excitation at the end of an action potential because the availability of sodium channels is too high (impaired inactivation) or too many sodium channels are open with a mild depolarization from the resting potential (enhanced activation).33,34 This ability of an altered sodium current to cause myotonia was first demonstrated by application of the voltage-gated sodium channel opener veratridine (a toxin that stabilizes the open state of the channel)44 in 1969, and, later by identifying a persistent tetrotoxin (TTX)-sensitive current that functions as a voltage-gated sodium channel blocker in muscle from patients with periodic paralysis (PP) with temperature sensitive myotonia.45,46
It is notable that mutations in SCN4A can cause a phenotype that varies from paralysis to increased excitability (myotonia). Mutations associated with prominent defects of inactivation (larger persistent inward sodium currents and those with disrupted slow inactivation) increase the susceptibility to PP from sustained depolarization of the resting potential, which inactivates the wild-type sodium channels and renders the fiber inexcitable.47,48 Conversely, sodium channel gain of function mutations that result in smaller persistent currents or only a transient defect (eg, inactivation that is too slow, but eventually complete) will increase fiber excitability and lead to myotonia.
Molecular genetic confirmation of mutations in the SCN4A gene encoding the voltage-gated skeletal muscle sodium channel alpha subunit was first reported in HyperPP in 1991,49,50 and in PMC51 and SCM52 in 1992. Functional expression studies53 and genetic cohort studies have confirmed the pathogenicity of around 20 autosomal dominant, missense mutations in SCN4A causing PMC or SCM (with a hot spot in exon 22 and 2454) and less than 10 autosomal dominant missense mutations causing HyperPP.5,6
4 |. GENERAL CLINICAL PRESENTATION
Symptom onset for patients with NDM is usually in the first decade, with a slightly lower mean age of onset in patients with SCN4A-related myotonia.7,8 While NDM is sometimes thought of as a static disorder, Trip et al. found that a majority of participants reported moderate worsening of symptoms over their lifetimes.8 The most common symptom in NDMs is muscle stiffness (myotonia), with this complaint being present in 100% of participants in one study.7 Leg muscle stiffness is more common in chloride channelopathies and facial muscle stiffness is more common in sodium channelopathies.7 The improvement of muscle stiffness with repeated activity, known as the warm-up phenomenon, is classically seen in MC, but is also seen in some patients with SCN4A-related myotonia.7 And while cold sensitivity is generally thought of as a feature of PMC, a majority of people with chloride channelopathies report this symptom as well.7 Other factors that have been reported to increase muscle stiffness are pregnancy or menstruation, dietary potassium (mainly in SCM), hunger, and emotional stress.7,54–57
In addition to muscle stiffness, other common symptoms include weakness, pain, and fatigue. Episodic muscle weakness, oftentimes triggered by cold or exercise, can occur with SCN4A mutations and usually lasts seconds to minutes, but can last up to 2 days.7,58,59 Conversely, those with recessive chloride channel myotonia often describe weakness lasting only a few seconds, specifically when initiating movement (“transient paresis”).58 While fixed weakness and myotonia are generally considered hallmarks for DM, fixed weakness can occasionally be seen in NDM and PP.7,54,60 For the majority of patients, the pattern of weakness or extra-muscular manifestations distinguish these disorders (see the Differential Diagnosis section). That said, there are isolated families described with SCN4A mutations and either distal weakness, or more profound proximal weakness.61,62 A proportion of patients with NDM have pain as well. Although it is generally more common in sodium channelopathies, in one study, pain was the most prominent symptom in approximately 15% of people independent of the genotype.7 Fatigue is also common, but it is usually not the most prominent symptom.7 The presence of pain and fatigue seem to have the greatest impact on quality of life measures.10
Childhood presentations of NDM are more diverse than those in adults, especially in children with SCN4A-related myotonia.63 While the majority of patients presented with limb myotonia, other presenting symptoms included eyelid myotonia, double or blurred vision, strabismus, and stridor or choking episodes. In addition, SCN4A neonates are at risk of life-threatening complications due to SNEL, which usually presents with hypertonia and laryngospasm causing stridor and apnea.23 In children with chloride channelopathies, the main symptom was leg myotonia, with one child having jaw myotonia causing difficulty swallowing. Leg myotonia was described by patients, parents, or clinicians as reduced running or skipping ability, frequent falls, or a “funny gait.” Some patients had contractures, and one of these patients developed progressive scoliosis requiring surgical intervention.63
Finally, anesthetic complications are well-described in both children and adults.8,64 These can come in the form of severe, generalized muscle spasms making intubation and mask ventilation difficult. For some people, this may even be the presenting symptom.65 In general, multi-systemic involvement, such as cardiac arrhythmias, cognitive dysfunction, respiratory muscle weakness, or gastrointestinal problems, are not a feature of NDM and should prompt one to consider DM as an alternative diagnosis. Some researchers have questioned whether SCN4A variants may sometimes be related to the development of Brugada syndrome or cardiac arrhythmogenesis, but more studies are needed.66 Table 1 lists important questions that should be asked as part of the patient’s history.
TABLE 1.
Summary of general medical history questions
What are your primary symptoms: stiffness, cramps, muscle weakness and/or pain?
|
When did your symptoms start?
|
| Have you experienced complications of general anesthesia? |
Family history:
|
| Are there any signs of multi-systemic complaints as seen in myotonic dystrophy in the patient or relatives? (eg, cardiac arrhythmia, sudden cardiac death, respiratory problems, gastrointestinal problems, ptosis, cataracts, cognitive problems, etc.) |
4.1 |. Physical examination
The neurological examination starts with the observation of the patient (and accompanying relatives) getting up from the chair in the waiting room, walking to the consultation room, and the strength and relaxation of the grip during a handshake. It is also necessary to note whether the patient has muscle hypertrophy, or looks like he or she exercises regularly (referred to as a Hercules-appearance in the literature). Muscle hypertrophy has been described as a typical feature of recessive MC, but can also be seen in those with a sodium channelopathy.7
Confirmation of clinical myotonia should be sought using myotonia bedside tests (Table 2). Action myotonia, which is commonly tested in handgrip and eyelid muscles, can be observed by watching for delayed muscle relaxation following 5–10 s of maximal muscle contraction.67 Handgrip myotonia is common to both genotypes, eyelid myotonia is more common in sodium channelopathies, and leg muscle myotonia is more common in chloride channelopathies.7 It is also valuable to evaluate for the warm-up phenomenon versus paradoxical myotonia, which may be a clue as to the type of channelopathy present. This is done by asking the patient to repeatedly tightly close and open their eyes or handgrip up to 5 times to determine whether the speed of relaxation improves (warm-up) or worsens (paramyotonia) with repetition. Delayed muscle relaxation can also be observed following mechanical stimulation of the muscle, called percussion myotonia. This is usually performed by using a reflex hammer to percuss over the thenar eminence or extensor muscles in the forearm and watching for a catch in the muscle relaxation. Other helpful signs are the presence of a transient paresis (for example, of the biceps brachii muscle on bedside testing, which is unique to chloride channelopathies, and the lid-lag sign which is more typical in sodium channelopathies.8,68
TABLE 2.
Myotonia bedside tests
| Percussion myotonia in thenar, forearm extensor, trapezius, quadriceps and tongue muscles |
| Action myotonia of handgrip, eyelid closure, quadriceps muscles (5- to 10-s maximal voluntary contraction - repeated five times to check for warm-up phenomenon or paradoxical myotonia) |
| Transient paresis test in biceps8 |
| Lid-lag sign and extra-ocular muscle myotonia causing short-term diplopia68 |
In general, weakness is more likely to be noted by patients than found on examination,7 possibly due to its episodic nature. One study found 17/32 PMC patients had episodic weakness but only 4/32 with fixed weakness, all at least Medical Research Council (MRC) grade 4/5.54 Trivedi et al. also found that weakness was overall mild in both genotypes and typically in a proximal distribution if present.7 Other less common findings that may be seen on examination, especially in children, are strabismus, scoliosis, or contractures.15,63,69
While there is phenotypic overlap between the sodium and chloride channelopathies, some of the findings on history, examination, and electrophysiological testing (see the Electrophysiological Evaluation section) are highly specific for a particular genotype. For example, the presence of paradoxical myotonia is close to 100% specific for SCN4A-related disorders and the presence of transient paresis was found to be 100% specific for recessive chloride channelopathies.7,8 While other features are not as specific, Table 3 lists potential distinguishing features between the two genotypes.
TABLE 3.
Clinical characteristics that may help distinguish between nondystrophic myotonia genotypes
| Sodium channelopathies | Chloride channelopathies |
|---|---|
| Clinical clues | |
| Lower mean age at onset (mean 5 years) | Higher mean age at onset (mean 10 years) |
| Worsening of stiffness after repetitive use/ muscle contractions (ie, paramyotonia) | Decrease of myotonia after repetitive use/muscle contractions (ie, warm-up phenomenon) |
| Presence of pain | Lack of pain |
| Presence of face stiffness | Presence of leg stiffness (eg, difficulty in standing up quickly, climbing stairs) |
| Episodic weakness | Transient paresis at the onset of activity (usually recessive disease) |
| Worsening of symptoms in the cold | |
| Examination | |
| Eyelid myotonia | Action/percussion myotonia in leg muscles |
| Increase of myotonia after repetitive contractions (ie, paramyotonia) | Decrease of myotonia after repetitive contractions (ie, warm-up phenomenon) |
| Transient paresis positive in biceps8 | |
| Electrophysiological findings | |
| Long duration (>2 s), high amplitude (>4 mV) myotonic potentials with a “slowly decelerating motorcycle” sound | Short duration (<1 s), low amplitude (<0.4 mV) myotonic potentials with a “dive bomber” sound75 |
| Interdischarge interval of myotonic potentials <30 ms | Interdischarge Interval of myotonic potentials >30 ms |
| Fournier Pattern I or III with short-exercise testing | Fournier Pattern II with short-exercising testing |
5 |. DIAGNOSIS
5.1 |. Diagnostic algorithm and criteria
Here, we propose simple diagnostic criteria to help in the clinical diagnosis and to serve as a framework to support inclusion in future clinical trials (Table 4). If there is a high clinical suspicion for NDM, we recommend starting the diagnostic evaluation with an EMG to confirm the presence of electrical myotonia (especially if clinical myotonia is not obvious) followed by confirmatory genetic testing (Figure 2). In most cases, testing for both CLCN1 and SCN4A mutations concurrently is suggested due to the large phenotypic overlap. If a pathogenic variant (or two pathogenic variants for BMC) is identified, then there is no need for further diagnostic testing, and this would be considered a definite diagnosis of NDM. If a variant of uncertain significance (VUS) is found, then this would be considered probable NDM, and if no variant is found, it would be considered possible NDM (Table 4). If genetic testing does not identify a known pathogenic mutation, or if genetic testing is unavailable, then further diagnostic work-up is recommended and is supportive.
TABLE 4.
Diagnostic criteria for nondystrophic myotonias
Presence of:
|
| Absence of: Signs and symptoms consistent with myotonic dystrophy or other potential causes of myotonia (eg, medications). |
| Definite NDM: Must have 1 and/or 2, and 3 |
| Probable NDM: Must have 1 and 2, and a VUS OR 1 or 2 are atypical and 3 |
| Possible NDM: Must have 1 and 2 |
NDM, nondystrophic myotonia; VUS, variant of uncertain significance.
FIGURE 2.
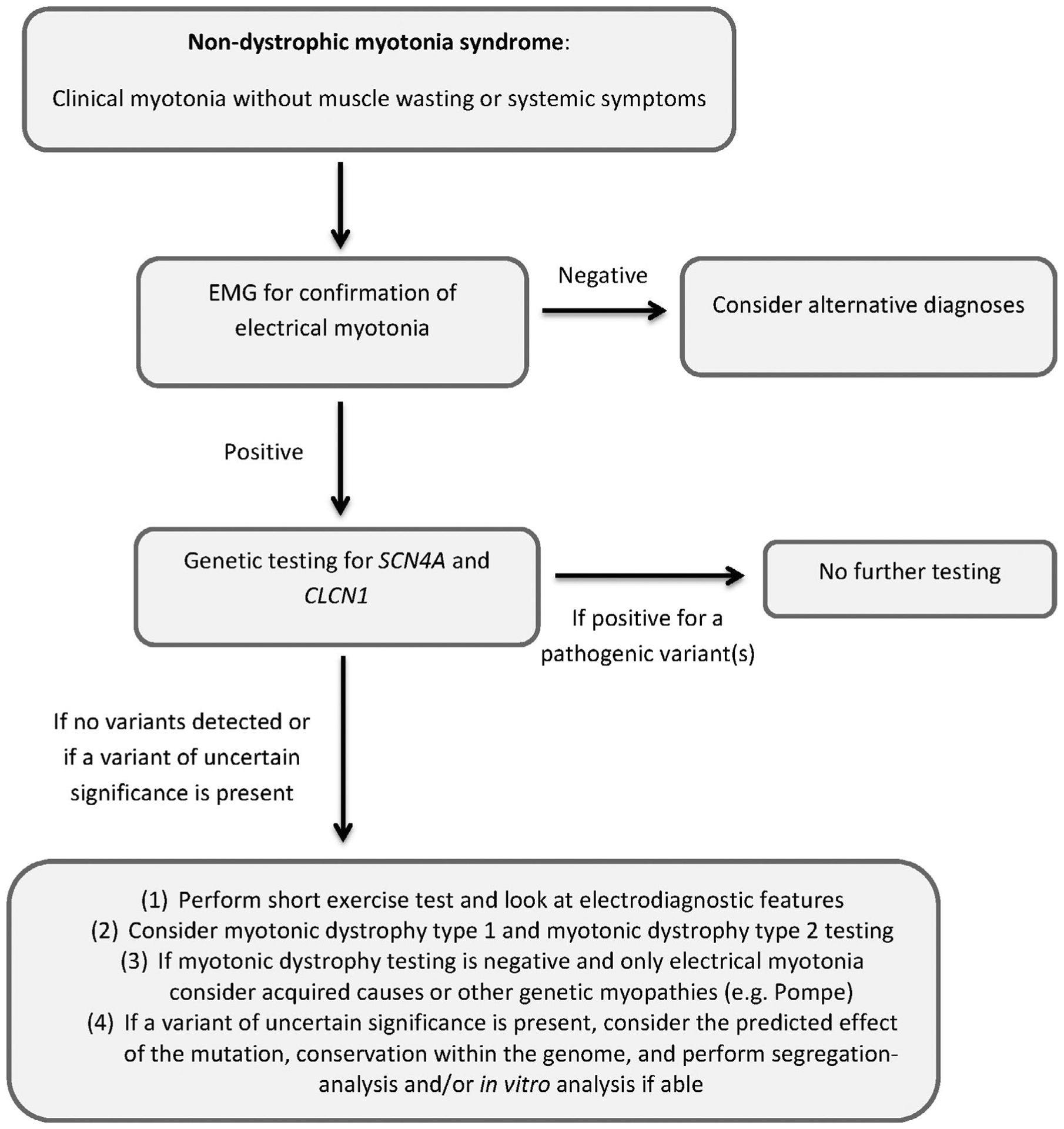
Diagnostic algorithm
Taken together with the history and examination, electrophysiological testing can help support the diagnosis of NDM, and even give clues as to the type of channelopathy. It is also important in this situation to exclude other disorders, especially DM (see more discussion in the Differential Diagnosis section). If a VUS is present, its pathogenicity can also be supported by the type of mutation and predicted effect on the channel, conservation within the genome and segregation-analysis. If available, in vitro analysis of the mutation can be completed as well.70
5.2 |. Genetic testing
There are limited data on detection rates of CLCN1 and SCN4A variants in patients for whom there is a high clinical suspicion of NDM. One group reported the false-negative rate to be as high as 20% in 2007.71 However, in a large cohort of Dutch families with suspected NDMs (54 families),70 in-tandem single gene sequencing analysis identified at least one variant in 100% of cases. The actual mutation detection yield was thought to be, at worst, 93% due to three recessive and three sporadic cases not yielding a second CLCN1 mutation.70 An overview of all reported mutations of SCN4A and CLCN1 is beyond the scope of this report but can be found at the LOVD (Leiden Open Variant Database).72,73
5.3 |. Electrophysiological evaluation
Needle EMG can be used to test for the presence of myotonic discharges (eg, electrical myotonia). Myotonic potentials are defined as repetitive discharges, firing at a rate of 20–80 Hz, in which the amplitude and frequency of the potentials wax and wane, creating a characteristic “dive bomber” sound.74 The individual potentials may resemble fibrillation potentials or positive sharp waves, but it is the repetitive firing and unique sound that distinguishes it as a myotonic potential.74 The sensitivity of myotonic discharges in NDMs and DM1 was found to be 100% if enough muscles were examined bilaterally (tibialis anterior, quadriceps, first dorsal interosseous, and biceps brachii; 10 insertions per muscle).75–77
In DM2, sensitivity of myotonic discharges using this protocol has been reported to be 100% in the largest cohort study,76 but reduced sensitivity is reported in case reports and a small cohort study.78,79 For these reasons, the negative predictive value of EMG in excluding a myotonic syndrome approaches 100%, but myotonia may be harder to find in DM2. However, the finding of myotonic discharges on needle EMG is not specific for a myotonic syndrome.
Myotonic discharges can also be found in other neuromuscular disorders such as Pompe disease (often isolated discharges in para-spinal muscles80), inflammatory myopathy, congenital myopathy (especially myofibrillar myopathy81), rippling muscle disease,82 and anti-MuSK myasthenia gravis (for complete overview see Hehir and Logigian83).84
Myotonia has also been found in severe hypothyroidism (although these pregenetic era patients might have had unrecognized DM2 in which hypothyroidism can elicit symptoms of myotonia)85 and after the use of certain drugs: 20,25-diazacholesterol, clofibrate, 2,4-dichlorophenoxyacetate, chloroquine, colchicine, and hydroxymethylgutaryl coenzyme A reductase inhibitors.86–93 However, many of these cases may actually be examples of “pseudomyotonia,” in which complex repetitive discharges may have been confused for myotonia, and clinically symptomatic myotonia is not usually present.74
The quantitative and qualitative differences in myotonic discharges may also be used to help discriminate between different disorders (see Table 3). For example, the duration of the first interdischarge interval of a myotonic discharge upon needle examination of the rectus femoris muscle can discriminate between sodium and chloride channelopathies with a discriminative power of >95% (where the interdischarge interval is less than 30 ms for sodium channelopathies and greater than 30 ms for chloride channelopathies).75 In addition, electrical myotonia differs between DM1 and DM2. With the exception of proximal leg muscles, electrical myotonia is more evocable in DM1 than DM2 and tends to be waxing and waning in DM1 but only waning in DM2.76
Finally, needle EMG can be used to identify patterns that rule out NDMs and may point toward another neuromuscular disorder. For example, myopathic changes make DM more likely, electrically silent cramps may point toward Brody disease, or a combination of myokymic and neuromyotonic discharges may indicate Isaacs’ syndrome.94 Exercise in combination with determination of compound muscle action potential amplitude or area can be helpful in situations where genetic testing is negative or indeterminate. The short exercise test (SET), first reported by Streib et al.95 is useful for differentiating types of NDMs from one another. It is performed by having the patient maximally contract a muscle (typically the abductor digiti minimi [ADM]) for 10–12 s followed by supramaximal (ulnar) nerve stimulation immediately after exercise and then every 10 s for 50 s.96 This is repeated three times in succession, and after cooling and rewarming, which can increase the sensitivity.97
An abnormal decrement is defined as a concordant reduction in amplitude and area of greater than 20%.98 Different patterns, called Fournier patterns (see Table 5), can be distinguished from a combination of needle-EMG and SET results. Pattern I is typical in PMC patients, in whom decrements are maximal after the third trial and pattern II is common in MC patients, particularly BMC patients, in whom the maximal decrement occurs after the first trial. Pattern III, which does not show any abnormal decrement, is typical for SCM patients (long exercise tests [LETs]) have been reported in both MC and PMC.98,99 The LET, first reported by McManis et al.,100 is performed by having the patient maximally contract the ADM for 5 min followed by supramaximal stimulation of the ulnar nerve every minute for 40 to 60 min. A positive test is defined by a reduction in the compound muscle action potential (CMAP) amplitude, typically during the postexercise period, by more than 40%. In MC and PMC patients, decrements of greater than 40% on this test have been seen, but the decrement occurred during the 5-min exercise instead of during the period of rest.98
TABLE 5.
Short exercise test - Fournier pattern description and sensitivity/specificity data
| Fournier et al.96 * (healthy controls n = 41, NDMs n = 30, PP n = 21) | Tan et al.98 ** (healthy controls n = 65, NDMs n = 47, PP n = 19) | ||
|---|---|---|---|
| Pattern I | Electrical myotonia and post-exercise myotonic potentials (PEMPs) with significant post-exercise decrease in CMAP amplitude that worsened with repeating exercise | PMC: 100% sens., spec. 100%*** | PMC: 100% sens., spec. 100%*** |
| Pattern II | Electrical myotonia with significant transient decreased CMAP amplitude after short exercise that disappeared with repeating exercise | MC: 83% sens., spec. 84% (8/51) | MC: 72% sens. and 100% spec. |
| Pattern III | Electrical myotonia not associated with significant post-exercise CMAP changes | SCM: 50% sens., spec. 74% (12/51) | SCM: 100% sens., spec. (?) |
NDMs, nondystrophic myotonias; PMC, paramyotonia congenita; MC, myotonia congenita; SCM, sodium channel myotonia; CMAP, compound muscle action potential; sens: sensitivity; spec, specificity.
Significance threshold defined by a mean ± 2 SD from healthy population outcomes.
Significance threshold defined by concordant amplitude and area (CAA) decrement >20%, includes refined exercise testing with pre-cooling and rewarming of the other hand.
Specificity for a certain phenotype is calculated as 1 – (% of patients within the cohort of skeletal muscle channelopathies with a positive test results and a different phenotype). In comparison with the healthy controls, specificity was found to be 100% in all cases (none of the healthy volunteers exhibited an abnormal Fournier pattern).
5.4 |. Differential diagnosis
An alternative diagnosis should be considered if the signs or symptoms are not typical of NDM or if the diagnosis is not confirmed by genetic testing. In a patient with clinical and/or electrical myotonia, it is important to consider DM. DM1 is much more common than NDM, and in some patients the muscle weakness and systemic signs may be subtle. Clues that suggest a diagnosis of DM include frontal balding, ptosis, temporal atrophy, cardiac arrhythmias, respiratory weakness, early cataracts, gastrointestinal or cognitive dysfunction, and progressive muscle weakness.101 In DM1, the muscle weakness usually occurs first in the neck flexors, finger flexors, and foot dorsiflexors, whereas in DM2, the muscle weakness is in proximal limbs. As noted above, proximal limb weakness can be seen in NDM, but it is uncommon and rarely worse than MRC grade 4. DM2 may be harder to distinguish from NDM, but, DM2 usually presents in adulthood, and episodic weakness is not a feature of DM2. Furthermore, pain was found to be the most prominent disease symptom in DM2, whereas complaints of muscle stiffness were less common.7 In a subset of DM2 patients, a CLCN1 or SCN4A mutation is found as a disease modifier making diagnosis more challenging.102–104
There are also other related disorders that need to be considered. If PP is the main symptom within the phenotype, then a primary PP may be the correct alternative diagnosis. On the other hand, if certain features are seen in addition to myotonia (or pseudomyotonia), such as mask-like facies, blepharospasm, and skeletal deformities, then Schwartz-Jampel syndrome should be considered.105,106
Laboratory analysis can also be helpful when the diagnosis is uncertain. Creatine kinase (CK) and thyroid function tests are not discriminatory but may help to suggest an alternative diagnosis. For example, CK in NDM can typically range from normal to moderately increased (1–3 × normal), but a very high CK should lead one to investigate other causes of muscle disease.7 In addition, other muscle disorders should be considered if electrical myotonia is present without clinical myotonia. Inflammatory myopathies have been reported to have electrical myotonia or pseudomyotonia,107 but these disorders are usually easily distinguished by the clinical history. Hereditary muscle disorders, such as Pompe disease, centronuclear and myofibrillar myopathies, have also been reported to sometimes have electrical myotonia (see the Electrophysiological Evaluation section for a more complete list).107 Other hereditary muscle disorders such as Brody myopathy (caused by mutations in ATP2A1)108 and rippling muscle disease (caveolinopathy caused by CAV3 mutations)109 may be difficult to distinguish clinically due to the similar symptoms of muscle stiffness, myalgia, cramps, and fatigue. If a hereditary myopathy is considered in the differential diagnosis, we recommend gene panel testing that includes these disorders.
Other nonmuscular etiologies should also be considered. As previously mentioned, severe hypothyroidism has been associated with electrical myotonia, so testing thyroid function is important.85 Electrical myotonia has also been reported secondary to some medications (see the Electrophysiological Evaluation section).86–93 Another neurologic disorder to consider is Isaacs’ syndrome, which is a peripheral nerve hyperexcitability disorder characterized by neuromyotonia and myokymia with symptoms of muscle stiffness, cramps, and fatigue. Neuromyotonic, rather than myotonic, potentials are seen on EMG, characterized by irregularly firing motor unit action potentials at a rate of 150–300 Hz. It is most commonly an autoimmune disorder caused by the presence of voltage-gated potassium channel antibodies, although mutations in KCNA1 can be responsible for the inherited form.110,111 Dystonia may also be considered as some of the symptoms may be similar, but the EMG should be normal.112
5.5 |. Muscle ultrasound and muscle MRI
Muscle ultrasound and muscle MRI are not currently part of the diagnostic algorithm, but may become so in the future, or may become an important part of future clinical trials. One study that evaluated muscle ultrasound in a NDM cohort found elevated echo intensities in all muscles except the rectus femoris as well as muscle hypertrophy in the arms.113 Muscle MRI evaluation in an NDM cohort showed hyperintensity within muscles on either T1-weighted or short-T1 inversion recovery (STIR) images in all patients. Edema was common in calf musculature especially in the medial gastrocnemius muscle (18/21 patients), where a fairly specific finding of a “central stripe” of STIR hyperintensity was observed in 10/11 CLCN1 patients and 3/10 SCN4A patients but no healthy volunteers.114
6 |. TREATMENT AND MANAGEMENT
See Table 6 for an overview of common medications used for the treatment of NDMs.
TABLE 6.
Common medications used for the treatment of nondystrophic myotonias
| Drug | Level of evidence | Regulatory Approval | Dosage | Side-effects | Monitoring | Other considerations |
|---|---|---|---|---|---|---|
| Mexiletine | Double-blind, placebo-controlled, randomized trials115,116 | EMA approval as orphan drug designation for NDMs | Start 150 mg BID with titration to 200 mg TID or occasionally 300 mg TID | GI discomfort, dizziness, tremor, ataxia | ECG, LFTs, consider CBC | If ECG abnormal or history of arrhythmia, consult with cardiology prior to use |
| Lamotrigine | Double-blind, placebo-controlled randomized trial117 | None | Start 25 mg daily with slow titration to 300 mg daily | Skin rash, headache, fatigue, nausea | LFTs, BUN/Cr | |
| Acetazolamide | Non-randomized, open-label trial58,124 | None | 125 mg BID with titration to 250 mg TID | Nephrolithiasis, paresthesias, rash, agranulocytosis, electrolyte abnormalities, GI discomfort | Basic metabolic panel (Na, K, CO2), LFTs, CBC | Do not use if sulfa allergy. Consider renal ultrasound monitoring if high risk of nephrolithiasis. |
| Ranolazine | Non-randomized, open-label trials119,120 | None | Start 500 mg BID then titrate to 1000 mg BID | Constipation, dizziness, headache, | EKG, BUN/Cr if renal impairment. | Do not use with simvastatin >20 mg. Limit dose to 500 mg with concurrent use of CYP3A4 inhibitors (e.g., diltiazem, verapamil) |
Note: Adapted by permission from Springer Nature: Neurotherapeutics, Phillips and Trivedi, 2018.58
EMA, European Medicines Agency; FDA, Food and Drug Administration; NDMs, nondystrophic myotonias; BID, twice daily; TID, three times daily; GI, gastrointestinal; ECG, electrocardiogram; LFTs, liver function tests; CBC, complete blood count; BUN, blood urea nitrogen; Cr, creatinine; Na, sodium; K, potassium; CO2, carbon dioxide.
6.1 |. Sodium channel blockers
Mexiletine, a class IB antiarrhythmic that works by enhancing fast inactivation of sodium channels, currently has the most evidence of effectiveness in the treatment of NDMs and received European marketing authorization through an orphan drug designation in 2018. Two independent, randomized, placebo-controlled trials have shown that mexiletine is effective in reducing patient reported muscle stiffness, weakness, tiredness, and pain.115,116 Mexiletine also improved quality of life scores and reduced clinical myotonia on examination. In addition to mexiletine, lamotrigine was also effective in reducing patient-reported measures of myotonia and improving quality of life scores in a single randomized, placebo-controlled trial.117 Similar to what has been recorded in the epilepsy literature, lamotrigine was well-tolerated. In the mexiletine trials,115,116 there was a significant difference in effectiveness in favor of the chloride channelopathy patients in comparison to the patients with a sodium channelopathy. However, the opposite was found in a long-term, retrospective clinical cohort.118
Ranolazine, a medication Food and Drug Adminstration (FDA) approved for chronic angina (European Medicines Agency [EMA] approval for add-on chronic angina treatment), is another sodium channel blocker that has recently shown effectiveness in small open-label trials.119,120 Instead of increasing fast-inactivation of sodium channels like mexiletine and lamotrigine, ranolazine enhances slow inactivation of sodium channels.121 There is also level 3 evidence for the use of other sodium channel blockers such as procainamide (the oral form is not available in the United States), flecainide, phenytoin, carbamazepine, and tocainide (withdrawn from the market).101,122 Lacosamide and rufinamide are sodium channel blockers with reported anti-myotonic efficacy in vitro, but have not been formally studied in patients.123
6.2 |. Other pharmacological treatment
Other types of medications, such as carbonic anhydrase inhibitors, antidepressants, and calcium channel blockers, have been tried in the treatment of myotonia. In a small open-label study of acetazolamide (a carbonic anhydrase inhibitor) performed before the availability of genetic testing, patients with MC had improvement in myotonia, but one of two PMC patients experienced severe weakness related to the treatment.124 And, as mentioned, one SCM phenotype is named for its responsiveness to acetazolamide.20 It remains unclear how acetazolamide reduces symptoms of myotonia. One study suggested that acetazolamide may increase the open probability of chloride channels at the resting potential, but this has not been confirmed with further studies.125 Also, in those with a SCN4A mutation and a HyperPP instead of PMC phenotype, acetazolamide or dichlorphenamide, a more potent carbonic anhydrase inhibitor, is often the treatment of choice to reduce paralytic attacks.27,126
Tricyclic antidepressants have also been tried for the treatment of myotonia with some success in DM1 patients,127,128 and may have had benefit in SCN4A patients as reported in a case series.129 Other medications like nifedipine, a calcium channel blocker, and taurine, an amino acid that that stabilizes muscle membranes, have also mainly been evaluated in small trials with DM1 patients with some improvement in myotonia.130,131 Botulinum toxin A has been tried in one case of MC but was not effective.132 Quinine has historically been used for the treatment of myotonia, but is no longer used due to concern for rare but severe hematological side effects.133
6.3 |. Treatment in children
No trials have been completed in children with NDM, but there are many case reports and case series using medications similar to those used in adults,134 including mexiletine, acetazolamide, carbamazepine, and dantrolene.135–137 One case series suggests acetazolamide as a possible first choice in children with both chloride and sodium channelopathies based on its safety and experience with the drug amongst neurologists, although with the available trial evidence in adults, mexiletine should probably be considered to be the first drug of choice for children with NDM too (expert opinion).134 Treatment of SNEL may require special consideration, such as the monitoring of serum drug levels. While mexiletine and carbamazepine have been used with some success,138 there is some clinical and in vitro evidence that flecainide may be most effective in this phenotype.139 A single case described flecainide-induced Brugada syndrome in an adult patient with SCN4A-related myotonia, suggesting some caution, although this might have been an unrelated co-morbidity.140
7 |. FUTURE DIRECTIONS IN TREATMENT
More treatment options are needed as some patients are “nonresponders” to certain medications and some patients continue to have disabling symptoms despite treatment.116 Furthermore, Trivedi et al. found that 39% of patients are not on anti-myotonic treatment,7 despite all patients reporting symptoms. It is not clear if this is because the symptoms were too mild for treatment; physicians were unaware of the possible treatment options; there was reluctance of some physicians as well as patients to start a “cardiac drug,” such as mexiletine; or because of medication-related problems, such as lack of efficacy, side effects, or insurance coverage issues. Future trials should help determine whether a combination of treatments can be more helpful than one medication alone as well as exploring new mechanisms for potential therapy. For example, further exploration of myotonia modifiers, such as calcium and magnesium, is one potential avenue43 as is further examination of ways to increase chloride conductance, which has proven to be a challenge thus far.141
One group is currently studying beta adrenergic drugs, which have been found to have an effect similar to mexiletine in a myotonic rat.142 This same group is also exploring a pharmacogenetics approach to find the best medication option for a particular mutation.143,144 From a methodological perspective, aggregated N-of-1 trials (ie, single patient multiple cross-over trials) can help to create the desired personalized treatment outcome estimates, with increased power due to the multiple cross-over design and use of a Bayesian hierarchical model, while also providing results on the (sub)group level(s).116
7.1 |. Behavioral modification and physical exercise
Behavioral modification is discussed with patients if specific triggers, such as cold or potassium ingestion, can be identified. Behavioral modification, in the form of diet or exercise changes, is especially important in those with a PP phenotype as detailed in a recent review.27 The effect of physical exercise on NDM has not been studied extensively. One study of six people with MC found that aerobic training was not an effective anti-myotonia treatment, but it did improve fitness and CK levels remained stable.145 In general, we tell our patients that physical exercise, especially aerobic exercise, such as swimming, bicycling, or walking is encouraged. If the patient is not weak, we tell people that resistance exercise is also not likely to be harmful.
7.2 |. Anesthetic considerations
All patients with NDM should be made aware of the potential anesthetic complications. The use of the depolarizing muscle relaxant succinylcholine should be avoided as there are many case reports of this inducing a myotonic crisis with severe generalized muscle stiffness.64,65 While there have been a few cases reports of malignant hyperthermia in patients with myotonia, it is not thought to be a considerable risk.146 We advise patients with NDM to carry a medical warning card or wrist band with information of the disease and drug contraindications.
7.3 |. Pregnancy
A majority of patients report worsening of symptoms during pregnancy,147 and this is likely exacerbated by having to stop medications. It may take some women months after the pregnancy to return to their baseline level of symptoms.147 Treatment of myotonia during pregnancy should only be considered if the patient’s symptoms are very severe due to the lack of safety data (ie, category C) on the risk of teratogenicity. However, there are case reports of mexiletine and acetazolamide being used safely in pregnancy (although not specifically for NDM).148,149 With regard to effects on the pregnancy itself, one study of 25 women with a total of 63 pregnancies found an increased rate of infertility and fetal distress, but overall pregnancy outcomes were benign.147 Still, due to the possibility of life threatening complications like SNEL in SCN4A newborns, it is recommended that mothers discuss this risk with their obstetrician when planning a pregnancy.
8 |. CONCLUSION
Due to its rarity, there have been relatively few clinical trials and natural history studies in patients with NDM. The trials that have been completed have been helpful in creating a better understanding of the disease spectrum and the impact it has on quality of life. Due to these studies, the large phenotypic overlap has been increasingly recognized, and now there is level I evidence for the use of two medications in NDM. Still, a treatment gap exists in this population, and many patients are left with disabling symptoms even on treatment. Future studies should help to address these gaps and lead to improved patient-specific treatment. Ultimately, disease modifying treatments in the form of gene therapy are likely to be investigated for NDM, and will hopefully lead to better patient outcomes as well.
FUNDING INFORMATION
B.F. research is funded by AFM Telethon. S.C.C. received support from the NIH (NIAMS AR42703). G.M. research is supported By FMM-Fondazione Malattie Miotoniche, Milan, Italy.
CONFLICT OF INTEREST
W.D.A. received research funding from Gilead Sciences for the clinical study of ranolazine as a treatment for myotonia. V.A.S. serves in Advisory Boards as a scientific consultant for Biogen, Avexis, Santhera, Sarepta, PTC, and Dyne. J.M.S. receives grant support from the NIH, FSHD Society, MDA, and Friends of FSH Research, and is a consultant or serves on advisory board for Fulcrum, Dyne, Sarepta, Strongbridge, Acceleron, and Genzyme. B.S., S.L., R.J.B., S.C.C., B.F., R.C.G., M.G.H., E.M., G.M., J.R.T., B.v.E., and S.V. have no relevant disclosures.
Abbreviations:
- ADM
abductor digiti minimi
- BMC
Becker myotonia congenita
- CMAP
compound muscle action potential
- CMS
congenital myasthenic syndrome
- DM
myotonic dystrophy
- DM1
myotonic dystrophy type 1
- DM2
myotonic dystrophy type 2
- EMA
European Medicines Agency
- EMG
electromyography
- FDA
Food and Drug Adminstration
- HyperPP
hyperkalemic periodic paralysis
- HypoPP
hypokalemic periodic paralysis
- LET
long exercise test
- MC
myotonia congenita
- MRC
Medical Research Council
- NaPIC
sodium persistent inward current
- NDM
nondystrophic myotonia
- PAM
potassium-aggravated myotonia
- PMC
paramyotonia congenita
- PP
periodic paralysis
- SCM
sodium channel myotonia
- SET
short exercise test
- SNEL
severe neonatal episodic laryngospasm
- STIR
short-T1 inversion recovery
- TMC
Thomsen myotonia congenita
- TTX
tetrodotoxin a voltage-gated sodium channel blocker
- VUS
variant of uncertain significance
Footnotes
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
REFERENCES
- 1.Koch MC, Steinmeyer K, Lorenz C, et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992; 257:797–800. [DOI] [PubMed] [Google Scholar]
- 2.McClatchey AI, Van den Bergh P, Pericak-Vance MA, et al. Temperature-sensitive mutations in the III-IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita. Cell. 1992;68:769–774. [DOI] [PubMed] [Google Scholar]
- 3.Strümpell A Tonische Krämpfe in willkürliche bewegten Muskeln (Myotonia congenita). Berl Klin Wochenchr. 1881;9:119–121. [Google Scholar]
- 4.Erb W Klinischen und Pathologisch-Anatomisches von der Thomsenschen Krankheit. Neurol Centralblb. 1885;13:289–194. [Google Scholar]
- 5.Horga A, Raja Rayan DL, Matthews E, et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. 2013;80:1472–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stunnenberg BC, Raaphorst J, Deenen JCW, et al. Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul Disord. 2018;28:402–407. [DOI] [PubMed] [Google Scholar]
- 7.Trivedi JR, Bundy B, Statland J, et al. Non-dystrophic myotonia: prospective study of objective and patient reported outcomes. Brain. 2013;136:2189–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trip J, Drost G, Ginjaar HB, et al. Redefining the clinical phenotypes of non-dystrophic myotonic syndromes. J Neurol Neurosurg Psychiatry. 2009;80:647–652. [DOI] [PubMed] [Google Scholar]
- 9.Statland JM, Wang Y, Richesson R, et al. An interactive voice response diary for patients with non-dystrophic myotonia. Muscle Nerve. 2011;44:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trip J, de Vries J, Drost G, Ginjaar HB, van Engelen BG, Faber CG. Health status in non-dystrophic myotonias: close relation with pain and fatigue. J Neurol. 2009;256:939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sansone VA, Ricci C, Montanari M, et al. Measuring quality of life impairment in skeletal muscle channelopathies. Eur J Neurol. 2012; 19:1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomsen J Tonische Krämpfe in willkürlich beweglichen Muskeln in Folge von ererbter psychischer Disposition. Arch Psychiatr Nervenkr. 1876;6:702–718. [Google Scholar]
- 13.Becker PE. Zur Frage der Heterogenie der eblichen Myotonien. Nevenartz. 1957;28:455–460. [PubMed] [Google Scholar]
- 14.Becker PE. Die Heterogenitat der Myotonien. In: Proc. 2nd Int. Congr. Human Genetics. Amsterdam: Excerpta Med, 1963, vol 3, p. 1547–1552. [Google Scholar]
- 15.Heatwole CR, Moxley RT III. The nondystrophic myotonias. Neurotherapeutics. 2007;4:238–251. [DOI] [PubMed] [Google Scholar]
- 16.Eulenberg A Über eine familiäre, durch 6 Generationen verfolgbare congenitaler Paramyotonia. Neurol Centralblb. 1886;12:265–272. [Google Scholar]
- 17.Cannon SC. Channelopathies of skeletal muscle excitability. Compr Physiol. 2015;5:761–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricker K, Moxley RT III, Heine R, Lehmann-Horn F. Myotonia fluctuans. A third type of muscle sodium channel disease. Arch Neurol. 1994;51:1095–1102. [DOI] [PubMed] [Google Scholar]
- 19.Colding-Jorgensen E, Duno M, Vissing J. Autosomal dominant monosymptomatic myotonia permanens. Neurology. 2006;67:153–155. [DOI] [PubMed] [Google Scholar]
- 20.Trudell RG, Kaiser KK, Griggs RC. Acetazolamide-responsive myotonia congenita. Neurology. 1987;37:488–491. [DOI] [PubMed] [Google Scholar]
- 21.Rudel R, Ricker K, Lehmann-Horn F. Genotype-phenotype correlations in human skeletal muscle sodium channel diseases. Arch Neurol. 1993;50:1241–1248. [DOI] [PubMed] [Google Scholar]
- 22.Ptacek LJ, Tawil R, Griggs RC, et al. Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology. 1994;44: 1500–1503. [DOI] [PubMed] [Google Scholar]
- 23.Matthews E, Guet A, Mayer M, et al. Neonatal hypotonia can be a sodium channelopathy: recognition of a new phenotype. Neurology. 2008;71:1740–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaharieva IT, Thor MG, Oates EC, et al. Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or ‘classical’ congenital myopathy. Brain. 2016;139:674–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsujino A, Maertens C, Ohno K, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci U S A. 2003;100:7377–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnold WD, Feldman DH, Ramirez S, et al. Defective fast inactivation recovery of Nav 1.4 in congenital myasthenic syndrome. Ann Neurol. 2015;77:840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Statland JM, Fontaine B, Hanna MG, et al. Review of the diagnosis and treatment of periodic paralysis. Muscle Nerve. 2018;57:522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hopkins PM. Skeletal muscle physiology. Contin Educ Anaesth Crit Care Pain. 2006;6:1–6. [Google Scholar]
- 29.Lipicky RJ, Bryant SH. Sodium, potassium, and chloride fluxes in intercostal muscle from normal goats and goats with hereditary myotonia. J Gen Physiol. 1966;50:89–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lipicky RJ, Bryant SH, Salmon JH. Cable parameters, sodium, potassium, chloride, and water content, and potassium efflux in isolated external intercostal muscle of normal volunteers and patients with myotonia congenita. J Clin Invest. 1971;50:2091–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furman RE, Barchi RL. The pathophysiology of myotonia produced by aromatic carboxylic acids. Ann Neurol. 1978;4:357–365. [DOI] [PubMed] [Google Scholar]
- 32.Adrian RH, Bryant SH. On the repetitive discharge in myotonic muscle fibres. J Physiol. 1974;240:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burge JA, Hanna MG. Novel insights into the pathomechanisms of skeletal muscle channelopathies. Curr Neurol Neurosci Rep. 2012;12: 62–69. [DOI] [PubMed] [Google Scholar]
- 34.Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999;79:1317–1372. [DOI] [PubMed] [Google Scholar]
- 35.Hawash AA, Voss AA, Rich MM. Inhibiting persistent inward sodium currents prevents myotonia. Ann Neurol. 2017;82:385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pusch M Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum Mutat. 2002;19:423–434. [DOI] [PubMed] [Google Scholar]
- 37.Pusch M, Steinmeyer K, Koch MC, Jentsch TJ. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the CIC-1 chloride channel. Neuron. 1995;15: 1455–1463. [DOI] [PubMed] [Google Scholar]
- 38.Saviane C, Conti F, Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J Gen Physiol. 1999;113:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vindas-Smith R, Fiore M, Vasquez M, et al. Identification and functional characterization of CLCN1 mutations found in nondystrophic myotonia patients. Hum Mutat. 2016;37:74–83. [DOI] [PubMed] [Google Scholar]
- 40.Lossin C, George AL Jr. Myotonia congenita. Adv Genet. 2008;63: 25–55. [DOI] [PubMed] [Google Scholar]
- 41.Tang CY, Chen TY. Physiology and pathophysiology of CLC-1: mechanisms of a chloride channel disease, myotonia. J Biomed Biotechnol. 2011;2011:685328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehmann-Horn F, Rudel R. Hereditary nondystrophic myotonias and periodic paralyses. Curr Opin Neurol. 1995;8:402–410. [DOI] [PubMed] [Google Scholar]
- 43.Skov M, Riisager A, Fraser JA, Nielsen OB, Pedersen TH. Extracellular magnesium and calcium reduce myotonia in ClC-1 inhibited rat muscle. Neuromuscul Disord. 2013;23:489–502. [DOI] [PubMed] [Google Scholar]
- 44.Ulbricht W The effect of veratridine on excitable membranes of nerve and muscle. Ergeb Physiol. 1969;61:18–71. [DOI] [PubMed] [Google Scholar]
- 45.Lehmann-Horn F, Rudel R, Dengler R, Lorkovic H, Haass A, Ricker K. Membrane defects in paramyotonia congenita with and without myotonia in a warm environment. Muscle Nerve. 1981;4:396–406. [DOI] [PubMed] [Google Scholar]
- 46.Lehmann-Horn F, Rudel R, Ricker K, Lorkovic H, Dengler R, Hopf HC. Two cases of adynamia episodica hereditaria: in vitro investigation of muscle cell membrane and contraction parameters. Muscle Nerve. 1983;6:113–121. [DOI] [PubMed] [Google Scholar]
- 47.Cannon SC. Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci. 2006;29:387–415. [DOI] [PubMed] [Google Scholar]
- 48.Cannon SC. Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol. 2018;246:309–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ptacek LJ, George AL Jr, Griggs RC, et al. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991;67: 1021–1027. [DOI] [PubMed] [Google Scholar]
- 50.Rojas CV, Wang JZ, Schwartz LS, Hoffman EP, Powell BR, Brown RH Jr. A Met-to-Val mutation in the skeletal muscle Na+ channel alpha-subunit in hyperkalaemic periodic paralysis. Nature. 1991;354:387–389. [DOI] [PubMed] [Google Scholar]
- 51.Ptacek LJ, George AL Jr, Barchi RL, et al. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron. 1992;8:891–897. [DOI] [PubMed] [Google Scholar]
- 52.Lerche H, Heine R, Pika U, et al. Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J Physiol. 1993;470:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang N, Ji S, Zhou M, et al. Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc Natl Acad Sci U S A. 1994;91:12785–12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matthews E, Tan SV, Fialho D, et al. What causes paramyotonia in the United Kingdom? Common and new SCN4A mutations revealed. Neurology. 2008;70:50–53. [DOI] [PubMed] [Google Scholar]
- 55.Statland J, Phillips L, Trivedi JR. Muscle channelopathies. Neurol Clin. 2014;32:801–815. x. [DOI] [PubMed] [Google Scholar]
- 56.Orrell RW, Jurkat-Rott K, Lehmann-Horn F, Lane RJ. Familial cramp due to potassium-aggravated myotonia. J Neurol Neurosurg Psychiatry. 1998;65:569–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Basu A, Nishanth P, Ifaturoti O. Pregnancy in women with myotonia congenita. Int J Gynaecol Obstet. 2009;106:62–63. [DOI] [PubMed] [Google Scholar]
- 58.Phillips L, Trivedi JR. Skeletal muscle channelopathies. Neurotherapeutics. 2018;15:954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fontaine B Periodic paralysis, myotonia congenita and sarcolemmal ion channels: a success of the candidate gene approach. Neuromuscul Disord. 1993;3:101–107. [DOI] [PubMed] [Google Scholar]
- 60.Miller TM, Dias da Silva MR, Miller HA, et al. Correlating phenotype and genotype in the periodic paralyses. Neurology. 2004;63: 1647–1655. [DOI] [PubMed] [Google Scholar]
- 61.Bednarz M, Stunnenberg BC, Kusters B, et al. A novel Ile1455Thr variant in the skeletal muscle sodium channel alpha-subunit in a patient with a severe adult-onset proximal myopathy with electrical myotonia and a patient with mild paramyotonia phenotype. Neuromuscul Disord. 2017;27:175–182. [DOI] [PubMed] [Google Scholar]
- 62.Taminato T, Mori-Yoshimura M, Miki J, et al. Paramyotonia congenita with persistent distal and facial muscle weakness: a case report with literature review. J Neuromuscul Dis. 2020;7(2):193–201. [DOI] [PubMed] [Google Scholar]
- 63.Matthews E, Silwal A, Sud R, et al. Skeletal muscle channelopathies: rare disorders with common pediatric symptoms. J Pediatr. 2017; 188:181–185.e186. [DOI] [PubMed] [Google Scholar]
- 64.Bandschapp O, Iaizzo PA. Pathophysiologic and anesthetic considerations for patients with myotonia congenita or periodic paralyses. Paediatr Anaesth. 2013;23:824–833. [DOI] [PubMed] [Google Scholar]
- 65.Farbu E, Softeland E, Bindoff LA. Anaesthetic complications associated with myotonia congenita: case study and comparison with other myotonic disorders. Acta Anaesthesiol Scand. 2003;47:630–634. [DOI] [PubMed] [Google Scholar]
- 66.Bissay V, Van Malderen SC, Keymolen K, et al. SCN4A variants and Brugada syndrome: phenotypic and genotypic overlap between cardiac and skeletal muscle sodium channelopathies. Eur J Hum Genet. 2016;24:400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matthews E, Fialho D, Tan SV, et al. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain. 2010;133:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stunnenberg BC, Drost G. Teaching video neuroimages: lid lag sign and diplopia in paramyotonia congenita. Neurology. 2014;83:e68. [DOI] [PubMed] [Google Scholar]
- 69.Du H, Grob SR, Zhao L, et al. Myotonia congenita with strabismus in a large family with a mutation in the SCN4A gene. Eye (Lond). 2012; 26:1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trip J, Drost G, Verbove DJ, et al. In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with nondystrophic myotonia. Eur J Hum Genet. 2008;16:921–929. [DOI] [PubMed] [Google Scholar]
- 71.Cleland JC, Logigian EL. Clinical evaluation of membrane excitability in muscle channel disorders: potential applications in clinical trials. Neurotherapeutics. 2007;4:205–215. [DOI] [PubMed] [Google Scholar]
- 72.Leiden Open Variation Database. https://databases.lovd.nl/shared/genes/SCN4A. Accessed April 13, 2020.
- 73.Leiden Open Variation Database. https://databases.lovd.nl/shared/genes/CLCN1. Accessed April 13, 2020.
- 74.Manousakis G, Al-Lozi M. Miller TM. AANEM Monogr: Differential diagnosis of myotonic disorders; 2012.
- 75.Drost G, Stunnenberg BC, Trip J, et al. Myotonic discharges discriminate chloride from sodium muscle channelopathies. Neuromuscul Disord. 2015;25:73–80. [DOI] [PubMed] [Google Scholar]
- 76.Logigian EL, Ciafaloni E, Quinn LC, et al. Severity, type, and distribution of myotonic discharges are different in type 1 and type 2 myotonic dystrophy. Muscle Nerve. 2007;35:479–485. [DOI] [PubMed] [Google Scholar]
- 77.Ghosh PS, Sorenson EJ. Use of clinical and electrical myotonia to differentiate childhood myopathies. J Child Neurol. 2015;30:1300–1306. [DOI] [PubMed] [Google Scholar]
- 78.Milone M, Batish SD, Daube JR. Myotonic dystrophy type 2 with focal asymmetric muscle weakness and no electrical myotonia. Muscle Nerve. 2009;39:383–385. [DOI] [PubMed] [Google Scholar]
- 79.Young NP, Daube JR, Sorenson EJ, Milone M. Absent, unrecognized, and minimal myotonic discharges in myotonic dystrophy type 2. Muscle Nerve. 2010;41:758–762. [DOI] [PubMed] [Google Scholar]
- 80.Hobson-Webb LD, Dearmey S, Kishnani PS. The clinical and electrodiagnostic characteristics of Pompe disease with post-enzyme replacement therapy findings. Clin Neurophysiol. 2011;122: 2312–2317. [DOI] [PubMed] [Google Scholar]
- 81.Hanisch F, Kraya T, Kornhuber M, Zierz S. Diagnostic impact of myotonic discharges in myofibrillar myopathies. Muscle Nerve. 2013;47: 845–848. [DOI] [PubMed] [Google Scholar]
- 82.Milone M, McEvoy KM, Sorenson EJ, Daube JR. Myotonia associated with caveolin-3 mutation. Muscle Nerve. 2012;45:897–900. [DOI] [PubMed] [Google Scholar]
- 83.Hehir MK, Logigian EL. Electrodiagnosis of myotonic disorders. Phys Med Rehabil Clin N Am. 2013;24:209–220. [DOI] [PubMed] [Google Scholar]
- 84.Joundi RA, Israelian G, Ghavanini A, Kassardjian CD. Myotonic discharges in anti-MuSK myasthenia. Can J Neurol Sci. 2018;45: 707–708. [DOI] [PubMed] [Google Scholar]
- 85.Venables GS, Bates D, Shaw DA. Hypothyroidism with true myotonia. J Neurol Neurosurg Psychiatry. 1978;41:1013–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blomberg LH. Dystrophia myotonica probably caused by chloroquine. Acta Neurol Scand Suppl. 1965;13(Pt 2):647–651. [DOI] [PubMed] [Google Scholar]
- 87.Dromgoole SH, Campion DS, Peter JB. Myotonia induced by clofibrate and sodium chlorophenoxy isobutyrate. Biochem Med. 1975; 14:238–240. [DOI] [PubMed] [Google Scholar]
- 88.Kwiecinski H Myotonia induced with clofibrate in rats. J Neurol. 1978;219:107–116. [DOI] [PubMed] [Google Scholar]
- 89.Kwiecinski H Myotonia induced by chemical agents. Crit Rev Toxicol. 1981;8:279–310. [DOI] [PubMed] [Google Scholar]
- 90.Pierno S, De Luca A, Tricarico D, et al. Potential risk of myopathy by HMG-CoA reductase inhibitors: a comparison of pravastatin and simvastatin effects on membrane electrical properties of rat skeletal muscle fibers. J Pharmacol Exp Ther. 1995;275:1490–1496. [PubMed] [Google Scholar]
- 91.Ramsey RB, McGarry JD, Fischer VW, Sarnat HB. Alteration of developing and adult rat muscle membranes by zuclomiphene and other hypocholesterolemic agents. Acta Neuropathol. 1978;44: 15–19. [DOI] [PubMed] [Google Scholar]
- 92.Rutkove SB, De Girolami U, Preston DC, et al. Myotonia in colchicine myoneuropathy. Muscle Nerve. 1996;19:870–875. [DOI] [PubMed] [Google Scholar]
- 93.Sonoda Y, Gotow T, Kuriyama M, Nakahara K, Arimura K, Osame M. Electrical myotonia of rabbit skeletal muscles by HMG-CoA reductase inhibitors. Muscle Nerve. 1994;17:891–897. [DOI] [PubMed] [Google Scholar]
- 94.Katirji B Peripheral nerve hyperexcitability. Handb Clin Neurol. 2019; 161:281–290. [DOI] [PubMed] [Google Scholar]
- 95.Streib EW, Sun SF, Yarkowski T. Transient paresis in myo-tonic syndromes: a simplified electrophysiologic approach. Muscle Nerve. 1982;5:719–723. [Google Scholar]
- 96.Fournier E, Arzel M, Sternberg D, et al. Electromyography guides toward subgroups of mutations in muscle channelopathies. Ann Neurol. 2004;56:650–661. [DOI] [PubMed] [Google Scholar]
- 97.Fournier E, Viala K, Gervais H, et al. Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol. 2006;60:356–365. [DOI] [PubMed] [Google Scholar]
- 98.Tan SV, Matthews E, Barber M, et al. Refined exercise testing can aid DNA-based diagnosis in muscle channelopathies. Ann Neurol. 2011;69:328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kuntzer T, Flocard F, Vial C, et al. Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve. 2000;23: 1089–1094. [DOI] [PubMed] [Google Scholar]
- 100.McManis PG, Lambert EH, Daube JR. The exercise test in periodic paralysis. Muscle Nerve. 1986;9:704–710. [DOI] [PubMed] [Google Scholar]
- 101.Heatwole CR, Statland JM, Logigian EL. The diagnosis and treatment of myotonic disorders. Muscle Nerve. 2013;47:632–648. [DOI] [PubMed] [Google Scholar]
- 102.Suominen T, Schoser B, Raheem O, et al. High frequency of co-segregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and Germany. J Neurol. 2008;255: 1731–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun C, Van Ghelue M, Tranebjaerg L, Thyssen F, Nilssen O, Torbergsen T. Myotonia congenita and myotonic dystrophy in the same family: coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin Genet. 2011;80:574–580. [DOI] [PubMed] [Google Scholar]
- 104.Bugiardini E, Rivolta I, Binda A, et al. SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord. 2015;25:301–307. [DOI] [PubMed] [Google Scholar]
- 105.Mathur N, Ghosh PS. Schwartz-Jampel Syndrome. Pediatr Neurol. 2017;68:77–78. [DOI] [PubMed] [Google Scholar]
- 106.Padmanabha H, Mathew T, Manjusha T. Schwartz-Jampel Syndrome Mimicking Myotonia Congenita. Neurol India. 2019;67(4):1160–1162. [DOI] [PubMed] [Google Scholar]
- 107.Miller TM. Differential diagnosis of myotonic disorders. Muscle Nerve. 2008;37:293–299. [DOI] [PubMed] [Google Scholar]
- 108.Braz L, Soares-Dos-Reis R, Seabra M, Silveira F, Guimaraes J. Brody disease: when myotonia is not myotonia. Pract Neurol. 2019;19: 417–419. [DOI] [PubMed] [Google Scholar]
- 109.Torbergsen T Rippling muscle disease: a review. Muscle Nerve Suppl. 2002;11:S103–S107. [DOI] [PubMed] [Google Scholar]
- 110.Sawlani K, Katirji B. Peripheral nerve hyperexcitability syndromes. Continuum (Minneap Minn). 2017;23:1437–1450. [DOI] [PubMed] [Google Scholar]
- 111.Ahmed A, Simmons Z. Isaacs syndrome: a review. Muscle Nerve. 2015;52:5–12. [DOI] [PubMed] [Google Scholar]
- 112.Bloem BR, Voermans NC, Aerts MB, Bhatia KP, van Engelen BG, van de Warrenburg BP. The wrong end of the telescope: neuromuscular mimics of movement disorders (and vice versa). Pract Neurol. 2016; 16:264–269. [DOI] [PubMed] [Google Scholar]
- 113.Trip J, Pillen S, Faber CG, van Engelen BG, Zwarts MJ, Drost G. Muscle ultrasound measurements and functional muscle parameters in non-dystrophic myotonias suggest structural muscle changes. Neuromuscul Disord. 2009;19:462–467. [DOI] [PubMed] [Google Scholar]
- 114.Morrow JM, Matthews E, Raja Rayan DL, et al. Muscle MRI reveals distinct abnormalities in genetically proven non-dystrophic myotonias. Neuromuscul Disord. 2013;23:637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Statland JM, Bundy BN, Wang Y, et al. Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: a randomized controlled trial. JAMA. 2012;308:1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Stunnenberg BC, Raaphorst J, Groenewoud HM, et al. Effect of mexiletine on muscle stiffness in patients with nondystrophic myotonia evaluated using aggregated N-of-1 trials. JAMA. 2018; 320:2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Andersen G, Hedermann G, Witting N, Duno M, Andersen H, Vissing J. The antimyotonic effect of lamotrigine in non-dystrophic myotonias: a double-blind randomized study. Brain. 2017;140:2295–2305. [DOI] [PubMed] [Google Scholar]
- 118.Suetterlin KJ, Bugiardini E, Kaski JP, et al. Long-term safety and efficacy of mexiletine for patients with skeletal muscle channelopathies. JAMA Neurol. 2015;72:1531–1533. [DOI] [PubMed] [Google Scholar]
- 119.Lorusso S, Kline D, Bartlett A, et al. Open-label trial of ranolazine for the treatment of paramyotonia congenita. Muscle Nerve. 2019;59: 240–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Arnold WD, Kline D, Sanderson A, et al. Open-label trial of ranolazine for the treatment of myotonia congenita. Neurology. 2017;89:710–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Novak KR, Norman J, Mitchell JR, Pinter MJ, Rich MM. Sodium channel slow inactivation as a therapeutic target for myotonia congenita. Ann Neurol. 2015;77:320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kwiecinski H, Ryniewicz B, Ostrzycki A. Treatment of myotonia with antiarrhythmic drugs. Acta Neurol Scand. 1992;86:371–375. [DOI] [PubMed] [Google Scholar]
- 123.Skov M, de Paoli FV, Nielsen OB, Pedersen TH. The anti-convulsants lacosamide, lamotrigine, and rufinamide reduce myotonia in isolated human and rat skeletal muscle. Muscle Nerve. 2017;56:136–142. [DOI] [PubMed] [Google Scholar]
- 124.Griggs RC, Moxley RT III, Riggs JE, Engel WK. Effects of acetazolamide on myotonia. Ann Neurol. 1978;3:531–537. [DOI] [PubMed] [Google Scholar]
- 125.Eguchi H, Tsujino A, Kaibara M, et al. Acetazolamide acts directly on the human skeletal muscle chloride channel. Muscle Nerve. 2006;34: 292–297. [DOI] [PubMed] [Google Scholar]
- 126.Sansone VA, Burge J, McDermott MP, et al. Randomized, placebo-controlled trials of dichlorphenamide in periodic paralysis. Neurology. 2016;86:1408–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Milner-Brown HS, Miller RG. Myotonic dystrophy: quantification of muscle weakness and myotonia and the effect of amitriptyline and exercise. Arch Phys Med Rehabil. 1990;71:983–987. [PubMed] [Google Scholar]
- 128.Antonini G, Vichi R, Leardi MG, Pennisi E, Monza GC, Millefiorini M. Effect of clomipramine on myotonia: a placebo-controlled, double-blind, crossover trial. Neurology. 1990;40:1473–1474. [DOI] [PubMed] [Google Scholar]
- 129.Bissay V, Keymolen K, Lissens W, Laureys G, Schmedding E, De Keyser J. Late onset painful cold-aggravated myotonia: three families with SCN4A L1436P mutation. Neuromuscul Disord. 2011;21: 590–593. [DOI] [PubMed] [Google Scholar]
- 130.Durelli L, Mutani R, Fassio F. The treatment of myotonia: evaluation of chronic oral taurine therapy. Neurology. 1983;33:599–603. [DOI] [PubMed] [Google Scholar]
- 131.Grant R, Sutton DL, Behan PO, Ballantyne JP. Nifedipine in the treatment of myotonia in myotonic dystrophy. J Neurol Neurosurg Psychiatry. 1987;50:199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dressler D, Adib SF. Botulinum toxin in myotonia congenita: it does not help against rigidity and pain. J Neural Transm. 2014;121: 531–532. [DOI] [PubMed] [Google Scholar]
- 133.Leyburn P, Walton JN. The treatment of myotonia: a controlled clinical trial. Brain. 1959;82:81–91. [DOI] [PubMed] [Google Scholar]
- 134.Markhorst JM, Stunnenberg BC, Ginjaar IB, Drost G, Erasmus CE, Sie LT. Clinical experience with long-term acetazolamide treatment in children with nondystrophic myotonias: a three-case report. Pediatr Neurol. 2014;51:537–541. [DOI] [PubMed] [Google Scholar]
- 135.Ohtaki E, Komori H, Yamaguchi Y, Matsuishi T. Successful dantrolene sodium treatment of a patient with myotonia congenita (Thomsen’s disease). Acta Paediatr Jpn. 1991;33:668–671. [DOI] [PubMed] [Google Scholar]
- 136.Ceccarelli M, Rossi B, Siciliano G, Calevro L, Tarantino E. Clinical and electrophysiological reports in a case of early onset myotonia congenita (Thomsen’s disease) successfully treated with mexiletine. Acta Paediatr. 1992;81:453–455. [DOI] [PubMed] [Google Scholar]
- 137.Sheela SR. Myotonia congenita: response to carbamazepine. Indian Pediatr. 2000;37:1122–1125. [PubMed] [Google Scholar]
- 138.Caietta E, Milh M, Sternberg D, et al. Diagnosis and outcome of SCN4A-related severe neonatal episodic laryngospasm (SNEL): 2 new cases. Pediatrics. 2013;132:e784–e787. [DOI] [PubMed] [Google Scholar]
- 139.Portaro S, Rodolico C, Sinicropi S, Musumeci O, Valenzise M, Toscano A. Flecainide-responsive myotonia permanens with SNEL onset: a new case and literature review. Pediatrics. 2016;137: pii: e20153289, e1–e6 [DOI] [PubMed] [Google Scholar]
- 140.Cavalli M, Fossati B, Vitale R, et al. Flecainide-Induced Brugada Syndrome in a patient with skeletal muscle sodium channelopathy: a case report with critical therapeutical implications and review of the literature. Front Neurol. 2018;9:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Imbrici P, Altamura C, Pessia M, Mantegazza R, Desaphy JF, Camerino DC. ClC-1 chloride channels: state-of-the-art research and future challenges. Front Cell Neurosci. 2015;9:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Desaphy JF, Costanza T, Carbonara R, Conte CD. In vivo evaluation of antimyotonic efficacy of beta-adrenergic drugs in a rat model of myotonia. Neuropharmacology. 2013;65:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.De Bellis M, Camerino DC, Desaphy JF. Toward precision medicine in myotonic syndromes. Oncotarget. 2017;8:14279–14280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Desaphy JF, Carbonara R, D’Amico A, et al. Translational approach to address therapy in myotonia permanens due to a new SCN4A mutation. Neurology. 2016;86:2100–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Andersen G, Lokken N, Vissing J. Aerobic training in myotonia congenita: effect on myotonia and fitness. Muscle Nerve. 2017;56:696–699. [DOI] [PubMed] [Google Scholar]
- 146.Parness J, Bandschapp O, Girard T. The myotonias and susceptibility to malignant hyperthermia. Anesth Analg. 2009;109:1054–1064. [DOI] [PubMed] [Google Scholar]
- 147.Snyder Y, Donlin-Smith C, Snyder E, Pressman E, Ciafaloni E. The course and outcome of pregnancy in women with nondystrophic myotonias. Muscle Nerve. 2015;52:1013–1015. [DOI] [PubMed] [Google Scholar]
- 148.Gregg AR, Tomich PG. Mexilitene use in pregnancy. J Perinatol. 1988;8:33–35. [PubMed] [Google Scholar]
- 149.Falardeau J, Lobb BM, Golden S, Maxfield SD, Tanne E. The use of acetazolamide during pregnancy in intracranial hypertension patients. J Neuroophthalmol. 2013;33:9–12. [DOI] [PubMed] [Google Scholar]