

Abstract
Cannabidiol (CBD), a principal phytocannabinoid constituent, has demonstrated antipsychotic properties in recent clinical trials. While it has also been suggested a promising candidate for the treatment of neurodegenerative disorders, it failed to demonstrate efficacy in cognitive impairments associated with schizophrenia as an add-on treatment (600 mg/day for 6 weeks) in 18 chronically ill patients co-treated with a variety of psychopharmacologic drugs. Here, we report on the results of parallel-group, active-controlled, mono-therapeutic, double-blind, randomized clinical trial (CBD-CT1; ClinicalTrials.gov identifier: NCT00628290) in 42 acute paranoid schizophrenic patients receiving either CBD (up to 800 mg/day) or amisulpride (AMI, up to 800 mg/day) for four weeks in an inpatient setting with neurocognition as a secondary objective. Twentynine patients (15 and 14 in the CBD and AMI group, respectively) completed two cognitive assessments at baseline and the end of the treatment period. We investigated the following cognitive domains: pattern recognition, attention, working memory, verbal and visual memory and learning, processing speed, and verbal executive functions. When applying the Bonferroni correction for multiple testing, p < 0.0004 would indicate statistical significance. There was no relevant difference in neurocognitive performance between the CBD and the AMI group at baseline, and we observed no post-treatment differences between both groups. However, we observed improvements within both groups from pre-to post-treatment (standardized differences reported as Cohen’s d) in visual memory (CBD: 0.49, p = 0.015 vs. AMI: 0.63, p = 0.018) and processing speed (CBD: 0.41, p = 0.004 vs. AMI: 0.57, p = 0.023). Furthermore, CBD improved sustained attention (CBD: 0.47, p = 0.013, vs. AMI: 0.52, p = 0.085), and visuomotor coordination (CBD: 0.32, p = 0.010 vs. AMI: 0.63, p = 0.088) while AMI led to enhanced working memory performance in two different paradigms (Subject Ordered Pointing Task–AMI: 0.53, p = 0.043 vs. CBD: 0.03, p = 0.932 and Letter Number Sequencing–AMI: 0.67, p = 0.017 vs. CBD: 0.08 p = 0.755). There was no relevant correlation between changes in neurocognitive parameters and psychotic symptoms or anandamide serum levels. This study shows that both CBD and AMI improve neurocognitive functioning with comparable efficacy in young and acutely ill schizophrenia patients via an anandamide-independent mechanism.
Keywords: cognition, neuropsychological function, cannabidiol, schizophrenia, endocannabinoids, RCT, human
Introduction
Schizophrenia is a complex psychiatric syndrome including positive symptoms (delusions, hallucinations, thought disorder) and negative symptoms (anhedonia, blunted affect, social withdrawal) as well as cognitive impairment (American Psychiatric Association, 2013). The disease concept has been originally described by Emil Kraepelin as “dementia praecox” or premature dementia. Originally, dementia was synonymous with insanity and not related to age, cognitive status, or reversibility (Adityanjee et al., 1999). In contrast, Kraepelin used it in the more modern sense of the word, including the cognitive decline he observed. Eugen Bleuler pointed to a temporary remission and even recovery of the “dementia praecox” syndrome he named “schizophrenia” (Bleuler, 1908). Nevertheless, more than a century later, the cognitive impairment in a large number of patients remains among the most difficult to influence aspects of schizophrenia.
The fundamental dimensions of cognitive deficits in schizophrenia encompass memory, attention, working memory, problem-solving, processing speed, and social cognition (Nuechterlein et al., 2004; Keefe and Fenton, 2007). Despite a considerable heterogeneity in cognitive symptoms, approximately 65–80% of patients show clinically significant impairments and perform one to two standard deviations (SD) below the population mean (Kremen et al., 2000; Keefe and Fenton, 2007; Mesholam-Gately et al., 2009; Uren et al., 2017; Islam et al., 2018; McCleery and Nuechterlein, 2019). However, it has been suggested that even those individuals who had been rated to perform as “within normal limits” exhibit a cognitive decline compared with what their cognitive functions would have been if they had never developed the illness (Kremen et al., 2000; Keefe and Fenton, 2007; Vaskinn et al., 2014). Another study reported that even patients with overall normal cognitive and intellectual functioning showed impairments in processing speed-dependent domains (Bechi et al., 2019).
Cognitive deficits are already profound early in the course of the illness (Riley et al., 2000; Mesholam-Gately et al., 2009), only modestly related to negative symptoms, essentially independent of positive symptom severity (Keefe et al., 2006), and exist already prior to the initiation of antipsychotic treatment (Saykin et al., 1994), indicating that they are not merely the result of other schizophrenia symptoms or psychotropic treatments. Furthermore, the presence of cognitive deficits or cognitive decline during adolescence has been found to predict the conversion to schizophrenia (Cannon et al., 2002; Reichenberg et al., 2005; Keefe et al., 2006; Keefe and Fenton, 2007; Seidman et al., 2016; Lam et al., 2018), supporting the view that significant cognitive deficits precede the onset of psychotic symptoms (McCleery and Nuechterlein, 2019).
The various cognitive deficits have been shown to contribute to functional outcomes, such as interpersonal relationships, social problem-solving, participation in recreational and community activities, occupational and vocational functioning, and self-care (Green et al., 2000; Bryson and Bell, 2003; Mohamed et al., 2008; Fervaha et al., 2014; Chang et al., 2016). Consequently, cognition is an important treatment target. Currently available antipsychotic medications can yield modest beneficial effects on cognitive functioning, although the findings have been inconsistent regarding whether atypical antipsychotics confer greater effects than typical antipsychotics (Woodward et al., 2005; Keefe et al., 2007; McCleery and Nuechterlein, 2019). Notably, detrimental effects of antipsychotics on cognition are also possible. This has been associated with very high dopamine D2 receptor occupancy level, very high dosing, polypharmacy, and concomitant use of anticholinergic medications (Hori et al., 2006; Sakurai et al., 2013; McCleery and Nuechterlein, 2019).
Due to the lack of marked cognitive benefits of standard antipsychotics, alternative pharmacological treatments with different mechanisms of action are currently investigated, including cholinergic agents, dopamine D1 agonists, and glutamatergic agents (Buchanan et al., 2007).
Another promising novel antipsychotic agent is cannabidiol (CBD), a major ingredient of Cannabis sativa. CBD has demonstrated antipsychotic properties in small case studies (Zuardi et al., 1995; Zuardi et al., 2006; Zuardi et al., 2009; Makiol and Kluge, 2019) and randomized, placebo-controlled clinical trials in acutely and non-acutely ill schizophrenia patients (Leweke et al., 2012; McGuire et al., 2018). Moreover, first placebo-controlled, double-blind studies showed that CBD might also have beneficial effects in individuals in a Clinical-High-Risk (CHR) mental state for psychosis (Bhattacharyya et al., 2018; Appiah-Kusi et al., 2020).
However, while CBD has also been suggested to be a promising therapeutic candidate for treating neurodegenerative diseases through multifaceted molecular mechanisms (Cassano et al., 2020), it failed to demonstrate efficacy in ameliorating cognitive impairments in schizophrenia patients. In a small double-blind, placebo-controlled, three-parallel-arm clinical trial with non-acutely ill schizophrenia patients, a single dose of 300 (N = 9) or 600 mg (N = 9) CBD did not improve cognitive performance–more precisely selective attention–compared to placebo (N = 10) (Hallak et al., 2010). Furthermore, in comparison to placebo (N = 18), a six-week treatment with CBD (600 mg/day; N = 18) did not improve the cognitive performance (assessed with the MATRICS Consensus Cognitive Battery) of stable antipsychotic-treated patients diagnosed with chronic schizophrenia (Boggs et al., 2018). At the same time, a double-blind, randomized, placebo-controlled, parallel-group clinical trial investigating the efficacy of a higher CBD dosage (1000 mg/day, over eight weeks) as an add-on to stable antipsychotic medication in sub-acute schizophrenia spectrum patients (n = 43), observed a slightly improved cognitive performance (Brief Assessment of Cognition in Schizophrenia (BACS) composite score and subdomain “executive functions”) compared to those who received placebo (n = 45) as a secondary outcome. Although these differences did not reach statistical significance, the motor speed improvements were significantly greater in the CBD than in the placebo group (McGuire et al., 2018).
In comparison to these previously published studies, the present study compared the effects of a mono-therapeutic approach with CBD or the second-generation antipsychotic AMI in earlier stages of schizophrenia on six neurocognitive domains (pattern recognition; sustained attention; working memory; verbal and visual memory and learning; processing speed; verbal executive functions) in 42 acute paranoid schizophrenic patients as a secondary objective. AMI has been chosen as a comparator because of its clear dopamine D2/3-receptor antagonistic mechanism of action. In contrast, CBD’s antipsychotic potential has been found to be substantially linked to an increase in anandamide levels (Leweke et al., 2012).
Methods
Subjects
This therapeutic-exploratory (phase II), double-blinded, monocenter, randomized, parallel-group, controlled clinical trial (RCT) of CBD vs. AMI (CBD-CT1; ClinicalTrials.gov. Identifier: NCT00628290) was approved by the Ethics Committee of the University of Cologne and the BfArM (Federal Institute for Drugs and Medical Devices). Initially, an independent psychiatrist assessed patients to confirm their ability to provide informed consent. After a detailed explanation of study procedures, written informed consent was obtained from each patient.
Details on the patient samples have been previously provided elsewhere (Leweke et al., 2012) and are summarized in Table 1. In brief, men and women aged 18–50 years and diagnosed with schizophrenia or schizophreniform psychosis, with a total Brief Psychiatric Rating Scale (BPRS) score ≥36 and a BPRS thought disorder score ≥12, were eligible to participate in the study. Exclusion criteria comprised a positive urine drug screening for illicit drugs, other relevant psychiatric disorders, treatment with a depot antipsychotic within three months before participation in the study, history of treatment resistance, a relevant and/or unstable medical condition, and insufficient contraception, pregnancy, or breast-feeding in female patients. The consort diagram (Figure 1) demonstrates the participants’ flow. Out of 42 inpatients with acute paranoid schizophrenia, 33 completed the study per protocol (participation in the study throughout the planned course). After a screening period of up to 7 days and a minimum period of three antipsychotic-free days (the vast majority of patients was antipsychotic-naïve or hospitalized for acute exacerbation after discontinuing antipsychotic treatment and therefore off antipsychotic well before inclusion in our study), patients were randomized (1:1) to receive either CBD or AMI starting with 200 mg per day each and increased stepwise by 200 mg per day to a daily dose of 200 mg four times daily (total 800 mg per day) each within the first week that was maintained for another three weeks. A reduction of each treatment to 600 mg per day was allowed for clinical reasons, such as unwanted side effects after week 2, which was the case for three patients in the CBD and five patients in the AMI arm. The assessment of the change of neurocognitive performance was a secondary objective of this RCT. Fifteen patients of the CBD and 14 patients of the AMI group were able to complete neuropsychological assessments prior to initiation of treatment (day -8 to -1) and on the last day of active treatment with either CBD or AMI (day 28). However, only 12 and 11 patients treated with CBD and AMI respectively completed all neurocognitive tests.
TABLE 1.
Characteristics of the patient sample.
| Full analyis set | Subset with full neurocognitive assessment | |||||
|---|---|---|---|---|---|---|
| CBD (n = 20) | AMI (n = 19) | CBD vs. AMI P-Value a | CBD (n = 15) | AMI (n = 14) | CBD vs. AMI P-Value a | |
| Demographic characteristics | ||||||
| Age, years (mean ± SD) | 29.7 ± 8.3 | 30.6 ± 9.4 | 0.966 | 28.8 ± 7.7 | 30.3 ± 9.7 | 0.844 |
| Male gender, count (%) | 15 (75.0) | 17 (89.5) | 0.407 | 12 (80.0) | 13 (92.9) | 0.598 |
| Baseline severity of illness scores, mean ± SD | ||||||
| PANSS Total | 91.2 ± 14.0 | 99.5 ± 17.1 | 0.736 | 89.9 ± 15.9 | 98.9 ± 16.7 | 0.347 |
| PANSS Positive | 24.6 ± 5.6 | 22.5 ± 6.2 | 0.205 | 23.5 ± 5.3 | 23.4 ± 6.3 | 0.678 |
| PANSS Negative | 23.7 ± 5.4 | 25.3 ± 5.6 | 0.573 | 23.8 ± 5.8 | 25.7 ± 6.1 | 0.511 |
| PANSS General | 42.9 ± 8.6 | 47.7 ± 11.4 | 0.155 | 42.5 ± 9.4 | 49.9 ± 11.1 | 0.063 |
| BPRS | 58.1 ± 9.7 | 57.7 ± 10.3 | 0.764 | 56.1 ± 9.8 | 59.4 ± 8.9 | 0.541 |
| CGI | 6.3 ± 0.7 | 6.8 ± 0.4 | 0.011 | 6.3 ± 0.6 | 6.7 ± 0.5 | 0.037 |
| Other, mean ± SD | ||||||
| Lorazepam, mg/day | 2.2 ± 1.6 | 4.2 ± 2.4 | 0.006 | 2.0 ± 1.7 | 3.8 ± 2.3 | 0.055 |
| SAS | 36.4 ± 7.7 | 36.9 ± 8.1 | 0.800 | 35.7 ± 6.8 | 37.6 ± 8.5 | 0.584 |
| EPS | 0.1 ± 0.2 | 0.0 ± 0.1 | 0.485 | 0.1 ± 0.2 | 0.0 ± 0.0 | 0.682 |
| Changes in PANSS after 28 days of treatment, mean ± SD; n [all changes significant compared to baseline, p < 0.001 in the full analysis set Leweke et al. (2012)] | ||||||
| PANSS Total | −30.5 ± 16.4; 17 | −30.1 ± 24.7; 18 | 0.843 | −31.3 ± 16.8 | −37.0 ± 21.4 | 0.332 |
| PANSS Positive | −9.0 ± 6.1; 17 | −8.4 ± 7.5; 18 | 0.519 | −8.9 ± 6.3 | −10.1 ± 7.3 | 0.903 |
| PANSS Negative | −9.1 ± 4.9; 17 | −6.4 ± 6.0; 18 | 0.234 | −9.6 ± 5.1 | −7.9 ± 5.7 | 0.527 |
| PANSS General | −12.5 ± 10.4, 17 | −15.3 ± 14.3; 18 | 0.457 | −12.8 ± 10.1 | −19.0 ± 13.0 | 0.159 |
CBD, cannabidiol; AMI, amisulpride; BPRS, brief psychiatric rating scale; CGI, clinical global impression scale; EPS, extrapyramidal symptoms rating scale; PANSS, positive and negative syndrome scale; SAS, social anxiety scale. Full analysis set (Leweke et al., 2012) and subset of patients who completed neuropsychological assessments prior to initiation of treatment. Please note, the p-values for changes in PANSS slightly differ from the ones given in the main article (Leweke et al., 2012), since therein results based on a mixed model for repeated measures with baseline adjustment are reported. However, both approaches support the same conclusions.
The Kruskal-Wallis test (continuous data) or Fisher’s exact (nominal data) test. Statistical significance between groups (p ≤ 0.05) is indicated in bold.
FIGURE 1.

CONSORT flow diagram (Leweke et al., 2012). Analyses sets: VfS, valid for safety; mITT, modified intention-to-treat and PP, per protocol.
Neurocognitive Assessment
The neurocognitive test battery took approximately 2 h to complete and was conducted by fully qualified neuropsychologists. Furthermore, training effects were diminished by using parallel versions of each test at t0 and t1. Premorbid verbal IQ was tested with the German version of the Multiple Choice Vocabulary Test (Lehrl, 1995), which is highly correlated with total IQ.
The test battery comprised 10 validated neuropsychological tests with good test-retest reliability (Delaney et al., 1992; Keefe et al., 2004; Ross et al., 2007; Nuechterlein et al., 2008) to assess the following neurocognitive domains:
(1) Pattern recognition. A computerized version of a visual backward masking task with letters F, H, or T as target stimuli and one of four masking conditions [random dot pattern or letter pattern masking stimulus after short (42 ms) or long (104 ms) interstimulus intervals] provided a measure of visual information-processing (number of hits). The session consisted of three blocks of 30 trials, each including six trials of each masking condition and six no-mask control trials presented in random order. To enhance reliability for the measurement of pattern recognition, aggregated scores across the different masking conditions were calculated.
(2) Attention. The Continuous Performance Test (identical pairs version (Cornblatt, 1996)) provided a measure of sustained attention. The signal detection parameter d’ was calculated across 150 trials with 4-digits stimuli (emphasis on left hemispheric processing) and 150 trials with meaningless symbols stimuli (emphasis on right-sided processing).
(3) Working memory. The Letter Number Sequencing (Gold et al., 1997) requires subjects to sort letters from numbers within a sequence of alternating letters and numbers read to them and recall the letters and numbers in ascending orders separately. During each trial of a computerized version of the Subject Ordered Pointing Task (Petrides and Milner, 1982), subjects had to point to 1 of 12 objects, and the relative positions of the objects varied randomly across trials. Across three sessions of 12 trials, the number of errors (pointing to an object already chosen on a previous trial) was calculated. During each trial of the Delayed Response Task (Park and Holzman, 1992; Pukrop et al., 2003) for spatial working memory, a black dot was presented for 200 ms at 1 of 16 possible positions of a circle followed by a specific delay (5 s or 15 s). During the delay period, subjects had to solve arithmetic distractor tasks. After the delay period, subjects were required to indicate the position of the previously presented dot on a touch-sensitive monitor to determine the Euclidean distance to the target. To enhance reliability for the measurement of the spatial working memory, aggregated scores across the two different delay periods were calculated.
(4) Verbal and visual memory and learning. The Auditory Verbal Learning Test (Lezak et al., 2012) provided measures for the immediate recall capabilities after one to five learning trials of word lists and delayed recall performance. A measure of visual memory was provided by the Rey-Osterrieth Complex Figure Test (Rey, 1941), calculating the delayed recall performance by a standardized scoring procedure.
(5) Processing speed. The Digit-Symbol-Test (Kaplan et al., 1991) and Trail-Making Test A and B (Reitan and Wolfson, 1985) provided measures for the speed of visual information-processing and visuomotor coordination. The ratio of TMT-B to TMT-A can be interpreted as an indicator of the cognitive component independent of the motor component.
(6) Verbal executive functions. A verbal fluency task (sum of four lexical and semantic category tasks) was used to measure verbal executive functions (Turek et al., 2017; Palsetia et al., 2018).
These domains of cognitive functioning are those found to be consistently impaired and related to outcome in schizophrenia and are assessed by the BACS as well (Keefe et al., 2004) that was not available at the time the trial started.
Determination of Anandamide Levels
For a subgroup of patients, serum anandamide levels were determined before (baseline) and after CBD (N = 14) or AMI (N = 8) treatment (day 28) by liquid chromatography/electrospray tandem mass spectrometry (LC/ESI-MS-MS) using a method following international guidelines and requirements for the validation of a method and the quantitative evaluation of the compounds as described previously in detail (Giuffrida et al., 2000; Schreiber et al., 2007; Leweke et al., 2012).
Statistical Analysis
Since observed distributions of all neuropsychological characteristics could be well approximated by normal distributions, parametric methods (from the t-test family) were used to evaluate differences in location. Due to the explorative character of the study, the type I error (alpha) was not adjusted for multiplicity. Thus, the results need to be interpreted carefully. When applying the Bonferroni correction, p < 0.0004 (i.e., 0.05/120) would indicate statistical significance. Throughout the manuscript, we added the Bonferroni-corrected p-values (pcorr) in square brackets. Alternatively, also based on given p values, readers may prefer to apply the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) to control the false discovery rate: The p values need to be ranked; find the maximum rank k with p(k) ≤i/120*0.05; reject all null hypotheses with p values of rank i≤k.
Standardized differences for neuropsychological performance changes from t0 to t1 in both groups are reported as Cohen’s d (within groups over time, i.e., standardized mean gain (t0 minus t1), and between groups, i.e., standardized mean difference (AMI minus CBD), respectively) (Lipsey and Wilson, 2001). Thus, in both cases, Cohen’s d was calculated based on the pooled standard deviation (either pooled over time or pooled over groups). Associations between neuropsychological performance and psychopathological symptoms were described by Pearson’s correlation coefficient. Moreover, for a subgroup of patients, serum anandamide levels were available (Leweke et al., 2012). Thus, associations between changes of the neurocognitive performance and serum anandamide levels were assessed by a median slope analysis (Wilcoxon signed-rank test of the null hypothesis that the distribution of slopes is symmetric about zero). This type of analysis is consistent with our previous analysis to assess the association of the change of anandamide levels in serum and the change in the PANSS total score (Leweke et al., 2012). Calculations were done with the software SPSS Statistics (IBM Corp., Armonk, NY, United States) and Stata/SE (StataCorp LLC, College Station, TX, United States).
Results
Both treatment groups improved on all neuropsychological functions from pre-to post-treatment (except for slight non-significant deteriorations of verbal working memory performance assessed by AVLT–delayed recall (p = 0.881 [pcorr = 1], d = -0.03) and the Letter Number Sequencing (p = 0.366 [pcorr = 1], d = 0.08) in the CBD group, and of working memory performance assessed by the delayed response task in the AMI group (p = 0.066 [pcorr = 1], d = -0.81); Table 2). In the AMI group, t-tests (Table 2) revealed improvements from t0 to t1 on the Letter Number Sequencing Test and Subject Ordered Point Task (both working memory tests), as well as the Rey-Osterrieth Complex Figure Test (ROFT, visual memory) and the Digit-Symbol Test (processing speed). Patients treated with CBD showed improvements on the Continuous Performance Test–symbol stimuli (sustained attention), the ROFT (visual memory), Digit-Symbol Test (processing speed), and Trail-Making Test B (visuomotor coordination). Effect sizes (Table 3'; Figure 2) for all improvements ranged from 0.21 (visuomotor coordination) to 0.67 (verbal working memory) in the AMI group and from 0.03 (working memory) to 0.49 (visual memory) in the CBD group. We did not find significant differences between treatment groups.
TABLE 2.
Changes in neurocognitive performance (raw score means ± SD) before (t0) and after (t1) intervention. Statistical significant changes (p ≤ 0.05) are indicated in bold.
| AMI | CBD | |||||
|---|---|---|---|---|---|---|
| t0 (mean ± SD; N) | t1 (mean ± SD; N) | paired t-test (t0-t1), [95% CI] | t0 (mean ± SD; N) | t1 (mean ± SD; N) | paired t-test (t0-t1) [95% CI] | |
| VBM %Hits | 71.79 ± 17.20; 14 | 80.34 ± 12.27; 14 | t(13) = −1.75, p = 0.105 [p corr = 1] | 70.57 ± 16.79; 15 | 77.15 ± 18.35; 15 | t(14) = −1.45, p = 0.170 [p corr = 1] |
| [−19.14, 2.04] | [−16.35, 3.19] | |||||
| CPT d’ figures | 1.04 ± 0.55; 13 | 1.32 ± 0.68; 13 | t(12) = −1.68, p = 0.119 [p corr = 1] | 1.49 ± 1.08; 15 | 1.73 ± 0.79; 15 | t(14) = −1.31, p = 0.213 [p corr = 1] |
| [−0.65, 0.09] | [−0.63, 0.152] | |||||
| CPT d’ symbols | 1.31 ± 0.84; 13 | 1.68 ± 0.75; 13 | t(12) = −1.88, p = 0.085 [p corr = 1] | 1.91 ± 0.82; 15 | 2.30 ± 0.85; 15 | t(14) = −2.85, p = 0.013 [p corr = 1] |
| [−0.80, 0.06] | [−0.69, −0.10] | |||||
| LNS-# correct | 14.36 ± 3.30; 11 | 16.36 ± 2.73; 11 | t(10) = −2.85, p = 0.017 [p corr = 1] | 16.23 ± 3.11; 13 | 15.92 ± 4.43; 13 | t(12) = 0.32, p = 0.755 [p corr = 1] |
| [−3.56, −0.44] | [−1.80, 2.41] | |||||
| SOPT # Errors | 5.75 ± 2.83; 12 | 4.33 ± 2.15; 12 | t(11) = 2.28, p = 0.043 [p corr = 1] | 4.58 ± 3.09; 12 | 4.50 ± 2.39; 12 | t(11) = 0.09, p = 0.932 [p corr = 1] |
| [0.05, 2.78] | [−2.02, 2.19] | |||||
| DRT Euclidian distance | 9.41 ± 18.76; 14 | 21.41 ± 11.08; 14 | t(13) = −2.00, p = 0.066 [p corr = 1] | 15.05 ± 23.47; 15 | 11.81 ± 16.06; 15 | t(14) = 0.60, p = 0.563 [p corr = 1] |
| [−24.93, 0.94] | [−8.49, 14.98] | |||||
| AVLT immediate recall # correct | 8.64 ± 3.63; 14 | 9.86 ± 4.11, 14 | t(13) = −1.30, p = 0.216 [p corr = 1] | 9.40 ± 3.54; 15 | 9.53 ± 3.09; 15 | t(14) = −0.15, p = 0.881 [p corr = 1] |
| [−3.23, 0.80] | [−2.02, 1.75] | |||||
| AVLT delayed recall # Correct | 8.07 ± 4.39; 14 | 9.29 ± 4.14; 14 | t(13) = −1.09, p = 0.296 [p corr = 1] | 8.80 ± 3.78; 15 | 8.67 ± 3.958; 15 | t(14) = 0.15, p = 0.881 [p corr = 1] |
| [−3.62, 1.20] | [−1.75, 2.02] | |||||
| ROFT delayed recall standardized score | 18.54 ± 7.19; 13 | 24.19 ± 8.83; 13 | t(12) = −2.74 p = 0.018 [p corr = 1] [−10.16, −1.15] | 21.14 ± 7.17; 14 | 24.68 ± 7.17; 14 | t(13) = −2.80 p = 0.015 [p corr = 1] |
| [−6.27, −0.80] | ||||||
| Digit-symbol coding # Correct | 42.46 ± 9.37; 11 | 46.36 ± 7.41; 11 | t(10) = −2.68, p = 0.023 [p corr = 1] | 52.15 ± 13.25; 13 | 57.46 ± 12.82; 13 | t(12) = −3.60, p = 0.004 [p corr = 0.480] |
| [−7.17, −0.65] | [−8.52, −2.10] | |||||
| TMT-B time in sec | 120.73 ± 64.31; 11 | 83.64 ± 27.78; 11 | t(10) = 1.89, p = 0.088 [p corr = 1] [−6.58, 80.76] | 88.79 ± 49.70; 13 | 69.31 ± 32.24; 13 | t(12) = 3.05, p = 0.010 [p corr = 1] |
| [5.55, 33.42] | ||||||
| Ratio TMT-B/TMT-A time in sec | 3.17 ± 1.19; 11 | 2.82 ± 1.38; 11 | t(10) = 0.65, p = 0.531 [p corr = 1] | 2.74 ± 0.94; 13 | 2.65 ± 0.61; 13 | t(12) = 0.36, p = 0.728 [p corr = 1] [0.65, 0.36] |
| [−0.84, 1.53] | ||||||
| Verbal fluency | 11.28 ± 4.16; 12 | 12.21 ± 2.63; 11 | t(10) = −0.92, p = 0.376 [p corr = 1] [−3.16, 1.29] | 11.90 ± 2.48; 12 | 12.64 ± 3.46; 12 | t(11) = −0.94, p = 0.366 [p corr = 1] |
| # Correct | [2.5, 0.98] | |||||
CBD, cannabidiol; AMI, amisulpride; AVLT, auditory verbal learning test; d′, signal detection parameter; DRT, delayed response task, LNS, letter number sequencing; ROFT, Rey-Osterrieth complex figure test; SOPT, subject ordered pointing task; TMT-A, trail-making test A; TMT-B, trail-making test B; VBM, visual backward masking. Descriptive statistics, and paired t-test results (intention-to-treat set). Improvements are indicated by negative t-values except for SOPT (#error), DRT (Euclidian distance), TMT-B, and ratio TMT-B/TMT-A (both time in s).
TABLE 3.
Effect size (Cohen’s d) for changes in neurocognitive test scores (t0-t1) and independent t-test results, to assess the equality of the effect sizes. Improvements are indicated by negative effect sizes except for SOPT (#error), DRT (Euclidian distance), TMT-B, and ratio TMT-B/TMT-A (both times in s).
| Cohen’s d | |||
|---|---|---|---|
| AMI d (t0 vs. t1) [95% CI] | CBD d (t0 vs. t1) [95% CI] | AMI vs. CBD (t0-t1) t(df), p, d [95% CI] | |
| VBM %hits | −0.59 [−1.23, 0.05] | −0.37 [−0.90, 0.15] | t(27) = −0.30, p = 0.770 [pcorr = 1], d = −0.11 [−0.84, 0.62] |
| CPT d’ figures | −0.55 [−1.11, 0.02] | −0.23 [−0.60, 0.13] | t(26) = −0.19, p = 0.850 [pcorr = 1], d = −0.07 [−0.81, 0.67] |
| CPT d’ symbols | −0.52 [−1.02, −0.02] | −0.47 [−0.83, −0.11] | t(26) = −0.10, p = 0.923 [pcorr = 1], d = 0.04 [−0.71, 0.78] |
| LNS # correct | −0.67 [−1.72, −0.17] | −0.08 [−0.40, 0.55] | t(22) = −1.87, p = 0.074 [pcorr = 1], d = −0.77 [−1.60, 0.07] |
| SOPT # errors | 0.53 [0.06, 1.01] | 0.03 [−0.64, 0.71] | t(22) = 1.17, p = 0.255 [pcorr = 1], d = 0.48 [−0.34, 1.28] |
| DRT mean Euclidian distance | −0.81 [−1.58, −0.03] | 0.16 [−0.36, 0.68] | t(27) = −1.88, p = 0.071 [pcorr = 1], d = −0.70 [−1.45, 0.06] |
| AVLT immediate recall # correct | −0.33 [−0.79, 0.11] | −0.04 [−0.56, 0.48] | t(27) = −0.85, p = 0.406 [pcorr = 1], d = −0.31 [−1.04, 0.42] |
| AVLT delayed recall # correct | −0.29 [−0.77, 0.19] | 0.03 [−0.41, 0.48] | t(27) = −0.96 p = 0.347 [pcorr = 1], d = −0.36 [−1.09, 0.38] |
| ROFT delayed recall standardized score | −0.63 [−1.14, −0.12] | −0.49 [−0.88, −0.10] | t(25) = −0.89 p = 0.383 [pcorr = 1], d = −0.34 [−0.54, 1.08] |
| Digit-symbol coding # correct | −0.57 [−1.01, −0.13] | −0.41 [−0.68, −0.14] | t(22) = 0.67 p = 0.511[pcorr = 1], d = 0.27 [−0.71, 0.78] |
| TMT-B time in s | 0.63 [−0.08, 1.34] | 0.32 [0.08, 0.56] | t(22) = 0.91 p = 0.371 [pcorr = 1], d = 0.37 [−0.44, 1.18] |
| Ratio TMT-B/TMT-A time in s | 0.21 [−0.56, 0.98] | 0.11 [−0.51, 0.73] | t(22) = 0.45 p = 0.655 [pcorr = 1], d = 0.19 [−0.62, 0.99] |
| Verbal fluency # correct | −0.34 [−0.89, 0.21] | −0.24 [−0.73, 0.26] | t(22) = −0.15 p = 0.880 [pcorr = 1], d = −0.06 [−0.86, 0.74] |
CBD, cannabidiol; AMI, amisulpride; AVLT, auditory verbal learning test; d′, signal detection parameter; CPT, continous performance task; DRT, delayed response task, LNS, letter number sequencing; ROFT, Rey-Osterrieth complex figure test; SOPT, subject ordered pointing task; TMT-A, trail-making test A; TMT-B, trail-making test B; VBM, visual backward masking.
FIGURE 2.
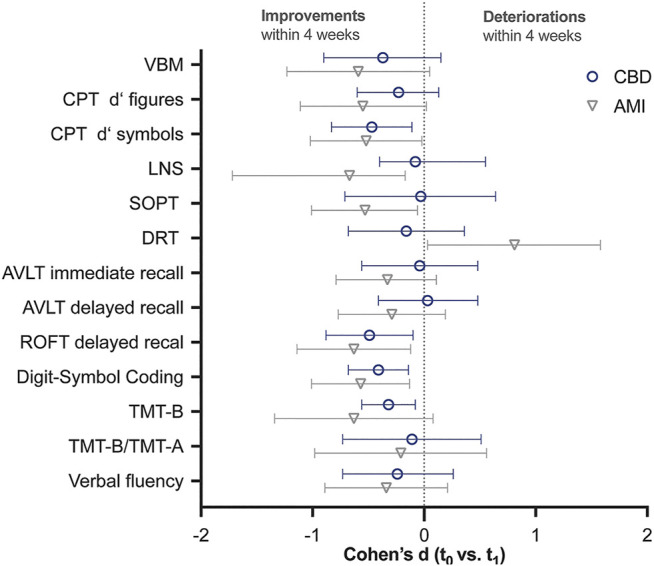
Effect size [Cohen’s d (95% CI)] for changes in neurocognitive test scores (t0-t1 mean). Cohen’s d for SOPT (#error), DRT (Euclidian distance), TMT-B, and ratio TMT-B/TMT-A (both times in s) have been inverted to allow for a better comparison. Thus, improvements of neurocognitive functioning within four weeks of treatment are indicated by negative effect sizes. 95% CI that do not contain 0, indicate significant improvements at the 5% level. CBD, cannabidiol; AMI, amisulpride; AVLT, auditory verbal learning test; d′, signal detection parameter; DRT, delayed response task, LNS, letter number sequencing; ROFT, Rey-Osterrieth complex figure test; SOPT, subject ordered pointing task; TMT-A, trail-making test A; TMT-B, trail-making test B; VBM, visual backward masking.
Changes in neurocognitive performance were not systematically correlated with psychopathological improvements (PANSS total, general, positive, and negative symptom scores). Only 4 out of 104 (13 neuropsychological and 4 psychopathological parameter per treatment group) correlation coefficients between difference scores became “significant” (note, that about 5 (i.e., 5%) coefficients can be expected by chance alone): In the AMI group, the Letter Number Sequencing performance correlated with PANSS total (r = -0.69; p = 0.026 [pcorr = 1], N = 10), and PANSS general scores (r = -0.81; p = 0.005 [pcorr = 0.600], N = 10), while in the CBD group the verbal fluency performance was significantly associated with PANSS total (r = -0.65; p = 0.024 [pcorr = 1], N = 12) and PANSS general (r = -0.61; p = 0.037 [pcorr = 1], N = 12). The association with PANSS positive did not reach significance (r = -0.52; p = 0.085 [pcorr = 1], N = 12).
In addition, changes in neurocognitive performance were also not systematically associated with changes in serum anandamide levels (Table 4). Solely improvements on the Continuous Performance Test–figure stimuli (sustained attention, median slope = 0.99, 95% CI [0.03, 4.21]; p = 0.046 [pcorr = 1]) and the Digit-Symbol Test (processing speed, median slope = 3.31, 95% CI [1.72, 8.95], p = 0.012 [pcorr = 1]) in the AMI and CBD group respectively, were associated with higher anandamide levels.
TABLE 4.
Association of change in serum anandamide levels and change in neurocognitive performance. Wilcoxon signed-rank test of the null hypothesis that the distribution of slopes is symmetric about zero. Statistical significant changes (p ≤ 0.05) are indicated in bold.
| Amisulpride | CBD | |||
|---|---|---|---|---|
| Slope (mean ± SD; N) | Z, P [P corr], median slope [95% CI] | Slope (mean ± SD; N) | Z, P [P corr], median slope [95% CI] | |
| VBM %hits | 40.10 ± 123.77; 6 | 0.11, 0.917 [1], 2.94 [−30.87, 148.23] | 15.36 ± 34.71; 11 | 1.16, 0.248 [1], 8.82 [−7.10, 42.98] |
| CPT d’ figures | 1.72 ± 2.41; 6 | 1.99, 0.046 [1], 0.99 [0.03, 4.21] | 0.26 ± 1.66; 11 | 0.80, 0.424 [1], 0.19 [−0.81, 1.43] |
| CPT d’ | 1.71 ± 3.81; 6 | 1.15, 0.249 [1], 0.36 [−0.19, 4.98] | 0.30 ± 2.02; 9 | 1.51, 0.131 [1], 0.24 [−0.21, 1.95] |
| LNS # correct | 6.15 ± 12.33; 6 | 1.48, 0.138 [1], 1.85 [−0.58, 17.38] | −0.12 ± 2.51; 9 | 0.00, 1,000 [1], 0.14 [−2.20, 2.20] |
| SOPT # errors | −0.23 ± 3.29; 7 | −0.37, 0.715 [1], 0.00 [−3.61, 3.45] | 1.47 ± 2.70; 11 | 1.72, 0.086 [1], 1.57 [−0.63, 3.33] |
| DRT mean euclidian distance | −21.11 ± 86.48; 6 | −0.31, 0.753 [1], −5.58 [−113.38, 53.76] | −15.10 ± 28.01; 11 | −1.25, 0.213 [1], −9.89 [−37.10, 2.78] |
| AVLT immediate recall # correct | 4.6 ± 11.84; 7 | 0.73, 0.465 [1], 0.00 [−4.23, 15.78] | 0.53 ± 4.93; 12 | −0.05, 0.959 [1], 0.00 [−1.56, 4.04] |
| AVLT delayed recall # correct | 5.85 ± 14.78; 7 | 0.41, 0.686 [1], 0.00 [−5.24, 22.10] | −1.33 ± 4.85; 11 | −1.07, 0.285 [1], −1.30 [−4.76, 1.65] |
| ROFT delayed recall standardized score | 22.15 ± 47.90; 6 | 0.11, 0.917 [1], 14.41 [−11.07, 78.03] | −1.22 ± 15.39; 11 | 0.45, 0.657 [1], 1.28 [−9.81, 6.52] |
| Digit-symbol coding # correct | 8.02 ± 17.04; 6 | 1.214, 0.225 [1], 3.61 [−3.45, 24.41] | 4.30 ± 4.31; 9 | 2.52, 0.012 [1], 3.31 [1.72, 8.95] |
| TMT-B time in s | −6.73 ± 175.76; 5 | −0.14, 0.893 [1], −4.19 [−286.24, 156.87] | −13.78 ± 30.78; 9 | −1.60, 0.110 [1], −14.90 [−37.32, 14.04] |
| Ratio TMT-B/TMT−A time in s | 0.43 ± 3.47; 5 | 0.41, 0.686 [1], 0.88 [−5.10, 3.99] | 0.44 ± 2.94; 9 | −0.53, 0.594 [1], −0.19 [−1.52, 3.67] |
| Verbal fluency # correct | −3.54 ± 12.32; 7 | −0.11, 0.917 [1], 0.14 [−18.94, 6.71] | 5.88 ± 14.20; 9 | 1.24, 0.214 [1], 1.26 [−0.30, 21.59 ] |
AVLT, auditory verbal learning test; d′, signal detection parameter; DRT, delayed response task, LNS, letter number sequencing; P, p-value; ROFT, rey-osterrieth complex figure test; SOPT, subject ordered pointing task; TMT-A, trail-making test A; TMT-B, trail-making test B; VBM, visual backward masking; Z, Wilcoxon signed-rank standardized test statistic.
Discussion
This study shows that both CBD and AMI improve neurocognitive functioning with comparable efficacy in young, acutely ill schizophrenia patients. Interestingly, AMI improved working and visual memory performance as well as the processing speed, while CBD treatment led to improvements in processing speed, visual memory, visuomotor coordination, and sustained attention. However, all effect sizes were similar for both treatment groups.
In consideration of previous studies showing limited (McGuire et al., 2018) to no (Boggs et al., 2018) effect of CBD on neurocognitive functioning as an add-on medication in sub-acutely and chronically ill schizophrenia patients, our results indicate that CBD may be more beneficial when administered during the earlier acute phases of the illness. Neuroprotective properties of CBD have been described earlier (Hampson et al., 1998), and more recent data suggests anti-inflammatory effects of CBD as well (Mori et al., 2017), potentially contributing to an effectiveness at earlier stages of the disease. In addition, the influence of an add-on treatment is not clear, and potential pharmacodynamic interactions need to be considered. McGuire et al. (McGuire et al., 2018) accepted a stable treatment with antipsychotics only and observed a trend in favor of CBD (at a dosage of 1 g per day) in improving cognitive symptoms as assessed by the Brief Assessment of Cognition in Schizophrenia (BACS), an instrument basically covering the cognitive domains assessed in our study. In the study of Boggs et al. (Boggs et al., 2018), in which no beneficial CBD effects were observed, the patients were allowed to take a much broader spectrum of concomitant medication, including antipsychotics, antidepressants, and mood stabilizers and combined administration of these drugs. In addition, the daily dosage of CBD was limited to 600 mg.
Interestingly, data on the effects of AMI on cognitive function in schizophrenia patients is limited to a few open-label studies and randomized controlled trials (Tyson et al., 2004; Wagner et al., 2005; Mortimer et al., 2007; Wang et al., 2008; Kumar and Chaudhury, 2014). Consistent with our findings, AMI ameliorated cognitive impairments in all studies. Furthermore, the reported effect size of 0.4 for the global cognitive index after an 8-weeks treatment (Wagner et al., 2005) is comparable to the median effect size of 0.53 observed in the current study after four weeks of AMI treatment.
It has been suggested that the combined serotonin (5-HT2A) and dopamine-2 (D2) receptor blockade of second-generation antipsychotics is relevant for their ameliorating effects on neurocognitive impairments (Wagner et al., 2005). However, AMI is a second-generation antipsychotic with almost no affinity to 5-HT2A receptors but a high affinity to block dopamine-3 (D3) receptors (Schoemaker et al., 1997). Nevertheless, AMI reduces cognitive impairments with at least similar efficacy as high 5-HT2A receptor affine second-generation antipsychotics such as olanzapine or risperidone (Tyson et al., 2004; Wagner et al., 2005; Tyson et al., 2006; Mortimer et al., 2007; Wang et al., 2008; Kumar and Chaudhury, 2014). Furthermore, significant improvements in attention (Wagner et al., 2005; Tyson et al., 2006), executive function (Wagner et al., 2005), and auditory verbal learning (Mortimer et al., 2007) were only observed in patients treated with AMI. These findings suggest that low or no 5-HT2A affinity may be more beneficial for cognition than high affinity (Tyson et al., 2004; Wagner et al., 2005). Although AMI and other second-generation antipsychotics have a considerable affinity to D3 receptors, little is known about the role of D3 receptor antagonism in ameliorating positive, negative, and cognitive symptoms (Gross and Drescher, 2012; Sokoloff and Le Foll, 2017). In preclinical studies, D3 receptor antagonists improved cognitive function, emotional processing, executive function, flexibility, and social behavior, but the few clinical studies with compounds of high affinity for D3 receptors and different degrees of selectivity over D2 receptors give only limited insight into the therapeutic potential of selective D3 antagonism (Gross and Drescher, 2012; Sokoloff and Le Foll, 2017).
Although current antipsychotics can yield modest beneficial effects on neurocognitive functioning (McCleery and Nuechterlein, 2019), cognitive impairments continue to pose a burden on patients. Therefore, novel compounds with a different mechanism of action are currently investigated, such as cholinergic agents, dopamine D1 agonists, glutamatergic agents (Buchanan et al., 2007), and CBD (Boggs et al., 2018). CBD seems to mediate its antipsychotic effects by modulating the endocannabinoid system (Leweke et al., 2012; Rohleder et al., 2016; Leweke et al., 2018). More precisely, its antipsychotic actions have been found to be related to increased levels of the endocannabinoid anandamide (Leweke et al., 2012), e.g., by blocking the anandamide degrading enzyme fatty acid amide hydrolase (FAAH) (Leweke et al., 2012) or the fatty amide binding proteins (FABPs) that transport anandamide to the FAAH enzyme (Elmes et al., 2015). Elevated anandamide levels can, in turn, interact with other neurotransmitter (e.g., dopamine) systems via cannabinoid type 1 receptors (CB1R) (Giuffrida et al., 1999; Giuffrida et al., 2004; Leweke, 2012), enhance glucose metabolism via peroxisome proliferator-activated receptor-γ (PPARγ) (Bouaboula et al., 2005) or modulate the immune function via cannabinoid type 2 receptors (CB2R). Other suggested pharmacological effects of CBD include: the activation of the vanilloid receptor 1 (TRPV1, transient receptor potential cation channel subfamily V member 1) (Bisogno et al., 2001; De Petrocellis et al., 2011), negative allosteric modulation of CB1R (Laprairie et al., 2015; Sabatucci et al., 2018), the facilitation of serotonergic neurotransmission via allosteric 5-HT1A receptor modulation (Rock et al., 2012; Hind et al., 2016; Sonego et al., 2016), and modulation of glucose homeostasis and inflammatory processes by PPARγ activation (Esposito et al., 2011; Hind et al., 2016; Sonego et al., 2018).
However, further research is needed to clarify which of these pharmacological mechanisms contribute to CBD’s beneficial effects on cognition in acutely ill patients. It may be that CBD reduces cognitive impairments by reducing the proposed synaptic dopaminergic excess indirectly via CB1R activation by anandamide (Giuffrida et al., 1999; Giuffrida et al., 2004; Leweke, 2012). In this study, the neurocognitive performance changes were not systematically associated with changes in serum anandamide levels, indicating that CBD’s effects on cognition are mediated via different mechanisms, in particular given the fact that in the same patients, the significant increase in serum anandamide levels has been shown to be significantly associated with clinical improvement (Leweke et al., 2012). It has been suggested that the stimulation of 5-HT1A receptors may improve cognition in schizophrenia (Meltzer and Sumiyoshi, 2008). Thus, the allosteric 5-HT1A receptor modulation by CBD may also be an additional relevant mechanism of action. This hypothesis is consistent with the preclinical finding that CBD attenuates cognitive impairments in a schizophrenia-like animal mode and that these effects can be blocked by a 5-HT1A receptor antagonist but not by CB1 and CB2 receptor antagonists (Rodrigues da Silva et al., 2020). Thus, the allosteric 5-HT1A receptor modulation by CBD may also be an additional relevant mechanism of action. Notably, none of these mechanisms alone or a combination of different mechanisms seem to be more effective than the D2/D3 receptor blockade by AMI, as the efficacy of CBD and AMI was comparable in our study.
Changes in neurocognitive performance were also not systematically correlated with psychopathological improvements. Only PANSS total and PANSS general score were associated with the performance in one of three working memory tests (Letter Number Sequencing) in the AMI group and with the verbal learning performance in the CBD group. This finding supports the view that cognitive deficits are not merely the result of other schizophrenia symptoms (Keefe et al., 2006). In particular, the independence of cognitive deficits from positive symptoms has been shown previously (Green, 1996; Mohamed et al., 1999; Hughes et al., 2003; Gladsjo et al., 2004; Wagner et al., 2005; Keefe et al., 2006). On the other hand, a relationship between negative symptom severity and cognitive performance has been observed (Mohamed et al., 1999; Gladsjo et al., 2004; Keefe et al., 2006; Üçok et al., 2020). Furthermore, improvements in negative symptoms have been found to be associated with amelioration of cognitive deficits in people with schizophrenia treated with olanzapine or AMI for eight weeks (Wagner et al., 2005). The authors suggested that the same mechanisms may partly mediate both improvements in negative symptoms and cognitive performance. However, this hypothesis is not supported by our data. It may be that the treatment duration was not long enough to detect an association in this group. Furthermore, as discussed above, our findings indicate that CBD affects psychopathology via an anandamide-dependent pathway (Leweke et al., 2012), while its effects on cognitive performance seem to be mediated by another mechanism. This hypothesis is supported by the absence of a systematic correlation between the changes in neurocognitive performance and psychopathological improvements.
Strengths and Limitations
The major strength of our study is the monotherapeutic, parallel-group design (Leweke et al., 2012). In contrast to previous add-on studies (Boggs et al., 2018; McGuire et al., 2018), our study design allows for investigating the therapeutic potential of the substance more precisely as no pharmacodynamic interactions need to be considered when assessing the effects of CBD. However, the lack of a placebo condition does not allow for an estimate of a potential placebo response to cognitive functioning. Nevertheless, these tests are generally considered robust for rater bias because of their objective character. While placebo-effects in cognitive trials in schizophrenia have been considered fairly small (Keefe et al., 2017), we cannot rule out practice effects on the improvements. Parallel versions of each cognitive test were used at t0 and t1, and all tests have been reported to have a good test-retest reliability and low item-specific learning. While we acknowledge the potential development of test-taking strategies and/or procedural learning as potential confounders in our trial, their contribution to the observed improvement in cognitive scores after four weeks of treatment is likely quite limited (Goldberg et al., 2010).
Further, the administration of lorazepam as a co-medication (up to 7.5 mg per day) may have influenced results as initial use of lorazepam was higher in the AMI than in the CBD group (Leweke et al., 2012) with a similar mean dosage at the end of the trial. This may have caused a cognitive improvement due to reduction in lorazepam favoring the AMI group, although the effect of short-term benzodiazepines on cognitive performance in schizophrenia is not well investigated (Baandrup et al., 2017; Fond et al., 2018). Furthermore, we included only acutely ill patients in earlier phases of the disease with a mean age of 29.7 ± 8.3 and 30.6 ± 9.4 years in the CBD and AMI group, respectively. In this group of patients, CBD seems to be more effective than in older sub-acutely (40.9 ± 12.5 years (McGuire et al., 2018)) and chronically (48.4 ± 9.3 years (Boggs et al., 2018)) ill patients. However, further studies investigating CBD’s therapeutic effects in first-episode psychosis or the prodromal phase are needed to confirm this hypothesis and investigate the contributing factors.
The current study is limited by the small sample size and comparatively short treatment duration. The actual sample size of n = 14–15 per treatment group is sufficient to detect large effect sizes (i.e., Cohen’s d) of 1.1 with a power of 80% at two-sided alpha 5%. However, the calculated 95% confidence intervals still give useful ranges for true differences that are compatible with the data obtained. Likewise, differences outside the intervals are not compatible with the data. Furthermore, as above mentioned, we did not make any multiplicity adjustments due to the exploratory character of the study, and none of the p-values would be significant after Bonferroni correction. Consequently, our exploratory finding that CBD improves neurocognitive functioning needs to be confirmed in a larger cohort of acutely ill schizophrenia patients (i.e., in a large RCT with neurocognitive functioning as primary objective). Furthermore, it needs to be investigated whether the maximal effect of CBD had already been achieved after four weeks of treatment or whether an extended treatment duration will lead to larger effects.
Conclusion
This exploratory study shows that both CBD and AMI improve neurocognitive functioning with comparable efficacy in younger and acutely ill schizophrenia patients. However, larger RCTs are needed to confirm this explorative finding. Furthermore, our data indicates that CBD may affect psychopathology and cognitive performance via different physiological mechanisms. While improvements in psychopathology were significantly associated with an increase in serum anandamide levels (Leweke et al., 2012), cognitive improvements (if at all present) seemed to be induced via anandamide-independent pathways. Several alternative mechanisms of action have already been suggested for CBD, including an allosteric 5-HT1A receptor modulation that may be relevant for CBD’s effects on neurocognitive functioning. However, the actual involvement of 5-HT1A receptor modulation and other postulated mechanisms of action need to be examined explicitly in future studies.
Acknowledgments
We thank Karsten Blume (neuropsychology) and Torsten Noack-Schoenborn (study assistance) for their invaluable support. We acknowledge the contribution of Profs. Frauke Schultze-Lutter and Stephan Ruhrmann to the protocol and design and of Drs. Christian Mauss, Brit M. Nolden, Tobias Buzello, Miriam A. Neatby and Anita Haensel to the execution of the clinical part of this study. Our special thanks go to Dr. Daniela Muhl (née Schreiber) for her skillful analysis of Fatty Acid Ethanol amides in serum. We are also grateful to Prof. Andrea Giuffrida for his fruitful input during the course of this study.
Data Availability Statement
The datasets presented in this article are not readily available because they are available only to approved collaborators and competent authorities. Requests to access the datasets should be directed to markus.leweke@zi-mannheim.de.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the University of Cologne, Cologne, Germany. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
FML conceived and designed the study with input from MH. FML, CG, and DK treated the majority of patients in the trial, and RP performed the neuropsychological experiments; RP, MH and CR analyzed the data with input from FML; FML and CR drafted the manuscript with input from RP and MH. All authors contributed to final manuscript preparation and have read and approved the final manuscript.
Funding
This study was supported by grants from the Stanley Medical Research Institute (FML) and the Koeln Fortune Program (FML). The funding sources had no influence on the design of the study or the analysis and interpretation of the results.
Conflict of Interest
FML and DK are shareholders of curantis UG (Ltd.).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Adityanjee, Aderibigbe Y. A., Theodoridis D., Vieweg W. V. R. (1999). Dementia praecox to schizophrenia: the first 100 years. Psychiatry Clin. Neurosci. 53 (4), 437–448. 10.1046/j.1440-1819.1999.00584.x [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders (DSM-5). 5 ed. Washington, DC: American Psychiatric Association Publishing, 991. [Google Scholar]
- Appiah-Kusi E., Petros N., Wilson R., Colizzi M., Bossong M. G., Valmaggia L., et al. (2020). Effects of short-term cannabidiol treatment on response to social stress in subjects at clinical high risk of developing psychosis. Psychopharmacology 237, 1121–1130. 10.1007/s00213-019-5442-610.1007/s00213-019-05442-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baandrup L., Fagerlund B., Glenthoj B. (2017). Neurocognitive performance, subjective well-being, and psychosocial functioning after benzodiazepine withdrawal in patients with schizophrenia or bipolar disorder: a randomized clinical trial of add-on melatonin versus placebo. Eur. Arch. Psychiatry Clin. Neurosci. 267 (2), 163–171. 10.1007/s00406-016-0711-8 [DOI] [PubMed] [Google Scholar]
- Bechi M., Spangaro M., Agostoni G., Bosinelli F., Buonocore M., Bianchi L., et al. (2019). Intellectual and cognitive profiles in patients affected by schizophrenia. J. Neuropsychol. 13 (3), 589–602. 10.1111/jnp.12161 [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodological) 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bhattacharyya S., Wilson R., Appiah-Kusi E., O’Neill A., Brammer M., Perez J., et al. (2018). Effect of cannabidiol on medial temporal, midbrain, and striatal dysfunction in people at clinical high risk of psychosis. JAMA Psychiatry 75 (11), 1107–1117. 10.1001/jamapsychiatry.2018.2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T., Hanuš L., De Petrocellis L., Tchilibon S., Ponde D. E., Brandi I., et al. (2001). Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 134 (4), 845–852. 10.1038/sj.bjp.0704327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleuler E. (1908). Die prognose der Dementia praecox (schizophreniegruppe). Allgemeine Z. für Psychiatrie psychisch-gerichtliche Medizin 65, 436–464. [Google Scholar]
- Boggs D. L., Surti T., Gupta A., Gupta S., Niciu M., Pittman B., et al. (2018). The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacology 235 (7), 1923–1932. 10.1007/s00213-018-4885-9 [DOI] [PubMed] [Google Scholar]
- Bouaboula M., Hilairet S., Marchand J., Fajas L., Fur G. L., Casellas P. (2005). Anandamide induced PPARγ transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol. 517 (3), 174–181. 10.1016/j.ejphar.2005.05.032 [DOI] [PubMed] [Google Scholar]
- Bryson G., Bell M. D. (2003). Initial and final work performance in schizophrenia:. J. Nervous Ment. Dis. 191 (2), 87–92. 10.1097/01.Nmd.0000050937.06332.3c [DOI] [PubMed] [Google Scholar]
- Buchanan R. W., Freedman R., Javitt D. C., Abi-Dargham A., Lieberman J. A. (2007). Recent advances in the development of novel pharmacological agents for the treatment of cognitive impairments in schizophrenia. Schizophrenia Bull. 33 (5), 1120–1130. 10.1093/schbul/sbm083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon M., Caspi A., Moffitt T. E., Harrington H., Taylor A., Murray R. M., et al. (2002). Evidence for early-childhood, pan-developmental impairment specific to schizophreniform disorder. Arch. Gen. Psychiatry 59 (5), 449–456. 10.1001/archpsyc.59.5.449 [DOI] [PubMed] [Google Scholar]
- Cassano T., Villani R., Pace L., Carbone A., Bukke V. N., Orkisz S., et al. (2020). From cannabis sativa to cannabidiol: promising therapeutic candidate for the treatment of neurodegenerative diseases. Front. Pharmacol. 11, 124. 10.3389/fphar.2020.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W. C., Hui C. L. M., Chan S. K. W., Lee E. H. M., Chen E. Y. H. (2016). Impact of avolition and cognitive impairment on functional outcome in first-episode schizophrenia-spectrum disorder: a prospective one-year follow-up study. Schizophrenia Res. 170 (2-3), 318–321. 10.1016/j.schres.2016.01.004 [DOI] [PubMed] [Google Scholar]
- Cornblatt B. A. (1996). Continuous performance test - identical pairs version computer software. New York: Author. [Google Scholar]
- De Petrocellis L., Ligresti A., Moriello A. S., Allarà M., Bisogno T., Petrosino S., et al. (2011). Effects of cannabinoids and cannabinoid-enrichedCannabisextracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 163 (7), 1479–1494. 10.1111/j.1476-5381.2010.01166.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney R., Prevey M. L., Cramer J., Mattson R. H., Group t. V. E. C. S. R. (1992). Test-retest comparability and control subject data for the rey-auditory verbal learning test and rey-osterrieth/taylor complex figures,. Arch. Clin. Neuropsychol. 7 (6), 523–528. 10.1093/arclin/7.6.52310.1016/0887-6177(92)90142-a [DOI] [PubMed] [Google Scholar]
- Elmes M. W., Kaczocha M., Berger W. T., Leung K., Ralph B. P., Wang L., et al. (2015). Fatty acid-binding proteins (FABPs) are intracellular carriers for d9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J. Biol. Chem. 290 (14), 8711–8721. 10.1074/jbc.M114.618447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G., Scuderi C., Valenza M., Togna G. I., Latina V., De Filippis D., et al. (2011). Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS One 6 (12), e28668. 10.1371/journal.pone.0028668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fervaha G., Foussias G., Agid O., Remington G. (2014). Motivational and neurocognitive deficits are central to the prediction of longitudinal functional outcome in schizophrenia. Acta Psychiatr. Scand. 130 (4), 290–299. 10.1111/acps.12289 [DOI] [PubMed] [Google Scholar]
- Fond G., Berna F., Berna F., Boyer L., Godin O., Brunel L., et al. (2018). Benzodiazepine long-term administration is associated with impaired attention/working memory in schizophrenia: results from the national multicentre FACE-SZ data set. Eur. Arch. Psychiatry Clin. Neurosci. 268 (1), 17–26. 10.1007/s00406-017-0787-9 [DOI] [PubMed] [Google Scholar]
- Giuffrida A., Leweke F. M., Gerth C. W., Schreiber D., Koethe D., Faulhaber J., et al. (2004). Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacol 29 (11), 2108–2114. 10.1038/sj.npp.1300558 [DOI] [PubMed] [Google Scholar]
- Giuffrida A., Parsons L. H., Kerr T. M., de Fonseca F. R., Navarro M., Piomelli D. (1999). Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat. Neurosci. 2 (4), 358–363. 10.1038/7268 [DOI] [PubMed] [Google Scholar]
- Giuffrida A., Rodríguez de Fonseca F., Piomelli D. (2000). Quantification of bioactive acylethanolamides in rat plasma by electrospray mass spectrometry. Anal. Biochem. 280 (1), 87–93. 10.1006/abio.2000.4509 [DOI] [PubMed] [Google Scholar]
- Gladsjo J. A., McAdams L. A., Palmer B. W., Moore D. J., Jeste D. V., Heaton R. K. (2004). A six-factor model of cognition in schizophrenia and related psychotic disorders: relationships with clinical symptoms and functional capacity. Schizophrenia Bull. 30 (4), 739–754. 10.1093/oxfordjournals.schbul.a007127 [DOI] [PubMed] [Google Scholar]
- Gold J. M., Carpenter C., Randolph C., Goldberg T. E., Weinberger D. R. (1997). Auditory working memory and Wisconsin Card Sorting Test performance in schizophrenia. Arch. Gen. Psychiatry 54 (2), 159–165. 10.1001/archpsyc.1997.01830140071013 [DOI] [PubMed] [Google Scholar]
- Goldberg T. E., Keefe R. S. E., Goldman R. S., Robinson D. G., Harvey P. D. (2010). Circumstances under which practice does not make perfect: a review of the practice effect literature in schizophrenia and its relevance to clinical treatment studies. Neuropsychopharmacol 35 (5), 1053–1062. 10.1038/npp.2009.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. F. (1996). What are the functional consequences of neurocognitive deficits in schizophrenia?. Am. J. Psychiatry 153 (3), 321–330. 10.1176/ajp.153.3.321 [DOI] [PubMed] [Google Scholar]
- Green M. F., Kern R. S., Braff D. L., Mintz J. (2000). Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the "right stuff"?. Schizophrenia Bull. 26 (1), 119–136. 10.1093/oxfordjournals.schbul.a033430 [DOI] [PubMed] [Google Scholar]
- Gross G., Drescher K. (2012). The role of dopamine D3 receptors in antipsychotic activity and cognitive functions. Handb Exp. Pharmacol. (213), 167–210. 10.1007/978-3-642-25758-2_7 [DOI] [PubMed] [Google Scholar]
- Hallak J. E. C., Machado-de-Sousa J. P., Crippa J. A. S., Sanches R. F., Trzesniak C., Chaves C., et al. (2010). Performance of schizophrenic patients in the Stroop Color Word Test and electrodermal responsiveness after acute administration of cannabidiol (CBD). Rev. Bras. Psiquiatr. 32 (1), 56–61. 10.1590/s1516-44462010000100011 [DOI] [PubMed] [Google Scholar]
- Hampson A. J., Grimaldi M., Axelrod J., Wink D. (1998). Cannabidiol and (-) 9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. 95 (14), 8268–8273. 10.1073/pnas.95.14.8268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hind W. H., England T. J., O'Sullivan S. E. (2016). Cannabidiol protects an in vitro model of the blood-brain barrier from oxygen-glucose deprivation via PPARγ and 5-HT1A receptors. Br. J. Pharmacol. 173 (5), 815–825. 10.1111/bph.13368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H., Noguchi H., Hashimoto R., Nakabayashi T., Omori M., Takahashi S., et al. (2006). Antipsychotic medication and cognitive function in schizophrenia. Schizophrenia Res. 86 (1-3), 138–146. 10.1016/j.schres.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Hughes C., Kumari V., Soni W., Das M., Binneman B., Drozd S., et al. (2003). Longitudinal study of symptoms and cognitive function in chronic schizophrenia. Schizophr Res. 59 (2-3), 137–146. 10.1016/s0920-9964(01)00393-0 [DOI] [PubMed] [Google Scholar]
- Islam M. A., Habtewold T. D., van Es F. D., Quee P. J., van den Heuvel E. R., Alizadeh B. Z., et al. (2018). Long-term cognitive trajectories and heterogeneity in patients with schizophrenia and their unaffected siblings. Acta Psychiatr. Scand. 138 (6), 591–604. 10.1111/acps.12961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E., Fein D., Morris R., Delis D. C. (1991). WAIS-R NI manual. San Antonio, TX: Psychological Corporation. [Google Scholar]
- Keefe R., Goldberg T. E., Harvey P. D., Gold J. M., Poe M. P., Coughenour L. (2004). The Brief Assessment of Cognition in Schizophrenia: reliability, sensitivity, and comparison with a standard neurocognitive battery. Schizophrenia Res. 68 (2-3), 283–297. 10.1016/j.schres.2003.09.011 [DOI] [PubMed] [Google Scholar]
- Keefe R. S. E., Bilder R. M., Davis S. M., Harvey P. D., Palmer B. W., Gold J. M., et al. (2007). Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch. Gen. Psychiatry 64 (6), 633–647. 10.1001/archpsyc.64.6.633 [DOI] [PubMed] [Google Scholar]
- Keefe R. S. E., Bilder R. M., Harvey P. D., Davis S. M., Palmer B. W., Gold J. M., et al. (2006). Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacol 31 (9), 2033–2046. 10.1038/sj.npp.1301072 [DOI] [PubMed] [Google Scholar]
- Keefe R. S. E., Davis V. G., Harvey P. D., Atkins A. S., Haig G. M., Hagino O., et al. (2017). Placebo response and practice effects in schizophrenia cognition trials. JAMA Psychiatry 74 (8), 807–814. 10.1001/jamapsychiatry.2017.1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe R. S. E., Fenton W. S. (2007). How should DSM-V criteria for schizophrenia include cognitive impairment?. Schizophrenia Bull. 33 (4), 912–920. 10.1093/schbul/sbm046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe R. S. E., Perkins D. O., Gu H., Zipursky R. B., Christensen B. K., Lieberman J. A. (2006). A longitudinal study of neurocognitive function in individuals at-risk for psychosis. Schizophrenia Res. 88 (1-3), 26–35. 10.1016/j.schres.2006.06.041 [DOI] [PubMed] [Google Scholar]
- Kremen W. S., Seidman L. J., Faraone S. V., Toomey R., Tsuang M. T. (2000). The paradox of normal neuropsychological function in schizophrenia. J. Abnormal Psychol. 109 (4), 743–752. 10.1037//0021-843x.109.4.74310.1037/0021-843x.109.4.743 [DOI] [PubMed] [Google Scholar]
- Kumar S., Chaudhury S. (2014). Efficacy of amisulpride and olanzapine for negative symptoms and cognitive impairments: an open-label clinical study. Ind. Psychiatry J. 23 (1), 27–35. 10.4103/0972-6748.144953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam M., Lee J., Rapisarda A., See Y. M., Yang Z., Lee S.-A., et al. (2018). Longitudinal cognitive changes in young individuals at ultrahigh risk for psychosis. JAMA Psychiatry 75 (9), 929–939. 10.1001/jamapsychiatry.2018.1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie R. B., Bagher A. M., Kelly M. E. M., Denovan-Wright E. M. (2015). Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 172 (20), 4790–4805. 10.1111/bph.13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrl S. (1995). Mehrfachwahl-wortschatz-intelligenztest: MWT-B [multiple Choice vocabulary test]. Ballingen: Perimed-spitta. [Google Scholar]
- Leweke F. M. (2012). Anandamide dysfunction in prodromal and established psychosis. Curr. Pharm. Des. 18 (32), 5188–5193. 10.2174/138161212802884843 [DOI] [PubMed] [Google Scholar]
- Leweke F. M., Mueller J. K., Lange B., Fritze S., Topor C. E., Koethe D., et al. (2018). Role of the endocannabinoid system in the pathophysiology of schizophrenia: implications for pharmacological intervention. CNS Drugs 32 (7), 605–619. 10.1007/s40263-018-0539-z [DOI] [PubMed] [Google Scholar]
- Leweke F. M., Piomelli D., Pahlisch F., Muhl D., Gerth C. W., Hoyer C., et al. (2012). Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2, e94. 10.1038/tp.2012.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezak M. D., Howieson D. B., Bigler E. D., Tranel D. (2012). Neuropsychological assessment. 5 ed. Oxford; New York: Oxford University Press. [Google Scholar]
- Lipsey M. W., Wilson D. B. (2001). Practical meta-analysis. Thousand Oaks, CA, USA; London, UK: Sage Publications, Inc. [Google Scholar]
- Makiol C., Kluge M. (2019). Remission of severe, treatment-resistant schizophrenia following adjunctive cannabidiol. Aust. N. Z. J. Psychiatry 53 (3), 262. 10.1177/0004867418815982 [DOI] [PubMed] [Google Scholar]
- McCleery A., Nuechterlein K. H. (2019). Cognitive impairment in psychotic illness: prevalence, profile of impairment, developmental course, and treatment considerations. Dialogues Clin. Neurosci. 21 (3), 239–248. 10.31887/DCNS.2019.21.3/amccleery [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire P., Robson P., Cubala W. J., Vasile D., Morrison P. D., Barron R., et al. (2018). Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicenter randomized controlled trial. Am. J. Psychiatry 175 (3), 225–231. 10.1176/appi.ajp.2017.17030325 [DOI] [PubMed] [Google Scholar]
- Meltzer H. Y., Sumiyoshi T. (2008). Does stimulation of 5-HT(1A) receptors improve cognition in schizophrenia?. Behav. Brain Res. 195 (1), 98–102. 10.1016/j.bbr.2008.05.016 [DOI] [PubMed] [Google Scholar]
- Mesholam-Gately R. I., Giuliano A. J., Goff K. P., Faraone S. V., Seidman L. J. (2009). Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology 23 (3), 315–336. 10.1037/a0014708 [DOI] [PubMed] [Google Scholar]
- Mohamed S., Paulsen J. S., O'Leary D., Arndt S., Andreasen N. (1999). Generalized cognitive deficits in schizophrenia. Arch. Gen. Psychiatry 56 (8), 749–754. 10.1001/archpsyc.56.8.749 [DOI] [PubMed] [Google Scholar]
- Mohamed S., Rosenheck R., Swartz M., Stroup S., Lieberman J. A., Keefe R. S. E. (2008). Relationship of cognition and psychopathology to functional impairment in schizophrenia. Am J. Psychiatry 165 (8), 978–987. 10.1176/appi.ajp.2008.07111713 [DOI] [PubMed] [Google Scholar]
- Mori M. A., Meyer E., Soares L. M., Milani H., Guimarães F. S., de Oliveira R. M. W. (2017). Cannabidiol reduces neuroinflammation and promotes neuroplasticity and functional recovery after brain ischemia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 75, 94–105. 10.1016/j.pnpbp.2016.11.005 [DOI] [PubMed] [Google Scholar]
- Mortimer A. M., Joyce E., Balasubramaniam K., Choudhary P. C., Saleem P. T., Group S. S. (2007). Treatment with amisulpride and olanzapine improve neuropsychological function in schizophrenia. Hum. Psychopharmacol. Clin. Exp. 22 (7), 445–454. 10.1002/hup.865 [DOI] [PubMed] [Google Scholar]
- Nuechterlein K. H., Barch D. M., Gold J. M., Goldberg T. E., Green M. F., Heaton R. K. (2004). Identification of separable cognitive factors in schizophrenia. Schizophrenia Res. 72 (1), 29–39. 10.1016/j.schres.2004.09.007 [DOI] [PubMed] [Google Scholar]
- Nuechterlein K. H., Green M. F., Kern R. S., Baade L. E., Barch D. M., Cohen J. D., et al. (2008). The MATRICS Consensus Cognitive Battery, part 1: test selection, reliability, and validity. Ajp 165 (2), 203–213. 10.1176/appi.ajp.2007.07010042 [DOI] [PubMed] [Google Scholar]
- Palsetia D., Chandrasekhar K., Reddy M. S., De Sousa A., Karia S. (2018). Executive function in patients with schizophrenia based on socio-occupational impairment: a cross-sectional study. Ind. Psychiatry J. 27 (2), 181–189. 10.4103/ipj.ipj_85_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S., Holzman P. S. (1992). Schizophrenics show spatial working memory deficits. Arch. Gen. Psychiatry 49 (12), 975–982. 10.1001/archpsyc.1992.01820120063009 [DOI] [PubMed] [Google Scholar]
- Petrides M., Milner B. (1982). Deficits on subject-ordered tasks after frontal- and temporal-lobe lesions in man. Neuropsychologia 20 (3), 249–262. 10.1016/0028-3932(82)90100-2 [DOI] [PubMed] [Google Scholar]
- Pukrop R., Matuschek E., Ruhrmann S., Brockhaus-Dumke A., Tendolkar I., Bertsch A., et al. (2003). Dimensions of working memory dysfunction in schizophrenia. Schizophrenia Res. 62 (3), 259–268. 10.1016/s0920-9964(02)00427-9 [DOI] [PubMed] [Google Scholar]
- Reichenberg A., Weiser M., Rapp M. A., Rabinowitz J., Caspi A., Schmeidler J., et al. (2005). Elaboration on premorbid intellectual performance in schizophrenia. Arch. Gen. Psychiatry 62 (12), 1297–1304. 10.1001/archpsyc.62.12.1297 [DOI] [PubMed] [Google Scholar]
- Reitan R. M., Wolfson D. (1985). The halstead-reitan neuropsychological test battery. Tucson, AZ: Neuropsychology Press. [Google Scholar]
- Rey A. A. (1941). L’examen psychologique dans les cas d’encephalopathie traumatique. Arch. Psychol. 28, 286–340. [Google Scholar]
- Riley E. M., McGovern D., Mockler D., Doku V. C. K., ÓCeallaigh S., Fannon D. G., et al. (2000). Neuropsychological functioning in first-episode psychosis - evidence of specific deficits. Schizophrenia Res. 43 (1), 47–55. 10.1016/s0920-9964(99)00177-2 [DOI] [PubMed] [Google Scholar]
- Rock E., Bolognini D., Limebeer C., Cascio M., Anavi-Goffer S., Fletcher P., et al. (2012). Cannabidiol, a non-psychotropic component of cannabis, attenuates vomiting and nausea-like behaviour via indirect agonism of 5-HT1A somatodendritic autoreceptors in the dorsal raphe nucleus. Br. J. Pharmacol. 165 (8), 2620–2634. 10.1111/j.1476-5381.2011.01621.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues da Silva N., Gomes F. V., Sonego A. B., Silva N. R. d., Guimarães F. S. (2020). Cannabidiol attenuates behavioral changes in a rodent model of schizophrenia through 5-HT1A, but not CB1 and CB2 receptors. Pharmacol. Res. 156, 104749. 10.1016/j.phrs.2020.104749 [DOI] [PubMed] [Google Scholar]
- Rohleder C., Müller J. K., Lange B., Leweke F. M. (2016). Cannabidiol as a potential new type of an antipsychotic. A critical review of the evidence. Front. Pharmacol. 7 (422), 422. 10.3389/fphar.2016.00422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross T., Hanouskova E., Giarla K., Calhoun E., Tucker M. (2007). The reliability and validity of the self-ordered pointing task. Arch. Clin. Neuropsychol. 22 (4), 449–458. 10.1016/j.acn.2007.01.023 [DOI] [PubMed] [Google Scholar]
- Sabatucci A., Tortolani D., Dainese E., Maccarrone M. (2018). In silico mapping of allosteric ligand binding sites in type-1 cannabinoid receptor. Biotechnol. Appl. Biochem. 65 (1), 21–28. 10.1002/bab.1589 [DOI] [PubMed] [Google Scholar]
- Sakurai H., Bies R. R., Stroup S. T., Keefe R. S. E., Rajji T. K., Suzuki T., et al. (2013). Dopamine D2 receptor occupancy and cognition in schizophrenia: analysis of the CATIE data. Schizophr Bull. 39 (3), 564–574. 10.1093/schbul/sbr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saykin A. J., Shtasel D. L., Gur R. E., Kester D. B., Mozley L. H., Stafiniak P., et al. (1994). Neuropsychological deficits in neuroleptic naive patients with first-episode schizophrenia. Arch. Gen. Psychiatry 51 (2), 124–131. 10.1001/archpsyc.1994.03950020048005 [DOI] [PubMed] [Google Scholar]
- Schoemaker H., Claustre Y., Fage D., Rouquier L., Chergui K., Curet O., et al. (1997). Neurochemical characteristics of amisulpride, an atypical dopamine D2/D3 receptor antagonist with both presynaptic and limbic selectivity. J. Pharmacol. Exp. Ther. 280 (1), 83–97. [PubMed] [Google Scholar]
- Schreiber D., Harlfinger S., Nolden B. M., Gerth C. W., Jaehde U., Schömig E., et al. (2007). Determination of anandamide and other fatty acyl ethanolamides in human serum by electrospray tandem mass spectrometry. Anal. Biochem. 361 (2), 162–168. 10.1016/j.ab.2006.11.027 [DOI] [PubMed] [Google Scholar]
- Seidman L. J., Shapiro D. I., Stone W. S., Woodberry K. A., Ronzio A., Cornblatt B. A., et al. (2016). Association of neurocognition with transition to psychosis. JAMA Psychiatry 73 (12), 1239–1248. 10.1001/jamapsychiatry.2016.2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff P., Le Foll B. (2017). The dopamine D3 receptor, a quarter century later. Eur. J. Neurosci. 45 (1), 2–19. 10.1111/ejn.13390 [DOI] [PubMed] [Google Scholar]
- Sonego A. B., Gomes F. V., Del Bel E. A., Guimaraes F. S. (2016). Cannabidiol attenuates haloperidol-induced catalepsy and c-Fos protein expression in the dorsolateral striatum via 5-HT1A receptors in mice. Behav. Brain Res. 309, 22–28. 10.1016/j.bbr.2016.04.042 [DOI] [PubMed] [Google Scholar]
- Sonego A. B., Prado D. S., Vale G. T., Sepulveda-Diaz J. E., Cunha T. M., Tirapelli C. R., et al. (2018). Cannabidiol prevents haloperidol-induced vacuos chewing movements and inflammatory changes in mice via PPARγ receptors. Brain Behav. Immun. 74, 241–251. 10.1016/j.bbi.2018.09.014 [DOI] [PubMed] [Google Scholar]
- Turek A., Machalska K., Chrobak A. A., Tereszko A., Siwek M., Dudek D. (2017). Speech graph analysis of verbal fluency tests distinguish between patients with schizophrenia and healthy controls. Eur. Neuropsychopharmacol. 27, S914–S915. 10.1016/s0924-977x(17)31626-7 [DOI] [Google Scholar]
- Tyson P. J., Laws K. R., Flowers K. A., Tyson A., Mortimer A. M. (2006). Cognitive function and social abilities in patients with schizophrenia: relationship with atypical antipsychotics. Psychiatry Clin. Neurosci. 60 (4), 473–479. 10.1111/j.1440-1819.2006.01534.x [DOI] [PubMed] [Google Scholar]
- Tyson P. J., Roberts K. H., Mortimer A. M. (2004). Are the cognitive effects of atypical antipsychotics influenced by their affinity to 5HT-2A receptors?. Int. J. Neurosci. 114 (6), 593–611. 10.1080/00207450490430552 [DOI] [PubMed] [Google Scholar]
- Uren J., Cotton S. M., Killackey E., Saling M. M., Allott K. (2017). Cognitive clusters in first-episode psychosis: overlap with healthy controls and relationship to concurrent and prospective symptoms and functioning. Neuropsychology 31 (7), 787–797. 10.1037/neu0000367 [DOI] [PubMed] [Google Scholar]
- Üçok A., Direk N., Kaya H., Çağlar N., Çıkrıkçılı U., Noyan H., et al. (2020). Relationship of negative symptom severity with cognitive symptoms and functioning in subjects at ultra‐high risk for psychosis. Early Intervention in Psychiatry. 10.1111/eip.13042 [DOI] [PubMed] [Google Scholar]
- Vaskinn A., Ueland T., Melle I., Agartz I., Andreassen O. A., Sundet K. (2014). Neurocognitive decrements are present in intellectually superior schizophrenia. Front. Psychiatry 5, 45. 10.3389/fpsyt.2014.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M., Quednow B. B., Westheide J., Schlaepfer T. E., Maier W., Kühn K.-U. (2005). Cognitive improvement in schizophrenic patients does not require a serotonergic mechanism: randomized controlled trial of olanzapine vs amisulpride. Neuropsychopharmacol 30 (2), 381–390. 10.1038/sj.npp.1300626 [DOI] [PubMed] [Google Scholar]
- Wang Y. T., Chiu N. Y., Jou S. H., Kuang Yang Y., Hui Lee I., Wang C. C., et al. (2008). Effects of amisulpride on the cognitive function of patients with schizophrenia who switched from risperidone. Int. J. Psychiatry Clin. Pract. 12 (3), 180–186. 10.1080/13651500701805727 [DOI] [PubMed] [Google Scholar]
- Woodward N. D., Purdon S. E., Meltzer H. Y., Zald D. H. (2005). A meta-analysis of neuropsychological change to clozapine, olanzapine, quetiapine, and risperidone in schizophrenia. Int. J. Neuropsychopharm. 8 (3), 457–472. 10.1017/s146114570500516x [DOI] [PubMed] [Google Scholar]
- Zuardi A., Crippa J., Hallak J., Pinto J., Chagas M., Rodrigues G., et al. (2009). Cannabidiol for the treatment of psychosis in Parkinson's disease. J. Psychopharmacol. 23 (8), 979–983. 10.1177/0269881108096519 [DOI] [PubMed] [Google Scholar]
- Zuardi A. W., Morais S. L., Guimarães F. S., Mechoulam R. (1995). Antipsychotic effect of cannabidiol. J. Clin. Psychiatry 56 (10), 485–486. [PubMed] [Google Scholar]
- Zuardi A. W., Hallak J. E. C., Dursun S. M., Morais S. L., Sanches R. F., Musty R. E., et al. (2006). Cannabidiol monotherapy for treatment-resistant schizophrenia. J. Psychopharmacol. 20 (5), 683–686. 10.1177/0269881106060967 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets presented in this article are not readily available because they are available only to approved collaborators and competent authorities. Requests to access the datasets should be directed to markus.leweke@zi-mannheim.de.