

Shigella causes millions of dysentery cases worldwide, primarily affecting children under 5 years old. Despite active research in developing vaccines and new antibiotics, relatively little is known about the variation of physiology or metabolism across multiple isolates.
KEYWORDS: Shigella flexneri, microaerobic metabolism, mixed acid fermentation, formate, acetate, acetyl-CoA
ABSTRACT
The instability of Shigella genomes has been described, but how this instability causes phenotypic differences within the Shigella flexneri species is largely unknown and likely variable. We describe here the genome of S. flexneri strain PE577, originally a clinical isolate, which exhibits several phenotypic differences compared to the model strain 2457T. Like many previously described strains of S. flexneri, PE577 lacks discernible, functional CRISPR and restriction-modification systems. Its phenotypic differences compared to 2457T include lower transformation efficiency, higher oxygen sensitivity, altered carbon metabolism, and greater susceptibility to a wide variety of lytic bacteriophage isolates. Since relatively few Shigella phages have been isolated on 2457T or the previously characterized strain M90T, developing a more universal model strain for isolating and studying Shigella phages is critical to understanding both phages and phage-host interactions. In addition to phage biology, the genome sequence of PE577 was used to generate and test hypotheses of how pseudogenes in this strain—whether interrupted by degraded prophages, transposases, frameshifts, or point mutations—have led to metabolic rewiring compared to the model strain 2457T. Results indicate that PE577 can utilize the less-efficient pyruvate oxidase/acetyl coenzyme A (acetyl-CoA) synthetase (PoxB/Acs) pathway to produce acetyl-CoA, while strain 2457T cannot due to a nonsense mutation in acs, rendering it a pseudogene in this strain. Both strains also utilize pyruvate-formate lyase to oxidize formate but cannot survive with this pathway alone, possibly because a component of the formate-hydrogen lyase (fdhF) is a pseudogene in both strains.
IMPORTANCE Shigella causes millions of dysentery cases worldwide, primarily affecting children under 5 years old. Despite active research in developing vaccines and new antibiotics, relatively little is known about the variation of physiology or metabolism across multiple isolates. In this work, we investigate two strains of Shigella flexneri that share 98.9% genetic identity but exhibit drastic differences in metabolism, ultimately affecting the growth of the two strains. Results suggest that additional strains within the S. flexneri species utilize different metabolic pathways to process pyruvate. Metabolic differences between these closely related isolates suggest an even wider variety of differences in growth across S. flexneri and Shigella in general. Exploring this variation further may assist in the development or application of vaccines and therapeutics to combat Shigella infections.
INTRODUCTION
Shigella flexneri is a causative agent of shigellosis, a type of bacillary dysentery. Shigella is endemic in numerous countries worldwide and contributes to the approximately 269.2 million cases of shigellosis per year (1). Dozens of S. flexneri serotypes and subtypes have been described, with each serotype determined by the lipopolysaccharide (LPS) O-antigen chemical structure (2, 3). These serotypes exhibit unique antigenic properties (4), and the genes responsible for determining serotype are often recombined and/or modified by lysogenic bacteriophages (3, 5), meaning serotype is a highly variable characteristic. Despite the large global burden of S. flexneri infections, relatively few strains have been thoroughly characterized; moreover, these strains represent an extremely small fraction of S. flexneri diversity (6). The model organism S. flexneri strain 2457T is serotype 2a—an antigenic type which is found worldwide—but belongs to only one of seven phylogenetic groups. Another commonly studied strain, S. flexneri M90T, is serotype 5a and belongs to a separate phylogenetic group with a narrower geographic range, primarily found in North America, Europe, and southern Asia (6). Besides these two strains, relatively little is known about other isolates or phylogenetic groups of the species.
PE577 is a serotype Y strain, with an O-antigen that is not decorated by glucosyl or acetyl moieties (3). This serotype serves as the most basic O-antigen structure, with other serotypes resulting from modification of the Y type. The strain was obtained from the Institute of Medical and Veterinary Science (now SA Pathology) in Adelaide, Australia, in 1988. Though originally a clinical isolate, it has lost its virulence plasmid since isolation. Most recently isolated S. flexneri-infecting bacteriophages infect serotype Y, with some exclusively infecting this serotype (7, 8). A majority of these novel phages were isolated using strain PE577; comparably few were detected when nonpathogenic derivatives of other model strains 2457T or M90T were used, even from the same environmental samples. This strain has thus been useful for assessing both abundance and diversity of Shigella phages in the environment. While strain PE577 has been used to isolate and study many bacteriophages, it had not been previously described or characterized genomically or physiologically. Establishing a model organism with such widespread bacteriophage susceptibility may therefore advance our understanding of phage-host interactions, including O-antigen recognition and other determinants of host recognition. As part of this goal, we sequenced the entire genome of strain PE577, including its chromosome and two small plasmids. In addition, Shigella is known to produce variable responses in the human host (9, 10). Some of this is likely due to serotype and antigenicity, which has been the primary focus of vaccine development (4); however, some of the variability may also be due to variance in Shigella physiology and metabolism. Therefore, we also performed metabolic analyses of S. flexneri PE577 compared to CFS100, an avirulent derivative of 2457T (11), to measure differences in carbon metabolism under aerobic or microaerobic conditions the bacteria may encounter while in the environment or moving through the human gut.
Unlike other genera of Enterobacteriaceae, Shigella species lack functional CRISPR and restriction-modification systems and are susceptible to parasitism by mobile genetic elements (12). This likely contributes to the ongoing degradation of Shigella genomes, along with selective pressure to eliminate antivirulence genes, which interfere with the pathogenic lifestyle of these bacteria (13–15). While Connor et al. (6) describe overall stable genomes outside these mobile elements, this work was based on large-scale whole-genome sequencing data from hundreds of isolates: functional analyses were understandably beyond the scope of the study. In addition to its greater susceptibility to bacteriophage infection from samples collected from various environments (7, 8), we describe here that PE577 exhibits several differences in cell physiology compared to its 2a counterpart, CFS100. These are exacerbated by microaerobic conditions and media where glucose is the primary carbon source compared to aerobic conditions and nutrient-rich media with a variety of carbon sources.
Through comparative genomics, we identified differences in genes involved in pyruvate catabolism between PE577 and CFS100. After determining the metabolic pathways in which these genes are involved, we examined the phenotypic effects of growing the two strains in a variety of conditions favored by alternate pathways. After growth, we measured specific metabolic products in spent media via high-pressure liquid chromatography (HPLC) to investigate which pathways were utilized under each condition. The combination of genetic and metabolic analyses described here indicate that PE577 utilizes the PoxB/Acs bypass to generate acetyl coenzyme A (acetyl-CoA), while CFS100 does not. This metabolic rewiring suggests drastic physiological changes can occur from small genomic variation over a relatively short evolutionary history. These results have implications for S. flexneri pathogenesis, treatment, vaccine development, and bacteriophage susceptibility—including the research and development of phage therapy or its application.
RESULTS
PE577 belongs to a different S. flexneri phylogenetic group than other model strains.
To begin comparing genetic changes between strains, the complete genome of S. flexneri strain PE577 was sequenced, including its 4.6-Mbp chromosome and two plasmids (Fig. 1). Individual characteristics and accession numbers of these are shown in Table 1, which can be cross-referenced using the GenBank accession numbers PRJNA533747 for the BioProject or SAMN12588114 for the BioSample.
FIG 1.

Genome maps and features of the S. flexneri PE577 chromosome and plasmids, with coding sequences (CDS), tRNA, rRNA, and GC characteristics colored as indicated. GC skew refers to the GC richness of a given region of the genome.
TABLE 1.
Properties of the Shigella flexneri PE577 chromosome and plasmids
Previous work has consolidated worldwide S. flexneri isolates into seven phylogenetic groups (PGs), which cluster by location and serotype (refer to Fig. 1 and supplemental material of Conner et al. [6]). Strains of serotype Y are primarily found in four of these groups—PG1, PG3, PG6, and PG7. These PGs can be further distinguished by the presence of serine protease autotransporter toxin genes sigA/sat and pic, the iron uptake sit and fec operons, the enterobactin ent operon, and the Shigella enterotoxin 1 (ShET1) genes set1A and set1B. Strain PE577 encodes all but the fec operon and ShET1 genes. Combined, these characteristics suggest that PE577 is a member of PG7, which is predominantly found in east and southeast Asia—consistent with PE577 originating in Australia. In contrast, the phylogenetic groups of 2457T and M90T are PG3 and PG5, respectively. Strains of PG3 were isolated from around the world, while PG5 is primarily composed of historical strains originating from North America and Europe. Strain PE577 therefore provides a third representative for comparing separate phylogenetic and geographic groups within the species. Since PE577 is the host to numerous bacteriophages, an analysis of prophage remnants was conducted using the PHAge Search Tool–Enhanced Released (PHASTER) search tool (16). Genome remnants from at least 11 phages are present and include identity to known phages such as iGifsy-1, Sf6, sal3, Stx2, N4, etc. Additionally, this region includes some structural genes of unknown origin. While the full genomes of these phages are incomplete, some genes remain intact. These are primarily limited to transposases, though some regions also include structural phage genes. These prophages are indeed defunct, as no phage particles were produced after induction (data not shown).
In addition to the chromosome, PE577 harbors two plasmids, characterized by similarities to ColE1 and pHS-2 (17, 18). The first of these encodes seven full-length proteins, including mobilization proteins in the mob/mbe operon. This plasmid has been found in numerous strains of enteric bacteria, where it frequently encodes additional transposable elements or antibiotic resistance genes (17). Thus far, at least among strains of S. flexneri, only the mobilization operon is present, suggesting the additional genes are either not retained or have not yet been transferred. Since this plasmid encodes the full-length exclusion protein MbeD, transferring a similar mob/mbe operon-encoding plasmid is unlikely. The second plasmid resembles the stably maintained pHS-2, which has been found in many Shigella and Escherichia coli isolates (19). It encodes two small full-length proteins, along with WzzpPHS2, a regulator of lipopolysaccharide O-antigen length (20, 21). The chromosomally encoded allele of fepE contains an internal stop codon, but the plasmid encodes a full-length homolog. This has been seen in other strains of S. flexneri, including 2457T (22, 23).
The chromosomes of PE577 and 2457T exhibit 98.9% average nucleotide identity, calculated as the query coverage multiplied by the percent identity, with only one large ∼1-megabase inversion. Compared to the alternative model strain M90T, PE577 shares an average nucleotide identity of 96.9%, with several rearrangements throughout the genome. Despite this overall close similarity, PE577 and CFS100 are distinct during routine laboratory growth; PE577 cultures contain fewer CFU per milliliter after overnight growth, and they grow poorly in microaerobic conditions or in minimal medium (Fig. 2). Combined, these qualities suggest systematic differences between these two strains, which may contribute to PE577’s enhanced susceptibility to bacteriophages. To measure these differences more quantitatively, specific properties were investigated in greater detail.
FIG 2.
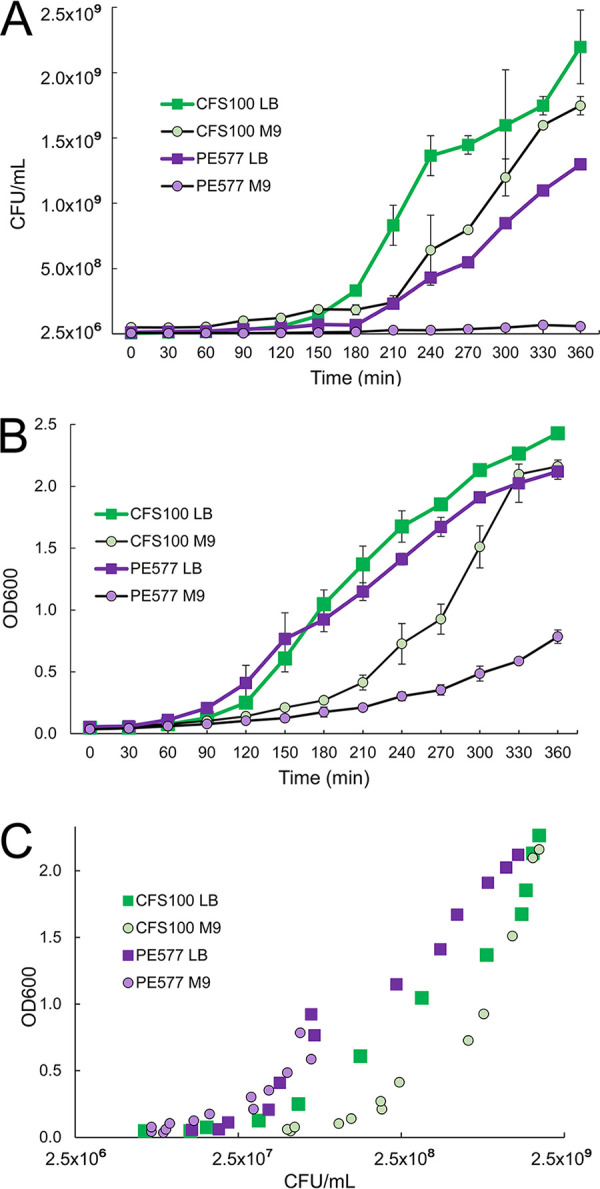
Growth of strains CFS100 and PE577 in nutrient-rich (LB) or nutrient-poor (M9) medium in aerobic conditions. (A and B) Measurements are based on CFU per milliliter over time or optical density at 600 nm (B). (C) The relationship between these two measurements.
PE577 exhibits metabolic differences compared to model strain 2457T.
To characterize PE577, general properties were first determined and compared to CFS100, an established model organism. CFS100 is an avirulent derivative of the model organism S. flexneri 2457T (11). First, overnight cell densities were measured in nutrient-rich or minimal medium and under aerobic or microaerobic conditions. Different levels of aeration were achieved by growing cultures in tubes with (i) loose caps and over 5× volume of headspace for good aeration versus (ii) tubes with screw caps and less than 1/5 volume of headspace for poor aeration (“microaerobic” conditions). Based on CFU per ml, PE577 grew to slightly lower density than CFS100 under nutrient-rich conditions, with the greatest decrease under microaerobic conditions (data not shown). In minimal medium, PE577 growth was significantly lower than that of CFS100 under both aerobic and microaerobic conditions. To further investigate the effect of nutrients on growth between these two strains, growth curves were conducted for 6 h under aerobic conditions, with measurements taken as CFU per milliliter and optical density at 600 nm (OD600). As shown in Fig. 2A, PE577 grows significantly more slowly than CFS100 in terms of CFU per milliliter in both types of medium, with hardly any growth in M9 minimal medium. In addition, while both strains showed initial lower OD600 readings in minimal medium, CFS100 ultimately returned to densities similar to growth in nutrient-rich medium (Fig. 2B). Conversely, PE577 density increased slightly over time but never reached levels of LB growth. Despite this, there was no clear change in the relationship between OD600 and CFU per milliliter for PE577 based on medium type (Fig. 2C). When the dimensions of individual cells from each strain were measured after overnight growth in LB or M9, PE577 was elongated compared to CFS100, though both cell types were longer in M9 than in LB (see Table S1 and Fig. S1 in the supplemental material).
To examine potential changes in physiology between these two strains in minimal medium, residual glucose was measured in medium after overnight growth. Up to 36.7% of the initial glucose was unused in PE577 cultures (2.94 ± 2.43 mM remaining of 12.87 mM starting glucose). While highly variable, this was contrasted with the residual glucose composition of spent CFS100 medium, which was consistently below the limit of detection after overnight growth. These results suggested an inefficiency or inconsistency in PE577 glucose catabolism under microaerobic conditions. To develop this hypothesis further, we specifically examined genes involved in glycolysis and fermentation. We created a comprehensive list of all pseudogenes found in PE577 (Table S2) and CFS100 (Table S3) and then analyzed them to see which pseudogenes were unique to each strain (Table 2).
TABLE 2.
Pseudogenes unique to each straina
| Gene name | Protein name or function | Complete in 2457T? | Complete in PE577? | Descriptionoir commentb |
|---|---|---|---|---|
| acs | Acetate-CoA ligase | No | Yes | Truncation in CFS100 (248 aa); full length is 652 aa |
| aes | Acetyl esterase | No | Yes | Truncation in CFS100 (272 aa); full length is 319 aa |
| ag43 | Autotransporter adhesin Ag43 | No | Yes | Truncation in CFS100 (297 aa); full length is 1,040 aa |
| arnA | Bifunctional UDP-4-amino-4-deoyl-l-arabinose formyltransferase/UDP glucuroncic acid oxidase | No | Yes | Truncation in CFS100 (258 aa); full length is 660 aa |
| chbC | PTS N,N′-diacetylchitobiose transporter subunit IIC | No | Yes | Truncation in CFS100 (63 aa); full length is 449 aa |
| fusA | Elongation factor G | No | Yes | Truncation in CFS100 very early in protein (11 aa); full length is 704 aa |
| glpD | Glycerol-3-phosphate dehydrogenase | No | Yes | Truncation in CFS100 (56 aa); full length is 501 aa |
| glpF | Glycerol uptake facilitator protein GlpF | No | Yes | Truncation in CFS100 (214 aa); full length is 281 aa |
| hexR | DNA-binding transcriptional regulator HexR | No | Yes | Truncation in CFS100 (273 aa); full length is 289 aa |
| mreC | Rod shape-determining protein MreC | No | Yes | Truncation in CFS100 (298 aa); full length is 367 aa |
| nimT | 2-Nitroimidazole transporter | No | NA | Completely absent in PE577 |
| rpoS | RNA polymerase sigma factor | No | Yes | Truncation in CFS100 (254 aa); full length is 330 aa |
| tcyJ | Cystine ABC transporter substrate-binding protein | No | Yes | Truncation in CFS100 (176 aa); full length is 266 aa |
| trbL | P-type conjugative transfer protein | No | Yes | Truncation in CFS100 (261 aa); full length is 475 aa |
| wcaC | Colonic acid biosynthesis glycosyltransferase | No | Yes | Truncation in CFS100 (202 aa); full length is 405 aa |
| yfdV | Transporter YfdV | No | Yes | Truncation in CFS100 (253 aa); full length is 314 aa |
| yijE | Cystine transporter YijE | No | Yes | Truncation in CFS100 (141 aa); full length is 301 aa |
| acnA | Aconitate hydratase AcnA | Yes | No | Truncation in PE577 (237 aa); full length is 891 aa |
| dmsA | Dimethylsulfoxide reductase subunit A | Yes | No | Truncation in PE577 (119 aa); full length is 814 aa |
| glcC | Transcriptional regulator GlcC | Yes | Yes | Same sequence for residues 1 to 222, frameshift resulted in last 35 aa being different |
| hycC | Formate hydrogenlyase subunit 3 | Yes | No | Truncation in PE577 (227 aa); full length is 500 aa |
| malZ | Maltodextrin glucoside | Yes | No | Truncation in PE577 (391 aa); full length is 605 aa |
| yrbL | PhoP regulatory network protein YrbL | Yes | No | Truncation in PE577 (95 aa); full length is 210 aa |
“Complete” implies a full-length gene, but potential differences are based solely on genomic information, and some genes may remain functional even if truncated in length. NA, not applicable. Shading indicates strains which have a pseduogene.
aa, amino acids.
From a comparison to the CFS100 parental 2457T genome (22), PE577 was found to encode a frameshifted, truncated version of HycC, which is a subunit of the formate hydrogenlyase complex (FHL) (Fig. 3A) (24). A 16-nucleotide insertion in the PE577 hycC gene results in a frameshift, causing a premature stop codon; the new coding sequence is for a 298-amino acid protein rather than the 597-amino acid HycC protein in CFS100 (Fig. 3B). The FHL complex is responsible for oxidizing formate produced by pyruvate-formate lyase (PFL), which converts pyruvate to formate and acetyl-coA (24, 25). While the better-characterized strains of S. flexneri encode full-length HycC, protein databases indicate that numerous other strains of S. flexneri encode truncated versions of HycC; for example, in a BLAST query using the PE577 protein sequence, 42 out of 89 S. flexneri-specific hits were similarly truncated at a range between 274 and 325 amino acids. While a complete hycC deletion mutant still exhibits functional FHL activity in vitro (25), the complex is unable to transport resulting protons out of the cell in vivo, resulting in an overall decreased rate of formate oxidation (26). From an analysis of single amino acid substitutions throughout the protein, only two changes, D354A and E391A, significantly reduced formate-dependent hydrogen production (26). The effect of a truncated version—where HycC lacks 6 C-terminal transmembrane helices of 13 total—has not been described.
FIG 3.

(A) Pathways of pyruvate oxidation in S. flexneri. (B) Comparison of genes involved in the PoxB/Acs bypass or in formate detoxification between PE577 and CFS100. In the latter, PoxB is grayed out because CFS100 encodes a truncated RpoS. Since RpoS controls poxB expression, this may affect protein abundance. Similarly, FdhF is grayed out because it is a pseudogene in both backgrounds, but a different formate dehydrogenase may associate with the FHL complex.
Since the HycC truncation in strain PE577 includes the region identified by Pinske and Sargent as important for FHL activity in vivo (26), we hypothesized that this strain would be unable to convert formate to carbon dioxide and hydrogen as effectively as CFS100. To test whether PE577 medium contains greater levels of formate compared to CFS100, single colonies of PE577 or CFS100 were grown in minimal medium where glucose was the primary carbon source and under conditions where oxygen was limited. After overnight incubation, the cells were then removed by centrifugation, and fermentation products in the supernatant were measured by HPLC. As shown in Fig. 4D, these measurements indicate that strain PE577 produces less formate than CFS100, with an average of 6.06 ± 1.34 mM formate for PE577 and 11.41 ± 0.35 mM formate for CFS100. These results nullified the hypothesis that the truncated HycC results in a buildup of formate, but the greater variation in formate production between biological replicates of PE577 suggests there may be some other effect. An additional confounding factor is that in E. coli, the canonical formate dehydrogenase associated with FHL is encoded by fdhF (26); however, this is a pseudogene in both S. flexneri strains PE577 and 2457T (Fig. 3; Tables S2 and S3), encoding only 79 amino acids. Whether Shigella can use a different formate dehydrogenase in conjunction with FHL or uses HycC in a different pathway or complex is unclear.
FIG 4.
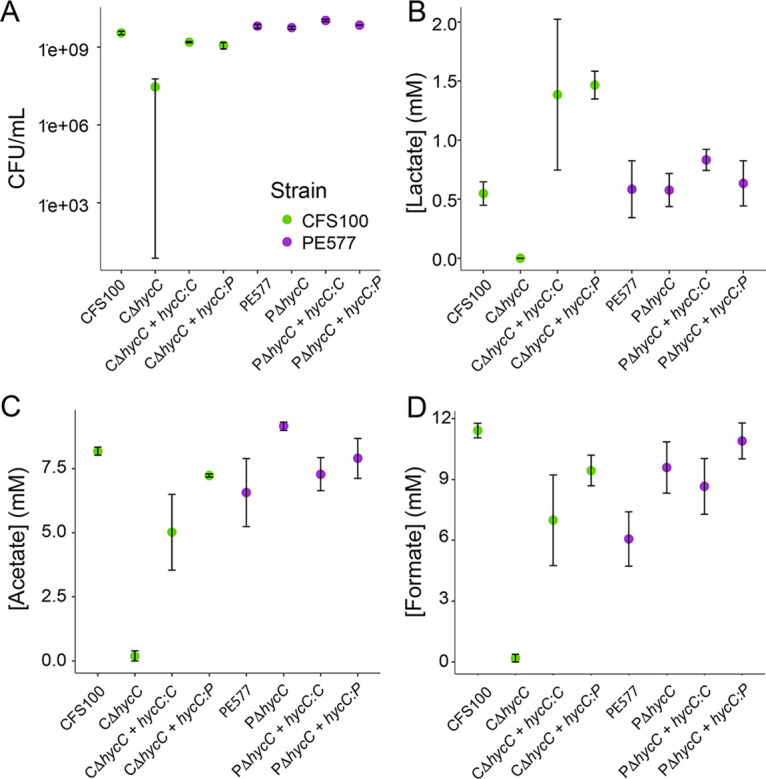
CFU per milliliter (A) and concentrations of lactate (B), acetate (C), and formate (D) in ΔhycC strains with and without complementation after overnight growth. The strain is indicated on the x axis. Error bars represent the standard error from three biological replicates.
PE577 and CFS100 use different mechanisms to generate acetyl-CoA in microaerobic conditions.
The variation in PE577 formate accumulation could be explained by two mechanisms: (i) the truncated HycC protein functions enough to allow the FHL complex to remain active, albeit at variable levels, or (ii) PE577 produces variable levels of formate, providing more flexibility to bypass the requirement for FHL activity. To investigate these possibilities, hycC was completely deleted in both strains, and the resulting knockouts were characterized. The PE577ΔhycC was indistinguishable from the parental PE577 strain in terms of CFU per milliliter and fermentation products (Fig. 4), whereas the CFS100ΔhycC mutant exhibited a severe growth defect and an altered fermentation profile relative to both wild-type strains. Complementing CFS100ΔhycC with either the truncated PE577 or full-length CFS100 hycC gene restored both the growth and metabolite profile of this strain (Fig. 4). These results suggest that (i) FHL is functional despite the fdhF pseudogene, (ii) CFS100 relies on FHL under microaerobic conditions, and (iii) PE577—despite having a functional HycC—does not require this pathway. Since overnight growth and metabolic products were not significantly different from each other, a measurable difference in HycC activity between the full-length and truncated versions seems unlikely.
As an alternative to PFL, pyruvate can be oxidized directly to acetyl-CoA by pyruvate dehydrogenase (PDH) or indirectly through an acetate intermediate via the ubiquinone-dependent pyruvate oxidase (PoxB). Acetate is then ligated to coenzyme A by acetate-CoA synthetase (Acs) to produce acetyl-CoA in an anaerobic PoxB/Acs bypass (Fig. 3A) (27, 28). Although PoxB expression is highest under aerobic conditions, it is still active under low-oxygen conditions (29). Generally, the PoxB/Acs bypass is regarded as less efficient than the PFL/FHL pathway during exponential growth and is instead only used to mediate the transition between exponential and stationary phases or during periods of slow growth (27, 29). Relying on this mechanism to produce acetyl-CoA avoids the production of formate and could explain some of the variation in PE577 formate accumulation.
An examination of the CFS100 genome revealed that the acs gene encodes a truncated protein of 248 amino acids, compared to the 652-amino-acid protein in PE577 (Fig. 3B). Based on protein database entries, truncated or partial Acs proteins are also found in other S. flexneri strains (10 of the top 37 high-scoring alignments), including numerous reference sequences. In addition, CFS100 also encodes a truncated RpoS protein predicted to lack the helix-turn-helix (HTH) motif. Since poxB transcription is mediated by RpoS, this may affect overall levels of PoxB.
To further explore pyruvate metabolism in both strains, we generated deletions of PFL (pflB), PDH (aceE), and the PoxB/Acs bypass and analyzed them in the same way as the HycC deletion strains (Fig. 5). We observed that both strains showed very similar phenotypes when pflB was deleted, with little impact on growth but the abolition of formate production, an increase in lactate production, and a decrease in acetate production. This suggests that both strains utilize PFL to oxidize pyruvate, but the activity is dispensable. Conversely, an aceE deletion produced drastically different phenotypes between the two strains, with only a small growth defect in PE577 compared to a greater than 4-log reduction in final cell density for CFS100. This is consistent with the lack of a functional acs in CFS100; PE577 has an additional route for pyruvate oxidation, making the aceE deletion less harmful, whereas CFS100 has no effective alternative. The poxB and acs deletions did not cause strong phenotypes in either strain, consistent with previous literature suggesting that this route plays a minor role in pyruvate oxidation under normal circumstances.
FIG 5.
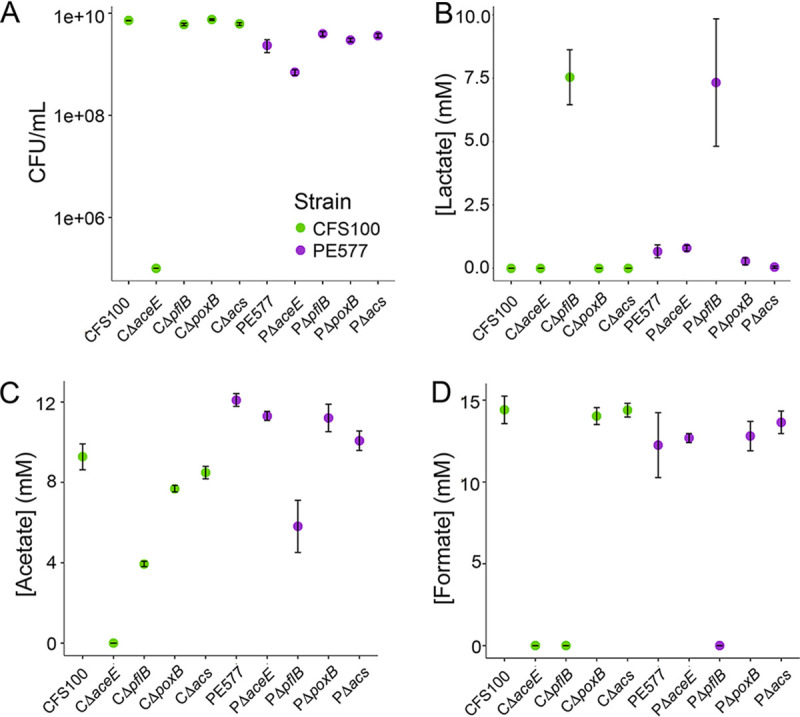
CFU per milliliter (A) and concentrations of lactate (B), acetate (C), and formate (D) in strains lacking various components of pyruvate oxidation pathways after overnight growth. The strain is indicated on the x axis. Error bars represent the standard error from three biological replicates.
DISCUSSION
Altered carbon flux from few genetic changes.
Despite the close genetic identity between the two strains, PE577 and CFS100, they exhibit significantly different phenotypes in terms of growth, morphology, and metabolism. Although these strains are distinct isolates and are therefore not isogenic, assigning specific genetic changes to phenotypic changes is difficult; however, the differences can nevertheless be informative. Based on our results, it appears that while PE577 and CFS100 primarily rely on PFL to convert pyruvate to acetyl-CoA in micro- or anaerobic conditions, PE577 is also capable of using the PoxB/Acs bypass to generate acetyl-CoA. Since this pathway is active under a different set of growth conditions and is overall a minor contributor to pyruvate oxidation, this could explain some of the variation observed in PE577 formate production. It is unclear how this rewiring evolved and whether the truncated HycC of the FHL complex arose before or after this switch.
Inefficient acetyl-CoA production by the PoxB/Acs bypass could also explain morphological differences between these two strains. For example, modest defects in cell division could explain the elongated phenotype of PE577 compared to that of CFS100, with this defect exacerbated under nutrient-limited conditions (30). In addition, as a critical component of fatty acid synthesis, reduced acetyl-CoA availability could limit the amount or the type of phospholipids available for the membrane. PE577 cells display “pointed” poles (see Fig. S1), which have been seen in E. coli cells lacking the phosphatidylethanolamine (PE) biosynthetic pathway (31). As a major component of the membrane, PE-deficient cells exhibit clear structural defects compared to wild-type cells, including increased production of outer membrane vesicles and detachment of the inner membrane from the outer membrane. These cells also show severely delayed growth in M9 medium, only entering exponential phase around 3 h (31), which is the same delay as PE577. Alternatively, additional changes in the genome could explain the altered morphology. For example, mreC encodes a rod shape-determining protein, and knockdowns have been shown to affect cell size and shape (32). The CFS100 genome encodes a truncated version of this protein (Table 2), which could lead to an overall rounder shape.
Implications for persistence and virulence.
Using 2457T, the parental strain of CFS100, it has been shown that formate induces expression of virulence genes icsA and ipaJ, both of which are encoded on the 200-kbp virulence plasmid (33). These gene products control cell-to-cell spread and modulate the host immune response, respectively (34–37). A mutant ΔpflB strain exhibits attenuated intracellular growth and intercellular movement, suggesting that S. flexneri-produced formate induces efficient expression of virulence genes (33). Exogenously applied formate can also induce genes, suggesting formate metabolism itself is not required for virulence; rather, when S. flexneri produces formate and releases it into the cell, it is the local increase in formate concentration that induces expression of virulence genes. Conversely, exogenously applied acetate has no apparent effect on virulence (33). In the intracellular environment, where pyruvate is readily accessible, S. flexneri has been shown to use the quicker pyruvate-to-acetate pathway, producing large amounts of acetate to facilitate rapid growth (38). Of note, these experiments were done using strain 2457T, which lacks acetyl-CoA synthetase and is therefore unable to convert acetate into acetyl-CoA.
Since strain PE577 appears to have an additional route to bypass formate production, other virulent strains with similar genome signatures—a truncated HycC and the presence of acetyl-CoA synthetase—may have similar metabolic rewiring. It is unclear whether a ΔpflB mutant in these backgrounds would affect virulence similarly to 2457T. Formate and acetate are known to influence pathogenicity of small intestine pathogens such as Salmonella enterica serovar Typhimurium (39, 40) and enteropathogenic E. coli (41). In S. enterica serovar Typhimurium, either acetate or formate can induce the expression of invasion genes (39, 40), while acetate is the primary signal for enteropathogenic E. coli adhesion and motility (41). In S. flexneri, there may be intraspecies variation for virulence gene induction, with some strains relying on formate and others relying on acetate, perhaps through the induction of different sets of virulence genes.
This may suggest that a group of closely related Shigella strains, with a range of genetic adaptations or in various stages of evolution, led to the observed variations in virulence. These physiological changes may facilitate bacteriophage infection, often leading to phage-mediated serotype conversion, further contributing to the genetic pool from which human-adapted virulence strains arise.
Establishing a model serotype Y strain.
In this work, we investigated genomic, physiological, and metabolic differences between strain PE577 and another model strain of S. flexneri, CFS100. The PE577 strain has been instrumental for analyzing the abundance and diversity of phages from the environment, in addition to characterizing numerous phages. While two model strains have already been described for S. flexneri, the variability between isolates warrants further study. We describe here significant differences between the physiology and metabolism of PE577 and CFS100 (avirulent 2457T), but it is unclear how widespread these differences are and whether these differences affect S. flexneri persistence in the environment or its virulence.
MATERIALS AND METHODS
Strains, plasmids, and media.
S. flexneri strain PE577 was a gift from R. Morona at the University of Adelaide. It was originally obtained from C. Murray at the Institute of Medical and Veterinary Science in South Australia by P. Manning at the University of Adelaide in December 1988. Strain CFS100 was a gift from I. Molineux at the University of Texas—Austin and has been described previously (11).
For PCR amplification, mutagenesis, and cloning, plasmids pKD4 (AddGene; catalog no. 45605) or pKD13 were used as templates for amplifying kanamycin cassettes; pKM208 (AddGene; catalog no. 13077) was used for recombination, and pGRB (AddGene; catalog no. 71539) was used for cloning. These plasmids have been previously described (42–44). To clone into pGRB, the gene of interest was amplified by PCR using primers that introduced homologous regions of the multi-cloning site. To linearize the plasmid, pGRB was digested with EcoRI and HindIII. The plasmid and PCR product were added to Gibson assembly master mix according to the manufacturer’s protocol. After assembly, the product was transformed, and colonies were recovered. The insert size was determined by agarose gel electrophoresis, and the sequence was confirmed by Sanger sequencing.
Minimal medium comprised 0.1 mM CaCl2, M9 salts, 0.2% glucose, and 1 mM MgSO4 and was supplemented with amino acids (0.2 mM histidine, 0.6 mM leucine, 0.3 mM methionine, 0.1 mM tryptophan), 0.3 μM thiamine HCl, and 0.1 mM niacin.
Growth curves and microscopy.
Growth curves were conducted under aerobic conditions, using 25 ml medium in Erlenmeyer flasks with shaking. Medium was inoculated using 2% vol/vol of an overnight culture grown in the same medium. Every 30 min, cell density was determined using the optical density at 600 nm, and an aliquot was taken to determine the CFU per milliliter. These experiments were conducted three times. To measure cell dimensions, 1-ml overnight cultures were centrifuged to concentrate cells and resuspended in 50 μl buffer (10 mM MgCl2, 10 mM Tris, pH 7.6) or water for cryo-electron microscopy. Small aliquots (∼5 μl) were then applied to R2/2 Quantifoil grids that had been glow discharged for 60 s in a Pelco EasyGlow discharge unit. Samples were plunge-frozen in liquid ethane using a Vitrobot Mark IV system under 100% humidity at 4°C with a blot force of 1 and blot time of 4 s. Cells were viewed in an FEI Talos Arctica microscope, with micrographs collected on a Ceta camera at ×11,000 nominal magnification (9.66 Å/pixel). Individual cell’s dimensions were measured using ImageJ and Adobe Photoshop.
DNA extraction and sequencing methods.
Chromosomal DNA was extracted from 5 ml of S. flexneri PE577 overnight culture using a standard phenol-chloroform protocol, followed by ethanol precipitation with the addition of 0.3 M sodium acetate. Purity was determined by measuring the absorbance at 230, 260, and 280 nm. Plasmid DNA was extracted from 50 ml of overnight culture using the ZymoPure II plasmid midiprep kit, with individual plasmids purified via gel electrophoresis. These were then subsequently extracted via Zymoclean gel or large fragment DNA recovery kits.
Separate sequencing technologies were used for the chromosome and two small plasmids. Strain PE577 lacks the 200-kbp virulence plasmid pWR100, which was verified by PCR using primers that target the virG (F, 5′-TTA TGG TGA GGG TGA TGG CG-3′; R, 5′-GGG ATT TTC CGT CCC AGC TA-3′) and virB (F, 5′-CCG GGG GCA GAT TTG TAT CA-3′; R, 5′-TGG TGG ATT TGT GCA ACG AC-3′) genes. Strain M90T, which contains pWR100, was used as a positive control. In addition, total plasmid isolated from 100 ml of overnight culture using a ZymoPURE II plasmid midiprep kit revealed only two small plasmids based on agarose gels and sequencing. The chromosome was sequenced using 250-bp paired-end reads on an Illumina HiSeq instrument at MicrobesNG, with a minimal coverage of 30×. These were then assembled into a single 4.6-Mbp contig using the A5 pipeline version 08-25-2016 (45). The plasmids were sequenced at the Massachusetts General Hospital Center for Computational and Integrative Biology (MGH CCIB) DNA Core, where they were assembled into 4.0-kbp, and 3.1-kbp contigs. The minimal coverage for each plasmid was 3,150× and 2,015×, respectively. Since all molecules are circular, the termini are based on reference sequences. Sequences were then annotated via the NCBI Prokaryotic Genome Annotation pipeline and submitted to GenBank (accession numbers CP042978.1 and CP042979.1 for plasmids and CP042980.1 for the chromosome).
Generation of mutants.
Competent PE577 and CFS100 cells harboring the pKM208 plasmid were generated by inoculating 100 ml of LB with 2 ml of overnight culture. Cells were grown with 4 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and 100 μg/ml ampicillin at 27.5°C to an OD600 of ∼0.4 and pelleted by centrifugation of 1,500 × g for 10 min at 4°C. The cells were resuspended in ice-cold water and centrifuged again at 1,500 × g for 10 min at 4°C. This pellet was gently resuspended in 5 ml of ST buffer (0.125% yeast extract, 0.25% tryptone, 10% PEG8000, 7% dimethyl sulfoxide [DMSO]) by swirling. The cells were subsequently pelleted by centrifuging at 1,500 × g for 15 min at 4°C, followed by a final resuspension in 2 ml of ST buffer by swirling. Then, 100-μl aliquots of cells were used for each transformation reaction.
PCR products containing the kanamycin cassette for recombination were generated by designing primers for two schemes. In the first, primers bind ∼500 bp upstream and downstream of the gene of interest (e.g., for hycC, LH1, 5′ GCG CGG CGA GAT AAT GTT GAC CTA ATT TTT CTT CAG ACA TGC TCA AAC 3′; LH2, 5′ GGC GTG TCG ATG AGT GTC GAA AAT GAC ATT TCA TCG GCA TGT TTT CG 3′). In the second scheme, one set of primers binds to either end of the antibiotic resistance cassette of pKD4 or pKD13 with ∼20 additional nucleotides matching the gene of interest (e.g., for hycC, Rxn1F, 5′ GGT TTG TCG CCG CCG CTG TTC TGT GTA GGC TGG AGC TGC TTC GAA GTT C 3′; Rxn1R, 5′ CCA CCA GTA CCG CCA GTT CAA CCA GCG CAA TTC CGG GGA TCC GTC GAC C 3′); the second set of primers matches the gene of interest, with ∼15 bp of overlapping sequence between the two sets of primers (Rxn2F, 5′ GCA ATT TCC CTG ATC AAT AGC GGC GTG GCA TGG TTT GTC GCC GCC GCT G 3′; Rxn2R, 5′ CAC TCA TTC TCA GGC TCC TCG TGA AAC AAT AAT CAC CAC CAG TAC CGC CAG TT 3′). After the second reaction, 10 μl of final product was added to 100 μl of competent cells and gently mixed by pipetting up and down, followed by incubation in ice for 5 min. After electroporation with a Bio-Rad Gene Pulser Xcell system, using a current of 2.5 kV in 0.2-cm cuvettes, cells were recovered in 1 ml of SOC medium at 30°C for 90 min. Mutant clones were then confirmed by colony PCR with primers that bind upstream and downstream of the gene of interest (LH1 and LH2, respectively).
Analysis of mixed acid fermentation products.
Minimal medium was inoculated with a single colony for each replicate, with three replicates per set. For aerobic conditions, cultures were grown in standard culture tubes with loose caps; for microaerobic conditions, screw-cap culture tubes were used with minimal headspace (<1 ml volume for 5 ml of culture). When stricter anaerobic conditions or alternative media were used, PE577 was unable to grow to measurable optical density or CFU per milliliter. Tubes were placed in a rotating wheel overnight. An aliquot of each culture was used to measure cell concentrations by colony counts. Cultures were then spun for 10 min at 8,000 × g to remove cells, and then supernatants were transferred to 2-ml glass HPLC tubes.
HPLC analysis was conducted as described in reference 46. Briefly, compounds of interest were separated using a 0.6-ml/min flow rate in 5 mM sulfuric acid with a 30-min run time. The eluent was prepared by diluting a 50% HPLC-grade sulfuric acid solution (Fluka) in Milli-Q water and degassing the solution at 37°C for 3 to 5 days before use. Compounds of interest were detected using a refractive index detector (Shimadzu, RID-20A) maintained at 50°C, using an Aminex HPX-87H column with a Micro Guard cation H+ guard (Bio Rad, Hercules, CA). Mixed standards were prepared for glucose, acetate, formate, and lactate at concentrations of 0.5, 0.75, 1, 2, 3, 5, and 10 mM. Standards and samples were maintained at 10°C with an autosampler (Shimadzu, SIL-20AHT) throughout the analysis.
Data availability.
Sequences annotated via the NCBI Prokaryotic Genome Annotation pipeline were submitted to GenBank (accession numbers CP042978.1 and CP042979.1 for plasmids and CP042980.1 for the chromosome).
Supplementary Material
ACKNOWLEDGMENTS
We thank Ian Molineux for providing strains of bacteria and for helpful discussions and Sherwood Casjens for helpful discussions. We also thank John Dover, Tori Brown, Ashleigh Bass, and Vanessa Eaton for optimizing M9 composition and initial growth characterization and the MSU RTSF Cryo-EM Facility for preparation and imaging of cells.
This work was funded by the National Institutes of Health (GM110185), the National Science Foundation CAREER Award 1750125, the J. K. Billman, Jr., MD, Endowed Research Professorship, and a Burroughs Wellcome Fund Award to K.N.P.; the National Science Foundation CAREER Award 1750785 and a Beckman Young Investigator award from the Arnold and Mabel Beckman Foundation to M.A.T.; and the Australian Research Council Discovery Project DP170104325 to R.M.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the funders.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.GBD 2016 Diarrhoeal Disease Collaborators. 2018. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis 18:1211–1228. 10.1016/S1473-3099(18)30362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sethuvel DPM, Ragupathi NKD, Anandan S, Veeraraghavan B. 2017. Update on: Shigella new serogroups/serotypes and their antimicrobial resistance. Lett Appl Microbiol 64:8–18. 10.1111/lam.12690. [DOI] [PubMed] [Google Scholar]
- 3.Knirel YA, Sun Q, Senchenkova SN, Perepelov AV, Shashkov AS, Xu J. 2015. O-antigen modifications providing antigenic diversity of Shigella flexneri and underlying genetic mechanisms. Biochemistry (Mosc) 80:901–914. 10.1134/S0006297915070093. [DOI] [PubMed] [Google Scholar]
- 4.Livio S, Strockbine NA, Panchalingam S, Tennant SM, Barry EM, Marohn ME, Antonio M, Hossain A, Mandomando I, Ochieng JB, Oundo JO, Qureshi S, Ramamurthy T, Tamboura B, Adegbola RA, Hossain MJ, Saha D, Sen S, Faruque AS, Alonso PL, Breiman RF, Zaidi AK, Sur D, Sow SO, Berkeley LY, O'Reilly CE, Mintz ED, Biswas K, Cohen D, Farag TH, Nasrin D, Wu Y, Blackwelder WC, Kotloff KL, Nataro JP, Levine MM. 2014. Shigella isolates from the global enteric multicenter study inform vaccine development. Clin Infect Dis 59:933–941. 10.1093/cid/ciu468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allison GE, Verma NK. 2000. Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol 8:17–23. 10.1016/s0966-842x(99)01646-7. [DOI] [PubMed] [Google Scholar]
- 6.Connor TR, Barker CR, Baker KS, Weill FX, Talukder KA, Smith AM, Baker S, Gouali M, Pham Thanh D, Jahan Azmi I, Dias da Silveira W, Semmler T, Wieler LH, Jenkins C, Cravioto A, Faruque SM, Parkhill J, Wook Kim D, Keddy KH, Thomson NR. 2015. Species-wide whole genome sequencing reveals historical global spread and recent local persistence in Shigella flexneri. Elife 4:e07335. 10.7554/eLife.07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doore SM, Schrad JR, Dean WF, Dover JA, Parent KN. 2018. Shigella phages isolated during a dysentery outbreak reveal uncommon structures and broad species diversity. J Virol 92:e02117-17. 10.1128/JVI.02117-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doore SM, Schrad JR, Perrett HR, Schrad KP, Dean WF, Parent KN. 2019. A cornucopia of Shigella phages from the Cornhusker State. Virology 538:45–52. 10.1016/j.virol.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Islam MM, Azad AK, Bardhan PK, Raqib R, Islam D. 1994. Pathology of shigellosis and its complications. Histopathology 24:65–71. 10.1111/j.1365-2559.1994.tb01272.x. [DOI] [PubMed] [Google Scholar]
- 10.Zaidi MB, Estrada-Garcia T. 2014. Shigella: a highly virulent and elusive pathogen. Curr Trop Med Rep 1:81–87. 10.1007/s40475-014-0019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marman HE, Mey AR, Payne SM. 2014. Elongation factor P and modifying enzyme PoxA are necessary for virulence of Shigella flexneri. Infect Immun 82:3612–3621. 10.1128/IAI.01532-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramanian S, Parent KN, Doore SM. 2020. Ecology, structure, and evolution of shigella phages. Annu Rev Virol 7:121–141. 10.1146/annurev-virology-010320-052547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maurelli AT. 2007. Black holes, antivirulence genes, and gene inactivation in the evolution of bacterial pathogens. FEMS Microbiol Lett 267:1–8. 10.1111/j.1574-6968.2006.00526.x. [DOI] [PubMed] [Google Scholar]
- 14.Maurelli AT, Fernandez RE, Bloch CA, Rode CK, Fasano A. 1998. “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc Natl Acad Sci U S A 95:3943–3948. 10.1073/pnas.95.7.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bliven KA, Maurelli AT. 2012. Antivirulence genes: insights into pathogen evolution through gene loss. Infect Immun 80:4061–4070. 10.1128/IAI.00740-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ares-Arroyo M, Bernabe-Balas C, Santos-Lopez A, Baquero MR, Prasad KN, Cid D, Martin-Espada C, San Millan A, Gonzalez-Zorn B. 2018. PCR-based analysis of ColE1 plasmids in clinical isolates and metagenomic samples reveals their importance as gene capture platforms. Front Microbiol 9:469. 10.3389/fmicb.2018.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rehel N, Szatmari G. 1996. Characterization of the stable maintenance of the Shigella flexneri plasmid pHS-2. Plasmid 36:183–190. 10.1006/plas.1996.0045. [DOI] [PubMed] [Google Scholar]
- 19.Adam T, Siewerdt R, Offermann I, Lang J, Tschape H, Sieper J, Graf B. 2003. Prevalence and molecular diversity of pHS-2 plasmids, marker for arthritogenicity, among clinical Escherichia coli Shigella isolates. Microbes Infect 5:579–592. 10.1016/s1286-4579(03)00093-5. [DOI] [PubMed] [Google Scholar]
- 20.Purins L, Van Den Bosch L, Richardson V, Morona R. 2008. Coiled-coil regions play a role in the function of the Shigella flexneri O-antigen chain length regulator WzzpHS2. Microbiology (Reading) 154:1104–1116. 10.1099/mic.0.2007/014225-0. [DOI] [PubMed] [Google Scholar]
- 21.Morona R, Purins L, Tocilj A, Matte A, Cygler M. 2009. Sequence-structure relationships in polysaccharide co-polymerase (PCP) proteins. Trends Biochem Sci 34:78–84. 10.1016/j.tibs.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Wei J, Goldberg MB, Burland V, Venkatesan MM, Deng W, Fournier G, Mayhew GF, Plunkett G, III, Rose DJ, Darling A, Mau B, Perna NT, Payne SM, Runyen-Janecky LJ, Zhou S, Schwartz DC, Blattner FR. 2003. Complete genome sequence and comparative genomics of Shigella flexneri serotype 2a strain 2457T. Infect Immun 71:2775–2786. 10.1128/iai.71.5.2775-2786.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong M, Payne SM. 1997. Effect of mutations in Shigella flexneri chromosomal and plasmid-encoded lipopolysaccharide genes on invasion and serum resistance. Mol Microbiol 24:779–791. 10.1046/j.1365-2958.1997.3731744.x. [DOI] [PubMed] [Google Scholar]
- 24.Sauter M, Bohm R, Bock A. 1992. Mutational analysis of the operon (hyc) determining hydrogenase 3 formation in Escherichia coli. Mol Microbiol 6:1523–1532. 10.1111/j.1365-2958.1992.tb00873.x. [DOI] [PubMed] [Google Scholar]
- 25.McDowall JS, Murphy BJ, Haumann M, Palmer T, Armstrong FA, Sargent F. 2014. Bacterial formate hydrogenlyase complex. Proc Natl Acad Sci U S A 111:E3948–E3956. 10.1073/pnas.1407927111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinske C, Sargent F. 2016. Exploring the directionality of Escherichia coli formate hydrogenlyase: a membrane-bound enzyme capable of fixing carbon dioxide to organic acid. Microbiologyopen 5:721–737. 10.1002/mbo3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdel-Hamid AM, Attwood MM, Guest JR. 2001. Pyruvate oxidase contributes to the aerobic growth efficiency of Escherichia coli. Microbiology (Reading) 147:1483–1498. 10.1099/00221287-147-6-1483. [DOI] [PubMed] [Google Scholar]
- 28.Wolfe AJ. 2005. The acetate switch. Microbiol Mol Biol Rev 69:12–50. 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang YY, Wang AY, Cronan JE, Jr. 1994. Expression of Escherichia coli pyruvate oxidase (PoxB) depends on the sigma factor encoded by the rpoS(katF) gene. Mol Microbiol 11:1019–1028. 10.1111/j.1365-2958.1994.tb00380.x. [DOI] [PubMed] [Google Scholar]
- 30.Westfall CS, Levin PA. 2018. Comprehensive analysis of central carbon metabolism illuminates connections between nutrient availability, growth rate, and cell morphology in Escherichia coli. PLoS Genet 14:e1007205. 10.1371/journal.pgen.1007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowlett VW, Mallampalli V, Karlstaedt A, Dowhan W, Taegtmeyer H, Margolin W, Vitrac H. 2017. Impact of membrane phospholipid alterations in Escherichia coli on cellular function and bacterial stress adaptation. J Bacteriol 199:e00849-16. 10.1128/JB.00849-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kruse T, Bork-Jensen J, Gerdes K. 2005. The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol Microbiol 55:78–89. 10.1111/j.1365-2958.2004.04367.x. [DOI] [PubMed] [Google Scholar]
- 33.Koestler BJ, Fisher CR, Payne SM. 2018. Formate promotes Shigella intercellular spread and virulence gene expression. mBio 9:e01777-18. 10.1128/mBio.01777-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ. 1989. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A 86:3867–3871. 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldberg MB, Theriot JA. 1995. Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc Natl Acad Sci U S A 92:6572–6576. 10.1073/pnas.92.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burnaevskiy N, Fox TG, Plymire DA, Ertelt JM, Weigele BA, Selyunin AS, Way SS, Patrie SM, Alto NM. 2013. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature 496:106–109. 10.1038/nature12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. 2015. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18:157–168. 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kentner D, Martano G, Callon M, Chiquet P, Brodmann M, Burton O, Wahlander A, Nanni P, Delmotte N, Grossmann J, Limenitakis J, Schlapbach R, Kiefer P, Vorholt JA, Hiller S, Bumann D. 2014. Shigella reroutes host cell central metabolism to obtain high-flux nutrient supply for vigorous intracellular growth. Proc Natl Acad Sci U S A 111:9929–9934. 10.1073/pnas.1406694111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawhon SD, Maurer R, Suyemoto M, Altier C. 2002. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol 46:1451–1464. 10.1046/j.1365-2958.2002.03268.x. [DOI] [PubMed] [Google Scholar]
- 40.Huang Y, Suyemoto M, Garner CD, Cicconi KM, Altier C. 2008. Formate acts as a diffusible signal to induce Salmonella invasion. J Bacteriol 190:4233–4241. 10.1128/JB.00205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang F, Yang L, Chang Z, Chang L, Yang B. 2018. Regulation of virulence and motility by acetate in enteropathogenic Escherichia coli. Int J Med Microbiol 308:840–847. 10.1016/j.ijmm.2018.0s7.010. [DOI] [PubMed] [Google Scholar]
- 42.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murphy KC, Campellone KG. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol 4:11. 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Lin Z, Huang C, Zhang Y, Wang Z, Tang YJ, Chen T, Zhao X. 2015. Metabolic engineering of Escherichia coli using CRISPR-Cas9 meditated genome editing. Metab Eng 31:13–21. 10.1016/j.ymben.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Tritt A, Eisen JA, Facciotti MT, Darling AE. 2012. An integrated pipeline for de novo assembly of microbial genomes. PLoS One 7:e42304. 10.1371/journal.pone.0042304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duhl KL, Tefft NM, TerAvest MA. 2018. Shewanella oneidensis MR-1 utilizes both sodium- and proton-pumping nadh dehydrogenases during aerobic growth. Appl Environ Microbiol 84:e00415-18. 10.1128/AEM.00415-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequences annotated via the NCBI Prokaryotic Genome Annotation pipeline were submitted to GenBank (accession numbers CP042978.1 and CP042979.1 for plasmids and CP042980.1 for the chromosome).