

Abstract
Background
Diffuse intrinsic pontine glioma (DIPG) is a devastating pediatric cancer with unmet clinical need. DIPG is invasive in nature, where tumor cells interweave into the fiber nerve tracts of the pons making the tumor unresectable. Accordingly, novel approaches in combating the disease are of utmost importance and receptor-driven cell invasion in the context of DIPG is under-researched area. Here, we investigated the impact on cell invasion mediated by PLEXINB1, PLEXINB2, platelet growth factor receptor (PDGFR)α, PDGFRβ, epithelial growth factor receptor (EGFR), activin receptor 1 (ACVR1), chemokine receptor 4 (CXCR4), and NOTCH1.
Methods
We used previously published RNA-sequencing data to measure gene expression of selected receptors in DIPG tumor tissue versus matched normal tissue controls (n = 18). We assessed protein expression of the corresponding genes using DIPG cell culture models. Then, we performed cell viability and cell invasion assays of DIPG cells stimulated with chemoattractants/ligands.
Results
RNA-sequencing data showed increased gene expression of receptor genes such as PLEXINB2, PDGFRα, EGFR, ACVR1, CXCR4, and NOTCH1 in DIPG tumors compared to the control tissues. Representative DIPG cell lines demonstrated correspondingly increased protein expression levels of these genes. Cell viability assays showed minimal effects of growth factors/chemokines on tumor cell growth in most instances. Recombinant SEMA4C, SEM4D, PDGF-AA, PDGF-BB, ACVA, CXCL12, and DLL4 ligand stimulation altered invasion in DIPG cells.
Conclusions
We show that no single growth factor-ligand pair universally induces DIPG cell invasion. However, our results reveal a potential to create a composite of cytokines or anti-cytokines to modulate DIPG cell invasion.
Keywords: cell invasion, chemokine/cytokine, DIPG, receptor
Key Points.
Multiple receptors are overexpressed at RNA and protein levels in DIPG patients.
Cell invasion activated in some DIPG cell lines upon chemoattractant stimulation.
SEMA4C, SEM4D, PDGF-AA, PDGF-BB, ACVA, CXCL12, and DLL4 induce cell invasion in DIPG.
Importance of the Study.
DIPG is a fatal pediatric brain tumor with radiotherapy as the only standard treatment option to date. DIPG is among the greatest unmet needs among childhood brain tumors, in part because DIPG tumor cells weave themselves into the neural fabric of the pons nerve cell tracts and because no treatments meaningfully work. Hence, development of unique approaches to overcome DIPG is quintessential. In this study, we examined multiple chemokines/receptors and their ligands that participate in cell invasion. This study investigates the creation of a composite of cytokines to selectively attract (and trap) DIPG tumor cells.
Diffuse intrinsic pontine gliomas (DIPG) are rare pediatric brain tumors consisting of about 10%–20% of all brain tumors in children.1,2 DIPG is the foremost cause of pediatric brain tumor deaths.3 DIPGs originate in the pons, which controls the essential biological functions such as breathing, blood pressure and heart rate,4 and infiltrate the brainstem2 and often metastasize to other parts of the brain such as subventricular region5 making surgical resection impossible.5,6 The onset of DIPG occurs between the ages of 4–11 years with an overall median survival of approximately 9–11 months and a 5-year survival rate of less than 1%.3,5 Unfortunately, the only palliative mode of treatment is fractionated radiation therapy (54–59 Gy over 30 fractions)6 with transient effect. With the advent of genomic and biochemical studies, availability of biopsy and autopsy samples from DIPG patients, and newly-developed DIPG animal models, new treatment avenues such as targeted therapies and immunotherapies are under extensive investigation; however, new strategies have not yet yielded breakthrough treatments.7,8 One of the main hindrances in effective drug delivery to the brain has been the blood–brain barrier.9 Hence, DIPG is one of the greatest unmet clinical needs among childhood brain tumors and has constantly challenged the scientific community to pose novel strategies to tackle the disease.
In past decades, an increasing number of published studies have demonstrated that overexpression of receptors/cytokines and their ligands is a common phenomenon in numerous human cancers of pancreas, cervix, and breast.10–12 Classically, the receptor/ligands axes participate in cell signaling to promote cell–cell communication, cell proliferation, and cell migration and invasion during mammalian development.13 However, aberrant expression of receptors/cytokines do play a key role in cell transformation and tumor formation and progression.11,13,14 While multiple studies have been conducted examining the role of growth factors/receptors/cytokines in adult brain cancer cell invasion,15,16 the cell growth and invasion capacity of receptors/ligands axes in DIPGs is still emerging.17 For example, certain factors such as HAS218,19 are intriguing. In this study, we shortlisted receptors/ligands that are overexpressed in various malignancies based on published cancer studies10,11,14,20–27 and investigated their roles in DIPG.
Specifically, we focused on PLEXINBs/SEMA4s subclasses of plexin/semaphorin signaling axes, platelet growth factor receptor (PDGFR)α, PDGFRβ, epithelial growth factor receptor (EGFR), activin receptor 1 (ACVR1), chemokine receptor 4 (CXCR4), NOTCH1 and the previously identified chemoattractant complex PTN, HSP90β, SPARC, and SPARC-L that are overexpressed in malignancies of breast, liver, and brain.10,11,13,22,23,26–32 Herein, we assessed gene and protein expression patterns of the receptors in tumor tissues and DIPG cell models, respectively. Next, we examined the cell growth properties of selected ligands in selected DIPG cell models followed by the invasion capabilities of DIPG cells upon ligand stimulation. Results provide evidence of elevated gene and protein expressions of PLEXINB2, PDGFRα, EGFR, ACVR1, CXCR4, and NOTCH1 in tumor tissues compared to control tissues. Results further revealed instances of cell line specific effects of ligands SEMAPHORIN4C (SEMA4C), SEMAPHORIN4D (SEM4D), platelet-derived growth factor (PDGF-A, PDGF-B and PDGF-C), and delta-like ligand 4 (DLL-4) on cell invasion in DIPG cell culture models.
Methods and Materials
Human DIPG Cell Culture
Patient-derived pediatric/adolescent (DIPG) cell lines/neurospheres and young adult cell lines (control) were used in this study. DNA fingerprinting via short tandem repeat analysis was performed routinely to authenticate all cell cultures used for experiments (Supplementary Table 1.1). Tumor cell culture SF-8628 was grown in Dulbecco Eagle Media (DMEM, Thermo Fisher Scientific (Thermo), cat#11995065) supplemented with 10% fetal bovine serum (FBS, Thermo, cat#26140079), 1% penicillin & streptomycin antibiotics (Thermo, cat#15140122) and 1% L-glutamine (Thermo, cat#25030081). CHLA-200 cell culture was grown in Iscove’s Modified Dulbecco’s Medium (Thermo, cat#12440061) with 10% FBS, 1% penicillin/streptomycin, 4 mM L-glutamine (Thermo, cat#25030081) and 1× insulin, transferrin and selenous acid (Corning Life Sciences, cat#41400045). DIPG 4, DIPG 13, DIPG 17, and DIPG 24 were maintained in Complete Tumor Stem Media (TSM)33,34 and the media composition is provided in Supplementary Tables 3.1 and 3.2. All cell cultures were maintained in an incubator at 37.0°C, 5% CO2, 20% O2.
Human DIPG Cell Lines Characteristics and the List of Receptors and Their Ligands
A complete description of DIPG cell lines used in the experiments is provided in Supplementary Table 1.2. Also, representative light microscopy images of the 5 cell lines most frequently used in this study are shown in Supplementary Figure S2. Supplementary Table 1.3 illustrates the list of ligands and their respective receptors used in this study. An expanded summary of the physiological and biological activity ranges of the recombinant proteins used in this study is shown in Supplementary Table 2.
RNA Sequencing
RNA sequencing data were accessed from a previous publication (dbgap ID: phs001526.v1.p1).34 DIPG tumor and their respective normal control samples (n = 18) were analyzed to measure RNA expression profiles.
Western Blot Analysis
Whole cell protein lysates were prepared using RIPA lysis buffer (Thermo, cat# 89901) supplemented with HALT protease/phosphatase inhibitor cocktail (Thermo, cat# 78440). The protein extracts were subjected to BCA protein assay to determine the concentration (Thermo, cat# 23227). Protein samples (20–30 μg) were resolved using (4%–20% or 7.5% or 10%) SDS-PAGE gels (Bio-Rad, cat#456–2023, cat# 456–1024, cat# 456–1033). Proteins were then transferred to Polyvinylidene difluoride (PVDF) membranes using the Mini-Transblot System (Bio-Rad, cat# 1658030). Membranes were subjected to blocking in 5% milk prepared in Tris-buffered saline with 0.1% Tween-20 for 2 h at room temperature. Membranes were incubated in primary antibodies overnight at 4°C. The primary antibodies used for this study are provided in Supplementary Table 4.1. After washing steps, the membranes were further incubated at room temperature for 1 h using Horseradish peroxidase (HRP) conjugated secondary antibodies: anti-mouse (Vector Laboratories, cat# PI-2000,), anti-rabbit (Vector Laboratories, cat# PI-1000). Immunoblots were subjected to Enhanced chemiluminescence (ECL) (Bio-Rad, cat# 1705061) and visualized using FluorChemQ imaging machine (Protein Simple).
Recombinant proteins used for cell viability and invasion assays are provided in Supplementary Table 4.2.
Cell Viability Assay
The DIPG cells were seeded post serum starvation in serum-free (SF-8628) and growth factor-free (GFs) media (DIPG 4, DIPG 13, DIPG 17, and DIPG 24). The dissociated cells were resuspended in recombinant ligands at various concentrations and were seeded in triplicates into 96-well plates (Thermo, cat#1360) at 500 cells/well (SF-8628) and 2000 cells per/well for DIPG 4, DIPG 13, DIPG 17, and DIPG 24 using multichannel pipette. The cells were placed in the incubator for 72 h and cell viability was measured using the CellTiter-Glo luminescent reagent (Promega, cat# G7570) in a BioTek plate reader. Experiments were conducted in triplicates.
Cell Invasion Assay
In order to determine the effect of chemoattractant on cell invasion toward serum/GF containing standard media or ligand (chemoattractant), cells were dissociated into single-cell suspensions using Trypsin-EDTA (Thermo, cat# 25300062) or TrypLE Express Enzyme (Thermo, cat# 12604-021). For all invasion assays, we used Biocoat Growth Factor Reduced Matrigel Invasion Chamber (Corning, cat# 354480), per manufacturer’s instructions. All experimental conditions were performed in triplicate. For SF-8628 cell line, 50,000 cells were resuspended in serum-free DMEM media and seeded into the transwell inserts on the top chamber. DIPG 4, DIPG 13, DIPG 17, and DIPG 24 cell lines cell were resuspended in TSM media containing B27-A and heparin but no GFs and seeded at 100,000 cells per well 16 h post serum/GFs starvation. The ligands (chemoattractants) were diluted in serum and GFs free media and plated to the bottom well transwell at 700 μL per well. In all the experiments, either (+) serum or (+) GFs were used as positive controls and the treatment groups were normalized to either (-) serum or (-) GFs controls.
The cell suspensions and the chemoattractant combinations were incubated for 72 h and assayed by a colorimetric crystal violet assay for quantification of invasion.33 Briefly, postincubation, the media from the upper chamber and lower chambers was aspirated and the noninvading cells were removed from the inner part of the insert using cotton swab. The invading cells that passed through the Matrigel-coated membrane were fixed with 4% paraformaldehyde followed by washing steps. Next, the cells were stained using 0.1% crystal violet dye in 10% methanol. The inserts were washed and air-dried before eluting the dye with 10% acetic acid. The dye intensity of the eluates was measured at a wavelength of 595 nm using a Biotek plate reader.
Statistical Analyses
The RNA-seq data were analyzed using the analysis of variance (ANOVA) or Kruskall–Wallis as appropriate. In invasion assays, to compare control and cytokine-treated groups, ANOVA followed by Dunnett’s multiple comparison tests was performed. We performed 2-tailed with a significance level of 5% using R1 software.35 All graphs were plotted using GraphPad Prism software.
Results
PLEXINBs and PDGFRs Are Differentially Expressed in DIPG Tumor Tissues and Cell Cultures
To quantify the overall gene expression of cell-surface receptors in human DIPG tissue samples, we referred to the RNA-sequencing data previously generated34 and compared the mRNA expression between DIPG tumor tissues (n = 18) with matched normal tissue samples (n = 18). PLEXINB1 gene expression was decreased in DIPG tumors comparison to their matched normal samples (Figure 1A), whereas PLEXINB2 mRNA levels were elevated in tumor samples versus normal samples, albeit not significant (Figure 1B). The gene expression in 2 of the 3 PLEXINB ligands, SEMA4D and SEMA4F, was significantly downregulated in tumor tissues versus normal tissues, while SEMA4C gene expression was increased (Figure 1C). The gene expression of platelet-derived growth factor receptors (PDGFR (α and β)) showed differing expression patterns. While PDGFRα expression was significantly upregulated in tumor tissues in comparison to the normal samples, PDGFRβ expression was downregulated in DIPG tumor tissues than the normal tissues (Figure 1D and E). While PDGF-A and PDGF-B ligands were downregulated in tumor tissues in comparison to normal tissues, PDGF-C mRNA level was upregulated (Figure 1F). Overall, the data reveal variation in the expression levels of PLEXINBs and PDGFRs in the DIPG tumor samples.
Figure 1.

Aberrant gene and protein expression pattern in DIPG tumor tissues cell lines. (A–C) Box and whisker plots show the relative gene expression of PLEXINB1 and PLEXINB2 receptors and their known ligands, SEMA4C, SEMA4D, and SEMA4F in normal and DIPG tumor samples. The mRNA expression is shown in reads per kilobase of exon per million reads mapped (RPKM). (D–F) mRNA expression of PDGFR (α and β) and their preferred ligands PDGF-AA, PDGF-BB, and PDGF-CC in both normal and DIPG tumor samples. (* denotes P < .05, and ** denotes P < .01). (G,H) Immunoblots of receptors, PLEXINB2, PLEXINB1, PDGFRα, and PDGFRβ show aberrant expression in DIPG tumor cell lines. CHLA 200 and SJ-GBM2 are non-DIPG cell lines and serve as controls. DIPG cells: DIPG 4, DIPG 6, DIPG 13, DIPG 17, DIPG 24, SF-8628, and VU-DIPG A. β-ACTIN and GAPDH were used as loading controls.
To evaluate the protein expression profile of the PLEXIN and PDGFR genes in cell line model systems, we conducted western blot analyses on whole cell lysates from DIPG cell lines. PLEXINB1 was robustly expressed in DIPG 17 and DIPG 24 only (Figure 1G). DIPG 6, DIPG 13, DIPG 17, and DIPG 24 showed elevated protein expression of PLEXIN-B2 versus DIPG 4, SF-8628, and VU-DIPG-A as depicted by a mature, processed alpha-subunit band and beta-subunit band (Figure 1G). Interestingly, all 3 cell lines expressing low PLEXINB2, occurred in the adherent cell lines DIPG 4, SF-8628, and VU-DIPG-A with the exception of DIPG 24. Thus, we speculate that PLEXINBs’ expression might be associated with the cell growth properties. Next, PDGFRα showed robust protein expressions in all cell lines with DIPG 6 demonstrating the highest expression. In contrast, PDGFRβ expression varied greatly between the cell lines while DIPG 4, SF-8628, and VU-DIPG-A showed a robust protein expression (Figure 1H). CHLA 200 and SJ-GBM2 cells expressing high protein levels are non-DIPG controls used for the immunoblot analyses. The results reveal the occurrence of similar trend for gene and protein expression of receptors, PLEXINBs and PDGFRs in DIPG tumor tissues and cell culture models.
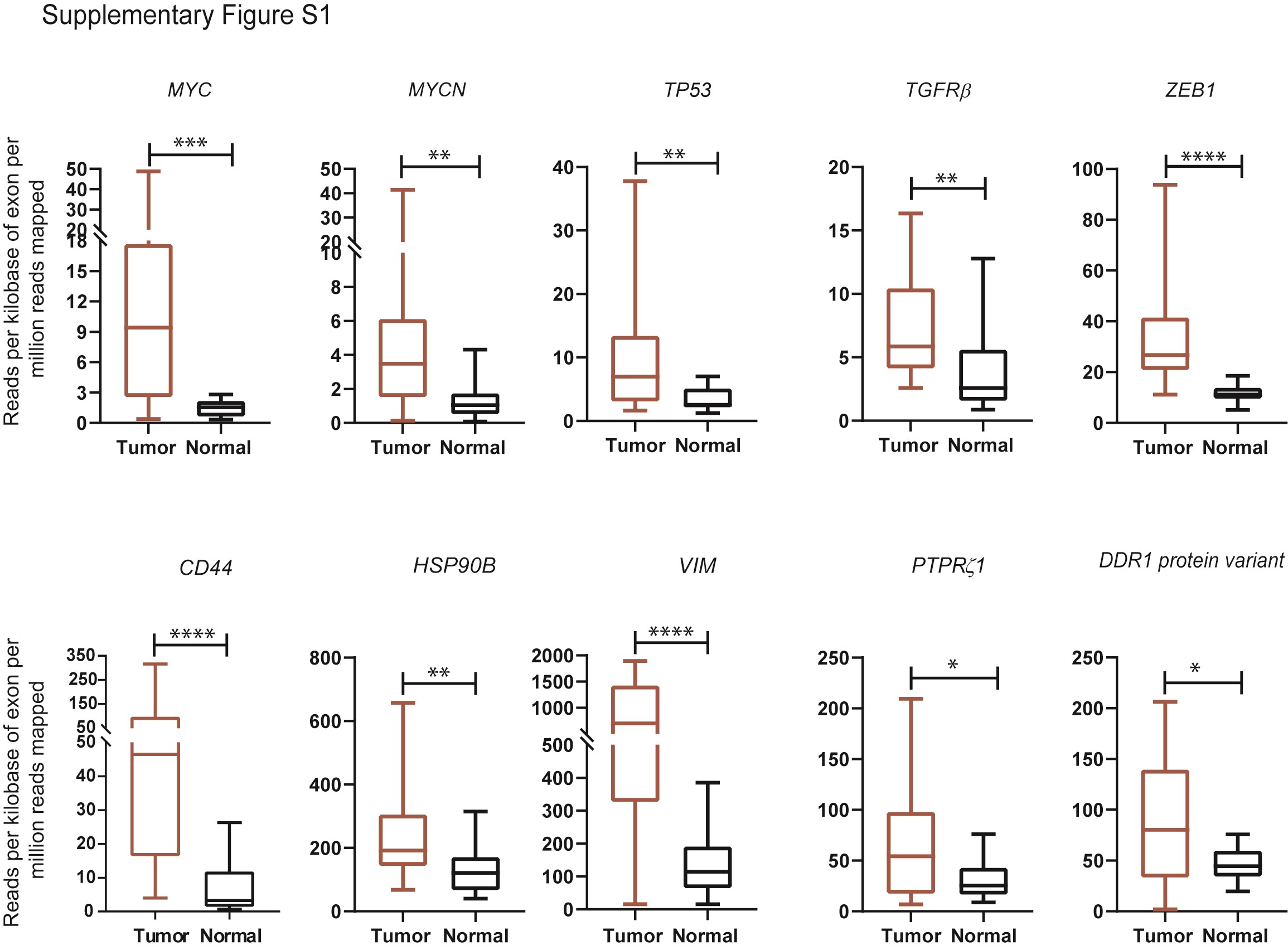
Next, we investigated gene and protein expression pattern of additional signaling receptors (Figure 2A and B). Notably, the expression of signaling receptors, EGFR, ACVR1, IL-13Rα2, CXCR4, and NOTCH1 were significantly upregulated in the DIPG tumor samples in comparison to the normal samples. The Low-density lipoprotein receptor-related protein 1 (LRP1) expression was also elevated, whereas tyrosine-protein kinase Met (c-MET) expression was lower in DIPG tumor tissues compared to the normal control samples. Hyaluronan synthase 2 (HAS2), the enzyme that metabolizes hyaluronan (HA) is shown to be elevated in diffusely infiltrating astrocytomas.36 Indeed, HAS2 was upregulated in the DIPG tumor samples versus normal control tissues (Figure 2A). We show additional genes that are significantly upregulated in DIPG tumor tissues over the normal samples (Supplementary Figure S1). Immunoblot analyses revealed consistent and robust expression for ACVR1, HAS2, IL-13Rα2, CXCR4, NOTCH1, and LRP1, whereas EGFR and c-MET revealed a stochastic expression pattern in the DIPG cell lines (Figure 2B). In recent years, somatic mutations in histone the H3 protein have been identified in gliomas and DIPGs. Particularly, H3F3A, H3.3 histone variant mutation is present in > 70% of all DIPGs.37 Consistent with the previous findings, all DIPG cell lines showed H3.3 elevation including DIPG 4 cell line which consists of H3.1K27M mutation.38 Taken together, we conclude that gene and protein expressions of the investigated receptors/cytokines, with some exceptions, are elevated in DIPG tumor tissues and cell culture models.
Figure 2.

Gene and protein expression of additional receptors in DIPG tumor tissues and cell lines. (A) RNA-sequencing analysis of multiple receptors showing the relative expression in normal and tumor tissue samples. Gene listed are, EGFR, ACVR1, HAS2, c-MET, IL-13Rα2, CXCR4, NOTCH1, and LRP1. The expression is depicted in RPKM. (* denotes P < .05, ** denotes P < .01, *** denotes P < .001, and **** denotes P < .0001). (B) Western blot analyses show the protein expression of all genes in panel A in multiple DIPG cell lines. Note: additional DIPG cell lines were used for the immunoblots. GAPDH and β-ACTIN were used as loading controls.
Cytokine Stimulation Induces Minimal Cell Growth in DIPG Cell Models
To assess the effect of cytokines/ligands on cell growth, we stimulated the DIPG cell lines with increasing concentrations of cytokines and measured cell viability after 72 h. As illustrated in Figure 3A, tested DIPG cell lines showed little to no cell growth upon SEMA4C and SEM4D receptor stimulation irrespective of PLEXINBs’ expression. In the tested low PDGFRα+ cell lines, SF-8628 and DIPG 13 showed the highest cell growth upon addition of the ligand PDGF-AA. Next, PDGF-BB showed slight overall elevation in cell growth among the tested cell lines, with the exception of DIPG 24. Cell stimulation with PDGF-CC, the ligand of PDGFRɑβ, showed elevated growth in DIPG 13 and DIPG 24 (Figure 3B). Nevertheless, the overall cell growth remained minimal to mild upon ligand stimulations in PDGFR expressing cells. We also investigated the cell viability effect of additional cytokines, that is, ACTIVIN A, EGF, PTN, CXCL12, IL-13, and DLL4 (Figure 3C–H) in DIPG cells. Among the ACVR1+ cell lines, DIPG 13 had elevated cell growth relative to other cell lines at higher ACVA stimulation (Figure 3C). In EGFR+ cell lines, SF-8628, DIPG 4, and DIPG 24, cell growth was minimal at lower EGF concentration (Figure 3D). Interestingly, as shown in Figure 3E–H, despite a robust protein expression of CXCR4, IL-13Rα2, and NOTCH1 receptors, the cell growth of SF-8628, DIPG 4, DIPG 13, DIPG 17, and DIPG 24 was affected minimally upon cell stimulation by CXCL12, IL-13, and DLL4 ligands. Thus, our data suggest that DIPG cell lines expressing elevated levels of receptors show minimal cell viability upon ligand stimulation with the exception of some cell lines such as DIPG 24, whereby cell growth is robustly elevated upon cell stimulation by PDGF-BB and PDGF-CC.
Figure 3.

Ligand stimulation of multiple cell-surface receptors have minimal cell viability effect in DIPG cell lines. (A–H) Selected DIPG cell lines were treated with recombinant ligands at increasing concentrations to activate their respective receptors. The ligands used are SEMA4C, SEMA4D, PDGF-AA, PDGF-BB, PDGF-CC, ACTIVIN A, EGF, PTN, CXCL12, IL-13, and DLL4. Note: DIPG cell lines used are SF-8628, DIPG 4, DIPG 13, DIPG 17, and DIPG 24 but not all cell lines are used for all ligand treatments. Cell viability values were normalized to untreated controls.
SEMAPHORINs and PDGFs Promote Cell Invasion in Multiple DIPG Cancer Cells
To determine the invasive capacity of DIPG cells upon cytokine/ligand induction, we used Matrigel-coated invasion chambers. We stimulated PLEXINB expressing DIPG cell lines using SEMA4C alone, SEMA4D alone, and the combination of SEMA4C and SEMA4D. Low PLEXINB1 and PLEXINB2 expressing cell line, SF-8628, showed a significant increase in cell invasion under all stimulation conditions in comparison to the untreated group (Figure 4A). Next, the low PLEXINB1 and low PLEXINB2 expressing cell lines, DIPG 4 and DIPG 13 revealed increased but statistically insignificant to no cell invasion at all treatment conditions (Figure 4A). DIPG 24, the cell line expressing high levels of both PLEXINB1 and PLEXINB2 showed increased but statistically insignificant DIPG cell invasion poststimulation. Overall, these results confirm that SEMA4C or SEMA4D alone can activate cell invasion similar to that of the combined ligand treatment in the tested cell lines irrespective of PLEXINBs’ expression.
Figure 4.

Increased cell invasion stimulated by PLEXIN and PDGFR ligands in multiple DIPG cell lines. (A) SF-8628, DIPG 4, DIPG 13, and DIPG 24 cell lines were used for the invasion assays. The recombinant ligands SEMA4C and SEMA4D were added individually or in combination at 200 ng/mL concentrations. (B) The recombinant ligands PDGF-AA, PDGF-BB, and PDGF-CC were added at 100 ng/mL concentrations in all the experiments. All experiments were performed in triplicates. The (+) serum and (+) growth factors (GF) data served as positive controls and (-) serum and (-) GF were used as negative controls. (* denotes P < .05, ** denotes P < .01, and *** denotes P < .001).
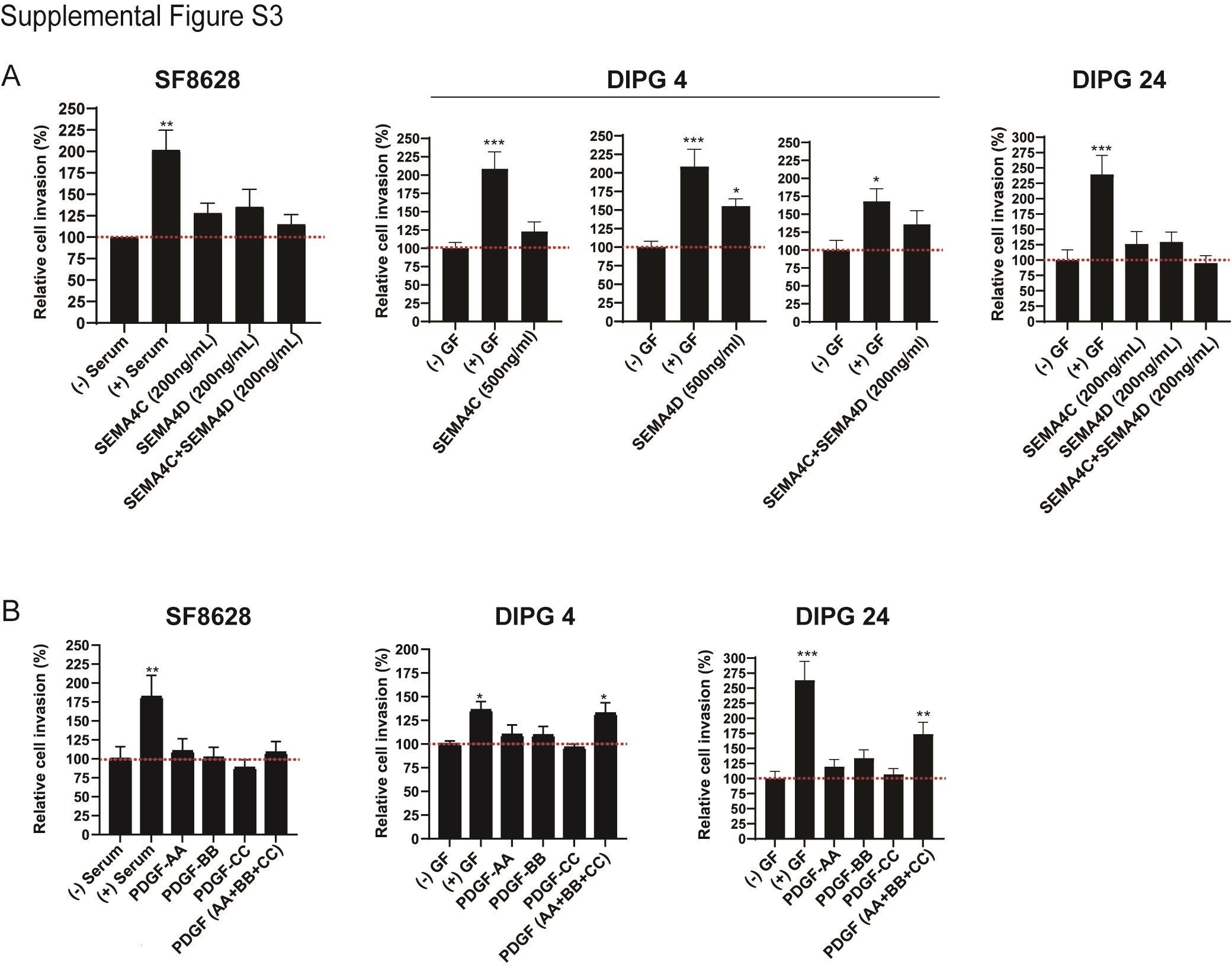
PDGF growth factors are essential in basic postnatal biological functions in cell growth via chemotaxis, cell division, and blood vessel formation.29,39 Mutation, amplification, deletion, and fusions in PDGF receptors and ligands have been reported in glioblastoma, pancreatic cancer,11 ovarian cancer,24 and DIPG.32 Next, we examined the invasion effect of DIPG cells following stimulation by PDGF receptor associated ligands, PDGF-AA, PDGF-BB, and PDGF-CC (Figure 4B and Supplementary Figure S3). Classically, PDGF-AA preferably binds to the alpha form of the PDGFRα 21,29,31; PDGF-BB binds to both alpha and beta21,31 receptors; and PDGF-CC binds to both alpha and beta forms of PDGFRs.21,29,31 In case of SF-8628, which expressed PDGFRα (mild) and PDGFRβ (high) proteins, no significant cell invasion occurred upon PDGF-AA alone stimulation, but cell invasion significantly increased upon PDGF-BB alone stimulation (Figure 4B). Notably, cell invasion in DIPG 4 (PDGFRα- and PDGFRβ+) was statistically significant upon stimulation by PDGF-AA alone, PDGF-BB alone and PDGF-CC alone. DIPG 13 and DIPG 24 (PDGFRα- and PDGFRβ-) revealed opposite cell invasion trends. Upon PDGF-AA alone, PDGF-BB alone and PDGF-CC alone stimulation, DIPG 13 showed no effect and DIPG 24 showed increased to significantly increased cell invasion. Interestingly, while stimulation by the combination of all 3 ligands in SF-8628, DIPG 4 and DIPG 24 increased invasion, the effect was comparable to that of PDGF-BB stimulation. However, in DIPG 13, the cell invasion capacity was not affected by combined ligand stimulation. Hence, these data suggest that PDGFR ligands PDGF-AA alone and PDGF-BB alone can stimulate cell invasion in DIPG cell culture models at varying capacity irrespective PDGFRs’ expression profile highlighting the possibility of DIPG cell line-dependent binding affinity of PGFRs/PDGFs. Supplementary Table 5 displays an overview of the selected DIPG cell lines, protein expression of the growth factors/cytokines, and the response to growth factors/cytokines.
ACVA, CXCL12, and DLL4 Ligands But Not EGF Stimulate Cell Invasion in a Patient-Specific Manner
Besides PLEXINBs and PDGFRs, our gene and protein expression analyses showed upregulation of additional receptors in the DIPG tumor tissues and cells. Hence, we sought to investigate the invasion capacity of the cytokines/ligands of EGFR, ACVR1, CXCR4, and NOTCH1 receptors in DIPG cancer cells. In ACVR1 expressing cell lines (DIPG 4, DIPG 24, and SF-8628), we found that ACVA stimulation increased (statistically insignificant) invasion in all cell lines compared to untreated control group (Figure 5B). In contrast, the EGFR expressing cell lines (SF-8628 and DIPG 24) had reduced cell invasion upon EGF stimulation (Figure 5A). CXCR4 expressing cell lines (SF-8628>DIPG 24) showed inverse influence on cell invasion post-CXCL12 stimulation, that is, the invasion activity was significantly increased in DIPG 24, whereas in SF-8628 invasion was lower than the control (Figure 5C). Lastly, the effect of DLL4 induced cell invasion in high to low NOTCH1 expressing cell lines, that is, DIPG 24 > DIPG 4 > SF-8628 showed increased invasion of all 3 cell lines with no statistical significance versus control. The invasion activity was greatest in SF-8628 followed by DIPG 4 and DIPG 24 cell lines. Notably, the invasion capacity of DIPG cells is inversely proportional to the protein expression of the receptors, ACVR1, CXCR4 and NOTCH1. These results suggest that multiple factors participate to promote cell invasion in DIPG tumor cells albeit at varying capacity.
Figure 5.

Invasion activity shown by multiple recombinant ligands in different DIPG cell lines. (A) Invasion in SF-8628 (100 ng/mL) and DIPG 4 (200 ng/mL) cell lines was unchanged or decreased following EGF stimulation. (B) ACVR stimulation by ACVA ligand (100 ng/mL) showed increased effects in SF-8628, DIPG 4, and DIPG 24 cell lines. (C) CXCR4 receptor stimulation by CXCL12 ligand (100 ng/mL) decreased invasiveness in SF-8628 but increased cell invasion in DIPG 24 cell line. (D) NOTCH1 receptor ligand, DLL4 showed increased invasion in all tested DIPG cell lines (SF-8628, DIPG 4, and DIPG 24) at 10 ng/mL concentration versus controls. All experiments were performed in triplicates. The (+) serum and (+) growth factors (GF) data served as positive controls and (-) serum and (-) GF were used as negative controls. (* denotes P < .05, ** denotes P < .01, and *** denotes P < .001).
Cell Line-Dependent Invasion Effect of Previously Identified PTN Chemoattractant Complex on DIPG Cell Lines
Qin et al. (2017) showed a novel chemoattractant complex secreted by neural precursor cells consisting of PTN, HSP90β, SPARC, and SPARC-L molecules. The study demonstrated that the combination of all 4 chemoattractants would activate the RhoA/ROCK signaling pathway and ultimately signal DIPG cells to invade to the subventricular zone.33 Hence, we wanted to explore the invasion capacity effect of these 4 cytokines in DIPG cell culture models. We assessed the invasion effect of PTN alone, HSP90β alone, SPARC alone, SPARC-L alone and the combination of all 4 molecules in SF-8628, DIPG 4, and DIPG 24 cell lines. (Note: the concentration used in our study is 500 ng/mL per molecule instead of 100 nM used by33, ie, 5.6 nM for HSP90β, 6.7 nM for SPARC-L, 14 nM for SPARC, and 27 nM for PTN). As shown in Figure 6, the invasion effect of the chemoattractant complex significantly increased in SF-8628 cells but not in DIPG 4 and DIPG 24. Interestingly, HSP90β alone significantly increased the invasion activity in SF-8628. However, this effect was not seen in DIPG 4 and DIPG 24. In both SF-8628 and DIPG 24 cell lines, a molecule alone was able to stimulate cell invasion greater than the untreated controls. However, DIPG 4 showed minimal change in invasion upon cell stimulation in all treatment groups in comparison to the untreated control. These findings suggest that the invasion capacity of the PTN complex varies between cell lines.
Figure 6.

Invasion stimulation by previously published33 chemoattractant complex in DIPG cell lines. The effect of either individual or the combination of each chemoattractant treatment was minimal in DIPG 4 cell lines compared to unstimulated control group. DIPG 24 showed comparable increased invasiveness upon individual and combination chemoattractant treatments in comparison to the untreated group. The (+) serum and (+) growth factors (GF) served as positive controls and (-) serum and (-) GF data were used as negative controls. (* denotes P < .05, ** denotes P < .01).
Discussion
Here, we demonstrate that receptor molecules, PLEXINB2, PDGFRs, EGFR, ACVR1, CXCR4, and NOTCH1 are highly expressed both transcriptionally and translationally in DIPG tumor tissues and in multiple DIPG cell lines, respectively. The cell growth assay confirmed minimal cell viability post cytokine-induction in the tested DIPG cell lines. Upon cytokine stimulation with SEMA4C, SEMA4D, PDGF-AA, PDGF-BB, ACVA, CXCL12, or DLL4, elevate cell invasion in DIPG cell lines with consistent trends across independent biological replicate. However, we observed experiment-to-experiment variation which could be due to confounding factors, namely, biological variance, the nature of the assay, and cell passage-dependent variation,40 but confirmatory experiments are required.
Previous findings have recognized that SEMA4C has a high affinity toward PLEXINB230 and PLEXINB1 preferentially binds to SEMA4D.41 Additionally, PLEXINB1/SEMA4D is overexpressed in other cancer types, such as breast, liver, and colon28,41,42 and has been shown to participate in tumor progression.42 Likewise, PLEXINB2/ SEMA4C signaling axis is involved in cell growth and cell invasion in breast cancer and glioblastoma.10,25,41,42 Our studies revealed that DIPG tumor tissues expressed overall lower gene levels of PLEXINB1 in comparison to PLEXINB2, and that the ligand (SEMA4C) was highly expressed in tumor tissues and not in SEM4D and SEMA4F. In invasion assays, we observed that SEMA4D increased invasive potential versus SEM4AC. This phenomenon might have occurred because of low endogenous PLEXINB1 expression in the DIPG tested. However, to precisely assess the potency of each ligand, knockdown studies or antibody inhibitory studies are required.
Our findings show that PDGF-AA and PDGF-CC increased invasion in some tumor cell lines is offset by the observation that PDGF-BB ligand potently enhanced cell invasion in all 4 DIPG cell lines. These findings are consistent with previous studies where PDGF-BB enhanced cell migration in comparison to PDGF-AA and PDGF-CC in breast cancer.43 Furthermore, overexpression of PDGF-BB ligand alone could contribute to the activation of PDGFR signaling pathways as alluded previously.44 Future studies require knockdown or antibody blocking/neutralizing experiments to elucidate the role of each ligand in DIPG cell invasion. Interestingly, EGFR showed high gene expression and varying protein levels in DIPG tissues and cells, which is on par with other cancer types.34,45 However, unlike other reported findings,27,46 our studies also revealed unchanged or decreased cell invasion upon EGF induction. Activin A receptor type I (ACVR1) binds to activin and is also a member of bone morphogenesis protein and transforming growth factor-β (TGF-β) family.47,48 ACVR1 participates in cell motility, cell viability, cell migration, and cell invasion in prostrate, ovarian, and breast cancers.49–51 Recently, ACVR1 has claimed to be a distinctive marker of DIPG whereby ACVR1 is recurrently mutated and may coordinate with H3.1K27M to enhance DIPG pathogenesis in the context of ACVR1 R206H mutation.1,26 The ACVR1 mutation status in each cell lines need to be studied in order to determine the effect of ACVR1 mutation on invasion in DIPG.
The receptor/ligand binding required optimum concentrations to induce biological effect. A study by Zhu et al. (2010) reported the FGFR/FGF signaling axes could only stimulate DNA synthesis at an optimum dose of 300 pg/mL of FGF but the higher concentration of 100 ng/mL or more had minimal effect.52 It is important to note that in our studies we exposed DIPG cells with supramaximal concentrations of the ligands/cytokines for stimulation purposes except for DLL4 ligands. Hence, additional experiments need to be performed at low ligand concentrations. A limitation of our study is that the cell lines might not be entirely representative of the in vivo tumor microenvironment for many reasons, including the lack of tumor cell heterogeneity.53
In summary, we performed analyses of receptor/ligand axes to determine RNA/protein expression and invasion profile in DIPG tissues and cell lines. We took a novel approach for assessing cancer cell invasion in DIPG cell culture models. We reveal that no single growth factor-ligand pair universally induces DIPG cell invasion. However, that it will be challenging to translate these results into effective therapy, but results offer the possibility that a compromise of cytokines could be created to selectively attract and trap DIPG cells and prevent them from distant infiltration.
Funding
This study was supported by Storm the Heavens Foundation in Memory of Philomena.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Michelle Monje and Dr. Suzanne Baker for generously sharing cell lines. Our sincere appreciation to our intern, Cynthia S. Martucci, who was funded by Pediatric Oncology Student Training (POST) grant program from Alex’s Lemonade Stand Foundation and Samuel Rasmussen, engineering fellow for helping us with technical support for the DIPG project.
Authorship Statement
Design: A.K., C.K. Reagents: N.E.B., A.K. Experiments: J.-A.K., A.K. Analysis: A.K., N.E.B., Q.L., J.E.K., C.K. Editing: A.K., C.K., J.-A.K., E.H., N.E.B.
Conflict of interest statement. C.K. has sponsored research agreements, industry research funding or research collaborations with Genentech/Roche, Novartis and Syndax Pharmaceuticals, and is co-founder of Artisan Biopharma and Tio Companies. The other authors have no conflicts of interest to declare.
References
- 1. Mathew RK, Rutka JT. Diffuse intrinsic pontine glioma: clinical features, molecular genetics, and novel targeted therapeutics. J Korean Neurosurg Soc. 2018;61(3):343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Panditharatna E, Yaeger K, Kilburn LB, Packer RJ, Nazarian J. Clinicopathology of diffuse intrinsic pontine glioma and its redefined genomic and epigenomic landscape. Cancer Genet. 2015;208(7-8):367–373. [DOI] [PubMed] [Google Scholar]
- 3. Donaldson SS, Laningham F, Fisher PG. Advances toward an understanding of brainstem gliomas. J Clin Oncol. 2006;24(8):1266–1272. [DOI] [PubMed] [Google Scholar]
- 4. Loveson KF, Fillmore HL. Intersection of brain development and paediatric diffuse midline gliomas: potential role of microenvironment in tumour growth. Brain Sci. 2018;8(11):e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caretti V, Bugiani M, Freret M, et al. . Subventricular spread of diffuse intrinsic pontine glioma. Acta Neuropathol. 2014;128(4):605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cohen KJ, Jabado N, Grill J. Diffuse intrinsic pontine gliomas-current management and new biologic insights. Is there a glimmer of hope? Neuro Oncol. 2017;19(8):1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plessier A, Le Dret L, Varlet P, et al. . New in vivo avatars of diffuse intrinsic pontine gliomas (DIPG) from stereotactic biopsies performed at diagnosis. Oncotarget. 2017;8(32):52543–52559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Welby JP, Kaptzan T, Wohl A, et al. . Current murine models and new developments in H3K27M diffuse midline gliomas. Front Oncol. 2019;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warren KE. Beyond the blood:brain barrier: the importance of central nervous system (CNS) Pharmacokinetics for the treatment of CNS tumors, including diffuse intrinsic pontine Glioma. Front Oncol. 2018;8:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gurrapu S, Pupo E, Franzolin G, Lanzetti L, Tamagnone L. Sema4C/PlexinB2 signaling controls breast cancer cell growth, hormonal dependence and tumorigenic potential. Cell Death Differ. 2018;25(7):1259–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taeger J, Moser C, Hellerbrand C, et al. . Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol Cancer Ther. 2011;10(11):2157–2167. [DOI] [PubMed] [Google Scholar]
- 12. Schrevel M, Gorter A, Kolkman-Uljee SM, Trimbos JB, Fleuren GJ, Jordanova ES. Molecular mechanisms of epidermal growth factor receptor overexpression in patients with cervical cancer. Mod Pathol. 2011;24(5):720–728. [DOI] [PubMed] [Google Scholar]
- 13. Cross M, Dexter TM. Growth factors in development, transformation, and tumorigenesis. Cell. 1991;64(2):271–280. [DOI] [PubMed] [Google Scholar]
- 14. Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda). 2010;25(2):85–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma P, Debinski W. Receptor-targeted glial brain tumor therapies. Int J Mol Sci. 2018;19(11):3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di Cintio F, Dal Bo M, Baboci L, De Mattia E, Polano M, Toffoli G. The molecular and microenvironmental landscape of Glioblastomas: implications for the novel treatment choices. Front Neurosci. 2020;14:603647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kluiver TA, Alieva M, van Vuurden DG, Wehrens EJ, Rios AC. Invaders exposed: understanding and targeting tumor cell invasion in diffuse intrinsic pontine glioma. Front Oncol. 2020;10:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maria BL, Gupta N, Gilg AG, et al. . Targeting hyaluronan interactions in spinal cord astrocytomas and diffuse pontine gliomas. J Child Neurol. 2008;23(10):1214–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L, Wang H, Xu M, et al. . Long noncoding RNA HAS2-AS1 promotes tumor progression in glioblastoma via functioning as a competing endogenous RNA. J Cell Biochem. 2020;121(1):661–671. [DOI] [PubMed] [Google Scholar]
- 20. Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J Exp Med. 1996;184(3):1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol Med. 2013;19(8):460–473. [DOI] [PubMed] [Google Scholar]
- 22. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Taylor KR, Mackay A, Truffaux N, et al. . Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46(5):457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Farooqi AA, Siddik ZH. Platelet-derived growth factor (PDGF) signalling in cancer: rapidly emerging signalling landscape. Cell Biochem Funct. 2015;33(5):257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Le AP, Huang Y, Pingle SC, et al. . Plexin-B2 promotes invasive growth of malignant glioma. Oncotarget. 2015;6(9):7293–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoeman CM, Cordero FJ, Hu G, et al. . ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat Commun. 2019;10(1):1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu Z, Jiang G, Blume-Jensen P, Hunter T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol. 2001;21(12):4016–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Conrotto P, Corso S, Gamberini S, Comoglio PM, Giordano S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene. 2004;23(30):5131–5137. [DOI] [PubMed] [Google Scholar]
- 29. Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15(4):197–204. [DOI] [PubMed] [Google Scholar]
- 30. Witherden DA, Watanabe M, Garijo O, et al. . The CD100 receptor interacts with its plexin B2 ligand to regulate epidermal γδ T cell function. Immunity. 2012;37(2):314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Demoulin JB, Essaghir A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev. 2014;25(3):273–283. [DOI] [PubMed] [Google Scholar]
- 32. Hoeman C, Shen C, Becher OJ. CDK4/6 and PDGFRA signaling as therapeutic targets in diffuse intrinsic pontine glioma. Front Oncol. 2018;8:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qin EY, Cooper DD, Abbott KL, et al. . Neural precursor-derived pleiotrophin mediates subventricular zone invasion by glioma. Cell. 2017;170(5):845–859.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berlow NE, Svalina MN, Quist MJ, et al. . IL-13 receptors as possible therapeutic targets in diffuse intrinsic pontine glioma. PLoS One. 2018;13(4):e0193565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. http://www.R-project.org/. [Google Scholar]
- 36. Valkonen M, Haapasalo H, Rilla K, Tyynelä-Korhonen K, Soini Y, Pasonen-Seppänen S. Elevated expression of hyaluronan synthase 2 associates with decreased survival in diffusely infiltrating astrocytomas. BMC Cancer. 2018;18(1):664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee S, Kambhampati M, Yadavilli S, et al. . Differential expression of Wilms’ tumor protein in diffuse intrinsic pontine glioma. J Neuropathol Exp Neurol. 2019;78(5):380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagaraja S, Vitanza NA, Woo PJ, et al. . Transcriptional dependencies in diffuse intrinsic pontine Glioma. Cancer Cell. 2017;31(5):635–652.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hannink M, Donoghue DJ. Structure and function of platelet-derived growth factor (PDGF) and related proteins. Biochim Biophys Acta. 1989;989(1):1–10. [DOI] [PubMed] [Google Scholar]
- 40. Calles K, Svensson I, Lindskog E, Häggström L. Effects of conditioned medium factors and passage number on Sf9 cell physiology and productivity. Biotechnol Prog. 2006;22(2):394–400. [DOI] [PubMed] [Google Scholar]
- 41. Conrotto P, Valdembri D, Corso S, et al. . Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood. 2005;105(11):4321–4329. [DOI] [PubMed] [Google Scholar]
- 42. Ch’ng ES, Kumanogoh A. Roles of Sema4D and Plexin-B1 in tumor progression. Mol Cancer. 2010;9:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Salha S, Gehmert S, Brébant V, et al. . PDGF regulated migration of mesenchymal stem cells towards malignancy acts via the PI3K signaling pathway. Clin Hemorheol Microcirc. 2018;70(4):543–551. [DOI] [PubMed] [Google Scholar]
- 44. Paugh BS, Zhu X, Qu C, et al. . Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73(20):6219–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12(8):553–563. [DOI] [PubMed] [Google Scholar]
- 46. Wells A. Tumor invasion: role of growth factor-induced cell motility. Adv Cancer Res. 2000;78:31–101. [DOI] [PubMed] [Google Scholar]
- 47. Attisano L, Wrana JL, Montalvo E, Massagué J. Activation of signalling by the activin receptor complex. Mol Cell Biol. 1996;16(3):1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu X, Nagarajan RP, Vale W, Chen Y. Phosphorylation regulation of the interaction between Smad7 and activin type I receptor. FEBS Lett. 2002;519(1-3):93–98. [DOI] [PubMed] [Google Scholar]
- 49. Craft CS, Romero D, Vary CP, Bergan RC. Endoglin inhibits prostate cancer motility via activation of the ALK2-Smad1 pathway. Oncogene. 2007;26(51):7240–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alarmo EL, Pärssinen J, Ketolainen JM, Savinainen K, Karhu R, Kallioniemi A. BMP7 influences proliferation, migration, and invasion of breast cancer cells. Cancer Lett. 2009;275(1):35–43. [DOI] [PubMed] [Google Scholar]
- 51. Herrera B, van Dinther M, Ten Dijke P, Inman GJ. Autocrine bone morphogenetic protein-9 signals through activin receptor-like kinase-2/Smad1/Smad4 to promote ovarian cancer cell proliferation. Cancer Res. 2009;69(24):9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu H, Duchesne L, Rudland PS, Fernig DG. The heparan sulfate co-receptor and the concentration of fibroblast growth factor-2 independently elicit different signalling patterns from the fibroblast growth factor receptor. Cell Commun Signal. 2010;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vinci M, Burford A, Molinari V, et al. . Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat Med. 2018;24(8):1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.